
Less may be more: an informed reflection on molecular descriptors for drug design and discovery†
Trent
Barnard
,
Harry
Hagan
,
Steven
Tseng‡
and
Gabriele C.
Sosso
 *
*
Department of Chemistry and Centre for Scientific Computing, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: g.sosso@warwick.ac.uk
First published on 8th November 2019
Abstract
The phenomenal advances of machine learning in the context of drug design and discovery have led to the development of a plethora of molecular descriptors. In fact, many of these “standard” descriptors are now readily available via open source, easy-to-use computational tools. As a result, it is not uncommon to take advantage of large numbers – up to thousands in some cases – of these descriptors to predict the functional properties of drug-like molecules. This “strength in numbers” approach does usually provide excellent flexibility – and thus, good numerical accuracy – to the machine learning framework of choice; however, it suffers from a lack of transparency, in that it becomes very challenging to pinpoint the – usually, few – descriptors that are playing a key role in determining the functional properties of a given molecule. In this work, we show that just a handful of well-tailored molecular descriptors may often be capable to predict the functional properties of drug-like molecules with an accuracy comparable to that obtained by using hundreds of standard descriptors. In particular, we apply feature selection and genetic algorithms to in-house descriptors we have developed building on junction trees and symmetry functions, respectively. We find that information from as few as 10–20 molecular fragments is often enough to predict with decent accuracy even complex biomedical activities. In addition, we demonstrate that the usage of small sets of optimised symmetry functions may pave the way towards the prediction of the physical properties of drugs in their solid phases – a pivotal challenge for the pharmaceutical industry. Thus, this work brings strong arguments in support of the usage of small numbers of selected descriptors to discover the structure–function relation of drug-like molecules – as opposed to blindly leveraging the flexibility of the thousands of molecular descriptors currently available.
 Gabriele C. Sosso | Gabriele C. Sosso is an Assistant Professor in Computational Physical Chemistry at the University of Warwick, UK. He earned his PhD in Nanostructures and Nanotechnologies in 2013 from the University of Milano-Bicocca, Italy. He then carried out post-doctoral research at ETH Zürich, Switzerland, and at University College London, UK. His research focuses on molecular simulations of disordered systems and phase transitions, from nano-cavities in supercooled liquids to the formation of ice in biological matter. Gabriele can be reached by email at E-mail: g.sosso@warwick.ac.uk. |
Design, System, ApplicationThis work provides evidence that when trying to predict the functional properties of relatively small data sets of drug-like molecules via machine learning, constructing and selecting a small set of carefully tailored molecular descriptors may offer equally or even more accurate results compared to the usage of large numbers of descriptors – a worrying trend in the recent literature. In particular, we introduce two simple and yet effective classes of descriptors that can unravel part of the structure–function relation we desperately need to understand – in order to achieve the truly rational design of the next generation of drugs. In addition, our descriptors pave the way toward predictive frameworks taking into account three-dimensional models of either crystalline and amorphous formulations as well – a pivotal challenge for the pharmaceutical industry. As such, our findings provide practical guidelines for the community working in the field of machine learning for drug design and discovery; in fact, we have made available via a public repository our computational framework, so as to make our work immediately leverageable by several research groups across the globe – thus supporting the collaborative quest toward a concrete impact of machine learning on the drug discovery and design pipeline. |
1 Introduction
In the last two decades, the pharmaceutical industry has invested enormously in machine learning (ML) as a tool to transform the current paradigm of drug design and discovery.1,2 Despite the fact that deep learning is considered by many to sit at the very top of the hype cycle,3 recent collaborative efforts between some of the major pharmaceutical companies4 indicate that there is a strong driving force to improve on the existing ML algorithms and thus deliver the next generation of drugs. One of the most important consequences of this ambition is the ever-increasing amount of experimental data that is being accumulated on the many functional properties and/or biomedical activities of drug-like molecules.5 In fact, the volume, as well as the quality of the experimental data available to us are and will still be the key ingredients of any ML framework we may think of developing.Perhaps unsurprisingly, such a tremendous industrial interest has also substantially boosted the academic progress in the field:6 in turn, this resulted in a sizeable contribution to the already fast-developing area of ML algorithms, many of which are now readily available via open source packages such as the Python-based scikit-learn.7 Similarly, a plethora of molecular descriptors have been devised and implemented within the past few years.8 These mathematical objects are essential to process the information about the molecular structures of interest into a form digestible by ML algorithms, and packages such as RDKit9 allow for access to an impressive number of them very easily indeed. Crucially, given a certain molecular dataset, the choice of the descriptors has almost always a much greater impact on the predictive power of a ML framework if compared to the influence of picking a certain ML algorithm – albeit advanced frameworks such as the SchNet approach of Schütt et al.10 or parallel multistream training11 have to potential to improve the state of the art even further.
While all the progress detailed above provides a great opportunity to involve more and more scientists into the field, and thus to boost the chance we have to make a concrete impact onto drug design and discovery, we believe that this ease-of-use in terms of descriptors may present a risk as well.
In fact, it is tempting, given the availability of so many different molecular descriptors, to leverage as many of them as possible: for instance, the DRAGON software12 can calculate more than 4800 descriptors.13 As such, this approach is not only incredibly simple these days, but it may also enhance the flexibility of the ML algorithm of choice, in that the more descriptors we add into the mix, the higher the chances to include those features that are actually of relevance to improve the predictive capabilities of the framework.14 However, this strategy suffers from at least two major issues: (1.) redundancy/correlation: the more descriptors we choose to use, the higher the chance they will feed similar if not identical information to the ML algorithm,15 with the risk of introducing artificial noise that can be detrimental to both the accuracy and the reliability of the predictive framework; (2.) lack of transparency:16,17 it becomes quite challenging to pinpoint the structural features that have the largest impact on the functional properties of interest. While from a purely practical perspective one may not care about this pitfall, understanding the structure–function relation is key to achieve the truly rational design of the novel generation of drugs.18
Both redundancy and lack of transparency can be mitigated by using feature selection19 and/or by optimising the parameters that often enter the formulation of advanced molecular descriptors. As many options to perform feature selection are presently easily accessible, we see no immediate reason not to leverage them anytime we choose to employ a whole array of different descriptors. An additional issue with the many molecular descriptors currently available is that the overwhelming majority of ML frameworks aim to predict the properties of actual drug formulations – typically, but not exclusively, in the form of crystalline solids – utilising as starting point the structure of a single molecule in vacuum.20–23 As a result, most of the molecular descriptors we have available at the moment cannot be used to tackle the complexity of actual three-dimensional molecular models of e.g. crystalline or amorphous drugs. We believe that taking into account these models, generated by means of e.g. molecular dynamics simulations, and developing descriptors specifically tailored to extract insight about important features such as inter-molecular interactions is a step the community needs to take in order to improve the accuracy and reliability of ML for drug design. Descriptors borrowed from materials science, and particularly from ML for the development of inter-atomic potentials such as the smooth overlap of atomic positions (SOAP)24 or the atom-centred symmetry functions25 descriptors may be of great help in this context.
In this work, we show that, in some cases, utilising just a handful (10–20) of carefully designed molecular descriptors may yield results comparable – or even better – than those obtained by using a large number (∼100) of what we are going to label as “standard” (STD) descriptors hereafter, i.e. those descriptors immediately available via packages such as RDKit. We find that this is especially true when dealing with small datasets containing 100–500 molecular structures, where the number of STD descriptors that we may want to use can get dangerously close to the number of data points we intend to feed into our ML framework – an obviously over-determined problem.
We wanted in particular to probe the predictive power of two different classes of descriptors: molecular cliques (cliques hereafter) and histograms of weighted atom-centred symmetry functions (H-wACSFs hereafter), which we have built starting from the work of Jin et al.26 and Gastegger et al.,27 respectively. Cliques exclusively probe the “chemistry” of the molecular species of interest, in that they offer insight into the molecular fragments present, with no information about the structure of the molecule as a whole. Conversely, H-wACSFs probe the molecular structure from multiple angles, and can be straightforwardly employed to deal with three-dimensional molecular models of drug formulations. The nature of cliques and H-wACSFs makes them perfectly suitable to exploit feature selection and optimisation, respectively. We find that a surprisingly small set of tailored descriptors, as obtained upon either feature selection (cliques) and optimisation (H-wACSFS), can provide results comparable, if not of better quality, than those we have obtained by employing large numbers of STD descriptors. While an analysis of the most relevant cliques obtained upon feature selection allows us to draw interesting conclusions about the influence of specific functional groups on biomedical activities of pharmaceutical interest such as human hepatocytes intrinsic clearance,28 the H-wACSFS offer a very convenient opportunity to bridge the ML gap from a single molecule in vacuum to 3D models of e.g. amorphous drugs. While an ongoing effort within our research group is probing the benefits of bringing together “chemistry and structure” by combining these two classes of descriptors, we have made available via a public GitHub repository29 the entirety of our ML framework, in an effort to promote transparency and cross-fertilisation between different groups.
The paper is organised as follows: in the Methods section we provide the details of the computational framework we have used, with particular emphasis on cliques and H-wACSFS descriptors. In the Results section we offer a comparative analysis of the results obtained via cliques and H-wACSFS against STD and discuss the impact of feature selection and optimisation. We conclude with an opinionated perspective on the future of molecular descriptors, in particular with respect to the prediction of the functional properties of solid-state drug formulations.
