
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in transition metal-free catalytic hydroelementation (E = B, Si, Ge, and Sn) of alkynes
Vitthal B. Saptal
a,
Ruibin Wang
a and
Sehoon Park
 *ab
*ab
aDepartment of Chemistry, Guangdong Technion Israel Institute of Technology, Guangdong 515063, China
bTechnion-Israel Institute of Technology, Technion City, 32000 Haifa, Israel. E-mail: sehoon.park@gtiit.edu.cn
First published on 7th December 2020
Abstract
Catalytic hydroelementation of alkynes mainly with hydroboranes and hydrosilanes gives a straightforward and atom-economical access to a wide range of vinylmetalloids, which are used as synthetically useful and/or reactive species in both synthetic and materials chemistry. Thus far, although numerous transition metal catalysts with well-defined ligand systems have been developed for alkyne hydroelementation, the employed catalysts are mainly based on expensive and potentially toxic metals such as Rh, Pt, and Ir, and their conventional inner-sphere hydride transfer pathways are susceptible to reaction systems, often making it difficult to control the selectivity. In this regard, transition metal-free catalysts for hydroelementation (E = B, Si, etc.) have intensively been reported as an alternative to the conventional metal catalytic regimes over the last decade. In this review, we describe the recent advances in transition metal-free catalytic procedures for alkyne hydroelementation using hydrides based on Si, B, Sn, and Ge with strong emphasis on the variation in the catalytic working mode depending on the intrinsic nature of the reaction systems.
 Vitthal B. Saptal | Vitthal B. Saptal received his MSc in Organic Chemistry from Swami Ramanand Teerth Marathwada University, Nanded in 2011. He received his Ph.D. degree (2018) in Chemistry from the Institute of Chemical Technology, Mumbai (Prof. B. M. Bhanage). Then he moved to Pohang University of Science and Technology (POSTECH), Pohang, S. Korea as a Postdoctoral Fellow. From October 2019 he moved to Guangdong Technion Israel Institute of Technology (GTIIT) as a Postdoctoral Fellow with a mentorship by Prof. Sehoon Park. His research interest is the synthesis of cutting-edge homogeneous and heterogeneous catalysts and their synthetic applications for organic transformations. |
 Ruibin Wang | Ruibin Wang was born in China in 1986. He received his Master's Degree (2011) in Medicine Chemistry from Sun Yat-sen University (Prof. Gui Lu). From February 2012 to July 2016, he worked at the Chinese Academy of Sciences as a Research Fellow. Then, he moved to Guangdong Shunde Industrial Design Institute to work as a Research Fellow (2016–2020). In early 2020, he started his PhD degree under the guidance of Prof. Sehoon Park at Guangdong Technion Israel Institute of Technology (GTIIT). His research is focused on the synthetic and mechanistic organometallic chemistry particularly involved in catalysis. |
 Sehoon Park | Sehoon Park was born in Seoul (Korea). He received his Ph.D. (2008) from the Tokyo Institute of Technology (Prof. Kohtaro Osakada). He then moved to the University of North Carolina at Chapel Hill to work as a Postdoctoral Fellow with Prof. Maurice Brookhart (2009–2012). From April 2013 to February 2019, he worked at the Institute for Basic Science (Korea) as a Senior Research Fellow with a mentorship by Prof. Sukbok Chang. In early 2019, he started his independent research as an Assistant Professor at Guangdong Technion Israel Institute of Technology (GTIIT). His research interests are synthetic and mechanistic organometallic chemistry. |
1. Introduction
Alkynes, a class of unsaturated hydrocarbons, are important platform chemicals for the synthesis of various functions.1–6 Among the organic transformations of alkynes, catalytic hydroelementation leading to a broad range of functionalized vinyl molecules is of particular interest since the resultant vinyl moieties have versatile reactivities as nucleophiles to diverse electrophilic counterparts in organic synthesis.7–16 Accordingly, B, Si, and Sn-based metalloid hydrides are widely employed and studied as a reducing agent in combination with transition metal catalysts for alkyne hydroelementation.17–25 This reaction involves both regio- and stereochemical considerations, where the regiochemistry defines the position of incorporating metalloids between two carbons of unsymmetrical alkynes (α vs. β), and the stereochemistry indicates the olefinic geometry of the resultant vinyl products (trans vs. cis or E vs. Z) (Scheme 1a). The possible selectivity outcomes are highly sensitive to the reaction components including metal, ligand, and/or metalloid hydride, which often render multiple catalytic pathways operative to bring about undesirable side products.26–35 There are mainly two types of inner-sphere pathways known for the transition metal-mediated alkyne hydroelementation, where the metal center initially undergoes oxidative addition to afford an active metal species possessing M–H and/or M–[E] bonds ([E] = metalloids), into which alkynes are inserted in a cis-manner to eventually deliver (E)-vinylmetalloids (Scheme 1b, left).18,25,26,28 On the other hand, the outer-sphere pathway involving hydride transfer from an outer-sphere of catalytic species is another important mechanism in the context of alkyne reduction, and the majority of these catalytic systems are based on Lewis acids. In the proposed mechanism, alkyne substrates react with either Lewis acid catalysts or highly electrophilic metalloid ions generated in situ to form zwitterion or vinyl carbocation intermediates, respectively. These intermediates are finally reduced via the outer-sphere hydride transfer to release the β-(Z)-vinyl products (Scheme 1b, right).36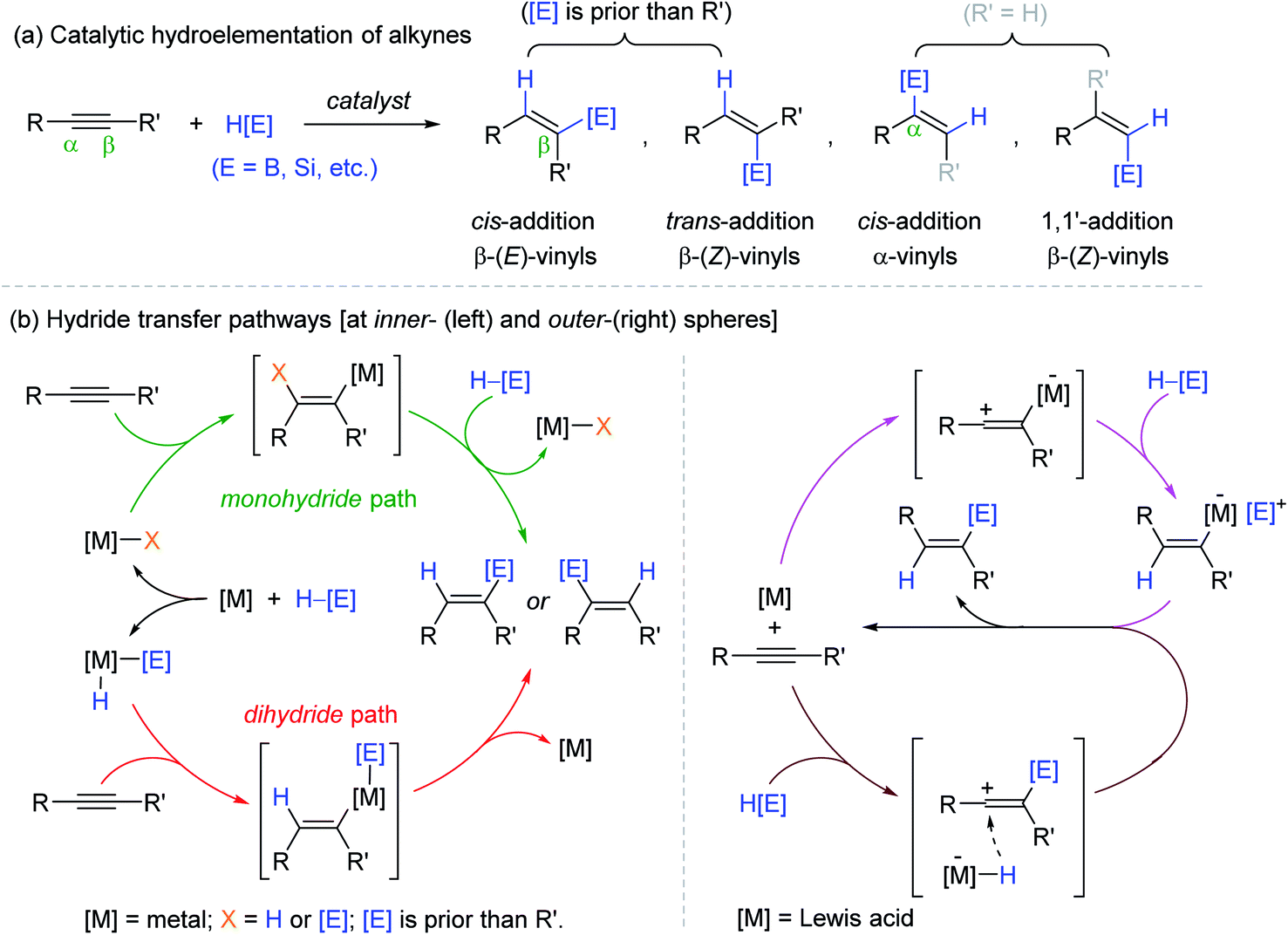 | ||
| Scheme 1 (a) Regio- and stereoselectivity and (b) general mechanisms of the catalytic hydroelementation of alkynes. | ||
Although numerous well-defined organometallic complexes are known to promote the selective alkyne reduction, this conventional procedure still has a range of cons in terms of practicality, namely the use of expensive metals (e.g. Ru, Rh, Pd, and Pt), instability of the metal complexes under ambient conditions, and difficult separation of the transition metals from crude reaction mixtures, which can be linked to toxicity in the pharmaceutical industry.37–40 In this regard, a significant number of transition metal-free, organo(metallic) catalyst systems for alkyne hydroelementation have recently been disclosed as a competent alternative by many research groups. However, to date, there have been no reviews documenting the transition metal-free catalytic procedures, whereas several reviews on the transition metal-catalyzed alkyne hydroelementation have been published.22,23,25,26,28,36 For example in 2000, Yamamoto reviewed the Lewis acid-promoted hydrometalation of unactivated alkynes, where various outer-sphere hydride transfer pathways were described.36 In 2005, Trost published a seminal review on transition metal-catalyzed alkyne hydrometallation with metalloid hydrides based on boron, silicon, and tin.28 In 2019, Alami and Provot reported a review on the hydrostannation of alkynes, focusing on the reaction mechanism and selectivity.23
In this review article, we aim to outline the recent examples of diverse transition metal-free catalysts ranging from Lewis acids (based on B, Mg, Al, Si, and P) and bases (based on Li, Mg, P, and K) to Brönsted acids (carboxylic acids) and a Brönsted base (NaOH) for the hydroelementation of alkynes with Si, B, Sn, and Ge-based hydrides (Scheme 2). The term “transition metal” in this review is defined as an element in the d-block of groups 3 to 12 in the periodic table.41,42 The representative catalytic working modes will be described by the type of catalysts in combination with hydride metalloids. In addition, the regio- and stereo-outcomes of the resultant vinylmetalloids will be discussed with emphasis on the hydride transfer pathway (inner- or outer-sphere mechanisms). The scope of the present review excludes Arase-Hoshi-type alkyne hydroboration involving RnBH3−n (n = 0, 1, 2) catalytic species,43–46 and transfer hydroelementation of alkynes using cyclohexa-1,4-diene-based metalloid hydride surrogates.47,48 This review covers the literature up to July, 2020.
 | ||
| Scheme 2 Transition metal-free catalysts for alkyne hydroelementation (this review). | ||
2. Hydrosilylation
The transition metal-catalyzed hydrosilylation of alkynes is one of the most utilized methods to synthesize a broad range of vinyl compounds since hydrosilanes are cheap, variable, and/or commercially accessible reducing agents.17–19 The majority of organometallic catalysts for alkyne hydrosilylation are based on precious metals, and their conventional inner-sphere pathways leading to cis-hydrosilylation are susceptible to the reaction system, usually giving a mixture of regioisomers. Accordingly, environmentally benign and selective transition metal-free catalysts can be a promising alternative to the conventional metal catalysts.In 1996, Yamamoto reported the electron-deficient Al(III)-catalyzed trans-hydrosilylation of alkynes (Scheme 3).49,50 This work represents the first transition metal-free catalytic hydrosilylation of alkynes. A broad range of terminal and internal alkynes reacted with Et3SiH at 0 °C in the presence of AlCl3 (1a) or EtAlCl2 (1a′) (20 mol%) to give the corresponding β-(Z)-vinylsilanes in good to high yields. Notably, the hydrosilylation of trimethylsilylacetylene yielded the α-vinylsilane exclusively. The reactions of unsymmetrical acetylenes led to a mixture of β- and α-vinylsilanes, and their ratios increased up to 7.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 depending on the differential steric effects of the alkyne substituents. It is interesting that Et2AlCl was an inactive catalyst possibly due to its low Lewis acidity, while 1a, which was insoluble in toluene, catalyzed the hydrosilylation in a heterogeneous manner. The scope of silanes was broad to include bulky silanes such as Ph3SiH. The present catalytic procedure was applicable to the hydrosilylative cyclization of 1,6-heptadiyne, affording the six-membered cyclization product.
:1 depending on the differential steric effects of the alkyne substituents. It is interesting that Et2AlCl was an inactive catalyst possibly due to its low Lewis acidity, while 1a, which was insoluble in toluene, catalyzed the hydrosilylation in a heterogeneous manner. The scope of silanes was broad to include bulky silanes such as Ph3SiH. The present catalytic procedure was applicable to the hydrosilylative cyclization of 1,6-heptadiyne, affording the six-membered cyclization product.
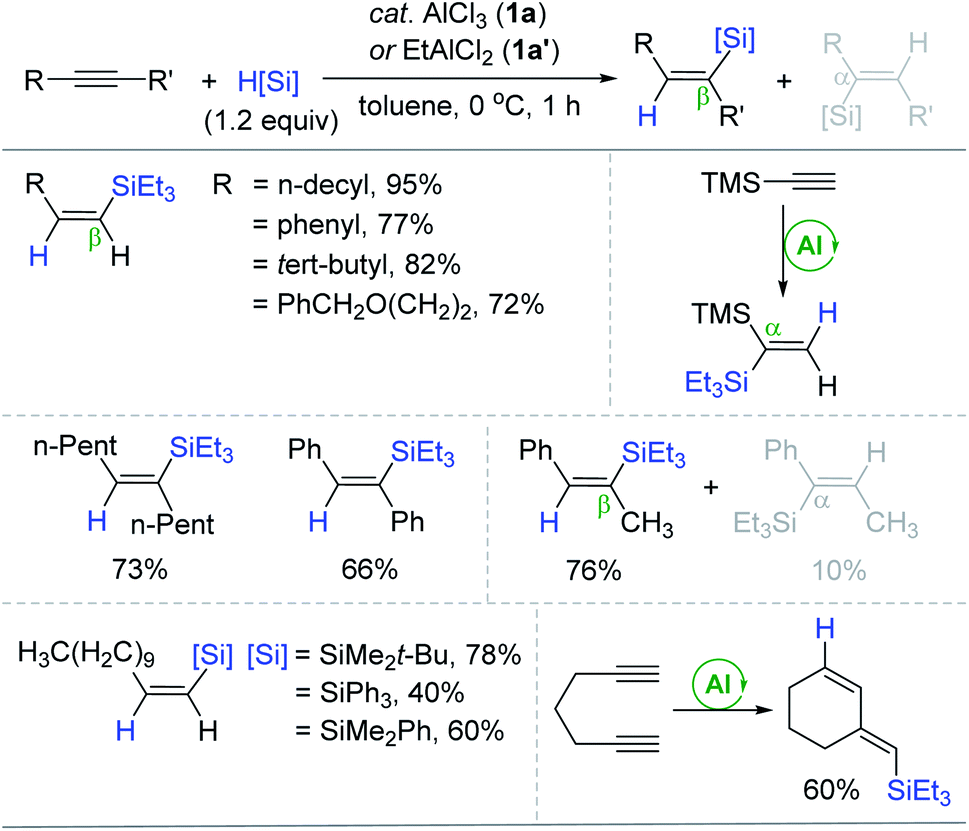 | ||
| Scheme 3 Al(III)-catalyzed trans-hydrosilylation of alkynes. TMS = trimethylsilyl. | ||
Based on the precedence of Lewis acid-catalyzed alkyne reduction,51–54 a reaction pathway for the Al-catalyzed alkyne hydrosilylation was proposed (Scheme 4). Initially, the Al catalyst (1a or 1a′) is assumed to react with an alkyne to form zwitterionic intermediate 1b, where the sp-hybridized vinyl carbocation is positioned β to the aluminum atom. The carbocation is likely reduced by a hydrosilane from the opposite side of the C(sp2)–Al moiety to generate aluminum ate-complex 1c. Then, 1c is suggested to be converted in a stereo-retentive manner to the (Z)-vinylsilane and the Al catalyst. This outer-sphere mechanism involving the vinyl carbocation intermediacy was corroborated by the α-selectivity observed in the hydrosilylation of trimethylsilylacetylene, involving an intermediate such as 1b′ under the β-silicon effect.
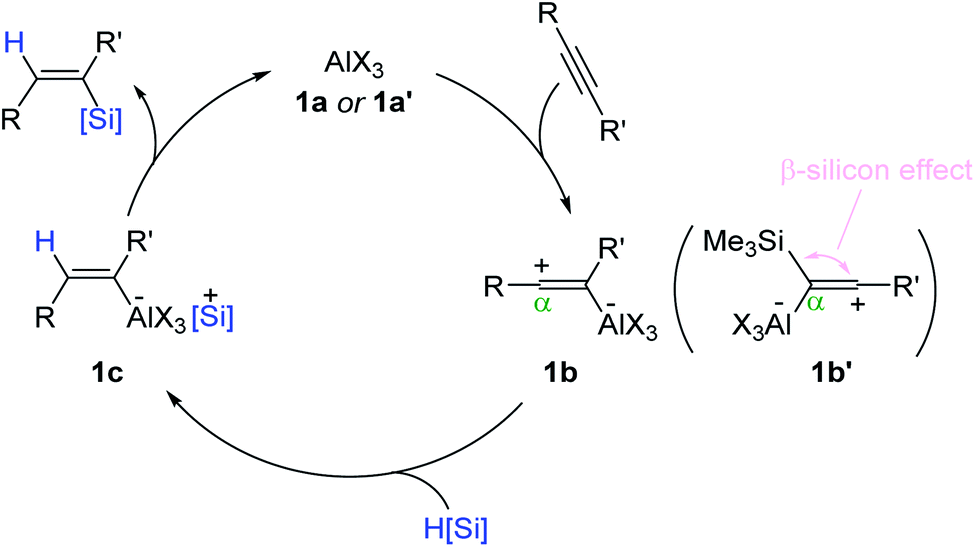 | ||
| Scheme 4 Proposed pathway for the Al-catalyzed trans-hydrosilylation of alkynes. | ||
Considering the preliminary result of the hydrosilylative cyclization of a diyne (vide supra),49,50 the Yamamoto group developed the 1a-catalyzed intramolecular hydrosilylation of alkynes bearing a hydrosilyl moiety to produce various silacycles (Scheme 5).55 A range of alkynes linked to an 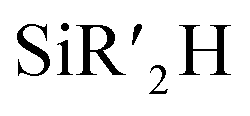 (R′ = Me, iPr, Ph) moiety by three methylene groups smoothly underwent cyclizative hydrosilylation in an endo-trans fashion in the presence of 1a (20 mol%) to produce the corresponding six-membered silacycles in 30–96% yield. Notably, the substituents iPr and Ph on the silicon retarded the cyclizative hydrosilylation to require a prolonged reaction time of up to 5 h. The phenyl-substituted alkynes possessing four to five methylene tethers were cyclized in an exo-mode to give the six- and seven-membered exo-silacycles, whereas the cyclization of substituted alkynes bearing a benzene ring spacer proceeded in an endo-mode to give the endo-trans products with seven to eight-membered ring sizes.
(R′ = Me, iPr, Ph) moiety by three methylene groups smoothly underwent cyclizative hydrosilylation in an endo-trans fashion in the presence of 1a (20 mol%) to produce the corresponding six-membered silacycles in 30–96% yield. Notably, the substituents iPr and Ph on the silicon retarded the cyclizative hydrosilylation to require a prolonged reaction time of up to 5 h. The phenyl-substituted alkynes possessing four to five methylene tethers were cyclized in an exo-mode to give the six- and seven-membered exo-silacycles, whereas the cyclization of substituted alkynes bearing a benzene ring spacer proceeded in an endo-mode to give the endo-trans products with seven to eight-membered ring sizes.
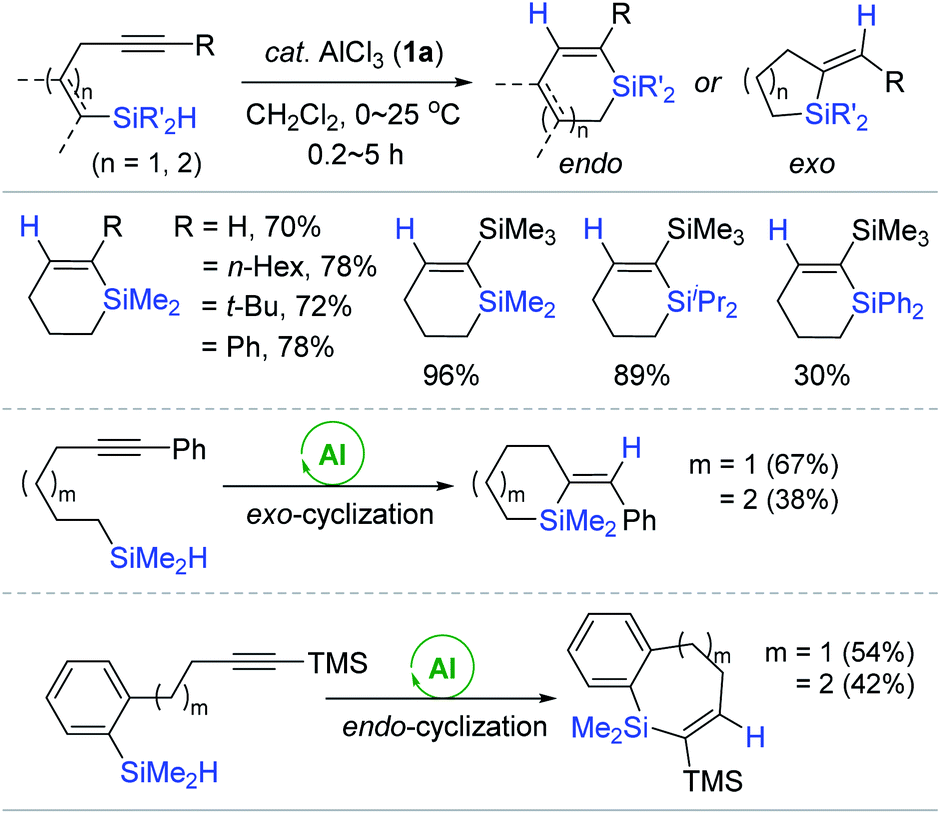 | ||
| Scheme 5 AlCl3-catalyzed intramolecular trans-hydrosilylation of alkynes possessing a hydrosilyl moiety. | ||
A reaction pathway for the Al-catalyzed regiodivergent cyclizative hydrosilylation of alkynes was proposed (Scheme 6). AlCl3 1a is assumed to initially interact with an alkynyl group to form a π-complex 1d. The species 1d, where the acetylenic bond is highly activated, is presumed to undergo intramolecular hydride transfer selectively to one of the acetylenic carbons depending on the identity of tethers, generating intermediate 1e or 1g bearing a C(sp2)–Al bond. Intermediates 1e and 1g are suggested to be cyclized to furnish the endo- and exo-silacycles, respectively with the regeneration of 1a.
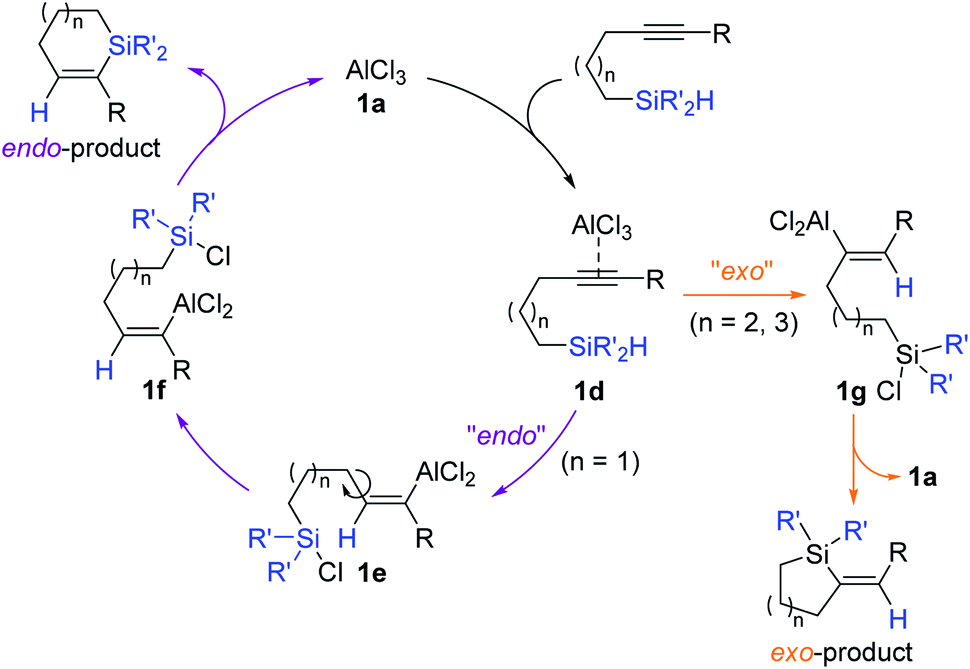 | ||
| Scheme 6 Proposed pathway for the Al-catalyzed regiodivergent cyclizative hydrosilylation of alkynes. | ||
Based on Yamamoto's Al catalysis using hydromonosilanes as a reducing agent, Tanaka conceived the AlCl3 (1a)-catalyzed hydrosilylation of alkynes with hydrooligosilanes H(SiMe2)nR; [n = 2–4, R = H, Me, Ph] as a reducing agent (Scheme 7).56 The hydrosilylation of internal and terminal alkynes with pentamethyldisilane (HSiMe2SiMe3, 1 equiv.) occurred at 0 to −42 °C to furnish the β-(Z)-vinylsilanes as the major product with small amounts of β-(E)- or α-vinylsilanes (∼5%). Interestingly, the reaction of phenylacetylene with HSiMe2SiMe3 not only gave the desired trans-addition products, but also 1,1,2-trisilyl-substituted ethylbenzene as a minor product (7–13%) depending on reaction temperature and concentration.
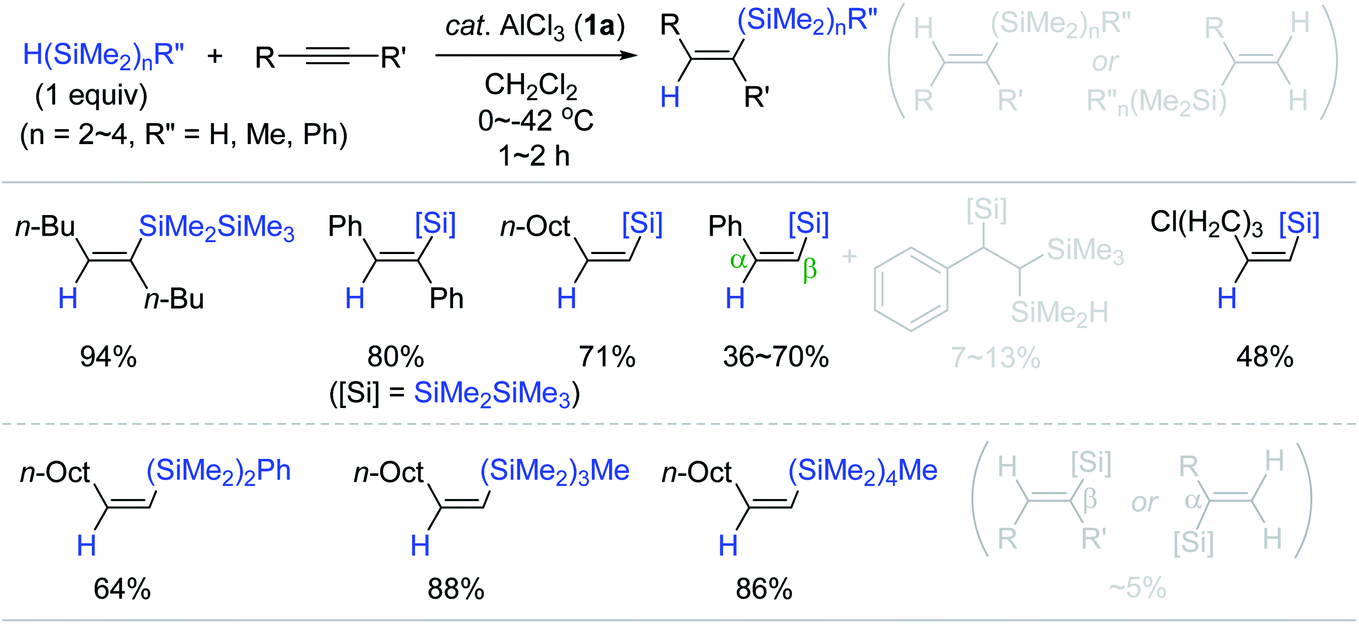 | ||
| Scheme 7 AlCl3-catalyzed trans-hydrosilylation of alkynes with monohydrooligosilanes. | ||
Next, α,ω-dihydrooligosilanes [H(SiMe2)nH (n = 2, 3)] were subjected to alkyne hydrosilylation and hydrosilylative polymerization of 1,7-octadiyne (Scheme 8). The reactions with 0.5 equivalents of α,ω-dihydrooligosilanes provided the corresponding β,β-(Z,Z)-bis-vinylsilanes in good yields. The reaction of 1,7-octadiyne with 1,3-dihydrohexamethyltrisilane in the presence of 1a (10 mol%) gave a mixture of oligomeric and polymeric materials [Mn = 3300 (Mw/Mn = 6.3)]. This Al catalysis was useful in modifying the backbone of poly(hydrosilylene)s. For example, poly(phenylsilylene) (Mw = 2700, Mw/Mn = 1.63) reacted with 1-decyne via multiple hydrosilylations to yield a polymer having decenyl pendants (Mw = 4260, Mw/Mn = 1.74).
