
Therapeutic peptide delivery via aptamer-displaying, disulfide-linked peptide amphiphile micelles†
Josiah D.
Smith‡
a,
Leah N.
Cardwell‡
b,
David
Porciani
bc,
Andrea
Nolla
a,
Brenna T.
Cornelison
a,
Megan C.
Schulte
a,
Fabio
Gallazzi
de,
Donald H.
Burke
abcf,
Mark A.
Daniels
bg and
Bret D.
Ulery
 *a
*a
aDepartment of Biomedical, Biological, and Chemical Engineering, University of Missouri, Columbia, MO, USA. E-mail: uleryb@missouri.edu
bDepartment of Molecular Microbiology and Immunology, University of Missouri, Columbia, MO, USA
cBond Life Science Center, University of Missouri, Columbia, MO, USA
dMolecular Interactions Core, University of Missouri, Columbia, MO, USA
eDepartment of Chemistry, University of Missouri, Columbia, MO, USA
fDepartment of Biochemistry, University of Missouri, Columbia, MO, USA
gDepartment of Surgery, University of Missouri, Columbia, MO, USA
First published on 7th October 2019
Abstract
Peptide amphiphile micelles (PAMs) are a powerful platform technology for improving the delivery of therapeutic and prophylactic peptides. While previous research has shown aptamer-displaying PAMs enhance cell association, transportation to intracellular targets still remains a substantial hurdle for these biomaterials. In this article, we detail our efforts to address this challenge through the creation of disulfide-linked peptide amphiphile (PAs). These molecules were found to self-assemble in water into PAMs for which lipidated DNA oligomers (i.e., antitail amphiphiles – AAs) could be entrapped and used to tether aptamers (Apt) to the nanoparticle surface. These Apt∼A/PAMs were physically characterized and evaluated for their blood-serum stability using fetal bovine serum exposure and glutathione reduction. To assess their enhanced intracellular delivery capacity and therapeutic functionality, PAMs bearing cell-penetrating peptide modified “Plenty of SH3 domains” scaffold protein competitive inhibitor (Tat-POSH) and B cell lymphoma targeting aptamer (C10.36) were incubated with Ramos cells, a non-Hodgkin lymphoma cell line. C10.36∼A/PAMs were found not only to be stable in blood-like conditions, but also to be capable of facilitating delivery of therapeutic Tat-POSH peptide to Ramos cells in vitro.
 Bret Ulery | Bret Ulery is an Assistant Professor of Biomedical, Biological, and Chemical Engineering and the Principal Investigator of the Biomodulatory Materials Engineering Laboratory at the University of Missouri. After earning undergraduate degrees in Chemical Engineering and Biochemistry from the University of Iowa in 2006, he conducted graduate research with Dr. Balaji Narasimhan at Iowa State University and received his Ph.D. in Chemical Engineering with a Graduate Minor in Immunobiology in 2010. He then spent four years as a postdoctoral researcher at the University of Connecticut Health Center and then at the University of Chicago. |
Design, System, ApplicationTargeted intracellular treatment delivery is of significant importance to achieving therapeutic efficacy for a number of drug products including small molecules, peptides, and proteins. In specific, materials-based vehicle design requires structural integrity when circulating in the body, highly specific association with the desired cell population of interest, and drug release inside the cell allowing it to carry out its bioactivity. Unfortunately, molecular design and engineering strategies that optimize one of these aspects can be detrimental to achieving another. With peptide amphiphile micelles, we previously found that the chemical modifications required to create stable nanoparticles with cell-specific aptamer targeting functionality limited their capacity to dissociate and deliver their bioactive peptide payload. To circumvent this issue, we have designed a peptide amphiphile with a disulfide linkage between its hydrophilic anti-cancer peptide and hydrophobic lipid. The structure of the micelle excitingly limited antioxidant-mediated cleavage at serum concentrations, but was highly sensitive at intracellular levels. This aspect turned out to be crucial in improving the anti-cancer effects of the micelle incorporated peptide on lymphoma cells beyond what could be achieved by any of the controls including continuous bombardment with the peptide for 24 hours. Future efforts will focus on further optimizing this novel product using an enzyme-sensitive linker (i.e. a cathepsin B cleavable peptide) and the inclusion of different cell penetrating peptides to increase cytosolic delivery, as this is the therapeutic site of action. |
Introduction
Non-Hodgkin lymphoma (NHL) is the seventh most common cancer and, due to recent advancements in front-line treatments, is associated with a 71% 5-year survival rate.1 Despite this improved overall survival rate, patients with refractory or relapsed NHL have a lower 5-year survival rate of 40–60%.2 The gold-standard treatment regimen for the most common subtype, B cell NHL, is R-CHOP, a combination of a B cell-specific monoclonal antibody, a steroid, and multiple conventional chemotherapeutic drugs.3 These chemotherapeutics are associated with many detrimental side effects such as nerve, bone, kidney, and endocrine system toxicity.4 They have even been found to directly cause a significant number of patient deaths.5 Newer therapeutics such as antibody-based and cell-based treatments face their own problems due to their significant life-long side-effects and the potential for cancer cell escape.6–9 Due to the recurrence risk of NHL and problems associated with newer treatments, there still exists a need for novel, effective therapeutics to treat this deadly cancer.Peptide-based therapeutics provide an exciting alternative option for NHL treatment10 due to a large, diverse set of potential biological targets and recently developed candidate peptides.11–13 In specific, a cell-penetrating peptide modified therapeutic peptide (Tat-POSH) has been shown capable of disrupting protein–protein interactions in hematological cancers, leading to cell death.14 Excitingly, this competitive inhibitor does not harm healthy lymphocytes.15,16 However, intracellular delivery of therapeutic peptides is a difficult challenge in vivo due to peptide degradation and clearance in the body.17 Nanoparticles are known to be capable of protecting peptides in vivo and facilitating directed delivery when combined with a targeting moiety.18,19 Micelles are a specific type of nanoparticle composed of self-assembled amphiphilic molecules. By lipidating therapeutic peptides, they can be transformed into peptide amphiphiles (PAs) which undergo hydrophobically-driven self-assembly into peptide amphiphile micelles (PAMs), nanoparticles composed mostly of the therapeutic peptide itself.
PAMs have been previously used to deliver a variety of bioactive peptides including immunomodulatory vasoactive intestinal peptide,20 group A streptococcal vaccine J8 peptide,21 and anti-cancer p5314–29 peptide.22 Recently, a PAM was developed containing a cathepsin B-mediated cleavable linker between the peptide and lipid that was capable of cellular internalization, component cleavage, and bioactive peptide release.23 An interesting novel PAM delivery device would be one that has similar post-internalization processability as those containing a cathepsin B sensitive linker, but employs a more ubiquitously cleavable linkage. One cleavage inducing target of interest could be glutathione, the primary antioxidant in the body, which is present in low concentrations in the blood, but much higher concentrations within cells.24 Glutathione is known for breaking disulfide bonds,25–27 which have been previously utilized to stabilize PAM cores.28–31 Therefore, disulfide-linked PAs have considerable potential for intracellular peptide therapeutic release applications.
In addition to nanoparticles protecting therapeutic peptide amphiphiles in vivo, they can also be modified with targeting moieties to direct their payload delivery to cells of interest. Aptamers are an exciting targeting technology composed of DNA or RNA designed to selectively bind a biological target which have been utilized to preferentially deliver nanoparticles to prostate membrane,32 gastric epithelium,33 and B cell leukemia and lymphoma.34,35 C10.36, a B cell lymphoma-specific aptamer developed via cell selection and validated in vitro by Opazo, et. al,34,36 has been used to target a payload to several different B cell leukemias.37 Modifying PAMs with C10.36 aptamer has the potential to facilitate cell-specific nanoparticle delivery to hematological cancer cells.
Here, we describe a novel, scalable synthesis route to produce a disulfide linkage between Tat-POSH peptide and a lipid yielding a disulfide-linked therapeutic PA (i.e., peptide-C_C-lipid), termed dPA, capable of self-assembling in water into dPAMs. Through micellar incorporation of oligonucleotide-amphiphile (A/dPAMs), C10.36 aptamer can be readily annealed to the dPAM surface yielding a product (C10.36∼A/dPAMs) with potential cell-specific targeting capabilities. Excitingly, the disulfide bond in this formulation is surprisingly stable in conditions that mimic serum. C10.36∼A/dPAMs mediate aptamer-specific cell interactions after ten minutes of incubation, mimicking possible in vivo scenarios. Post-incubation micellar fluorescence dequenching indicates the disassembly of the nanomaterial in vitro over time, making the disulfide bond available for cleavage and releasing Tat-POSH peptide. Finally, peptide-specific NHL toxicity was seen with minimal vehicular or control-peptide toxicity. These exciting results speak to the potential for aptamer-displaying micelles to achieve cell-specific delivery and leverage disulfide bonds to facilitate therapeutic peptide release to enhance NHL treatment outcomes.
