
Controllable and stable organometallic redox mediators for lithium oxygen batteries
Won-Jin
Kwak†
 a,
Atif
Mahammed
b,
Hun
Kim
a,
Trung Thien
Nguyen
a,
Zeev
Gross
*b,
Doron
Aurbach
*c and
Yang-Kook
Sun
*ad
a,
Atif
Mahammed
b,
Hun
Kim
a,
Trung Thien
Nguyen
a,
Zeev
Gross
*b,
Doron
Aurbach
*c and
Yang-Kook
Sun
*ad
aDepartment of Energy Engineering, Hanyang University, Seoul 04763, Republic of Korea. E-mail: yksun@hanyang.ac.kr
bSchulich Faculty of Chemistry, Technion-Israel Institute of Technology, Haifa 3200008, Israel. E-mail: chr10zg@technion.ac.il
cDepartment of Chemistry and BINA (BIU Institute of Nano-technology and Advanced Materials), Bar-Ilan University, Ramat-Gan 5290002, Israel. E-mail: doron.aurbach@biu.ac.il
dDepartment of Chemical Engineering, Hanyang University, Seoul 04763, Republic of Korea
First published on 29th August 2019
Abstract
The use of electrocatalysis in lithium–oxygen batteries is mandatory for reducing the over-potentials of the oxygen evolution reaction (OER), below the levels that endanger the anodic stability of the electrolyte solutions and the carbon electrodes. The most effective catalysts for the OER are solubilized redox mediators that may be oxidized at relatively low potentials, but still capable of oxidizing Li2O2 back to molecular oxygen. Since for the effective and long-term utilization of redox mediators in lithium–oxygen cells a clear evaluation of their stability is essential, we have developed a useful methodology for that purpose. This revealed, quite surprisingly, that most commonly used redox mediators are unstable in lithium–oxygen cells, even under argon atmosphere and without being in contact with Li anodes. Using the abovementioned methodology for evaluating efficiency, we now introduce corrole-chelated metal complexes as stable redox mediators in lithium oxygen batteries. This was achieved by taking advantage of the facile methods for introducing changes in the corrole ligands and by choosing properly the central transition metal cation, two aspects that allow for adjusting the redox properties of the metal complexes for the operative voltage window. We outline further directions and believe that this work will promote optimized selection of redox mediators for lithium–oxygen batteries.
New conceptsRechargeable lithium–oxygen (Li–O2) batteries are very important due to their very high theoretical energy density. If they will work properly, they will be able to rival internal combustion engines in terms of energy density and cost, thus revolutionizing electro-mobility and ground transportation. The operation of rechargeable Li–O2 batteries depends crucially on the reversible formation/decomposition of Li2O2 at the cathode upon discharge/charge cycling. One of the greatest challenges with these systems is their recharge, i.e. decomposing Li2O2, at low enough potentials, that do not endanger the stability of the electrolyte solution and the carbon cathode. Since Li2O2 is electrically insulating, its decomposition requires a too high over-potential – above 1 V even at low rates. Consequently, it mandatory to use redox mediators (RMs) in solution phase in Li–O2 batteries is mandatory. Their appropriate selection is critically important. We seek for RMs that are oxidized on the cathodes of Li – ion cells low enough potentials and then these RMs oxidize the Li2O2 from solution phase. Unfortunately, many RMs suggested and tested so far suffer from intrinsic instability problems. We developed a systematic methodology to select them properly. Here we introduce corrole-chelated metal complexes as effective and intrinsically stable RMs for Li–O2 batteries. The properties of these RMs can be controlled by the nature of their central metallic and the corrole structure. By a systematic study we were able to find a new, intrinsically stable RM for Li–O2 cells – corrole-chelated copper complex and to prove its superiority over benchmark systems. We anticipate that this study will promote a rational design for the development of most suitable redox mediators for active metal (Li, Na) oxygen batteries. |
Introduction
Lithium-ion rechargeable batteries are the most common energy sources for today's portable electronics such as laptops, smartphones, and camcorders, for which the practically attainable specific energy density of about 100–150 W h kg−1 is enough to fulfil the main requirements of this market. But electric cars require 300–400 W h kg−1 with an output power density of about 2000 W h kg−1, much above the performances of even the most advanced Li-ion technology (lithium iron phosphate). Considering that the maximum theoretical gravimetric energy density values of lithium–air batteries can reach up to 1000–2000 W h kg−1 with respect to the mass of the active components (electrodes, electrolyte solution) they were proposed a few years ago as possible power sources for applications in which very high energy density power sources are required.1 Properly working rechargeable lithium–oxygen (Li–O2) batteries may rival internal combustion engines (ICE) in terms of gravimetric specific energy density. Their successful development could hence promote the electro-mobility revolution and commence a wide and extensive use of electric vehicles propelled by electrochemical power sources. Despite intensive work on these systems by most prominent research groups throughout the world for more than a decade, this battery technology has not yet flourished due to a plethora of intrinsic problems, among which the two major problems are: (a) very limited stability upon cycling even at limited capacity (i.e. pronounced capacity fading); and (b) low energy efficiency due to huge hysteresis in the discharge (oxygen reduction reaction – ORR) and charge (oxygen evolution reaction – OER) potentials.2,3 Systematic studies uncovered, beyond trivial stability and durability problems related to the Li anodes in Li–O2 cells, severe stability problems at the cathode side that are not related to the Li anode in these cells.4 It actually became clear that all polar aprotic solvents tested to date are not stable toward side reactions with the oxygen reduction species formed in solution: peroxide and superoxide (solution phase) moieties (very strong bases and nucleophiles) in the presence of Li ions. The latter species can be considered as hard electrophiles whose presence facilitates many options for reactions between the basic and nucleophilic reduced oxygen species and all kinds of non-aqueous solvents.4 Another stability problem of these cells is the high anodic over-potential that may be required to oxidize Li-peroxide (the major discharge product of the ORR) back to molecular oxygen, because of its electrical insulating nature. This issue is critically important in high specific capacity cells, in which thick Li-peroxide deposits are formed upon discharge (in purpose). The high over-potentials required for fully oxidizing thick Li-peroxide deposits during the OER, endanger the stability of both the carbon cathodes and the electrolyte solutions. Hence, effective electro-catalysis in Li–O2 cells is mandatory and one suggested solution is to use solid catalysts.5 However, solid catalysts can also accelerate the decomposition of the electrolyte solution and may not be effective when the Li-peroxide precipitates as thick deposits on the cathode surface.6 Thereby, a better option is the use of homogeneous catalysis in Li–O2 cells by redox mediators (RMs) in solution phase. Indeed, RMs have been actively discussed and widely studied as soluble catalysts in Li–O2 cells.7–11 The RMs used are moieties with redox activity at low enough potentials, much below the anodic stability limit of the cells components, that are easily oxidized upon charging (on bare, unblocked cathode's sites). Their oxidized form then oxidizes the Li-peroxide deposits back to O2. Each RM has its unique oxidation–reduction potential, which can decompose Li2O2 at lower potential through chemical reactions during charge. Unlike solid catalysts, capable of exerting a catalytic effect only at contact points with both the cathode surface and the solid Li2O2 deposits, RMs decompose the Li2O2 deposits at their contact with the solution phase. Recent reports provide improved insight regarding the reaction mechanisms of RMs in Li–O2 batteries;12–17 and also for the kinetics of Li2O2 decomposition by different classes of RMs.18 Very little is however known about the intrinsic stability of the most explored RMs, although this aspect may safely be considered as the most important factor. In our work we assess stability of RMs upon voltammetric cycling of their solutions (DEGDME/LiTFSI) in Li–O2 cells under inert atmosphere (argon), while having no contact with the reactive Li anodes in the cells (using be-compartments cells). If their CVs remain invariant upon prolonged cycling, they can be defined as stable RMs.Most of the relevant literature has focused on the side reactions of RMs with the Li metal anode as a main cause of deterioration of RMs activity in Li–O2 cells.19 However, there might be intrinsic stability limitations of RMs in Li–O2 cells beyond the trivial concerns related to detrimental interactions with the Li anodes. It is hence important to conduct very systematic studies of Li–O2 cells in which the Li anode is fully isolated from the cathode side, so there are no detrimental “cross talks” between the cathode and the anode sides. Only by using such cells it is possible to find and confirm intrinsically stable components for Li–O2 cells.