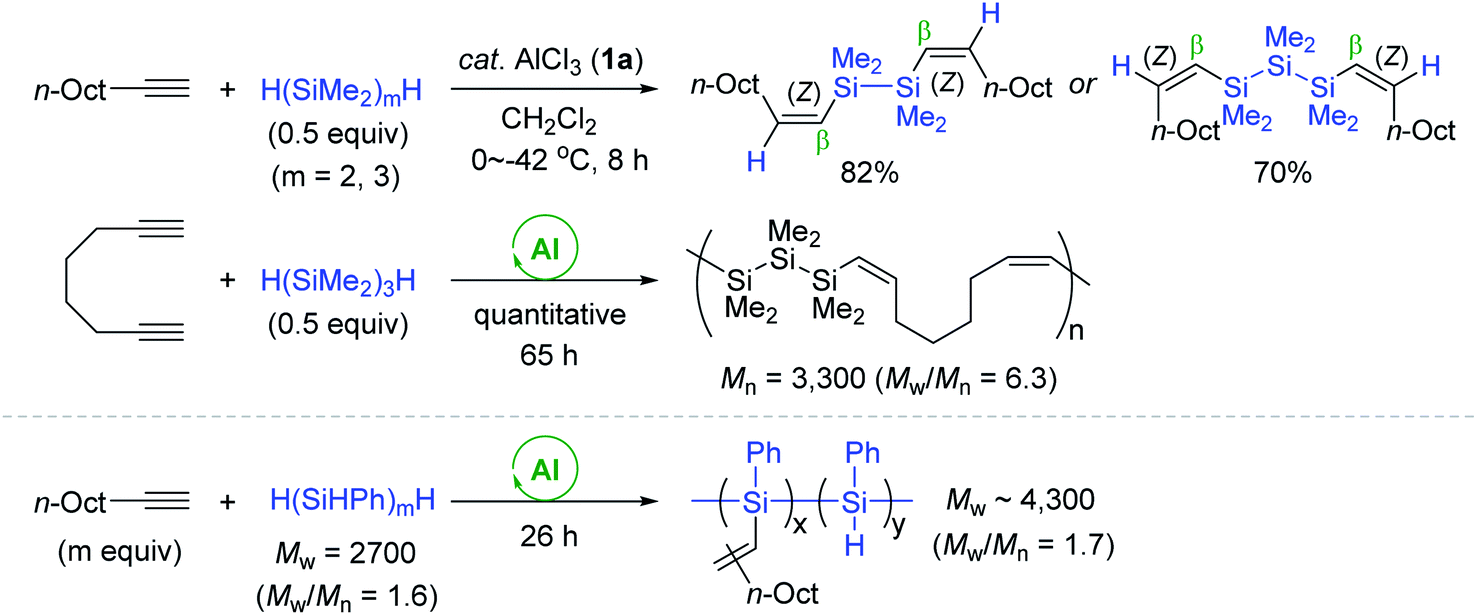 | ||
| Scheme 8 AlCl3-catalyzed trans-hydrosilylation of alkynes with polyhydrosilanes. | ||
Porous silicon, which has a high surface area morphology of hydride-terminated silicon, exhibits photoluminescence upon UV light irradiation, and has wide applications such as electroluminescent displays,57 photodetectors,58 and chemical/biosensing.59–61 However, silicon materials often suffer from corrosion and instability in highly oxidative and basic media, which are needed for practical applications.62,63
Considering this, Buriak et al. modified the surface of porous silicon via Al (1a′) catalysis in an attempt to strengthen its chemical stability (Scheme 9).64,65 The hydrosilylation of 1-dodecyne occurred on the porous silicon surface at 25 °C upon the addition of EtAlCl2 (1a′) to allow the clean incorporation of dodecenyl groups on the silicon surface. Under similar conditions, a range of alkynes bearing various functionalities were hydrosilylated on the surface, resulting in the corresponding alkenyl group-terminated surfaces. It is noteworthy that excess EtAlCl2 (1a′) was required for the hydrosilylation of alkynes bearing Lewis basic functional groups, such as cyano and ester groups. The incorporated alkenyl moieties on the surface were characterized via FT-IR and NMR spectroscopy, while elemental analysis and secondary ionization mass spectrometry (SIMS) depth profiling indicated a consistent level of alkenyl group incorporated throughout the porous silicon.
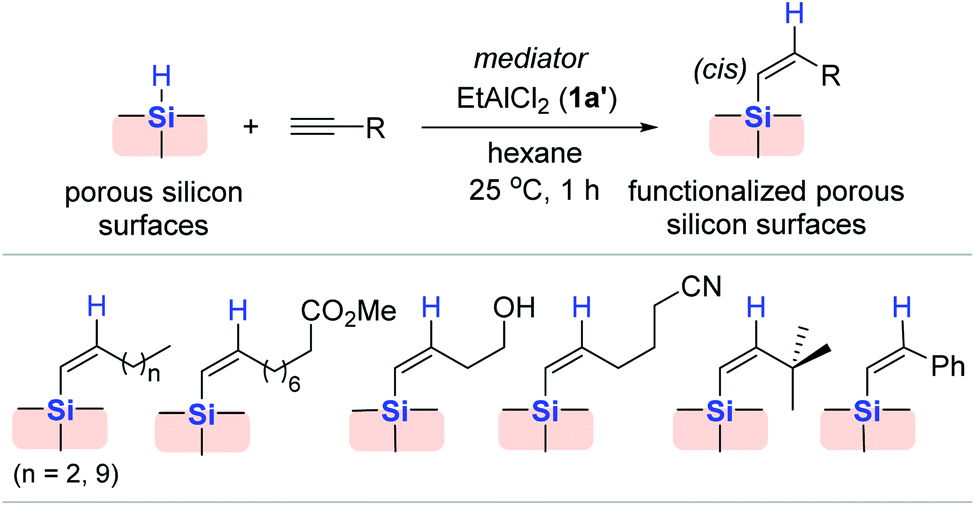 | ||
| Scheme 9 EtAlCl2-mediated hydrosilylation of various alkynes on porous silicon surfaces. | ||
Although the work by Buriak et al. showcased silicon surface modification via Al-mediated alkyne hydrosilylation, this process required a stoichiometric amount of EtAlCl2 1a′. Accordingly, Veinot and coworkers conceived to apply BH3·THF (2) as a catalyst for the hydrosilylative modification of the surface of hydride-terminated silica nanocrystals (H-SiNC).66 This work represents the first borane-catalyzed surface modification of nanoscale silicon. The hydrosilylation of terminal alkynes on H-SiNC occurred at 25 °C in the presence of 2 (2.5 mol%) to yield the alkenyl-functionalized SiNC (Akn-SiNC) (Scheme 10). The degree of SiNC surface coverage was found to decrease with an increase in the chain length of the alkyne substrates, and terminal alkynes were more reactive than internal alkynes in terms of surface functionalization probably due to steric hindrance. It is noteworthy that the catalytic activity was dependent on the size of SiNC, where the hydrosilylation of larger H-SiNC (d = 7.5 nm) required 12 h, whereas the same reaction on smaller SiNC (d = 3.6 nm) was faster and was completed within 0.5 h.
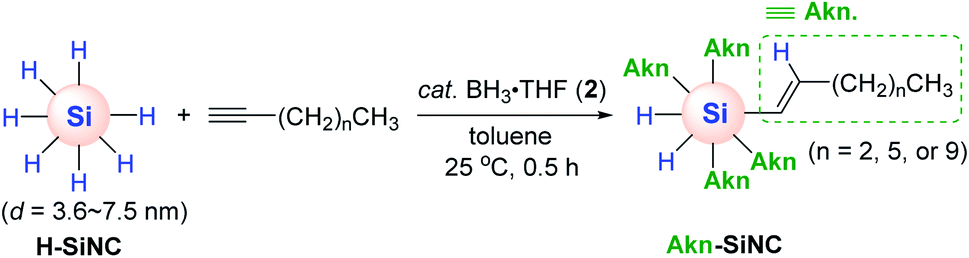 | ||
| Scheme 10 BH3-catalyzed hydrosilylation of alkynes on silicon nanocrystals. | ||
Two reaction pathways were proposed for the 2-catalyzed alkyne hydrosilylation on the surface of H-SiNC (Scheme 11). One possible mechanism (path A) involves the syn-addition of 2, followed by a metathesis process with the Si–H moiety on SiNC (Scheme 11, left). The alternative pathway (path B) is assumed to form zwitterionic vinyl carbocation 2b in the initial step upon the reaction of alkyne with 2. Subsequently, 2b is suggested to be reduced with H-SiNC to generate a vinyl borohydride having a silylium ion site (2c). Finally, 2c is likely converted to Akn-SiNCs with the liberation of 2 (Scheme 11, right).
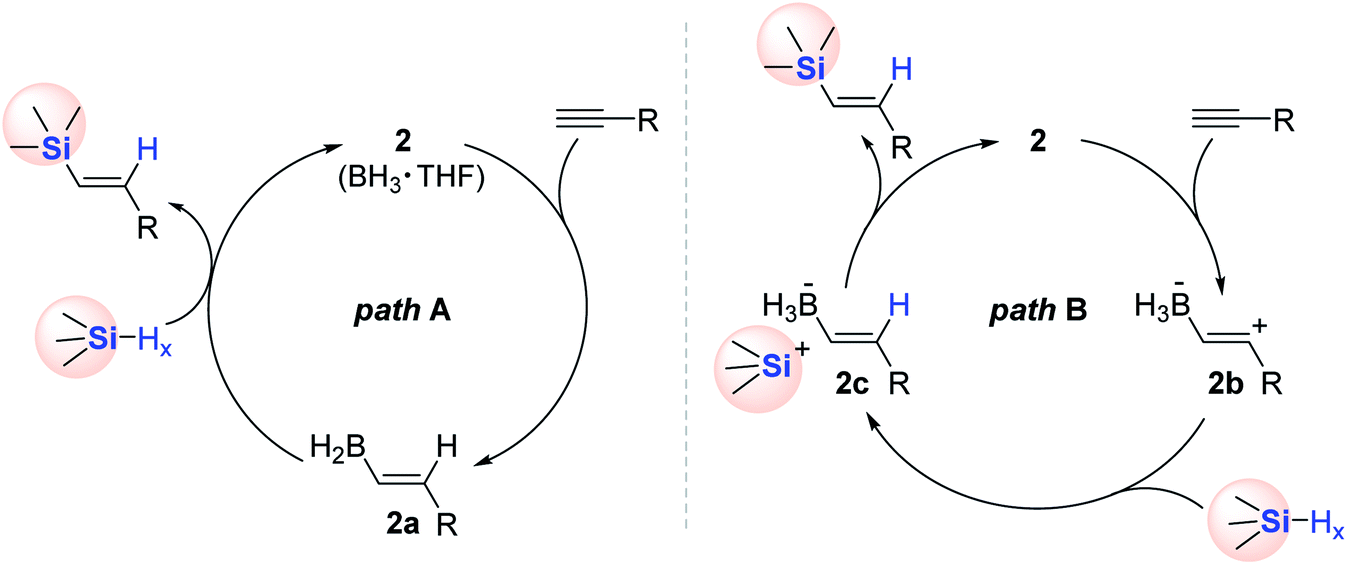 | ||
| Scheme 11 Two possible pathways for the BH3-catalyzed alkyne hydrosilylation on H-SiNC. | ||
As a continuing effort for the catalytic surface modification of silicon, Rieger and Venoit examined various transition metal-free Lewis acids as alkyne hydrosilylation catalysts to modify the surface of hydride-terminated silicon nanosheets (H-SiNS) (Scheme 12).67 The hydrosilylation of 1-decyne on the surface of H-SiNS proceeded at 25 °C with the Lewis acid catalysts (0.7 mol%) to provide the decenyl-functionalized silicon nanosheets. Less bulky catalysts such as BH3 (2) and BF3 were more active towards hydrosilylation than the bulky ones [tris(pentafluorophenyl)alane (ACF, 1′) and B(C6F5)3 2′]. Considering the poor reactivity of 2′ in the case of porous silicon64,65 and silicon nanocrystals,66 the observed catalytic reactivity of B(C6F5)3 2′ with H-SiNS suggests two insights: (i) the hydride donor ability of H-SiNS is higher than the precedent silicon materials, and (ii) the reaction proceeds via an outer-sphere mechanism involving a silylium ion. It is interesting to see that the Lewis acidity of the catalysts (e.g. BF3 vs. BH3·SMe2) did not influence the degree of functionalization, implying that the steric effect of the catalyst is a major factor in determining its reactivity with H-SiNS.
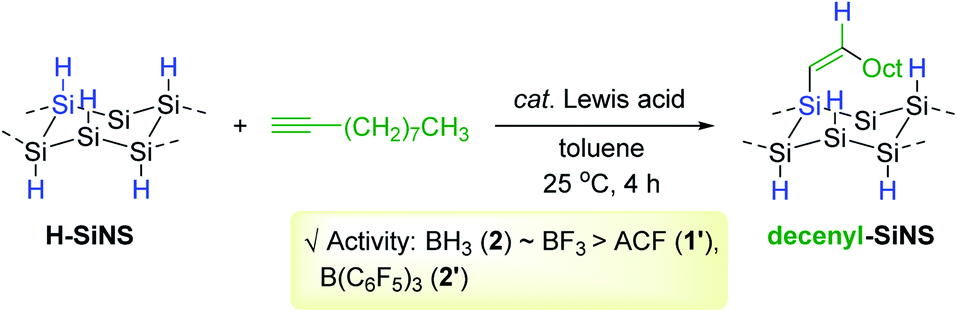 | ||
| Scheme 12 Transition metal-free Lewis acid-catalyzed hydrosilylation of 1-decyne on silicon nanosheets. | ||
Based on experimental observations, a borane-catalyzed outer-sphere hydrosilylation mechanism was proposed (Scheme 13). Borane is assumed to initially form an adduct with a hydrosilane on the surface (3), which generates a silylium ion site, at which 1-decyne reacts to generate a vinyl carbocation (3a) bearing a borohydride anion (HBR3−). Then 3a likely undergoes hydride transfer from the borohydride to yield decenyl-SiNS with the regeneration of active site 3.
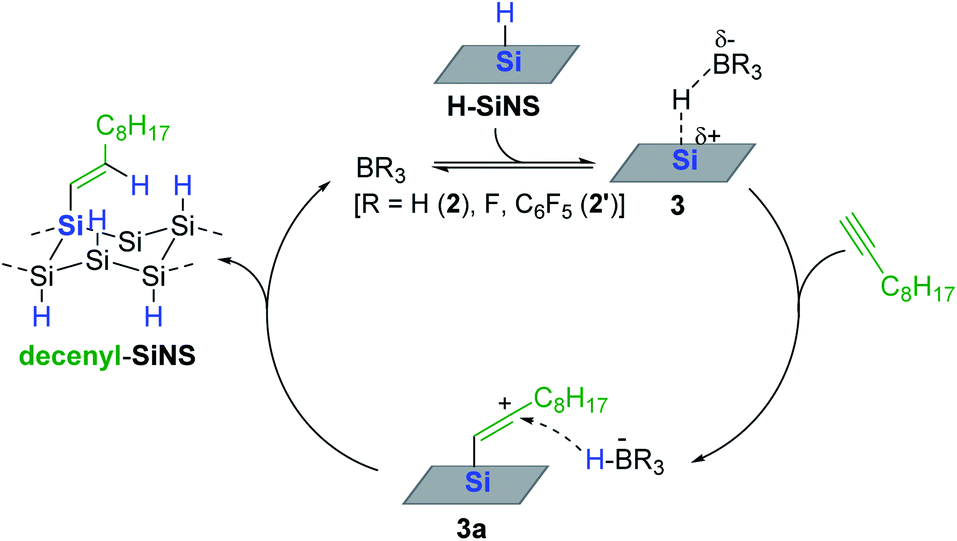 | ||
| Scheme 13 Proposed pathway for the Lewis acidic borane-catalyzed alkyne hydrosilylation on H-SiNS. | ||
Motivated by the precedent Lewis acidic Al-catalyzed alkyne hydrosilylation, Chang et al. developed for the first time the B(C6F5)3 (2′)-catalyzed hydrosilylation of internal ynamides, furnishing a broad range of β-silyl (Z)-enamides (Scheme 14).68 The hydrosilylation of ynamides having various substituents in the β-position occurred at 60 °C in the presence of 2′ (3 mol%) with Et2SiH2 (2 equiv.). Variation of the N-substituents in ynamides from N-methyl to -phenyl or -cycloalkyl, and from N-tosyl to -mesyl groups did not hamper the reactivity and selectivity (52–81%; Z/E, >20:1). This catalysis was also operative with other hydrosilanes except bulky tertiary silanes such as iPr3SiH (not shown).
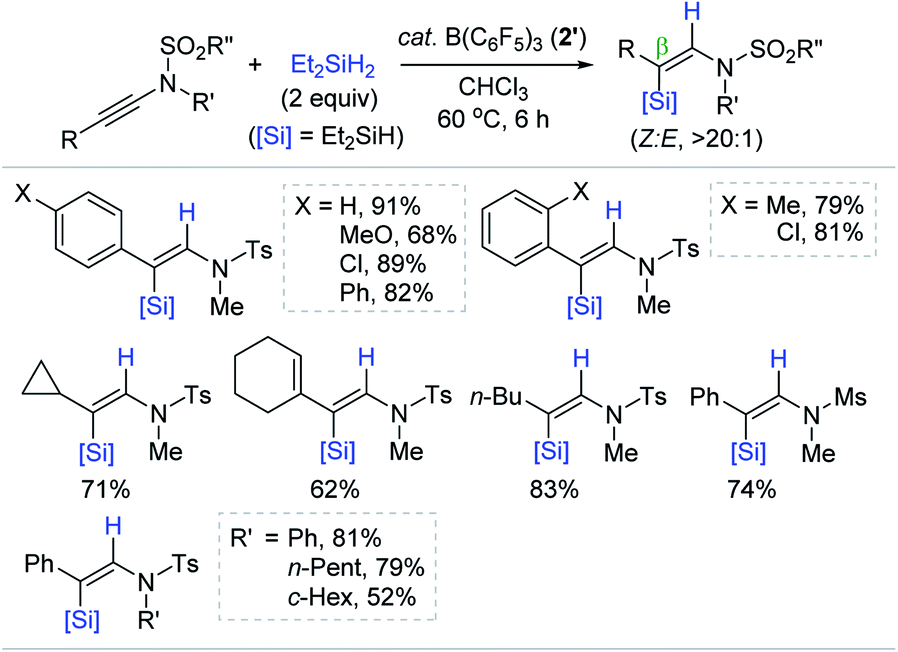 | ||
| Scheme 14 B(C6F5)3-catalyzed trans-hydrosilylation of internal ynamides. Ms = methanesulfonyl. | ||
Based on computational studies, a catalytic working mode was proposed (Scheme 15). The initial step is presumed to transfer a silylium ion from the B(C6F5)3-silane adduct 2′a to the β-carbon of an ynamide to form a ketene iminium bearing a HB(C6F5)3− (2′b). Subsequently, 2′b undergoes hydride attack by the borohydride from the opposite side of the silyl group, leading to a (Z)-β-silyl enamide. Density functional theory (DFT) calculations suggested that the anti-addition path of a hydrosilane is preferred by 3.1 kcal mol−1 over the syn-addition path, which is consistent with the observed high (Z)-stereoselectivity. The origin of the differential activation energy (ΔΔG‡ = 3.1 kcal mol−1) was attributed to the β-silicon effect, resulting in geometrical bias between the same and opposite sides from the silyl group in 2′b.
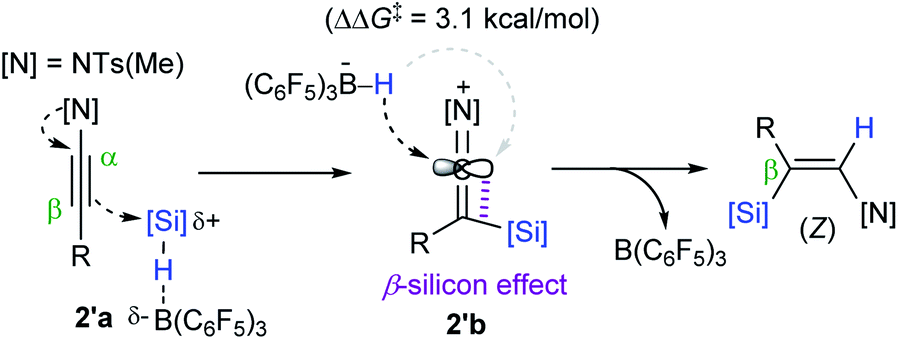 | ||
| Scheme 15 Proposed working mode for the B(C6F5)3-catalyzed hydrosilylation of internal ynamides. | ||
Following the Chang's report, Shen reported a similar outer-sphere trans-hydrosilylation of internal thioalkynes catalyzed by B(C6F5)3 2′ (Scheme 16).69 The hydrosilylation of phenyl(1-heptynyl)sulfane cleanly proceeded at 60 °C with 2′ (1.5 mol%) to give the β-(Z)-silylated vinyl phenyl sulfide in 85% yield in 12 h. Under the borane catalytic regime, a broad range of 1°, 2°, and 3° hydrosilanes were viable for the (Z)-selective hydrosilylation reactions. It is noteworthy that the reaction with bulky silanes required a greater catalyst loading (up to 10 mol%) and/or increased silane quantities (up to 3 equiv.) to achieve reasonable yields of the silylated vinyl sulfides. The borane catalyst was proven to be applicable for double hydrosilylation with PhSiH3 furnishing the β-silylated bis-vinyl sulfide as a single (Z,Z)-isomer in good yield.
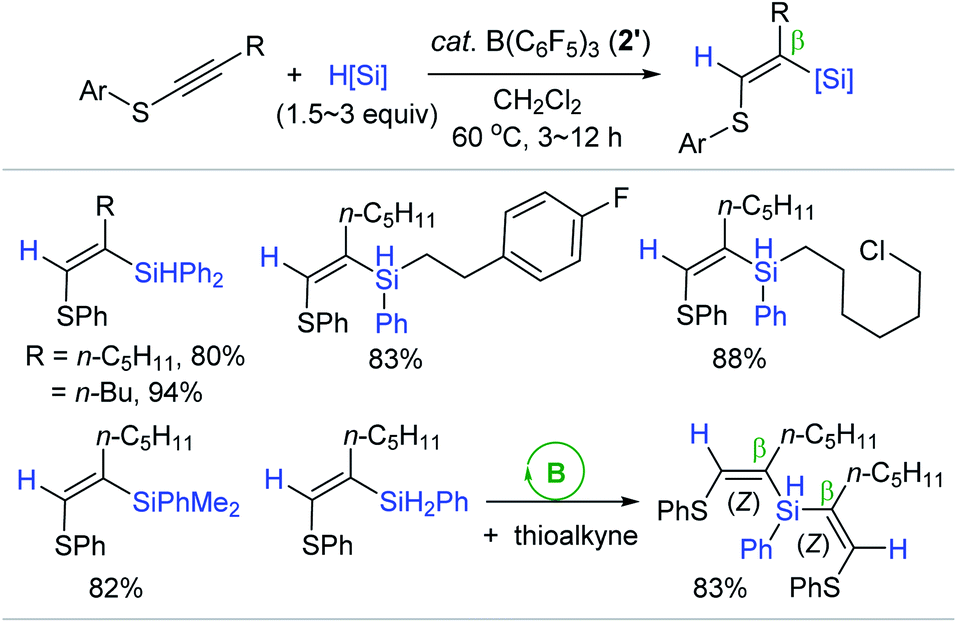 | ||
| Scheme 16 B(C6F5)3-catalyzed trans-hydrosilylation of internal thioalkynes. | ||
In 2014, Ingleson et al. utilized B(C6F5)3 (2′) as a catalyst for the synthesis of silaindenes from aryl-alkynes via a cascade hydrosilylation (Scheme 17).70 For example, the reaction of 1-phenyl-1-butyne with Ph2SiH2 in the presence of 2′ (5 mol%) at 60 °C gave the vinyl silane, which was fully converted to the corresponding silaindene via an intramolecular sila-Friedel–Crafts reaction upon the addition of 2,6-dichloropyridine (Cl2-py, 1 equiv.). The added Cl2-py was presumed to serve as a Lewis base for the deprotonation of the presupposed Wheland intermediate.
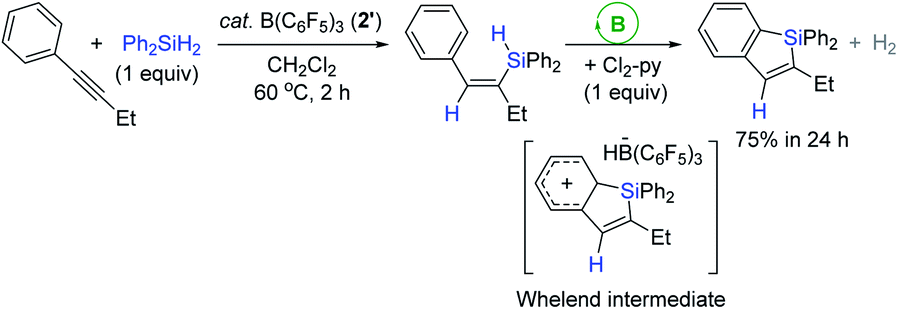 | ||
| Scheme 17 B(C6F5)3-catalyzed one-pot, two-step synthesis of a silaindene from an internal alkyne. Cl2-py = 2,6-dichloropyridine. | ||
Inspired by Ingleson's Lewis acid-mediated intramolecular sila-cyclization, Kawashima et al. developed an intramolecular alkyne hydrosilylation route, leading to silaindenes from 2-alkynylphenylsilane derivatives using trityl tetrakis(pentafluorophenyl)borate ([Ph3C]+[B(C6F5)4]− 4) as an initiator (Scheme 18a).71 A substoichiometric amount of 4 (1 mol%) initiated the sila-cyclization of a series of [2-(trimethylsilylethynyl)phenyl]silanes at 25 °C to provide the trimethylsilyl-substituted silaindenes in 34–81% yield within 1 h. It is noteworthy that this catalysis was susceptible to the substituents at the silicon center of the substrates, where the methyl- or phenyl moiety induced low to moderate yields (34–55%), whereas a bulky diisopropyl silyl group gave a higher yield of the silaindene of up to 75%. Interestingly, replacing the alkynyl TMS group with a butyl gave no hydrosilylation, suggesting that the alkynyl TMS moiety is essential for the catalytic turnover and serves as a neighboring group participating in the stabilization of a presumed vinyl carbocation by the β-silicon effect.72–74
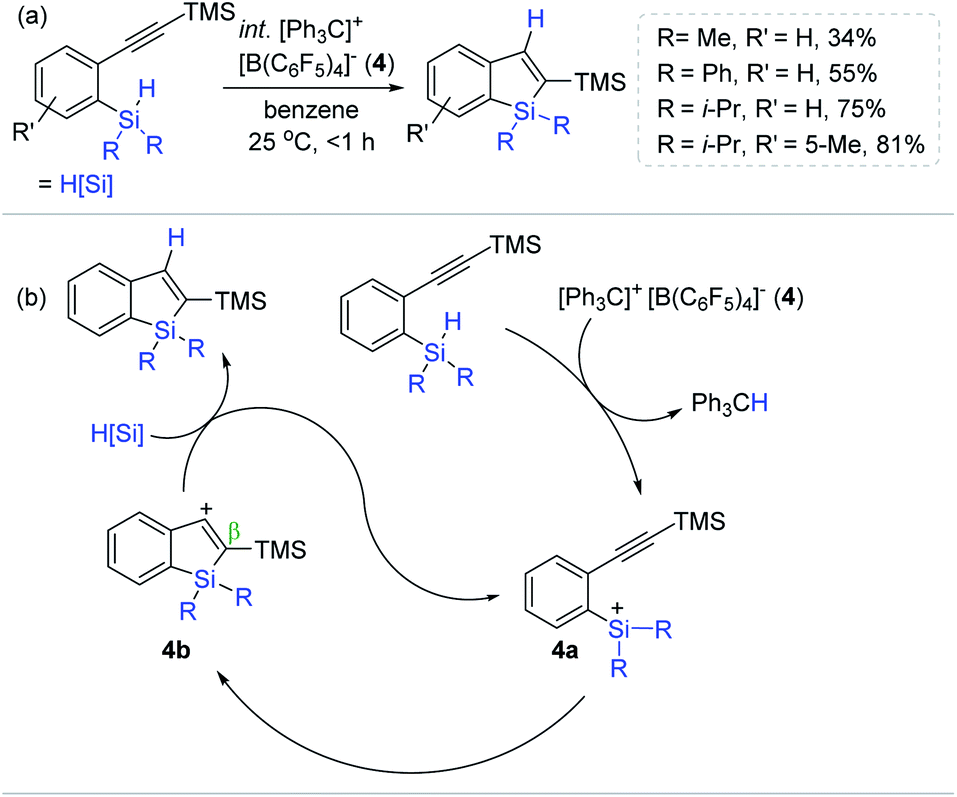 | ||
| Scheme 18 (a) Trityl borate salt-initiated intramolecular sila-cyclization of alkynes and (b) their catalytic pathway. | ||
A prototypical silylium ion-involved, outer-sphere mechanism of the trityl borate-initiated sila-cyclization was proposed (Scheme 18b). Specifically, 4 initially abstracts a hydride from the Si–H group of the substrates to generate active species 4a, which intramolecularly reacts with an adjacent alkynyl unit to form ethenyl carbocation 4b. This cationic intermediate is suggested to be stabilized by the (pre)installed silyl groups (β-silicon effect). The catalytic cycle is assumed to be closed upon hydride reduction by another hydrosilyl alkyne substrate, accompanied by the liberation of the silaindene product and the active catalyst 4a.