Methods
Component synthesis strategy
| Peptide amphiphile | Use |
|---|---|
| Tat-Scram-C_C-KPalm2 | Structural studies |
| FAM-Tat-Scram-C_C-KPalm2 | Stability control |
| FAM-Tat-Scram-C_C-K(TAMRA)KPalm2 | FRET |
| Tat-Scram-C_C-K(TAMRA)KPalm2 | Stability, cell interaction & death |
| Tat-POSH-C_C-KPalm2 | Structural studies |
| FAM-Tat-POSH-C_C-KPalm2 | Stability control |
| FAM-Tat-POSH-C_C-K(TAMRA)KPalm2 | FRET |
| Tat-POSH-C_C-K(TAMRA)KPalm2 | Stability, cell interaction & death |
Aptamer-displaying, disulfide-linked PAM production
Each micelle formulation was produced as previously described.37,40 Briefly, dPA and AA working solutions in ddH2O were mixed and lyophilized overnight (Labconco FreeZone 4.5). The resulting powder pellet was dissolved in 50 μL of 50/50 methanol/chloroform (MeOH/CHCl3) and dried under a nitrogen stream for ∼10 minutes. The film formed was then dried for 1 hour under hard vacuum using a Welch 8912 direct drive pump to remove trace solvent. The material was then dissolved in 50 μL of phosphate buffered saline (PBS – 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4 at a pH 7.4) aided by 5 seconds of sonication using a probe tip sonifier (VWR Branson 250) at a power setting of 1. To create aptamer-modified micelles (Apt∼A/dPAMs), aptamer stock solution was added to the A/dPAMs in solution at a 3/1 Apt/AA molar ratio. As magnesium ions (Mg2+) ions are important for maintaining appropriate aptamer structure, 6.25 μL of 100 mM MgCl2 was also included along with 4 μL of 10 mM N-acetyl glutamic acid to charge match the PA. The resulting solution was annealed by heating to 90 °C for 5 minutes on an aluminum block heater followed by cooling to room temperature over 45 minutes (Thermolyne 17600). Samples were then stored at 4 °C in the dark until used further. All micelle samples were processed identically (i.e., lyophilization, thin film drying, sonication, and annealing) regardless of their constituents. The final concentration of each component in stock Apt∼A/dPAM solutions were 100 μM dPA, 2 μM aptamer, and 0.67 μM AA. Micelle solutions throughout this manuscript are described by their dPA concentration, with other constituents' concentrations following this defined ratio. Each formulation (dPAM A/dPAM, C10.36∼A/dPAM) was produced PA, with the peptide denoted within the name (e.g. C10.36∼A/dTat-POSHAM) and any fluorophore specifically denoted within the experiment description. Scheme 1 is a cartoon of the proposed Apt∼A/dPAM structure.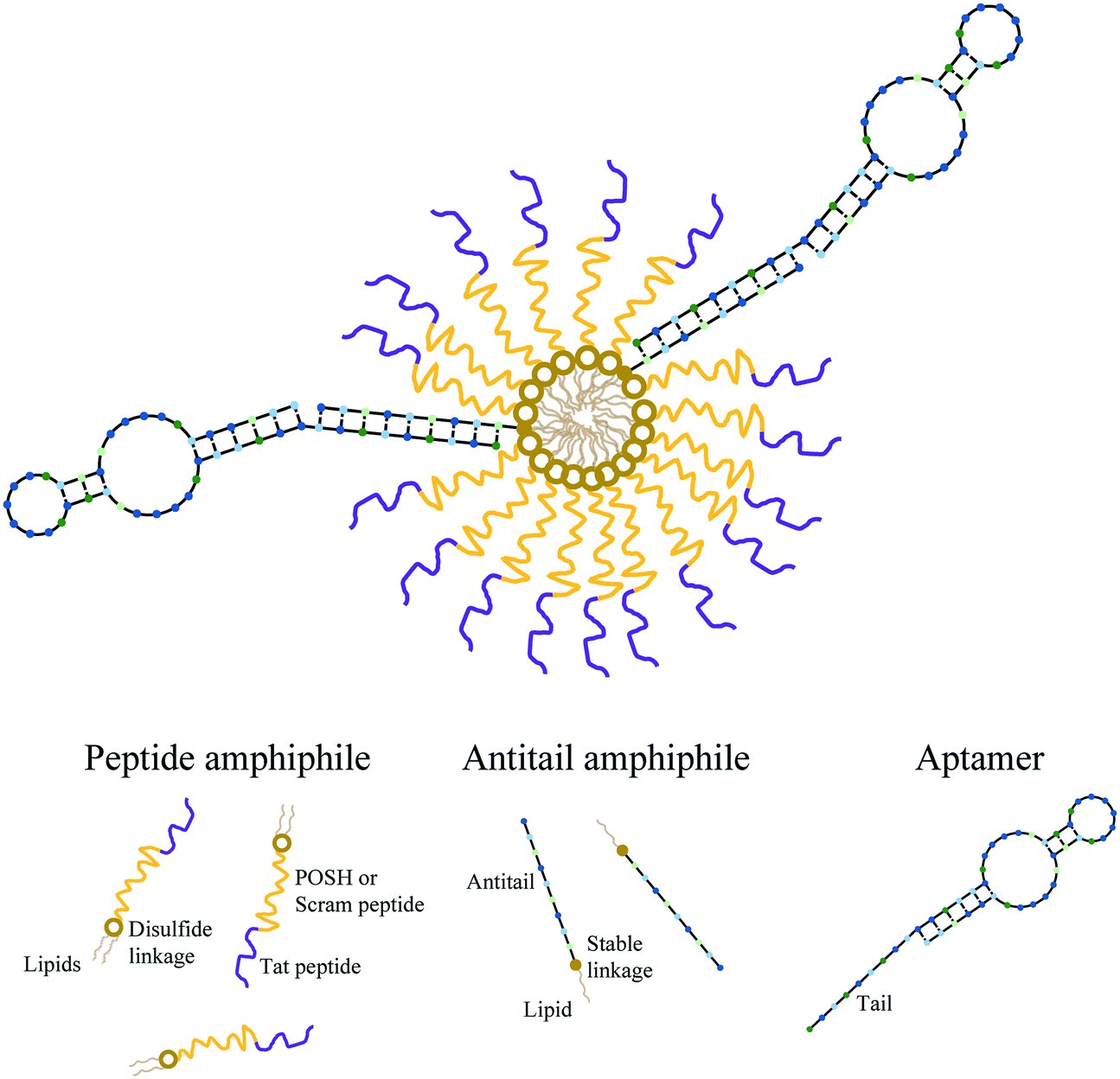 | ||
| Scheme 1 Apt∼A/dPAM structure. Therapeutic, disulfide linked peptide amphiphiles (dPAs) self-assemble into micelles (dPAMs) by hiding their lipids internally in the core and displaying their peptides externally in the corona. Antitail amphiphiles (AAs) are incorporated into the micelle via core hydrophobic interactions (A/dPAMs). The outwardly facing antitail amphiphile and aptamer are able to be readily annealed together (Apt∼A/dPAMs). The hollow circles linking peptide and lipid components represent a disulfide bond whereas solid circles represent stable thiol–maleimide linkages. In a specific example, a formulation containing dTat-POSH peptide amphiphile and C10.36 aptamer is referred to as C10.36∼A/dTat-POSHAM. | ||
Apt∼A/dPAM physical characterization
 | ||
| Scheme 2 Peptide amphiphile primary structure. FAM-Tat-POSH-C_CK(TAMRA)KPalm2 is shown with the green, pink, and yellow circles representing FAM, TAMRA, and disulfide linkage, respectively. The bend in the peptide around the three proline amino acids is a 2-D representation of a possible secondary structure. | ||
Apt∼A/dPAM cleavage
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 diluted glutathione stock solution (50 μM) and the total volume brought to 125 μL with PBS, resulting in final concentrations of 10 μM micelles with 0 μM, 5 μM, or 5 mM glutathione. These were incubated in a 96-well black plate in a humidified incubator held at 37 °C until analyzed at each time point. Fluorescent spectrometry measurements were taken at excitation/emission values of 492/522, 543/570, and 492/570 nm. Additionally, spectra were taken for 475/500–650 nm, measuring emission every 5 nm. Measurement time points included 0, 1, 2, 3, 6, 12, 24, 36, 48, and 72 hours.
:1000 diluted glutathione stock solution (50 μM) and the total volume brought to 125 μL with PBS, resulting in final concentrations of 10 μM micelles with 0 μM, 5 μM, or 5 mM glutathione. These were incubated in a 96-well black plate in a humidified incubator held at 37 °C until analyzed at each time point. Fluorescent spectrometry measurements were taken at excitation/emission values of 492/522, 543/570, and 492/570 nm. Additionally, spectra were taken for 475/500–650 nm, measuring emission every 5 nm. Measurement time points included 0, 1, 2, 3, 6, 12, 24, 36, 48, and 72 hours.
Apt∼A/dPAM biological functionality
000 in 25 μL 10% FBS supplemented RPMI) were added to each well of a 96 well plate. Each sample was treated with salmon sperm DNA (ssDNA, 5 μL of 10 mg mL−1) and formulation (20 μL of 25 μM), yielding final concentrations of 1 mg mL−1 and 10 μM, respectively. Following 10 minutes of incubation, 100 μL of 10% FBS supplemented PBS was added to the cells before they were centrifuged for 3 minutes at 1400 RPM with a Thermo Sorvall Legend R+. The solution was removed, the cell pellets were resuspended in 150 μL of 10% FBS supplemented PBS, and the samples were centrifuged again at the same conditions. After the solution was drawn off, 150 μL of 10% FBS supplemented PBS was used to resuspend the cell pellets to allow for their analysis by flow cytometry using a BD LSRFortessa X-20. TAMRA fluorescence was monitored from the PE channel from 50000 cells per sample and data were analyzed using FlowJo software. Live versus dead lymphocytes were identified via forward- and side-scatter measurements (Fig. S3†). Data are reported as the mean fluorescence intensity (MFI).
