
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHepaChip-MP – a twenty-four chamber microplate for a continuously perfused liver coculture model†
Marius
Busche
*a,
Olena
Tomilova
a,
Julia
Schütte
a,
Simon
Werner
a,
Meike
Beer
a,
Nicola
Groll
a,
Britta
Hagmeyer
a,
Michael
Pawlak
a,
Peter D.
Jones
 a,
Christian
Schmees
a,
Holger
Becker
b,
Juliane
Schnabel
b,
Karsten
Gall
c,
Roland
Hemmler
c,
Madlen
Matz-Soja
de,
Georg
Damm
f,
Simon
Beuck
g,
Tobias
Klaassen
g,
Jana
Moer
h,
Anett
Ullrich
h,
Dieter
Runge
h,
Katja
Schenke-Layland
aijk,
Rolf
Gebhardt
del and
Martin
Stelzle
*a
a,
Christian
Schmees
a,
Holger
Becker
b,
Juliane
Schnabel
b,
Karsten
Gall
c,
Roland
Hemmler
c,
Madlen
Matz-Soja
de,
Georg
Damm
f,
Simon
Beuck
g,
Tobias
Klaassen
g,
Jana
Moer
h,
Anett
Ullrich
h,
Dieter
Runge
h,
Katja
Schenke-Layland
aijk,
Rolf
Gebhardt
del and
Martin
Stelzle
*a
aNMI Natural and Medical Sciences Institute, University of Tübingen, Reutlingen, Germany. E-mail: martin.stelzle@nmi.de
bMicrofluidic ChipShop GmbH, Jena, Germany
cIonovation GmbH, Bissendorf, Germany
dSection of Hepatology, Clinic and Polyclinic for Gastroenterology, Hepatology, Infectiology, Pneumology, University Clinic Leipzig, Leipzig, Germany
eRudolf-Schönheimer-Institute of Biochemistry, Leipzig University, Leipzig, Germany
fDepartment of Hepatobiliary Surgery and Visceral Transplantation, University Hospital, Leipzig University, Leipzig, Germany
gA & M Labor fuer Analytik und Metabolismusforschung Service GmbH, Bergheim, Germany
hPRIMACYT Cell Culture Technology GmbH, Schwerin, Germany
iDepartment of Women's Health, Research Institute for Women's Health, Eberhard Karls University Tübingen, Tübingen, Germany
jCluster of Excellence iFIT (EXC 2180) “Image-Guided and Functionally Instructed Tumor Therapies”, Eberhard Karls University Tübingen, Germany
kDepartment of Medicine/Cardiology, Cardiovascular Research Laboratories (CVRL), University of California (UCLA), Los Angeles, CA, USA
lInViSys-Tübingen GbR, Leipzig, Germany
First published on 14th July 2020
Abstract
HepaChip microplate (HepaChip-MP) is a microfluidic platform comprised of 24 independent culture chambers with continuous, unidirectional perfusion. In the HepaChip-MP, an automated dielectrophoresis process selectively assembles viable cells into elongated micro tissues. Freshly isolated primary human hepatocytes (PHH) and primary human liver endothelial cells (HuLEC) were successfully assembled as cocultures aiming to mimic the liver sinusoid. Minimal quantities of primary human cells are required to establish micro tissues in the HepaChip-MP. Metabolic function including induction of CYP enzymes in PHH was successfully measured demonstrating a high degree of metabolic activity of cells in HepaChip-MP cultures and sufficient sensitivity of LC-MS analysis even for the relatively small number of cells per chamber. Further, parallelization realized in HepaChip-MP enabled the acquisition of dose–response toxicity data of diclofenac with a single device. Several unique technical features should enable a widespread application of this in vitro model. We have demonstrated fully automated preparation of cell cultures in HepaChip-MP using a pipetting robot. The tubeless unidirectional perfusion system based on gravity-driven flow can be operated within a standard incubator system. Overall, the system readily integrates in workflows common in cell culture labs. Further research will be directed towards optimization of media composition to further extend culture lifetime and study oxygen gradients and their effect on zonation within the sinusoid-like microorgans. In summary, we have established a novel parallelized and scalable microfluidic in vitro liver model showing hepatocyte function and anticipate future in-depth studies of liver biology and applications in pre-clinical drug development.
Introduction
Drug-induced liver injury represents a major cause for the failure of drug candidates.1 Current test systems including cell culture and animal models offer insufficient predictivity of drug-induced hepatotoxicity.2–6 Fundamentally new models are needed to address this challenge.7 Consensus exists that these models should i) use human cells (PHH or induced pluripotent stem cell (iPSC)-derived hepatocyte-like cells), ii) exhibit organ-like morphology, iii) provide an organ-specific microenvironment including appropriate extracellular matrix and non-parenchymal cells, and iv) allow for continuous perfusion to enable the establishment of proper chemical gradients within the culture.8–10Numerous in vitro models developed to mimic liver function have provided important insights into the biological function and physiology.2,11–19 The addition of growth factors and medium supplements as well as coculture of feeder cells or non-parenchymal cells with primary hepatocytes have been shown to extend the useful culture time and to maintain liver-specific functions of the hepatocytes.2,11–16 Also, culture in a three-dimensional (3D) environment stabilize hepatic functions, but still show a delayed dedifferentiation.17 Spheroid-based models have been shown to exhibit higher sensitivity to toxic substances than two-dimensional (2D) models,18,19 which has been comprehensively reviewed.20–22 These reviews highlight organ-on-chip systems, which add perfusion and associated physiological shear stress to the culture systems. The latter aids in maintaining hepatocytes in their differentiated state. Dash et al. showed enhanced albumin and urea production as well as enhanced expression and activity of CYP enzymes of primary hepatocytes under shear stress.23 This underlines the importance of a 3D culture and the presence of shear stress to achieve a truly in vivo-like physiology within an in vitro system of the liver.
Employing microfluidic technologies has enabled long-term coculture of hepatocytes with non-parenchymal cells or feeder cells under dynamic conditions in 3D.27–30 In these systems, cells exhibited gene expression patterns that suggest a more physiological state. Clearance of xenobiotic substances as well as responses after drug treatment compared favorably to in vivo data. In particular, enhanced metabolic activity was detected31–33 even for compounds normally metabolized slowly.34 Reduced albumin production was observed upon treatment with bosentan at a lower dose of the drug.35 Also, cell response towards hormones was recapitulated in a more in vivo like manner36 in perfused systems when compared to static 2D models.
While these systems have demonstrated the power of microfluidic technology to mimic the in vivo situation more closely than possible with static models, certain shortcomings hampering widespread application remain to be overcome.
Mostly, devices with single or only a few micro tissues have been demonstrated.40 However, parallelization represents a critical prerequisite to gaining robust and statistically valid data sets in a cost-effective manner. Unfortunately, this has been demonstrated by only a few systems so far. Kim et al. introduced the platform of InSphero with eleven parallelized cultures, each comprising a row of six spheroids.41 Jang et al. used the Mimetas OrganoPlate with 40 independent chambers to differentiate HepaRG cells and analyzed their function.42 However, both systems use tilting platforms to generate bidirectional flow, which prohibits the development of physiological gradients of substances inside the chamber. Domansky et al. developed a system that used integrated pneumatic pumps on a device in multiwell plate format comprising 12 chambers to coculture primary rat hepatocytes and liver sinusoidal endothelial cells (LSECs).43
Perfusion is often achieved by applying flow using syringe pumps. While this enables precise control of flow rate, establishing proper fluidic connections tends to be complex and prone to the introduction of air bubbles. Bubbles can damage micro tissues or interrupt flow and therefore must be avoided. Also, large scale parallelization is not feasible utilizing these systems.
Many devices have been manufactured from polydimethylsiloxane (PDMS) by soft lithography. PDMS provides for favorable biocompatibility, transparency and relative ease of manufacturing of individual devices. However, PDMS also is known to absorb hydrophobic compounds.44–46 Reliable dose–response data often cannot be obtained in such systems, given the inherently large surface-to-volume ratio of microfluidic devices and the hydrophobicity of many drug candidates.
Often, organ-on-chip cultures do not resemble the actual 3D morphology of the organ tissue but rather represent perfused 2D or randomly organized 3D cellular arrangements. While immobilization of cells within gels enables arrangement in a 3D structure, gels may influence diffusion within the microtissue. This also applies to the assembly of cells on membranes with properties differing from physiological tissue.
Therefore, the objective of this work was the development of a parallelized, microfluidic platform for cocultures of hepatocytes and endothelial cells resembling liver sinusoid morphology. Metabolic activity as well as toxicological response to a known compound were to be assessed as a proof of sufficient sensitivity and compatibility of the used assays and physiologic cell function.
To enable integration into common cell culture workflows, we designed a plate following the SBS-standard 96-well plate format with 24 independent microorgans. We chose a cyclic olefin polymer (COP) as a material with excellent optical transparency and low absorbance of compounds. This enables the use of conventional microscopy and state-of-the-art biochemical assays. Further, COP allows for seamless upscaling of manufacturing by micro-injection molding. A high degree of parallelization along with ease-of-use was achieved by continuous, tubeless, gravity-driven, unidirectional flow at physiological rates.
In addition, liver cells were to be assembled in elongated structures closely mimicking the physiological dimension with respect to length and width of the natural human liver sinusoid. Only viable cells were actively assembled by dielectrophoretic forces generated by integrated electrodes.
A complete workflow from priming of the microplate to cell assembly was realized using a pipettor system. A miniaturized, 24-channel, incubator-compatible pump system was developed which can be used to continuously transfer culture media from outlet to inlet reservoirs thus enabling constant reperfusion.
In summary, we present an automated system allowing for parallelized culture of cells mimicking the liver sinusoid both with respect to physical dimensions as well as function. In particular, we demonstrate compatibility with common biological workflows in terms of usability and readout methods.
Materials & methods
Reagents were purchased from Merck KGaA (Darmstadt, Germany) unless stated otherwise.i) Microplate manufacture
HepaChip microplates (HepaChip-MPs) were jointly developed and are now manufactured by microfluidic ChipShop (Jena, Germany).The HepaChip-MP (Fig. 1) consists of cyclic olefin polymer (COP) and was manufactured by micro injection molding. The mold was generated using ultra-precision micro milling. Prior to bonding, electrodes were fabricated by evaporation of titanium (10 nm) and gold (approx. 100 nm) through a shadow mask. These electrodes are intended for generating a high-frequency electric field within the cell chambers for dielectrophoretic assembly of cells onto the assembly ridges (see Fig. 1). Prior to bonding the assembly ridges were illuminated through a shadow mask by short wavelength (185 nm) UV light using a low-pressure mercury lamp (Heraeus Noblelight, Hanau, Germany, NIQ lamp, quartz tube, 5 W lamp), thereby introducing acid groups on the assembly ridges.47 Finally, microplates were bonded with a COP foil (188 μm thickness). Openings in the foil were positioned to enable electrical access to the electrode connection pads on the bottom face of the microplate.
