
Metal single-atom coordinated graphitic carbon nitride as an efficient catalyst for CO oxidation†
Shiyan
Wang
a,
Jiaqi
Li
b,
Qiang
Li
*a,
Xiaowan
Bai
a and
Jinlan
Wang
 *a
*a
aSchool of Physics, Southeast University, Nanjing 211189, China. E-mail: qiang.li@seu.edu.cn; jlwang@seu.edu.cn
bCollege of Chemistry and Chemical Engineering, Hunan University, Changsha, Hunan 410082, China
First published on 15th November 2019
Abstract
Single-atom catalysts (SACs) often present outstanding activity due to their high ratio of low-coordinated metal atoms and can be applied to the activation of strong chemical bonds such as C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O. Herein, we investigate the potential usage of a single-atom catalyst, in which isolated cobalt atoms are supported on porous graphitic carbon nitride (Co/g-C3N4), for CO oxidation. Based on the adsorption/co-adsorption energies of O2, CO, 2O2, CO + O2 and 2CO, the screening criteria and the reaction mechanisms of CO oxidation, including the Eley–Rideal, New Eley–Rideal, Langmuir–Hinshelwood, and termolecular Eley–Rideal mechanisms, are established and compared. In particular, the energy barriers of the rate-limiting steps for the CO oxidation process by all possible reaction pathways are in a range from 0.21 to 0.59 eV, suggesting that the Co/g-C3N4 catalyst can boost CO oxidation at low temperature. Moreover, the preparation of the SAC (Co/g-C3N4) by using CoCl2 as an appropriate metal precursor and the stability (up to 600 K) are evaluated by ab initio molecular dynamics simulations. The high stability and excellent activity of the Co/g-C3N4 SAC for CO oxidation offer a high possibility of clean energy production.
O. Herein, we investigate the potential usage of a single-atom catalyst, in which isolated cobalt atoms are supported on porous graphitic carbon nitride (Co/g-C3N4), for CO oxidation. Based on the adsorption/co-adsorption energies of O2, CO, 2O2, CO + O2 and 2CO, the screening criteria and the reaction mechanisms of CO oxidation, including the Eley–Rideal, New Eley–Rideal, Langmuir–Hinshelwood, and termolecular Eley–Rideal mechanisms, are established and compared. In particular, the energy barriers of the rate-limiting steps for the CO oxidation process by all possible reaction pathways are in a range from 0.21 to 0.59 eV, suggesting that the Co/g-C3N4 catalyst can boost CO oxidation at low temperature. Moreover, the preparation of the SAC (Co/g-C3N4) by using CoCl2 as an appropriate metal precursor and the stability (up to 600 K) are evaluated by ab initio molecular dynamics simulations. The high stability and excellent activity of the Co/g-C3N4 SAC for CO oxidation offer a high possibility of clean energy production.
1. Introduction
The catalytic oxidation of carbon monoxide (CO) has attracted great interest due to its importance in various industrial1,2 and environmental applications,3,4 both from a fundamental point of view and for practical applications, e.g., removal of CO contamination from H2-rich fuel gases for ammonia synthesis and low temperature fuel cell applications or reducing CO emission in exhaust gases from catalytic converters for cars, and serving as key prototype reactions for the design of heterogeneous catalysts.5Over the past few years, SACs have become a hot topic due to their distinct advantages of maximized atomic utilization, uniform active sites with excellent stability and selectivity, and much improved catalytic activity over conventional metal nanoparticles.6–10 Generally, metals are dispersed finely on a support with a high surface area for the efficient use of catalytically active components, which maximizes the metal atom efficiency. To date, various SACs6,7,11,12 have been successfully synthesized with high activity for catalytic CO oxidation reactions. However, the synthesis of SACs with high stability and cost-efficiency remains a huge challenge. In 2011, Zhang and coworkers successfully uniformly deposited single Pt atoms on an iron oxide surface, which showed excellent stability and activity for CO oxidation with experimental and theoretical elucidation.6 Inspired by such an important study, various types of single metal atoms supported on Al2O3,13 CeO2,14–16 MgAl2O4,17 and AuTi2O3–6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 catalysts have been reported to be active for low-temperature CO oxidation. Additionally, it is not limited to successful stabilization of metal single atoms on a metal oxide surface, but can be developed further and made applicable to 2D materials based SACs. For instance, Zhang et al.19 demonstrated that various metal single atoms anchored on Ti2CO2 were oxidized by CO via Eley–Rideal (ER) and Langmuir–Hinshelwood (LH) mechanisms, and titanium atoms showed the best performance. Du et al.20 reported MoS2 as an excellent substrate for fabricating SACs, which can effectively oxidize CO with an energy barrier of the rate determining state of only 0.40 eV.
18 catalysts have been reported to be active for low-temperature CO oxidation. Additionally, it is not limited to successful stabilization of metal single atoms on a metal oxide surface, but can be developed further and made applicable to 2D materials based SACs. For instance, Zhang et al.19 demonstrated that various metal single atoms anchored on Ti2CO2 were oxidized by CO via Eley–Rideal (ER) and Langmuir–Hinshelwood (LH) mechanisms, and titanium atoms showed the best performance. Du et al.20 reported MoS2 as an excellent substrate for fabricating SACs, which can effectively oxidize CO with an energy barrier of the rate determining state of only 0.40 eV.
In recent years, porous 2D materials have been used as potential substrates to anchor metal single atoms in catalysis.21–44 For example, SACs supported on graphdiyne, β12 boron monolayers, h-BN and C2N could serve as low-cost but highly efficient catalysts for CO2 reduction,21–25 hydrogen evolution reduction,26–30 N2 reduction,27,31–35 CO oxidation,36–39 and oxygen reduction/evolution reactions29,40–43 from previous experimental and theoretical studies. Recently, highly active and stable single-atom Cu supported by a 2D metal–organic framework (Cu/UiO-66) catalyst has also been reported, which showed an excellent selectivity of about 100% for CO oxidation.44
In particular, the excellent wide-bandgap semiconductor electro-/photo-catalyst 2D porous material of g-C3N4 exhibits high chemical stability and an appealing catalytic capability for splitting of water into hydrogen and oxygen.45–50 Moreover, large periodic holes of g-C3N4 provide sufficient space as an excellent substrate for supporting catalysts. Thus, the introduction of SACs can effectively adjust the electronic structures, and the synergistic effect between SACs and g-C3N4 plays a crucial role in improving the catalytic activity of g-C3N4. Previous literature reports have confirmed that g-C3N4 can serve as an excellent support catalyst, and the coordination of single-atom Co and g-C3N4 shows a remarkable performance toward both the oxygen reduction and evolution reactions, which is even better than those of benchmark Pt and other carbon nitride-based electrocatalysts.51–53 Inspired by the exciting experimental discovery, it is natural to ask: (I) whether Co/g-C3N4 also faces CO poisoning as found in Pt-based or other electrocatalysts?54 In other words, whether the CO oxidation process on Co/g-C3N4 can be realized at low temperature? (II) If so, what is its mechanism? In this work, we systematically evaluated that the CO oxidation reaction on Co/g-C3N4 occurred through the ER, NER, TER and LH mechanisms, and found that the CO oxidation process can be realized with the energy barriers of the rate-limiting steps in a range from 0.21 to 0.59 eV, avoiding the CO poisoning of the catalyst. Thus, with the excellent oxygen reduction/evolution reactions and CO oxidation properties, Co/g-C3N4 can be further developed as a multi-functional catalyst for fuel cells.