Considering the above conclusion, we have developed bi-compartments Li–O2 cells in which the Li metal anode compartment is chemically isolated from the cathode side by a selective ceramic membrane that can transfer only Li ions.20 Our systematic study of the most commonly used RMs, including TEMPO, TTF, DMPZ, LiI and LiBr,21 clearly revealed that all of them suffer from stability problems that are not related to side reactions with Li metal anodes. Cycling the same cells used as Li–O2 cells (same cathodes and separated/isolated Li anodes) with the same glyme based electrolyte solutions which includes these 5 RMs under argon atmosphere within the relevant potential range, clearly shows that these RMs are unstable. Their redox response decays upon repeated voltammetric cycling.21 Their instability may become higher under oxygen atmosphere, during ORR and OER upon cycling in bi-compartments Li–O2 cells.21 That study clearly proved that both established and newly proposed RMs for Li–O2 batteries shall be tested in bi-compartments cells under argon as a mandatory step for their appropriate selection.
In light of our finding about the instability and possible parasitic reactions of several widely studied RMs in Li–O2 batteries,21,22 we now introduce a series of corrole-chelated metal complexes23–26 as potential RMs for Li–O2 batteries. As described further, the properties of these core–shell materials are adjustable via both the metallic core and the organic shell. Both the chosen transition metal in the core and the exact structure of the corrole ligands (shell) determine the redox potentials of these compounds. The structure of the corrole ligands in turn may affect very considerably the stability of these RMs. For instance, RMs with fluorinated corroles may be more stable in Li–O2 cells than the reference compounds.
This study examined several types of corrole-chelated metal complexes as RMs for Li–O2 cells, in comparison with several previously studied RMs. We show that this class of RMs in which the core redox centers (transition metal cations) are surrounded by organic ligands and whose redox properties are adjustable through both the core and the shell, may provide intrinsically stable RMs for Li–O2 cells. The main goal of the work was a preliminary selection and assessment of suitable new RMs belonging to the transition metals (TM) corrole complex materials, for Li–O2 cells. The criterion was a stable behaviour upon cycling in the bi-compartments cells we developed, under argon within the potential domain of their redox activity. Major advantages of TM corroles is their outstanding hydrolytic stability and the opportunity of easy tuning of redox potential by proper choice of the chelated metal ion and substitution on the corrole. Then we examined to what extent RMs demonstrating stable behaviour under argon, improve the behaviour of Li–O2 cells compared to reference and benchmark Li–O2 systems.
Results and discussion
Fig. 1 provides very clearly the starting point and the incentive for the work reported herein. It shows voltammetric studies of solutions containing three representative RMs: TTF, DMPZ and TEMPO (Fig. 1a) in the newly developed bi-compartments cells,20 under argon atmosphere. The solutions selected for this study are composed of diglyme (DEGDME) and LiN(SO2CF3)2 (LiTFSI), which are not perfectly stable in Li–O2 cells because the solvent is attacked by the oxygen reduction products formed upon ORR.6 They are nevertheless widely used in these systems because there are no better options yet and they are actually been considered as the least reactive among all other polar aprotic solutions of relevance to Li–O2 cells. Most important for the current study is fact that any possible instability of the RMs due to reactions with the Li anodes is fully omitted, since the Li anodes are isolated and there is no contact between them and the RMs in the bi-compartment cells we used. Indeed, the 1 m (molal) LiTFSI/DEGDME solutions were first confirmed to be fully stable under argon in the cathode compartment, at anodic potentials below 4.2 V vs. Li, which is quite sufficient for Li–O2 cells in which RMs are used. The voltammetric charts in Fig. 1 are striking, showing that all the three well-studied RMs are not stable, within the potential window relevant to ORR and OER, even under argon and while being isolated from the Li – metal anodes. Although the stability issues of these RMs were previously discussed,20 the new results enforce the conclusion that a search for more stable RMs may be considered as mandatory for making progress in this field.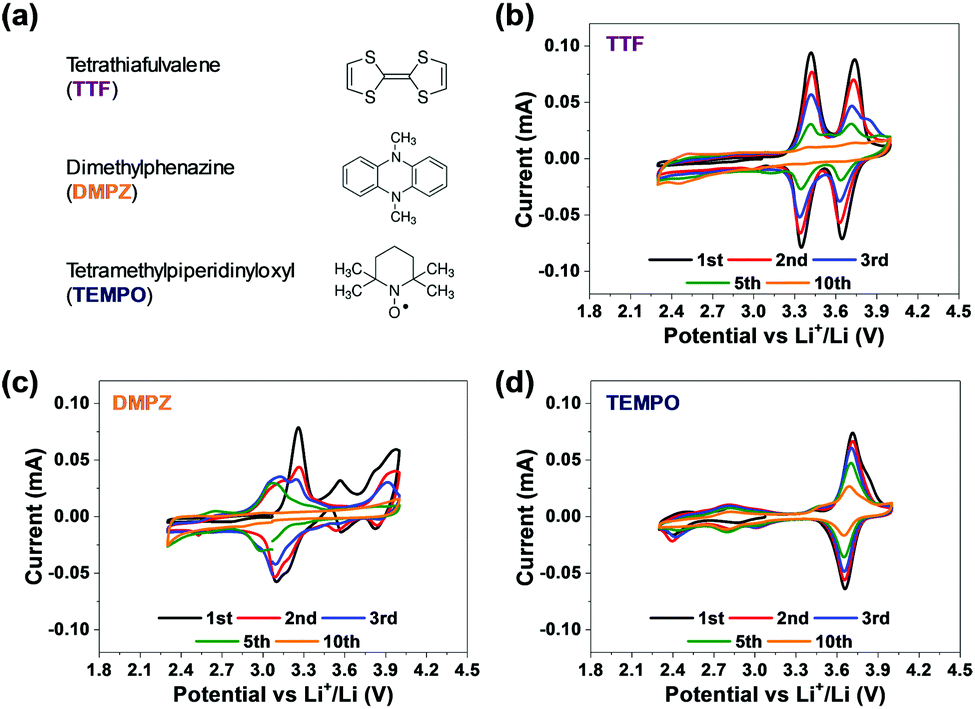 | ||
| Fig. 1 (a) The molecular structure of representative RMs (TTF, DMPZ, and TEMPO) and the (b–d) cyclic voltammogram of bi-compartment cells (with SE) for 10 cycles under Ar atmosphere using 1 m LiTFSI/DEGDME solutions with 0.02 m of (b) TTF, (c) DMPZ, and (d) TEMPO (scan rate: 0.1 mV s−1, voltage range: 2.3–4.0 V). | ||
Organometallic complexes, in which the redox center is composed of a transition metal cation that is chelated and stabilized by non-innocent ligands, are attractive RMs for Li–O2 batteries. In this study we have chosen to examine the potential of metallocorroles (Fig. 2a) due to their following features: (a) hydrolytic stability that exceeds by far that of other metal–ligand complexes;23,27 (b) proven excellence as electrocatalysts for both reduction (protons, carbon dioxide, dioxygen, hydrogen peroxide)28–31 and oxidations (water and organic compounds);32–34 and (c) the easy tuning of their redox potentials via the identity of the metal ion and changes on the macrocyclic ligand.35–37 The currently investigated series is composed of three metal ions – Co, Mn, and Cu, chelated by two corroles that differ in the number of strongly electron-withdrawing F atoms: fifteen for the M(tpfc) and six for the M(tdfc) complexes. The first cyclic voltammetric charts of these six complexes (Fig. 2b, measured under argon in the bi-compartments cells) disclose the effect of both the metals and the corroles on the redox potentials: shifted positively in the order of Mn > Co > Cu and M(tpfc) > M(tdfc) for any of the metals.