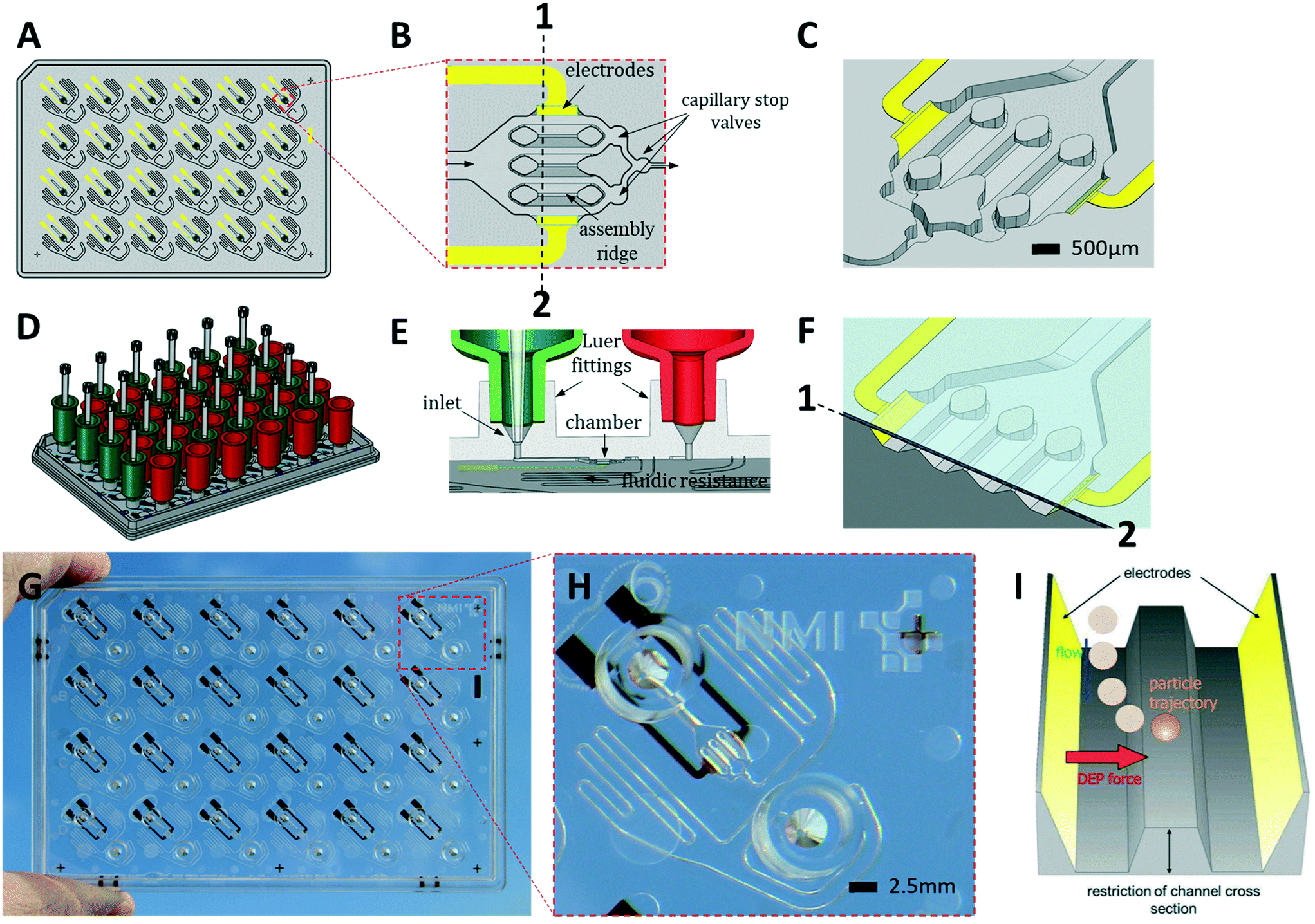 | ||
| Fig. 1 The HepaChip-MP design. A) Layout of the microplate made of COP (grey) in ANSI/SBS format with electrodes (yellow). B) Close-up of an individual chamber showing electrodes at the periphery of the chamber, three assembly ridges and capillary stop valves for bubble-free filling. The direction of flow is from left to right. C) 3D view of a chamber. D) HepaChip-MP shown with tanks (reservoirs) on inlets (green) and outlets (red) as well as pipette tips reaching into the tanks. E) Tank connectors fit into Luer fittings. Gravity-driven flow may be bypassed by a pipette positioned in the conical inlet, thus tightly sealing and enabling defined flow for process steps such as cell assembly or fast media exchange. F) A cross-section along 1–2 in (B) including the cover foil which leaves a gap of about 50 μm height above the assembly ridges where cell aggregates mimicking sinusoids are formed. G) HepaChip-MP made by micro injection molding of COP and evaporation of gold electrodes. H) Close-up of an individual unit showing Luer fittings, cell chamber, electrodes, and meandering fluid resistor. I) Schematic illustration of the dielectrophoretic (DEP) force which draws cells onto the assembly ridges. | ||
ii) Peripheral instrumentation
A CyBio FeliX pipetting system (Analytic Jena AG, Fig. 2B) was modified to hold a dielectrophoresis (DEP) fixture (Fig. 2C) for the HepaChip-MP comprising LED illumination and electrical connectors for the electrodes located on the bottom face of the microplate. The metallic encasing provides electromagnetic shielding of the high frequency electric fields applied during DEP assembly of cells. Openings on the bottom of the DEP fixture allow optical access of cell chambers by an imaging system to monitor chamber priming and cell assembly. Two different pipetting heads can be attached automatically by program control and either hold 24 or 4 pipetting tips. Batch programs for automated procedures were implemented to render robust and reproducible operation of the HepaChip-MP system.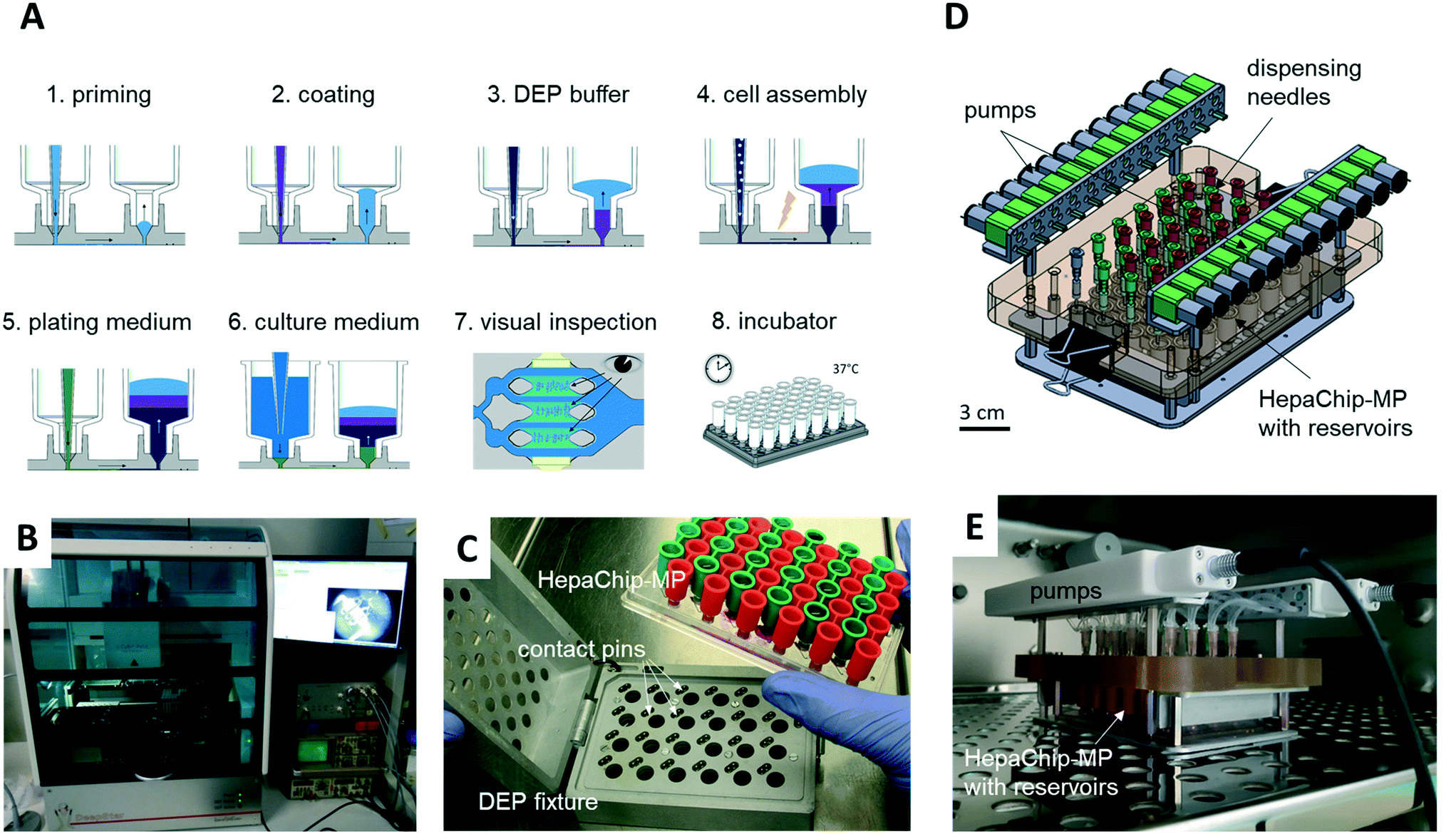 | ||
| Fig. 2 Automated workflow and perfusion system for cell culture in HepaChip-MP. A) Schematic depiction of the workflow. Each process step except for the incubation is performed by a pipetting robot (B). During automated chamber priming, cell assembly and medium exchange, the HepaChip-MP is mounted in the DEP fixture (C), which establishes electrical contact between the integrated electrodes and a high-frequency generator to arrange cells by dielectrophoresis. D) The pump system is compatible with microfluidic microplates with fluid ports in the 96-well pattern and enables re-circulation by pumping medium from the outlet reservoirs (red) to the inlet reservoirs (green). The system comprises 24 peristaltic pumps and mounts directly on the HepaChip-MP. E) The pump system may be operated within an incubator. | ||
iii) Automated priming, functionalization, cell assembly and cell culture workflow in HepaChip-MP
Chambers were primed with Pluronic F127 in HEPES, pH 7.4. In order to coat collagen on the assembly ridges a solution of Pluronic F127 (1 mg mL−1) and collagen (100 μg mL−1) in HEPES, pH 4, was pumped through the chambers. The collagen selectively binds to areas previously activated by UV illumination (see Fig. S2†). At the same time, non-activated, hydrophobic surfaces were coated by a monolayer of Pluronic to reduce cell adhesion and substance adsorption (see Fig. S6†). The procedure has been reported in detail previously.47 Chambers were incubated with collagen solution overnight at room temperature under slow perfusion by filling the inlets with 500 μL of collagen/Pluronic solution.After removing the collagen solution from inlet and outlet, the culture chambers were filled with low conductivity DEP medium (see v) to allow for positive DEP of cells. The cells were suspended in DEP medium immediately prior to the assembly routine (see v). The pipetting robot infused the cell suspension at 4 μL min−1, while a high-frequency electric field was applied across the electrodes (80 Vpp, 360 kHz for hepatocytes, 80 Vpp, 620 kHz for endothelial cells) to assemble the cells.