2. Computational methods
Spin-polarized calculations were performed by using plane-wave density functional theory (DFT) as implemented in the Vienna Ab Initio Simulation Package (VASP) code.55,56 The electronic exchange–correlation energy was calculated using the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA).57,58 The projector augmented wave (PAW) method was used to describe the ionic cores.59 The kinetic energy cutoff for the plane-wave basis set was chosen to be 450 eV. The vacuum space was 20 Å, which should be sufficient to avoid interactions between periodic images.51,60,61 Supercells consisting of 2 × 2 × 1 g-C3N4 unit cells were used and the Brillouin zones were sampled using a Monkhorst–Pack k-point mesh with a 3 × 3 × 1 k-point grid.51,62 The convergence criteria were set to 10−5 eV and −0.01 eV Å−1 for the energy and the forces on each atom, respectively.63To assess the thermodynamic stability of Co/g-C3N4, ab initio molecular dynamics (AIMD) simulations were performed. The atomic charge transfer in different systems was evaluated by Bader's charge analysis.64 To investigate the kinetic processes of O2 activation and CO oxidation, the climbing image nudged elastic band method65 (CI-NEB) was employed to identify the minimum energy pathways, and each transition state structure was further confirmed by vibrational analysis. Further calculation details of the binding energies and the activation energy barriers are given in the ESI.†
3. Results and discussion
3.1. The stability and electronic structures of the Co/g-C3N4 catalyst
To find the most favorable deposition sites for the single-atom catalysts, three possible Co locations on a 2 × 2 × 1 g-C3N4 supercell were considered, as shown in Fig. 1a. After geometry optimization, Co connected with the two adjacent pyridinic-N atoms from two separate triazine units, forming a CoN3C2 ring (Fig. 1b). The Co atom exhibits a short bond length with N of 1.95 Å and the strongest bonding of 3.25 eV, which are much larger than those of Pd and Pt embedded in g-C3N460 (2.17 and 2.95 eV, respectively). Moreover, the mobility of the Co atom needs to overcome a rather big energy barrier of 1.98 eV, as shown in Fig. S1.† The large diffusion barrier indicates that it is difficult for the isolated Co atom to form clusters on g-C3N4. Moreover, the relative energy (ΔE) of two cobalt atoms scattered at two sites (Fig. S2a†) is lower than that of cobalt dimers adsorbed at one site (Fig. S2b and c†) of g-C3N4, suggesting that isolated distribution is more preferred compared with forming clusters. Furthermore, we calculated the energy barrier of a Co dimer dissociated into two separated Co monomers at two pore sites of g-C3N4, which is energetically favorable, as shown in Fig. S3.† A moderate energy barrier of 0.58 eV implies that the relocation of the Co dimer into two pore sites of g-C3N4 readily occurs under ambient conditions.
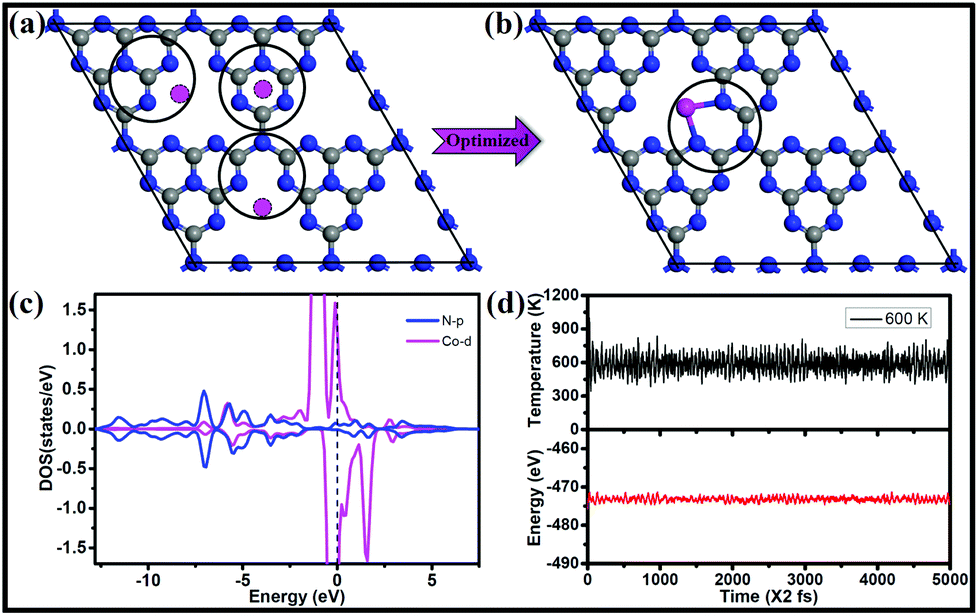 | ||
| Fig. 1 (a) Schematic of adsorption of Co atoms on hexagonal holes, bridge sites, and pentagonal holes of g-C3N4 and (b) the most stable adsorption structure is illustrated in the right panel where the Co atom is connected to two adjacent pyridinic-N atoms from two separate triazine units, forming a CoN3C2 ring on g-C3N4. (c) The density of states (DOS) and (d) variations of temperature and energy fluctuations with time progress for AIMD simulations of Co/g-C3N4. The simulation is run at 600 K for 10 ps with a time step of 2 fs. Color scheme: gray, blue and magenta represent C, N and Co atoms, respectively. | ||
The electronic structure and bonding features are analyzed by means of density of states (DOS), which can provide a fundamental understanding of the interaction between the single Co atom and the substrate as well as the activity. As shown in Fig. 1c, strong coupling between the d-orbitals of Co and the p-orbitals of N is observed in the energy range from −7.5 to 2.5 eV, indicating the strong interaction between Co and the g-C3N4 substrate. The DOS of Co/g-C3N4 shows a higher peak at the Fermi energy, which is attributed to the change in the electronic structure of the outermost Co modified by g-C3N4, indicating high activity of supported Co on g-C3N4. In addition, significant amounts of electrons (0.84 e− based on Bader charge analysis) are transferred from the Co atom to the g-C3N4 sheet. The stability of Co/g-C3N4 was further evaluated by performing AIMD simulations, in which the time step was set as 2 fs for a total period of 10 ps. As shown in Fig. S4,† the energy fluctuations with time evolution oscillates near the equilibrium state and the snapshot of the atomic configuration remains very well at 400 K. Even when the temperature increases to 600 K (Fig. 1d), there is still no significant distortion of the geometric structure (Fig. S5†), indicating the high thermodynamic stability of Co/g-C3N4.