2 Methods
In this section, we describe the main features of the computational framework we have employed. We start by providing essential information about the molecular datasets we have used. We then discuss the details of the descriptors we have used, with special emphasis on cliques and H-wACSFS. The methods we have employed for feature selection (cliques) and optimisation (H-wACSFS) will also be discussed, together with a brief description of the specific ML algorithms we have chosen.2.1 Molecular datasets
We have taken into account three different molecular datasets:• Lipophilicity [Lipo]: this dataset is publicly available via the moleculenet.ai project.30 It contains ∼4000 molecular structures as SMILES strings31 and their corresponding lipophilicity,32 measured experimentally as octanol/water distribution coefficients (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D at pH 7.4). In the context of pharmaceuticals, the lipophilicity of a certain drug provides a measure of its affinity for a lipid environment – thus including the cellular membrane. It is a majorly important biophysical target, as it affects the pharmacokinetic and the absorption of many drugs formulations.
D at pH 7.4). In the context of pharmaceuticals, the lipophilicity of a certain drug provides a measure of its affinity for a lipid environment – thus including the cellular membrane. It is a majorly important biophysical target, as it affects the pharmacokinetic and the absorption of many drugs formulations.
• Hepatocytes [Hepa]: this dataset has been provided to us by AstraZeneca – it is not included in the MSDE_Sosso_alpha GitHub repository.29 It contains ∼400 molecular structures as SMILES strings and their corresponding human hepatocytes intrinsic clearance (clint),28 measured experimentally as log(volume/time). Clint values quantify the ability of the human liver (particularly of the hepatocytes cells that constitute more than half of it) to remove a given drug: as the liver plays a very important role in dictating drug metabolism in our bodies, clint values are considered as crucial biological targets for drug design. We note that this is a very “challenging” dataset, in that it combines a small number of data points with an exceptionally complex biomedical activity.
• Amorphous [Amo]: this is a dataset we have recently put together from literature data (ref. 19, 33 and 34) about the functional properties of amorphous drugs. It contains the structures of ∼150 molecules as SMILES strings and the glass transition temperature Tg of their corresponding amorphous phases. Tg is a key property in the context of amorphous formulations21,35,36 in that (i.) it affects the propensity of the system to form a disordered solid as opposed to a crystal in the first place and; (ii.) it correlates to a good extent with the physical stability of the amorphous phase, which needs to not re-crystallize over the typical timescales involved with the shelf-life of a marketed pharmaceutical. In here, we move our first steps toward the prediction of such an important feature by focusing on single molecular species only – though it would be desirable to consider the actual three-dimensional models of the amorphous phases. Much as the Hepa dataset, the Amo dataset is quite a challenging one, combining a very small number of data points with a solid-state property.
2.2 Descriptors
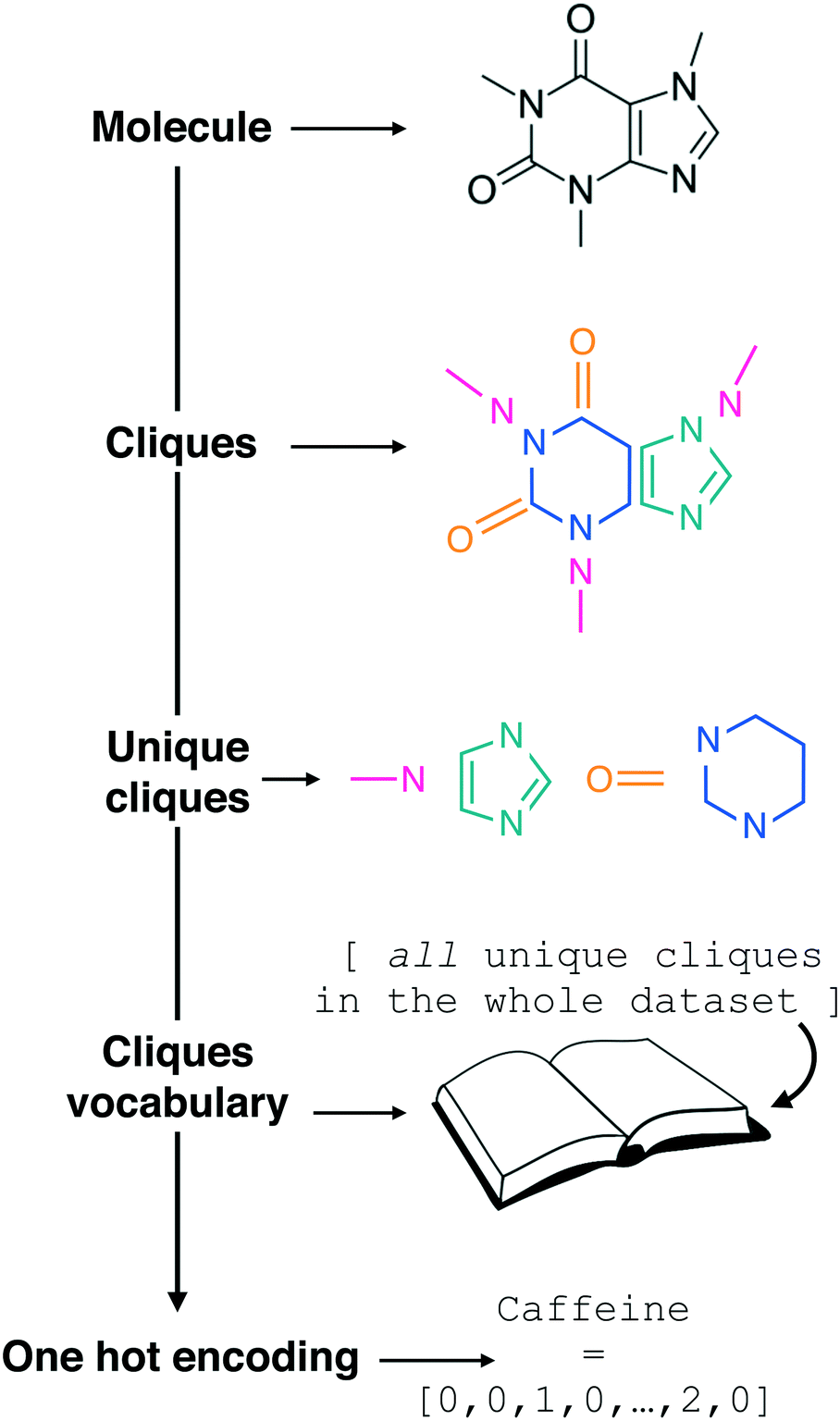 | ||
| Fig. 1 Constructing the molecular cliques descriptor. In line with the work of Jin et al.,26 a given molecular structure (we started from SMILES strings) is decomposed in molecular fragments known in graph theory as “cliques”. All the Nclq unique cliques across the entire molecular dataset are then indexed and collected into a single cliques vocabulary. Each molecule in the dataset can thus be represented by means of one hot encoding as a Nclq-long vector with each i-th element equal to the number of occurrences the i-th clique appears in the molecule. Following an analogy with natural language processing, we are treating molecular fragments as words that we can combine together into sentences, i.e. molecules. Note the transparency of this descriptor, which requires as a starting point the molecular graph only and it does not include any information about the connectivity of the molecular fragments. | ||
In the context of natural language processing, we are thus treating the clique vocabulary as a “bag of words” to form sentences – i.e. molecules, in a similar fashion to the “bag of bonds” descriptor explored in e.g.ref. 40. As the meaning of a given sentence may usually be determined to a good extent from its word content alone (i.e. without considering syntax), we are assuming that the presence of the cliques alone, without any information about the order by which they appear in a given molecular structure, would be enough to allow us to establish a structure–function relation between SMILES strings and the functional property of interest. It is thus reasonable to treat the cliques as a descriptor that is looking exclusively at the “chemistry” of the molecules, in that it highlights the presence or absence of specific molecular fragments and/or functional groups as opposed to the overall structure, albeit information about the size of the molecule is indirectly contained into the cliques vector. As we shall see in the Results section, this incredibly simple descriptor possesses a surprising predictive power, and it lends itself to feature selection in a very straightforward manner.