Details of the chamber design resulting in an inhomogeneous electric field distribution, including multiphysics simulations of electric fields and DEP forces on cells have been reported previously.48 A video showing cell assembly by DEP is in the ESI† (Video S2).
After completion of the assembly process, the high-frequency voltage was turned off. DEP medium was rinsed out of the cell culture chamber by perfusion with cell culture medium. Cells not assembled over the assembly ridge but attached to the periphery of the cell aggregate were thereby also rinsed out of the chamber. Finally, cells that were rinsed into the outlet were removed by pipetting. Fluid ports were routinely checked for the presence of cells prior to any assays to prevent interference by residual cells located in either the inlet or outlet. Shear forces in a HepaChip chamber were calculated by multiphysics simulation and have been reported previously.49
iv) Perfusion system
A perfusion system was built to enable continuous reperfusion of cell culture medium (Fig. 2D and E). It comprises 24 miniature peristaltic pumps (Takasago Electric Inc., Nagoya, Japan) with cannulas reaching into inlet and outlet reservoirs on the microplate. These pumps transfer medium from the outlet to the inlet reservoir, in this way maintaining a defined pressure difference and flow rate. The absence of a pressure-tight fluidic connection between the perfusion system and the microfluidic device ensures that flow is completely gravity driven. This design furthermore eliminates the risk of bubbles, as any air possibly introduced in the tubing will not be pumped into the microfluidic device. The pumps are operated every 150 min for 4 min to transfer medium perfused during this period (approx. 75 to 100 μL) back to the inlet reservoir. The level difference and flow rate can be adjusted by varying the vertical position of the cannula which draws medium out of the outlet reservoir.The HepaChip-MP also may be operated as stand-alone device which indeed was the mode of use routinely employed in this study. With inlet reservoirs filled with approximately 900 μL, continuous flow at rates decreasing from about 60 μL h−1 to 5 μL h−1 can be maintained over a period of at least 12 h (Fig. S1†), after which time period the medium can be re-perfused manually or by use of a pipetting robot.
v) Cell culture and media formulations
Fresh and cryopreserved primary human hepatocytes (PHH) (Donor BHuf16068, Cytes Biotechnologies, Barcelona) as well as cryopreserved primary human liver endothelial cells (HuLEC) were used in our experiments.Fresh PHH and HuLEC were isolated from liver tissue samples by a two-step EDTA/collagenase perfusion technique as described elsewhere.50 Liver tissue samples were obtained from macroscopically healthy tissue that remained from resected human liver of patients with primary or secondary liver tumors or benign local liver diseases. Informed consent of the patients for the use of tissue for research purposes was obtained according to the ethical guidelines of Leipzig University Hospital (ethics vote 006/17-ek). Fresh hepatocytes were stored on ChillProtect Plus and shipped overnight on ice.
The appropriate HuLEC cell number for cryopreservation was centrifuged at 300 × g for 4 min at 4 °C and the HuLEC pellet was re-suspended in a cryopreservation medium consisting of 70% ChillProtec Plus medium (Biochrom, Berlin, Germany), 20% FBS and 10% dimethyl sulfoxide (DMSO; Carl Roth, Karlsruhe, Germany) whereby a maximum concentrations of 2–10 × 106 HuLEC per mL medium are recommended. The resulting cell suspension was transferred into CryoPure vials. The vials were inverted briefly, placed as soon as possible in a Mr. Frosty Freezing Container (Thermo Fisher Scientific, Waltham, USA) and transferred to −80 °C for at least 24 h. Cryopreserved cells were shipped on dry ice overnight.
Cryopreserved PHH were thawed using a thawing kit (Primacyt) according to instructions and cell number and viability were determined with the trypan blue exclusion test. Next, 3–4 million cells were centrifuged at 4 °C and 50 × g for 5 min. Cells were re-suspended in 2 mL of DEP medium. DEP stock solution was prepared by dissolving 1 g glucose, 57.9 mg sodium pyruvate, 28.4 mg CaCl2 and 24.6 mg MgSO4 in 100 mL diH2O. To obtain the final DEP medium, 10 mL of DEP stock was diluted in 100 mL diH2O, 9.5 g saccharose was added and the medium was sterilized by filtration. After re-suspending cells in DEP medium, the suspension was placed in the pipetting robot and used for cell assembly in the HepaChip-MP.
Fresh PHH used in HepaChip-MP cultures were washed twice with ice-cold PBS (centrifugation 50 × g, 5 min, 4 °C). Subsequently, cells were re-suspended in culture medium and cell number and viability were determined with the trypan blue exclusion test. For culturing PHH different medium formulations were used. Medium 1 denotes Williams E (with GlutaMAX; Thermo Fisher Scientific) supplemented with 10% FBS, 15 mM HEPES (Thermo Fisher Scientific), 1 mM Na-pyruvate, 1% non-essential amino acids (Thermo Fisher Scientific), 1 μg mL−1 dexamethasone, 1 μg mL−1 human insulin and primocin (Invivogen, San Diego, CA, USA). Medium 2 was composed of medium 1 plus 0.55 μg mL−1 human transferrin, 0.5 ng mL−1 Na-selen and 5.35 μg mL−1 linoleic acid. After determination of cell number and viability, 3–4 million cells were centrifuged at 4 °C and 50g for 5 min and re-suspended in 2 mL of DEP medium. Then, the cell suspension was placed in the pipetting robot and for cell assembly in the HepaChip-MP.
For co-culture experiments, HuLEC were assembled at day 1 after assembly of hepatocytes. Cryopreserved HuLEC were transferred to 10 mL prewarmed EGM2 (Promocell, Heidelberg, Germany) and then centrifuged at 300g for 5 min at room temperature. Cells were re-suspended in EGM2 and cell number and viability were determined by trypan blue exclusion test. HuLEC concentration employed for assembly in the HepaChip-MP was 0.5 × 106 cells per ml.
For spheroid cultures, cryopreserved PHH from a single donor (BHuf16068, Cytes Biotechnologies, Barcelona) were thawed following the distributor's manual. Cells were re-suspended in 3D-HMM (Primacyt) and cell number and viability were determined with the trypan blue exclusion test. Cells were seeded into ULA U-bottom 96-well plates (Greiner Bio-One, Kremsmünster, Austria) in 70 μL 3D-HMM at a density of 2500 cells per well. To aid sedimentation of cells and formation of spheroids, plates were centrifuged once at 500 × g for 2 min. Spheroids were cultured at 37 °C with 5% CO2 for 6 days until a spheroid with regular shape was formed. On day 3 fresh 3D-HMM (30 μL) was added to each well.
Induction of cytochrome P450 (CYP) enzymatic activity was performed in 3D spheroid cultures and HepaChip-MP cultures in the presence of 3D-HMM medium (Primacyt). For the coculture of PHH and HuLEC, medium 2 and EGM2 were used at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. For all other experiments medium 2 was used if not stated differently.
:1. For all other experiments medium 2 was used if not stated differently.
vi) Assays
| Antigen | Fluorescent label | Vendor | Catalog# | Host species | Reactivity | Dilution |
|---|---|---|---|---|---|---|
| CD31 | Alexa Fluor 488 | Abcam | ab215911 | Mouse | Human | 1:500 |
| CK18 | Alexa Fluor 647 | Abcam | ab206269 | Rabbit | Human | 1:500 |
In HepaChip-MP cultures, the induction of CYP enzymes was started at day 2 of culture and carried out for 72 h by perfusing medium containing 20 μM rifampicin. Thereafter, solutions of the CYP substrate in the respective cell culture medium were perfused for 5 h through the chamber (50 μM diclofenac for CYP2C9; 15 μM midazolam for CYP3A4). Sample volumes of approx. 150 μL were obtained. After transferring the sample to tubes, 15 μL of 10% acetic acid were added and samples were stored at −80 °C until analysis by liquid chromatography-mass spectrometry (LC-MS).
For induction experiments in spheroid cultures, medium was removed from wells and spheroids were treated with 70 μL of 20 μM rifampicin for 72 h starting on day 6, whereby the induction solution was replaced every day. At day 9, spheroids were washed twice with PBS and incubated with 90 μL substrate solution (50 μM diclofenac for CYP2C9; 15 μM midazolam for CYP3A4) for 5 h. After incubation with substrates, the supernatant of two wells was pooled (final volume 180 μL), and the samples were transferred into tubes containing 18 μL of 10% acetic acid in 3D-HMM. Samples were stored at −80 °C until analysis by LC-MS.
For cell lysis in spheroid cultures, spheroids were washed once with PBS and incubated with 0.1 M NaOH for 15–20 min. Cell lysates of two spheroids were pooled and stored at −80 °C until protein analysis.
Samples from HepaChip-MP and spheroid cultures were analyzed by a BCA-protein assay kit for low concentrations (Abcam, ab207002) according to its instructions.
Preparation of stock and working solutions, calibration standards and QC samples. Stock solution of 4′-hydroxydiclofenac (HDC) and the respective internal standard (4′-hydroxydiclofenac-13C6; HDC-13C6) were prepared by accurate weighing of the reference item and dissolution in a mixture of dimethylsulfoxide and acetonitrile (7 + 3, v + v) to yield a final concentration of 1 mg mL−1. A commercially available, certified stock solution of 1 mg mL−1 in methanol was used for HMDZ and its internal standard (1′-hydrixymidazolam-d4; HDMZ-d4). Working solutions for spiking of matrix samples were prepared by serial dilution of the stock solutions in a mixture of water and methanol (1 + 1, v + v).
Calibration standards were prepared freshly on each day of analysis by the addition of 10 μL of spiking solution to 100 μL of pre-acidified blank matrix (neat cell culture medium, i.e. 3D-HMM, +10% aqueous acetic acid, 100 + 10, v + v). QC samples in pre-acidified blank matrix were prepared in bulk in advance and stored at ≤−18 °C until use.
Sample processing. Fifty (50) μL of pre-acidified sample (blank matrix, calibration standard, QC, or cell culture incubate) were pipetted into a preconditioned OASIS HLB μElution plate (96 well format, Waters, Eschborn, Germany) and mixed with 50 μL of water + formic acid (100 + 1, v + v) and 10 μL of internal standard working solution (conc. of HDC-13C6 and HMDZ-d4, and approx. 250 ng mL−1). After washing of the loaded sample with water + formic acid (100 + 1, v + v), the analytes were eluted with 50 μL of acetonitrile + water + formic acid (90 + 10 + 1, v + v + v) into a 96 well collection plate. The eluate was concentrated under a gentle stream of nitrogen at approx. 40 °C for 10 min and the residue was reconstituted in 35 μL of acetonitrile + water + formic acid (20 + 80 + 1, v + v + v). The plate was sealed and 15 μL of the processed sample were injected for LC-MS analysis.