3.2 Adsorption of reaction species on the Co/g-C3N4 catalyst
It is important to investigate the interactions of adsorbates and the catalyst and gain fundamental insights into the catalytic performance. Thus, we first study the adsorption/co-adsorption of O2 and CO on Co/C3N4, and then explore their corresponding reaction mechanisms. As shown in Fig. 2a, O2 prefers to adsorb at the Co-Top site with the O–O bond parallel to the g-C3N4 sheet forming two Co–O bonds of 1.80 Å. In contrast to the O–O bond length (dO–O) of free O2 (1.23 Å), the dO–O of the adsorbed O2 molecule is elongated by about 0.15 Å on Co/g-C3N4 (1.38 Å). The adsorption energy of 2.12 eV is accompanied by a large amount of charge transfer (0.32 electrons) from Co/g-C3N4 to O2. Moreover, the 1π and 5σ orbitals of O2 at the energy region of −12.5 eV to the Fermi level well overlap with Co 3d and O2-2π* with a higher peak appearing around the Fermi level (Fig. 2b), suggesting the strong interaction between the Co atom and O2 molecule. In summary, the enlarged O–O bond, the charge transfer and the strong hybridization of Co 3d and O2 2p states demonstrate that the O2 molecule is activated on Co/g-C3N4.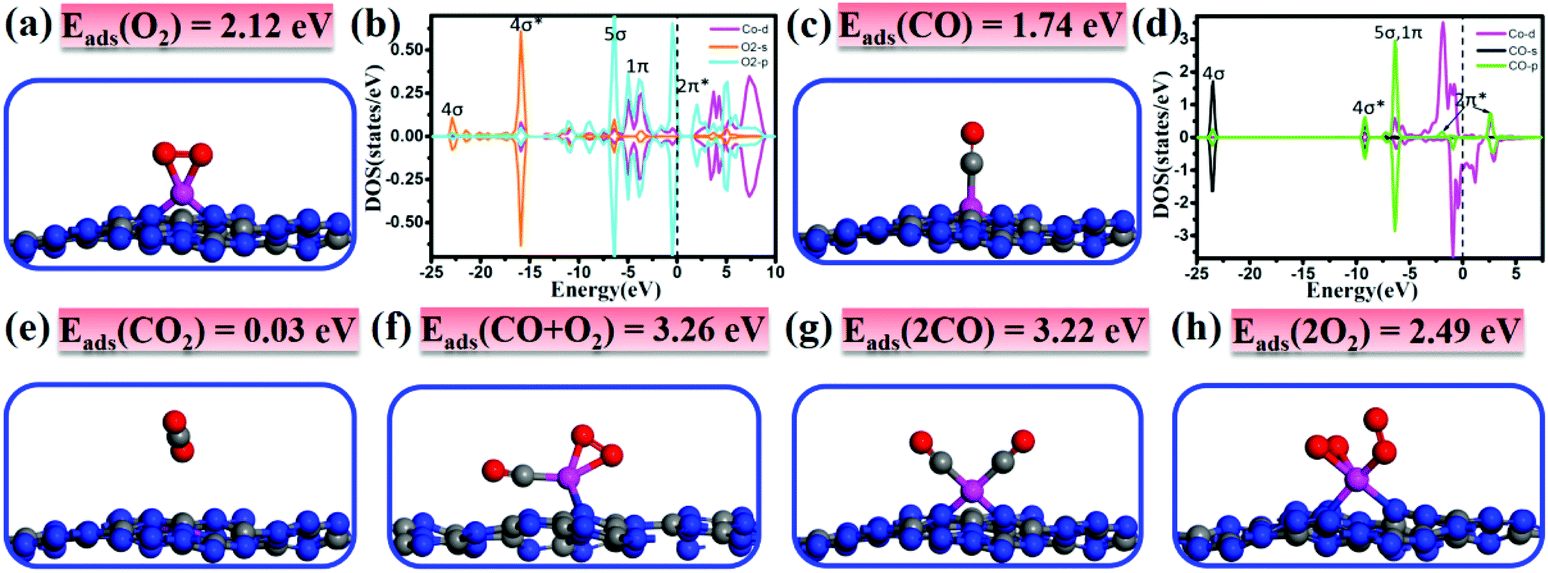 | ||
| Fig. 2 Adsorption structures and energies of (a) O2, (c) CO, (e) CO2, (f) CO + O2, (g) 2CO and (h) 2O2 on Co/g-C3N4. DOS of (b) O2 and (c) CO adsorbed on Co/g-C3N4 are also shown in the figure. Color scheme: red represents O atoms. | ||
The most stable CO adsorption configuration and corresponding DOS are presented in Fig. 2c and d, respectively. It is found that the CO molecule prefers to adsorb vertically at the Co-Top site by forming Co–C bonds of 1.76 Å and the C–O bond is elongated as compared with the free CO molecule (1.21 vs. 1.14 Å). The adsorption energy is 1.74 eV, which is smaller than that of O2 adsorption, suggesting that the adsorption of O2 is slightly more preferable than that of CO. The Bader charge analysis shows that the adsorbed CO transferred 0.16 electrons to Co/g-C3N4, which is in line with the result that the 4σ*, 5σ and 1π orbitals of CO in the energy range of −10 to −5 eV are hybridized with the Co 3d states. Upon CO adsorption, CO-2π* orbitals are also partially occupied because some electrons transfer from Co to CO-2π*. The charge transfer and the strong hybridization of the Co atom and CO molecule lead to the elongation of the C–O bond from 1.14 to 1.21 Å.
As the final product of CO oxidation, CO2 is physisorbed on Co/g-C3N4 with a rather small adsorption energy of 0.03 eV, as shown in Fig. 2e. The long distance of the Co–O bond (∼3.00 Å) and the weak interaction together with the negligible charge transfer between CO2 and the support indicate that the formed CO2 species would be spontaneously released.