Statistical analysis
JMP software (SAS) was used to make comparisons between experimental groups. Specifically, Tukey's HSD tests were completed to determine statistical differences between each group in multiple pair-wise comparisons with the α value set at 0.05 for each comparison. For each plot, groups that possess different letters have statistically significant differences in mean.Results
Disulfide-linked PA, AA, and aptamer readily self-assemble, inducing peptide conformational changes and yielding small, anionic micelles
While aptamers can be annealed to A/PAMs,40 the capacity to fabricate composite micelles from a large, complex, cationic PA (Tat-POSH-C_CKPalm2) has not been previously shown. Using the aptamer-modifying micelle preparation protocol, C10.36 aptamer, antitail amphiphile, and Tat-POSH-C_CKPalm2 were composited together. The formulation was found to form micelles (C10.36∼A/dTat-POSHAMs) with a CMC of ∼0.18 μM as evidenced by an increase in fluorescence (Fig. 1(a)) due to DPH molecular stacking in the hydrophobic micellar core. The presence of the aptamer was found to have a negligible effect on micelle formation as dPAMs and A/dPAMs were found to have very similar CMCs (i.e., 0.16 μM). All formulations possessed limited variance in CMC values, indicating consistency among the batches tested. To ensure that aptamer was actually complexed to the micelle instead of non-specifically associated, a FRET study was conducted to determine physical proximity of 3:1 Cy3-Apt:AF488-labeled AA (Fig. 1(b)). A significant decrease in donor emission (518 nm) and a slight increase in acceptor emission (575 nm) for the matched tail/antitail formulation (i.e., C10.36∼A/dTat-POSHAMs) compared to the unmatched tail/antitail formulation (i.e., sctC10.36∼A/dTat-POSHAMs) confirms specificity and control over aptamer–micelle complexation.
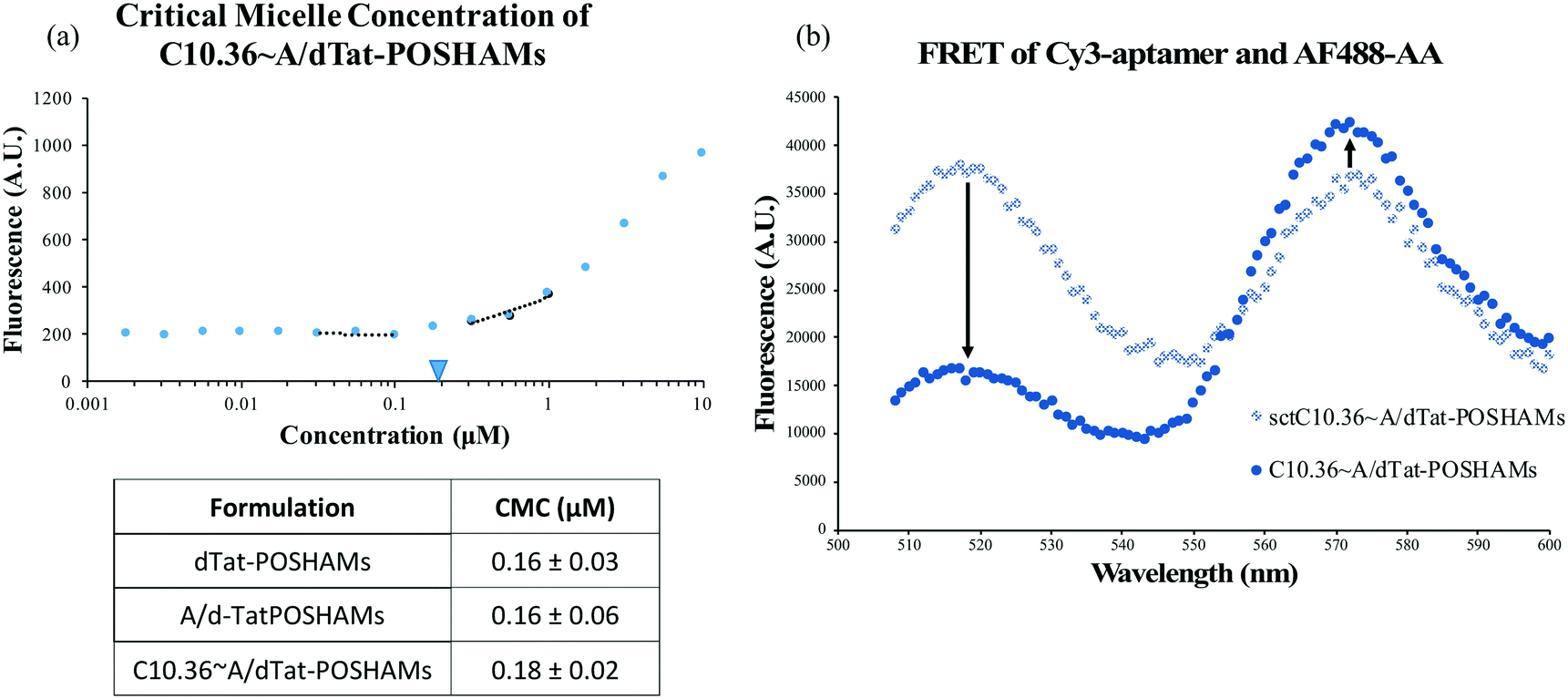 | ||
| Fig. 1 Characterization of micelle self-assembly. (a) The CMC of each formulation was evaluated. Low CMCs in the sub-micromolar range were found for each sample type. These values were very consistent between batches. (b) FRET between Cy3-aptamer and AF488-labeled antitail indicates that aptamer successfully anneals to the AA in the context of the micelle. | ||
To determine the effect that micellization and aptamer annealing have on peptide secondary structure, micelle size, and micelle surface charge, circular dichroism (CD), transmission electron microscopy (TEM), and zeta potential were evaluated. CD spectra showed that peptide conformation changed significantly with micellization (i.e., Tat-POSH-C compared to dTat-POSHAMs) and was further altered by the inclusion of AA, G24A, or C10.36 (Fig. 2(a)). Additionally, using a poly(lysine) CD model for correlating spectral data to secondary structure, micellization was found to increase β-sheet conformation from 36% to 54% and decrease random coil from 64% to 47% for Tat-POSH-C compared to C10.36∼A/dTat-POSHAMs (Table S2†). TEM micrographs of each formulation were taken to evaluate micelle size using particle analysis software (Bruker ESPRIT 2.0), with the original and processed micrographs of C10.36∼A/dTat-POSHAMs shown, along with size distributions (Fig. 2(b) and (c)). Analyses of A/dTat-POSHAMs and dTat-POSHAMs are shown in Fig. S4.† TEM size measurements confirmed the presence of nanoparticles for each formulation, with a number average diameter of ∼10–12 nm and a size range of ∼6–20 nm. Zeta potential measurements confirmed the presence of nanoparticles as well as aptamer association for C10.36∼A/dTat-POSHAMs as shown by the decrease in zeta potential from ∼18 mV to ⋍45 mV (Table 2).