LC-MS method. LC-MS analysis was performed on an 1100 Series high-performance liquid chromatography system (Agilent Technologies, Waldbronn, Germany) coupled to a TSQ Vantage mass spectrometer (Thermo Fisher Scientific, Dreieich, Germany). Chromatographic separation was achieved on a Reprospher 100 C18-DE analytical column (2.0 × 50 mm, 1.8 μm particle size; Dr. Maisch, Ammerbruch, Germany) equipped with a Security Guard cartridge precolumn (C18, 4 × 2.0 mm; Phenomenex, Aschaffenburg, Germany), maintained at 40 °C. Mobile phases consisted of 10 mM ammonium carbamate in water (A) and in a mixture of methanol and acetonitrile (80 + 20; B). The following gradient elution was carried out at a constant flow rate of 250 μL min−1 [% A (min)]: 99 (0.0), 99 (0.3), 30 (4.0), 20 (6.0), 0 (6.01), 0 (7.0), 99 (7.01), 99 (10.0). The MS was operated in positive ESI mode (HESI II probe), applying a spray voltage of 4500 V and a vaporizer temperature of 200 °C. Argon was used as collision gas.
Protonated analytes were detected after collision induced dissociation (collision gas: argon) in multiple reaction monitoring mode using the following ion transitions (m/z–m/z, collision energy [V], S-lens [V]): 312.0–230.0, 35, 80 (HDC), 318.0–236.0, 35, 80 (HDC-13C6), 342.0–324.0, 21, 130 (HMDZ), 346.0–328.0, 21, 130 (HMDZ-d4).
Quantification and analytical acceptance criteria. The concentrations of HDC and HDMZ in cell culture incubations were determined using the corresponding calibration functions with weighting of the measured results (weighting 1/conc2). HDC-13C6 and HMDZ-d4 were used as respective internal standards. The chromatograms were recorded and integrated in peak area mode by use of the LCquan software (Thermo Fisher Scientific, Dreieich, Germany). Quantification of the analytes was conducted in accordance with the current guidelines on bioanalytical method validation (EMA guideline and FDA guidance for industry).
The calibration range was set from 0.200 ng mL−1 (lower limit of quantification, LLOQ) to 200 ng mL−1 (upper limit of quantification, ULOQ). An explorative calibration standard at 0.100 ng mL−1 was analyzed within each run and included into regression analysis, if higher sensitivity was required. The volume of the acetic acid solution added to each individual sample was corrected during quantification.
One set of calibration standards (including one blank sample and zero calibration standard each) was analyzed at the start of the analytical run and one at the end. Each set of calibration standards consisted of at least eight non-zero standards distributed over the calibration range. Calibration standards were excluded from linear regression if the accuracy of an individual standard was outside the limits of ±20% (±25% at the LLOQ). No more than 25% of the calibration standards were excluded from each series of calibration standards. At least one sample at the LLOQ and the ULOQ level had to meet the specifications.
At least six QC samples (three different levels as duplicates) were analyzed within each run. At least 67% of all QCs per run and at least 50% of the QCs of one concentration level had to meet the acceptance criteria for QC samples (bias of each individual QC sample concentration of max. ±20%).
In order to determine track viability, a resazurine assay was carried out on day 1, day 3 and day 6 of culture in the HepaChip-MP as described above. A stock solution of resazurine in medium 2 was prepared, aliquoted and stored at 4 °C until the respective day of experiment. As a negative control, cells were treated with 2 mM diclofenac starting at day 2 of culture.
vii) Statistics
Values are given as mean and standard deviation (mean ± SD). As the results presented here are intended to demonstrate various assays in this new platform, the results include technical rather than biological replicates. The results of the induction experiment of CYP2C9 and CYP3A4 were analyzed for statistical significance by an unpaired t-test. For the cytotoxicity test, a one-way ANOVA with post hoc Tukey was carried out. Statistics software GraphPad Prism (GraphPad Software, USA, California, San Diego) was used. P-Values are provided in the figure description.Results
1. Hepachip-MP microfluidic design
The HepaChip-MP was designed at NMI and manufactured by microfluidic ChipShop in conformity with ANSI/SBS standards for outer dimensions and position of fluid ports (Fig. 1). Each HepaChip-MP has 24 culture chambers (Fig. 1B and C), which contain three assembly ridges with dimensions similar to the human liver sinusoid (approx. 30 cells or 900 μm in length52) on which cells can adhere. The chamber is 4.3 mm long, 2.3 mm wide and has a height of 0.19 mm. The resulting chamber volume is approximately 0.85 μl. As fluid enters through the inlet and flows along the assembly ridges, capillary stop valves sequentially arranged within the chamber ensure reproducible priming and efficiently prevent trapping of bubbles (Video S1†).53 Electrodes on the chamber sides are employed to generate high frequency electric fields across the assembly ridges for assembly of cells by DEP. Maximum field strengths are achieved in the vertical gaps between the assembly ridges and the cover (Fig. 1C). Electrical field distribution, hydrodynamic flow and cell trajectories resulting thereof have been reported previously.48Both inlets and outlets have Luer fittings to enable versatile connections. Tanks attached to these Luer fittings (Fig. 1D) enable continuous gravity-driven flow when the inlet (green) and outlet tanks (red) are filled to different levels. A fluid resistor (width: 100 μm, length: approx. 140 mm) at the outlet of the culture chamber defines a flow rate ranging from approximately 1 to 0.1 μL min−1 over a period of 12 h (Fig. S1†).
Conical structures at the bottom of the Luer fittings allow for pressure-tight connection of pipette tips (Fig. 1E). This mode of operation is used during priming and cell assembly to achieve flow rates which are constant or different from those that can be achieved by gravity-driven flow. This mode allows infusion of minute amounts of fluids such as staining solutions or test samples at any time, temporarily suspending the flow of medium contained in the tank. Thus, automated assays such as viability tests or immunohistochemical staining may be performed. Upon retraction of the pipette tip, gravity-driven medium flow resumes without delay.
We designed the fluid resistance such as to enable in vivo-like perfusion. To this end, the number of cells in the chamber and their oxygen consumption was considered. As gas permeability of COP is quite small54 diffusion of oxygen through the polymer may be neglected. The tank volume of 900 μL allows continuous perfusion for at least 12 h without user intervention or the perfusion system. With 500–1000 cells per chamber consuming 0.3–8 fmol min−1 per cell,55–57 this flow rate was expected to provide sufficient oxygen while allowing for the development of an appropriate oxygen gradient along the sinusoid structures. Perfusion by gravity-driven flow was measured with medium at 37 °C (Fig. S1†). As expected, the flow rate decreases over time as levels within the inlet and outlet tank equilibrate. This perfusion mode was employed in the experiments described in this study.
However, to also enable constant flow rates, we developed an incubator-compatible perfusion system (Fig. 2D and E) which can be used to continually pump media from outlet reservoirs back into the inlet reservoirs, thus maintaining constant pressure differences and flow rates. This device enables long-term perfusion without user attention and thus can enhance overall system usability.
2. Process development and cell assembly
The priming of chambers was evaluated. Only chambers completely filled without any air bubbles were considered usable. Priming with a pipetting rate of 1.11 μL min−1 properly filled an average >80% of the chambers.Selective coating of only the assembly ridges with ECM protein by perfusion of a collagen I/Pluronic F127 mixture was confirmed using collagen labelled with rhodamine (RITC). The selective deposition of RITC-labelled collagen on assembly ridges was verified by fluorescence microscopy (Fig. S2†).
The fraction of cells assembled on the assembly ridges vs. cells passing through the chamber strongly depends on the flow rate as reported previously.48 Lower flow rate results in lower hydrodynamic drag forces and higher efficiency of cell assembly by DEP. However, cells can sediment and clog the microfluidic channels if flow rates are too low. The pipettor provides a higher degree of precision and thus reproducibility than manual pipetting. A video showing the automated priming and cell assembly process is provided in the ESI† (Video S1).
Cells assembled on the assembly ridges adhere due to the collagen I coating and the reduced shear forces present in this region of the chamber.48 In contrast, higher hydrodynamic forces wash out cells which may have been collected incorrectly in the chamber (Fig. 4).
3. Liver cultures in HepaChip-MP
PHH monoculture. All experiments reported here were carried out using PHH. Cell culture experiments using the HepaChip-MP confirmed the desired formation of elongated structures on the assembly ridges. Cells adhered to the assembly ridges and started to build compact structures within two days (Fig. 4). Fig. S3† shows images obtained from a larger number of chambers of several HepaChip-MP demonstrating reproducibility of cell assembly on the assembly ridges. The development of compact structures can be observed from the dense grouping of cells on the assembly ridges. In addition, a barrier-like structure can be seen at the interface between the microtissue and the flow channel. At DIV5, these structures became more compact and shorter and broadened. This process continued until day 12 of culture. The viability of cells up until day 8 was confirmed by calcein staining (Fig. 4). Cells which are initially not part of the compact structure are flushed out or integrated into the cell structures (Fig. 4, day 0 to day 5).
Unexpectedly, cells assembled on the outermost assembly ridges (denoted by “L” and “R” in Fig. 4), were observed to collectively migrate over time towards the periphery of the chamber. As a minute, previously unnoticed cross flow was suspected to be responsible, the movement of fluorescent microparticles (diameter 5 μm) under flow in the chamber was studied. Indeed, movement of particles indicated flow perpendicular to the chamber axis, including crossflow traversing the left and right assembly ridges (Fig. S4†). Hydrodynamic forces of this flow pattern, as minute as they may be considering small pressure and flow rates present in the chamber during culture, may lead to the dislocation of the assembled cells. Additional study in future experiments may guide a redesign of the culture chamber to ensure flow strictly parallel to the assembly ridges.
To track viability of PHH cultured in HepaChip-MP over a culture period of 6 days, a resazurine assay was carried out on days 1, 3 and 6 of culture (Fig. 3). Non-treated PHH show constant viability over the duration of the experiment (green lines). PHH treated with 2 mM diclofenac at day 2 show RFU values declining to about 40–70% of the initial values (red lines).