The most stable adsorption structure of atomic O on Co/g-C3N4 is presented in Fig. S6,† in which atomic O prefers to adsorb on the top site of Co. Compared with that of O2 adsorption (Fig. 2a), the adsorption energy of atomic O is reduced to 1.37 eV and the Co–O bond is elongated to 1.84 Å. The adsorbed O atom is negatively charged by 0.23 electrons, indicating the formation of the O˙− species. The O˙− species is believed to be more reactive than O2 and would present great activity to oxidize CO.
In order to investigate the reaction mechanism by which CO oxidation proceeds on the catalyst surface, it is also necessary to study the capture performance (co-adsorption of the reactants). Note that the co-adsorption energy of CO + O2 (Fig. 2f) and 2CO (Fig. 2g) is significantly larger than the individual Eads of CO or O2 (3.26 and 3.22 vs. 1.74 or 2.12 eV), and larger than that of 2O2 (Fig. 2h) as well (2.49 eV). The large adsorption energies and the small adsorption energy difference between CO and O2 may facilitate the catalytic reaction of CO oxidation and decrease the reaction barriers accordingly.
According to the adsorption/co-adsorption energies of O2, CO, 2O2, CO + O2 and 2CO, the screening criteria and reaction mechanisms of CO oxidation based on SACs are thus proposed. As shown in Scheme 1: (I) the adsorption of O2 is slightly more preferable than that of CO, in which the ER mechanism (i.e., the reactant CO molecule approaching an already activated O2) would take place. (II) The co-adsorption of O2 and CO is stronger than the isolate O2 adsorption (low CO partial pressure), and the LH mechanism is preferred. (III) As two CO molecules are pre-adsorbed on the catalyst surface (high CO partial pressure), a TER mechanism, in which a free O2 molecule can be activated by two co-adsorbed CO molecules, can dominate the CO oxidation process.66,67
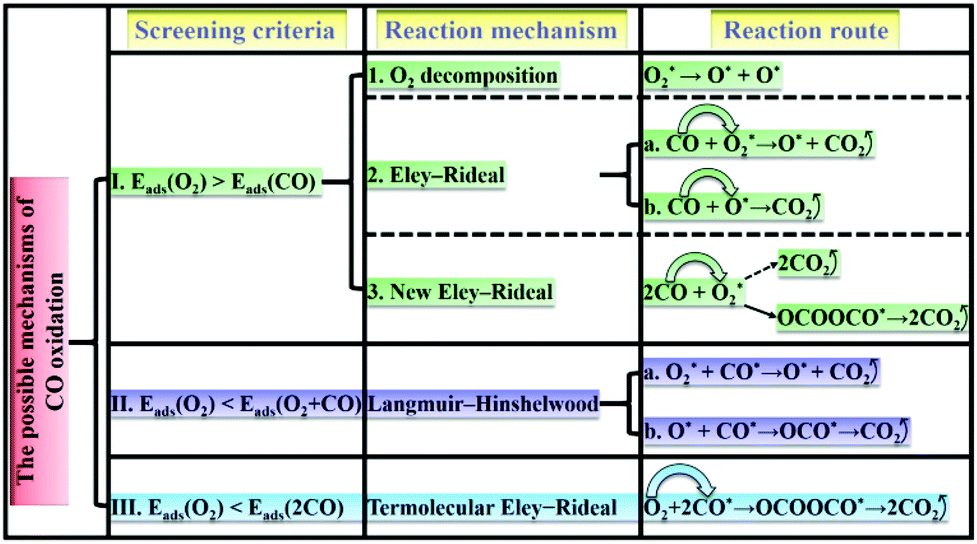 | ||
| Scheme 1 The screening criteria, reaction mechanism and reaction route of CO oxidation. | ||
3.3 CO oxidation on the Co/g-C3N4 catalyst
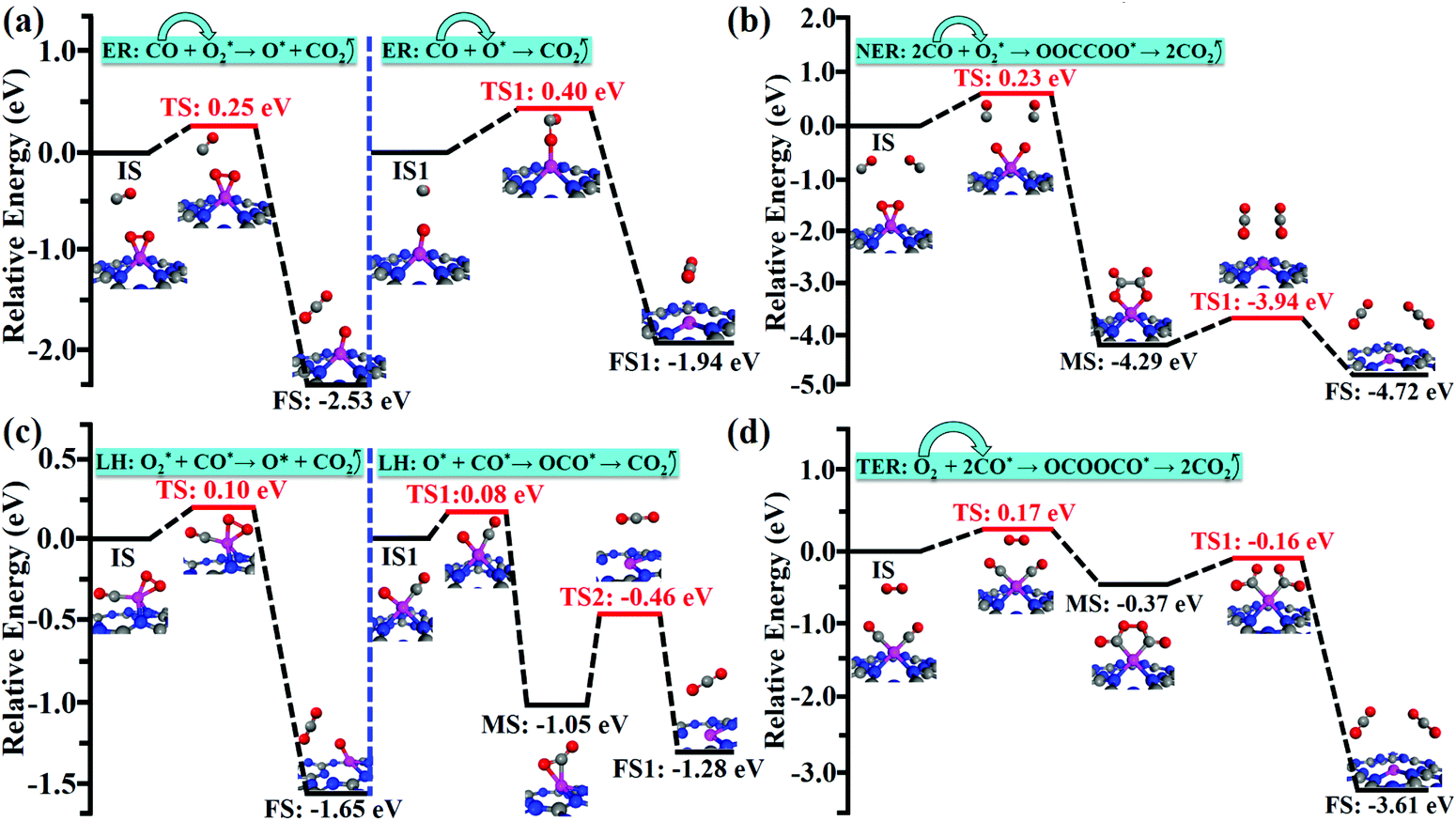 | ||
| Fig. 3 Minimum energy profiles and the configurations of different states including the IS, TS, MS and FS for CO oxidation on Co/g-C3N4. (a) ER mechanism: CO + O2* → CO2 + O* and CO + O* → CO2, (b) NER mechanism: 2CO + O2* → OOCCOO* → CO2, (c) LH mechanism: CO* + O2* → CO2 + O* and CO* + O* → OCO* → CO2, and (d) TER mechanism: O2 + 2CO* → OCOOCO* → 2CO2. Here, * denotes a catalytically active site. | ||
Compared with the reaction process of CO + O2* → CO2 + O* in the ER mechanism, the first CO2 molecule is easier to be released via the LH mechanism of O2* + CO* → CO2 + O* (0.25 vs. 0.10 eV). However, CO could readily react with an unreacted O atom for releasing the second CO2 molecule via the ER mechanism (CO + O* → CO2), which is superior to the LH mechanism (O* + CO* → OCO* → CO2) on the Co/g-C3N4 catalyst (0.40 vs. 0.59 eV). After releasing two CO2 molecules, the Co/g-C3N4 catalyst could be recovered for a new cycle of CO oxidation.