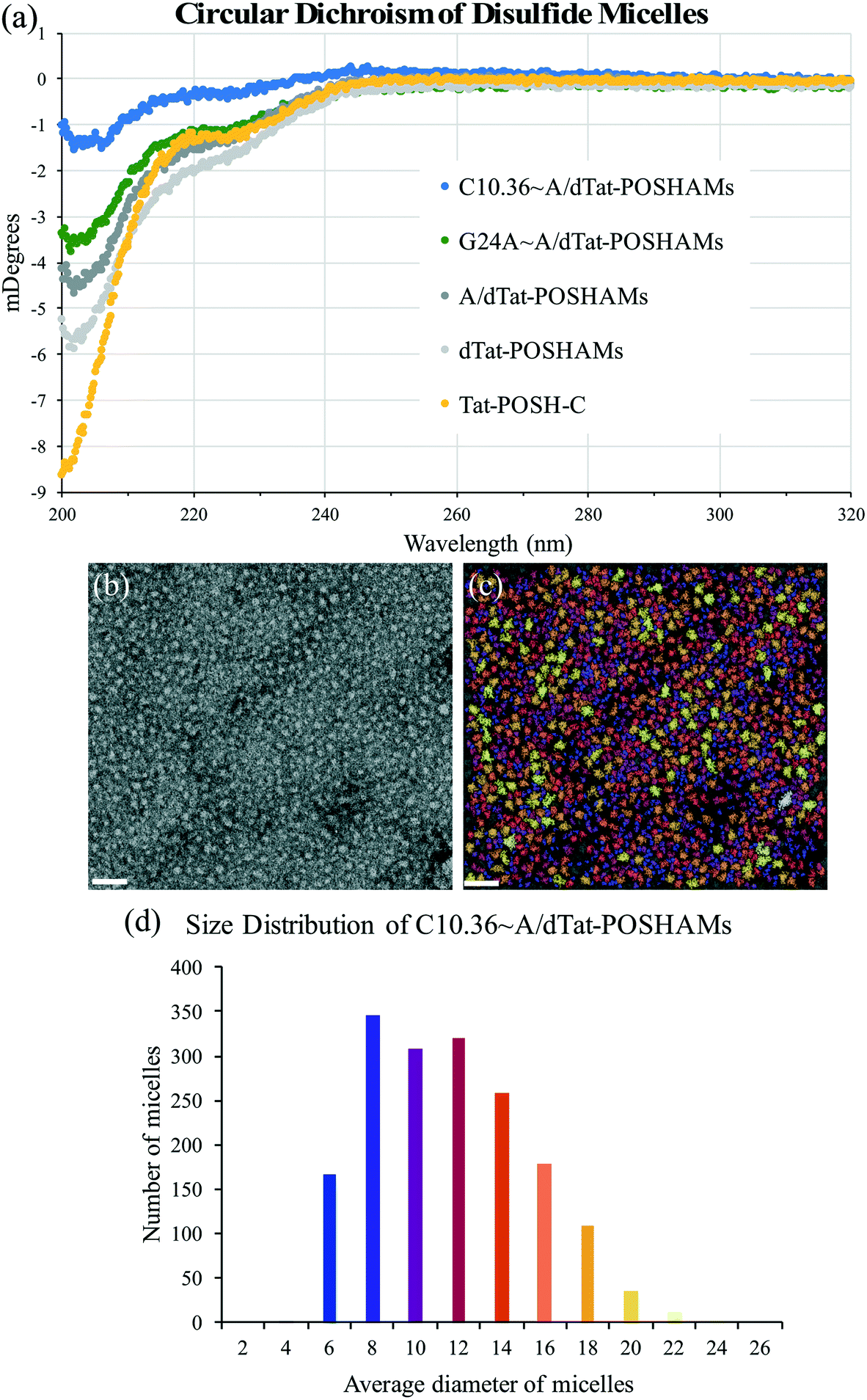 | ||
| Fig. 2 Micelle structural evaluation. (a) CD spectra show a dramatic change in peptide secondary structure when lipid was attached (i.e., for dTat-POSHAMs, A/dTat-POSHAMs, and Apt∼A/dTat-POSHAMs). (b) and (c) TEM evaluation of C10.36∼A/dTat-POSHAMs revealed homogeneous micelles similar in size. Further TEM micrograph analysis provides a method to better analyze micelle size, with the original on the left and overlay of original and particle map on the right. The scale bar for each micrograph is 50 nm. (d) The histogram generated from the micrograph analysis shows a tight window of C10.36∼A/dTat-POSHAMs particle size. | ||
| Disulfide formulation | TEM diameter (nm) | Zeta potential (mV) |
|---|---|---|
| dTat-POSHAMs | 12 ± 5 | 18.4 ± 6.1 |
| A/dTat-POSHAMs | 10 ± 3 | 20.1 ± 10.0 |
| C10.36∼A/dTat-POSHAMs | 12 ± 5 | −44.9 ± 5.2 |
Aptamer-displaying, disulfide-linked micelles are stable in serum-like conditions, but unstable in intracellular-like conditions
To evaluate the long-term impact of blood-serum like conditions on micellar disassembly, formulations were incubated in 10% and 80% FBS over 72 hours. During dissociation, PA-tethered TAMRA is de-quenched due to a decrease in micelle core-mediated molecular packing. As shown in Fig. 3, all micelles were found to slowly disassemble in the presence of FBS regardless of chemistry. The dissociation half-life (i.e., time required to achieve 50% of final fluorescence) was found to be ∼5.0 hours in 10% FBS and ∼4.2 hours in 80% FBS for C10.36∼A/dTat-ScramAMs, which agrees with previous data generated with a similar micellar system.40 Half-lives of A/dTat-ScramAMs and dTat-ScramAMs were found to be ∼1.3 hours and ∼1.3 hours in 10% FBS and ∼5.7 hours and ∼8.0 hours in 80% FBS, respectively. The differences observed in the half-life for each formulation at the two different concentrations is believed to be an effect of micelle/serum protein interactions. The increases in half-life for aptamer-free formulations in 80% FBS may be due to FBS proteins interacting with the micelles in a way that inhibits their disassembly.41 For the aptamer containing formulation, this stabilization effect may be mitigated by surface aptamer participation in DNA/micelle association and nanoparticle disassembly.42,43 Interestingly, each formulation slowly approached steady-state fluorescence over 72 hours though the magnitude of the values was different for each formulation. Additionally, TEM analysis found that C10.36∼A/dTat-ScramAMs were still intact after 1 hour incubation in 10% FBS at 37 °C (Fig. S5†), directly confirming the structural stability of these micelles in serum.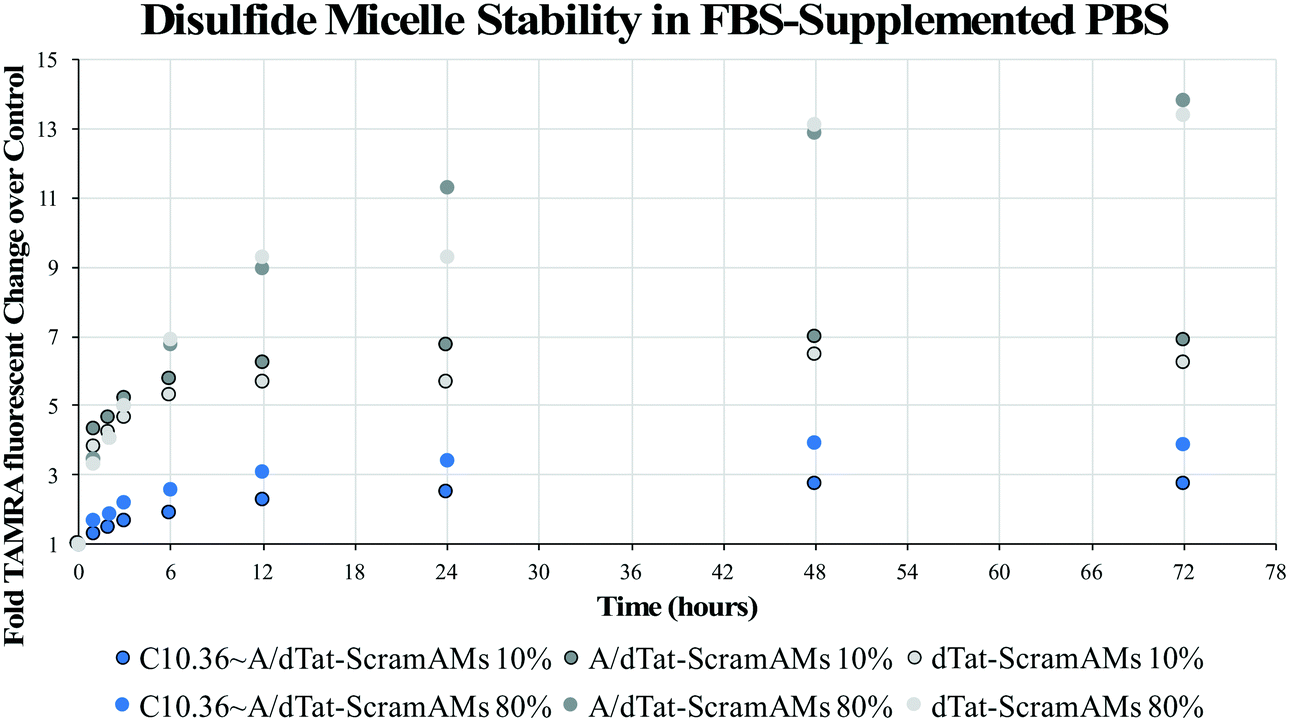 | ||
| Fig. 3 FBS-mediated micelle disassembly. Micelles were incubated at 37 °C in 10% or 80% FBS for 72 hours with PA-associated fluorescence evaluated as an assessment of the magnitude of micelle disassembly. Data are plotted as fold TAMRA fluorescence change from each formulation incubated in FBS-free PBS. C10.36∼A/dTat-ScramAMs disassembled slightly more slowly in 10% than 80% FBS, with a disassembly half-life of ∼5.0 and ∼4.2 hours, respectively. | ||
To determine the stability of the disulfide bonds present in dPAMs under reducing conditions similar to those found in vivo, formulations were exposed to glutathione, a natural antioxidant and thiol reducing agent present inside cells (5 mM) and at much lower concentrations in serum (5 μM).44–46 As dPAs possess both FAM and TAMRA, FRET, or the lack thereof, can be used as a reporter for disulfide presence or cleavage. The disulfide bonds of C10.36∼A/dScramAMs were confirmed to slowly cleave in glutathione in PBS in a concentration-dependent manner (Fig. S6†). To further assess biologically-relevant bond sensitivity, C10.36∼A/dScramAMs were incubated in 80% FBS supplemented PBS with glutathione. As shown in Fig. 4(a), micelles exposed to intracellular glutathione levels showed a much more rapid rise in FAM fluorescence (525 nm) than those subjected to serum glutathione levels indicating more rapid disulfide cleavage. In Fig. 4(b), the long-term change in donor fluorescence indexed against no glutathione exposure indicates that within an hour the 5 mM glutathione treatment group reaches maximum fluorescence that then slowly decreases over time likely due to fluorophore photobleaching. The 5 μM glutathione treatment group slowly increased in donor fluorescence, reaching the same level as the 5 mM glutathione group nearly 36 hours of incubation.