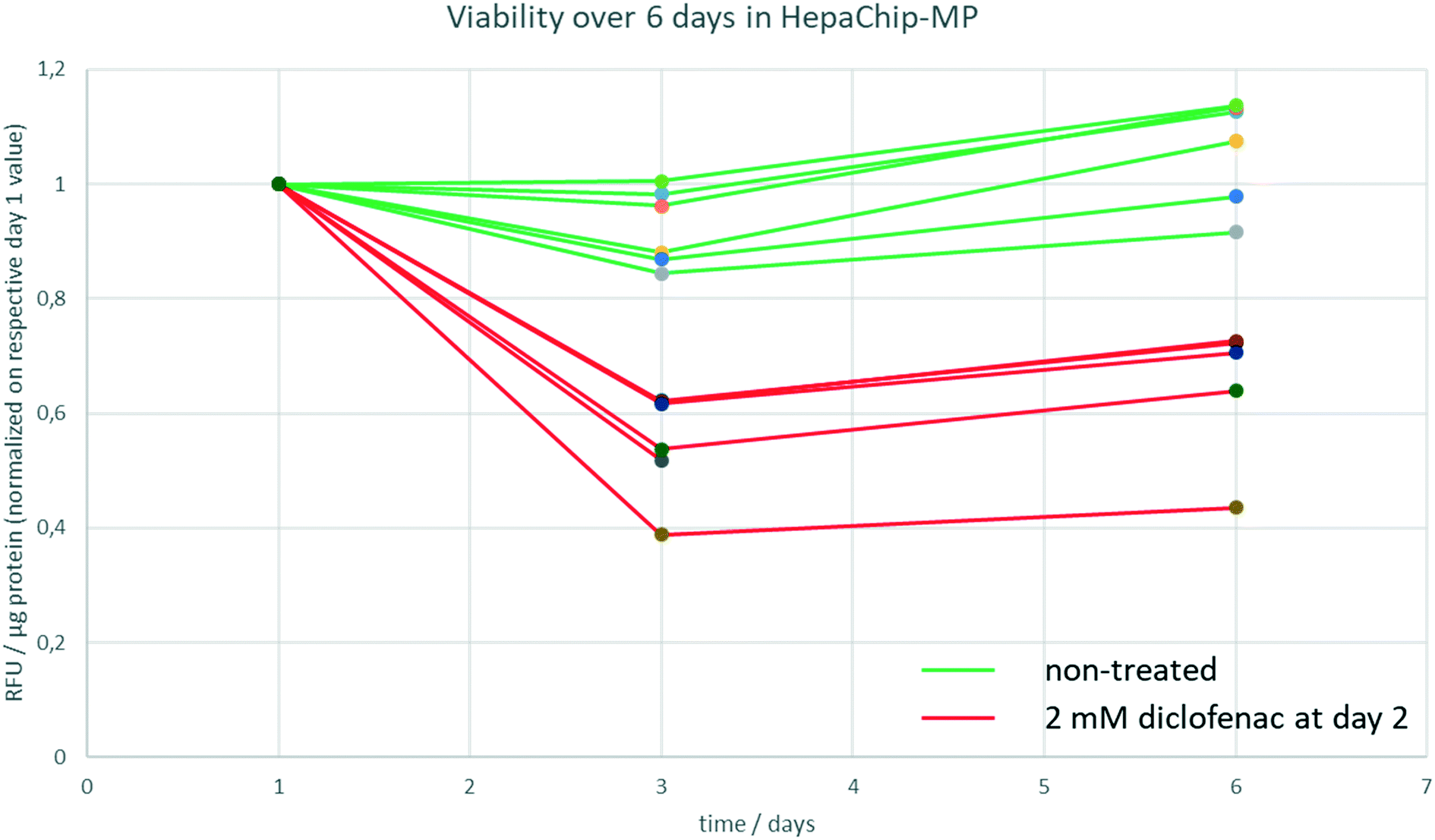 | ||
| Fig. 3 Viability of PHH during 6 days in HepaChip-MP. The viability was measured by a resazurine assay at day 1, 3 and 6 of culture. Non-treated PHH show no significant change of RFU values during 6 days of culture (green lines) whereas the RFU values of PHH treated with 2 mM diclofenac decline to 40–70% (red lines). Six chambers of a single HepaChip-MP of both non-treated and treated samples are shown. | ||
Basal CYP activities during culture. Basal activities of CYP2C9 and CYP3A4 of cultures of fresh PHH (donor: female, 55 years, hemangioma) were evaluated on day 3 and day 6 in the HepaChip-MP with two different cell culture media (Table 2). CYP2C9 was equally active at day 3 and 6 in medium 1 (ratio of 1.05 ± 0.18) and slightly more active at day 6 in medium 2 (ratio of 1.38 ± 0.14). CYP3A4 activity was higher on day 6 vs. day 3 in either medium 1 (ratio of 1.39 ± 0.21) or medium 2 (ratio of 1.62 ± 0.05).
| Ratio day6/day3 | HDC | HMDZ |
|---|---|---|
| Medium 1 | 1.05 ± 0.18 | 1.39 ± 0.21 |
| Medium 2 | 1.38 ± 0.14 | 1.62 ± 0.05 |
CYP activity in HepaChip-MP in comparison to 3D-spheroid culture. We evaluated xenobiotic metabolism of PHH in the HepaChip-MP and in spheroids as a benchmark system (Fig. 5). Induction was carried out by incubation with rifampicin for 72 h. The activity of CYP3A4 and CYP2C9 was measured by substrate turnover after 5 h of substrate incubation using LC-MS analysis of metabolites in the perfusates. In spheroids, induced activity vs. basal activity was 6.6-fold higher for CYP3A4 and 2.1-fold higher for CYP2C9. In the HepaChip-MP cultures, induced activity was 3.4-fold higher for CYP3A4 and 3.3-fold higher for CYP2C9. The measured conversion rates of CYP2C9 were of the same order of magnitude in both the spheroid and HepaChip-MP. The conversion rates observed for CYP3A4 were more than 20-fold lower in HepaChip-MP than in the spheroid model, while induction ratios were similar.
Discussion
Here, we have demonstrated a perfusable liver-on-a-chip platform in SBS/ANSI microplate format which enables i) active assembly and coculture of PHH and HuLEC; ii) continuous unidirectional gravity-driven flow; iii) integration in common testing workflows and iv) automation using comprehensive instrumentation (pipetting robot, perfusion system). We expect that this platform will facilitate the use of an organ-on-chip system by end users without particular proficiency in microfluidic technology. Demonstrative experiments confirmed important biological functions and assays, namely xenobiotic metabolism (via LC/MS) including induction, viability assay, assessment of drug-induced toxicity and immunostaining of marker proteins.While we regard the HepaChip-MP model as an important step towards fully workflow-compatible microphysiological systems, our study also shows that certain improvements will be required to fully attain this goal. These will be subject to further research and development as outlined below.
Chamber priming and parallelization
Priming successfully fills more than 80% of chambers without air bubbles, enabling 20 independent experiments with the same cell preparation on a single microplate. Working with primary cells with high donor variability requires such parallelization to reproducibly obtain valid data. While successful priming demonstrates the proper function of capillary stop valves in the chamber,48 optimization of the process and possibly of the design of the capillary stop features is required to attain success rates approaching 100%.A single HepaChip-microplate can acquire a complete dose–response curve. Robustness of operation is attained by process automation using a pipetting robot. Open access of the medium reservoirs enables handling comparable to standard microplates. These features will facilitate the integration of microfluidic microplates such as the HepaChip-MP into common pharmaceutical and biological workflows.
Technical and biological features
In contrast to other in vitro systems, HepaChip-MP has distinct advantages with respect to usability and biologic mimicry. While microfluidic microplate platforms using a rocker to induce gravity-driven perfusion this flow is bi-directional.41,42 In contrast, HepaChip-MP cultures are unidirectionally perfused to enable generation of in vivo-like chemical gradients and a more in vivo-like shear stress along micro tissues. The HepaChip-MP platform can be handled like a common microplate while microfluidic models with chip-integrated peristaltic pumps require connection to external periphery.60Most importantly, the assembly of cells by DEP exclusively selects viable cells. Non-viable cells do not maintain the difference in intra- and extracellular conductivity which is fundamental in DEP for the generation of a dipole moment.48 As a consequence, non-viable cells simply pass through the chamber. Furthermore, DEP allows sequential assembly of multiple cell types to mimic physiological structures. This paradigm of controlled assembly of cells into micro tissues in specifically designed cell chambers can be generalized to other organ systems and we present liver sinusoids in the HepaChip-MP as a representative example (Fig. 4).
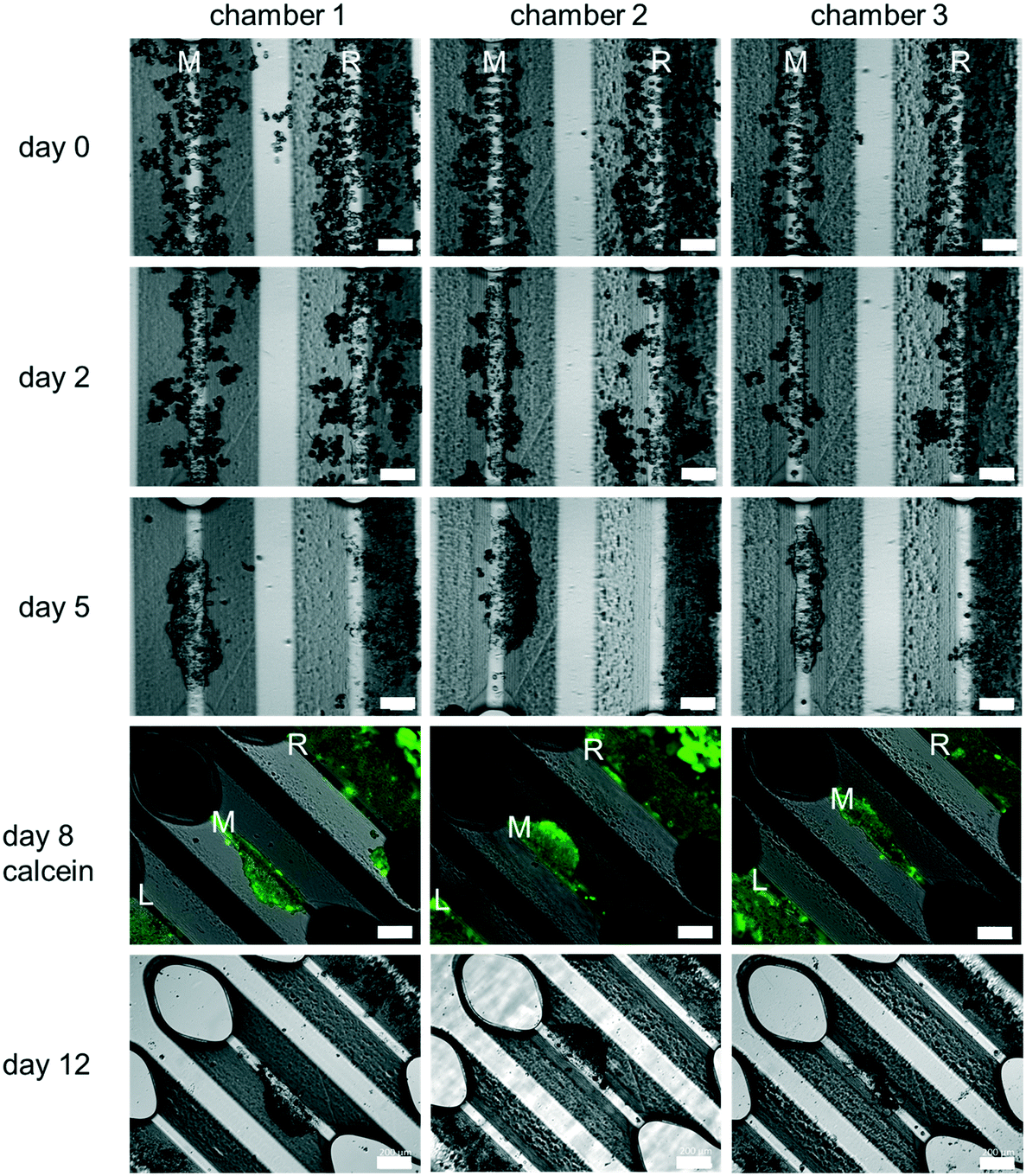 | ||
| Fig. 4 Representative microscopic images obtained from three different cultivation chambers showing cultures of fresh PHH in the HepaChip-MP over a period of twelve days. Images from day 0 to 5 show the middle assembly ridge (M) and the right assembly ridge (R). Images on day 8 and twelve (rotated by 45 degrees) show the middle assembly ridge (M), the right assembly ridge (R) and a part of the left assembly ridge (L). The cells can be assembled on the assembly ridges (day 0) and a compact elongated structure forms there within 2 days. After 5 days the cells on the middle assembly ridge are still in a compact elongated but more contracted structure. The cells on the right assembly ridge collectively migrate to the right. All cells are still viable on day 8 as shown with calcein staining. Throughout the culture, the cells on the middle assembly ridge are contracting to form a more spheroid-like structure (scale bar day 0, day 2 and day 5: 300 μm; scale bar day 8 and day 12: 200 μm, n = 8). | ||
The feasibility of controlled assembly of cells into micro tissues in the desired structure is a general advantage of microfluidic systems when compared to spheroid models. In spheroids, cells are assembled randomly in spherical structures. Hepatocytes in the HepaChip-MP are assembled in compact elongated structures (Fig. 4), which resembles the structure of the liver sinusoid more closely than spheroid structures and perfused monolayers. We have observed that initially elongated structures contract over time (Fig. 4). Cell–cell interactions may become stronger than their adhesion (via collagen coating) on the assembly ridges. Further experiments will investigate alternative ECM coatings to influence cell adhesion and long-term development of the organoid structure and function.