Compared with the ER, NER and LH mechanisms, the TER mechanism is preferable on Co/g-C3N4. The high spin density of the single Co atom during the CO oxidation process via the TER mechanism (Fig. S8†) is regarded as a key factor for the higher activity of Co/g-C3N4 SACs.
Moreover, the rate-limiting-steps of CO oxidation of Co/g-C3N4 are compared with those of metal single-atom supported catalysts (Table 1). Overall, the energy barriers with TER mechanisms on Co/g-C3N4 are lower than those on other substrates such as C2N,37h-BN,38 graphene,70 Mo2CO2,71 Ti2CO2,19 SnO2,72 and FeOx,6 suggesting that Co/g-C3N4 catalysts have better tolerance against CO poisoning.
| Substrate | Barriermechanism (eV) | Substrate | Barriermechanism (eV) |
|---|---|---|---|
| Co/g-C3N4 (this work) | 0.21TER, 0.35NER, 0.40ER, 0.59LH | Ti/Ti2CO219 |
0.25ER, 0.35LH |
| Cr/g-C2N37 | 0.59ER | Pt/Mo2CO271 |
1.39LH, 0.49TER |
| Mn/g-C2N37 | 0.64ER | Pt/graphene70 | 0.77ER, 0.59LH |
| V/g-C2N37 | 1.01ER | Pt/SnO272 |
0.51ER |
| g-C3N4/Pt(111)75 | 1.31ER, 0.77LH | Pt/FeOX6 |
0.79LH |
| Co/h-BN38 | 0.52ER | Co/FeOX76 |
0.45LH |
Based on the formula of the Sabatier rate73 of CO2 formation described in the computational details, there are six parameters that determine the kinetics: Eads (CO), Eads (O2), Eads (CO + O2), Eads (CO + CO), EIS and ETS. In order to find the independent variables characterizing the SAC in the microkinetic model, it is necessary to associate the adsorption energies of reactants with relative energies of the different states along the MEPs. Therefore, we focus on finding the Brønsted–Evans–Polanyi (BEP) linear correlation between EIS and ETS on Co/g-C3N4 from the adsorption energy of CO, O2, O, CO + O2, O + CO, and CO + CO. The ETS as a function of the EIS is shown in Fig. 4, with the data (Table S1†) fitted by using linear regression. ETS can be correlated with EIS using the following formula
| ETS = 0.99 × EIS − 0.24 (R2 = 0.98). |
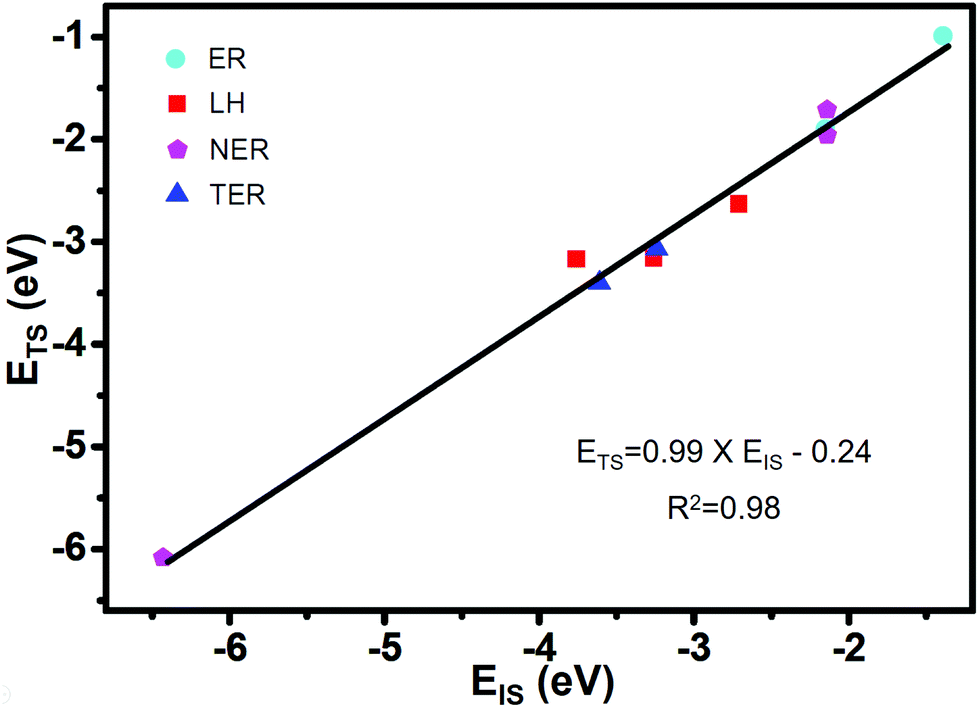 | ||
| Fig. 4 BEP linear correlations between the initial-state energies (EIS) and transition-state energies (ETS) for CO oxidation on Co/g-C3N4 catalysts via different mechanisms of ER, NER, LH and TER. EIS corresponding to the adsorption energy of CO, O2, O, CO + O2, O + CO and 2CO. | ||
Our results indicate the BEP relationships that have been established for different SACs,74 which can also describe CO oxidation via different mechanisms of ER, NER, LH and TER on Co/g-C3N4. Thus, the idea of Sabatier analysis may be applied to identify activity trends with different CO oxidation mechanisms on SACs.