In 2013, Stephan et al. synthesized an electrophilic organofluorophosphonium salt [(C6F5)3PF]+[B(C6F5)4]− 5 through the reaction of a difluorophosphorane (C6F5)3PF2 with the salt reported by Lambert et al., [Et3Si]+[B(C6F5)4]−.75 The highly electrophilic P(V) center in 5 was capable of abstracting a fluoride anion from fluoroalkanes to generate (C6F5)3PF2 and a carbocation. This carbocation was reduced by a hydrosilane to release an alkane with the regeneration of Lambert's salt for catalytic turnover (Scheme 19a). Based on the observed high electrophilicity of 5, Stephan et al. further applied 5/silane as a catalytic system for the hydrosilylation of alkynes.76 The hydrosilylation of 1-decyne with Et3SiH occurred at 25 °C by 5 (1.5 mol%) to quantitatively afford the β-cis-vinylsilane within 1 h, suggesting the anti-1,2-addition of hydrosilanes to an alkyne. DFT calculations suggested that the LUMO of 5 contains the σ* orbital oriented opposite to the P–F bond to promote the formation of η1-silane adduct 5a (ΔH = −15.2 kcal mol−1), which was regarded as an active catalytic species and indeed observed by 1H NMR (Scheme 19b).
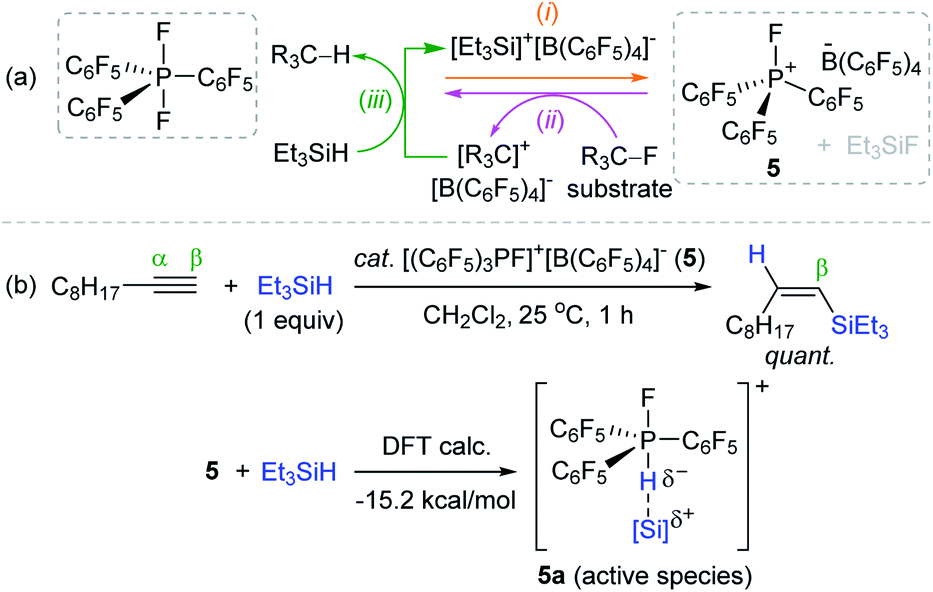 | ||
| Scheme 19 (a) Generation of an electrophilic phosphonium salt and (b) its catalytic application for alkyne hydrosilylation. | ||
Although the electrophilic phosphonium salt 5 displayed unique Lewis acidic catalytic reactivity, the fluoroarene substituents in 5 limit structural variations. Considering this, Stephan et al. reported a new synthetic strategy, leading to a highly electrophilic phosphonium salt that does not bear electron-deficient aryl groups. Specifically, NHC-ligated cationic phosphine(III) 6a is oxidized by XeF2 to a P(V) cationic difluorophosphorane 6b, which subsequently undergoes fluoride abstraction by Lambert's salt to eventually afford a fluorophosphonium dication supported by an NHC ligand [(NHC)PFPh2]2+[B(C6F5)4]2− (6) (Scheme 20).77 To demonstrate the Lewis acidity of 6 in catalytic reactions, Stephan et al. performed the hydrosilylation of 1,2-diphenylacetylene in the presence of a catalytic amount of 6, giving the (Z)-vinylsilane in excellent yield at 45 °C within 24 h.
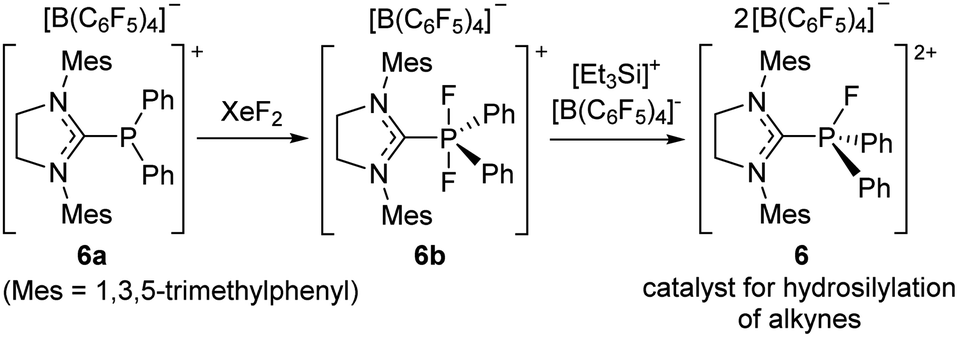 | ||
| Scheme 20 Synthetic route to a dicationic imidazolium–phosphonium salt 6 as a hydrosilylation catalyst. | ||
Pentacoordinate hydrosilicates in situ-generated from hydrosilanes and Lewis base activators are well-known catalytic species for the reduction of carbonyl functions,78,79 and alkynylsilanes have been utilized for the alkynylation of carbonyls and imines via the corresponding hypervalent silicate intermediates.80–84 Based on this background, Lee developed a KOt-Bu (7)-catalyzed tandem alkynylation and hydrosilylative cyclization of carbonyls with alkynylhydrosilanes (Scheme 21).85 A range of aldehydes and ketones reacted with alkynylsilanes at 25 °C by catalyst 7 (10 mol%), providing diverse oxasilacyclopentenes in 48–87% yield within 20 min. This catalysis was highly sensitive to electronic and steric variations in substrates, where less bulky and electron-neutral or -deficient aryl-substituted alkynylsilanes were only viable for the tandem transformation.
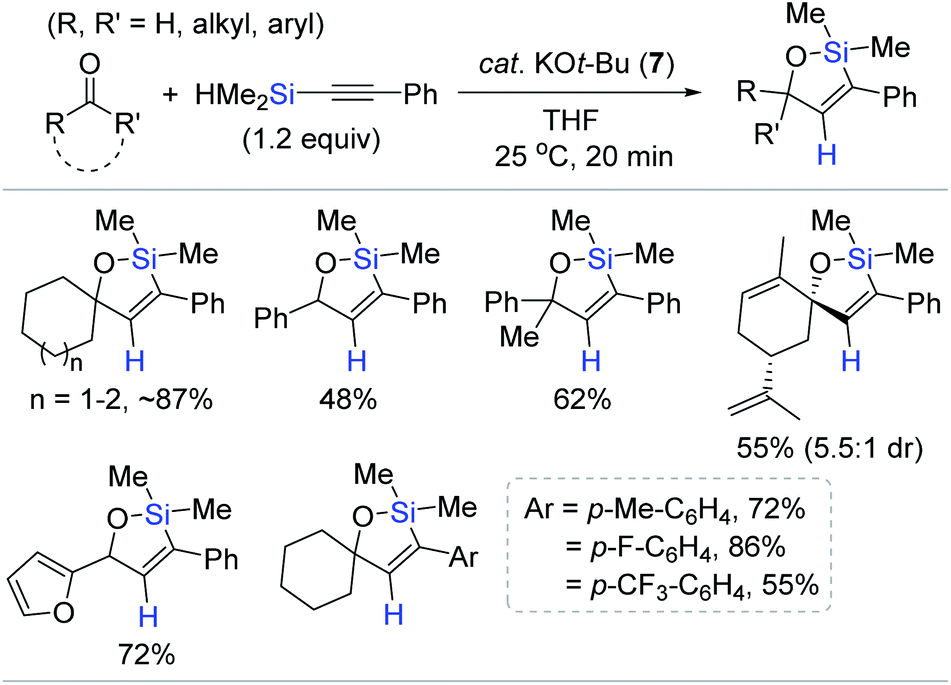 | ||
| Scheme 21 KOt-Bu-promoted tandem alkynylation of carbonyls and intramolecular trans-hydrosilylation. | ||
The key steps for the 7-catalyzed tandem alkynylation and hydrosilylation sequence were proposed (Scheme 22). Initially, tert-butoxide attacks the silicon center of an alkynylsilane to form a pentavalent silicate intermediate 7a, which transfers the alkynyl group to the carbonyl to generate an activated hydridosilyl ether 7b. This species is assumed to undergo intramolecular trans-hydrosilylation over the triple bond to liberate an oxasilacyclopentene. A cross-over experiment using two similar alkynylsilanes suggested a naked alkynyl anionic species is involved in the alkynylation step for ketones.86–89
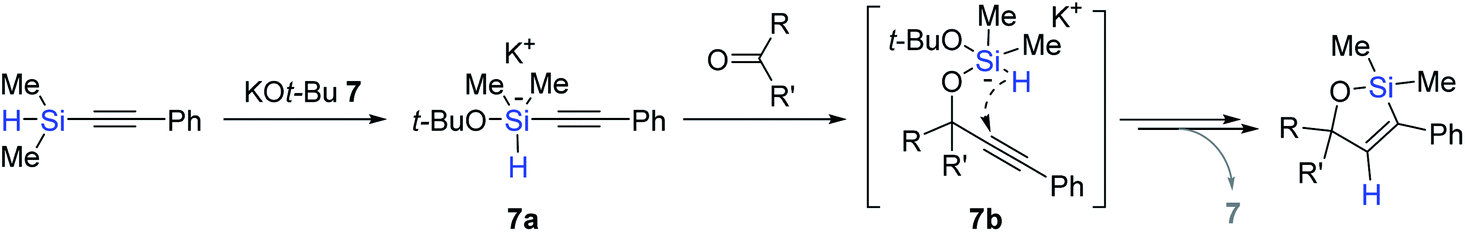 | ||
| Scheme 22 Proposed key steps for the KOt-Bu-catalyzed tandem alkynylation and hydrosilylation. | ||
Recently, a series of Al complexes with various β-diketiminate [ArNacNac![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH{C(Me)NAr}2] ligands have emerged as competent catalysts for hydroelementation of unsaturated functions.90–92 Additionally, neutral ArNacNacAl complexes [e.g. DIPPNacNacAl(OTf)H, DEPNacNacAlH2; DIPP = 2,6-i-Pr2C6H3, DEP = 2,6-Et2C6H3] were reported to catalyze the hydroboration of alkynes and carbonyls,90,91 but the cationic ArNacNacAl complexes remained unexplored. In this regard, Nikonov et al. prepared the cationic DIPPNacNacAlH complex (8) through the reaction of DIPPNacNacAlH2 with [Ph3C]+[B(C6F5)4]− (4), and examined its catalytic activity towards the alkyne hydrosilylation.93 The reactions of a few alkyne substrates with Et3SiH proceeded via the action of 8 (5 mol%) with high conversions to afford the vinylsilanes, albeit in low yields (7–29%) (Scheme 23). The observed low yields were ascribed to the facile redistribution of hydrosilanes by 8. Although the exact catalytic working mode of 8 was unclear, the experimental observations implied that the cationic Al center in 8 serves as a Lewis acid for the activation of Et3SiH.
CH{C(Me)NAr}2] ligands have emerged as competent catalysts for hydroelementation of unsaturated functions.90–92 Additionally, neutral ArNacNacAl complexes [e.g. DIPPNacNacAl(OTf)H, DEPNacNacAlH2; DIPP = 2,6-i-Pr2C6H3, DEP = 2,6-Et2C6H3] were reported to catalyze the hydroboration of alkynes and carbonyls,90,91 but the cationic ArNacNacAl complexes remained unexplored. In this regard, Nikonov et al. prepared the cationic DIPPNacNacAlH complex (8) through the reaction of DIPPNacNacAlH2 with [Ph3C]+[B(C6F5)4]− (4), and examined its catalytic activity towards the alkyne hydrosilylation.93 The reactions of a few alkyne substrates with Et3SiH proceeded via the action of 8 (5 mol%) with high conversions to afford the vinylsilanes, albeit in low yields (7–29%) (Scheme 23). The observed low yields were ascribed to the facile redistribution of hydrosilanes by 8. Although the exact catalytic working mode of 8 was unclear, the experimental observations implied that the cationic Al center in 8 serves as a Lewis acid for the activation of Et3SiH.
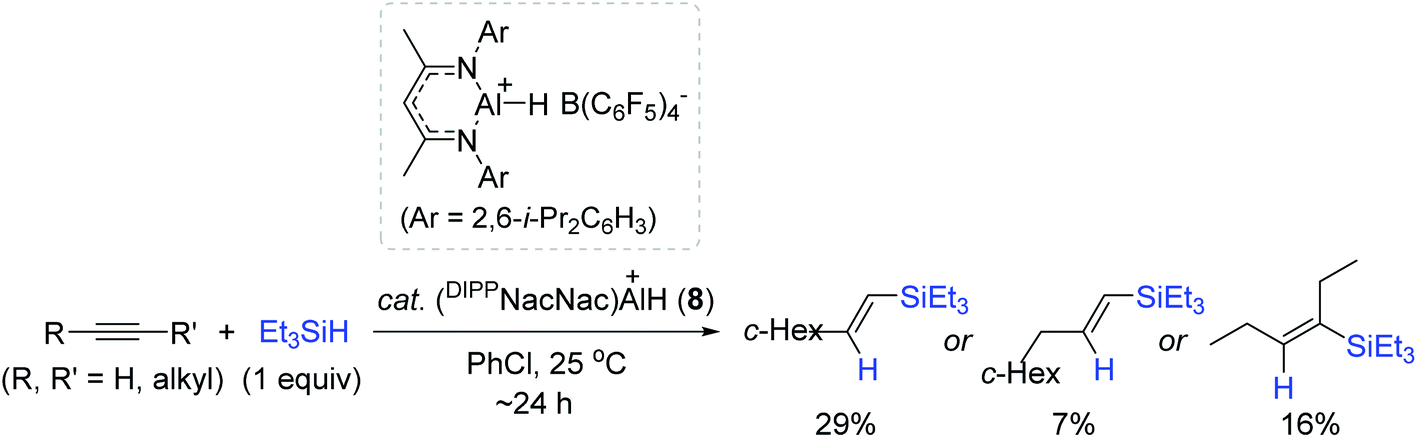 | ||
| Scheme 23 Cationic Al(III)-catalyzed hydrosilylation of alkynes. | ||
Similar to the Nikonov's work, Hill et al. reported a neutral NacNac-Mg hydride dimer (DIPPNacNacMgH)2 (9) as a catalyst for alkyne hydrosilylation.94 The reaction of diphenylacetylene with PhSiH3 slowly proceeded to give the β-(E)-vinylsilane in 95% yield in 30 days (Scheme 24). Unlike Al (8) catalysis under the Lewis acid-activation mode,93 its isoelectronic monomeric magnesium hydride 9′ was found to work via the classical inner-sphere pathway, involving alkyne insertion, followed by a metathesis reaction with a hydrosilane.
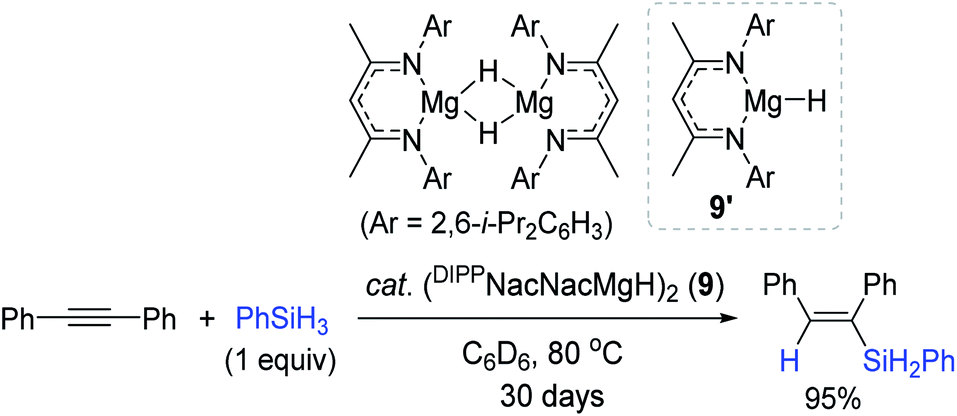 | ||
| Scheme 24 Mg(II)-catalyzed cis-hydrosilylation of diphenylacetylene. | ||
Recently, Fritz-Langhals et al. reported the catalytic use of a silyliumylidene cation 10 [Cp*Si:+B(C6F5)4−; Cp* = pentamethylcyclopentadienyl] having a vacant coordination site.95 The hydrosilylations of some terminal alkynes proceeded in the presence of 10 (0.04–0.1 mol%) at 25–50 °C to bring about a complex mixture of the corresponding vinylsilanes (cis/trans) and alkynylsilanes in poor yields, while in the case of ethynyltrimethylsilane, it underwent both hydrosilylation and redistribution reactions to give various vinylsilanes as a mixture (Scheme 25). As similar as the borane-silane adduct 2′a,68–70 the η1-silane adduct (10a) was proposed to act as an active catalyst for the outer-sphere hydrosilylation of alkynes. In fact, the η1-Si–H coordination to the cationic Si center at 10a was evidenced by the coalescence of the Si–H signal in the time-dependent 1H NMR spectra.
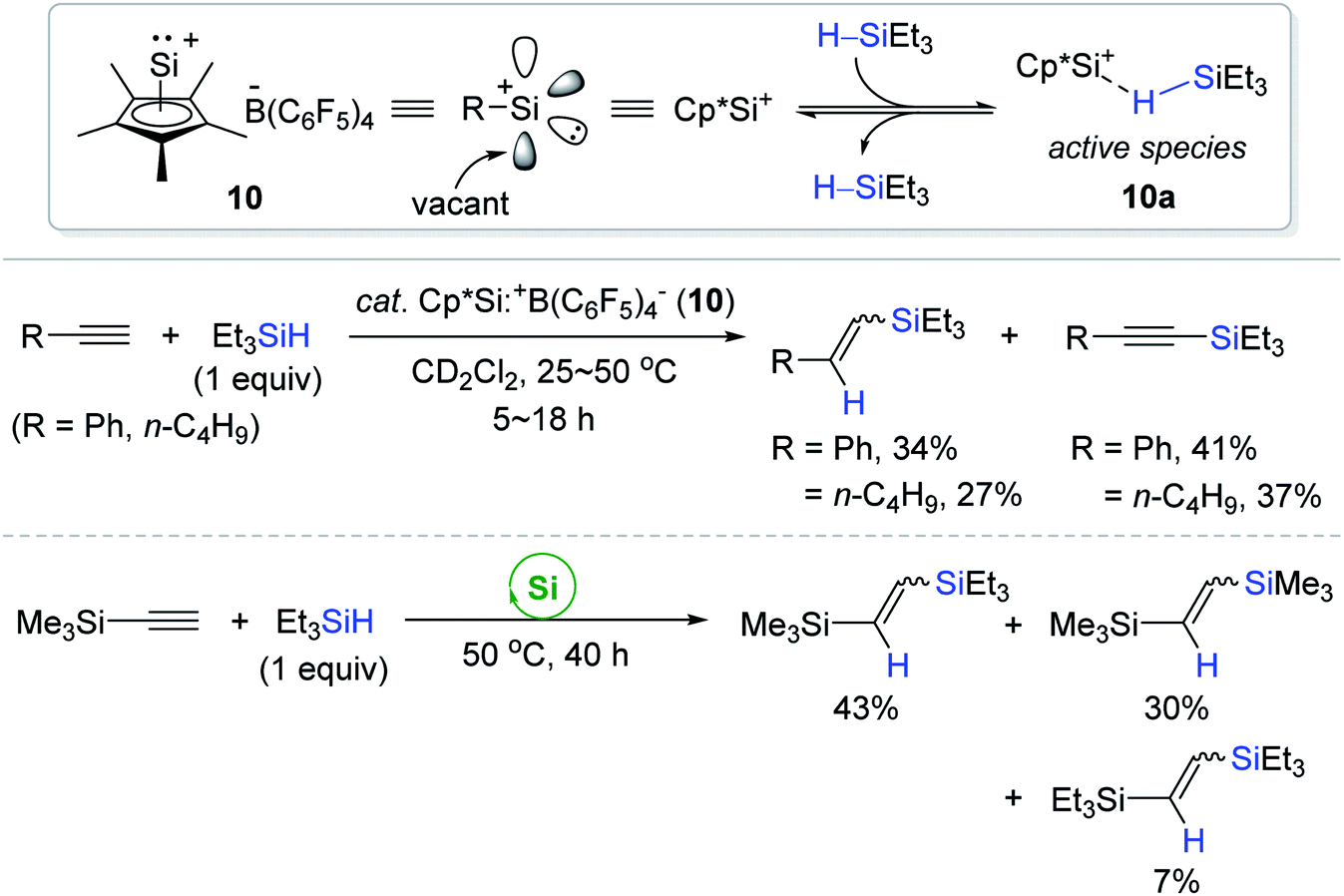 | ||
| Scheme 25 Cp*Si:+-catalyzed hydrosilylation of alkynes. Cp* = pentamethylcyclopentadienyl. | ||
3. Hydroboration
Vinylboranes and vinylboronate esters have been widely utilized as coupling reagents and/or intermediates in the context of synthetic organic chemistry to construct new bonds of C–C, C–N, and C–O on the vinyl skeleton.12,14,16 Hydroboration of alkynes is the most straightforward method to access the vinylborane reagents, while their regio- and stereoselectivity (α/β and E/Z issues) are variable and closely related to the possible catalytic working modes.20–22,28 Conventional transition metal catalytic systems tend to yield (E)-vinyl products via the syn-addition of a B–H moiety (cis-hydroboration) at an inner-sphere of the active catalytic species, whereas metal-catalyzed trans-hydroboration affording (Z)-vinyl products is relatively less documented.With this background, Ingleson et al. reported the B(C6F5)3 2′-catalyzed trans-hydroboration of terminal alkynes (Scheme 26).96 Their work represents the first transition metal-free trans-hydroboration of alkynes. The hydroboration process was proposed to involve an electrophilic borenium cation possessing a borohydride HB(C6F5)3− (11) as the active species, which was generated in situ upon the 1:1 reaction of I-DCDM-9-BBN(H) (11a) [(I-DCDM = 1,3-dimethyl-4,5-dichloroimidazolylidene; 9-BBN(H) = 9-borabicyclo3.3.1]nonane) and 2′. Consistent with 11 as the presumed active species, the stoichiometric reaction of 11, 11a, and 1-pentyne gave the corresponding β-trans-hydroboration product with the constant observation of 11 (Scheme 26a). Having this stoichiometric insight, the hydroboration of a range of terminal alkynes with 11a as a reducing agent was conducted in the presence of 2′ (5 mol%) to reveal that the reaction was completed within 10 min at 20 °C to afford the cis-vinylboranes in 56–97% yield (Scheme 26b). Although most of the internal alkynes remained inert, the nucleophilic internal alkyne 4-(1-propynyl)-N,N-dimethylaniline reacted with 11a under the B(C6F5)3 catalytic regime to yield the corresponding vinylborane in 70% in 3 h.
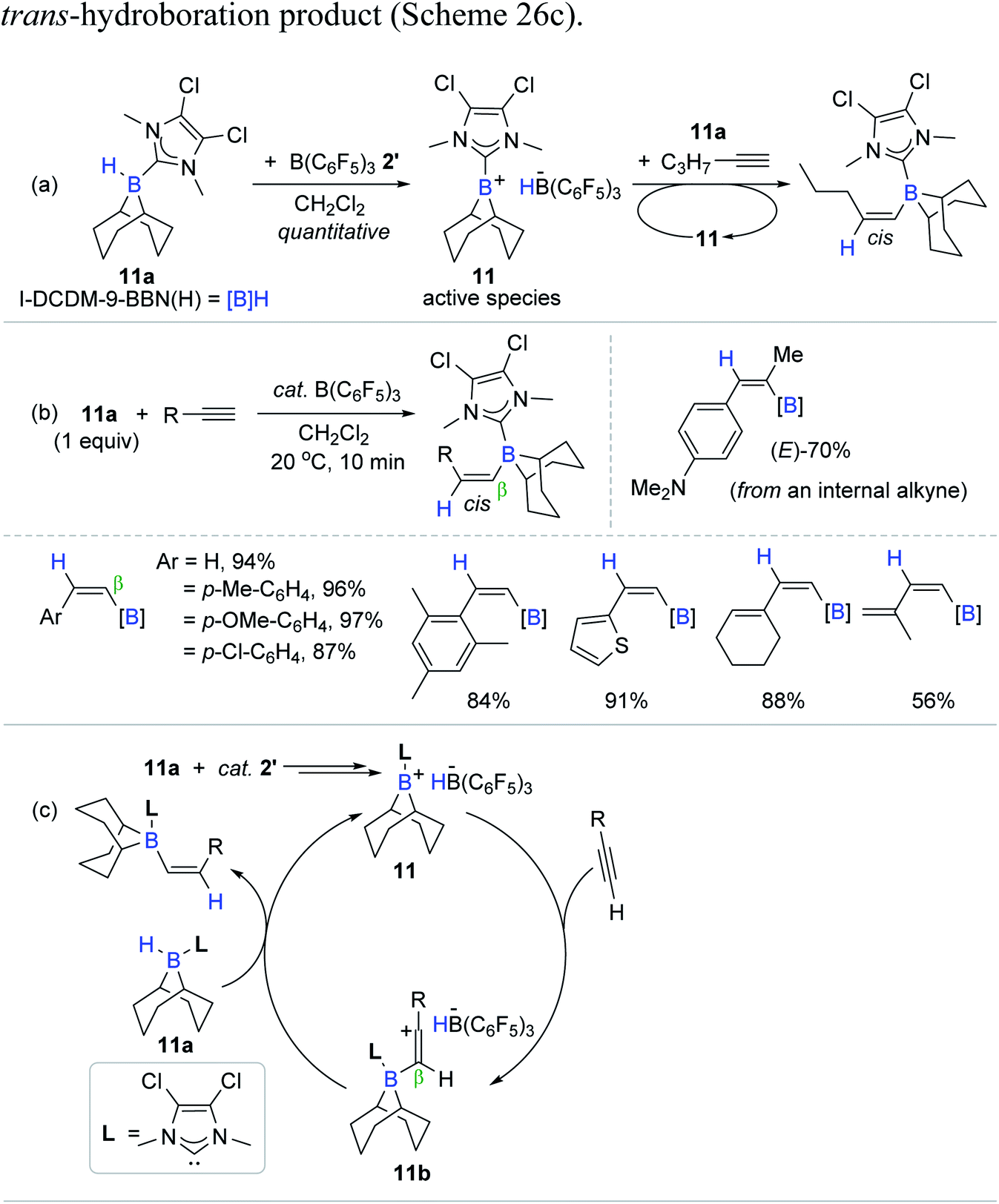 | ||
| Scheme 26 (a) Stoichiometric and (b) catalytic trans-hydroboration of alkynes mediated by B(C6F5)3, and (c) its plausible catalytic pathway. | ||
A deuterium labelling study and stoichiometric and catalytic reactions supported a stepwise ionic hydroboration mechanism, involving the β-addition of a borenium cation,97,98 leading to a vinyl carbocation intermediate 11b, followed by hydride transfer mainly from 11a to the less hindered face of 11b to release the trans-hydroboration product (Scheme 26c).