In terms of mimicking the liver sinusoid in all its complexity, there is still a long way to go.20,21In vivo, there are additional cell types present when compared to current test systems including HepaChip-MP. Cell functionality, protein expression and physical dimensions as well as the microenvironment differ from model systems.20,21 Some in vitro models include non-parenchymal cells and thereby achieve promising results in terms of improved long term culture with hepatocyte specific functionality, disease modelling and drug response.2,17,30 However, these models do not reproduce the dimensions and physiologic microenvironment of the liver sinusoid.
Therefore, the cell culture chamber of the HepaChip-MP was designed to enable the cultivation of hepatocytes along with non-parenchymal cells in elongated cell aggregates with physical dimensions of the liver sinusoid (see Results section and ref. 52). Also, cells are in direct contact to the perfused medium as no hydrogel or membrane is required for the assembly of cells. In this study we also demonstrate seeding and coculture of HuLEC and PHH in the HepaChip-MP (Fig. 7). In future experiments, additional non-parenchymal cell types will be included in order to reach at a relevant cellular complexity as reported previously,2,17,30 yet cultured in cell aggregates mimicking the physical dimensions of the liver sinusoid more closely.
Migration of organoids to the periphery
The observation of migration of cells on the outer assembly ridges towards the periphery suggests that cells may be sensitive to even minute, yet continuous hydrodynamic forces which they experience due to the design of the cell culture chamber (see Fig. S4†). This finding will inform future designs of flow chambers.Metabolic activity
So far, PHH cultured in the HepaChip-MP were shown to exhibit xenobiotic metabolism over at least six days (Table 2). This is an important feature in model systems for toxicity testing and drug development since many substances are detoxified or activated by CYP enzymes of PHH. Induction of CYP2C9 and CYP3A4 in the HepaChip-MP (Fig. 5) indicates physiological enzyme regulation, which is also an important feature of a liver model.8,61 Future experiments will analyze the stability of CYP activity and induction over time. Other authors demonstrated the possibility to measure CYP activity of PHH in vitro co-cultured with non-parenchymal cells over 14 to 90 days.2,27,62–64 It was also shown that the coculture of PHH with primary Kupffer cells does not influence the CYP activity.65 Consequently, the coculture of PHH with HuLEC and stellate cells in HepaChip-MP is expected to enhance CYP activity even in future long term cultures.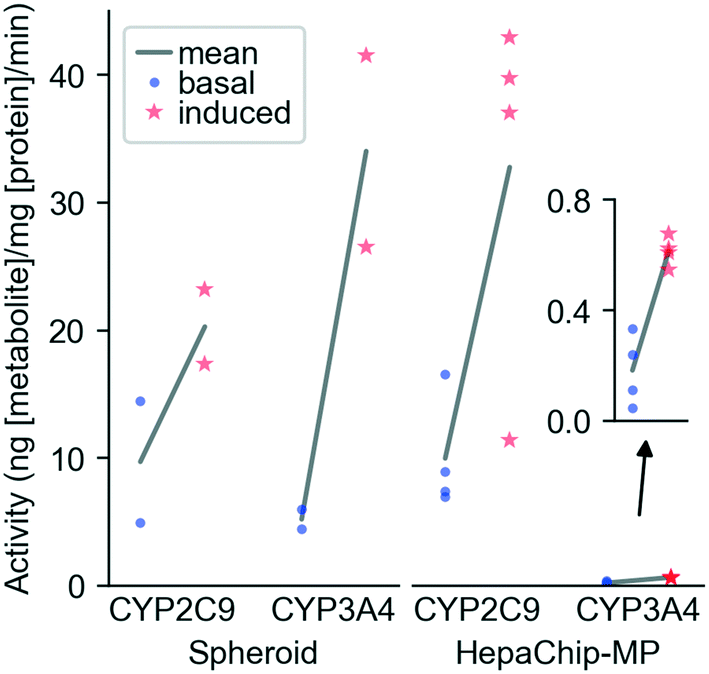 | ||
| Fig. 5 CYP induction in spheroids and HepaChip-MP. Primary human hepatocytes (PHH) (donor BHuf 16068) were cultured in vitro and CYP activity was induced with 20 μM rifampicin from day 2 to day 5 (HepaChip-MP) or day 6 to day 9 (3D spheroid cultures). Basal and induced CYP activities were determined after 5 h of substrate incubation with spheroids and HepaChip-MP cultures. Data represent the metabolite formation normalized to protein content. For the spheroids, each incubation condition included four spheroids of which two were pooled for metabolite and protein analysis as technical replicates. In case of the HepaChip-MP cultures, samples from four to five cell culture chambers (technical replicates) were analyzed for each condition. Means of values obtained from basal and induced activity of cells were analyzed by an unpaired t-test for each enzyme and in vitro system. P-Values are the following. CYP2C9 spheroid: 0.1985; CYP3A4 spheroid: 0.0618; CYP2C9 HepaChip-MP: 0.0236; CYP3A4 HepaChip-MP: 0.0008. | ||
Our experiments of CYP induction demonstrate that the HepaChip-MP allows quantification of physiological liver cell function and enzyme activity despite the relatively low cell count per chamber. This enables the efficient use of primary cells which are costly and have limited availability. Analysis of cell behavior using the HepaChip-MP nevertheless requires sufficiently sensitive assays.
When compared to 3D spheroids, HepaChip-MP cultures showed similar basal CYP2C9 activity. In contrast to a report by Foster et al. that basal CYP2C9 activity in spheroids was more than 20 times higher than in a microfluidic liver model with hepatocytes and non-parenchymal cells.66 To this end, the potential influence of non-parenchymal cells on metabolic activity remains to be evaluated in the HepaChip-MP as well as in our spheroid model. We observed basal CYP3A4 activity approximately 25 times higher in spheroids than in HepaChip-MP cultures. Foster et al. observed 60-times-higher CYP3A4 activity in spheroids compared to a microfluidic liver model.66 These findings stand in contrast to results of Vinci et al. who found an upregulation of CYP expression as well as CYP activity of CYP2C9, CYP3A4, and other CYP enzymes under flow conditions.67 However, Vinci et al. only evaluated the influence of medium flow on the cells. They used exactly the same culture conditions (seeding, plates, model system) whereas Foster et al. and the present study investigated two different model systems (3D spheroid and microfluidic chip). In spheroids and microfluidic liver models, cell assembly and microenvironment around the cells also differ, which may explain the differences between the cited reports.