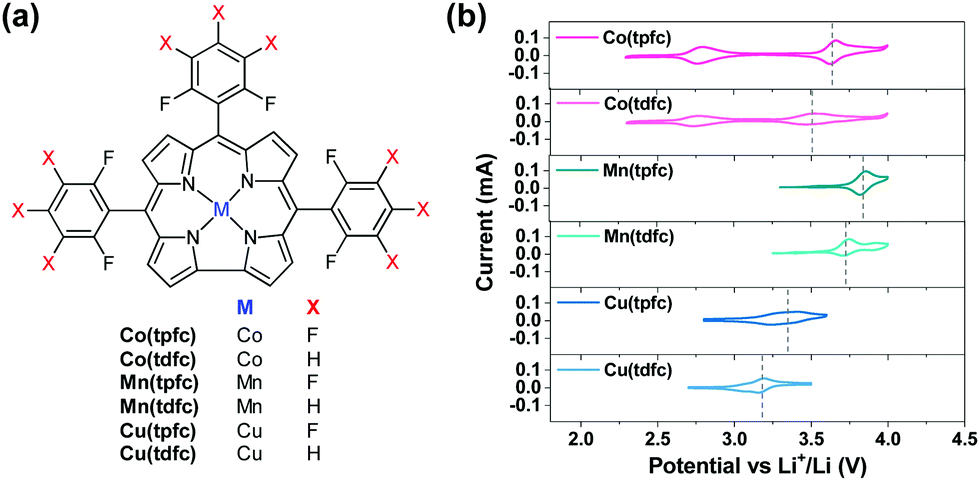 | ||
| Fig. 2 (a) The molecular structures of corrole-based organometallic RMs (ORMs) and (b) cyclic voltammograms of bi-compartments cells at the first cycles under Ar atmosphere using 1 m LiTFSI/DEGDME solutions with 0.02 m of ORMs (scan rate: 0.1 mV s−1, voltage ranges are controlled for each ORM). | ||
Based on the extensive knowledge regarding redox processes of metallocorroles in other solvents and vs. Ag/AgCl references, the single redox process seen here for the Cu and Mn corroles between 2.3–4.0 V vs. Li+/Li may safely be attributed to Cu3+/Cu2+ and Mn4+/Mn3+ couples, respectively.38–40 The Co corroles displays two redox potentials in this electrochemical window, Co3+/Co2+ and Co4+/Co3+, as observed in previous studies that focused on their role as electrocatalysts for water oxidation.41,42 The more positive redox potentials of the more heavily fluorinated corroles is also consistent with expectations,43 but it is still remarkable to note that the redox potentials of these RMs may be increased by about 150 mV by a very remote change on the supporting ligand.
The conclusions regarding the suitability for Li–O2 cells are quite clear: (a) the oxidation potential of the two Mn corroles might be too high; (b) the same may hold for the upper oxidation potential of the heavier fluorinated Co corrole, Co(tpfc); (c) the lower redox potential activity of the Co corroles (the Co3+/Co2+ couple of <2.7 V) is also not suitable for oxidizing effectively Li-peroxide to molecular oxygen upon charging (OER); and (d) the Cu corroles appear to be the most interesting RMs in this group. They are the only one to fulfill the most important requirement: redox activity in the electrochemical window of the Li–O2 cells.
The next step was to examine the six metallocorroles in DEGDME/LiTFSI solutions in bi-compartments cells – without contact with Li metal anodes, under argon atmosphere rather than aerobic conditions and at potential ranges relevant to Li–O2 batteries – by applying 50 consecutive cycles (at 0.1 mV s−1). The reference experiment, a cell without RM in solution, is shown in Fig. 3a, while the voltammograms of Fig. 3b–g were obtained for cells containing the corrole-based RMs. Comparison to the charts shown in Fig. 1 clearly uncovers a higher intrinsic stability of the metallocorroles compared to TTF, TEMPO and DMPZ; and the data of Fig. 3 further discloses this to be in the order of Co ≪ Mn < Cu, with Cu(tpfc) and Cu(tdfc) displaying unmatched performances. The latter complex, whose potential was lowest, was also examined under oxygen atmosphere (Fig. 3h), at conditions that include/exclude the formation/decomposition of Li2O2, achieved by controlling the voltage range at 2.3–3.6 V and 2.9–3.6 V, respectively. These experiments uncover Cu(tdfc) as the highest stability RM with the lowest redox potential that we have measured or aware of. This is consistent with corrole-chelated copper complexes being outstandingly stable in both the di- and trivalent oxidation states.44
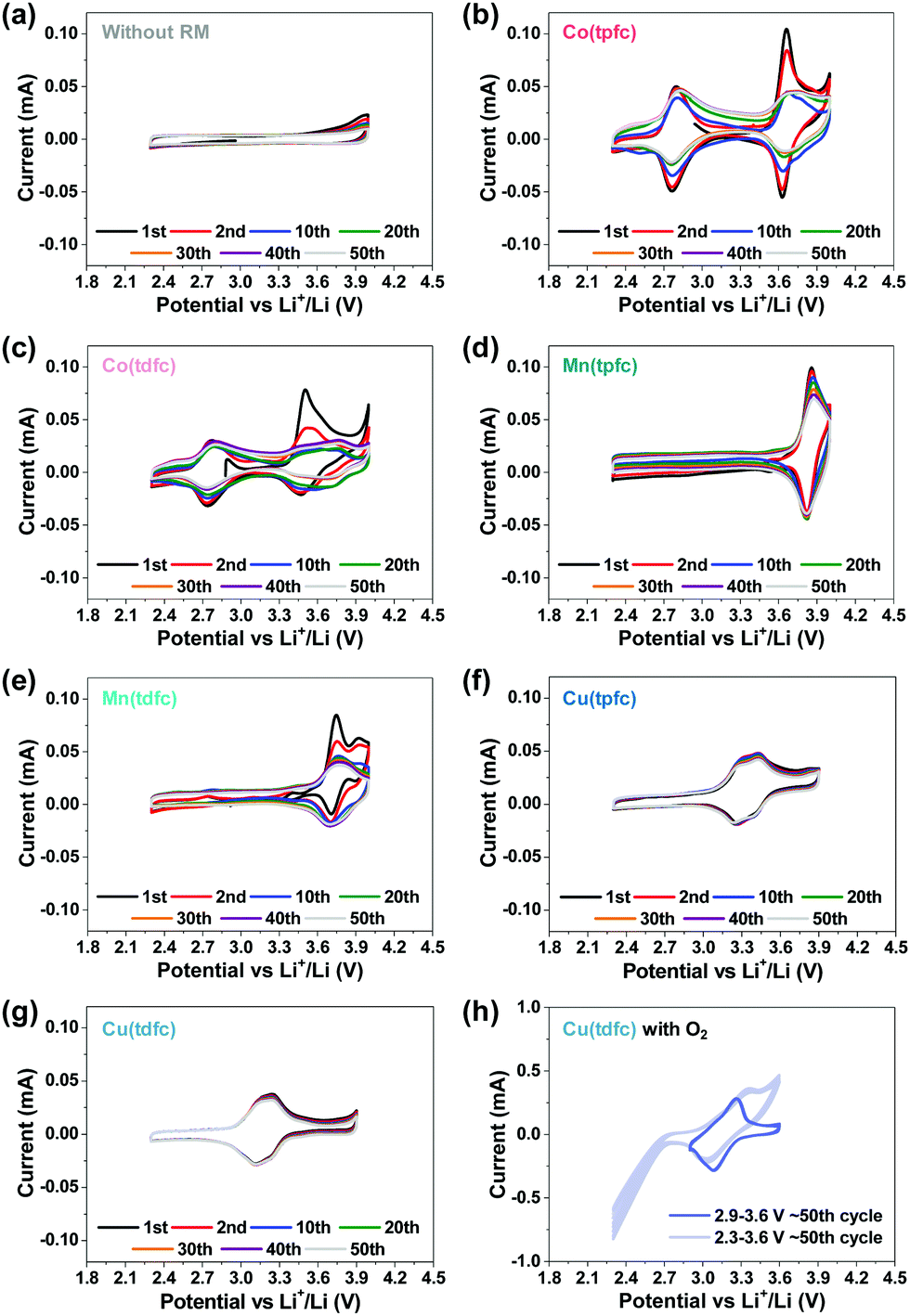 | ||
| Fig. 3 Cyclic voltammograms of bi-compartment cells (with SE) for 50 cycles using 1 m LiTFSI/DEGDME solutions (scan rate: 0.1 mV s−1, voltage range: 2.3–4.0 V). Charts (a)–(g) relate to cells under argon atmosphere while chart (h) relate to a cell under oxygen atmosphere. Chart (a) reflects the reference experiment – no RM is present in solutions. The rest of the charts relate to cells containing 0.02 m of ORMs. Charts (b) and (c), (d) and (e), (f) and (g), relate to RMs with Co, Mn and Cu, respectively. Charts (b), (d) and (f) relate to corrole rings containing pentafluorophenyl ligands while charts (c), (e) and (g) relate to corrole rings with difluorophenyl ligands. Chart (h) relates to a solution containing 0.02 m Cu (tdfc) under O2 atmosphere, displaying two sets of voltammograms with or without Li2O2 formation and decomposition by controlling the voltage range (3.6–2.3 V and 3.9–3.6 V) at 1.0 mV s−1. | ||