3.4 The feasibility of Co/g-C3N4 in experiment
A wet chemical method is a suitable method to achieve high dispersion of single atoms, which can solve the problem of the possibility of experimental synthesis of SACs. Using CoCl2 as an appropriate metal precursor, the feasibility of experimental synthesis of Co/g-C3N4 is verified by calculating the energy profile of the synthetic route (Fig. 5a) and simulating the synthesis process by AIMD at 350 k (Fig. 5c). The formation of Co/g-C3N4 was simulated by using the following synthetic route (Fig. 5a).| CoCl2 + * → *CoCl2 (step 1) |
| *CoCl2 + 2H3O + 2e− → *Co(HCl)2 + 2H2O (step 2) |
| *Co(HCl)2 → *Co + 2HCl (step 3) |
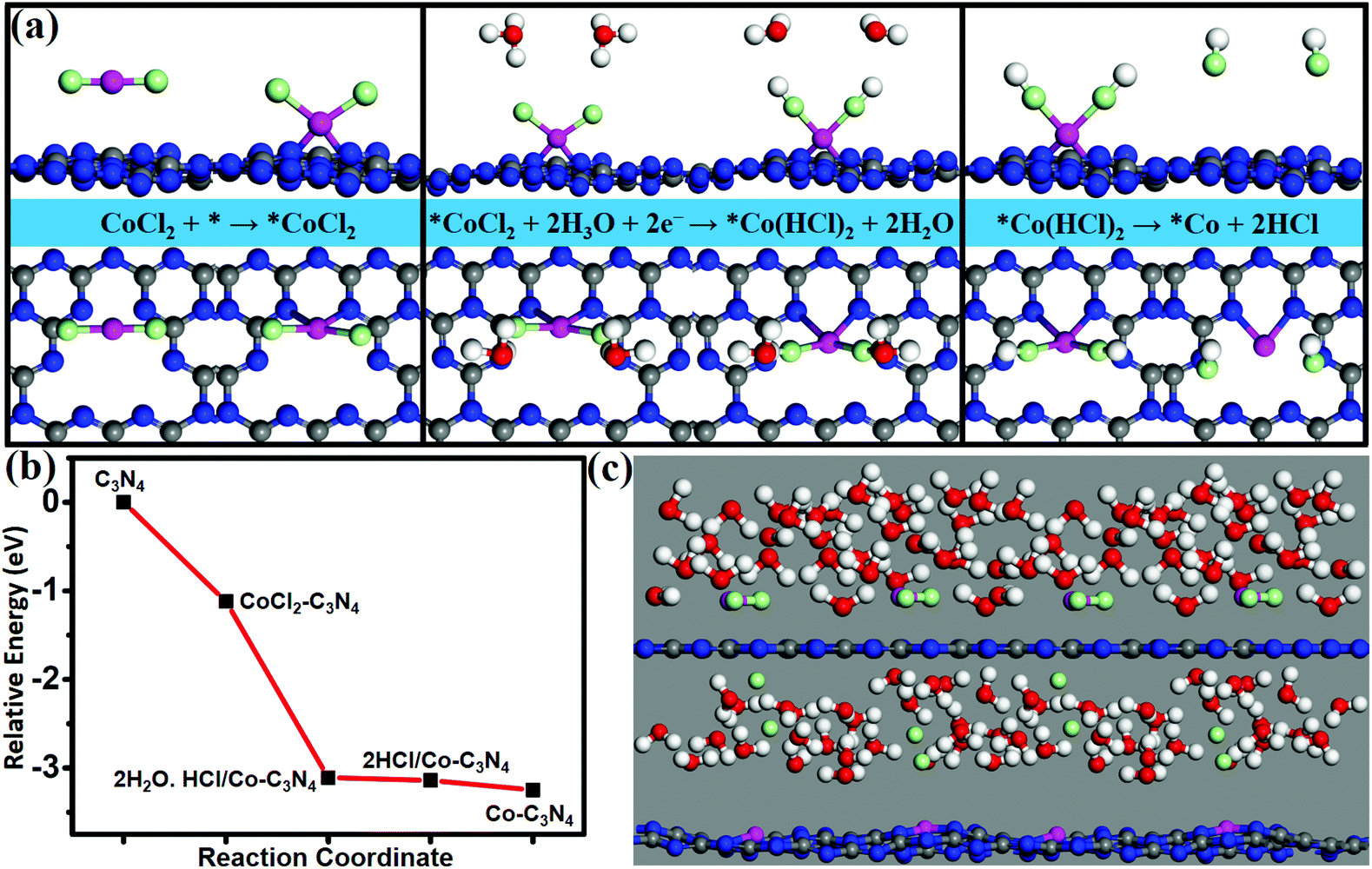 | ||
| Fig. 5 (a) Designed synthetic route for Co/g-C3N4. Here * denotes g-C3N4. (b) Energy profile of the proposed synthetic route. (c) Snapshots of atomic configurations of AIMD simulations for the synthetic process of Co/g-C3N4. The upper panel shows the initial structure, and the lower panel shows the final structure. The model consists of a 2 × 2 × 1 supercell of g-C3N4, two CoCl2 molecules, and 25H2O molecules. The simulation is run at 350 K for 2 ps with a time step of 1 fs. Color scheme: white and pale green represent H and Cl atoms, respectively. | ||
Adsorption of CoCl2 on a g-C3N4 monolayer is the first step with an adsorption energy of 1.12 eV. In the second step, H+ ions in the H3O+ groups approach Cl− ions in the adsorbed CoCl2*, and then H3O+ groups dissociate to form H–Cl bonds and produce H2O molecules; such a process is spontaneous and barrierless. Finally (in step 3), the HCl groups can be easily released from the surface with a binding energy as low as 0.11 eV, resulting in the formation of Co/g-C3N4. All the reaction steps can easily occur since they are spontaneous (Fig. 5b). In our AIMD simulations (Fig. 5c), CoCl2 in the solution is observed to adsorb on g-C3N4, and afterward the Cl− ions are desorbed, and the ultimate formation of the Co/g-C3N4 system occurs within 2 ps at 350 K. Thus, the highly stable and efficient Co/g-C3N4 catalysts may be synthesized by using CoCl2 as an appropriate metal precursor.