Assessment of cell vitality after drug treatment
Feasibility of measuring cell vitality after drug treatment using HepaChip-MP was demonstrated with a resazurin (alamarBlue) assay (Fig. 6). Using cells from a single donor yielded a dose–response curve of cell vitality after treatment with diclofenac. While low vitality at high concentrations (1500 μM) was observed, cells showed an increased vitality at intermediate concentrations (150 and 450 μM).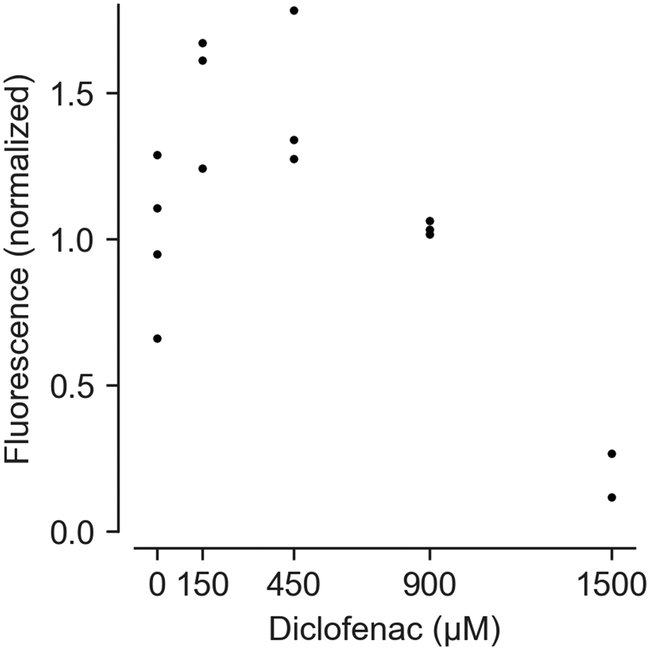 | ||
| Fig. 6 Assessment of dose-dependent effects of diclofenac on PHH vitality. Cells from a male donor (63 years, cholangiocellular carcinoma) were cultivated in the HepaChip-MP. Fluorescence values from the resazurin assay were normalized vs. protein mass and the control without diclofenac. Three to four chambers were analyzed for 0–900 μM diclofenac; two chambers were analyzed for 1500 μM diclofenac. Individual data points of technical replicates are shown. Test for statistical significance by ANOVA showed statistical significant differences between the different conditions (p-value = 0.0005). Post hoc comparisons using Tukey's showed statistical significant differences between 0 and 1500 μM (p-value = 0.0118), 150 and 1500 μM (p-value = 0.0005), 450 and 1500 μM (p-value = 0.0006) as well as 900 and 1500 μM diclofenac (p-value = 0.0123). | ||
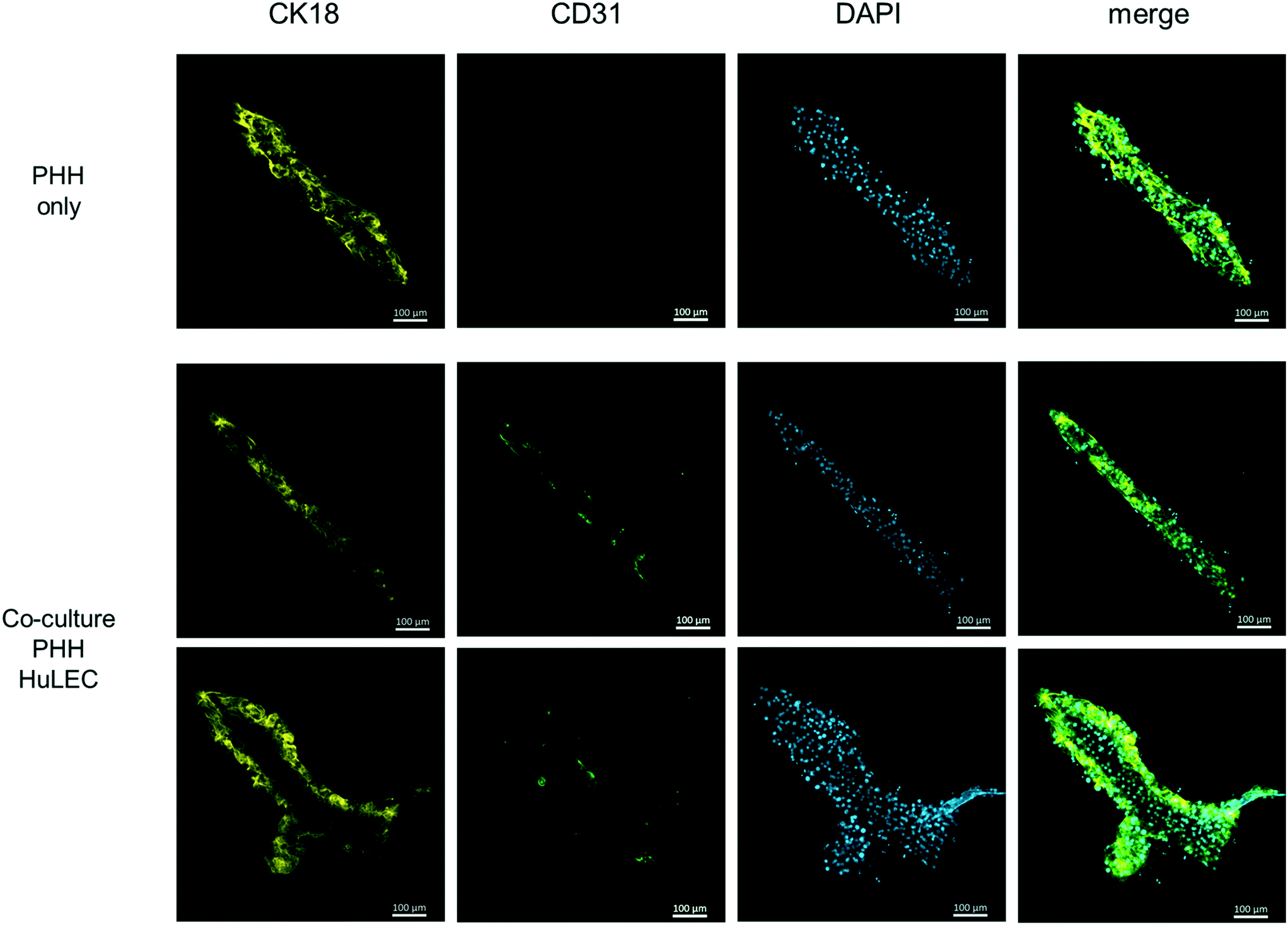 | ||
| Fig. 7 Fluorescence micrographs of a monoculture of cryopreserved PHH (first row) and cocultures with cryopreserved HuLEC (second and third rows) at day 3 of culture. CK18 indicates PHH (yellow) and CD31 labels endothelial cells (green). DAPI staining shows cell nuclei. The monoculture used medium 2. The coculture used a 1:1 mixture of medium 2 and EGM2. These images of single assembly ridges are representative of at least three chambers for each condition. Scale bar: 100 μm. | ||
At high concentrations, diclofenac induces oxidative stress leading to cell damage and death.68,69 However, non-monotonic dose–response curves in resazurin assays can result from changed NADPH production rates. Cells attempt to counteract oxidative stress with reduced glutathione (GSH), the regeneration of which requires NADPH.70 NADPH produced to regenerate GSH also reduces resazurine. This can lead to apparent vitality above 100% as seen with 150 and 450 μM diclofenac.
While this interference is undesirable, an advantage of the resazurin assay is that it may be carried out at multiple time points during culturing. Such results can be complemented by end-point viability assays.
Manufacturing and availability of organ-on-chip systems
Adoption of organ-on-chip systems requires scalable fabrication and the availability of devices for validation of results. Microinjection molding enables mass production of devices with consistent properties, in comparison with methods based on soft lithography or photolithography. Use of COP achieves good optical properties and low substance adsorption, as shown by a panel of hydrophobic drugs (Fig. S6†).Future research will investigate the predictivity of the HepaChip-MP with comprehensive tests of substances with known effects. Also, we will attempt to establish and measure the oxygen gradient inside the culture chambers and assess a zonation24–26,37–39 of protein expression. Further, the influence of endothelial or stellate cells on hepatocyte function will be analyzed in cocultures. Finally, the optimization of medium composition to attain long-term stable cultures and the establishment of disease models will be undertaken.
Conclusions
We have designed, fabricated and evaluated a microfluidic model of the liver in a parallelized and scalable format. Towards industrial relevance, we focused on injection molding of COP microfluidics in the ANSI/SBS microplate format. The formation of functional structures resembling liver sinusoids demonstrated the successful active assembly of cocultures in 3D culture chambers. Evaluation of physiological function confirmed both the vitality and specific enzyme activity of sinusoidal structures as well as the compatibility of this microfluidic model with established cell-based assays. The HepaChip-MP has significant potential as an in vitro liver model, and furthermore presents a new technological approach to create organ-on-a-chip models of additional organ systems.Author contributions
M. S., H. B., R. G. conceived the concept of HepaChip-MP. H. B., J. S. developed chip fabrication technology. B. H. performed multiphysics simulation and fluidic design. S. W. contributed to chip design and periphery instrumentation. M. P. contributed to the multiwellplate design of the chip. M. Bu., O. T., J. S., M. Be. conducted cell culture experiments with Hepachip-MP. S. B., T. K. performed LC/MS analysis of samples. K. G., R. H. developed the automated pipetting system. A. U., J. M., and D. R. developed and evaluated a spheroid model and conducted benchmarking experiments and contributed to discussion and manuscript writing. G. D., M. M. provided human hepatocytes and non-parenchymal cells. M. S., R. G., C. S., N. G., K. S. guided research. M. S., M. Bu, P. J., C. S. wrote the manuscript.Conflicts of interest
H. B. is CSO of microfluidic ChipShop GmbH, which developed HepaChip-MP jointly with NMI and currently manufactures these devices. M. S., J. S., B. H., and S. W. are inventors in patents covering HepaChip-MP technology.Acknowledgements
We thank Prof. Dr. med Daniel Seehofer and his team of the Department of Hepatobiliary Surgery and Visceral Transplantation, Leipzig University for liver tissue samples. Funding for this research provided (in part) by the German ministry for education and research (BMBF) through grants no. 01GG0729, 031A121, 031B0481 is gratefully acknowledged.References
- M. D. Leise, J. J. Poterucha and J. A. Talwalkar, Drug-induced liver injury, Mayo Clin. Proc., 2014, 89(1), 95–106 CrossRef CAS PubMed.
- R. Kostadinova, et al., A long-term three dimensional liver co-culture system for improved prediction of clinically relevant drug-induced hepatotoxicity, Toxicol. Appl. Pharmacol., 2013, 268(1), 1–16 CrossRef CAS PubMed.
- M. Chen, J. Borlak and W. Tong, High lipophilicity and high daily dose of oral medications are associated with significant risk for drug-induced liver injury, Hepatology, 2013, 58(1), 388–396 CrossRef CAS PubMed.
- N. Kaplowitz, Avoiding idiosyncratic DILI: Two is better than one, Hepatology, 2013, 58(1), 15–17 CrossRef PubMed.
- S. Babai, L. Auclert and H. Le-Louët, Safety data and withdrawal of hepatotoxic drugs, Therapie, 2018 DOI:10.1016/j.therap.2018.02.004 , in press.
- L. Kuna, et al., Models of Drug Induced Liver Injury (DILI) - Current Issues and Future Perspectives, Curr. Drug Metab., 2018, 19(10), 830–838 CrossRef CAS PubMed.
- V. Y. Soldatow, et al., In vitro models for liver toxicity testing, Toxicol. Res., 2013, 2(1), 23–39 CrossRef CAS PubMed.
- A. R. Baudy, et al., Liver microphysiological systems development guidelines for safety risk assessment in the pharmaceutical industry, Lab Chip, 2020, 20(2), 215–225 RSC.
- K. Tetsuka, M. Ohbuchi and K. Tabata, Recent Progress in Hepatocyte Culture Models and Their Application to the Assessment of Drug Metabolism, Transport, and Toxicity in Drug Discovery: The Value of Tissue Engineering for the Successful Development of a Microphysiological System, J. Pharm. Sci., 2017, 106(9), 2302–2311 CrossRef CAS PubMed.
- A. Poloznikov, et al., In vitro and in silico liver models: Current trends, challenges and opportunities, ALTEX, 2018, 35(3), 397–412 CrossRef PubMed.
- S. R. Khetani and S. N. Bhatia, Microscale culture of human liver cells for drug development, Nat. Biotechnol., 2008, 26(1), 120–126 CrossRef CAS PubMed.
- J.-B. Ferrini, et al., Long-term primary cultures of adult human hepatocytes, Chem.-Biol. Interact., 1997, 107, 31–45 CrossRef CAS PubMed.
- A. Ullrich, et al., Long term cultures of primary human hepatocytes as an alternative to drug testing in animals, ALTEX, 2009, 26(4/09), 295–302 CrossRef PubMed.
- D. R. Berger, et al., Enhancing the functional maturity of induced pluripotent stem cell-derived human hepatocytes by controlled presentation of cell-cell interactions in vitro, Hepatology, 2015, 61(4), 1370–1381 CrossRef CAS PubMed.
- S. March, et al., Micropatterned coculture of primary human hepatocytes and supportive cells for the study of hepatotropic pathogens, Nat. Protoc., 2015, 10(12), 2027–2053 CrossRef CAS PubMed.
- M. Zhou, et al., Long-term maintenance of human fetal hepatocytes and prolonged susceptibility to HBV infection by co-culture with non-parenchymal cells, J. Virol. Methods, 2014, 195, 185–193 CrossRef CAS PubMed.
- C. C. Bell, et al., Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease, Sci. Rep., 2016, 6, 25187 CrossRef CAS PubMed.
- Y. Edling, et al., Increased sensitivity for troglitazone-induced cytotoxicity using a human in vitro co-culture model, Toxicol. In Vitro, 2009, 23(7), 1387–1395 CrossRef CAS PubMed.
- W. R. Proctor, et al., Utility of spherical human liver microtissues for prediction of clinical drug-induced liver injury, Arch. Toxicol., 2017, 91(8), 2849–2863 CrossRef CAS PubMed.