![[r with combining macron]](https://www.rsc.org/images/entities/i_char_0072_0304.gif) j – i| distances (radial SFs) or θijk angles (angular SFs) between pairs or triplets of atoms – up to a certain cutoff radius Rc. The interested reader can find a thorough introduction to SFs in ref. 25. Here, we have used as radial SFs:
j – i| distances (radial SFs) or θijk angles (angular SFs) between pairs or triplets of atoms – up to a certain cutoff radius Rc. The interested reader can find a thorough introduction to SFs in ref. 25. Here, we have used as radial SFs: | (1) |
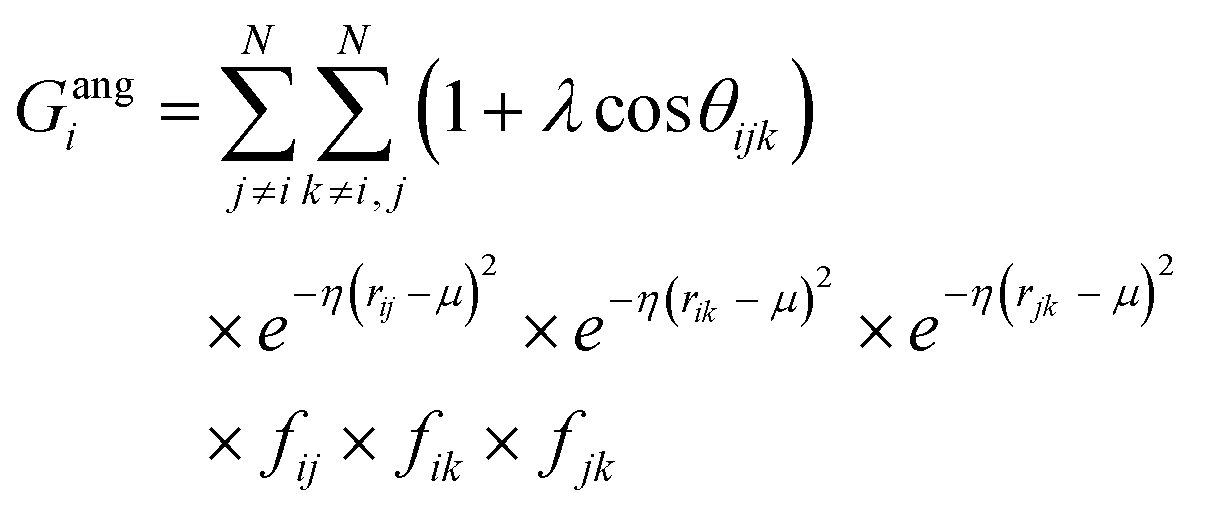 | (2) |
 | (3) |
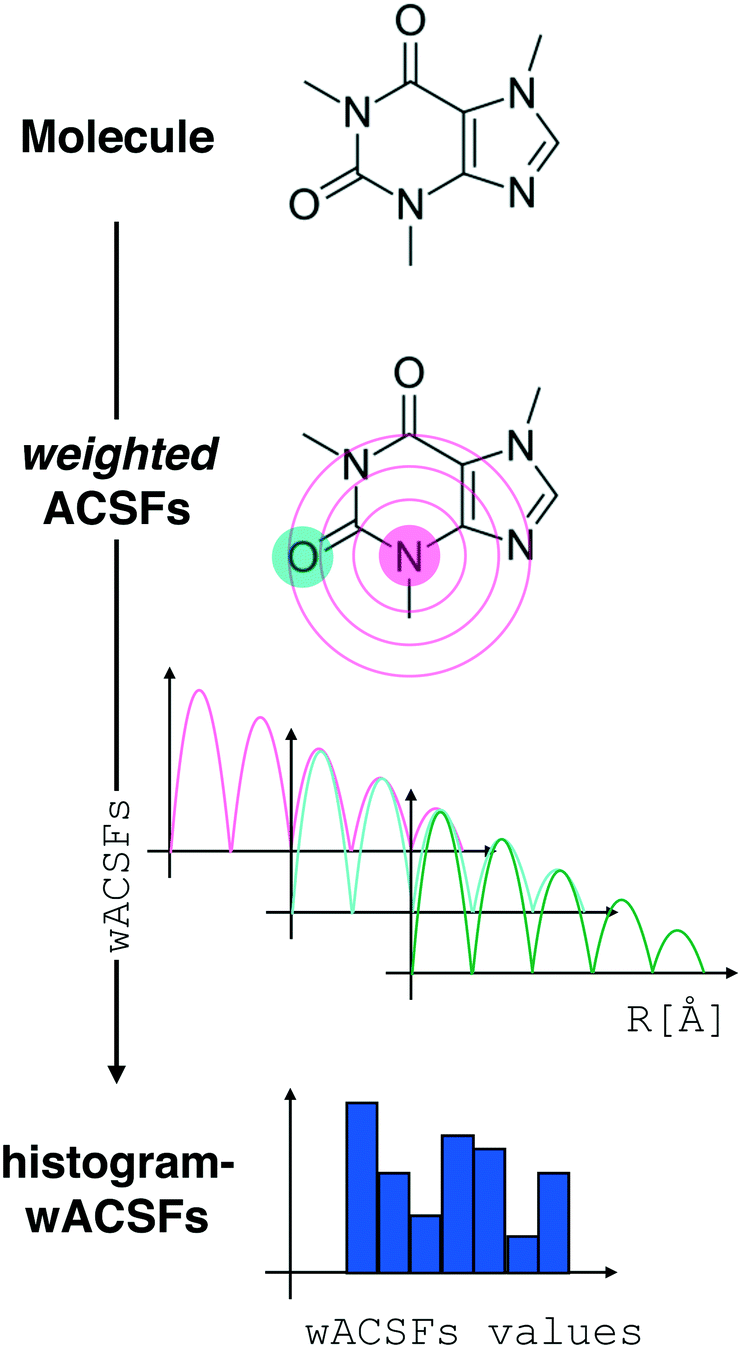 | ||
| Fig. 2 Constructing the H-wACSFs descriptor. A three-dimensional conformer (ideally, an ensemble of them) has to be generated for each molecule. Then, in line with the work of Behler,41 radial and angular symmetry functions are computed by sitting on each atom within the molecule and calculating the value of (usually Gaussian) functions that depends on either rij = |j – i| distances (radial SFs) or θijk angles (angular SFs) between pairs or triplets of atoms – up to a certain cutoff radius Rc. In principle, different sets of symmetry functions are needed for each combination of elements in a given molecule. Gastegger et al. have recently27 introduced a weighting scheme that substantially reduces the number of functions needed to encode the structure of multi component systems such as drug-like molecules. As molecules with different numbers of atoms and or elements are characterised by different number of symmetry functions, we regularise these features by building histograms of weighted atomic symmetry functions. Each molecule can then be represented by a vector with as many elements as the bins chosen to build said histogram: low and higher number of bins thus provide more or less coarse-grained representations of the molecular structure. Note that this descriptor can be straightforwardly applied to three-dimensional models of crystalline or amorphous drugs – a major challenge laying ahead. | ||
Two sets of angular symmetry functions were calculated, one set with λ = 1, the other with λ = −1. Values for μ and η are determined by the number of SFs N used and the cutoff radius. For N SFs, the value of μ for function n is given by:
| μ = 0.5 + (n − 1)∇r | (4) |
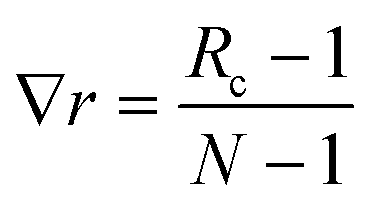 | (5) |
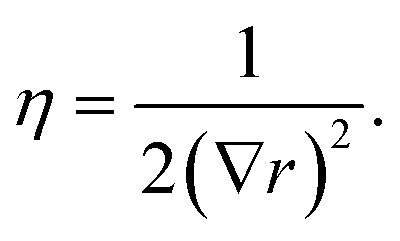 | (6) |
Crucially, the original formulation of SFs41 required a distinct set of SFs for each combination of the different elements in a given molecule. While this is a perfectly sensible option in most materials science applications, where the number of elements involved is usually well below five (in fact, it is incredibly challenging to build ML-based interatomic potential for multi-component systems42,45,46), in the context of drug design and discovery a molecular dataset may very well contain more than ten elements, which leads to a huge number of SFs. Gastegger et al. have recently devised27 a clever workaround to this issue by introducing so-called weighted SFs such as:
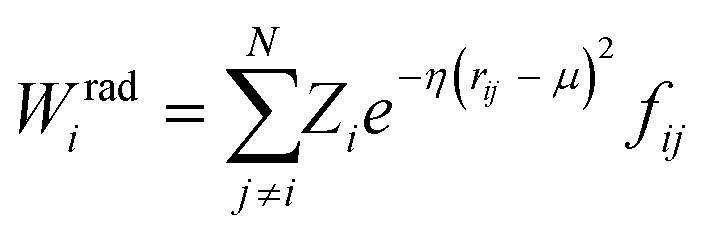 | (7) |
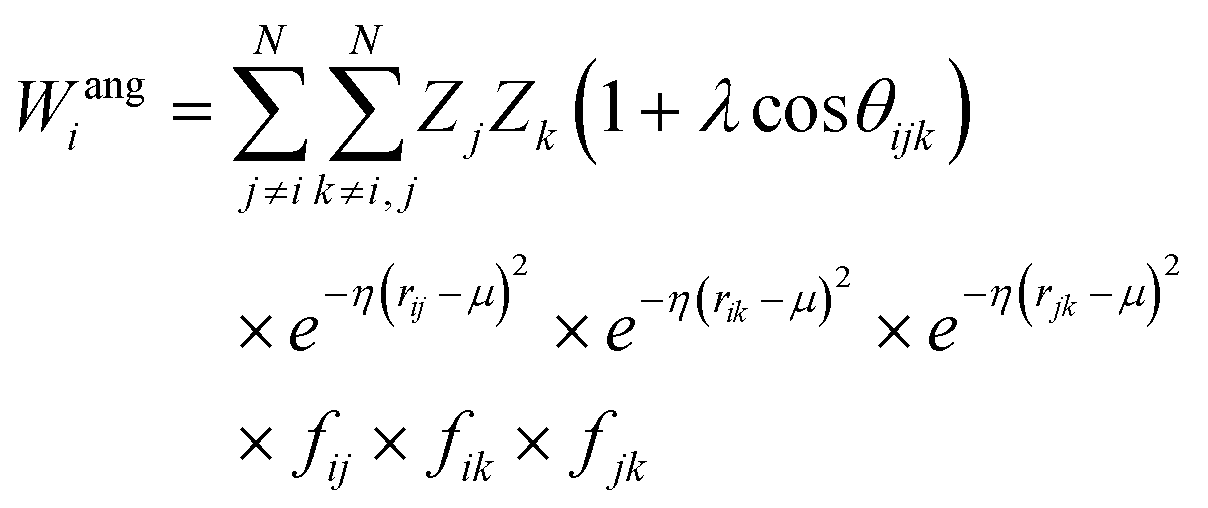 | (8) |
Even weighted SFs, however, suffer from an issue of consistency, in that molecules with different elements and/or number of atoms are characterised by different numbers of SFs. As a result, the SFs vectors we would like to use as inputs for our ML algorithms are not of the same length. This problem may be circumvented in several ways. As a start, if one seeks to predict a functional property that can be written as the sum of atomic contributions, the original approach of Behler and Parrinello41 can be straightforwardly used. However, while one can think of some thermodynamic quantities such as energy or enthalpy as additive, functional properties or biomedical activities can often not be treated as such.