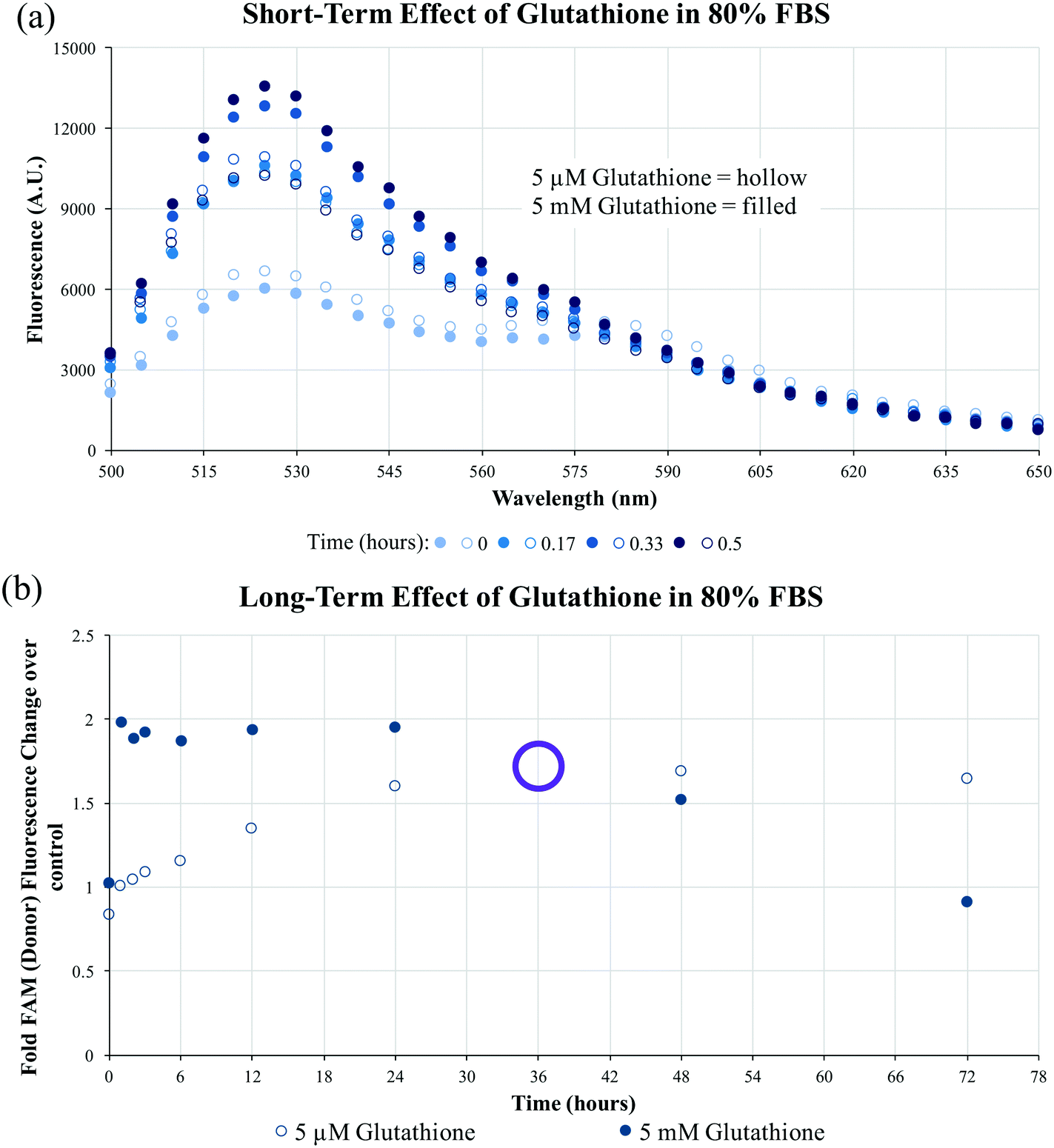 | ||
| Fig. 4 FBS and glutathione-mediated disulfide bond cleavage. (a) Disulfide micelle peptide-lipid separation was monitored by FRET-based fluorescent measurements for 30 minutes in the presence of 5 μM or 5 mM glutathione aided, 80% FBS supplemented PBS. Treatment with 5 mM glutathione caused a more rapid increase in FAM fluorescence than 5 μM glutathione having reached a maximum value in only 30 minutes. (b) The serum glutathione level (5 μM) induced a steady increase in disulfide micelle fluorescence indexed against no glutathione exposure which finally achieved similar FRET-determined disulfide cleavage as intracellular glutathione level (5 mM) at ∼36 hours (hollow purple circle). | ||
Aptamer specificity drives micelle interactions with human B cell lymphoma resulting in peptide-mediated cell death
Micelles were incubated with Ramos lymphoma cells to probe the impact that surface-displayed aptamer and C10.36 specificity played on targeted cell delivery. Cells were incubated with 10 μM of one of the Tat-POSH dPA micelle formulations (i.e., dTat-POSHAMs, A/dTat-POSHAMs, scApt∼dTat-POSHAMs, G24A∼A/dTat-POSHAMs, and C10.36∼A/dTat-POSHAMs) for 10 minutes, after which non-associated micelles were washed off the cells using multiple-rounds of rinsing and centrifugation. Cells were then either immediately analyzed for their fluorescence by flow cytometry or cultured for a total of 24 hours before assessed. TAMRA, was used as the reporter for cell association, as it was found to be more sensitive than FAM for all groups, and specifically shown for C10.36∼A/dTat-POSHAMs to be similar to expected changes associated with micellar disassembly (Fig. S7†). This behavior is likely attributable to significant initial quenching of TAMRA in the micelle core and the sensitivity of FAM to reductive bleaching. TAMRA mean fluorescence intensities (MFIs) were determined for all POSH dPA micelle formulations and indexed against Ramos cell autofluorescence at 24 hours (Fig. 5). Interestingly, all fluorescence values are similar, indicating that neither the presence of aptamer nor the choice of aptamer used impacted the ability for PAMs to associate with Ramos cells.Each formulation was also evaluated for its capacity to induce peptide-mediated toxicity of Ramos cells. Similar to the association study, cells were incubated with 10 μM of one of the aforementioned Tat-POSH dPA micelle formulations, their Tat-Scram dPA analogs, or control peptides. After 10 minutes, cells were washed as previously described to remove unassociated materials and cultured for an additional 24 hours. Cell death was evaluated via 7AAD+ permeation, a death stain and indicator of late apoptosis, and verified by side-scatter versus forward-scatter gating.
Pulsatile exposure (i.e., 10 minutes) of Tat-POSH-C to Ramos cells was found not to enhance cell death. In contrast, continuous exposure (i.e., 24 hours, no cell rinsing step) improved cell death, although this increase was not statistically significant above control treatments (Fig. 6). Similarly, micelles with and without non-specific aptamer annealing (i.e., dTat-POSHAMs and G24A∼A/dTat-POSHAMs) showed modest, yet statistically insignificant, increases in cell death. Ramos cells were excitingly found to succumb to exposure with C10.36∼A/dTat-POSHAMs with statistical significance over all groups except continuous exposure to Tat-POSH-C. All formulations containing the Tat-Scram control peptide were found to have caused cell death similar to background indicating the observed results were Tat-POSH peptide specific.
Discussion
Three central challenges in nanoparticle-based therapeutic peptide delivery are physicochemical stability in the biological environment, targeting the desired cell population, and successfully releasing bioactive peptide.18 While PAMs are useful for extending peptide biological lifetimes19 and some PAs have been found to be inherently therapeutic,47,48 many PAs are not biologically active, requiring release of the peptide payload for them to exert their bioactivity. In the interest of better releasing bioactive peptide from Apt∼A/PAMs, a cleavable disulfide linker between the peptide and lipid was developed and used. Due to the facile lability of disulfide bonds in biological systems, these were introduced near the lipid core with the intention of preventing unwanted enzymatic access49 and delaying peptide release until after cellular internalization. The peptides used in this study, Tat-POSH-C and Tat-Scram, require rigorous synthetic measures to obtain high purities, rendering solid-phase methods of the entire disulfide PA formation untenable. Instead, a three-step approach was adopted in which peptide and activated lipid were synthesized separately and then chemoselectively conjugated. The resulting disulfide-linked PAs were purified by LC/MS, providing highly pure products (Fig. S1 and S2†).Due to the bioactive peptide chosen, a primary concern was whether physically stable, controlled micelles could be formed that incorporate both a cationic peptide (either Tat-POSH-C or Tat-Scram-C) and anionic DNA aptamer. Prior to this work, it was unknown whether hydrophobically-driven self-assembly and nucleic acid annealing would be able to overcome electrostatic interactions among constitutive components. In the process of constructing Apt∼A/dPAMs, noticeable aggregation took place after aptamer addition, resulting in the formation of a turbid solution with large, sedimentary particles (data not shown). However, after annealing, the solution became clear, indicating that initial aggregation could be overcome through nucleic acid hybridization without impacting micellization (Fig. 1(a)). Additional evidence for this controlled assembly after annealing was demonstrated through FRET between C10.36 and AA (Fig. 1(b)) indicating that oligonucleotide base-pair interactions annealed C10.36 to the micelle surface. This result suggests that the Apt∼A/dPAM platform could be utilized to achieve targeted delivery of a wide variety of hydrophilic peptides regardless of their charge.