Solutions that experienced 50 cycles under oxygen atmosphere as in Fig. 3h were examined by UV-vis spectroscopy (Fig. 4a), not only for gaining further confidence in the stability of the catalysts, but also a more advanced understanding regarding their mechanism of action. For the latter aspect, the spectra of both neutral Cu(tdfc) and its reduced form [Cu(tdfc)]− were recorded in ethanol solution (Fig. 4b): the spectrum of the copper(III) corrole is characterized by a maximum absorbance at 406 nm, which upon reduction to copper(II) shifts to 429 nm. Investigation of the working solutions was performed by disassembling the cycled bi-compartment cells and diluting the solutions containing [CuII(tdfc)]− from the cathode side with pure DEGDME for the spectral studies. The top spectrum of Fig. 4a, of a pristine solution before the experiments, discloses that the major species therein is copper(II) with its red-shifted maximum. The spectra change to a much shorter wavelength maximum upon polarization of the cell with the solution therein to 3.6 V clearly signifies the conversion of [CuII(tdfc)]− to its oxidized state CuIII(tdfc) as shown in Fig. 4b. The most important finding is that the UV-vis spectra of the solutions examined (in their non-oxidized state) after being cycled under oxygen between 3.6 V and either 2.9 or 2.6 V were identical to that of the pristine uncycled solution (Fig. 4a). This verifies very well the stability of Cu(tdfc) in Li–O2 batteries and also identifies the catalytically relevant oxidation states of this RM. Altogether, the data is fully consistent with the reaction mechanism depicted in Fig. 4c. The Cu(tdfc) RM is synthesized and is introduced into the cells in its neutral (oxidized) form Cu3+(tdfc), in which it is stable and relatively easy for handling. A first discharge process of the Li–O2 cell reduces both oxygen and this RM to its reduced form, namely Li+–[Cu2+(tdfc)]−. Upon charging, the reduced RM is being oxidized and then help in a chemical oxidation of the Li2O2 deposits. The charging process leaves this RM in its reduced state which is not affected further by the subsequent discharge process (follows by the charging step in which Li+–[Cu2+(tdfc)]− is being oxidized to Cu3+(tdfc) which further oxidizes the Li2O2 deposits etc.).
 | ||
| Fig. 4 UV-vis spectra of (a) the electrolyte solutions containing the neutral form [Cu3+(tdfc)] (red trace) after oxidation up to 3.6 V and the reduced form [Li–Cu2+(tdfc)] (black trace) and (light and dark blue traces) after the two sets of tests reported in Fig. 3h (50 cycles under oxygen in the bi-compartments cells with or without Li2O2 formation). The CV tests ended with the RMs at the reduced state (the cells were in the discharged state) and (b) ethanolic solutions of Cu3+(tdfc) and [Cu2+(tdfc)−] (obtained via reduction of the former moiety by sodium dithionite). (c) The reaction mechanism of RM in Li–O2 cell, which is consistent with all the data presented herein. | ||
Experiments in which Li–O2 cells (bi-compartment cells, oxygen atmosphere) were cycled more than 100 times also revealed that cells containing Cu(tdfc) display high stability during prolonged cycling. The results we obtained clearly show that systematic studies allowed us to identify a new and very promising RM, which we anticipate encouraging further studies aimed at discovering more macrocyclic metal complexes as RMs for Li–O2 cells.