4. Conclusions
In summary, we have demonstrated that a g-C3N4 supported metal single-atom catalyst (Co/g-C3N4) exhibits excellent stability and catalytic activity for CO oxidation computationally. The stability of the SAC is guaranteed by the strong binding strength between metals and the substrate, the high diffusion barrier of the metals, and excellent thermal stability. The superior catalytic activity of Co/g-C3N4 is verified by its considerably low kinetic energy barriers for all possible reaction mechanisms, ranging from 0.21 to 0.59 eV. In addition, the results also show that Co supported on g-C3N4 shows higher activity for CO oxidation than on other substrates such as h-BN and graphene, and other kinds of carbon nitrides, MXenes and metal oxides. Based on the adsorption and co-adsorption energies of CO, O2, 2O2, CO + O2 and 2CO, the BEP relationships have been established for different reaction mechanisms on the Co/g-C3N4 catalyst, which can quantitatively assess the catalytic activity trends and provide guidelines for designing new SACs. Therefore, this work not only spans the non-noble metal single atom catalysts, but also provides useful insights and guidance for the design and development of novel low-cost and efficient SACs for many other important reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Natural Science Foundation of China (21525311, 21773027 and 21703032). We thank the Big Data Center of Southeast University for providing the facility support for the numerical calculations in this paper.References
- J. Kua and W. A. Goddard, J. Am. Chem. Soc., 1999, 121, 10928–10941 CrossRef CAS.
- J. Saavedra, T. Whittaker, Z. Chen, C. J. Pursell, R. M. Rioux and B. D. Chandler, Nat. Chem., 2016, 8, 584–589 CrossRef CAS PubMed.
- M. V. Twigg, Appl. Catal., B, 2007, 70, 2–15 CrossRef CAS.
- M. Shelef and R. W. McCabe, Catal. Today, 2000, 62, 35–50 CrossRef CAS.
- W. Lubitz and W. Tumas, Chem. Rev., 2007, 107, 3900–3903 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634 CrossRef CAS PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. Pereira Hernández, Y. Wang and A. K. Datye, Science, 2016, 353, 150 CrossRef CAS PubMed.
- B. Zhang, H. Asakura, J. Zhang, J. Zhang, S. De and N. Yan, Angew. Chem., Int. Ed., 2016, 55, 8319–8323 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- W. Zhang and W. Zheng, Adv. Funct. Mater., 2016, 26, 2988–2993 CrossRef CAS.
- J. Liu, ACS Catal., 2017, 7, 34–59 CrossRef CAS.
- M. Moses-DeBusk, M. Yoon, L. F. Allard, D. R. Mullins, Z. Wu, X. Yang, G. Veith, G. M. Stocks and C. K. Narula, J. Am. Chem. Soc., 2013, 135, 12634–12645 CrossRef CAS PubMed.
- B. Qiao, J. Liu, Y.-G. Wang, Q. Lin, X. Liu, A. Wang, J. Li, T. Zhang and J. Liu, ACS Catal., 2015, 5, 6249–6254 CrossRef CAS.
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard, L. Kovarik, A. K. Datye and Y. Wang, Science, 2017, 358, 1419–1423 CrossRef CAS PubMed.
- C. Wang, X.-K. Gu, H. Yan, Y. Lin, J. Li, D. Liu, W.-X. Li and J. Lu, ACS Catal., 2016, 7, 887–891 CrossRef.
- Y. Lu, J. Wang, L. Yu, L. Kovarik, X. Zhang, A. S. Hoffman, A. Gallo, S. R. Bare, D. Sokaras, T. Kroll, V. Dagle, H. Xin and A. M. Karim, Nat. Catal., 2018, 2, 149–156 CrossRef.
- J.-J. Chen, X.-N. Li, Q. Chen, Q.-Y. Liu, L.-X. Jiang and S.-G. He, J. Am. Chem. Soc., 2019, 141, 2027–2034 CrossRef CAS PubMed.
- X. Zhang, J. Lei, D. Wu, X. Zhao, Y. Jing and Z. Zhou, J. Mater. Chem. A, 2016, 4, 4871–4876 RSC.
- C. Du, H. Lin, B. Lin, Z. Ma, T. Hou, J. Tang and Y. Li, J. Mater. Chem. A, 2015, 3, 23113–23119 RSC.
- C. Yan, H. Li, Y. Ye, H. Wu, F. Cai, R. Si, J. Xiao, S. Miao, S. Xie, F. Yang, Y. Li, G. Wang and X. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC.
- W. Bi, X. Li, R. You, M. Chen, R. Yuan, W. Huang, X. Wu, W. Chu, C. Wu and Y. Xie, Adv. Mater., 2018, 30, 1706617 CrossRef PubMed.
- C. Zhang, S. Yang, J. Wu, M. Liu, S. Yazdi, M. Ren, J. Sha, J. Zhong, K. Nie, A. S. Jalilov, Z. Li, H. Li, B. I. Yakobson, Q. Wu, E. Ringe, H. Xu, P. M. Ajayan and J. M. Tour, Adv. Energy Mater., 2018, 8, 1703487 CrossRef.
- C. Ling, Q. Li, A. Du and J. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 36866–36872 CrossRef CAS PubMed.
- X. Cui, W. An, X. Liu, H. Wang, Y. Men and J. Wang, Nanoscale, 2018, 10, 15262–15272 RSC.
- J. Mahmood, S.-M. Jung, S.-J. Kim, J. Park, J.-W. Yoo and J.-B. Baek, Chem. Mater., 2015, 27, 4860–4864 CrossRef CAS.
- L. Hui, Y. Xue, H. Yu, Y. Liu, Y. Fang, C. Xing, B. Huang and Y. Li, J. Am. Chem. Soc., 2019, 141, 10677–10683 CrossRef CAS PubMed.
- Y. Xue, B. Huang, Y. Yi, Y. Guo, Z. Zuo, Y. Li, Z. Jia, H. Liu and Y. Li, Nat. Commun., 2018, 9, 1460 CrossRef PubMed.