The secondary structures of therapeutic peptides are known to impact their bioactivities.50–52 For the peptides explored in this research, Tat has been shown to intercalate into the cell membrane when in the random coil configuration53 whereas the POSH peptide is predicted to be β-sheet within the confines of its parent protein.15,16,54,55 As Tat-POSH-C was found to possess both random coil and β-sheet content (i.e., 64% and 36%, respectively – Table S2†), conjugating these together does not appear to have negatively impacted the secondary structure of either component. PA micellization is known to alter the secondary structure of incorporated peptides,56,57 which could potentially be further modified due to the presence of disulfide linkages, antitail amphiphile, and annealed aptamer. Micellization did yield a modest shift in Tat-POSH-C peptide secondary structure (i.e., dTat-POSHAMs – 52%/48% random coil/β-sheet – Table S2†) which may be due to micelles acting as an artificial tertiary structure confining POSH peptides near one another and allowing them to participate in intermolecular hydrogen bonding.58,59 Though a change was observed, both desired random coil and β-sheet content were still present. Interestingly, neither the presence of antitail amphiphile nor aptamer actually impacted the secondary structure of micelle-associated Tat-POSH-C beyond what could be attributed to micellization (Table S2†).
Having established that C10.36∼A/dPAMs are small and anionic nanoparticles, ideal for long-term trafficking in the blood (Fig. 2, Table 2),60–62 micelle physical stability and disulfide bond chemical stability were evaluated using TAMRA-labeled and FRET-pair labeled dPAs, respectively. In aggregate, the experimental results indicate that despite FBS-mediated disassembly (Fig. 3) and glutathione-based antioxidant sensitivity (Fig. S6†) over time, C10.36∼A/dPAMs are quite stable in blood-serum like conditions for at least several hours (Fig. S5† and 4). Importantly, when exposed to the intracellular glutathione concentration in FBS, the disulfide bond was found to readily reduce, reaching maximum cleavage as measured by FRET in under 1 hour (Fig. 4).
Given the promising physicochemical characteristics of C10.36∼A/dPAMs, the bioactivity of this novel technology was evaluated in vitro. After pulsed incubation and prolonged co-culture of Ramos cells with various micelle formulations, all cells were found to have similar quantities of associated fluorescently-labeled dPA regardless of the presence or lack of C10.36 aptamer (Fig. 5). In the literature, PAMs have been shown to intercalate into cell membranes due to their hydrophobic lipid content,63,64 a dynamic process which may be enhanced by the presence of cell penetrating peptides like Tat. As micelle-driven and Tat-mediated interactions with cells are non-specific, it appears that these forces were the primary drivers of cell association over aptamer specificity. It should be noted that due to the constraints of the experiment (i.e., including only Ramos cells), the capacity for C10.36 to preferentially associate A/dPOSHAMs with their desired target over off-target cells was not evaluated and is an area of potential future study. Though the presence of C10.36 aptamer did not enhance micelle–cell interactions, Ramos sensitivity to POSH-based cytotoxicity was found to be highly peptide and aptamer specific (Fig. 6). Modest increases in Ramos cell death similar to continuous Tat-POSH-C presence were achieved through pulsatile exposure to cell-associating micelles, though all results were statistically insignificant over background. When cell-specific C10.36 aptamer was annealed to the micelles, the resulting formulation possessed considerable anti-cancer behavior, killing nearly half of all living Ramos cells present. This observation indicates that although C10.36 does not improve micelle–cell association, it clearly impacts localized dPA delivery. As POSH inhibition must be carried out in the cytosol,14–16 proper internalization and processing of Tat-POSH-C is a fundamental requirement of any therapeutic vehicle. Recently published research has shown that C10.36 binds Ramos surface spliceosomal complex which then facilitates its intracellular trafficking36 providing possible insight into the cancer cell cytotoxicity observed with C10.36∼A/dTat-POSHAMs. Interestingly, C10.36 may synergistically impact treatment toxicity as this aptamer has been reported to cause Ramos cell necrosis.36
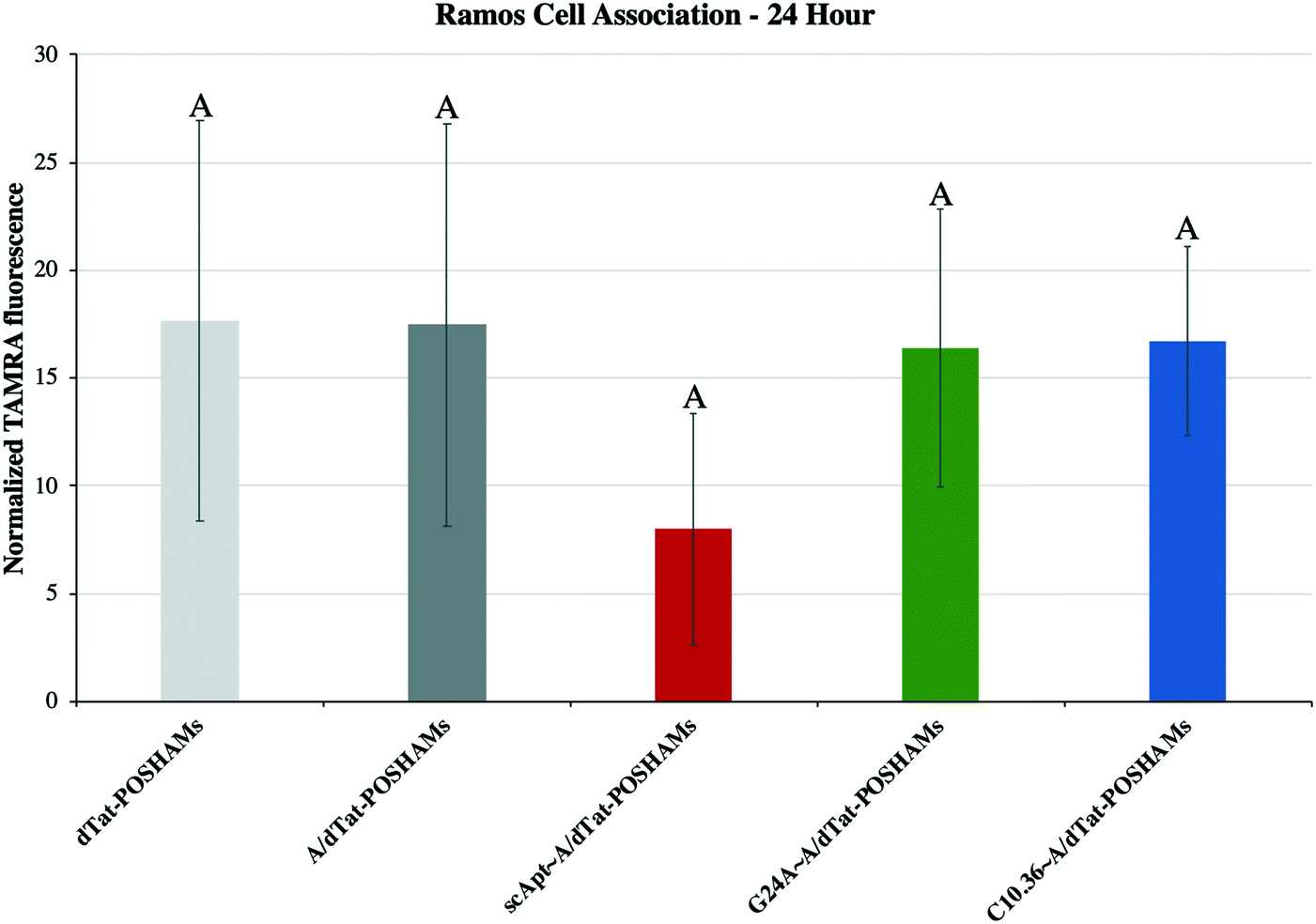 | ||
| Fig. 5 Cellular interaction of micelles with Ramos cells. Each experimental group was evaluated after a 10-minute incubation with Ramos cells, rinse, and 24-hour culture. No differences were seen between groups, independent of aptamer inclusion (Tukey HSD, α = 0.05). | ||
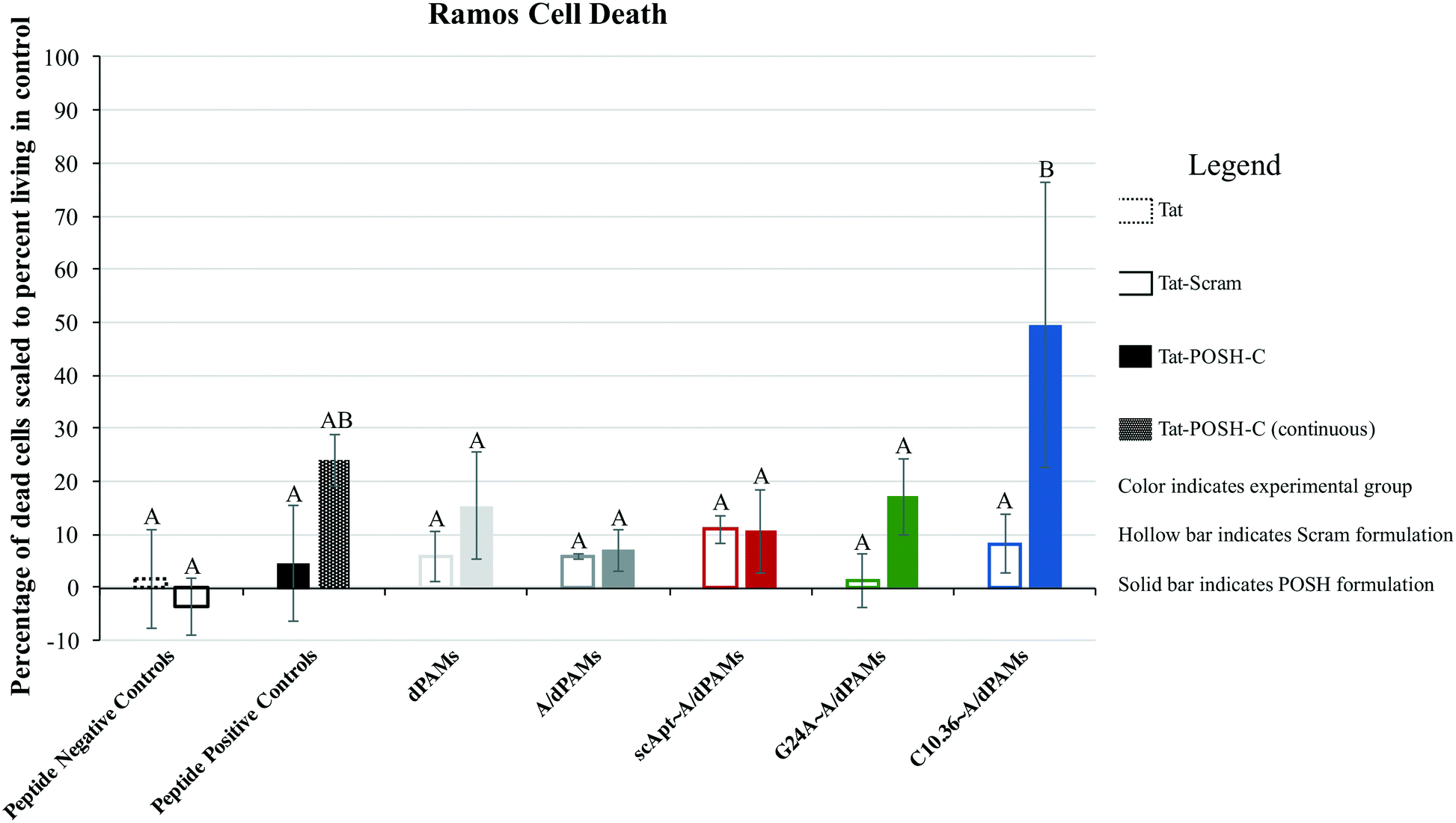 | ||
| Fig. 6 Tat-POSH-C peptide-mediated Ramos cell toxicity. All formulations were incubated for 10 minutes, rinsed, and cultured for 24 hours, except Tat-POSH-C (continuous) which was not rinsed. Cell death was then investigated using flow cytometry employing 7-AAD+ staining with data indexed against the population of untreated living Ramos cells. Cell death was found to be aptamer and peptide-specific. Groups with different letters have statistically different means (Tukey HSD, α = 0.05). | ||