Considering that Li–O2 cells filled with ethereal solutions cannot demonstrate full stability (vide supra), it still allows for appreciation of if and how efficiently new RMs affect the stability relative to reference and benchmark systems. The above claims regarding the superiority of the copper corrole were hence further substantiated through a rigorous performance comparison. This is illustrated in Fig. 5, which compares bi-compartments Li–O2 cells (the cathode compartment under oxygen atmosphere) with DEGDME/LiTFSI solutions with either no RM (reference) or TEMPO (as benchmark RM) or Cu(tdfc). The first voltage profiles of Li–O2 cells with Cu(tdfc) and RM-free cells in galvanostatic cycling experiments in which the capacity was not limited (i.e. the discharge process was terminated when the voltage started to decline because the cathode became fully isolated due to full coverage by precipitation of insulating Li-peroxide, initially nearly 10 hours per a single process) are shown in Fig. 5a. This demonstrates that the charging processes (OER) of cells containing Cu(tdfc) removes nearly all the Li2O2 deposits formed upon the prolonged ORR, while without the RM the charging process is very short: the overvoltage values increased to high values before removing even half of the Li2O2 deposited during the ORR. Fig. 5b shows typical voltage profiles of the same cells, but with a limited capacity, which serves well for demonstrating the pronouncedly lower charging (OER) voltage profiles of the cells containing Cu(tdfc). Consecutive voltage profiles of Li–O2 cells containing either Cu(tdfc) or TEMPO were measured during prolonged galvanostatic cycling experiments (limited capacity as indicated, 10 h per process, 0.32 mA h cm−2) and are shown in Fig. 5c and d, respectively. The superiority of Cu(tdfc) is easiest appreciated by Fig. 5e, in which these results are presented as graphs of areal capacity vs. cycle number: these values are only slightly improved with TEMPO as RM, while truly (relatively) high stability is apparent with Cu(tdfc). All these prolonged cycling tests demonstrated clearly the advantage of Cu(tdfc) as a RM in bi-compartments cells in which the contribution of the RMs to stability is emphasized due to the total elimination of the negative effect of the Li anode and detrimental cross fluxes of species between the anode and cathode sides. Cells with TEMPO could survive 10–20 cycles more than cells cycled without it, but the change in their voltage profiles becomes drastic after 80 cycles (Fig. 5d), while cells with Cu(tdfc) survived more than 150 cycles. The voltage profiles of cycled cells with Cu(tdfc) presented in Fig. 5c require discussion. While the capacity of these cells is well retained during more than 150 cycles (the best cycle life we have recorded with any Li–O2 cells operated in similar conditions) the voltage profiles change during cycling and the charging voltage that enable to maintain the constant capacity increases gradually. This finding seems apparently to contradict the claims about the high stability of cells cycled with this RM. However, the answer is simple: the use of suitable (even excellent) RM solves only stability problems related to the OER (charging) steps. All of these systems suffer from additional stability problems related to the ORR (discharge) steps during which highly reactive oxygen reduction species in solution phase can attack the DEGDME molecules as explained in the introduction section above. These inevitable side reactions change the cathode's surface due to deposits of side products that cannot be detected by SEM and even spectroscopy (we detected them only by MALDI)4 and lead to the higher over-potentials required for any charge transfer from the electrode to solution phase as cycling of these electrodes (and the accompanied side reactions) progresses. Yet, despite this change it appears that the presence of the Cu(tdfc) as a RM in the cells facilitates the OER and thus extends the cells’ cycle life.
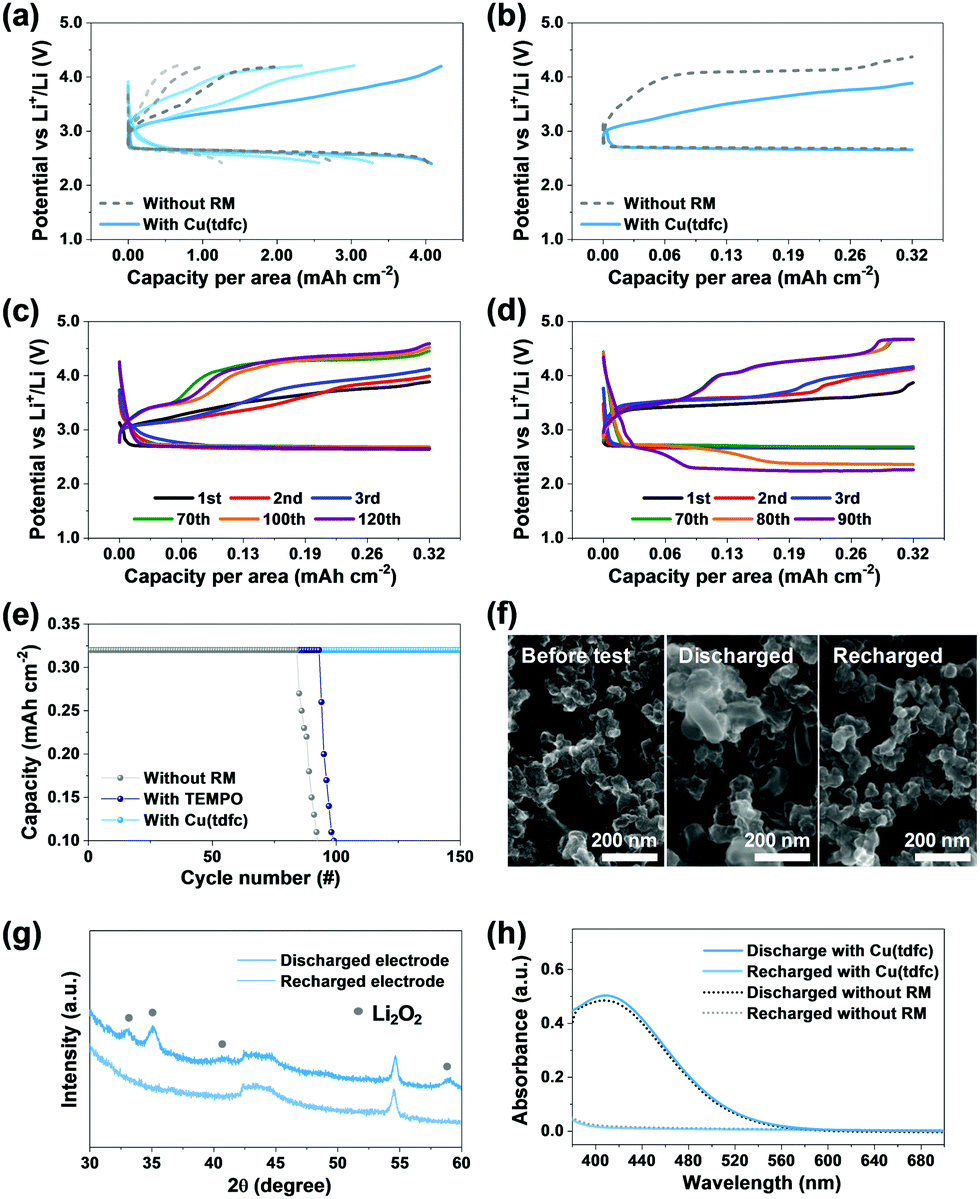 | ||
| Fig. 5 (a) Cycling data of bi-compartments Li–O2 cells with or without Cu(tdfc) during galvanostatic discharge and recharge without capacity limit for 3 cycles. (b) Voltage profiles of same cells with constant capacity of 0.032 mA cm−2 (10 h per process). Cycling data of bi-compartments Li–O2 cells during galvanostatic cycling tests with constant capacity of 0.032 mA cm−2 (10 h per process), using 1 m LiTFSI/DEGDME electrolyte solutions containing 0.02 m of (c) Cu(tdfc) and (d) TEMPO. (e) Results of prolonged cycling tests – cells capacity vs. cycle number without RM, with TEMPO, and with Cu(tdfc). (f) SEM and (g) XRD data of discharged and recharged cathodes from cells containing Cu(tdfc). A typical SEM image of a cathode before testing is presented as well for comparison. (h) UV-vis of solutions that contained titration products of Li2O2 extracted from discharged and recharged cathodes that underwent ORR with or without Cu(tdfc), as fully described in the experimental section. | ||