Here, we have decided to build histograms of weighted-SFs (H-wACSFs): by binning the values of all the weighted SFs for each molecule, we obtained a representation which is independent from the number of atoms in a given molecule. While the number of bins is one of the parameters we seek to optimise (see the following section), broadly speaking low and high numbers of bins provide more or less coarse-grained representations of the molecular structure. This interesting feature can be easily leveraged in the context of three-dimensional models of crystalline or amorphous drugs – where we believe that materials science-inspired descriptors such as H-wACSFs could deliver important contributions.
As the starting point for our H-wACSFs sets we have chosen the following parameter values: NRad = NAng = 8, RRadc, RAngc = 20 and NH-bins = 10, where NRad, NAng, RRadc, RAngc and NH-bins stand for the number of radial SFs, the number of angular SFs, the cutoff radius for the radial SFs, the cutoff radius for the angular SFs and the number of bins we have used to build the histograms, respectively.
2.3 Machine learning algorithms
In terms of the specific ML algorithm, we have been experimenting with multiple options, including neural networks, Gaussian processes and random forests. We have found that the choice of the ML algorithm has very little impact on our results. The numbers reported in the Results section have been obtained by using feed-forward neural networks, built using the Keras API47 with Tensorflow48 as a backend. The descriptors and the target properties for each dataset (Lipo, Hepa and Amo, see above) have been pre-processed by scaling them between zero and one and by removing the mean and scaling to unit variance, respectively. In terms of the neural networks optimisation, a simple parameter space grid search optimisation has been employed, taking into consideration different neural networks architectures (in terms of number of layers and nodes), different activation functions, and different solvers for the optimisation of the weights. Further details are included in the MSDE_Sosso_alpha GitHub repository.29As many as 300 epochs have been accumulated for each combinations of these parameters. The “optimal” number of epochs was determined according to an early stopping criterion based on the mean square error relative to the test set. 80% and 20% of the datasets have been randomly selected as training and test data, respectively, according to a k-fold cross validation49 procedure with k = 5 which allowed us to reliably assess the average performance of each neural network architecture. The “best” model was then selected and used to compute the results reported in section 3. We note that we have intentionally avoided to remove zero and near-zero variance features from our sets of descriptors: this is a practice commonly encountered in the recent literature50 which is based on the assumption that said features are simply non-informative – if anything, they present a risk for numerical errors within the machine learning algorithm of choice. However, we found that this is not always the case: as discussed in the ESI,† the removal of e.g. zero and near-zero features can even result in a loss of accuracy in some cases. In fact, a similar argument holds for the removal of highly correlated features, as discussed in detail in the ESI.† We also note that while it is certainly possible to leverage more advanced techniques (e.g. some form of ensemble learning51) to improve the accuracy of these algorithms, we have focused in here to provide a rather unbiased picture of the performance of the different descriptors under consideration. As a result, the numerical quality of our results is on average not very impressive, albeit we envisage that both the Hepa and Amo datasets will probably provide a tough challenge in terms of accuracy for more advanced ML frameworks as well.
2.4 Feature selection and optimisation
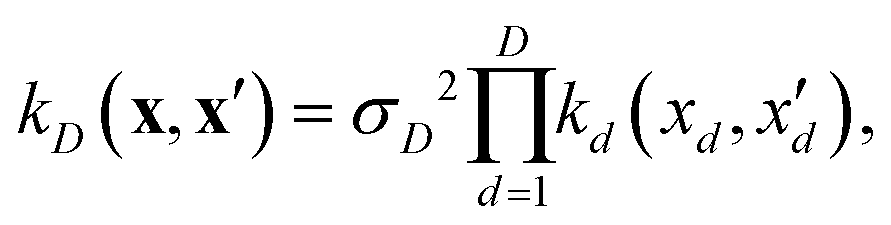 | (9) |
If, for sake of simplicity, one chooses the ubiquitous radial basis function (RBF) kernel, one obtains:
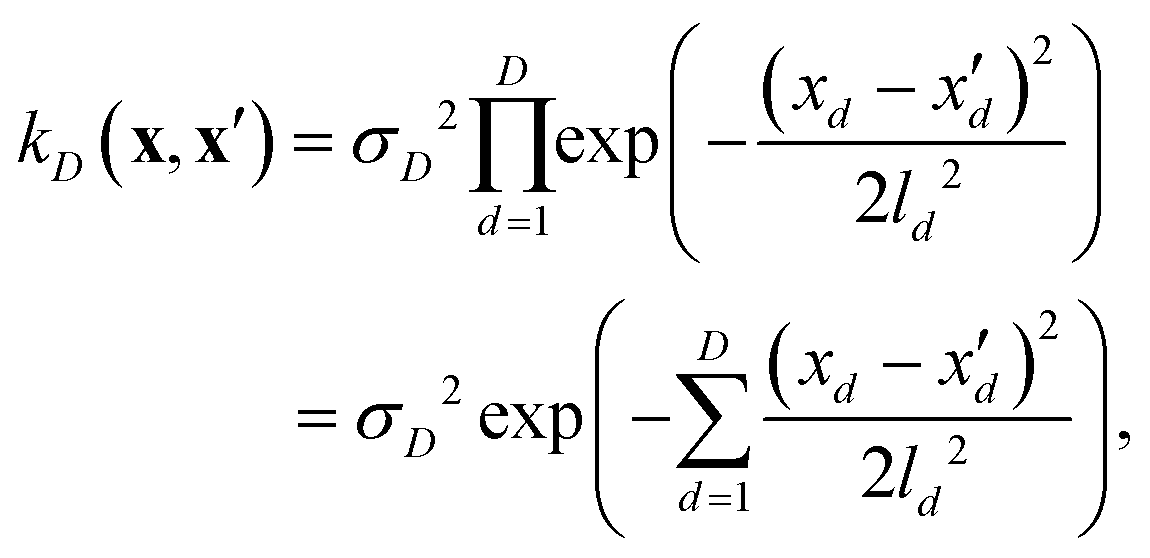 | (10) |
The expression in eqn (10) is known as the squared exponential kernel with automatic relevance determination (SE-ARD) or simply the ARD kernel. As each dimension of the input – i.e. each clique – is characterised by its own length-scale ld, upon e.g. regression, the magnitude of ld for the i-th kernel provides a measure of the importance of the clique in predicting the target property of interest. Specifically, small and large values of ld indicate high and low relevance of the corresponding clique, respectively. We have used the GPy53 package to implement this approach.
Though it has been shown that SE-ARD kernels can successfully remove irrelevant input dimensions,54 we have found that their usage led to rather inconsistent outcomes, with the value of the length-scale characteristic of a given clique fluctuating substantially depending on a particular training-test split (see the MSDE_Sosso_alpha GitHub repository29). To an extent, this is expected, particularly in the case of the Hepa and Amo datasets, where the small number of data points implies that different cliques may play different roles in specific training-test splits. Nevertheless, it would be obviously desirable to extract solid trends across different splits. We have found that achieving consistency is possible, but it does require extensive testing in terms of setting the initial values as well as the low/high boundaries for the different length-scales, and substantial statistics has to be accumulated with respect to different training-test splits.