- V. M. Lauschke, et al., Novel 3D Culture Systems for Studies of Human Liver Function and Assessments of the Hepatotoxicity of Drugs and Drug Candidates, Chem. Res. Toxicol., 2016, 29(12), 1936–1955 Search PubMed.
- J. Deng, et al., Engineered Liver-on-a-Chip Platform to Mimic Liver Functions and Its Biomedical Applications: A Review, Micromachines, 2019, 10(10), 676 Search PubMed.
- J. H. Lee, K. L. Ho and S. K. Fan, Liver microsystems in vitro for drug response, J. Biomed. Sci., 2019, 26(1), 88 CrossRef PubMed.
- A. Dash, et al., Hemodynamic flow improves rat hepatocyte morphology, function, and metabolic activity in vitro, Am. J. Physiol., Cell Physiol., 2013, 304(11), C1053–C1063 CrossRef CAS PubMed.
- F. T. Lee-Montiel, et al., Control of oxygen tension recapitulates zone-specific functions in human liver microphysiology systems, Exp. Biol. Med., 2017, 242(16), 1617–1632 CrossRef CAS PubMed.
- J. W. Allen and S. N. Bhatia, Formation of steady-state oxygen gradients in vitro: application to liver zonation, Biotechnol. Bioeng., 2003, 82(3), 253–262 CrossRef CAS PubMed.
- J. Hellkamp, et al., Modulation by oxygen of the glucagon-dependent activation of the phosphoenolpyruvate carboxykinase gene in rat hepatocyte cultures, Eur. J. Biochem., 1991, 198, 635–639 CrossRef CAS PubMed.
- L. Prodanov, et al., Long-term maintenance of a microfluidic 3D human liver sinusoid, Biotechnol. Bioeng., 2016, 113(1), 241–246 CrossRef CAS PubMed.
- Y. B. Kang, et al., Liver sinusoid on a chip: Long-term layered co-culture of primary rat hepatocytes and endothelial cells in microfluidic platforms, Biotechnol. Bioeng., 2015, 112(12), 2571–2582 CrossRef CAS PubMed.
- M. Yamada, et al., Controlled formation of heterotypic hepatic micro-organoids in anisotropic hydrogel microfibers for long-term preservation of liver-specific functions, Biomaterials, 2012, 33(33), 8304–8315 CrossRef CAS PubMed.
- L. Vernetti, et al., Functional Coupling of Human Microphysiology Systems: Intestine, Liver, Kidney Proximal Tubule, Blood-Brain Barrier and Skeletal Muscle, Sci. Rep., 2017, 7, 42296 CrossRef CAS PubMed.
- R. Baudoin, et al., Evaluation of a liver microfluidic biochip to predict in vivo clearances of seven drugs in rats, J. Pharm. Sci., 2014, 103(2), 706–718 CrossRef CAS PubMed.
- J. M. Prot, et al., A cocktail of metabolic probes demonstrates the relevance of primary human hepatocyte cultures in a microfluidic biochip for pharmaceutical drug screening, Int. J. Pharm., 2011, 408(1–2), 67–75 CrossRef CAS PubMed.
- C. Ma, et al., On-Chip Construction of Liver Lobule-like Microtissue and Its Application for Adverse Drug Reaction Assay, Anal. Chem., 2016, 88(3), 1719–1727 CrossRef CAS PubMed.
- E. Novik, et al., A microfluidic hepatic coculture platform for cell-based drug metabolism studies, Biochem. Pharmacol., 2010, 79(7), 1036–1044 CrossRef CAS PubMed.
- K. J. Jang, et al., Redproducing human and cross-species drug toxicities using a Liver-Chip, Sci. Transl. Med., 2019, 11, eaax5516 CrossRef CAS PubMed.
- R. Gebhardt and D. Mecke, Permissive Effect of Dexamethasone on Glucagon Induction of urea-cycle enzymes in perifused, Eur. J. Biochem., 1978, 97, 6 Search PubMed.
- P. P. C. Poyck, et al., Expression of Glutamine Synthetase and Carbamoylphosphate Synthetase I in a Bioartificial Liver: Markers for the Development of Zonation in vitro, Cells Tissues Organs, 2008, 188(3), 259–269 CrossRef CAS PubMed.
- W. J. McCarty, O. B. Usta and M. L. Yarmush, A Microfabricated Platform for Generating Physiologically-Relevant Hepatocyte Zonation, Sci. Rep., 2016, 6, 26868 CrossRef CAS PubMed.
- Y. B. A. Kang, et al., Metabolic Patterning on a Chip: Towards in vitro Liver Zonation of Primary Rat and Human Hepatocytes, Sci. Rep., 2018, 8(1), 8951 CrossRef PubMed.
- A. Junaid, et al., An end-user perspective on Organ-on-a-Chip: Assays and usability aspects, Curr. Opin. Biomed. Eng., 2017, 1, 15–22 CrossRef.
- J. Y. Kim, et al., 96-well format-based microfluidic platform for parallel interconnection of multiple multicellular spheroids, J. Lab. Autom., 2015, 20(3), 274–282 CrossRef CAS PubMed.
- M. Jang, et al., Differentiation of the human liver progenitor cell line (HepaRG) on a microfluidic-based biochip, J. Tissue Eng. Regener. Med., 2019, 13(3), 482–494 CrossRef CAS PubMed.
- K. Domansky, et al., Perfused multiwell plate for 3D liver tissue engineering, Lab Chip, 2010, 10(1), 51–58 RSC.
- N. Li, M. Schwartz and C. Ionescu-Zanetti, PDMS compound adsorption in context, J. Biomol. Screening, 2009, 14(2), 9 Search PubMed.
- M. W. Toepke and D. J. Beebe, PDMS absorption of small molecules and consequences in microfluidic applications, Lab Chip, 2006, 6(12), 1484–1486 RSC.
- B. J. van Meer, et al., Small molecule absorption by PDMS in the context of drug response bioassays, Biochem. Biophys. Res. Commun., 2017, 482(2), 323–328 CrossRef CAS PubMed.
- J. Schütte, et al., A method for patterned in situ biofunctionalization in injection-molded microfluidic devices, Lab Chip, 2010, 10, 2551–2558 RSC.
- F. Holzner, et al., Numerical modelling and measurement of cell trajectories in 3-D under the influence of dielectrophoretic and hydrodynamic forces, Electrophoresis, 2011, 32(17), 2366–2376 CrossRef CAS PubMed.
- B. Hagmeyer, et al., Tailoring microfluidic systems for organ-like cell culture applications using multiphysics simulations, Proc. SPIE, 2013, 8615 DOI:10.1117/12.2002475.
- V. Kegel, et al., Protocol for Isolation of Primary Human Hepatocytes and Corresponding Major Populations of Non-parenchymal Liver Cells, J. Visualized Exp., 2016, 109, e53069 Search PubMed.
- S. J. Fey and K. Wrzesinski, Determination of drug toxicity using 3D spheroids constructed from an immortal human hepatocyte cell line, Toxicol. Sci., 2012, 127(2), 403–411 CrossRef CAS PubMed.
- H. F. Teutsch, The modular microarchitecture of human liver, Hepatology, 2005, 42(2), 317–325 CrossRef PubMed.
- B. Hagmeyer, F. Zechnall and M. Stelzle, Towards plug and play filling of microfluidic devices by utilizing networks of capillary stop valves, Biomicrofluidics, 2014, 8(5), 056501 CrossRef CAS PubMed.
- C. J. Ochs, et al., Oxygen levels in thermoplastic microfluidic devices during cell culture, Lab Chip, 2014, 14(3), 459–462 RSC.
- A. P. Schmeisch, et al., Zonation of the metabolic action of vasopressin in the bivascularly perfused rat liver, Regul. Pept., 2005, 129(1), 233–243 CrossRef CAS PubMed.
- R. E. McClelland, J. M. MacDonald and R. N. Coger, Modeling O2 transport within engineered hepatic devices, Biotechnol. Bioeng., 2003, 82(1), 12–27 CrossRef CAS PubMed.
- R. D. Guarino, et al., Method for determining oxygen consumption rates of static cultures from microplate measurements of pericellular dissolved oxygen concentration, Biotechnol. Bioeng., 2004, 86(7), 775–787 CrossRef CAS PubMed.
- J. Poisson, et al., Liver sinusoidal endothelial cells: Physiology and role in liver diseases, J. Hepatol., 2017, 66(1), 212–227 CrossRef CAS PubMed.
- E. Pfeiffer, et al., Featured Article: Isolation, characterization, and cultivation of human hepatocytes and non-parenchymal liver cells, Exp. Biol. Med., 2015, 240(5), 645–656 CrossRef CAS PubMed.
- I. Maschmeyer, et al., A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents, Lab Chip, 2015, 15(12), 2688–2699 RSC.
- K. M. Olsavsky Goyak, E. M. Laurenzana and C. J. Omiecinski, Hepatocyte differentiation, Methods Mol. Biol., 2010, 640, 115–138 CrossRef PubMed.
- K. Zeilinger, et al., Three-dimensional Co-culture of primary human liver cells in bioreactors for in vitro drug studies: Effects of the initial cell quality on the long-term maintenance of hepatocyte-specific functions, Altern. Lab. Anim., 2002, 30, 525–538 CrossRef CAS PubMed.
- K. Zeilinger, et al., Scaling down of a clinical three-dimensional perfusion multicompartment hollow fiber liver bioreactor developed for extracorporeal liver support to an analytical scale device useful for hepatic pharmacological in vitro studies, Tissue Eng., Part C, 2011, 17(5), 549–556 CrossRef CAS PubMed.
- D. G. Nguyen, et al., Bioprinted 3D Primary Liver Tissues Allow Assessment of Organ-Level Response to Clinical Drug Induced Toxicity In Vitro, PLoS One, 2016, 11(7), e0158674 CrossRef PubMed.
- T. V. Nguyen, et al., Establishment of a hepatocyte-kupffer cell coculture model for assessment of proinflammatory cytokine effects on metabolizing enzymes and drug transporters, Drug Metab. Dispos., 2015, 43(5), 774–785 CrossRef CAS PubMed.
- A. J. Foster, et al., Integrated in vitro models for hepatic safety and metabolism: evaluation of a human Liver-Chip and liver spheroid, Arch. Toxicol., 2019, 93(4), 1021–1037 CrossRef CAS PubMed.
- B. Vinci, et al., Modular bioreactor for primary human hepatocyte culture: medium flow stimulates expression and activity of detoxification genes, Biotechnol. J., 2011, 6(5), 554–564 CrossRef CAS PubMed.
- C. Haritha, et al., Oxidative Stress Induced by Diclofenac Alone and under the Influence of Certain Variables in Broilers, Toxicol. Int., 2010, 17(1), 27–29 CrossRef CAS PubMed.
- H. Li, et al., Cyclooxygenase 2-selective and nonselective nonsteroidal anti-inflammatory drugs induce oxidative stress by up-regulating vascular NADPH oxidases, J. Pharmacol. Exp. Ther., 2008, 326(3), 745–753 CrossRef CAS PubMed.
- J. Pizzorno, Glutathione!, Integr. Med., 2014, 13(1), 8–12 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc00357c |
| This journal is © The Royal Society of Chemistry 2020 |