Analyses that addressed the major cathodic process in the cells containing Cu(tdfc) were also performed. The XRD patterns of cathodes taken from such a cell after discharge and recharge processes clearly display the typical patterns of Li2O2 and their absence (Fig. 5f), as expected from the relatively smooth cycling behaviour of these cells. Similar conclusions were deduced from the SEM images of Fig. 5g, which clearly show deposits of Li2O2 on a cathode after discharge and their absence on a recharged cathode (nearly a pristine morphology). To understand whether the presence of Cu(tdfc) interferes somehow with the Li2O2 formation during discharge, titration of discharged electrodes taken from cell using electrolytes with or without Cu(tdfc) were conducted and were monitored by UV spectroscopy. The results presented in Fig. 5h (see description of the entire analysis in the experimental section) reveal that the two spectra are nearly overlapping, which suggests that the Li2O2 formation/precipitation reaction in Li–O2 batteries is preserved in cells containing Cu(tdfc) as the RM. It can be concluded that Cu (tdfc) assists in the decomposition of Li2O2 as RM and does not induce any further side reactions. We conclude that the systematic study reported herein with Li–O2 cells containing transition metal (Co, Mn, Cu) chelated by corroles as RMs, led to a true discovery. The lead member of this family, namely Cu(tdfc), serves as a very suitable RM in Li–O2 cells, due to a quite ideal redox potential and a unique intrinsic stability that is absent in most of the RMs that were studied so far in Li–O2 cells. It is important to note that this study is far from being completed. After discovering herein the new promising RM from the complex TM corroles family, more optimization is required (e.g. choice of other glyme solvents, Li salt, RM concentration, trying other TM corrole type RMs) in order to maximize capacity and rate capability. Also, a further hard analytical work that will explain all the possible side reactions in these cells is interesting (all much beyond the scope of this work).
Conclusions
DEGDME/LiTFSI solutions were determined to be unstable in Li–O2 cells at the cathode side, even when contact with Li metal was prevented by using bi-compartment cells, due to reactions with partially reduced oxygen species (peroxide and superoxide moieties in the presence of Li cations). Hence the cycle life of these cells is limited anyway due to unavoidable side reactions during ORR. In the absence of RMs the charging reaction (OER) may require too high over-potentials that may lead to anodic deterioration of the ethereal solvents and detrimental oxidation of the carbonaceous cathode components, what further shortens pronouncedly the cycle life of these cells. In the presence of RMs in solutions, the anodic deterioration may be completely avoided, so the cycle life of Li–O2 cells can be pronouncedly extended, compared to reference cells that do not include RMs, undergoing the same cycling protocol. Considering the hypothesis that RMs can prevent cycle life shortening mechanisms upon charging (during the OER), some most commonly used RMs were investigated and shown to be unstable even in the absence of oxygen. This emphasized the need for new RMs and initiated the introduction of transition metal (Co, Mn, Cu) complexes of fluorinated corroles (two levels of fluorination for further affecting the redox potentials) as potential candidates. Their evaluation was carried out by very systematic studies, in bi-compartment cells. Simple cyclic voltammetric measurements of cells under argon enabled a primary evaluation of redox activity and stability in voltage ranges relevant for Li–O2 batteries. This was followed by a full outline of the investigations that discovered Cu(tdfc) as a promising organometallic RM for Li–O2 cells. We trust that the findings presented in this study will encourage further work on corrole-based RMs, as their properties can be easily controlled and tuned by synthetic modifications.Experimental
Materials
Diethylene glycol dimethyl ether (DEGDME, 99.5%), bis(trifluoromethane)sulfonimide lithium salt (LiTFSI, 99.95%), TTF (tetrathiafulvalene, 97%), TEMPO (tetramethylpiperidinyloxyl, 98%), and DMPZ (dimethylphenazine, 97%) were purchased from Sigma Aldrich.The free-base corroles 5,10,15-tris(pentafluorophenyl)corrole (H3(tpfc)) and 5,10,15-tris(2,6-difluorophenyl)corrole (H3(tdfc)) were prepared according to reported procedures.45–47 The metallocorroles Co(tpfc), Mn(tpfc), Cu(tpfc), and Mn(tdfc) were synthesized following the same protocol as in previously published reports.39,43,48,49
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexanes:pyridine (1:4:0.01) as eluent. Pure crystalline material (40 mg, 75% yield) was obtained after recrystallization in CH2Cl2–hexanes in the presence of 0.1% pyridine. 1H NMR (400 MHz, C6D6) δ = 9.23 (d, J = 4.2 Hz, 2H), 9.17 (d, J = 4.4 Hz, 2H), 8.98–8.95 (m, 4H), 7.14–6.99 (m, 7H), 6.98–6.90 (m, 2H), 4.68 (br s, 2H), 4.17 (br s, 4H), 1.57 (br s, 4H) ppm. 19F NMR (377 MHz, C6D6) δ = −109.17 (t, J = 6.0 Hz, 2F), −109.36 (t, J = 6.0 Hz, 4F) ppm. UV-vis (CH2Cl2, 1% pyridine): λmax (ε) [nm (M−1 cm−1)] = 415 (61000), 439 (88000), 581 (21000) and 606 (36000). HR-MS (APCI negative mode) for C37H17F6N4Co [M − 2pyridine]−: m/z = 690.0689 (calculated), 690.0693 (observed).
:1) as eluent (20 mg, 46% yield). 1H NMR (400 MHz, CDCl3) δ = 7.88 (br s, 2H), 7.44–7.35 (m, 3H), 7.32 (d, J = 3.84 Hz, 2H), 7.17 (d, J = 3.68 Hz, 2H), 7.06–6.98 (m, 6H), 6.96 (d, J = 4.44 Hz, 2H) ppm. 19F NMR (377 MHz, C6D6) δ = −108.84 (t, J = 5.9 Hz, 4F), −109.80 (t, J = 6.4 Hz, 2F) ppm. UV-vis (CH2Cl2): λmax (ε) [nm (M−1 cm−1)] = 407 (78000), 540 (8100) and 620 (4600). HR-MS (APCI positive mode) for C37H17F6N4Cu [M]−: m/z = 694.0653 (calculated), 694.0628 (observed).