Instead, we have explored the possibility of using the intrinsic ability of random forests (RFs) to provide a measure of importance for each clique via a measure called the mean decrease in impurity (MDI).55 An RF uses an impurity function i(τ) as a criterion for how to best split the dataset at each node τ such that similar target values will be in the same set.56 In general, the impurity function for RF regression is the variance;56 however, for illustrative purposes, we consider the simplest regression problem, one of binary classification, which utilises the Gini impurity function:
| i(τ) = 1 − p12 − p02, | (11) |
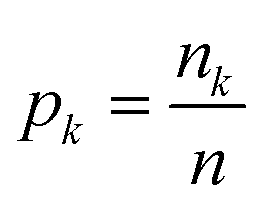 is the fraction of the nk samples of class k = {0, 1} out of n samples at node τ, to measure how well a potential split at each node τ within the binary trees T will separate the data.57 A decrease in i(τ) or Δi resulting from a split that sends a sample point to two sub-nodes, τl and τr, by a threshold tθ on feature θ is defined as:
is the fraction of the nk samples of class k = {0, 1} out of n samples at node τ, to measure how well a potential split at each node τ within the binary trees T will separate the data.57 A decrease in i(τ) or Δi resulting from a split that sends a sample point to two sub-nodes, τl and τr, by a threshold tθ on feature θ is defined as:| Δi(τ) = i(τ) − pli(τl) − pri(τr), | (12) |
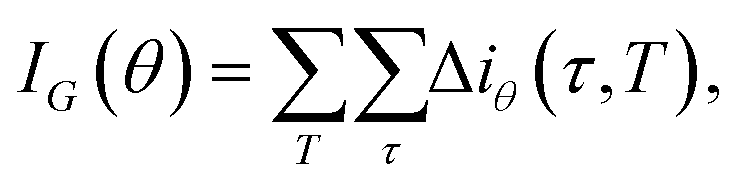 | (13) |
This strategy is easily implemented through the use of standard random forests algorithms. We have used the RandomForestRegressor model from the scikit-learn7 package. Contrary to the Gaussian processes approach described above, we have found that the MDI values corresponding to the different cliques are very consistent throughout different training-test splits – as discussed further in the Results section. Once the MDI for each clique has been reliably assessed, we sort all the cliques in our vocabulary according to their importance; at this point, one has to choose a threshold above which a certain clique is considered to be “important enough”. While in principle this is a parameter that can be optimised by means of a simple grid search, we have found for all the datasets under consideration that rather natural thresholds can be easily found – see the MSDE_Sosso_alpha GitHub repository29 for further details. The selected subset of cliques is then used to re-train a neural network following the same basic optimisation procedure detailed above.
GAs are a metaheuristic based around the principles of natural selection and evolution.60 An initial population is randomly generated where each individual in the population represents a solution to the problem. At each generation of the algorithm the “fittest” individuals “breed” with a subset of the remaining population, the offspring from this process then goes on to form the population for the next generation. There is also a chance for each individual to mutate, theoretically preventing the optimisation from converging on a local maxima.
In the case of our SFs, an initial population of 12 was used where each individual was comprised of 5 genes representing NRad, NAng, RRadc, RAngc and B. The fitness of each individual was calculated by generating the SFs with the appropriate parameters and training a NN using these, the negative MSE was used as the fitness score. Each of the three fittest individuals were then selected to breed with one of the remaining nine individuals with whom they would produce four offspring. Each of the offspring's genes had a 50% chance of being from each parent and there was a 50% chance that one of the genes (randomly selected) would mutate to a random value. This process was repeated for 20 generations and the individual with the best fitness in the entire history was selected as the parameters to the optimised SFs – see the MSDE_Sosso_alpha GitHub repository29 for further details. The results from this process are given in Table 3. Note that is perfectly possible to apply feature selection strategies to descriptors such as H-wACSFs as well: possible options include CUR decomposition and farthest point sampling, as recently demonstrated by Imbalzano et al.61
3 Results
The overall performance of the three classes of descriptors discussed in the previous section is summarised in Table 1: STD, cliques and H-wACSFs refers to the ∼100 “standard” RDKit descriptors, the vocabularies of molecular cliques and the histograms of weighted atom-centred symmetry functions, respectively. We report the mean squared error (MSE) and the Pearson correlation coefficient (PCC)62 for both the training and the test sets; averages and uncertainties (included as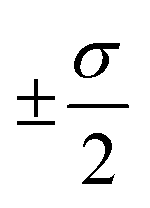 ) have been obtained according to the cross-validation procedure detailed in section 2.3. Detailed predictions for selected molecular structures can be found in the ESI.†
) have been obtained according to the cross-validation procedure detailed in section 2.3. Detailed predictions for selected molecular structures can be found in the ESI.†
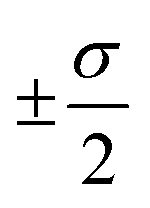 . Cliques [FS] and H-wACSFs [GAs] refer to the results obtained for cliques upon feature selection (the numbers in square brackets specify the number of selected cliques) and H-wACSFs upon optimisation, respectively. See text for further details about both datasets and descriptors
. Cliques [FS] and H-wACSFs [GAs] refer to the results obtained for cliques upon feature selection (the numbers in square brackets specify the number of selected cliques) and H-wACSFs upon optimisation, respectively. See text for further details about both datasets and descriptors
| MSE | |||||
|---|---|---|---|---|---|
| STD | Cliques | Cliques [FS] | H-wACSFs | H-wACSFs [GAs] | |
| Lipo | 0.198 ± 0.098 (0.682 ± 0.023) | 0.412 ± 0.016 (0.950 ± 0.019) | 0.690 ± 0.005 (1.032 ± 0.040) [15] | 0.889 ± 0.020 (0.939 ± 0.022) | 0.746 ± 0.019 (0.920 ± 0.031) |
| Hepa | 0.253 ± 0.063 (0.413 ± 0.059) | 0.176 ± 0.007 (0.317 ± 0.029) | 0.125 ± 0.005 (0.304 ± 0.028) [18] | 0.590 ± 0.055 (1.238 ± 0.171) | 0.314 ± 0.010 (0.350 ± 0.037) |
| Amo | 0.460 ± 0.127 (0.806 ± 0.171) | 0.130 ± 0.009 (0.950 ± 0.360) | 0.497 ± 0.029 (0.994 ± 0.167) [13] | 0.362 ± 0.041 (1.348 ± 0.465) | 0.124 ± 0.019 (0.838 ± 0.084) |
| PCC | |||||
|---|---|---|---|---|---|
| STD | Cliques | Cliques [FS] | H-wACSFs | H-wACSFs [GAs] | |
| Lipo | 0.933 ± 0.003 (0.737 ± 0.019) | 0.859 ± 0.003 (0.623 ± 0.010) | 0.727 ± 0.003 (0.554 ± 0.020) [15] | 0.336 ± 0.011 (0.273 ± 0.020) | 0.503 ± 0.020 (0.0327 ± 0.013) |
| Hepa | 0.687 ± 0.043 (0.295 ± 0.031) | 0.731 ± 0.012 (0.359 ± 0.054) | 0.826 ± 0.007 (0.450 ± 0.041) [18] | 0.641 ± 0.035 (0.148 ± 0.033) | 0.417 ± 0.037 (0.136 ± 0.077) |
| Amo | 0.873 ± 0.008 (0.637 ± 0.058) | 0.935 ± 0.007 (0.400 ± 0.218) | 0.733 ± 0.015 (0.349 ± 0.111) [13] | 0.802 ± 0.028 (0.261 ± 0.101) | 0.936 ± 0.009 (0.497 ± 0.134) |
Concerning the Lipo dataset, STD outperform both cliques and H-wACSFs. The latter are clearly not very suitable to deal with this particular dataset. As discussed in further detail below, this was expected, given the nature of the target property to be predicted. On the other hand, by using the full set of cliques (i.e. without feature selection) one can achieve results of similar quality to those obtained via the STD – quite impressive, considering how basic the cliques descriptors are. Upon feature selection, specifically utilising only 15 cliques (out of 246), the performance of the cliques degrades further; however, being able to retain some predictive capabilities using 15 molecular fragments is indicative of the potential of this descriptor.
In fact, the cliques consistently outperform the STD in the case of both the Hepa and the Amo dataset: we remind the reader that while the Lipo dataset is relatively large (∼4000 molecules), the Hepa and particularly the Amo dataset are quite small (∼400 and ∼150 molecules, respectively). Interestingly, in the case of the Hepa dataset, using just the most relevant (according to the MDI-based feature selection procedure discussed in sec. 2) 18 cliques (out of the 132 contained in the full set) results in even better outcomes compared to what we have obtained for the full set of cliques, as illustrated in Fig. 3. This is an impressive result: just 18 molecular components appear to capture some of the structure–function relation at the heart of a complex biomedical activity such as human hepatocytes intrinsic clearance. As detailed in Table 2, these 18 cliques are characterised by an MDI about one order of magnitude larger compared to that of the least important cliques. We also note that the RF-based feature selection procedure we have used is capable to assign MDIs characterised by very small uncertainties, thus making the selection process quite reliable indeed. Amongst these 18 cliques we find molecular components such as (in SMILES notation) CC, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C1CCCCC1 (cyclohexane) and C1CCCCC1 (benzene) which are ubiquitous in small drug-like molecules: in fact, they possess quite high MDI scores for the Lipo and Amo datasets as well. On the contrary, we also find cliques whose role in the context of human hepatocytes intrinsic clearance is perhaps not immediately obvious: CF, CS and C1CSCN1/C1NCCS1 (2,3/4,5-dihydrothiazole).
O, C1CCCCC1 (cyclohexane) and C1CCCCC1 (benzene) which are ubiquitous in small drug-like molecules: in fact, they possess quite high MDI scores for the Lipo and Amo datasets as well. On the contrary, we also find cliques whose role in the context of human hepatocytes intrinsic clearance is perhaps not immediately obvious: CF, CS and C1CSCN1/C1NCCS1 (2,3/4,5-dihydrothiazole).