Taken together, the physical and chemical stability of, aptamer specificity of, and therapeutic effect from C10.36∼A/dTat-POSHAMs indicate that this technology has promising potential for in vivo delivery of therapeutic Tat-POSH-C to kill NHL.
Conclusion
Intracellular delivery of therapeutic peptides remains a difficult obstacle to overcome. Novel approaches have been developed, including the incorporation of cell penetrating peptides and direct association of a targeting moiety. Nanoparticles are an attractive solution for the intracellular delivery of cargo as they can protect peptide payloads against enzymatic degradation and host clearance as well as provide opportunities for surface association of targeting moieties. In particular, micelles are a modular and capable nanoparticle capable of facile incorporation of both peptide payloads and targeting aptamers. The peptide amphiphile micelles produced in this work were found to be small spherical particles able to stably associate with cells, making them attractive for targeted delivery via the vasculature. When coupled with a cell-specific and internalizing aptamer, the resulting micelles (i.e., C10.36∼A/dTat-POSHAMs) induced significant peptide-based toxicity in human lymphoma cells. Due to the highly modular and flexible nature of this technology, further enhancement of peptide bioactivity and aptamer specificity can be achieved by altering either component. Future steps for this project include employing dual-fluorophore FRET dPA further to understand how Apt∼A/dPAMs are being processed by cells; utilizing a less cationic, liposome-penetrating sequence in the place of Tat to improve aptamer specificity; and exploring alternative biologically sensitive linkers to achieve the more desirable peptide release kinetics after cell internalization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grant funds from the MU Research Board and BioNexus KC and by start-up funds from the University of Missouri. We would like to thank the staff at the MU Electron Microscopy Core facility, especially DeAna Grant, for their assistance on this project. Additionally, we thank Dr. Krishna Sharma for his help in completing the CD experiments, especially with operating the instrument.Notes and references
- SEER Cancer Statistics Review, 1975-2016, ed. N. Howlader, A. M. Noone, M. Krapcho, D. Miller, A. Brest, M. Yu, J. Ruhl, Z. Tatalovich, A. Mariotto, D. R. Lewis, H. S. Chen, E. J. Feuer and K. A. Cronin, National Cancer Institute, Bethesda, MD, https://seer.cancer.gov/csr/1975_2016/, based on November 2018 SEER data submission, posted to the SEER web site, April 2019 Search PubMed.
- S. Gangatharan and J. Kuruvilla, Transfus. Apher. Sci., 2013, 49, 72–79 CrossRef PubMed.
- American Cancer Society, Cancer Facts & Figures 2019, American Cancer Society, Atlanta, GA, 2019 Search PubMed.
- K. Schmiegelow, K. Muller, S. S. Mogensen, P. R. Mogensen, B. O. Wolthers, U. K. Stoltze, R. Tuckuviene and T. Frandsen, F1000Research, 2017, 6, 444 Search PubMed.
- B. Lund, A. Asberg, M. Heyman, J. Kanerva, A. Harila-Saari, H. Hasle, S. Soderhall, O. G. Jonsson, S. Lydersen, K. Schmiegelow and H. Nordic Society of Paediatric and Oncology, Pediatr. Blood Cancer, 2011, 56, 551–559 CrossRef.
- C. E. Annesley and P. Brown, Ther. Adv. Hematol., 2015, 6, 61–79 CrossRef CAS PubMed.
- F. Huguet and S. Tavitian, Expert Opin. Emerging Drugs, 2017, 22, 107–121 CrossRef CAS PubMed.
- S. S. Neelapu, F. L. Locke, N. L. Bartlett, L. J. Lekakis, D. B. Miklos, C. A. Jacobson, I. Braunschweig, O. O. Oluwole, T. Siddiqi, Y. Lin, J. M. Timmerman, P. J. Stiff, J. W. Friedberg, I. W. Flinn, A. Goy, B. T. Hill, M. R. Smith, A. Deol, U. Farooq, P. McSweeney, J. Munoz, I. Avivi, J. E. Castro, J. R. Westin, J. C. Chavez, A. Ghobadi, K. V. Komanduri, R. Levy, E. D. Jacobsen, T. E. Witzig, P. Reagan, A. Bot, J. Rossi, L. Navale, Y. Jiang, J. Aycock, M. Elias, D. Chang, J. Wiezorek and W. Y. Go, N. Engl. J. Med., 2017, 377, 2531–2544 CrossRef CAS.
- C. L. Batlevi, E. Matsuki, R. J. Brentjens and A. Younes, Nat. Rev. Clin. Oncol., 2016, 13, 25–40 CrossRef CAS PubMed.
- R. J. Boohaker, M. W. Lee, P. Vishnubhotla, J. M. Perez and A. R. Khaled, Curr. Med. Chem., 2012, 19, 3794–3804 CrossRef CAS PubMed.
- R. J. Leeman-Neill and G. Bhagat, Expert Opin. Ther. Targets, 2018, 22, 143–152 CrossRef CAS.
- V. Y. Yazbeck and S. Grant, Expert Opin. Invest. Drugs, 2015, 24, 965–979 CrossRef CAS.
- C. Y. Cheah, N. H. Fowler and M. L. Wang, Ann. Oncol., 2016, 27, 778–787 CrossRef CAS.
- L. N. Cardwell, Y. Guan, E. Teixeiro and M. A. Daniels, POSH scaffold complex necessary for survival of broad range of B cell leukemia, In Preparation Search PubMed.
- C. A. Cunningham, K. M. Knudson, B. J. Peng, E. Teixeiro and M. A. Daniels, Eur. J. Immunol., 2013, 43, 3361–3371 CrossRef CAS.
- C. A. Cunningham, L. N. Cardwell, Y. Guan, E. Teixeiro and M. A. Daniels, J. Immunol., 2016, 196, 4003–4013 CrossRef CAS.
- C. H. Niu and Y. Y. Chiu, J. Pharm. Sci., 1998, 87, 1331–1334 CrossRef CAS.
- J. Zhang, S. S. Desale and T. K. Bronich, Ther. Delivery, 2015, 6, 1279–1296 CrossRef CAS.
- H. Acar, J. M. Ting, S. Srivastava, J. L. LaBelle and M. V. Tirrell, Chem. Soc. Rev., 2017, 46, 6553–6569 RSC.