:hexanes:pyridine (1:4:0.01) as eluent. Pure crystalline material (40 mg, 75% yield) was obtained after recrystallization in CH2Cl2–hexanes in the presence of 0.1% pyridine. 1H NMR (400 MHz, C6D6) δ = 9.23 (d, J = 4.2 Hz, 2H), 9.17 (d, J = 4.4 Hz, 2H), 8.98–8.95 (m, 4H), 7.14–6.99 (m, 7H), 6.98–6.90 (m, 2H), 4.68 (br s, 2H), 4.17 (br s, 4H), 1.57 (br s, 4H) ppm. 19F NMR (377 MHz, C6D6) δ = −109.17 (t, J = 6.0 Hz, 2F), −109.36 (t, J = 6.0 Hz, 4F) ppm. UV-vis (CH2Cl2, 1% pyridine): λmax (ε) [nm (M−1 cm−1)] = 415 (61000), 439 (88000), 581 (21000) and 606 (36000). HR-MS (APCI negative mode) for C37H17F6N4Co [M − 2pyridine]−: m/z = 690.0689 (calculated), 690.0693 (observed).
:1) as eluent (20 mg, 46% yield). 1H NMR (400 MHz, CDCl3) δ = 7.88 (br s, 2H), 7.44–7.35 (m, 3H), 7.32 (d, J = 3.84 Hz, 2H), 7.17 (d, J = 3.68 Hz, 2H), 7.06–6.98 (m, 6H), 6.96 (d, J = 4.44 Hz, 2H) ppm. 19F NMR (377 MHz, C6D6) δ = −108.84 (t, J = 5.9 Hz, 4F), −109.80 (t, J = 6.4 Hz, 2F) ppm. UV-vis (CH2Cl2): λmax (ε) [nm (M−1 cm−1)] = 407 (78000), 540 (8100) and 620 (4600). HR-MS (APCI positive mode) for C37H17F6N4Cu [M]−: m/z = 694.0653 (calculated), 694.0628 (observed).
The solvents were purified by distillation and further dried over activated molecular sieves until the final water content was <10 ppm. The lithium salt was dried in a vacuum oven for 5 d at 120 °C. The moisture content of the solvents and electrolytes was measured by Karl–Fischer titration (Mettler–Toledo) without exposure. For the cathodes, gas-diffusion layer sheets (GDL, SGL, 39 BC) were punched into circular pieces of 1.4 cm in diameter and were dried at 180 °C under vacuum for 3 d. Glass fibers (GF/C, Whatman) were used as the separator after vacuum drying for 3 d at 180 °C. 1 m LiTFSI in DEGDME with 0.02 m of RMs was used as the electrolyte solution. Molal concentration (molality) was used for calculation of exact amount of Li salt and RMs. Li metal foil (thickness: 200 μm) was purchased from Honjo Chemical and used as the counter and reference electrodes (CE and RE).
Cell assembly
The home-made type Li–O2 cells were assembled with the dried cathodes, dried glass fiber (GF/C, Whatman, 180 °C under vacuum for 3 d), lithium metal anode (100 μm, Honjo), solid electrolyte (LICGC, Ohara), and different electrolyte solutions in an Ar filled glove box (water and oxygen contents were less than 0.1 ppm). The configuration of the bi-compartments cells was already described and reported.20,21 After the cell assembly, the Li–O2 cells were stabilized in an O2 atmosphere (1.0 bar) for 1 h before the relevant electrochemical tests. Closed coin cells without O2, for the experiments under pure Ar atmosphere, were fabricated with same procedure under an Ar-filled glove box.Characterization
Electrochemical tests were conducted using a VMP3 potentiostat (Biologic Instruments) set for cyclic voltammetric measurements with scan rate 0.1 mV s−1 in different voltage windows which are provided in each profile, and galvanostatic cycling with a current density of 0.1 mA and time limit of 5 h in the voltage window of 2.0–4.8 V.The evaluation of the products developed on the carbon electrodes after ORR (discharge) and OER (recharge) was carried out using field-emission scanning electron microscopy (FE-SEM, SUPRA 55VP, Carl Zeiss) and high-resolution X-ray diffraction (HR-XRD, 9 kW, SmartLab, Rigaku) with a Cu-Kα radiation source within a 2θ range of 30.0–60.0° at a scan rate of 1° min−1. All the analyses of the electrodes were performed after a careful washing procedure with pure DEGDME solvent. Then, the samples were dried in an Ar-filled glove box and wrapped in a vacuum sealed pack to prevent exposure in air during transfer to the analytical instruments for characterization.
UV-vis absorption spectra were recorded on a Cary 50 UV-vis spectrophotometer (Varian). The presence of Li2O2 was confirmed by UV-vis spectrometry via a titration method using titanium oxysulfate (TiOSO4). The discharged and recharged working electrodes were introduced into the titration solution (2% TiOSO4 in 0.1 M H2SO4) and were shaken for 30 s. The concentration of the yellowish complex [Ti(O2)]2+ thus formed which is proportional to the Li2O2 concentration, was measured by analyzing the UV-vis absorption spectra. Solution containing the electrodes after discharge (containing Li2O2) showed the expected UV spectrum (chart 4 h) while solutions which were in contact with electrodes after charging did not show that spectral response.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Human Resources Development programme (No. 20184010201720) of a Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant, funded by the Ministry of Trade, Industry and Energy of the Korean government and this work was also supported by the Global Frontier R&D Programme (NRF-2013M3A6B1078875) on the Center for Hybrid Interface Materials (HIM), by the Ministry of Science, ICT & Future Planning. DA and ZG acknowledge support from the Israel Committee of High Education in the framework of the INREP consortium.Notes and references
- Rechargeable Lithium Batteries, ed. M. Bini, D. Capsoni, S. Ferrari, E. Quartarone, P. Mustarelli and A. A. Franco, Woodhead Publishing, 2015, pp. 1–17 Search PubMed.
- B. D. McCloskey, A. Speidel, R. Scheffler, D. C. Miller, V. Viswanathan, J. S. Hummelshøj, J. K. Nørskov and A. C. Luntz, J. Phys. Chem. Lett., 2012, 3, 997–1001 CrossRef CAS.
- D. Aurbach, B. D. McCloskey, L. F. Nazar and P. G. Bruce, Nat. Energy, 2016, 1, 16128 CrossRef CAS.
- V. Etacheri, D. Sharon, A. Garsuch, M. Afri, A. A. Frimer and D. Aurbach, J. Mater. Chem. A, 2013, 1, 5021–5030 RSC.
- R. Black, J. H. Lee, B. Adams, C. A. Mims and L. F. Nazar, Angew. Chem., Int. Ed., 2013, 52, 392–396 CrossRef CAS.
- D. Sharon, D. Hirsberg, M. Salama, M. Afri, A. A. Frimer, M. Noked, W. J. Kwak, Y. K. Sun and D. Aurbach, ACS Appl. Mater. Interfaces, 2016, 8, 5300–5307 CrossRef CAS PubMed.
- Y. Chen, S. A. Freunberger, Z. Peng, O. Fontaine and P. G. Bruce, Nat. Chem., 2013, 5, 489 CrossRef CAS PubMed.
- B. J. Bergner, A. Schürmann, K. Peppler, A. Garsuch and J. Janek, J. Am. Chem. Soc., 2014, 136, 15054–15064 CrossRef CAS PubMed.
- T. Liu, M. Leskes, W. Yu, A. J. Moore, L. Zhou, P. M. Bayley, G. Kim and C. P. Grey, Science, 2015, 350, 530–533 CrossRef CAS PubMed.
- H. D. Lim, B. Lee, Y. Zheng, J. Hong, J. Kim, H. Gwon, Y. Ko, M. Lee, K. Cho and K. Kang, Nat. Energy, 2016, 1, 16066 CrossRef CAS.