With interest in the synthesis of boron-containing heterocycles,99–103 Stephan et al. previously reported a synthetic route to obtain unique B–N heterocycles via “click reactions” of boron-azides with (phospha)alkynes.104 In a related effort, in 2016, Stephan et al. reported the stoichiometric reactions of alkynylphosphines with azido-phosphine-borane adducts, forming a series of PNP species [representatively, t-Bu2P(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)NP(BH3)iPr2 (12)].105 Interestingly, species 12 underwent an intramolecular trans-hydroboration at the phosphaalkynyl unit by the catalytic action of 2′ (10 mol%) to liberate the corresponding PNPCB heterocycle in 64% yield (Scheme 27). The reaction pathway leading to PNPCB from 12 was proposed to involve a borenium cation bearing HB(C6F5)3− (12a) as the key intermediate. This outer-sphere mechanism was corroborated by the trans-added B–H moiety on PNPCB, as also shown in the report by Ingleson et al.96
CPh)NP(BH3)iPr2 (12)].105 Interestingly, species 12 underwent an intramolecular trans-hydroboration at the phosphaalkynyl unit by the catalytic action of 2′ (10 mol%) to liberate the corresponding PNPCB heterocycle in 64% yield (Scheme 27). The reaction pathway leading to PNPCB from 12 was proposed to involve a borenium cation bearing HB(C6F5)3− (12a) as the key intermediate. This outer-sphere mechanism was corroborated by the trans-added B–H moiety on PNPCB, as also shown in the report by Ingleson et al.96
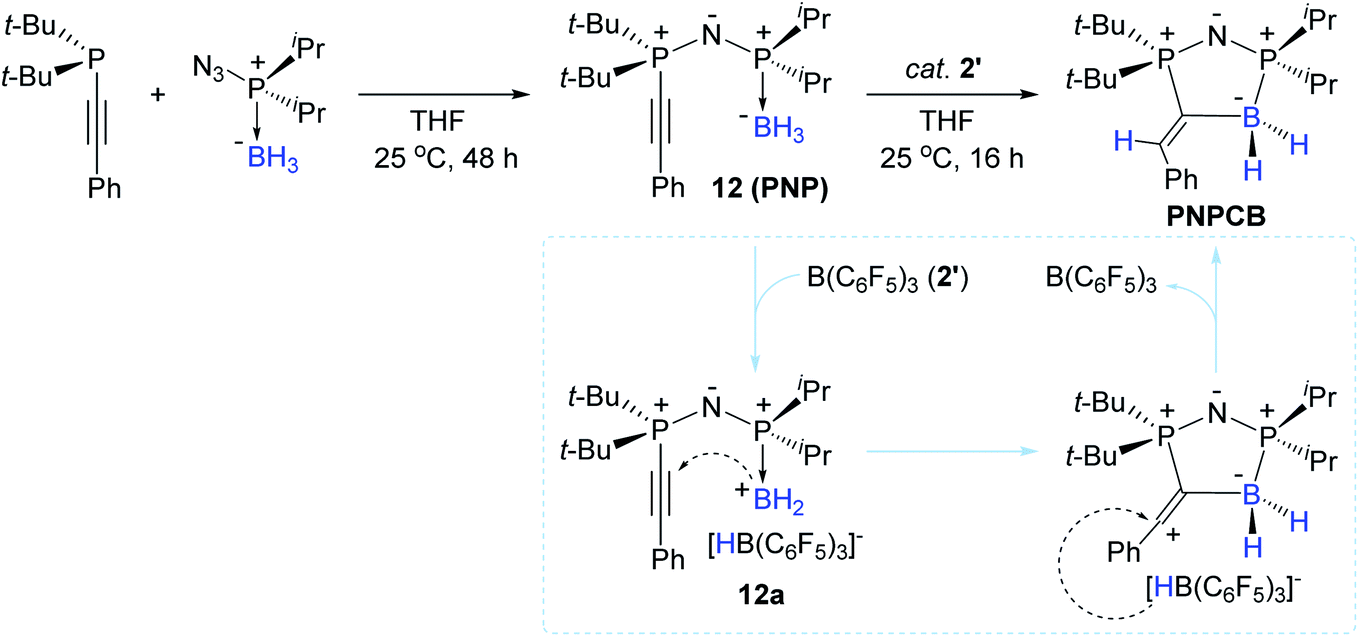 | ||
| Scheme 27 B(C6F5)3-catalyzed intramolecular trans-hydroboration of alkynylphosphines leading to B–P heterocycles. | ||
Having sought a highly Lewis acidic, but functional group tolerant boron-based reduction catalyst, the Oestreich group previously synthesized tris[3,5-bis(trifluoromethyl)phenyl]borane as a catalyst for the hydroboration of alkenes and imines, and demonstrated its superior catalytic activity over the typical borane catalyst B(C6F5)3 2′.106,107 Motivated by Oestreich's work, the Melen group applied tris(2,4,6-trifluorophenyl)borane 13 as a mild Lewis acid catalyst for the hydroboration of alkynes.108 It is noteworthy that the 1:1 reaction of 2′ and an alkyne led to 1,1-carboboration, whereas this carboboration did not occur with 13. The hydroboration of a range of alkynes smoothly proceeded at 60 °C in the presence of 13 (2 mol%) to provide a range of β-vinyl boranes with good functional group tolerance (Scheme 28). It is intriguing to see that the alkenyl geometry of the resultant vinylboronate esters was determined to be a trans or (Z)-geometry, which was opposite to the stereo-outcome (cis or E) shown in Ingleson's reaction system.96 Next, in an attempt to improve the catalytic efficiency, Melen et al. carried out the same hydroboration reaction under microwave irradiation conditions with a tris(3,4,5-trifluorophenyl)borane catalyst (13′).109 The hydroboration was completed within 1.5 h under the microwave conditions (180 °C, 20 atm) to afford the trans or (Z)-products in good to high yields.
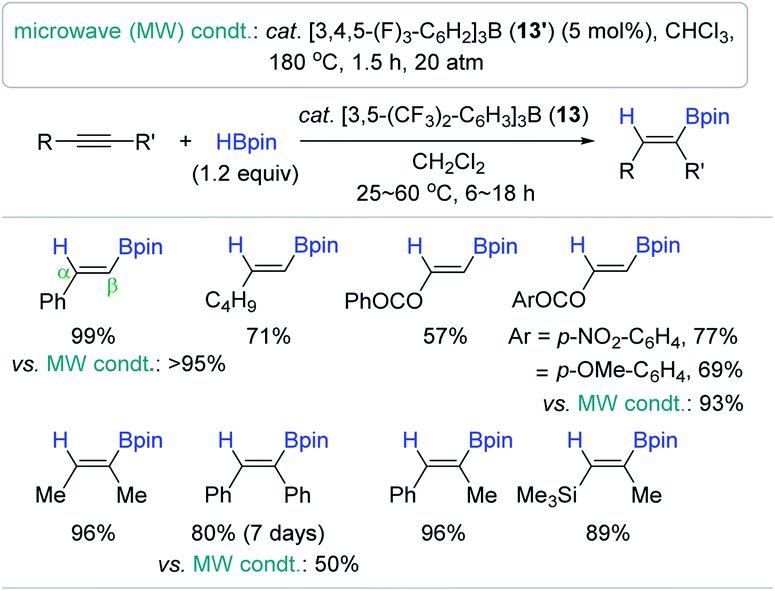 | ||
| Scheme 28 Lewis acidic borane-catalyzed cis-hydroboration of alkynes under conventional and microwave conditions. | ||
With an interest in the selective synthesis of 1,1-diborylalkenes110–114 via the direct hydroboration of alkynylboronates, Jin and coworkers found that carboxylic acids were capable of catalyzing the targeted hydroboration reaction (Scheme 29).115 This work represents the first direct hydroboration of alkynylboronates, and the first alkyne hydroboration catalyzed by carboxylic acids. A range of carboxylic acids with a pKa of 4.2–5.0 worked well as the catalyst. The hydroboration of 2-phenyl-1-ethynylboronic acid pinacol ester with HBpin (5 equiv.) in the presence of 4-(dimethylamino)benzoic acid (14) (5 mol%) gave the 1,1-diborylalkene in quantitative yield in 12 h. Under these conditions, various alkynylboronates, and common terminal and internal alkynes were smoothly reduced with exclusive regio (β)- and stereoselectivity (trans or Z), and functional group compatibility with polar groups. Although the working mode of 14 remained unclear, a tentative pinacolboronate ester generated from 14 and HBpin was suggested as a key catalytic species for this hydroboration.116–118
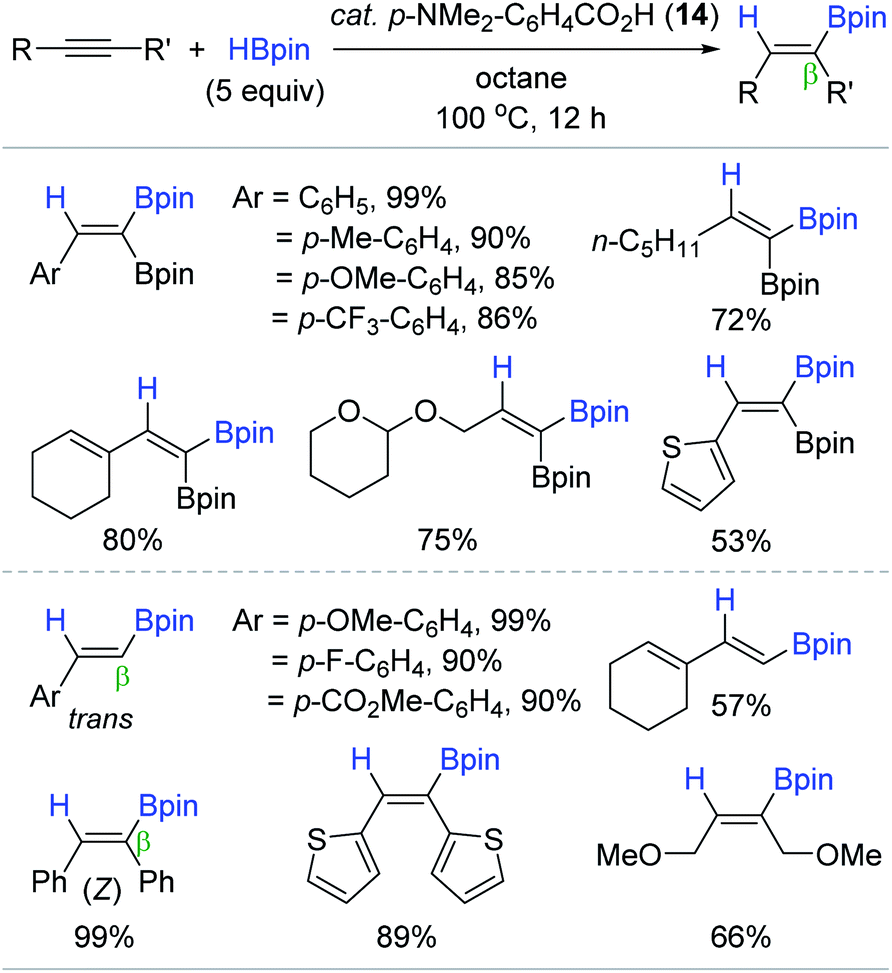 | ||
| Scheme 29 Benzoic acid-catalyzed cis-hydroboration of alkynes. | ||
Highly motivated by Jin's molecular benzoic acid catalyst, Jones et al. applied poly(vinyl benzoic acid) [poly(vba)] 15 as a heterogeneous catalyst for the hydroboration of alkynes (Scheme 30).119 Phenylacetylene was cleanly hydroborated in the presence of 15 (5 mol%) to give the corresponding trans-2-styryl-Bpin in quantitative yield in 16 h. A variety of terminal alkynes were viable, in which aryl-substituted terminal alkynes were more reactive than the alkyl-substituted alkynes toward hydroboration. However, unlike the catalyst system of 14, the reactivity of internal alkynes with 15 was poor under the conditions presumably due to their steric effect, limiting access to the catalytic site on the polymer. Catalyst 15 was sparsely soluble in octane solvent, and thus was readily recycled up to the 3rd catalytic batch, maintaining its catalytic activity.
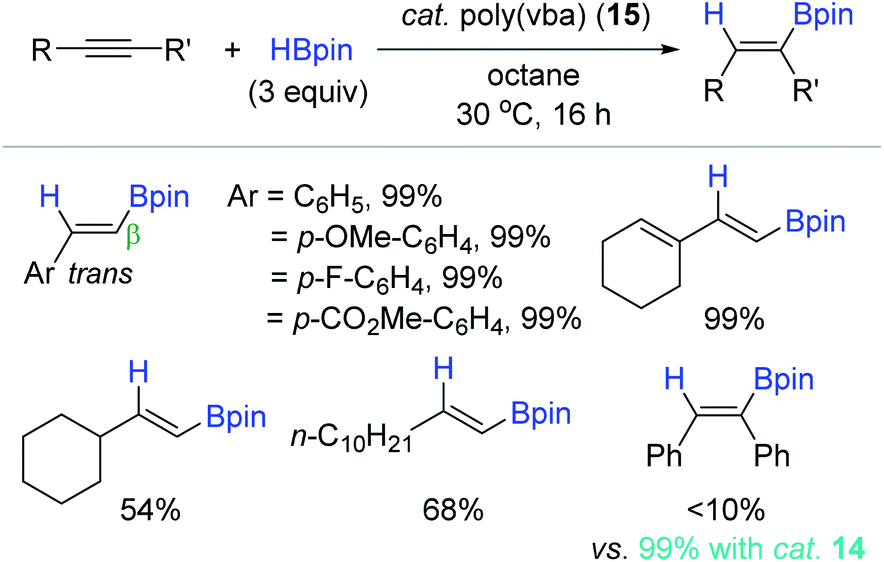 | ||
| Scheme 30 Poly(vinyl benzoic acid)-catalyzed cis-hydroboration of alkynes. | ||
Based on the experimental mechanistic observations (1°-order in [15] and [HBpin], and fractional-order in [alkyne]; kinetic isotope effect KIE (kH/kD) ∼ 0.5), a hypothetical reaction pathway was depicted, as shown in Scheme 31. The initial step is assumed to involve the interaction between a proton of a carboxylic acid and an alkyne to induce polarization over the alkyne unit. An HBpin bound at another acid site has close contact with the polarized alkyne moiety, where the turnover-limiting syn-addition of H–Bpin via a four-membered transition state occurs to release the β-trans-vinylborane with the regeneration of the catalytic site of 15.
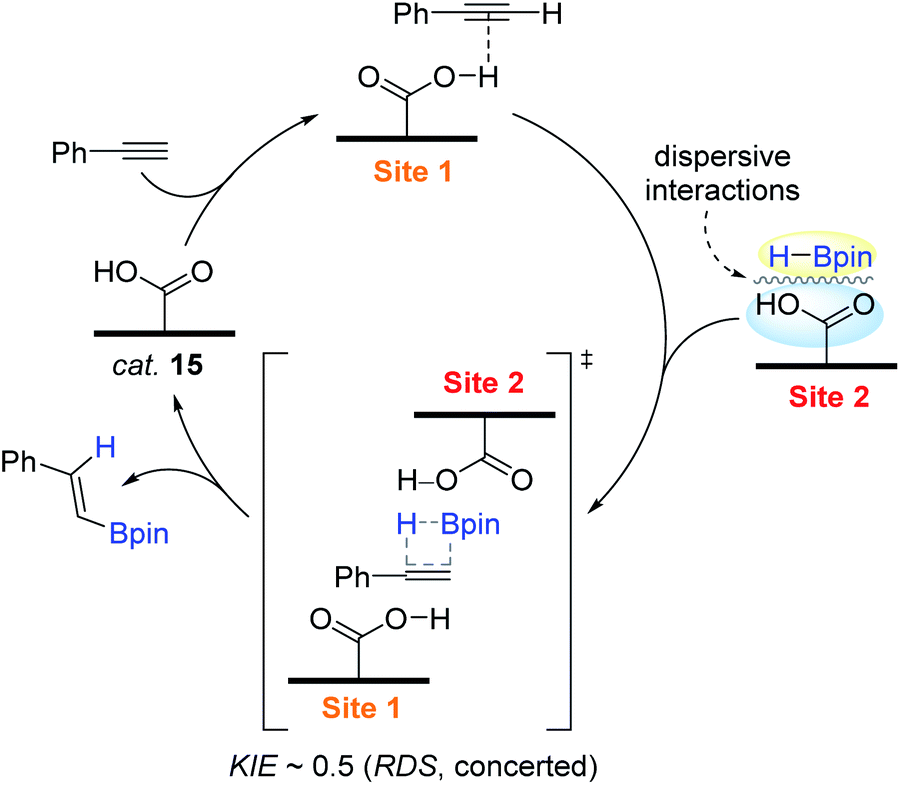 | ||
| Scheme 31 Proposed pathway for the poly(vba)-catalyzed hydroboration of terminal alkynes. | ||
In addition to these Brönsted acid catalysts, Liu and Zhao reported the NaOH (16)-initiated hydroboration of terminal alkynes with HBpin.120 This work is the first example of Brönsted base-catalyzed alkyne hydroboration. The preliminary NBO (natural bond orbital) calculations suggested that the HBpin-NaOH adduct would enhance its hydride character (hydricity) relative to free HBpin,121,122 which was envisioned to facilitate the hydroboration process. Encouraged by this preliminary theoretical insight, a range of simple terminal alkynes were subjected to catalytic conditions [cat. NaOH (8 mol%), HBpin (1.4 equiv.), C6D6, 100 °C, 6 h] to reveal that the trans-vinyl products were obtained in moderate to excellent yields via the syn-addition of H–Bpin (Scheme 32). Notably, the product yield significantly decreased when the phenyl ring possessed an ortho-substituent. The stoichiometric reaction of HBpin with NaOH implied that various borohydride species including BH3–NaOH could be responsible for the observed catalytic activity, as supported by the reports of Clark123 and Fu,124 where BH3-base adducts generated from the reaction with NaOt-Bu or N,N-dimethylacetamide were observed.
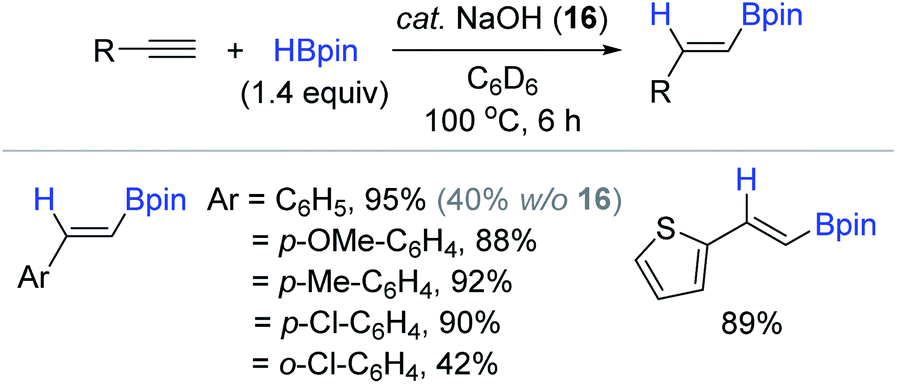 | ||
| Scheme 32 NaOH-catalyzed cis-hydroboration of terminal arylalkynes. | ||
Recently, Ohmiya and Sawamura documented the phosphine-catalyzed trans-addition of boron-containing interelement compounds across the C–C triple bond in alkynyloates, in which the electrophilic boron moiety was installed onto the electropositive carbon β to the carbonyl group to form a C(sp2)–B bond.125 As a continuing work, the Sawamura group succeeded in the phosphine-catalyzed trans-hydroboration of alkynoic acid derivatives, providing the β-(E)-alkenylboronates (Scheme 33).126 Among various tertiary phosphines, the less bulky and electron-donor PBu3 (17) or PMe3 (17′) (2.5–20 mol%) showed the best catalytic performance with anti-selectivity. The reaction of a broad range of β-substituted alkynoates, alkynylamides, or alkynylazoles with HBpin afforded the β-borylated (E)-vinyl products in 54–99% yield at 25–60 °C, and various reducible groups were tolerant. It is noteworthy that the β-alkyl substituents on the alkyne substrates led to lower anti-selectivity, while primary and secondary alkynylamides were barely reactive.
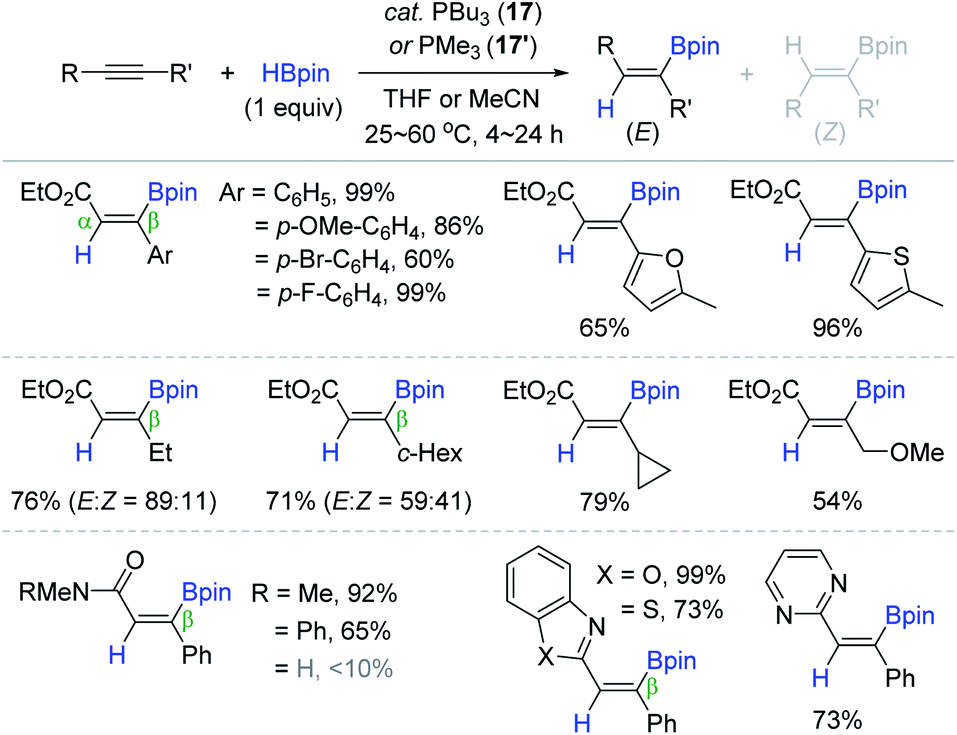 | ||
| Scheme 33 Trialkylphosphine-catalyzed trans-hydroboration of alkynoic acid derivatives. | ||
Based on their earlier work,125,127,128 the catalytic pathway for the phosphine-catalyzed trans-hydroboration was proposed (Scheme 34). Initially, the alkyne is assumed to undergo conjugate addition of the phosphine (17 or 17′) to the electropositive β-carbon to form zwitterionic allene intermediate 17a. The intramolecular hydride transfer to the allenyl central carbon in 17a generates ylide intermediates 17b and 17c. The ylide carbon in 17c is presumed to attack the proximal boron atom to form the corresponding cyclic borate 17d. Finally, the phosphine catalyst is assumed to be regenerated with the release of the anti-hydroboration product upon concerted B–X (X = O, N) bond cleavage associated with the electron rearrangement on 17d. When the Lewis acidity of a boron center in 17d is lowered by β-alkyl-substituents, 17d can be isomerized to 17d′ to eventually bring about a mixture of anti/syn-products.
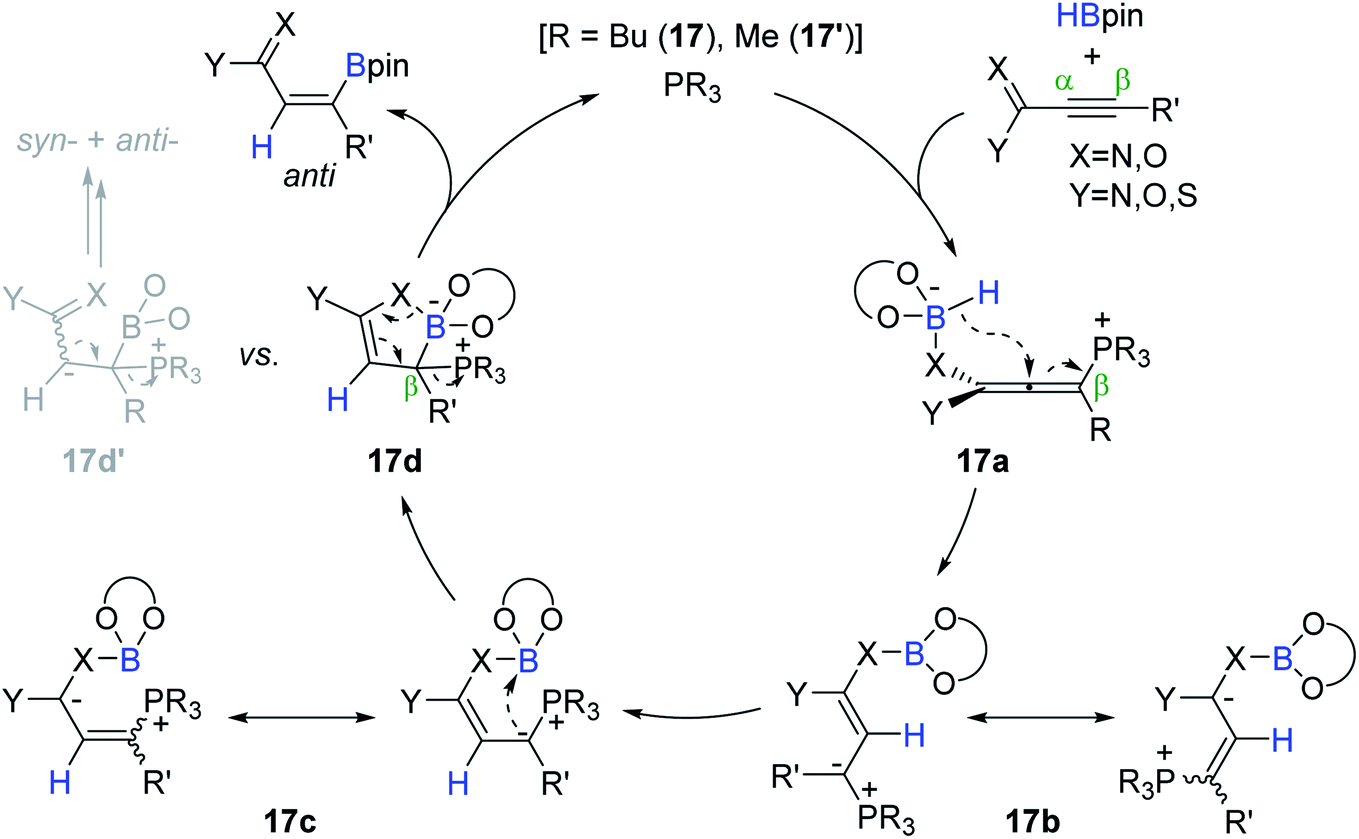 | ||
| Scheme 34 Proposed pathway for the phosphine-catalyzed hydroboration of alkynoic acid derivatives. | ||
Shortly after Sawamura's work, Santos et al. also reported the 17-catalyzed trans-hydroboration of alkynoate esters and alkynylamides.129 They reported similar reaction features to that observed previously. For example, the alkyl-substituted alkynoates at the β-position were converted to the β-borylacrylates with decreased (E)-selectivity, and the secondary alkynylamides were unreactive within the phosphine catalytic system.