- C. Ling, L. Shi, Y. Ouyang, X. C. Zeng and J. Wang, Nano Lett., 2017, 17, 5133–5139 CrossRef CAS PubMed.
- C. Hu, H. Liu, Y. Liu, J.-F. Chen, Y. Li and L. Dai, Nano Energy, 2019, 103874 CrossRef CAS.
- C. Choi, S. Back, N.-Y. Kim, J. Lim, Y.-H. Kim and Y. Jung, ACS Catal., 2018, 8, 7517–7525 CrossRef CAS.
- X. F. Li, Q. K. Li, J. Cheng, L. Liu, Q. Yan, Y. Wu, X. H. Zhang, Z. Y. Wang, Q. Qiu and Y. Luo, J. Am. Chem. Soc., 2016, 138, 8706–8709 CrossRef CAS PubMed.
- C. Ling, Y. Ouyang, Q. Li, X. Bai, X. Mao, A. Du and J. Wang, Small Methods, 2018, 1800376 Search PubMed.
- J. Zhao and Z. Chen, J. Am. Chem. Soc., 2017, 139, 12480–12487 CrossRef CAS PubMed.
- H.-R. Zhu, Y.-L. Hu, S.-H. Wei and D.-Y. Hua, J. Phys. Chem. C, 2019, 123, 4274–4281 CrossRef CAS.
- D. W. Ma, T. Li, Q. Wang, G. Yang, C. He, B. Ma and Z. Lu, Carbon, 2015, 95, 756–765 CrossRef CAS.
- D. W. Ma, Q. Wang, X. Yan, X. Zhang, C. He, D. Zhou, Y. Tang, Z. Lu and Z. Yang, Carbon, 2016, 105, 463–473 CrossRef CAS.
- S. Lin, X. Ye, R. S. Johnson and H. Guo, J. Phys. Chem. C, 2013, 117, 17319–17326 CrossRef CAS.
- G. Xu, R. Wang, Y. Ding, Z. Lu, D. Ma and Z. Yang, J. Phys. Chem. C, 2018, 122, 23481–23492 CrossRef CAS.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang, H. Sun, P. An, W. Chen, Z. Guo, C. Lee, D. Chen, I. Shakir, M. Liu, T. Hu, Y. Li, A. I. Kirkland, X. Duan and Y. Huang, Nat. Catal., 2018, 1, 63–72 CrossRef CAS.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Nat. Catal., 2018, 1, 339–348 CrossRef CAS.
- T. He, S. K. Matta, G. Will and A. Du, Small Methods, 2019, 1800419 CrossRef.
- X. Zhang, A. Chen, Z. Zhang, M. Jiao and Z. Zhou, J. Mater. Chem. A, 2018, 6, 11446–11452 RSC.
- A. M. Abdel-Mageed, B. Rungtaweevoranit, M. Parlinska-Wojtan, X. Pei, O. M. Yaghi and R. J. Behm, J. Am. Chem. Soc., 2019, 141, 5201–5210 CrossRef CAS PubMed.
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609–1612 CrossRef CAS.
- X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen, Y. Hou, X. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680–1681 CrossRef CAS PubMed.
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS PubMed.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- X. Chen, J. Zhang, X. Fu, M. Antonietti and X. Wang, J. Am. Chem. Soc., 2009, 131, 11658–11659 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S. Z. Qiao, J. Am. Chem. Soc., 2017, 139, 3336–3339 CrossRef CAS PubMed.
- W. Niu, K. Marcus, L. Zhou, Z. Li, L. Shi, K. Liang and Y. Yang, ACS Catal., 2018, 8, 1926–1931 CrossRef CAS.
- Y. Wu, C. Li, W. Liu, H. Li, Y. Gong, L. Niu, X. Liu, C. Sun and S. Xu, Nanoscale, 2019, 11, 5064–5071 RSC.
- M. Montano, K. Bratlie, M. Salmeron and G. A. Somorjai, J. Am. Chem. Soc., 2006, 128, 13229–13234 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Gao, Y. Jiao, E. R. Waclawik and A. Du, J. Am. Chem. Soc., 2016, 138, 6292–6297 CrossRef CAS PubMed.
- Z. Chen, J. Zhao, C. R. Cabrera and Z. Chen, Small Methods, 2018, 1800368 Search PubMed.
- C. Ling, X. Niu, Q. Li, A. Du and J. Wang, J. Am. Chem. Soc., 2018, 140, 14161–14168 CrossRef CAS PubMed.
- C. Liu, Q. Li, C. Wu, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, J. Am. Chem. Soc., 2019, 141, 2884–2888 CrossRef CAS PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- C. Liu, Y. Tan, S. Lin, H. Li, X. Wu, L. Li, Y. Pei and X. C. Zeng, J. Am. Chem. Soc., 2013, 135, 2583–2595 CrossRef CAS PubMed.
- K. Mao, L. Li, W. Zhang, Y. Pei, X. C. Zeng, X. Wu and J. Yang, Sci. Rep., 2014, 4, 5441 CrossRef CAS PubMed.
- S. L. Li, H. Yin, X. Kan, L. Y. Gan, U. Schwingenschlogl and Y. Zhao, Phys. Chem. Chem. Phys., 2017, 19, 30069–30077 RSC.
- F. Li and Z. Chen, Nanoscale, 2018, 10, 15696–15705 RSC.
- Y. Tang, Z. Yang and X. Dai, Phys. Chem. Chem. Phys., 2012, 14, 16566–16572 RSC.
- C. Cheng, X. Zhang, M. Wang, S. Wang and Z. Yang, Phys. Chem. Chem. Phys., 2018, 20, 3504–3513 RSC.
- S. Li, Z. Lu, Z. Yang and X. Chu, Sens. Actuators, B, 2014, 202, 83–92 CrossRef CAS.
- T. Bligaard, J. K. Nørskov, S. Dahl, J. Matthiesen, C. H. Christensen and J. Sehested, J. Catal., 2004, 224, 206–217 CrossRef CAS.
- H. Xu, C.-Q. Xu, D. Cheng and J. Li, Catal. Sci. Technol., 2017, 7, 5860–5871 RSC.
- S. Wang, Y. Feng, M. Yu, Q. Wan and S. Lin, ACS Appl. Mater. Interfaces, 2017, 9, 33267–33273 CrossRef CAS PubMed.
- F. Li, Y. Li, X. C. Zeng and Z. Chen, ACS Catal., 2014, 5, 544–552 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr07726j |
| This journal is © The Royal Society of Chemistry 2020 |