- W. J. Kwak, D. Hirshberg, D. Sharon, M. Afri, A. A. Frimer, H. G. Jung, D. Aurbach and Y. K. Sun, Energy Environ. Sci., 2016, 9, 2334–2345 RSC.
- B. J. Bergner, C. Hofmann, A. Schürmann, D. Schröder, K. Peppler, P. R. Schreinerb and J. Janek, Phys. Chem. Chem. Phys., 2015, 17, 31769–31779 RSC.
- B. J. Bergner, M. R. Busche, R. Pinedo, B. B. Berkes, D. Schröder and J. Janek, ACS Appl. Mater. Interfaces, 2016, 8, 7756–7765 CrossRef CAS.
- C. Yang, J. Han, P. Liu, C. Hou, G. Huang, T. Fujita, A. Hirata and M. Chen, Adv. Mater., 2017, 29, 1702752 CrossRef.
- V. Pande and V. Viswanathan, ACS Energy Lett., 2017, 2, 60–63 CrossRef CAS.
- P. P. Bawol, P. Reinsberg, C. J. Bondue, A. A. Abd-El-Latif, P. Königshoven and H. Baltruschat, Phys. Chem. Chem. Phys., 2018, 20, 21447–21456 RSC.
- Y. Ko, H. Park, B. Lee, Y. Bae, S. K. Park and K. Kang, J. Mater. Chem. A, 2019, 7, 6491–6498 RSC.
- Y. Chen, X. Gao, L. R. Johnson and P. G. Bruce, Nat. Commun., 2018, 9, 767 CrossRef.
- S. Ha, Y. Kim, D. Koo, K. H. Ha, Y. Park, D. M. Kim, S. Son, T. Yim and K. T. Lee, J. Mater. Chem. A, 2017, 5, 10609–10621 RSC.
- W. J. Kwak, H. G. Jung, D. Aurbach and Y. K. Sun, Adv. Energy Mater., 2017, 7, 1701232 CrossRef.
- W. J. Kwak, H. Kim, H. G. Jung, D. Aurbach and Y. K. Sun, J. Electrochem. Soc., 2018, 165, A2274–A2293 CrossRef CAS.
- W. J. Kwak, H. Kim, Y. K. Petit, C. Leypold, T. T. Nguyen, N. Mahne, P. Redfern, L. A. Curtiss, H. G. Jung, S. M. Borisov, S. A. Freunberger and Y. K. Sun, Nat. Commun., 2019, 10, 1380 CrossRef PubMed.
- I. Aviv and Z. Gross, Chem. Commun., 2007, 1987–1999 RSC.
- R. D. Teo, J. Y. Hwang, J. Termini, Z. Gross and H. B. Gray, Chem. Rev., 2017, 117, 2711–2729 CrossRef CAS.
- Y. Fang, Z. Ou and K. M. Kadish, Chem. Rev., 2017, 117, 3377–3419 CrossRef CAS.
- M. König, F. Faschinger, L. M. Reith and W. Schöfberger, J. Porphyrins Phthalocyanines, 2016, 20, 96–107 CrossRef.
- G. Pomarico, D. Monti, M. Bischetti, A. Savoldelli, F. R. Fronczek, K. M. Smith, D. Genovese, L. Prodi and R. Paolesse, Chem. – Eur. J., 2018, 24, 8438–8446 CrossRef CAS.
- A. Mahammed, B. Mondal, A. Rana, A. Dey and Z. Gross, Chem. Commun., 2014, 50, 2725–2727 RSC.
- J. Grodkowski, P. Neta, E. Fujita, A. Mahammed, L. Simkhovich and Z. Gross, J. Phys. Chem. A, 2002, 106, 4772–4778 CrossRef CAS.
- A. Friedman, L. Landau, S. Gonen, Z. Gross and L. Elbaz, ACS Catal., 2018, 8, 5024–5031 CrossRef CAS.
- A. Mahammed and Z. Gross, Chem. Commun., 2010, 46, 7040–7042 RSC.
- D. K. Dogutan, R. McGuire and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 9178–9180 CrossRef CAS.
- A. Mahammed, H. B. Gray, A. E. Meier-Callahan and Z. Gross, J. Am. Chem. Soc., 2003, 125, 1162–1163 CrossRef CAS.
- M. J. ZdillaMahdi and M. Abu-Omar, Inorg. Chem., 2008, 47, 10718–10722 CrossRef.
- A. Mahammed, K. Chen, J. Vestfridc, J. Zhao and Z. Gross, Chem. Sci., 2019, 10, 7091–7103 RSC.
- J. F. B. Barata, M. G. P. M. S. Neves, M. A. F. Faustino, A. C. Tomé and J. A. S. Cavaleiro, Chem. Rev., 2017, 117, 3192–3253 CrossRef CAS.
- B. J. Brennan, Y. C. Lam, P. M. Kim, X. Zhang and G. W. Brudvig, ACS Appl. Mater. Interfaces, 2015, 7, 16124–16130 CrossRef CAS.
- C. M. Lemon, M. Huynh, A. G. Maher, B. L. Anderson, E. D. Bloch, D. C. Powers and D. G. Nocera, Angew. Chem., Int. Ed., 2016, 55, 2176–2180 CrossRef CAS.
- I. Luobeznova, L. Simkhovich, I. Goldberg and Z. Gross, Eur. J. Inorg. Chem., 2004, 1724–1732 CrossRef CAS.
- G. Golubkov, J. Bendix, H. B. Gray, A. Mahammed, I. Goldberg, A. J. DiBilio and Z. Gross, Angew. Chem., Int. Ed., 2001, 40, 2132–2134 CrossRef CAS.
- K. Sudhakar, A. Mahammed, N. Fridman and Z. Gross, Dalton Trans., 2019, 48, 4798–4810 RSC.
- A. Mahammed, M. Botoshansky and Z. Gross, Dalton Trans., 2012, 41, 10938–10940 RSC.
- A. Kumar, I. Goldberg, M. Botoshansky, Y. Buchman and Z. Gross, J. Am. Chem. Soc., 2010, 132, 15233–15245 CrossRef CAS PubMed.
- Y. Fang, Z. Ou and K. M. Kadish, Chem. Rev., 2017, 117, 3377–3419 CrossRef CAS PubMed.
- B. Koszarna and D. T. Gryko, J. Org. Chem., 2016, 71, 3707–3717 CrossRef.
- C. Blumenfeld, K. J. Fisher, L. M. Henling, R. H. Grubbs, H. B. Gray and S. C. Virgil, Eur. J. Inorg. Chem., 2015, 3022–3025 CrossRef CAS.
- Z. Gross, N. Galili and I. Saltsman, Angew. Chem., Int. Ed., 1999, 38, 1427–1429 CrossRef CAS PubMed.
- A. Mahammed, I. Giladi, I. Goldberg and Z. Gross, Chem. – Eur. J., 2001, 7, 4259–4265 CrossRef CAS.
- Z. Gross, G. Golubkov and L. Simkhovich, Angew. Chem., Int. Ed., 2000, 39, 4045–4047 CrossRef CAS PubMed.
Footnote |
| † Present address: Energy & Environment Directorate, Pacific Northwest National Laboratory, Richland, Washington 99354, USA. |
| This journal is © The Royal Society of Chemistry 2020 |