Together with the transition metal-free organocatalysts for the hydroboration of alkynes, environmentally benign and commercially useful main-group catalysts based on Al,90,130–139 Li,138–142 and Mg143,144 have been recently developed as alternatives to transition metal catalysts.145 In 2016, Roesky et al. reported the cis-hydroboration of alkynes catalyzed by a neutral DEPNacNacAlH2 (18).90 This work represents the first hydroboration reaction catalyzed by a Group 13-based complex. A broad range of terminal alkynes reacted with HBpin in CDCl3 in the presence of 18 (3 mol%) to afford the β-trans-vinylboronate esters in good yields in 12 h. Notably, the hydroboration of ethynylcyclopropane was sluggish (82% in 32 h in neat), and the reaction of internal alkynes did not proceed at all. Similar to Roesky's work, Ma and Yang prepared DEPNacNac-ligated Al and Sn complexes (AlMe2 18′, AlEt2 18′′, SnCl 19, and SnCl3 19′) and demonstrated their good catalytic efficiency towards the cis-hydroboration of terminal alkynes (Scheme 35a).130
 | ||
| Scheme 35 (a) DEPNacNac-ligated Al or Sn complex-catalyzed cis-hydroboration of terminal alkynes and (b) proposed catalytic working mode of DEPNacNac-ligated AlH2. | ||
Based on DFT calculations, Roesky et al. proposed a catalytic pathway for the Al dihydride 18-mediated hydroboration of phenylacetylene (Scheme 35b). The key step is suggested to involve the turnover-liming, dehydrogenative σ-bond metathesis between the Al–H in 18 and PhCC(sp)–H bonds, leading to aluminum acetylide 18a. The alkynyl unit in 18a is assumed to undergo syn-addition of HBpin to generate the alkenyl aluminium hydride 18b, which likely reacts with the second molecule of phenylacetylene to release the (E)-vinylborane with the regeneration of the 18a active species. Although the catalytic working modes of 18′, 18′′, 19, and 19′′ remain unknown, the almost same yields by these catalysts indicate their similar reactivities towards alkyne hydroboration.130
Inspired by Roesky's NacNac-ligated Al(III) dihydride 18, Nembenna et al. recently synthesized a bidentate bis-guanidinate-ligated Al(III) dihydride complex 20 as a multifunctional catalyst for the reduction of unsaturated functions including alkynes.131 The reactions of various terminal alkynes with HBpin (1 equiv.) at 60 °C produced the β-trans-vinylboronate esters in 40–82% yield over 12 h in neat conditions (Scheme 36a). Notably, catalyst 20 enabled the hydroboration of internal alkynes, giving the (Z)-vinyl products in 30–40% yield, which could not be realized with the catalytic system of 18.90 The working mode of 20 in the hydroboration of terminal alkynes is assumed to involve aluminum acetylide species 20a, which is the same as that proposed by Roesky.90 However, the mechanism of the internal alkyne hydroboration is presumed to be associated with insertion of the alkyne into the Al–H bond of 20 as the key step to form aluminium vinyl intermediate 20b, as previously proposed by Cowley and Thomas.132 Subsequently, 20b reacts with HBpin via transmetallation to liberate the (Z)-vinylboronate ester and active catalyst 20 (Scheme 36b). In fact, 20a and 20b were observed in the stoichiometric reactions of 20 with phenylacetylene and 1-phenyl-1-propyne, respectively.
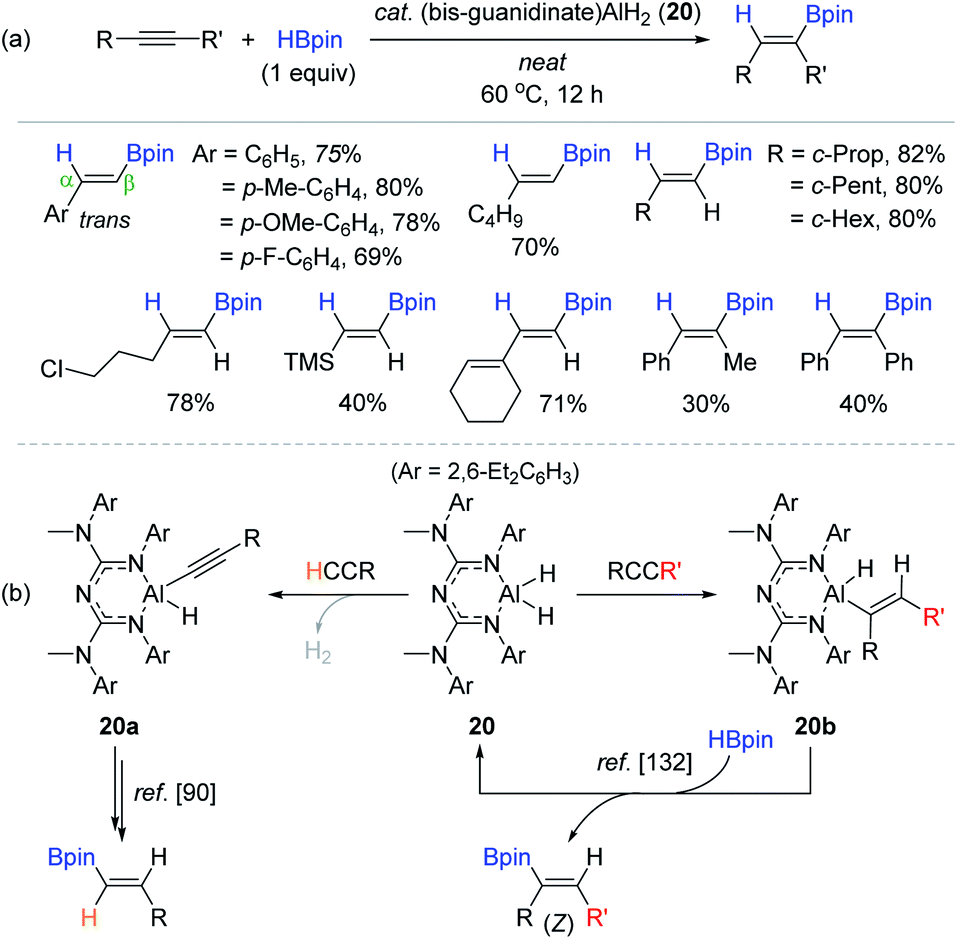 | ||
| Scheme 36 (a) Bis-guanidinate-ligated Al dihydride-catalyzed cis-hydroboration of alkynes and (b) its dual catalytic working modes depending on the type of alkyne substrate (terminal vs. internal). | ||
As another example of the N,N-bidentate ligand-based Al complex, Panda et al. synthesized an aluminum dimethyl complex supported by a functionalized amidophosphine ligand [κ2-{Ph2P(Se)NCH2(C5H4N)}AlMe2, 21]146 and applied it as a precatalyst for hydroboration of terminal alkynes (Scheme 37).133 A range of terminal alkynes possessing electronically variable and/or reducible functional moieties were chemoselectively reduced with HBpin to the corresponding anti-Markovnikov alkenyl boronate esters with exclusive trans-selectivity under mild conditions [cat. 21 (3 mol%), toluene, 25 °C, 8 h]. The trans selectivity as the outcome of the syn-addition of HBpin into the alkyne was further confirmed in the hydroboration of 4-bromo-phenylacetylene-d1. As proposed by Thomas and Cowley,132 and Nembenna,131 the 21-catalyzed alkyne hydroboration proceeds via a “hydroalumination-metathesis” mechanism, where an aluminum dihydride (21a) and an aluminium vinyl hydride (21b) are regarded as the key species.
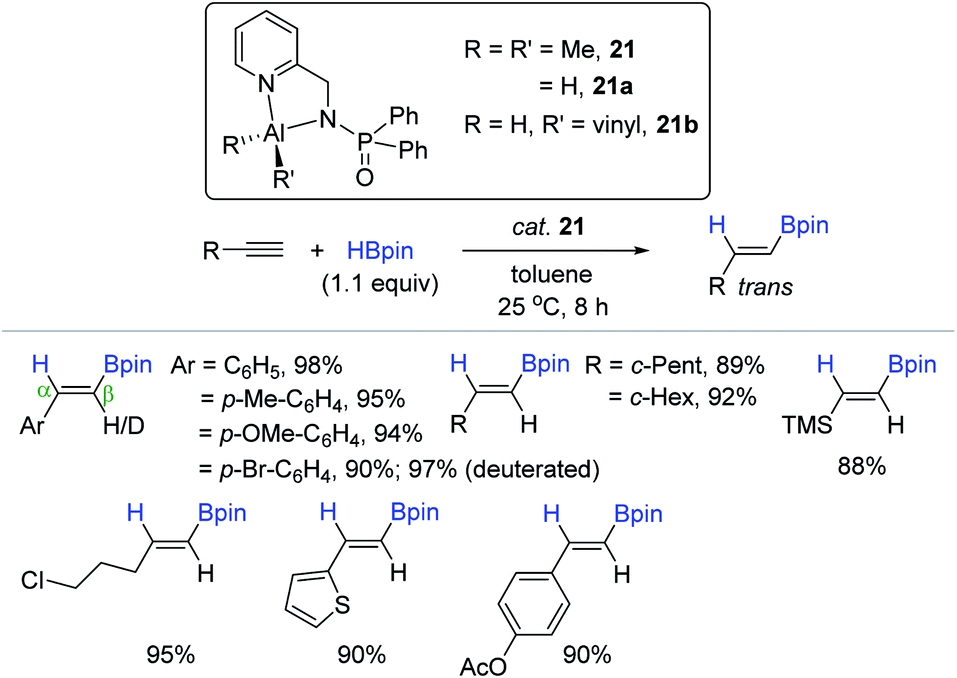 | ||
| Scheme 37 N,N-Bidentate Al(III) complex-catalyzed cis-hydroboration of terminal alkynes. | ||
Together with a range of bidentate Al hydroboration catalysts,90,130,131,133 Cowley recently communicated a series of new aluminium dihydride complexes with bidentate N,P-ligands (22, 22′, and 22′′) as the alkyne hydroboration catalyst.134 These dihydride species (10 mol%) were found to catalyze the hydroboration of phenylacetylene at 110 °C to give the trans-vinylboronate ester with excellent β-selectivity in good yields. It is noteworthy that catalyst 22′′ exhibited a slightly lower catalytic activity (53%) than that (79–83%) of 22–22′. This lower activity was ascribed to the reversible phosphine slip on 22′′ involving an isomer of κ2-N,N geometry (κ2-N,N 22′′) during the catalysis (Scheme 38).
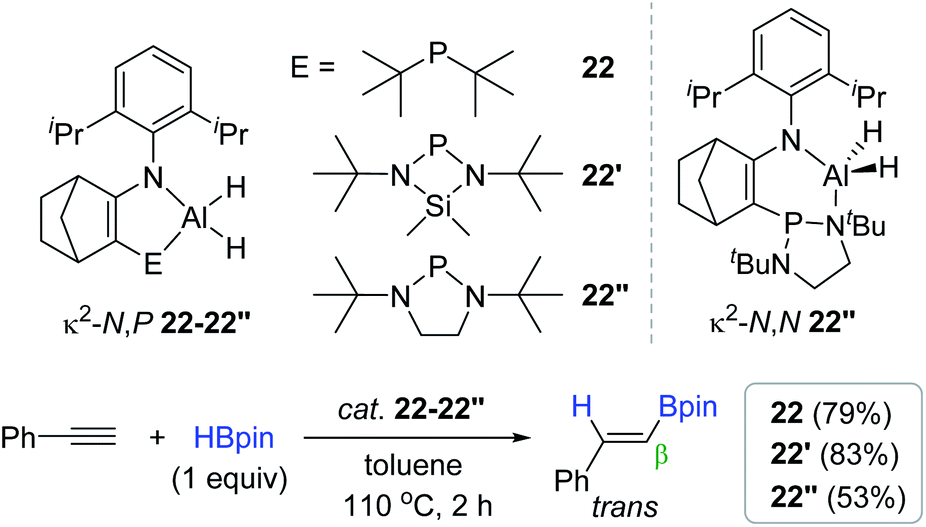 | ||
| Scheme 38 N,P-Bidentate Al(III) complex-catalyzed cis-hydroboration of phenylacetylene. | ||
Inoue synthesized a monodentate N-heterocyclic imine-stabilized dimeric aluminium dihydride [MesLNAlH2]2 23 (MesLN = 1,3-dimesityl-imidazolin-2-imino, Mes = 2,4,6-trimethylphenyl) and utilized it as a catalyst for the hydroboration of alkynes. The reaction of electron-neutral or -rich terminal arylalkynes proceeded with good efficiency in the presence of 23 (4 mol%) at 80 °C to afford the β-trans-vinylboronate esters, while an electron-deficient arylalkyne was slowly converted to the desired vinyl product (40% in 40 h) (Scheme 39).135 In the proposed catalytic cycle, 23 is assumed to be transformed to an alkynyl aluminium species, which is similar to Roesky's catalytic working mode.90
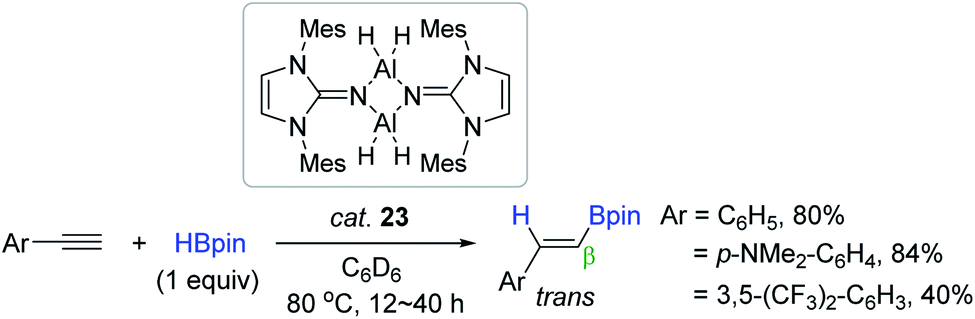 | ||
| Scheme 39 N-heterocyclic imine-ligated Al dihydride-catalyzed cis-hydroboration of terminal alkynes. | ||
Dub and coworkers were interested in terpyridine (tpy)-based redox active Al complexes147 and finally serendipitously synthesized a tridentate tpy-ligated aluminium dialkyl complex (24)136 that uniquely featured a zwitterionic Meisenheimer-type nature.148 The stoichiometric reaction of 24 with 2 equivalents of HBpin at 25 °C generated complex 24a with the byproduct (pinBCH2SiMe3) formation (Scheme 40a). Based on this result and computational studies, species 24a was assumed to be converted to the aluminium (di)hydride species (24b–c) via consecutive metathesis processes of 24a with excess HBpin, which was active species for hydroboration of alkynes. Indeed, 24 was shown to serve as a precatalyst for the cis-hydroboration of alkynes at 80 °C, providing a range of β-trans- or (Z)-vinylboronate esters in 84–96% yield with remarkable functional group tolerance. It needs to be emphasized that precatalyst 24 displayed a TON of 910, the highest among the Al-catalyzed alkyne hydroboration reactions. The hydroboration of alkyne was suggested to proceed via hydroalumination-metathesis processes involving alkenyl aluminium intermediate 24d (Scheme 40b).
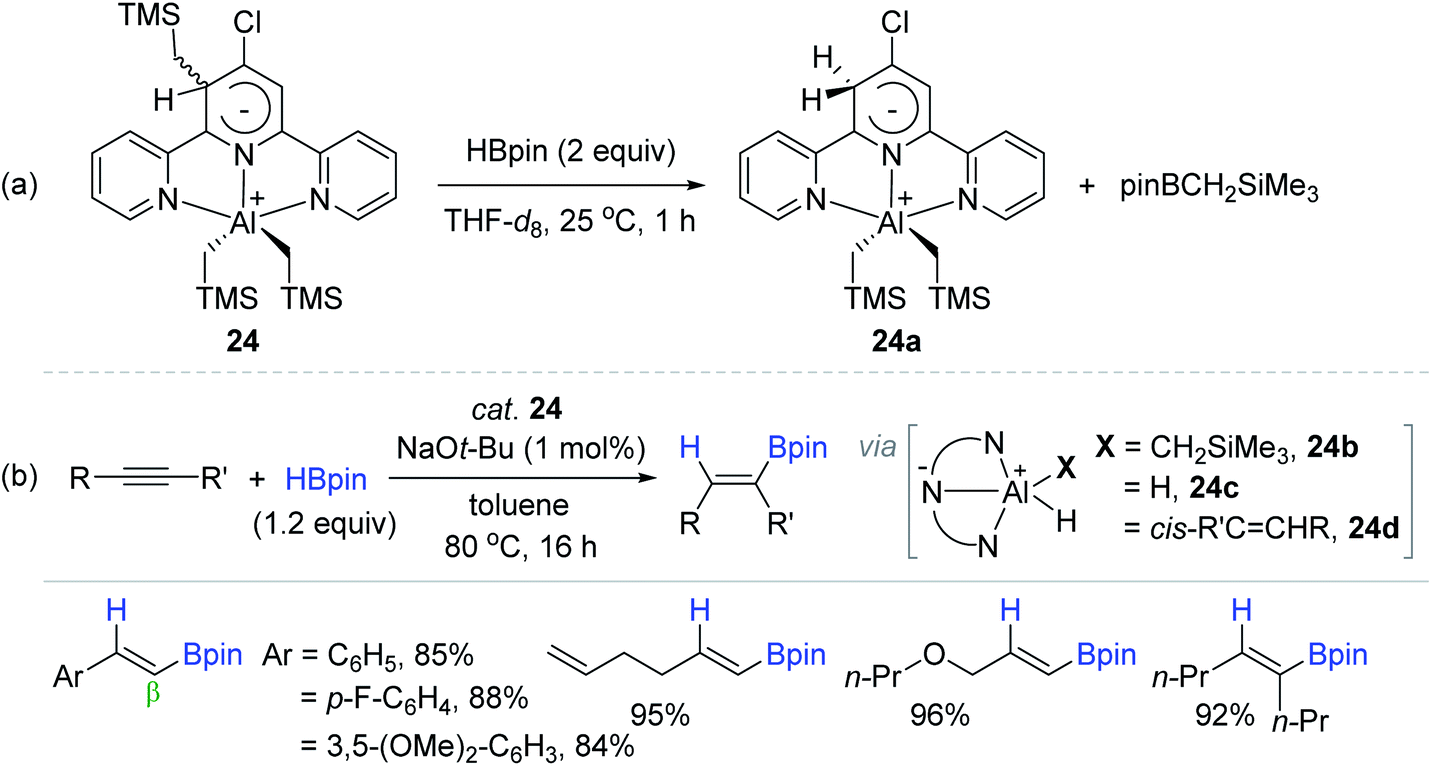 | ||
| Scheme 40 (a) Stoichiometric reaction of the zwitterionic Meisenheimer Al(III) complex with HBpin, and (b) its catalytic scope towards the cis-hydroboration of alkynes. tpy = terpyridine. | ||
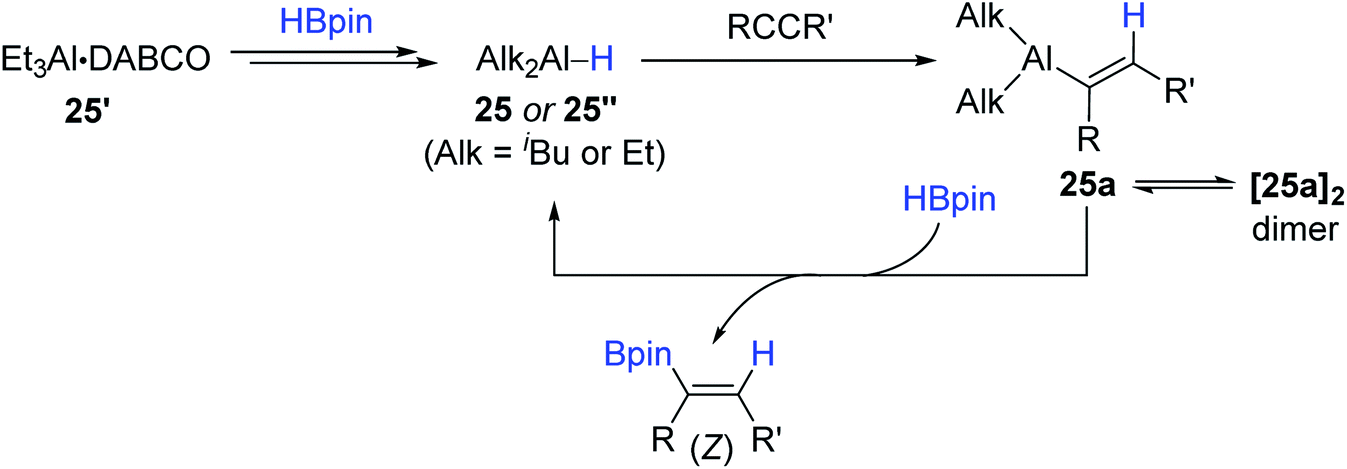
Considering the practicality of the Al catalytic procedure, Thomas and Cowley developed a more convenient aluminium catalytic system for alkyne hydroboration by employing the commercially available diisobutylaluminium hydride [DIBAL, (iBu2AlH)2] 25 and bench-stable Et3Al·DABCO 25′ (DABCO = 1,4-diazabicyclo[2.2.2]octane) as (pre)catalysts.132 It is noteworthy that these (pre)catalysts required no external ligand(s) in accomplishing the anti-Markovnikov (β), cis-selective hydroboration. Although the hydroboration occurred at 110 °C higher than Roesky's condition (30 °C), the substrate scope was broad, including various terminal and (non)symmetrical internal alkynes with moderate to high catalytic efficiency (Scheme 41).
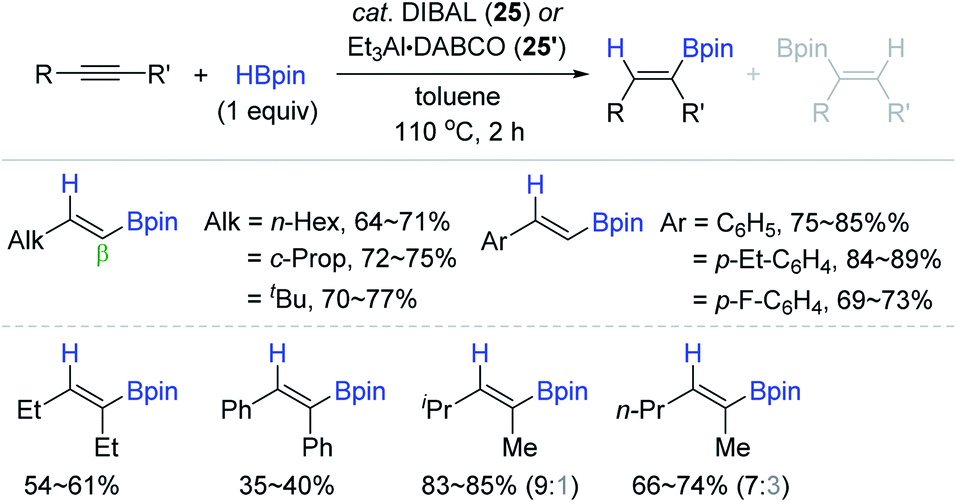 | ||
| Scheme 41 External ligand-free Al(III)-catalyzed cis-hydroboration of alkynes. DIBAL = diisobutylaluminium hydride. | ||
A plausible mechanism for the 25- or 25′-catalyzed alkyne hydroboration was proposed (Scheme 42). Initially, the aluminium hydride, which can be 25 or a hydridoalane 25′′, is assumed to undergo the anti-Markovnikov-selective insertion of an alkyne to form alkenyl aluminum species 25a, which reacts with HBpin via a turnover-limiting σ-bond metathesis to deliver the trans- or (Z)-hydroboration products and regenerate the active catalyst 25 or 25′′. In fact, the stoichiometric reaction of an isolated dimer [25a]2 with HBpin gave the borylated alkene, and 25a turned out to work as a catalyst for the alkyne hydroboration, corroborating the alkyne hydroalumination–σ-bond metathesis mechanism. DABCO as a base on 25′ was suggested to play a role in inhibiting the decomposition of the aluminium catalyst, as evidenced by the outcome from the stoichiometric reaction of Et3Al, HBpin, and DABCO.
 | ||
| Scheme 42 Proposed pathway for the external ligand-free Al(III)-catalyzed cis-hydroboration of alkynes. | ||
Following the work by Thomas and Cowley,132 Shi et al. recently reported the cis-hydroboration of alkynylsilanes catalyzed by the commercially available and external ligand-free AlMe3 (26). The reaction occurred at 80 °C in the presence of 26 (20 mol%) to produce a range of β-(Z)-alkenes bearing both C(sp2)–Si and C(sp2)–B moieties in moderate to high yields in 24 h (Scheme 43).137 HAlMe2 (26a) as an active catalyst for the alkyne hydroboration was suggested to be generated in situ via an σ-bond metathesis reaction between 26 and HBpin.
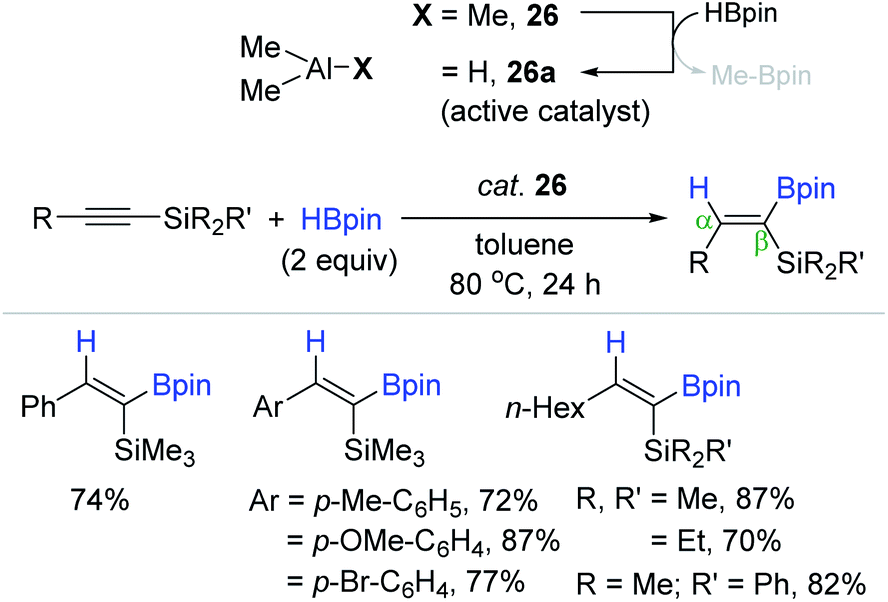 | ||
| Scheme 43 AlMe3-catalyzed cis-hydroboration of alkynylsilanes. | ||
With interest in the possible influence of the lithium element within bimetallic Al–Li catalysts,149–151 Mulvey compared the catalyst reactivity for the alkyne hydroboration between neutral monometallic Al complexes (27 and 27′) and their corresponding anionic bimetallic lithium aluminates (28 and 28′) (Scheme 44a).138 Under identical catalytic conditions [cat. Al (10 mol%), toluene-d8, 110 °C, 2 h], three types of simple alkynes were examined for hydroboration, revealing that the lithium aluminates 27′ and 28′ exhibited superior reactivity to phenylacetylene and 1-phenyl-propyne, respectively, when compared in parallel to their counterparts of monometallic aluminium catalysts (27 and 28). However, catalyst 28 was found to be more active than 28′ in the hydroboration of diphenylacetylene. The lower reactivity of 28′ was attributed to the steric repulsion between the isobutyl moiety on 28′ and phenyl group on the substrate (Scheme 44b).
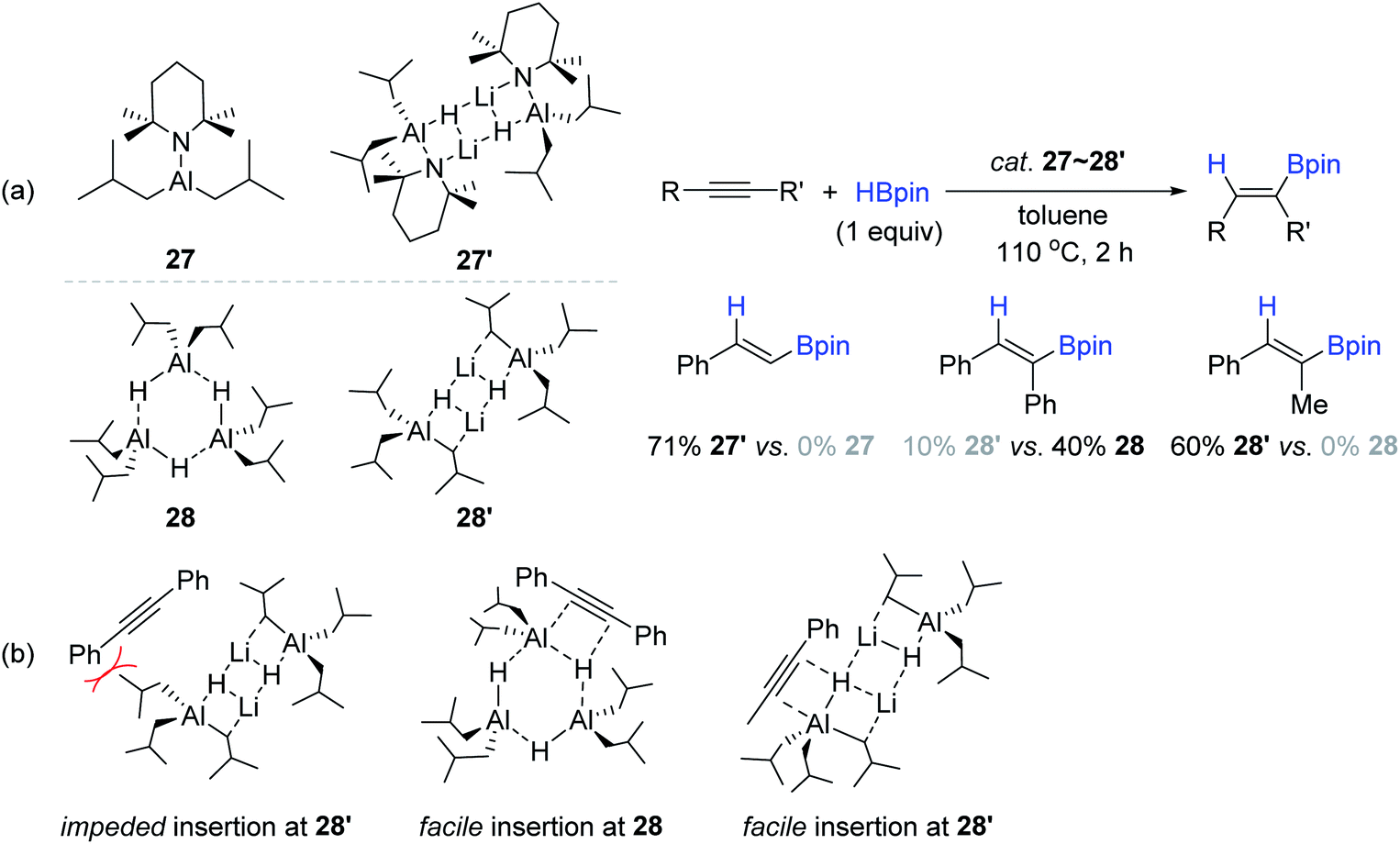 | ||
| Scheme 44 (a) Neutral or anionic Al(III)-catalyzed cis-hydroboration of alkynes, and (b) comparative insertion modes of alkynes into the Al–H bonds. | ||
Based on the report by Mulvey, the An group successfully applied lithium diisobutyl-tert-butoxyaluminum hydride (LDBBA, 29) as a bimetallic lithium aluminate catalyst for the hydroboration of alkynes.139 The reactions of a range of alkynes occurred in neat conditions at 50 °C with catalyst 29 (5 mol%) to furnish the corresponding β-trans-alkenyl boronates in 46–98% yield (Scheme 45). Electron-deficient or internal alkynes were less reactive relative to electron-rich or terminal alkynes, giving moderate yields of the desired vinyl products. It is noteworthy that 29 displayed higher hydroboration activity compared to a series of neutral or anionic Al-based (pre)catalysts including DIBAL 25 and SDBBA (sodium diisobutyl-tert-butoxyaluminum hydride). As proposed by the Roesky group,90 the catalytic pathway was suggested to involve the dehydrogenative C–H alumination of a terminal alkyne to generate an acetylide aluminium species (29a) in the initiation step.