- R. Zhang, C. N. Leeper, X. Wang, T. A. White and B. D. Ulery, Biomater. Sci., 2018, 6, 1717–1722 RSC.
- J. C. Barrett, B. D. Ulery, A. Trent, S. Liang, N. A. David and M. V. Tirrell, ACS Biomater. Sci. Eng., 2017, 3, 144–152 CrossRef CAS PubMed.
- D. Missirlis, D. V. Krogstad and M. Tirrell, Mol. Pharmaceutics, 2010, 7, 2173–2184 CrossRef CAS PubMed.
- H. Acar, R. Samaeekia, M. R. Schnorenberg, D. K. Sasmal, J. Huang, M. V. Tirrell and J. L. LaBelle, Bioconjugate Chem., 2017, 28, 2316–2326 CrossRef CAS PubMed.
- H. J. Forman, H. Zhang and A. Rinna, Mol. Aspects Med., 2009, 30, 1–12 CrossRef CAS.
- S. Cerritelli, D. Velluto and J. A. Hubbell, Biomacromolecules, 2007, 8, 1966–1972 CrossRef CAS.
- P. Zhong, J. Zhang, C. Deng, R. Cheng, F. Meng and Z. Zhong, Biomacromolecules, 2016, 17, 3602–3608 CrossRef CAS PubMed.
- P. Ju, J. Hu, F. Li, Y. Cao, L. Li, D. Shi, Y. Hao, M. Zhang, J. He and P. Ni, J. Mater. Chem. B, 2018, 6, 7263–7273 RSC.
- Y. Li, K. Xiao, J. Luo, W. Xiao, J. S. Lee, A. M. Gonik, J. Kato, T. A. Dong and K. S. Lam, Biomaterials, 2011, 32, 6633–6645 CrossRef CAS PubMed.
- Y. Oe, R. J. Christie, M. Naito, S. A. Low, S. Fukushima, K. Toh, Y. Miura, Y. Matsumoto, N. Nishiyama, K. Miyata and K. Kataoka, Biomaterials, 2014, 35, 7887–7895 CrossRef CAS PubMed.
- Y. J. Cheng, A. Q. Zhang, J. J. Hu, F. He, X. Zeng and X. Z. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 2093–2103 CrossRef CAS PubMed.
- D. Wu, Y. Li, J. Shen, Z. Tong, Q. Hu, L. Li and G. Yu, Chem. Commun., 2018, 54, 8198–8201 RSC.
- O. C. Farokhzad, J. Cheng, B. A. Teply, I. Sherifi, S. Jon, P. W. Kantoff, J. P. Richie and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6315–6320 CrossRef CAS PubMed.
- X. Zhang, J. Zhang, Y. Ma, X. Pei, Q. Liu, B. Lu, L. Jin, J. Wang and J. Liu, Int. J. Biochem. Cell Biol., 2014, 46, 1–8 CrossRef CAS.
- F. Opazo, L. Eiden, L. Hansen, F. Rohrbach, J. Wengel, J. Kjems and G. Mayer, Mol. Ther.--Nucleic Acids, 2015, 4, e251 CrossRef CAS.
- J. D. Smith, L. N. Cardwell, D. Porciani, J. A. Nguyen, R. Zhang, F. Gallazzi, R. R. Tata, D. H. Burke, M. A. Daniels and B. D. Ulery, Phys. Biol., 2018, 15, 065006 CrossRef.
- S. S. Tonapi, V. Pannu, J. E. Duncan, M. Rosenow, A. Helmstetter, D. Magee, Q. Zhang, T. T. Tinder, M. Richards, D. D. Halbert, M. Famulok, D. Spetzler, M. R. Miglarese, H. A. O'Neill and G. Mayer, Cell Chem. Biol., 2019, 26(5), 756–764 CrossRef CAS PubMed.
- D. Porciani, L. N. Cardwell, K. D. Tawiah, K. K. Alam, M. J. Lange, M. A. Daniels and D. H. Burke, Nat. Commun., 2018, 9, 2283 CrossRef.
- J. I. Finneman and M. J. Pozzo, J. Pept. Sci., 2012, 18, 511–518 CrossRef CAS.
- D. Porciani, G. Signore, L. Marchetti, P. Mereghetti, R. Nifosi and F. Beltram, Mol. Ther.--Nucleic Acids, 2014, 3, e144 CrossRef CAS.
- J. D. Smith, L. N. Cardwell, D. Porciani, J. A. Nguyen, F. Gallazzi, R. R. Tata, D. H. Burke, M. A. Daniels and B. Ulery, Phys. Biol., 2018, 15(6), 065006 CrossRef.
- M. Suvarna, S. Dyawanapelly, B. Kansara, P. Dandekar and R. Jain, ACS Appl. Nano Mater., 2018, 1, 5524–5535 CrossRef CAS.
- H. Malonga, J. F. Neault, H. Arakawa and H. A. Tajmir-Riahi, DNA Cell Biol., 2006, 25, 63–68 CrossRef CAS.
- H. Liu, K. D. Moynihan, Y. Zheng, G. L. Szeto, A. V. Li, B. Huang, D. S. Van Egeren, C. Park and D. J. Irvine, Nature, 2014, 507, 519–522 CrossRef CAS.
- F. Michelet, R. Gueguen, P. Leroy, M. Wellman, A. Nicolas and G. Siest, Clin. Chem., 1995, 41, 1509–1517 CAS.
- D. P. Jones, J. L. Carlson, V. C. Mody, J. Cai, M. J. Lynn and P. Sternberg, Free Radical Biol. Med., 2000, 28, 625–635 CrossRef CAS PubMed.
- A. Meister and M. E. Anderson, Annu. Rev. Biochem., 1983, 52, 711–760 CrossRef CAS PubMed.
- L. S. Dalboge, S. L. Pedersen, S. B. van Witteloostuijn, J. E. Rasmussen, K. T. Rigbolt, K. J. Jensen, B. Holst, N. Vrang and J. Jelsing, J. Pept. Sci., 2015, 21, 85–94 CrossRef CAS.
- J. Lau, P. Bloch, L. Schaffer, I. Pettersson, J. Spetzler, J. Kofoed, K. Madsen, L. B. Knudsen, J. McGuire, D. B. Steensgaard, H. M. Strauss, D. X. Gram, S. M. Knudsen, F. S. Nielsen, P. Thygesen, S. Reedtz-Runge and T. Kruse, J. Med. Chem., 2015, 58, 7370–7380 CrossRef CAS.
- M. Karimi, M. T. Ignasiak, B. Chan, A. K. Croft, L. Radom, C. H. Schiesser, D. I. Pattison and M. J. Davies, Sci. Rep., 2016, 6, 38572 CrossRef CAS.
- T. T. Bhattacharjee, M. L. Castilho, I. R. de Oliveira, V. P. S. Jesus, K. C. Hewitt and L. Raniero, Biochim. Biophys. Acta, Gen. Subj., 2018, 1862, 495–500 CrossRef CAS.
- M. Garton, S. Nim, T. A. Stone, K. E. Wang, C. M. Deber and P. M. Kim, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1505–1510 CrossRef CAS.
- N. G. O. Junior, M. H. Cardoso, E. S. Candido, D. van den Broek, N. de Lange, N. Velikova, J. M. Kleijn, J. M. Wells, T. M. B. Rezende, O. L. Franco and R. de Vries, Sci. Rep., 2018, 8, 11127 CrossRef.
- E. Eiriksdottir, K. Konate, U. Langel, G. Divita and S. Deshayes, Biochim. Biophys. Acta, 2010, 1798, 1119–1128 CrossRef CAS.
- N. Tapon, K. Nagata, N. Lamarche and A. Hall, EMBO J., 1998, 17, 1395–1404 CrossRef CAS.
- A. Musacchio, M. Noble, R. Pauptit, R. Wierenga and M. Saraste, Nature, 1992, 359, 851–855 CrossRef CAS.
- D. Missirlis, M. Farine, M. Kastantin, B. Ananthanarayanan, T. Neumann and M. Tirrell, Bioconjugate Chem., 2010, 21, 465–475 CrossRef CAS.
- R. Zhang, L. D. Morton, J. D. Smith, F. Gallazzi, T. A. White and B. D. Ulery, ACS Biomater. Sci. Eng., 2018, 4, 2330–2339 CrossRef CAS.
- J. C. Stendahl, M. S. Rao, M. O. Guler and S. I. Stupp, Adv. Funct. Mater., 2006, 16, 499–508 CrossRef CAS.
- K. Sato, W. Ji, L. C. Palmer, B. Weber, M. Barz and S. I. Stupp, J. Am. Chem. Soc., 2017, 139, 8995–9000 CrossRef CAS PubMed.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657–3666 CrossRef CAS.
- E. Frohlich, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef.
- X. Duan and Y. Li, Small, 2013, 9, 1521–1532 CrossRef CAS.
- D. Missirlis, T. Teesalu, M. Black and M. Tirrell, PLoS One, 2013, 8, e54611 CrossRef CAS.
- D. Missirlis, H. Khant and M. Tirrell, Biochemistry, 2009, 48, 3304–3314 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: dPA synthesis schemes and figures, flow cytometry gating strategies, micelle structural and stability analyses, FRET formulation fluorescence during cell treatment, and tables including DNA sequences and CD spectra analysis. See DOI: 10.1039/c9me00092e |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2020 |