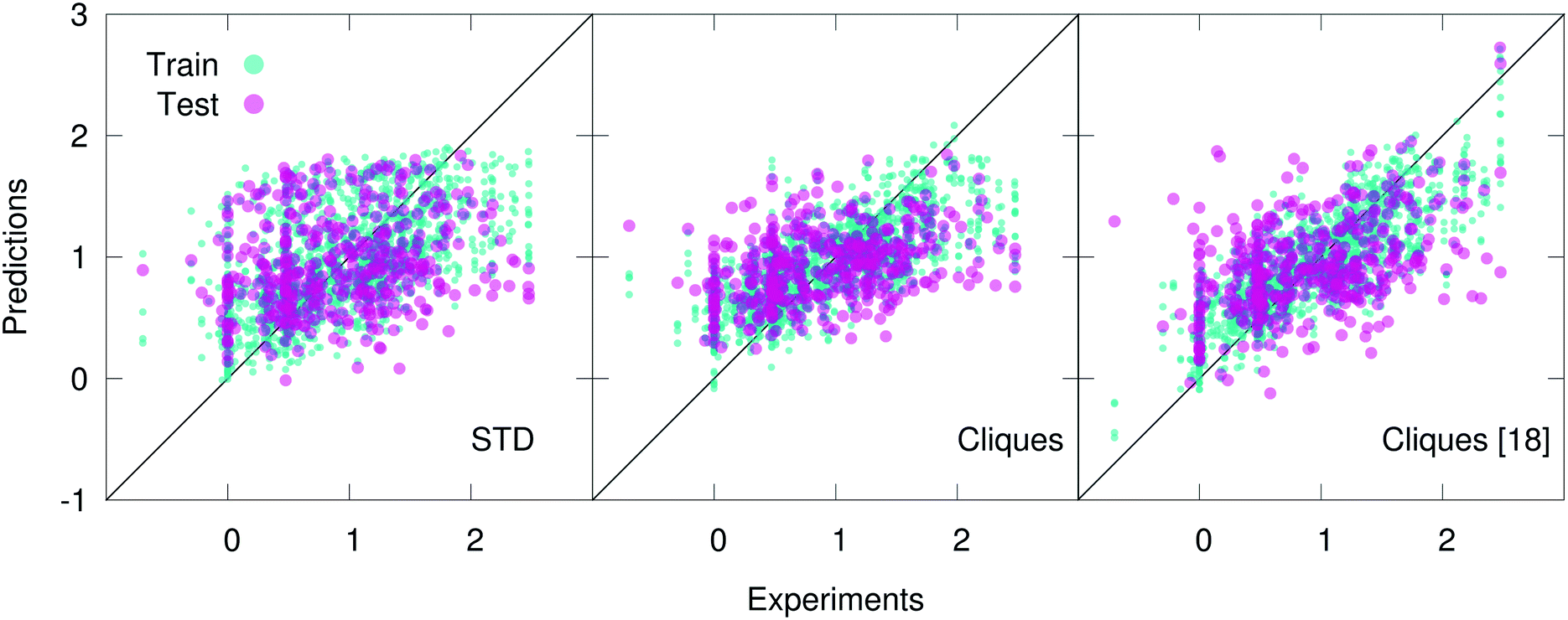 | ||
| Fig. 3 Scatter plots of the predicted vs. experimental values (scaled according to the pre-processing strategy detailed in section 2.3) of human hepatocytes intrinsic clearance for the hepatocytes dataset, using ∼100 “standard” RDKit descriptors (STD), the full vocabulary of molecular cliques (Cliques), and just 18 out of 132 cliques (Cliques [18]) according to the outcomes of the feature selection procedure discussed in section 2.4. The results obtained for five different training-test splits are plotted on the same graph, which thus contains 406 × 5 = 2030 points. Note the improvement of the predictions upon the cliques feature selection. | ||
| Feature selection – cliques | ||
|---|---|---|
| Hepatocytes dataset | ||
| Smiles | MDI (mean) | MDI (σ) |
| CC | 0.082263 | 0.002642 |
| CO | 0.069692 | 0.002545 |
| CN | 0.069352 | 0.001979 |
| C | 0.054925 | 0.002775 |
|
C1CCCCC1 |
0.052196 | 0.002532 |
|
CO |
0.032964 | 0.001487 |
| CF | 0.031491 | 0.002122 |
|
C1CNCCC1 |
0.030531 | 0.005510 |
|
C1COCCC1 |
0.028628 | 0.003882 |
| C1COCCN1 | 0.027860 | 0.002575 |
| C1CCNCC1 | 0.025989 | 0.002891 |
| CCl | 0.025489 | 0.001000 |
|
C1CSCC1 |
0.024680 | 0.003132 |
| C1CCCCC1 | 0.021090 | 0.002438 |
| CS | 0.017693 | 0.001977 |
| C1CNCCN1 | 0.017380 | 0.002165 |
|
C1CSCN1 |
0.017038 | 0.002653 |
|
C1NCCS1 |
0.013932 | 0.001524 |
| C1CNSC1 | 0.015341 | 0.003452 |
| C#N | 0.013333 | 0.001248 |
| [...] | ||
| C1CCOCC1 |
0.005135 | 0.000685 |
| C1CNC1 | 0.005111 | 0.001257 |
| C1CNCN1 | 0.004771 | 0.000744 |
| C1CCNCC1 |
0.004578 | 0.000439 |
| C1CCCCC1 |
0.004489 | 0.000649 |
The situation is slightly different in the case of the Amo dataset: while using the full set of cliques results in a substantial improvement with respect to the STD outcomes, using 13 out of 87 cliques (according to the results of feature selection) worsens the numerical accuracy of our prediction. Nonetheless, this small set of cliques provides predictive capabilities of the same quality of STD – i.e. using 13 molecular components gives similar results to those obtained by using ∼100 different descriptors. Appropriately, our findings suggest that molecular cliques may represent, despite their simplicity, an interesting way forward to identify structural patterns of interest in the context of drug design and discovery.
As opposed to cliques, which captures the main elements of the chemistry of the system, H-wACSFs provide information about the whole molecular structure. Thus, it is reasonable to expect them to perform their best when deployed to predict target properties largely determined by structure as opposed to chemistry. Indeed, we find that H-wACSFs score best when applied on the Amo dataset, where the property we seek to predict is the Tg of amorphous drugs. Using the non-optimised values of the H-wACSFs parameters NRad, NAng, RRadc, RAngc and B (see Table 3), we obtain a marginal improvement in the MSE when compared to the STD results (see Table 1), but also a significantly worse value for the PCC, as evident from Fig. 4. However, upon the optimisation of the above mentioned parameters via the genetic algorithms discussed in section 2.4, we obtain a significant improvement of our predictions across all metrics, as illustrated in Fig. 4. It is interesting to note that the optimised parameters obtained for the three different datasets (see Table 3) vary significantly, with no robust trends emerging – the potential benefits of introducing constraints within our genetic algorithms would be addressed in future work.