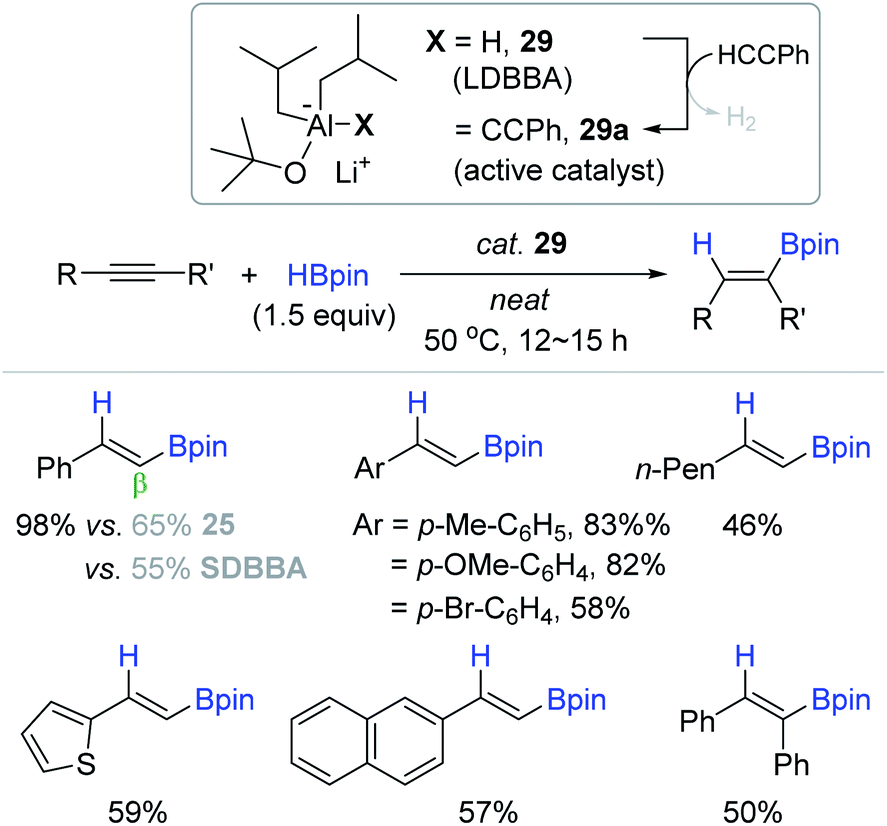 | ||
| Scheme 45 Anionic Al(III)-catalyzed cis-hydroboration of alkynes. LDBBA = lithium diisobutyl-tert-butoxyaluminum hydride; SDBBA = sodium diisobutyl-tert-butoxyaluminum hydride. | ||
Recently, the groups of Xue140 and Xu141 independently demonstrated that highly basic and commercially available n-BuLi (30) catalyzes the cis-hydroboration of terminal and internal alkynes to produce the β-trans-alkenyl boronic esters (Scheme 46). Under the n-BuLi (30) catalytic system, terminal alkynes were hydroborated with HBpin in neat conditions at 25 °C, while the hydroboration of (un)symmetrical internal alkynes occurred at 130 °C to give a mixture of regio- or stereoisomers of the vinyl products. It is interesting to see that the regioselectivity was improved from 3:1 to 50:1 with an increase in the steric difference between the two substituents on the unsymmetrical internal alkynes.
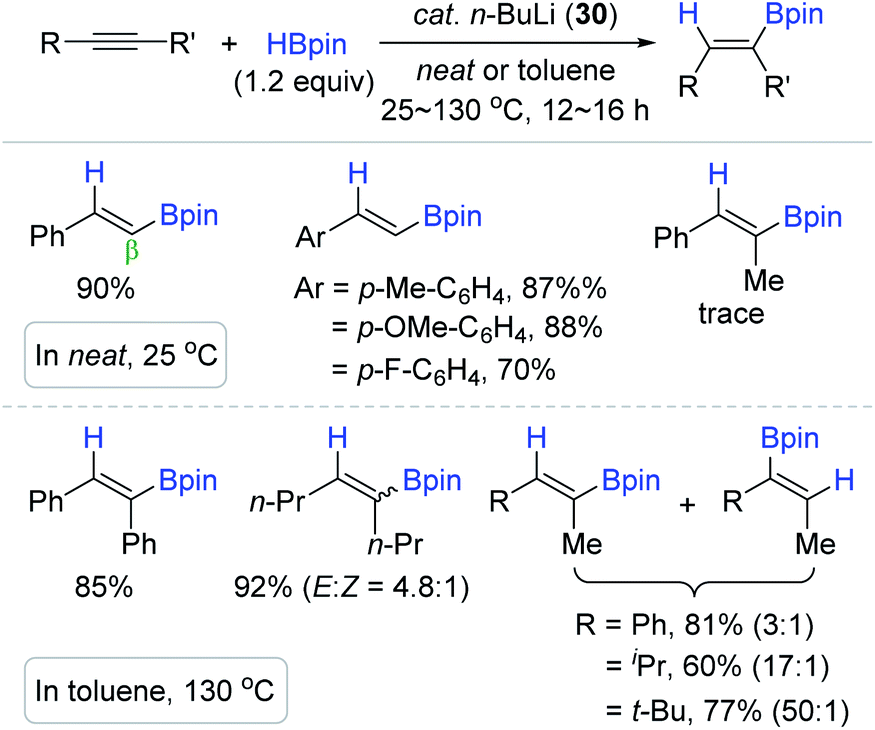 | ||
| Scheme 46 n-BuLi-catalyzed cis-hydroboration of alkynes. | ||
Although n-BuLi (30) worked as a hydroboration catalyst, its instability and inferior functional group compatibility were problematic. In this regard, Sen et al. applied sterically protected lithium complexes [2,6-ditertbutyl phenolatelithium (31) and 1,1′-dilithioferrocene (32)] as the alkyne hydroboration catalyst (Scheme 47).142 The reactions proceeded at 100 °C with 2 mol% of catalyst 31 or 32 to provide the β-trans-alkenyl boronate esters in good to high yields in 12 h. However, internal alkynes were relatively less reactive (up to ∼50% yield), and halogen substituents on the alkyne substrates partially underwent lithium–halide exchange, resulting in a decrease in catalytic efficiency.
 | ||
| Scheme 47 Sterically demanding Li complex-catalyzed cis-hydroboration of alkynes. | ||
Based on DFT calculations, the 31-mediated reaction mechanism was proposed (Scheme 48). Initially, a lithium cation in 31 weakly coordinates to the oxygen atom of HBpin to form adduct 31a, which is calculated to be more stable than lithium borate salt 31a′. The polarized H–B bond in 31a is presumed to insert into the alkyne in an anti-Markovnikov manner via transition state 31b (ΔG‡ = 35.4 kcal mol−1) to deliver the alkenyl boronate ester with the regeneration of 31. This step is regarded as the rate-determining step (RDS).
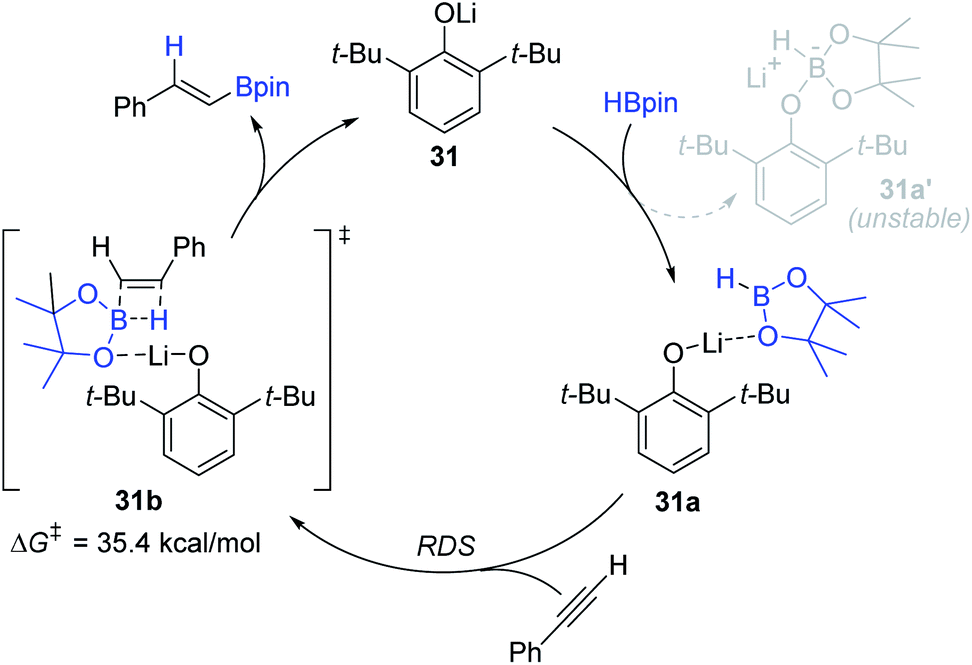 | ||
| Scheme 48 Proposed pathway for the Li-catalyzed hydroboration of alkynes. | ||
In parallel with aluminium-based catalysts, a few Mg complexes were shown to catalyze the cis-hydroboration of alkynes.143,144 In 2018, Ma et al. synthesized a dimeric Mg(I) complex with unsymmetrical ArNacNac ligands as a hydroboration catalyst (33).143 Notably, the dimeric Mg(I) complex 33 was prepared through the reduction of ArNacNacMg(II) halide with excess sodium metal. Also, 33 (5 mol%) with HBpin (1.5 equiv.) was found to enable hydroboration of terminal alkynes, producing the β-trans-alkenyl boronates in excellent yields at 110 °C. However, the dimeric Mg(I) with the symmetrical ligand (DIPPNacNac) displayed a slightly lower catalytic performance in the hydroboration of phenylacetylene [88% (not-shown) vs. 99% 33] (Scheme 49). This work represents the first Mg(I)-catalyzed hydroboration of alkynes.
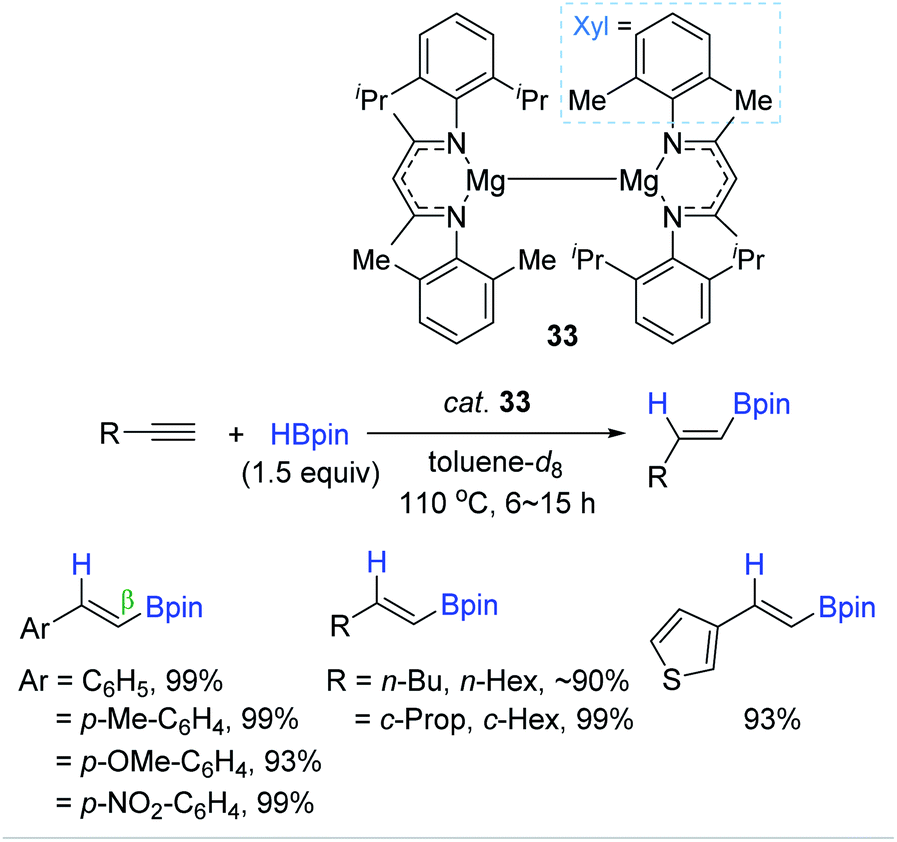 | ||
| Scheme 49 Unsymmetrical NacNac-ligated dimeric Mg(I)-catalyzed cis-hydroboration of terminal alkynes. | ||
Based on experimental observations in combination with computational studies, a catalytic pathway for the 33-promoted hydroboration of terminal alkynes was proposed (Scheme 50). Specifically, 33 is assumed to react with HBpin to lead to a dimeric Mg boryloxide complex (33a).152 Indeed, 33a was isolated as crystals during the catalysis. Species 33a is subsequently transformed to monomeric Mg hydride 33b with HBpin. Subsequently, 33b as the active catalyst likely undergoes turnover-liming syn-addition of the alkyne (R = Ph, ΔG‡ = 31.3 kcal mol−1) to form 33c, which is further metathesized with HBpin to release the trans-vinyl product (R = Ph, ΔG‡ = 27.8 kcal mol−1) with the regeneration of 33b.
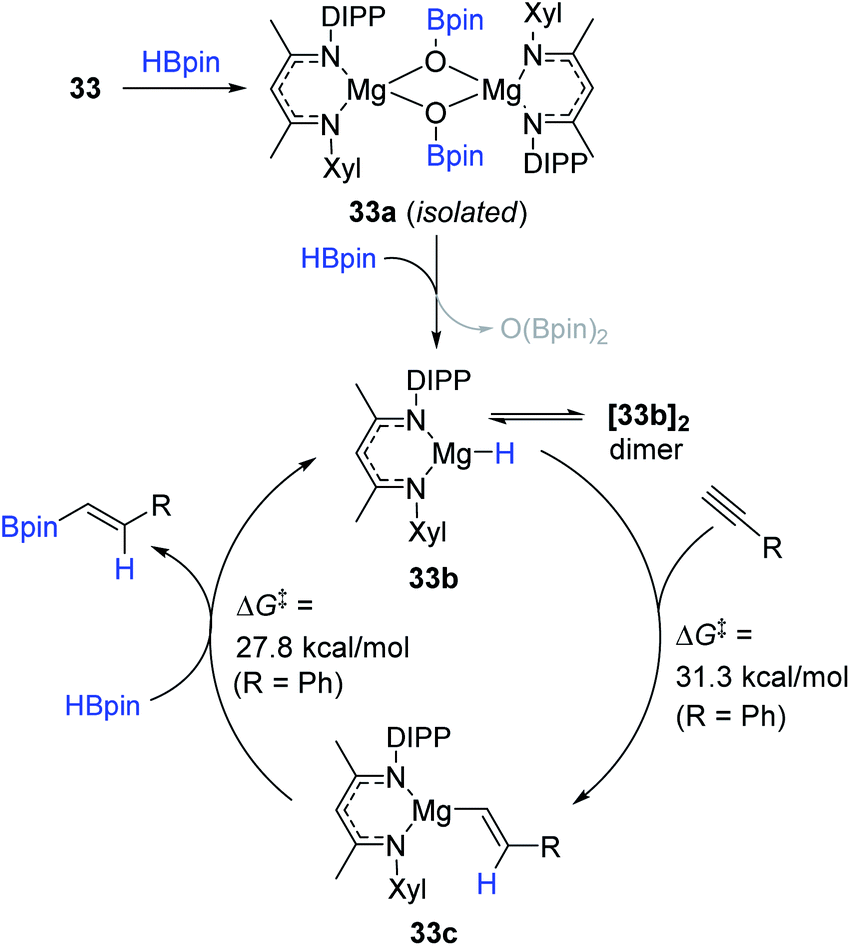 | ||
| Scheme 50 Proposed pathway for the Mg(I)-catalyzed hydroboration of terminal alkynes. | ||
Motivated by Ma's work, where the Mg(II) hydride was regarded as the active catalytic species, Rueping et al. reported a simpler and external ligand-free Mg catalyst [MgBu2 (34)] for alkyne hydroboration.144 Precatalyst 34 (7–10 mol%) with HBpin catalyzed the hydroboration of a broad range of alkynes at 80 °C with wide functional compatibility to alkoxy, amino, halide, alkenyl, and hydroxy groups, giving the β-trans-vinylboronate esters in good to high yields (Scheme 51). As observed in the lithium catalysis,141 the regioselectivity in the case of unsymmetrical internal alkynes was improved from 1.5:1 up to >99:1 by increasing the differential steric effect of the substituents (R and R′).
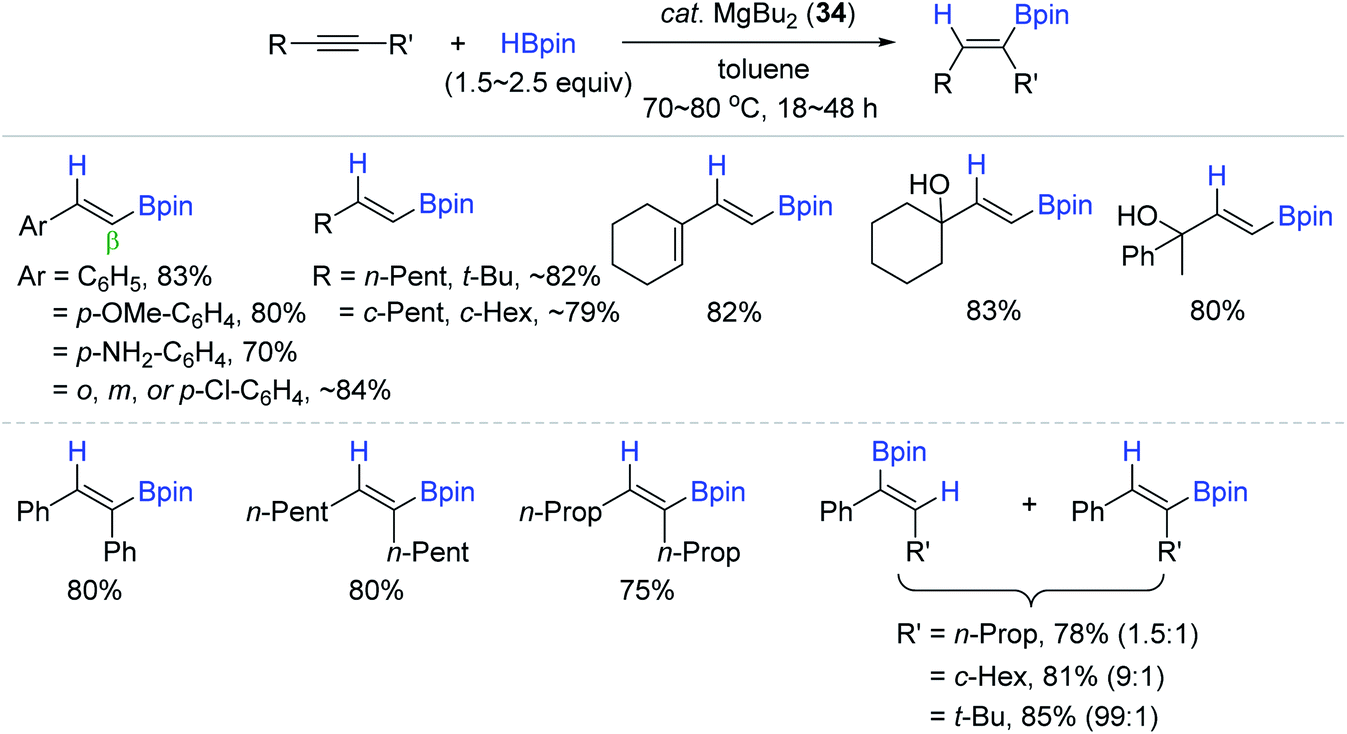 | ||
| Scheme 51 MgBu2-catalyzed cis-hydroboration of alkynes. | ||
Based on stoichiometric reactions and DFT calculations, a catalytic cycle for 34-promoted alkyne hydroboration was proposed (Scheme 52). Initially, MgBu2 in the presence of HBpin likely prefers to be BuMgH (34a) over an alkynyl MgBu (34a′), as predicted by the computational study (R = Ph, ΔΔG‡ = 16.5 kcal mol−1). Indeed, the stoichiometric reaction of 34 and excess HBpin was found to generate 34a. Specifically, 34a is assumed to undergo β-selective, syn-addition of an alkyne to afford Mg-vinyl intermediate 34b. The next step is suggested to involve the inner-sphere nucleophilic migration of the vinyl moiety to the coordinated HBpin to form zwitterionic intermediate 34c, which is subsequently isomerized to BuMg–H species 34d via hydride migration from the boron to magnesium centers. These inner-sphere migration processes were calculated to require activation energy of less than 10 kcal mol−1, and thus are highly facile (R = Ph). The vinyl product is suggested to be liberated from 34d with the regeneration of 34a.
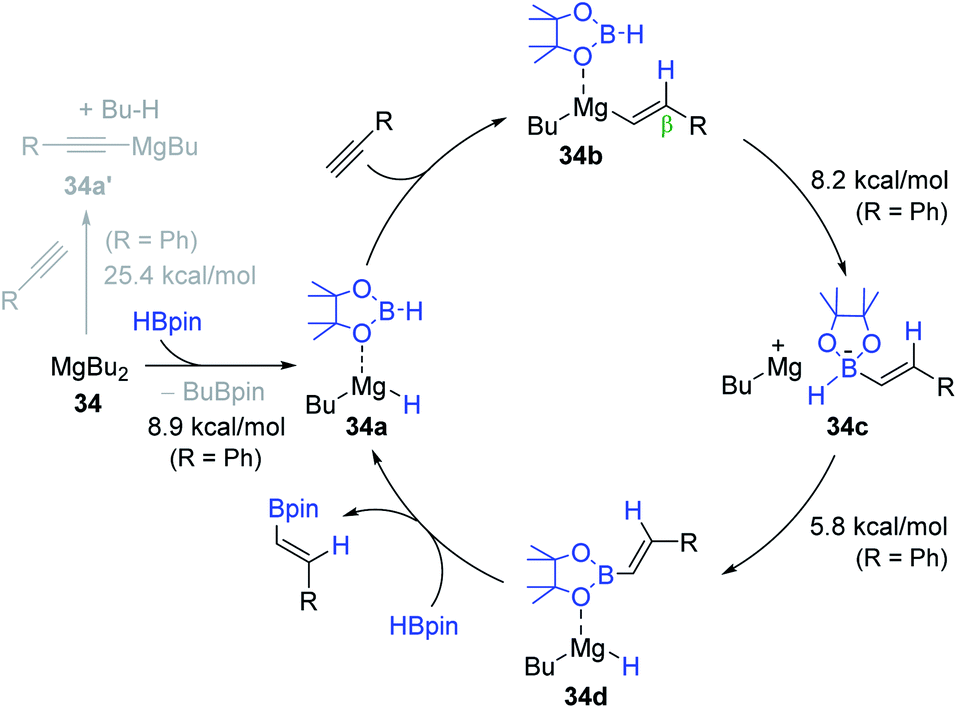 | ||
| Scheme 52 Proposed pathway for the MgBu2-mediated hydroboration of terminal alkynes. | ||
Recently, Thomas et al. re-investigated the alkyne hydroboration promoted by a series of nucleophiles with strong focus on the elucidation of the dominant active catalytic species.153 Surprisingly, the results suggested that several nucleophilic (pre)catalysts promote the decomposition of HBpin to generate BH3 and its borohydride species in situ, which are potentially active species for the alkyne hydroboration. Among the tested (pre)catalysts, particularly NaOt-Bu, MgBu2 (34), and n-BuLi (30) indeed turned out to serve as a promoter only for the HBpin decomposition, not for the main hydroboration process.
4. Hydrostannation and hydrogermylation
Alkenylstannes are valuable synthetic intermediates, allowing mild cross-coupling reactions and leading to C(sp2)–C(sp2) bonds with high functional group tolerance,7,15 and thus are applied in the synthesis of densely functionalized (un)natural compounds.154–158 In this regard, the transition metal-catalyzed hydrostannation of alkynes providing alkenylstannes has been extensively studied to date,23–25 while the transition metal-free catalysts have been relatively less highlighted, and thus are detailed in this section (vide infra). In 1998, Yamamoto et al. reported the B(C6F5)3 2′-catalyzed hydrostannation of a range of terminal and internal alkynes with Bu3SnH, in situ generated from Bu3SnCl and Et3SiH in one pot. This study represents the first transition metal-free hydrostannation of alkynes. The reactions proceeded at −35–25 °C to give the β-cis-vinylstannes in 70–90% yield via the trans-addition of an Sn–H bond (Scheme 53).159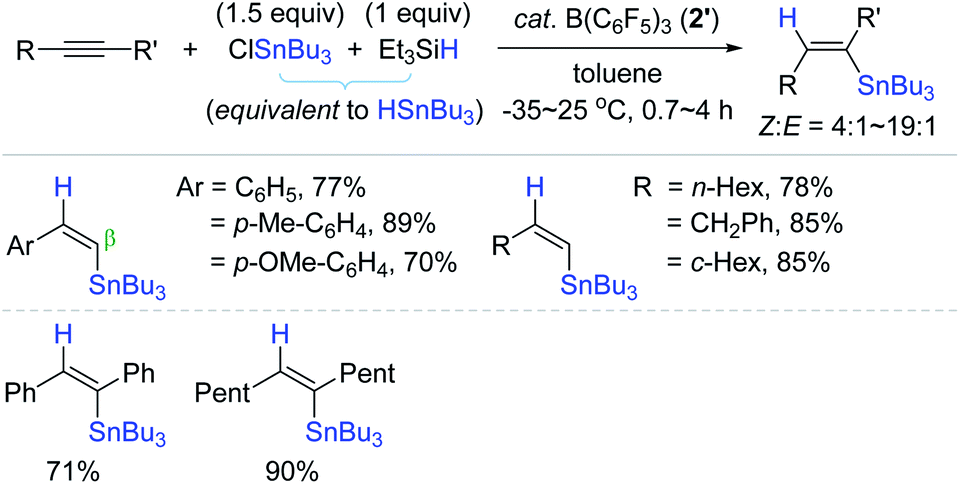 | ||
| Scheme 53 B(C6F5)3-catalyzed trans-hydrostannation of alkynes. | ||
The Maleczka group previously reported the in situ reduction of organotin halides to organotin hydrides involving an organotin oxide intermediate in the presence of aqueous Na2CO3 and PMHS (polymethylhydrosiloxane).160–162 Based on this and Yamamoto's protocol,159 Maleczka et al. developed a one-pot allylation-hydrostannation sequence for the reduction of alkynals, in which tin halide as the initial product was reduced to an organotin hydride, and this tin hydride was subsequently consumed for the hydrostannation of the alkyne unit on the allylated intermediate in the catalytic system of 2′/PMHS (Scheme 54).163 Under the optimal conditions [(i) BF3·OEt2 and allylstannes (1 equiv.), toluene, −35 °C, ∼1.5 h and (ii) B(C6F5)3 (20 mol%), PMHS (2 equiv.), ∼3 days], the sequential reduction of a series of alkynals was performed, demonstrating that this tandem reaction is highly susceptible to both steric and electronic factors. For example, an electron-donating or any substituent close to the aldehyde moiety on the aromatic alkynal substrates halted or slowed down the desired one-pot process, while aliphatic alkynals were less reactive compared to aromatic alkynals, requiring 3 days to achieve ∼40% yield.
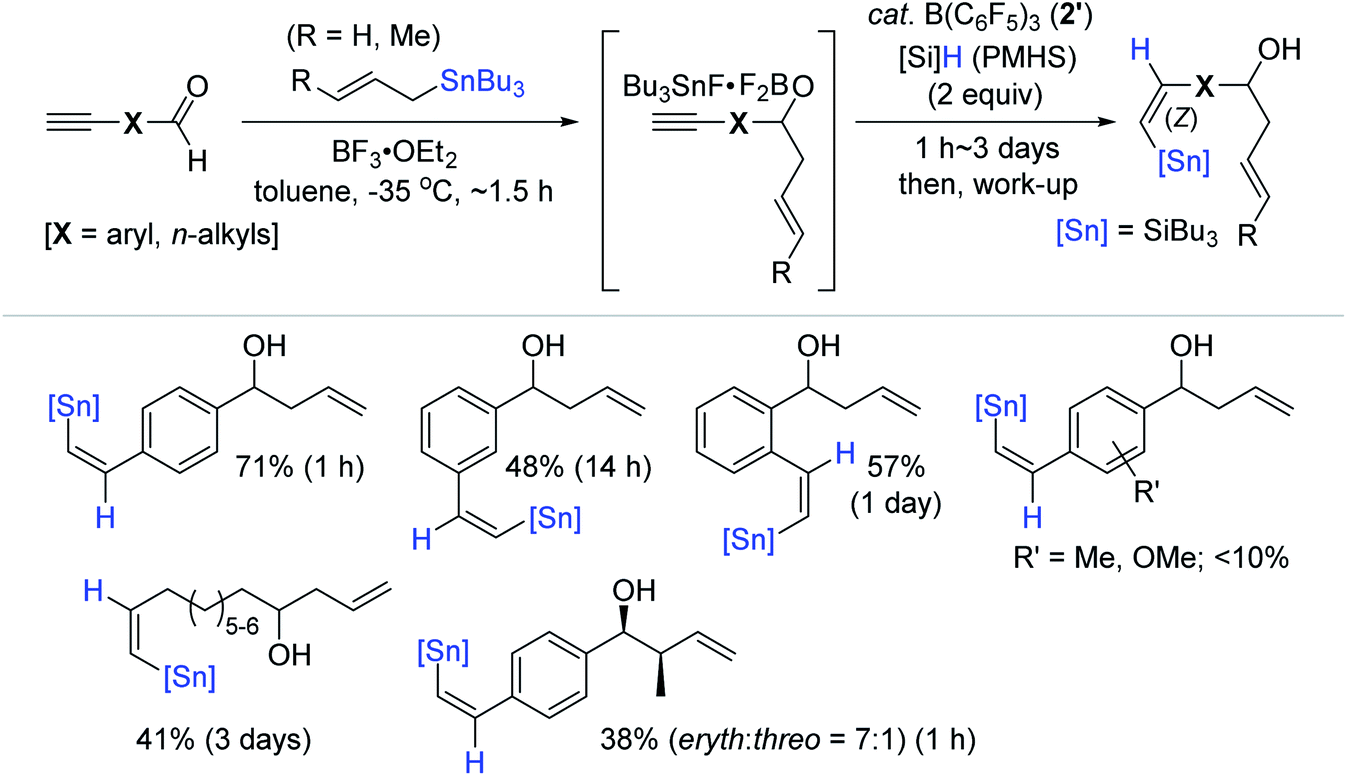 | ||
| Scheme 54 B(C6F5)3-mediated one-pot allylation-hydrostannation of alkynals. | ||
Based on the NMR-monitoring of the reaction progress, a catalytic pathway for the tandem reductive transformation of alkynals was proposed (Scheme 55). Initially, the aldehyde likely forms an adduct with BF3·OEt2, which rapidly undergoes allylation by tributyl(allyl)tin (119Sn δ −18) to form a boryl ether complexed with a stannyl cation species (35a, 119Sn δ 156).164–166 Subsequently, 35a is suggested to react with the B(C6F5)3-PMHS adduct to generate tributyltin hydride (Bu3SnH; 119Sn δ −88),167 which is then trans-added to the alkyne moiety on 35a by the action of 2′ to eventually yield the allylation-hydrostannation (Z)-product (119Sn δ −56).
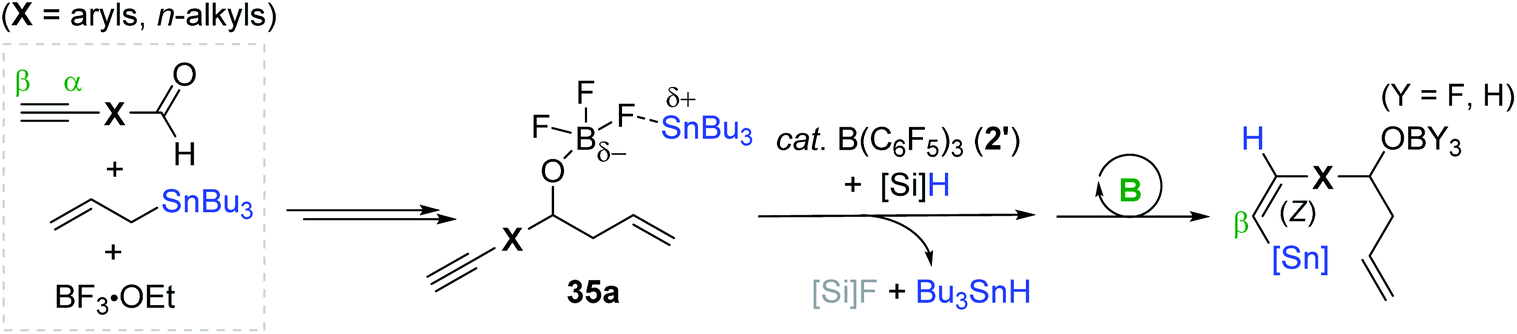 | ||
| Scheme 55 Proposed reaction pathway for the B(C6F5)3-mediated allylation-hydrostannation of alkynals. | ||
Based on Yamamoto's hydrostannation procedure,159 Organ et al. conceived to hydrostannylate complex internal alkynes using catalyst 2′, and found that B(C6F5)3 2′ enabled the regio- and stereoselective hydrostannation of densely functionalized, internal propargylic alcohol derivatives (Scheme 56).168 This work represents the first metal-free catalytic hydrostannation of propargylic alcohols. The reaction of 1-tert-butyldimethylsilyloxy-2-butene with Bu3SnH (2 equiv.) smoothly proceeded at 50 °C in toluene in the presence of B(C6F5)3 2′ (20 mol%) to quantitatively give the β-(Z)-vinylstannane. However, the reactions of hindered alkynes occurred only in THF solvent in high yields rather than in toluene. The observed solvent-specificity of the B(C6F5)3 catalysis was explained by the fact that THF, as a coordinating solvent, assists in stabilizing the stannyl cation (in situ generated) by forming a solvated adduct [Bu3Sn·THF]+ HB(C6F5)3− (35b),169,170 whereas less bulky alkynes in toluene can directly coordinate to the stannyl cation to form adduct 35c. Indeed, 35b was found to be quantitatively generated from the reaction of 2′ with excess Bu3SnH in C6D6/THF (1:1).
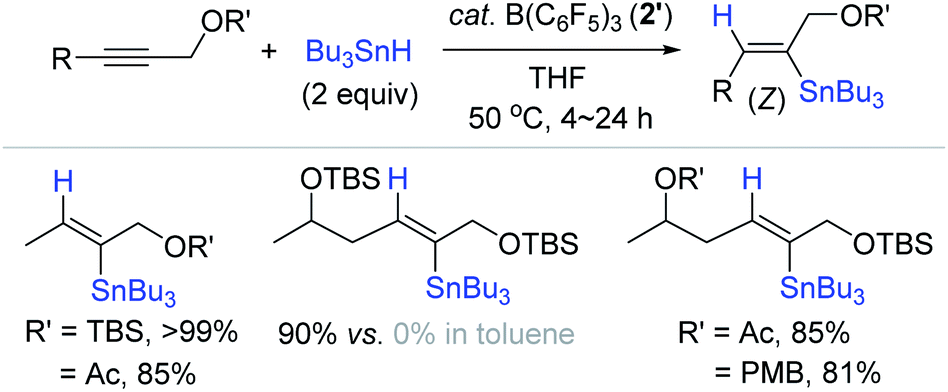 | ||
| Scheme 56 B(C6F5)3-catalyzed trans-hydrostannation of internal propargylic alcohol derivatives. TBS = tert-butyldimethylsilyl, Ac = acyl, PMB = p-methoxybenzyl. | ||
An outer-sphere ionic pathway was proposed for the B(C6F5)3-catalyzed alkyne hydrostannation (Scheme 57). Species 35b or 35c is presumed to insert into the β-position of the alkyne molecule to form a vinylcation bearing a HB(C6F5)3− anion (35d), which is reduced by “Bu3SnH” not HB(C6F5)3− to deliver the hydrostannation product with the regeneration of the stannyl cation. It is noteworthy that the stoichiometric reaction of 35b with 1-tert-butyldimethylsilyloxy-2-butene in the presence of Bu3SnH (2 equiv.) gave the (Z)-vinylstanne in quantitative yield, but no hydrostannation product was observed in the absence of Bu3SnH, corroborating the role of HB(C6F5)3− and Bu3SnH as a spectator counterion and a hydride donor, respectively.171–173
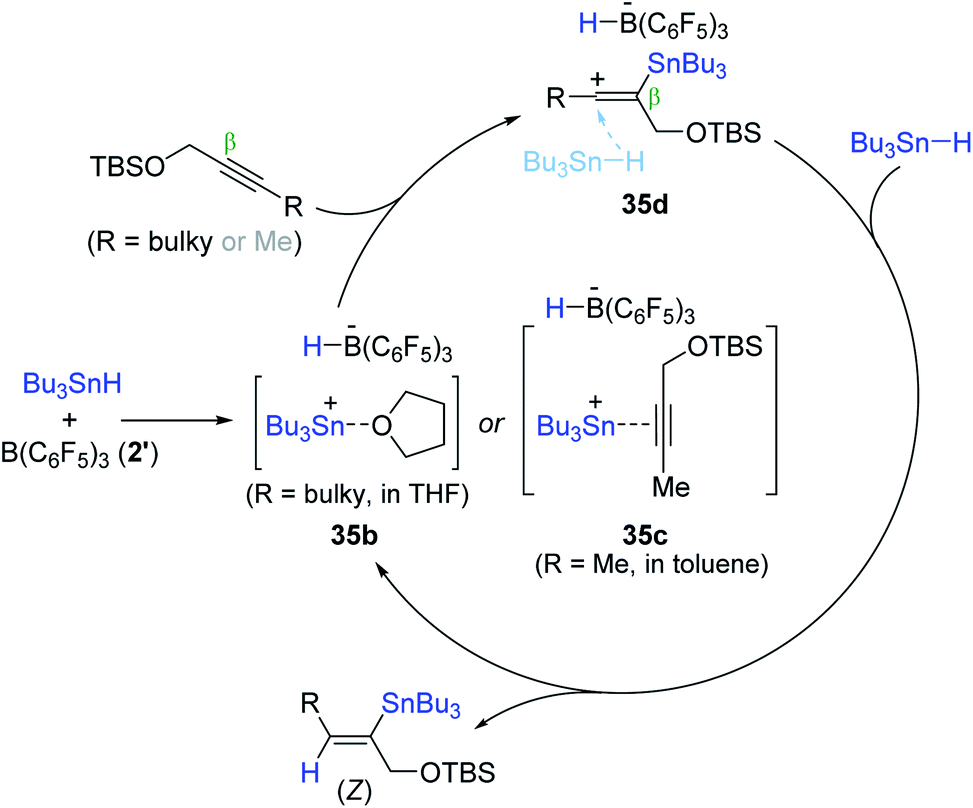 | ||
| Scheme 57 Proposed pathway for the B(C6F5)3-catalyzed hydrostannation of propargylic alcohol derivatives. | ||
Based on Yamamoto and Organ's outer-sphere hydrostannation reactions,159,168 Oestreich et al. reported the catalytic system of [Ph3C]+[B(C6F5)4]− (4)/Bu3SnH, generating a catalytically competent stannyl cation species in situ for the trans-hydrostannation of alkynes.174 The hydrostannation of phenylacetylene derivatives proceeded under mild conditions [cat. 4 (1 mol%), pentane, 0–25 °C, 1–24 h] to furnish the desired β-vinylstannes in 74–96% yield with cis-selectivity ranging from 87% to 95% (Scheme 58). It is interesting that propiolic acid methyl ester cleanly reacted with α-regioselectivity. This catalytic system was well-tolerant of halogen, alkoxy, and ester groups, whereas an amino substituent deactivated the catalyst. Only few alkyl-substituted terminal alkynes were cleanly converted to the corresponding β-cis-vinylstannes. Some of the hindered alkynes underwent the undesirable dehydrogenative C–H stannylation (ca. 10–40%). Interestingly, the C–H stannylation products were not observed under the catalytic system of B(C6F5)3, 2′ (vide supra).159 Internal alkynes were also reduced with excellent (Z)-selectivity, and the regioselectivity in the case of unsymmetrical internal alkynes possessing a phenyl group was well controlled to be β-preferred.
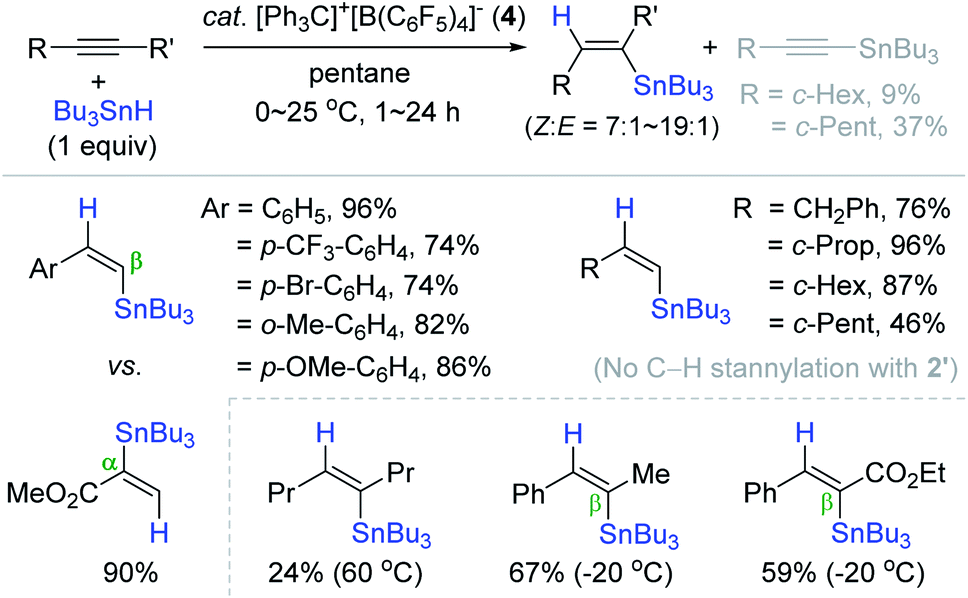 | ||
| Scheme 58 Trityl borate salt-catalyzed trans-hydrostannation of alkynes. | ||
Recently, Reuping et al. successfully expanded the scope of MgBu2 (34) catalysis from hydroboration144 to the hydrostannation of alkynes (Scheme 59).175 A range of (un)symmetrical internal alkynes were cleanly reduced with Bu3SnH at 50 °C in the presence of 34 (10 mol%) to the β-vinylstannes in 70–98% yield with high (Z)-selectivity as the outcome of the anti-addition of H–SnBu3. Intriguingly, an internal alkyne bearing an NMe2 substituent was reduced in the opposite manner of regio- and stereoselectivity to afford the corresponding α-(E)-vinylstanne. Unlike the case of internal alkynes, the hydrostannation of a broad range of terminal alkynes produced the β-trans-products in high yields under similar conditions [cat. 34 (7 mol%), toluene, 70 °C, 16 h]. This indicated that the initially formed cis-hydrostannation product is isomerized to the trans-vinylstannes during the Mg catalysis. Indeed, 34 was found to catalyze the isomerization of cis-vinylstannes to trans-vinylstannes presumably via reversible vinylstanne/organomagnesium exchange, as reported by Still and Seyferth.176,177 In addition to catalyst 34, DIPPNacNacMgBu, 9′′, as a catalyst was shown to improve the β-selectivity up to 12:1 for the hydrostannation of particularly terminal propargylic alcohols, in which the bulky N-aryl ligand on 9′′ was assumed to protect the active Mg center from coordination of the hydroxyl moiety on the substrate.178,179
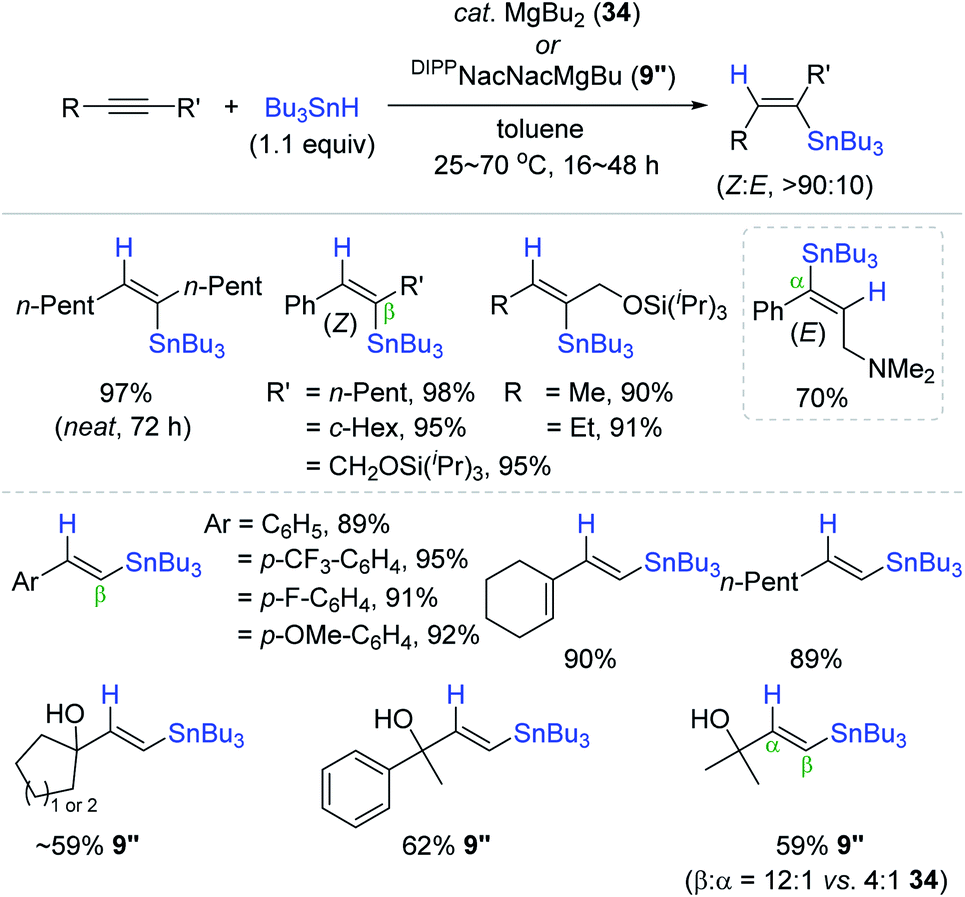 | ||
| Scheme 59 Alkyl Mg(II)-catalyzed hydrostannation of alkynes. | ||
Based on a series of control experiments, a reaction pathway for the Mg(II)-catalyzed hydrostannation of alkynes was proposed (Scheme 60). The present catalysis is suggested to operate via the outer-sphere hydride transfer pathway180–182 rather than an inner-sphere mechanism involving a Mg(II) hydride species.144 Indeed, a Mg hydride (BuMgH) was barely formed in the 1:5 reaction of 34 and Bu3SnH, not supporting the inner-sphere path. Species 34 is assumed to activate either Bu3SnH or the alkyne to form a vinyl carbocation as the key intermediate(s) (34e or 34f/34f′), which is supposed to undergo hydride reduction by the Bu2MgH anion or Bu3SnH to finally deliver the (Z)-vinylstanne. The resultant (Z)-product is likely isomerized to the more thermodynamically stable (E)-isomer in the hydrostannation of terminal alkynes.
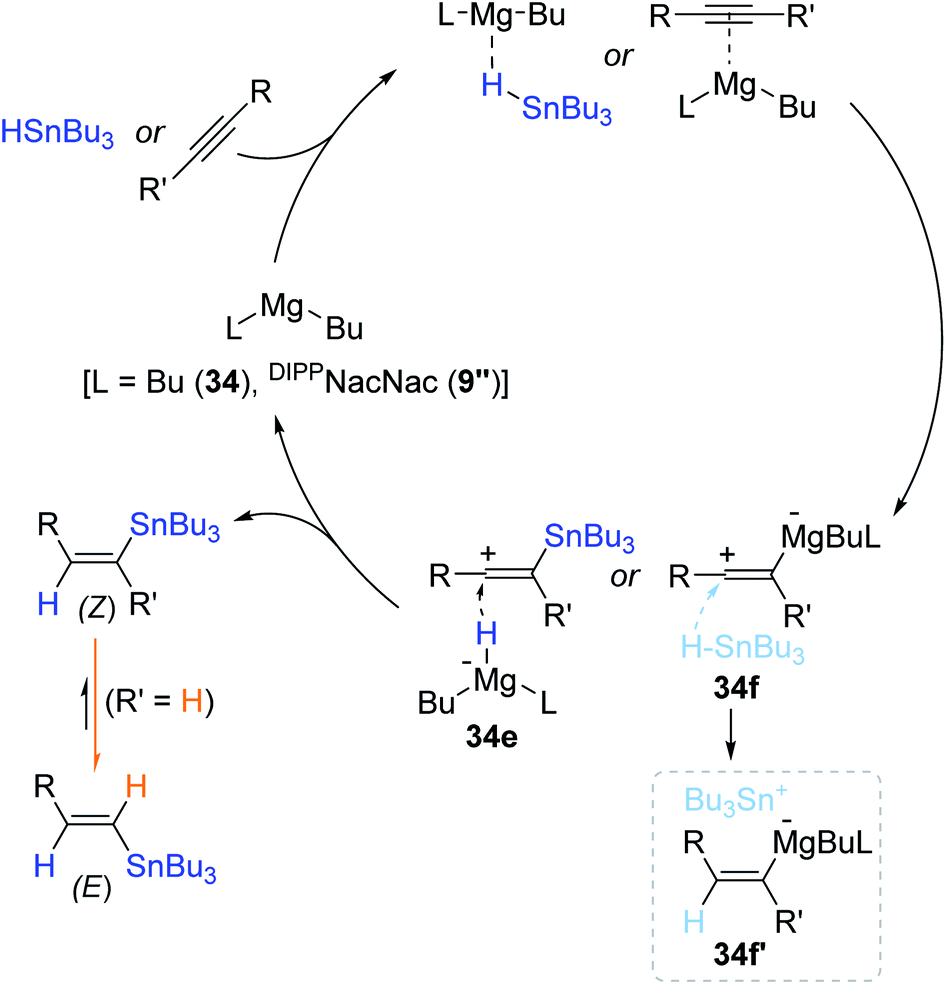 | ||
| Scheme 60 Proposed outer-sphere mechanism of the alkyl Mg(II)-catalyzed hydrostannation of alkynes. | ||
In 2005, Gevorgyan et al. communicated the B(C6F5)3 2′-catalyzed trans-hydrogermylation of alkynes with trialkylgermanes.183 This work represents the first transition metal-free hydrogermylation of alkynes. The hydrogermylation worked over a broad range of unsymmetrical internal alkynes to provide the β-(Z)-vinylgermanes in excellent yields at 25 °C (Scheme 61). It is interesting that electronically-bias internal alkynes dictated the insertion of a germyl moiety exclusively into the position β to a more e−-rich aryl group. This catalysis exhibited functional group tolerance to various reducible groups, which was not observed in the B(C6F5)3-catalyzed hydrosilylation181 and hydrostannation168,175 reactions. It is striking that substituted propiolates were reduced in an exclusively cis-fashion to give the corresponding (E)-vinylgermanes, in all high yields under the same conditions. The observed (E)-selectivity in the reaction of propiolates implied a mechanistic path different from the prototypical B(C6F5)3-mediated outer-sphere trans-hydroelementation.68,96
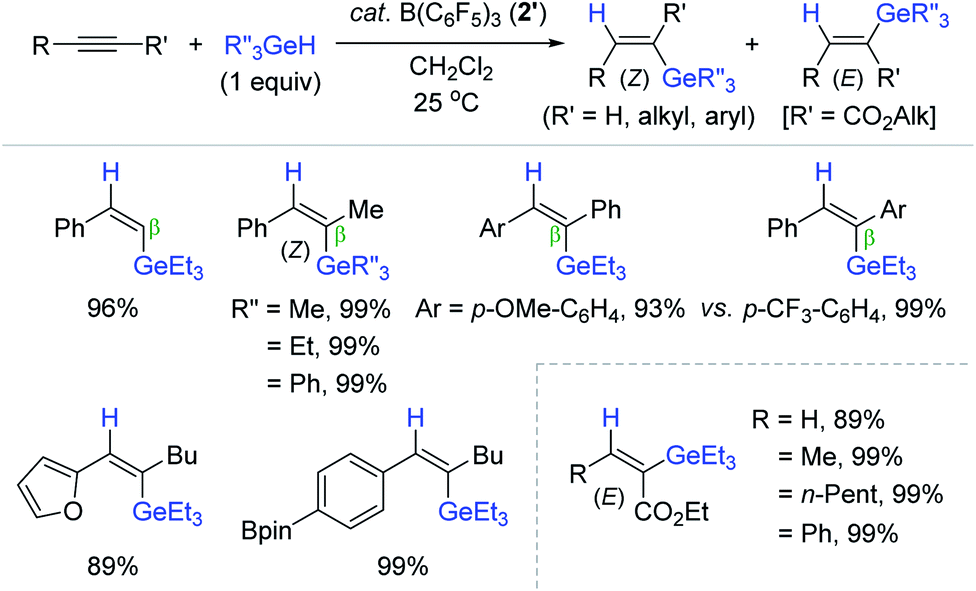 | ||
| Scheme 61 B(C6F5)3-catalyzed stereodivergent hydrogermylation of alkynes. | ||
A reaction pathway for the cis-hydrogermylation of propiolates was proposed (Scheme 62). Initially, B(C6F5)3 is assumed to bind the carbonyl moiety of the propiolate to form zwitterionic complex 2′e, where the carbocation is reduced by a hydrogermane184 to generate allenolate 2′f possessing a germylium ion. Subsequently, 2′f is suggested to combine the germylium species from the less congested face cis to H50 to deliver the cis-hydrogermylation product (Scheme 62).
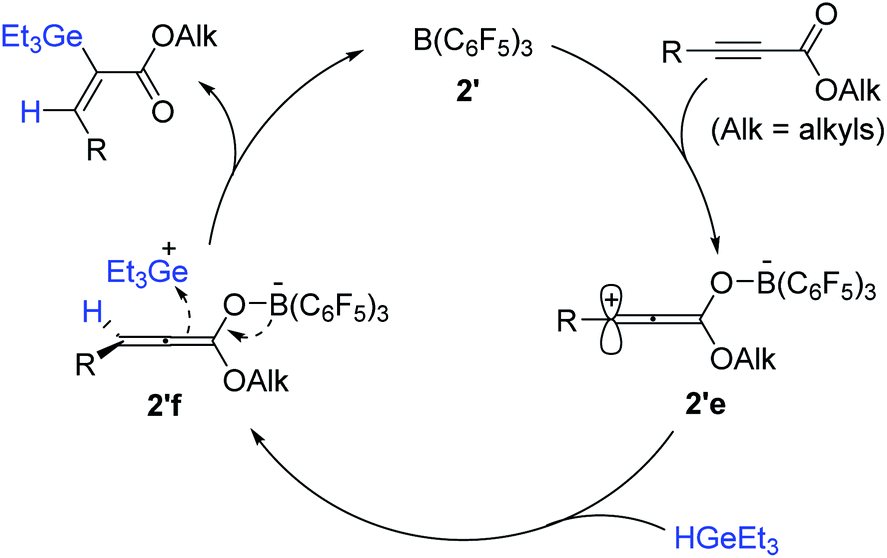 | ||
| Scheme 62 Proposed pathway for the B(C6F5)3-catalyzed cis-hydrogermylation of propiolates. | ||
5. Summary and outlook
This review described a broad range of transition metal-free molecular catalysts for the hydroelementation of terminal and internal alkynes with metalloid hydrides based on Si, B, Sn, and Ge, straightforwardly providing an array of functionalized vinyl compounds. Starting from Yamamoto's work using AlCl3 as the first transition metal-free Lewis acid catalyst for the alkyne hydrosilylation in 1996, a handful of research groups including Buriak, Veinot, Ingleson, Stephan, Nikonov, and Hill successfully applied their catalysts for alkyne hydrosilylation in solution or on the surface. In parallel with hydrosilylation, a range of Lewis acid and neutral complexes based on Al and Mg have been utilized as hydroboration catalysts mainly by the groups of Ingleson, Stephan, Melen, Roesky, Cowley, Ma, and Reuping, while several types of simple (Lewis) bases have been demonstrated to mediate the hydroboration of alkynes with high regio- and stereoselectivity over the last decade. The hydrostannation catalyzed by Lewis acids was recently reported by the groups of Maleczka, Organ, Oestreich, and Reuping, whereas only one report on the hydrogermylation was published by Gevorgyan et al.Overall, the viable metal-free catalysts for alkyne hydroelementation are highly diverse to include Lewis acids based on B, Al, Si, and P, and Lewis bases based on Li, Mg, P, and K, as well as Brönsted acids (carboxylic acids) and a Brönsted base (NaOH). Most of the Lewis acid catalysts are proposed to operate via an outer-sphere ionic mechanism, where the catalysts activate either the alkyne substrates or metalloid hydrides to form a vinyl carbocation as the key intermediate, which is reduced via the trans-oriented hydride transfer from the outer-sphere of the catalytic species to eventually deliver the hydroelementation products with high cis-selectivity. In contrast with the Lewis acid catalysis, a range of neutral hydride complexes based on Al and Mg, which are often in situ generated, catalyze particularly the hydroboration of alkynes via the inner-sphere mechanism involving alkyne insertion into an M–H bond (M = Al, Mg) or the syn-addition of HBpin into an alkyne unit of the alkynyl metal species to produce the trans-borylated vinyl compounds. Unlike alkyne hydrosilylation and hydroboration, the hydrostannation and hydrogermylation of alkynes known thus far are proposed to proceed via the outer-sphere mechanism by Lewis acid catalysts. It is interesting to compare that a neutral Mg(II) complex is supposed to serve as a Lewis acid catalyst for the outer-sphere alkyne hydrostannation through the activation of tin hydrides or alkyne molecules, whereas the same Mg complex is assumed to be readily converted to the magnesium hydride to catalyze the inner-sphere hydrosilylation and hydroboration reactions.