| Optimisation – H-wACSFs | ||||
|---|---|---|---|---|
| Non-optimised | Lipo | Hepa | Amo | |
| N Rad | 8 | 3 | 14 | 22 |
| N Ang | 16 | 14 | 8 | 10 |
| R Radc (Å) | 20 | 2 | 21 | 7 |
| R Angc (Å) | 20 | 21 | 12 | 2 |
| B | 10 | 16 | 19 | 12 |
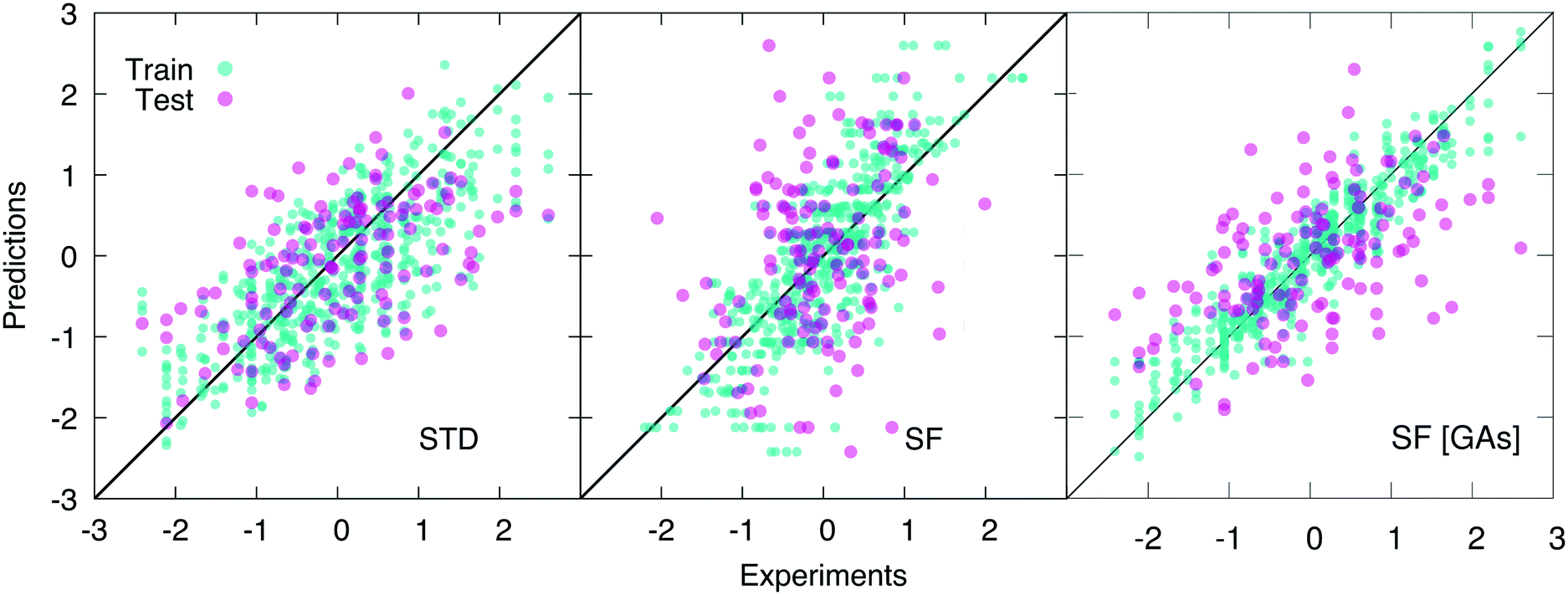 | ||
| Fig. 4 Scatter plots of the predicted vs. experimental values (scaled according to the pre-processing strategy detailed in section 2.3) of the glass transition temperature Tg for the amorphous dataset, using ∼100 “standard” RDKit descriptors (STD), H-wACSFs (SF), and H-wACSFs optimised according to the genetic algorithms-based procedure describe in section 2.4 (SF [GAs]). The results obtained for five different training-test splits are plotted on the same graph, which thus contain 132 × 5 = 660 points. Note the improvement of the predictions upon the H-wACSFs optimisation. | ||
For the Hepa and Amo datasets, where the H-wACSFs have outperformed STD, the genetic algorithms seem to have emphasised the connectivity of the molecular network as opposed to geometry of the specific conformers, in that NRad ∼ 2NAng. As discussed in section 2.3, the procedure we have used to generate said conformers is very basic, and as such, we expect the angular contributions to H-wACSFs to feature more prominently for ensembles of thoroughly (e.g. from first principles) optimised molecular conformers, and even more so in the case of actual three-dimensional models of either crystalline or amorphous drugs. Further support to this hypothesis comes from the fact that H-wACSFs did not perform especially well in the case of the Hepa dataset, where upon optimisation, we obtained results of similar, but not better quality when compared to the STD descriptors. In contrast to the Amo dataset, the Hepa dataset – and in fact, the Lipo dataset as well – seeks to predict a target property which may very well be ruled chiefly by chemistry as opposed to structure. Further evidence supporting this claim is provided in the ESI,† where we have built a classification model for the Tox21 dataset63 – a very well-known dataset including as many as twelve different toxicity targets of biological relevance for drug design. While the distinction between cliques and H-wACSFs is not absolute in this respect (the cliques hold some structural information, and the H-wACSFs indirectly contains information about all cliques), we believe there is scope to bring the two classes of descriptors together, thus combining chemistry and structure – within a reasonably small number of descriptors, as opposed to harnessing the whole array of STD currently available.
Overall, our results are suggestive of the fact that while for relatively large datasets there might be value in trying to take advantage of the many descriptors readily available via open source computational packages, for small datasets containing hundreds of molecular structures, one might very well obtain better results by using just a handful of carefully crafted descriptors. In this work, we focused on cliques and H-wACSFs, but countless other options are obviously available. Despite the still limited scope of our investigation, we feel confident in saying that feature selection and optimisation should be treated as a necessary step of any ML algorithm for drug design and discovery, much as data pre-processing – as opposed to be considered as optional possibilities. We also note that many datasets of interest to the pharmaceutical companies are very limited in size: the Hepa dataset considered in here is just one example, but broadly speaking it is still challenging, despite the speed with which the field is progressing, to collect large amounts of experimental measurements of complex biomedical activities. While it should be very clear at this point in time that no universal recipe exists that can provide a general-purpose framework to treat any given dataset, we believe this is yet another reason to be selective with respect to the choice of molecular descriptors.
4 Conclusions
The number of readily available molecular descriptors to be employed in the context of machine learning for drug design and discovery is growing at a spectacular rate. As such, one may be tempted to leverage as many of these descriptors as possible to increase the flexibility and the accuracy of the machine learning framework of choice. In this work, we have provided evidence that while this “strength in numbers” strategy may be rewarding when dealing with relatively larger datasets, in the case of small datasets containing only hundreds of molecular structures one might – potentially – obtain better numerical accuracy and – certainly – a deeper insight into the structure–function relation.In particular, we have explored the predictive potential of two classes of descriptors derived from the work of Jin et al.26 and Gastegger:27 vocabularies of molecular cliques (cliques) and histograms of weighted atomic-centred symmetry functions (H-wACSFs). While the former capture the “chemistry” of a given molecular species, the latter offer information about the whole structure of the molecule. When deployed to predict the functional properties or biomedical activities of two small molecular datasets, cliques and H-wACSFs descriptors give results of similar quality to those obtained by using ∼100 “standard” descriptors (STD) available via the RDKit package.
Importantly, upon feature selection (cliques) and optimisation (H-wACSFs) we were able to even outperform in some cases the STD results: we find that using as few as ∼15 cliques (i.e. molecular fragments) as descriptors one can retain, in some cases even improve, the numerical accuracy of the machine learning framework of choice, all the while gaining valuable insight into those structural features that play a key role in determining the target properties of interest. Similarly, the optimisation of the some of the parameters entering the formulation of H-wACSFs led to substantial improvement with respect to numerical accuracy, particularly when trying to predict solid-state functional properties such as the glass transition temperature.
While most would agree that designing a set of “universal” molecular descriptors might not ever be possible, we believe that an effort to limit the number of descriptors is a necessary step toward making machine learning for drug design and discovery more transparent. Even when dealing with large datasets, feature selections and/or optimisation should be seen as a mandatory step within the computational pipeline, much as data pre-processing, as opposed to an optional possibility. This is especially true given the multitude of easily accessible computational tools presently at our disposal. The case of the cliques descriptors offer a prime example, in that its intrinsic simplicity has the potential to provide clear indication about the relevance of specific molecular fragments.
Overall, we feel that while there is obvious practical value in striving for numerical accuracy, the ultimate goal of machine learning in the context of drug design and discovery should be to unravel the complexity of the structure–function relation that rules the macroscopic properties of interest to the pharmaceutical industry. In this respect, a major pitfall of the current paradigm is that we often try to predict solid-state properties (e.g. the solubility of a crystalline drug, or the physical stability of an amorphous drug) by looking at the structure of single molecules in vacuum.
We believe that taking into account actual three-dimensional models of either crystalline or amorphous drugs may very well be the next step the community has to take, and as such we need to devise molecular descriptors that will be able to capture the complexity of e.g. inter-molecular interactions. Materials science-inspired descriptors such as the H-wACSFs probed in this work may provide valuable contributions, and we are planning to bring together “chemistry and structure” by combining cliques and H-wACSFs to deliver a more general set of descriptors equally capable to tackle single molecules as well as molecular solids.
Contributions
G. C. S. conceived the research. T. B., H. H. and S. T. implemented the methodology and contributed to the analysis of the results: they all contributed to this work in equal measure, and are thus alphabetically listed within the authors' list. All the authors contributed to the interpretation of the results and to the writing of the manuscript as well.Data availability
With the sole exception of the hepatocytes dataset (given to use by AstraZeneca plc), the data and the software supporting the findings of this study are freely available via the public MSDE_Sosso_alpha GitHub repository.29Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the EPSRC and MRC Centre for Doctoral Training in Mathematics for Real-World Systems, funded by the UK Engineering and Physical Sciences Research Council (grant EP/L015374/1) and the University of Warwick. We gratefully acknowledge the high-performance computing facilities provided by the Scientific Computing Research Technology Platform at the University of Warwick. We are indebted to Claus Bendtsen, Ola Engkvist and Anders Broo (AstraZeneca plc) for their support and insights throughout the development of this work.References
- J. Vamathevan, D. Clark, P. Czodrowski, I. Dunham, E. Ferran, G. Lee, B. Li, A. Madabhushi, P. Shah and M. Spitzer, et al. , Nat. Rev. Drug Discovery, 2019, 1 Search PubMed.
- K.-K. Mak and M. R. Pichika, Drug Discovery Today, 2019, 24, 773–780 CrossRef PubMed.
- 5 Trends Emerge in the Gartner Hype Cycle for Emerging Technologies, 2018, http://www.gartner.com/smarterwithgartner/5-trends-emerge-in-gartner-hype-cycle-for-emerging-technologies-2018/ Search PubMed.
- Pharma groups combine to promote drug discovery with AI|Financial Times, https://www.ft.com/content/ef7be832-86d0-11e9-a028-86cea8523dc2 Search PubMed.
- Y. Hu and J. Bajorath, Future Sci. OA, 2017, 3, FSO179 CrossRef PubMed.
- H. Chen, O. Engkvist, Y. Wang, M. Olivecrona and T. Blaschke, Drug Discovery Today, 2018, 23, 1241–1250 CrossRef PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and E. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- R. Todeschini, V. Consonni, R. Mannhold, H. Kubinyi and G. Folkers, Molecular Descriptors for Chemoinformatics, Wiley, 2009 Search PubMed.
- RDKit, http://www.rdkit.org/ Search PubMed.
- K. T. Schütt, H. E. Sauceda, P.-J. Kindermans, A. Tkatchenko and K. Mueller, J. Chem. Phys., 2017, 148, 241722 CrossRef PubMed.
- A. Singraber, T. Morawietz, J. Behler and C. Dellago, J. Chem. Theory Comput., 2019, 15, 3075–3092 CrossRef CAS.
- A. Mauri, V. Consonni, M. Pavan and R. Todeschini, MATCH, 2006, 56, 237–248 CAS.
- I. Olier, N. Sadawi, G. R. Bickerton, J. Vanschoren, C. Grosan, L. Soldatova and R. D. King, Mach. Learn., 2018, 107, 285–311 CrossRef.
- A. Bender, J. L. Jenkins, J. Scheiber, S. C. K. Sukuru, M. Glick and J. W. Davies, J. Chem. Inf. Model., 2009, 49, 108–119 CrossRef CAS PubMed.
- M. Dehmer, F. Emmert-Streib and S. Tripathi, PLoS One, 2013, 8, e83956 CrossRef PubMed.
- S. Lapuschkin, S. Wäldchen, A. Binder, G. Montavon, W. Samek and K.-R. Müller, Nat. Commun., 2019, 10, 1–8 CrossRef CAS PubMed.
- D. Castelvecchi, Nature, 2016, 538, 20 CrossRef CAS PubMed.
- J. Drews, Science, 2000, 287, 1960–1964 CrossRef CAS.
- A. Alzghoul, A. Alhalaweh, D. Mahlin and C. A. Bergström, J. Chem. Inf. Model., 2014, 54, 3396–3403 CrossRef CAS.
- D. Mahlin and C. A. Bergström, Eur. J. Pharm. Sci., 2013, 49, 323–332 CrossRef CAS.
- A. Alzghoul, A. Alhalaweh, D. Mahlin and C. A. S. Bergström, J. Chem. Inf. Model., 2014, 54, 3396–3403 CrossRef CAS PubMed.
- A. Alhalaweh, A. Alzghoul, W. Kaialy, D. Mahlin and C. A. S. Bergström, Mol. Pharmaceutics, 2014, 11, 3123–3132 CrossRef CAS PubMed.
- C. A. S. Bergström and P. Larsson, Int. J. Pharm., 2018, 540, 185–193 CrossRef.
- S. De, A. P. Bartók, G. Csányi and M. Ceriotti, Phys. Chem. Chem. Phys., 2016, 18, 13754–13769 RSC.
- J. Behler, J. Chem. Phys., 2011, 134, 074106 CrossRef.
- W. Jin, R. Barzilay and T. Jaakkola, 2018, arXiv:1802.04364 [cs, stat].
- M. Gastegger, L. Schwiedrzik, M. Bittermann, F. Berzsenyi and P. Marquetand, J. Chem. Phys., 2018, 148, 241709 CrossRef CAS.
- C. Lu, P. Li, R. Gallegos, V. Uttamsingh, C. Q. Xia, G. T. Miwa, S. K. Balani and L.-S. Gan, Drug Metab. Dispos., 2006, 34, 1600–1605 CrossRef CAS.
- G. C. Sosso, Less may be more: an informed reflection on molecular descriptors for drug design and discovery: gcsosso/MSDE_Sosso_alpha, 2019, https://github.com/gcsosso/MSDE_Sosso_alpha, original-date: 2019–08-16T09:23:18Z Search PubMed.
- Datasets, http://moleculenet.ai/datasets-1.
- E. Anderson, G. Veith and D. Weininger, Environmental Research Laboratory-Duluth. Report No. EPA/600/M-87/021, 1987 Search PubMed.
- E. Rutkowska, K. Pajak and K. Jóźwiak, Acta Pol. Pharm., 2013, 70, 3–18 CAS.
- A. Alhalaweh, A. Alzghoul, W. Kaialy, D. Mahlin and C. A. Bergström, Mol. Pharmaceutics, 2014, 11, 3123–3132 CrossRef CAS.
- D. Mahlin and C. A. Bergström, Eur. J. Pharm. Sci., 2013, 49, 323–332 CrossRef CAS PubMed.
- E. O. Kissi, G. Kasten, K. Löbmann, T. Rades and H. Grohganz, Mol. Pharmaceutics, 2018, 15, 4247–4256 CrossRef CAS PubMed.
- M. Rams-Baron, Amorphous drugs: benefits and challenges, Springer Berlin Heidelberg, New York, NY, 2018 Search PubMed.
- S. Riniker and G. A. Landrum, J. Chem. Inf. Model., 2015, 55, 2562–2574 CrossRef CAS PubMed.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- P. Gramatica, QSAR Comb. Sci., 2006, 25, 327–332 CrossRef CAS.
- C. R. Collins, G. J. Gordon, O. A. von Lilienfeld and D. J. Yaron, J. Chem. Phys., 2018, 148, 241718 CrossRef PubMed.
- J. Behler and M. Parrinello, Phys. Rev. Lett., 2007, 98, 146401 CrossRef PubMed.
- G. C. Sosso, V. L. Deringer, S. R. Elliott and G. Csányi, Mol. Simul., 2018, 44, 866–880 CrossRef CAS.
- A. Singraber, T. Morawietz, J. Behler and C. Dellago, J. Phys.: Condens. Matter, 2018, 30, 254005 CrossRef PubMed.
- J. Li, K. Song and J. Behler, Phys. Chem. Chem. Phys., 2019, 21, 9672–9682 RSC.
- F. C. Mocanu, K. Konstantinou, T. H. Lee, N. Bernstein, V. L. Deringer, G. Csányi and S. R. Elliott, J. Phys. Chem. B, 2018, 122, 8998–9006 CrossRef CAS.
- V. Quaranta, J. Behler and M. Hellström, J. Phys. Chem. C, 2019, 123, 1293–1304 CrossRef CAS.
- F. Chollet, et al., Keras, 2015, https://keras.io Search PubMed.
- M. Abadi, A. Agarwal, P. Barham, E. Brevdo, Z. Chen, C. Citro, G. S. Corrado, A. Davis, J. Dean, M. Devin, S. Ghemawat, I. Goodfellow, A. Harp, G. Irving, M. Isard, Y. Jia, R. Jozefowicz, L. Kaiser, M. Kudlur, J. Levenberg, D. Mané, R. Monga, S. Moore, D. Murray, C. Olah, M. Schuster, J. Shlens, B. Steiner, I. Sutskever, K. Talwar, P. Tucker, V. Vanhoucke, V. Vasudevan, F. Viégas, O. Vinyals, P. Warden, M. Wattenberg, M. Wicke, Y. Yu and X. Zheng, TensorFlow: Large-Scale Machine Learning on Heterogeneous Systems, 2015, https://www.tensorflow.org/, Software available from tensorflow.org Search PubMed.
- K. A. Ross, in Encyclopedia of Database Systems, ed. L. Liu and M. T. Özsu, Springer US, Boston, MA, 2009, pp. 301–304 Search PubMed.
- M. Kuhn and K. Johnson, Applied Predictive Modeling, Springer-Verlag, New York, 2013 Search PubMed.
- S. Raschka, Python Machine Learning, Packt Publishing, 2015 Search PubMed.
- D. Duvenaud, H. Nickisch and C. E. Rasmussen, Proceedings of the 24th International Conference on Neural Information Processing Systems, USA, 2011, pp. 226–234 Search PubMed.
- GPy, GPy: A Gaussian process framework in python, http://github.com/SheffieldML/GPy, since 2012 Search PubMed.
- C. K. Williams and C. E. Rasmussen, Advances in neural information processing systems, 1996, pp. 514–520 Search PubMed.
- L. Breiman, Consistency for a simple model of random forests, technical report, 2004 Search PubMed.
- G. Biau and E. Scornet, Test, 2016, 25, 197–227 CrossRef.
- B. H. Menze, B. M. Kelm, R. Masuch, U. Himmelreich, P. Bachert, W. Petrich and F. A. Hamprecht, BMC Bioinf., 2009, 10, 213 CrossRef.
- L. Breiman, Mach. Learn., 2001, 45, 5–32 CrossRef.
- G. Louppe, 2014, arXiv preprint arXiv:1407.7502.
- C. Darwin, On the origin of species, 1859, Routledge, 2004 Search PubMed.
- G. Imbalzano, A. Anelli, D. Giofré, S. Klees, J. Behler and M. Ceriotti, J. Chem. Phys., 2018, 148, 241730 CrossRef.
- K. Pearson, Proc. R. Soc. London, 1895, 58, 240–242 CrossRef.
- A. Mayr, G. Klambauer, T. Unterthiner and S. Hochreiter, Front. Environ. Sci., 2016, 3, 80 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9me00109c |
| ‡ Present address: Department of Engineering Technology, University of Twente, De Horst 2, 7522 LW Enschede, The Netherlands. |
| This journal is © The Royal Society of Chemistry 2020 |