Despite the intensive surge in the study of environmentally and economically benign transition metal-free catalysts for the hydroelementation of alkynes, there are still some critical issues to be addressed as follows to compete with the traditional transition metal catalysis: (i) relatively lower turnover numbers, (ii) inferior regioselectivity in the case of unsymmetrical internal alkynes, (iii) limited mechanistic insights with a lack of characterized key catalytic intermediates, and (iv) facile disproportionation of metalloid hydrides such as HBpin. Thus, considering the limitations of the current metal-free catalytic systems, well-defined and robust complexes of alkaline or main group elements that are supported by finely tunable and strongly coordinating phosphine or NHC ligands can be next-generation catalysts for alkyne hydroelementation. Within the development of catalytic systems, robust chelation structures are supposed to suppress the decomposition of catalysts and/or metalloid hydrides to lead to improved catalytic efficiency, while tunable electronic and steric effects in the ligand are envisaged to enable regio- or stereodivergent hydroelementation with a broad substrate scope, including the challenging unsymmetrical internal alkynes. We hope that this review will draw significant interest from the community of organometallic catalysis and inspire synthetic chemists to develop elegant transition metal-free catalytic systems for the divergent hydroelementation of readily accessible olefins as a common chemical feedstock.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Li Ka Shing Foundation (2020LKSFG05A) in China. The authors also thank Yuhang Zhang for his comments on this manuscript.Notes and references
- P. J. Stang and F. Diederich, Modern acetylene chemistry, John Wiley & Sons, 2008 Search PubMed.
- R. A. Raphae, Acetylene compounds in organic synthesis, Academic Press, New York, 1955 Search PubMed.
- V. V. Voronin, M. S. Ledovskaya, A. S. Bogachenkov, K. S. Rodygin and V. P. Ananikov, Molecules, 2018, 23, 2442 CrossRef.
- A. M. Haydl, B. Breit, T. Liang and M. J. Krische, Angew. Chem., Int. Ed., 2017, 56, 11312–11325 CrossRef CAS.
- J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165–4179 RSC.
- B. A. Trofimov, Curr. Org. Chem., 2002, 6, 1121–1162 CrossRef CAS.
- M. Pereyre, J.-P. Quintard and A. Rahm, Tin in organic synthesis, Butterworth-Heinemann, 2013 Search PubMed.
- E. W. Colvin, Silicon reagents in organic synthesis, Academic Press, 1988 Search PubMed.
- W. P. Weber, Silicon reagents for organic synthesis, Springer, Berlin, 1983 Search PubMed.
- K. Indukuri, L. Cornelissen and O. Riant, Synthesis, 2016, 48, 4400–4422 CrossRef CAS.
- B. Hu and S. G. DiMagno, Org. Biomol. Chem., 2015, 13, 3844–3855 RSC.
- A. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC.
- E. Langkopf and D. Schinzer, Chem. Rev., 1995, 95, 1375–1408 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508–524 CrossRef.
- H. C. Brown, Pure Appl. Chem., 1976, 47, 49–60 CAS.
- B. Marciniec, Hydrosilylation: a comprehensive review on recent advances, Springer Berlin, 2009 Search PubMed.
- D. S. W. Lim and E. A. Anderson, Synthesis, 2012, 44, 983–1010 CrossRef CAS.
- B. Marciniec, Silicon Chem., 2002, 1, 155–174 CrossRef CAS.
- J. Carreras, A. Caballero and P. J. Pérez, Chem.–Asian J., 2019, 14, 329–343 CrossRef CAS.
- R. Barbeyron, E. Benedetti, J. Cossy, J.-J. Vasseur, S. Arseniyadis and M. Smietana, Tetrahedron, 2014, 70, 8431–8452 CrossRef CAS.
- I. Beletskaya and A. Pelter, Tetrahedron, 1997, 53, 4957–5026 CrossRef CAS.
- M. Alami, A. Hamze and O. Provot, ACS Catal., 2019, 9, 3437–3466 CrossRef CAS.
- H. Yoshida, Synthesis, 2016, 48, 2540–2552 CrossRef CAS.
- N. D. Smith, J. Mancuso and M. Lautens, Chem. Rev., 2000, 100, 3257–3282 CrossRef CAS.
- I. P. Beletskaya, C. Nájera and M. Yus, Chem. Rev., 2018, 118, 5080–5200 CrossRef CAS.
- T. G. Frihed and A. Fürstner, Bull. Chem. Soc. Jpn., 2016, 89, 135–160 CrossRef CAS.
- B. M. Trost and Z. T. Ball, Synthesis, 2005, 2005, 853–887 CrossRef.
- W. J. Jang, W. L. Lee, J. H. Moon, J. Y. Lee and J. Yun, Org. Lett., 2016, 18, 1390–1393 CrossRef CAS.
- B. Sundararaju and A. Fürstner, Angew. Chem., Int. Ed., 2013, 52, 14050–14054 CrossRef CAS.
- H. Katayama, K. Taniguchi, M. Kobayashi, T. Sagawa, T. Minami and F. Ozawa, J. Organomet. Chem., 2002, 645, 192–200 CrossRef CAS.
- S. E. Denmark and W. Pan, Org. Lett., 2001, 3, 61–64 CrossRef CAS.
- Y. Maruyama, K. Yamamura, I. Nakayama, K. Yoshiuchi and F. Ozawa, J. Am. Chem. Soc., 1998, 120, 1421–1429 CrossRef CAS.
- R. S. Tanke and R. H. Crabtree, J. Am. Chem. Soc., 1990, 112, 7984–7989 CrossRef CAS.
- I. Ojima, N. Clos, R. J. Donovan and P. Ingallina, Organometallics, 1990, 9, 3127–3133 CrossRef CAS.
- N. Asao and Y. Yamamoto, Bull. Chem. Soc. Jpn., 2000, 73, 1071–1087 CrossRef CAS.
- C.-J. Li, Chem, 2016, 1, 423–437 CAS.
- J. L. Tucker, Org. Process Res. Dev., 2010, 14, 328–331 CrossRef CAS.
- C.-J. Li and B. M. Trost, Proc. Natl. Acad. Sci., 2008, 105, 13197–13202 CrossRef CAS.
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS.
- R. H. Petrucci, W. S. Harwood, G. F. Herring and J. D. Madura, General chemistry : principles and modern applications, Pearson Prentice Hall, Upper Saddle River; New Jersey, 2007 Search PubMed.
- C. Housecroft; and A. G. Sharpe, Inorganic Chemistry, 2nd edn, 2005 Search PubMed.
- E. Nieto-Sepulveda, A. D. Bage, L. A. Evans, T. A. Hunt, A. G. Leach, S. P. Thomas and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2019, 141, 18600–18611 CrossRef CAS.
- K. Shirakawa, A. Arase and M. Hoshi, Synthesis, 2004, 2004, 1814–1820 CrossRef.
- M. Hoshi, K. Shirakawa and A. Arase, Chem. Commun., 1998, 1225–1226 RSC.
- A. Arase, M. Hoshi, A. Mijin and K. Nishi, Synth. Commun., 1995, 25, 1957–1962 CrossRef CAS.
- S. Keess and M. Oestreich, Chem. Sci., 2017, 8, 4688–4695 RSC.
- M. Oestreich, Angew. Chem., Int. Ed., 2016, 55, 494–499 CrossRef CAS.
- T. Sudo, N. Asao, V. Gevorgyan and Y. Yamamoto, J. Org. Chem., 1999, 64, 2494–2499 CrossRef CAS.
- N. Asao, T. Sudo and Y. Yamamoto, J. Org. Chem., 1996, 61, 7654–7655 CrossRef CAS.
- E. Yoshikawa, V. Gevorgyan, N. Asao and Y. Yamamoto, J. Am. Chem. Soc., 1997, 119, 6781–6786 CrossRef CAS.
- V. Gevorgyan, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 1997, 62, 2963–2967 CrossRef CAS.
- N. Asao, E. Yoshikawa and Y. Yamamoto, J. Org. Chem., 1996, 61, 4874–4875 CrossRef CAS.
- N. Asao, Y. Matsukawa and Y. Yamamoto, Chem. Commun., 1996, 1513–1514 RSC.
- T. Sudo, N. Asao and Y. Yamamoto, J. Org. Chem., 2000, 65, 8919–8923 CrossRef CAS.
- N. Kato, Y. Tamura, T. Kashiwabara, T. Sanji and M. Tanaka, Organometallics, 2010, 29, 5274–5282 CrossRef CAS.
- V. V. Doan and M. J. Sailor, Science, 1992, 256, 1791–1792 CrossRef CAS.
- M. Sailor, J. Heinrich and J. Lauerhaas, in Stud. Surf. Sci. Catal., Elsevier Science, New York, 1996, vol. 103 Search PubMed.
- A. Janshoff, K.-P. S. Dancil, C. Steinem, D. P. Greiner, V. S. Y. Lin, C. Gurtner, K. Motesharei, M. J. Sailor and M. R. Ghadiri, J. Am. Chem. Soc., 1998, 120, 12108–12116 CrossRef CAS.
- V. S.-Y. Lin, K. Motesharei, K.-P. S. Dancil, M. J. Sailor and M. R. Ghadiri, Science, 1997, 278, 840–843 CrossRef CAS.
- J. Harper and M. J. Sailor, Anal. Chem., 1996, 68, 3713–3717 CrossRef CAS.
- L. T. Canham, Adv. Mater., 1995, 7, 1033–1037 CrossRef CAS.
- K. H. Li, C. Tsai, S. Shih, T. Hsu, D. Kwong and J. Campbell, J. Appl. Phys., 1992, 72, 3816–3817 CrossRef CAS.
- J. M. Buriak, M. P. Stewart, T. W. Geders, M. J. Allen, H. C. Choi, J. Smith, D. Raftery and L. T. Canham, J. Am. Chem. Soc., 1999, 121, 11491–11502 CrossRef CAS.
- J. M. Buriak and M. J. Allen, J. Am. Chem. Soc., 1998, 120, 1339–1340 CrossRef CAS.
- T. K. Purkait, M. Iqbal, M. H. Wahl, K. Gottschling, C. M. Gonzalez, M. A. Islam and J. G. Veinot, J. Am. Chem. Soc., 2014, 136, 17914–17917 CrossRef CAS.
- T. Helbich, A. Lyuleeva, P. Marx, L. M. Scherf, T. K. Purkait, T. F. Fässler, P. Lugli, J. G. Veinot and B. Rieger, Adv. Funct. Mater., 2017, 27, 1606764 CrossRef.
- Y. Kim, R. B. Dateer and S. Chang, Org. Lett., 2017, 19, 190–193 CrossRef CAS.
- Y. Zhang, Y. Chen, Z. Zhang, S. Liu and X. Shen, Org. Lett., 2020, 22, 970–975 CrossRef CAS.
- L. D. Curless and M. J. Ingleson, Organometallics, 2014, 33, 7241–7246 CrossRef CAS.
- H. Arii, K. Nakabayashi, K. Mochida and T. Kawashima, Molecules, 2016, 21, 999 CrossRef.
- K. Müther, J. Mohr and M. Oestreich, Organometallics, 2013, 32, 6643–6646 CrossRef.
- K. Müther and M. Oestreich, Chem. Commun., 2011, 47, 334–336 RSC.
- J. B. Lambert, Y. Zhao and H. Wu, J. Org. Chem., 1999, 64, 2729–2736 CrossRef CAS.
- C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374–1377 CrossRef CAS.
- M. Perez, L. J. Hounjet, C. B. Caputo, R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 18308–18310 CrossRef CAS.
- M. H. Holthausen, M. Mehta and D. W. Stephan, Angew. Chem., Int. Ed., 2014, 53, 6538–6541 CrossRef CAS.
- R. R. Holmes, Chem. Rev., 1996, 96, 927–950 CrossRef CAS.
- C. Chuit, R. J. P. Corriu, C. Reye and J. C. Young, Chem. Rev., 1993, 93, 1371–1448 CrossRef CAS.
- R. B. Lettan and K. A. Scheidt, Org. Lett., 2005, 7, 3227–3230 CrossRef CAS.
- G. A. Kraus and J. Bae, Tetrahedron Lett., 2003, 44, 5505–5506 CrossRef CAS.
- J. E. Baldwin, G. J. Pritchard and R. E. Rathmell, J. Chem. Soc., Perkin Trans. 1, 2001, 2906–2908 RSC.
- J. Busch-Petersen, Y. Bo and E. J. Corey, Tetrahedron Lett., 1999, 40, 2065–2068 CrossRef CAS.
- I. Kuwajima, E. Nakamura and K. Hashimoto, Tetrahedron, 1983, 39, 975–982 CrossRef CAS.
- S. V. Maifeld and D. Lee, Org. Lett., 2005, 7, 4995–4998 CrossRef CAS.
- B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2004, 126, 13942–13944 CrossRef CAS.
- L. A. Aronica, F. Morini, A. M. Caporusso and P. Salvadori, Tetrahedron Lett., 2002, 43, 5813–5815 CrossRef CAS.
- M. E. Jung and G. Piizzi, J. Org. Chem., 2002, 67, 3911–3914 CrossRef CAS.
- I. Fleming, T. W. Newton, V. Sabin and F. Zammattio, Tetrahedron, 1992, 48, 7793–7802 CrossRef CAS.
- Z. Yang, M. Zhong, X. Ma, K. Nijesh, S. De, P. Parameswaran and H. W. Roesky, J. Am. Chem. Soc., 2016, 138, 2548–2551 CrossRef CAS.
- Z. Yang, M. Zhong, X. Ma, S. De, C. Anusha, P. Parameswaran and H. W. Roesky, Angew. Chem., Int. Ed., 2015, 54, 10225–10229 CrossRef CAS.
- L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031–3066 CrossRef CAS.
- K. Jakobsson, T. Chu and G. I. Nikonov, ACS Catal., 2016, 6, 7350–7356 CrossRef CAS.
- L. Garcia, C. Dinoi, M. F. Mahon, L. Maron and M. S. Hill, Chem. Sci., 2019, 10, 8108–8118 RSC.
- E. Fritz-Langhals, Org. Process Res. Dev., 2019, 23, 2369–2377 CrossRef CAS.
- J. S. McGough, S. M. Butler, I. A. Cade and M. J. Ingleson, Chem. Sci., 2016, 7, 3384–3389 RSC.
- M. A. Dureen, C. C. Brown and D. W. Stephan, Organometallics, 2010, 29, 6594–6607 CrossRef CAS.
- M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 8396–8397 CrossRef CAS.
- D. N. Lastovickova and C. W. Bielawski, Organometallics, 2016, 35, 706–712 CrossRef CAS.
- L. Xie, J. Zhang and C. Cui, Chem. - Eur. J., 2014, 20, 9500–9503 CrossRef CAS.
- H. Asakawa, K.-H. Lee, Z. Lin and M. Yamashita, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- T. K. Wood, W. E. Piers, B. A. Keay and M. Parvez, Chem. - Eur. J., 2010, 16, 12199–12206 CrossRef CAS.
- E. R. Abbey, L. N. Zakharov and S.-Y. Liu, J. Am. Chem. Soc., 2008, 130, 7250–7252 CrossRef CAS.
- R. L. Melen, A. J. Lough and D. W. Stephan, Dalton Trans., 2013, 42, 8674–8683 RSC.
- L. Fan and D. W. Stephan, Dalton Trans., 2016, 45, 9229–9234 RSC.
- Q. Yin, Y. Soltani, R. L. Melen and M. Oestreich, Organometallics, 2017, 36, 2381–2384 CrossRef CAS.
- Q. Yin, S. Kemper, H. F. Klare and M. Oestreich, Chem. - Eur. J., 2016, 22, 13840–13844 CrossRef CAS.
- J. R. Lawson, L. C. Wilkins and R. L. Melen, Chem. - Eur. J., 2017, 23, 10997–11000 CrossRef CAS.
- J. L. Carden, L. J. Gierlichs, D. F. Wass, D. L. Browne and R. L. Melen, Chem. Commun., 2019, 55, 318–321 RSC.
- M. Shimizu, I. Nagao, S.-i. Kiyomoto and T. Hiyama, Aust. J. Chem., 2012, 65, 1277–1284 CrossRef CAS.
- J. Takaya and N. Iwasawa, ACS Catal., 2012, 2, 1993–2006 CrossRef CAS.
- J. Takaya, N. Kirai and N. Iwasawa, J. Am. Chem. Soc., 2011, 133, 12980–12983 CrossRef CAS.
- M. Shimizu, C. Nakamaki, K. Shimono, M. Schelper, T. Kurahashi and T. Hiyama, J. Am. Chem. Soc., 2005, 127, 12506–12507 CrossRef CAS.
- T. Hata, H. Kitagawa, H. Masai, T. Kurahashi, M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2001, 40, 790–792 CrossRef CAS.
- H. E. Ho, N. Asao, Y. Yamamoto and T. Jin, Org. Lett., 2014, 16, 4670–4673 CrossRef CAS.
- Z. Zhang, P. Jain and J. C. Antilla, Angew. Chem., Int. Ed., 2011, 50, 10961–10964 CrossRef CAS.
- D. B. Collum, S.-C. Chen and B. Ganem, J. Org. Chem., 1978, 43, 4393–4394 CrossRef CAS.
- H. C. Brown and B. S. Rao, J. Am. Chem. Soc., 1960, 82, 681–686 CrossRef CAS.
- C. B. Hoyt, M. L. Sarazen and C. W. Jones, J. Catal., 2019, 369, 493–500 CrossRef CAS.
- Y. Wu, C. Shan, J. Ying, J. Su, J. Zhu, L. L. Liu and Y. Zhao, Green Chem., 2017, 19, 4169–4175 RSC.
- J. H. Docherty, J. Peng, A. P. Dominey and S. P. Thomas, Nat. Chem., 2017, 9, 595–600 CrossRef CAS.
- J. M. Farrell, R. T. Posaratnanathan and D. W. Stephan, Chem. Sci., 2015, 6, 2010–2015 RSC.
- I. P. Query, P. A. Squier, E. M. Larson, N. A. Isley and T. B. Clark, J. Org. Chem., 2011, 76, 6452–6456 CrossRef CAS.
- C. E. Garrett and G. C. Fu, J. Org. Chem., 1996, 61, 3224–3225 CrossRef CAS.
- K. Nagao, H. Ohmiya and M. Sawamura, J. Am. Chem. Soc., 2014, 136, 10605–10608 CrossRef CAS.
- K. Nagao, A. Yamazaki, H. Ohmiya and M. Sawamura, Org. Lett., 2018, 20, 1861–1865 CrossRef CAS.
- K. Nagao, H. Ohmiya and M. Sawamura, Org. Lett., 2015, 17, 1304–1307 CrossRef CAS.
- A. Yamazaki, K. Nagao, T. Iwai, H. Ohmiya and M. Sawamura, Angew. Chem., Int. Ed., 2018, 57, 3196–3199 CrossRef CAS.
- R. Fritzemeier, A. Gates, X. Guo, Z. Lin and W. L. Santos, J. Org. Chem., 2018, 83, 10436–10444 CrossRef CAS.
- Y. Ding, X. Liu, X. Ma, Y. Liu, M. Zhong, W. Li, Z. Yang and Y. Yang, J. Organomet. Chem., 2018, 868, 55–60 CrossRef CAS.
- N. Sarkar, S. Bera and S. Nembenna, J. Org. Chem., 2020, 85, 4999–5009 CrossRef CAS.
- A. Bismuto, S. P. Thomas and M. J. Cowley, Angew. Chem., Int. Ed., 2016, 55, 15356–15359 CrossRef CAS.
- A. Harinath, I. Banerjee, J. Bhattacharjee and T. K. Panda, New J. Chem., 2019, 43, 10531–10536 RSC.
- R. L. Falconer, G. S. Nichol and M. J. Cowley, Inorg. Chem., 2019, 58, 11439–11448 CrossRef CAS.
- D. Franz, L. Sirtl, A. Pöthig and S. Inoue, Z. Anorg. Allg. Chem., 2016, 642, 1245–1250 CrossRef CAS.
- G. Zhang, J. Wu, H. Zeng, M. C. Neary, M. Devany, S. Zheng and P. A. Dub, ACS Catal., 2018, 9, 874–884 CrossRef.
- F. Li, X. Bai, Y. Cai, H. Li, S.-Q. Zhang, F.-H. Liu, X. Hong, Y. Xu and S.-L. Shi, Org. Process Res. Dev., 2019, 23, 1703–1708 CrossRef CAS.
- V. A. Pollard, M. A. Fuentes, A. R. Kennedy, R. McLellan and R. E. Mulvey, Angew. Chem., Int. Ed., 2018, 57, 10651–10655 CrossRef CAS.
- A. K. Jaladi, H. Kim, J. H. Lee, W. K. Shin, H. Hwang and D. K. An, New J. Chem., 2019, 43, 16524–16529 RSC.
- D. Yan, X. Wu, J. Xiao, Z. Zhu, X. Xu, X. Bao, Y. Yao, Q. Shen and M. Xue, Org. Chem. Front., 2019, 6, 648–653 RSC.
- Z.-C. Wang, M. Wang, J. Gao, S.-L. Shi and Y. Xu, Org. Chem. Front., 2019, 6, 2949–2953 RSC.
- M. K. Bisai, S. Yadav, T. Das, K. Vanka and S. S. Sen, Chem. Commun., 2019, 55, 11711–11714 RSC.
- J. Li, M. Luo, X. Sheng, H. Hua, W. Yao, S. A. Pullarkat, L. Xu and M. Ma, Org. Chem. Front., 2018, 5, 3538–3547 RSC.
- M. Magre, B. Maity, A. Falconnet, L. Cavallo and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 7025–7029 CrossRef CAS.
- K. Revunova and G. Nikonov, Dalton Trans., 2015, 44, 840–866 RSC.
- A. Harinath, J. Bhattacharjee and T. K. Panda, Adv. Synth. Catal., 2019, 361, 850–857 CrossRef CAS.
- L. A. Berben, Chem. - Eur. J., 2015, 21, 2734–2742 CrossRef CAS.
- G. A. Artamkina, M. P. Egorov and I. P. Beletskaya, Chem. Rev., 1982, 82, 427–459 CrossRef CAS.
- V. A. Pollard, S. A. Orr, R. McLellan, A. R. Kennedy, E. Hevia and R. E. Mulvey, Chem. Commun., 2018, 54, 1233–1236 RSC.
- L. E. Lemmerz, R. McLellan, N. R. Judge, A. R. Kennedy, S. A. Orr, M. Uzelac, E. Hevia, S. D. Robertson, J. Okuda and R. E. Mulvey, Chem. - Eur. J., 2018, 24, 9940–9948 CrossRef CAS.
- Y. Zhang, J. Wei, W.-X. Zhang and Z. Xi, Inorg. Chem., 2015, 54, 10695–10700 CrossRef CAS.
- Y. Yang, M. D. Anker, J. Fang, M. F. Mahon, L. Maron, C. Weetman and M. S. Hill, Chem. Sci., 2017, 8, 3529–3537 RSC.
- A. D. Bage, T. A. Hunt and S. P. Thomas, Org. Lett., 2020, 22, 4107–4112 CrossRef CAS.
- G. W. O'Neil, A. M. Craig, J. R. Williams, J. C. Young and P. C. Spiegel, Synlett, 2017, 28, 1101 CrossRef.
- D. Mailhol, J. Willwacher, N. Kausch-Busies, E. E. Rubitski, Z. Sobol, M. Schuler, M.-H. Lam, S. Musto, F. Loganzo and A. Maderna, J. Am. Chem. Soc., 2014, 136, 15719–15729 CrossRef CAS.
- M. Dieckmann, M. Kretschmer, P. Li, S. Rudolph, D. Herkommer and D. Menche, Angew. Chem., Int. Ed., 2012, 51, 5667–5670 CrossRef CAS.
- J. Gagnepain, E. Moulin and A. Fürstner, Chem. - Eur. J., 2011, 17, 6964–6972 CrossRef CAS.
- A. Fürstner, J.-A. Funel, M. Tremblay, L. C. Bouchez, C. Nevado, M. Waser, J. Ackerstaff and C. C. Stimson, Chem. Commun., 2008, 2873–2875 RSC.
- V. Gevorgyan, J.-X. Liu and Y. Yamamoto, Chem. Commun., 1998, 37–38 RSC.
- W. P. Gallagher, I. Terstiege and R. E. Maleczka Jr, J. Am. Chem. Soc., 2001, 123, 3194–3204 CrossRef CAS.
- R. E. Maleczka Jr and W. P. Gallagher, Org. Lett., 2001, 3, 4173–4176 CrossRef CAS.
- R. E. Maleczka Jr and I. Terstiege, J. Org. Chem., 1998, 63, 9622–9623 CrossRef.
- B. Ghosh, M. D. R. I. Amado-Sierra, D. Holmes and R. E. Maleczka Jr, Org. Lett., 2014, 16, 2318–2321 CrossRef CAS.
- W. Plass and J. G. Verkade, Inorg. Chem., 1993, 32, 5153–5159 CrossRef CAS.
- D. Dakternieks and H. Zhu, Organometallics, 1992, 11, 3820–3825 CrossRef CAS.
- S. S. Al-Juaid, S. M. Dhaher, C. Eaborn, P. B. Hitchcock and J. D. Smith, J. Organomet. Chem., 1987, 325, 117–127 CrossRef CAS.
- A. G. Davies, Organotin Chemistry, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed.
- M. S. Oderinde and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 9834–9837 CrossRef CAS.
- J. Lambert, Y. Zhao and S. Zhang, J. Phys. Org. Chem., 2001, 14, 370 CrossRef CAS.
- J. B. Lambert and B. Kuhlamann, J. Chem. Soc., Chem. Commun., 1992, 931–932 RSC.
- G. E. Keck and D. Krishnamurthy, J. Org. Chem., 1996, 61, 7638–7639 CrossRef CAS.
- N. Asao, J.-X. Liu, T. Sudoh and Y. Yamamoto, J. Org. Chem., 1996, 61, 4568–4571 CrossRef CAS.
- N. Asao, J.-X. Liu, T. Sudoh and Y. Yamamoto, J. Chem. Soc., Chem. Commun., 1995, 2405–2406 RSC.
- F. Forster, V. M. Rendón López and M. Oestreich, Organometallics, 2018, 37, 2656–2659 CrossRef CAS.
- M. Magre, M. Szewczyk and M. Rueping, Org. Lett., 2020, 22, 1594–1598 CrossRef CAS.
- W. C. Still, J. Am. Chem. Soc., 1978, 100, 1481–1487 CrossRef CAS.
- D. Seyferth and M. A. Weiner, J. Am. Chem. Soc., 1962, 84, 361–364 CrossRef CAS.
- M. B. Rice, S. L. Whitehead, C. M. Horvath, J. A. Muchnij and R. E. Maleczka Jr, Synthesis, 2001, 2001, 1495–1504 Search PubMed.
- H. Zhang, F. Guibé and G. Balavoine, J. Org. Chem., 1990, 55, 1857–1867 CrossRef CAS.
- M. Rubin, T. Schwier and V. Gevorgyan, J. Org. Chem., 2002, 67, 1936–1940 CrossRef CAS.
- D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS.
- D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS.
- T. Schwier and V. Gevorgyan, Org. Lett., 2005, 7, 5191–5194 CrossRef CAS.
- H. Mayr and N. Basso, Angew. Chem., Int. Ed. Engl., 1992, 31, 1046–1048 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |