
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel nickel nanoparticles stabilized by imidazolium-amidinate ligands for selective hydrogenation of alkynes†
Angela M.
López-Vinasco
 a,
Luis M.
Martínez-Prieto
*ab,
Juan M.
Asensio
a,
Pierre
Lecante
c,
Bruno
Chaudret
a,
Juan
Cámpora
d and
Piet W. N. M.
van Leeuwen
*a
a,
Luis M.
Martínez-Prieto
*ab,
Juan M.
Asensio
a,
Pierre
Lecante
c,
Bruno
Chaudret
a,
Juan
Cámpora
d and
Piet W. N. M.
van Leeuwen
*a
aLPCNO; Laboratoire de Physique et Chimie des Nano-Objets, UMR5215 INSA-CNRS-UPS, Institut National des Sciences Appliquées-Toulouse, 135, Avenue de Rangueil, F-31077 Toulouse, France. E-mail: vanleeuw@insa-toulouse.fr
bITQ, Instituto de Tecnología Química, CSIC-Universitat Politécnica de València, Avda. Los Naranjos S/N, 46022, Valencia, Spain. E-mail: luismiguel.martinez@csic.es
cCEMES (Centre d'Elaboration de Matériaux et d'Etudes Structurales), CNRS, 29 Rue J. Marvig, F-31055 Toulouse, France
dIIQ, Instituto de Investigaciones Químicas, CSIC-Universidad de Sevilla, C/Américo Vespucio, 49, 41092 Sevilla, Spain
First published on 4th December 2019
Abstract
The main challenge in the hydrogenation of alkynes into (E)- or (Z)-alkenes is to control the selective formation of the alkene, avoiding the over-reduction to the corresponding alkane. In addition, the preparation of recoverable and reusable catalysts is of high interest. In this work, we report novel nickel nanoparticles (Ni NPs) stabilized by three different imidazolium-amidinate ligands (ICy·(Ar)NCN; L1: Ar = p-tol, L2: Ar = p-anisyl and L3: Ar = p-ClC6H4). The as-prepared Ni NPs were fully characterized by (HR)-TEM, XRD, WASX, XPS and VSM. The nanocatalysts are active in the hydrogenation of various substrates. They present a remarkable selectivity in the hydrogenation of alkynes towards (Z)-alkenes, particularly in the hydrogenation of 3-hexyne into (Z)-3-hexene under mild reaction conditions (room temperature, 3% mol Ni and 1 bar H2). The catalytic behaviour of Ni NPs was influenced by the electron donor/acceptor groups (–Me, –OMe, –Cl) in the N-aryl substituents of the amidinate moiety of the ligands. Due to the magnetic character of the Ni NPs, recycling experiments were successfully performed after decantation in the presence of an external magnet, which allowed us to recover and reuse these catalysts at least 3 times preserving both activity and chemoselectivity.
Introduction
The selective hydrogenation of alkynes into (E)- or (Z)-alkenes is an important synthesis method in fine chemistry and the polymerization industry.1 The classical catalysts for this transformation are based on passivated, supported Pd catalysts.2 However, they often involve the use of toxic lead salts as a catalyst inhibitor. The most widely used one industrially is Lindlar's catalyst (Pd/CaCO3 poisoned with Pb(OAc)2).3 This catalyst has shown good selectivity in the semi-hydrogenation of simple internal alkynes, but it exhibits some drawbacks such as poor selectivity in the semi-hydrogenation of monosubstituted alkynes or the partial isomerization of (Z)-alkenes to (E)-alkenes for the disubstituted ones.1c,3c Therefore, the development of environmentally friendly, chemoselective and recyclable catalysts based on earth-abundant and low-cost metals such as nickel is highly desirable. In this respect, metallic nanoparticles (MNPs) are good alternatives,4 because they present the advantages of heterogeneous and homogeneous catalysts such as good activity and selectivity, as well as the possibility to separate and reuse the catalyst at the end of the reaction.5 Many recent NP catalysts for semi-hydrogenation reactions are based on the noble metal Pd.6 Nevertheless, well-defined nickel nanoparticles (Ni NPs) have been also successfully applied to the selective hydrogenation of alkynes into alkenes.7 In addition, Ni NPs have shown better catalytic activity than conventional heterogeneous catalysts because they have higher active surfaces areas. Ni NPs also present magnetic properties, which could be exploited to recover them from the reaction medium after precipitation in the presence of an external magnet. Indeed, magnetic recovery has emerged as a robust, efficient and fast catalyst separation technique.8 Thus, the intrinsic magnetic properties of Ni NPs, which facilitate their recycling, together with their potential as catalysts in hydrogenation reactions, makes them good candidates for catalytic semi-hydrogenation reactions in an environmentally friendly way.It has been already shown in the literature that the catalytic properties of MNPs can be tuned by the use of polymeric stabilizers, surface active agents, solid supports, but notably by ligands, which in turn also provide stability and solubility to the MNPs.9 Imidazolium-amidinate ligands demonstrated to be a good stabilizer for Ru NPs10 and Pt NPs,11 enabling the synthesis of very small nanoparticles but until now they have never been used on nickel NPs. Due to the zwitterionic structure of the imidazolium-amidinate ligands, the nitrogen atoms exhibit a large electron-donor capability and coordinate strongly to transition metals without adding electric charges to the system. Furthermore, a large influence of the substituents of the ligands on catalytic activity of the Pt NPs in the hydrogenation of activated ketones was found.11 Here, we describe novel Ni NPs stabilized by three different imidazolium-amidinate ligands, ICy–(Ar)NCN (L1: Ar = p-tol, L2: Ar = p-anisyl and L3: Ar = p-ClC6H4). The three systems were fully characterized by multiple techniques ((HR)TEM, XRD, WAXS, AAS, XPS and VSM). In this contribution, we explore the three catalytic systems containing modified ligands in various hydrogenation reactions, paying special attention to the semi-hydrogenation of 3-hexyne, and we investigate the recyclability of such systems by utilizing their magnetic properties.
Results and discussion
Synthesis and characterization of Ni@L nanoparticles
Ni NPs were prepared from the Ni precursor bis(1,5-cyclooctadiene)nickel(0), [Ni(COD)2], which was decomposed in THF at 70 °C under 3 bar H2 in the presence of 0.2 equivalents of the corresponding ligand. Three different imidazolium-amidinate compounds were employed as stabilizing ligands, L1 = p-tol, L2 = p-anisyl and L3 = p-ClC6H4, leading to Ni@L1, Ni@L2 and Ni@L3 nanoparticles, respectively (Fig. 1, top). TEM micrographs of Ni@L1 and Ni@L2 revealed the formation of spherical Ni NPs of similar sizes (ca. 2.8 nm), independently of the electron donor/acceptor character of the N-aryl substituent in the amidinate ligands (Fig. 1a and b). However, the electron-poor system for Ni@L3 was composed by slightly bigger (3.4 (2.0) nm) and considerably more polydisperse NPs (Fig. 1c and S1 of ESI†). High-resolution TEM (HRTEM) images of Ni@L1 showed the presence of crystalline fcc-Ni NPs (face centred cubic), which is characteristic of metallic nickel (Fig. S2 of ESI†). Also, Fourier analysis applied to this image showed reflections of the (11−1), (0−11) and (0−11) atomic planes.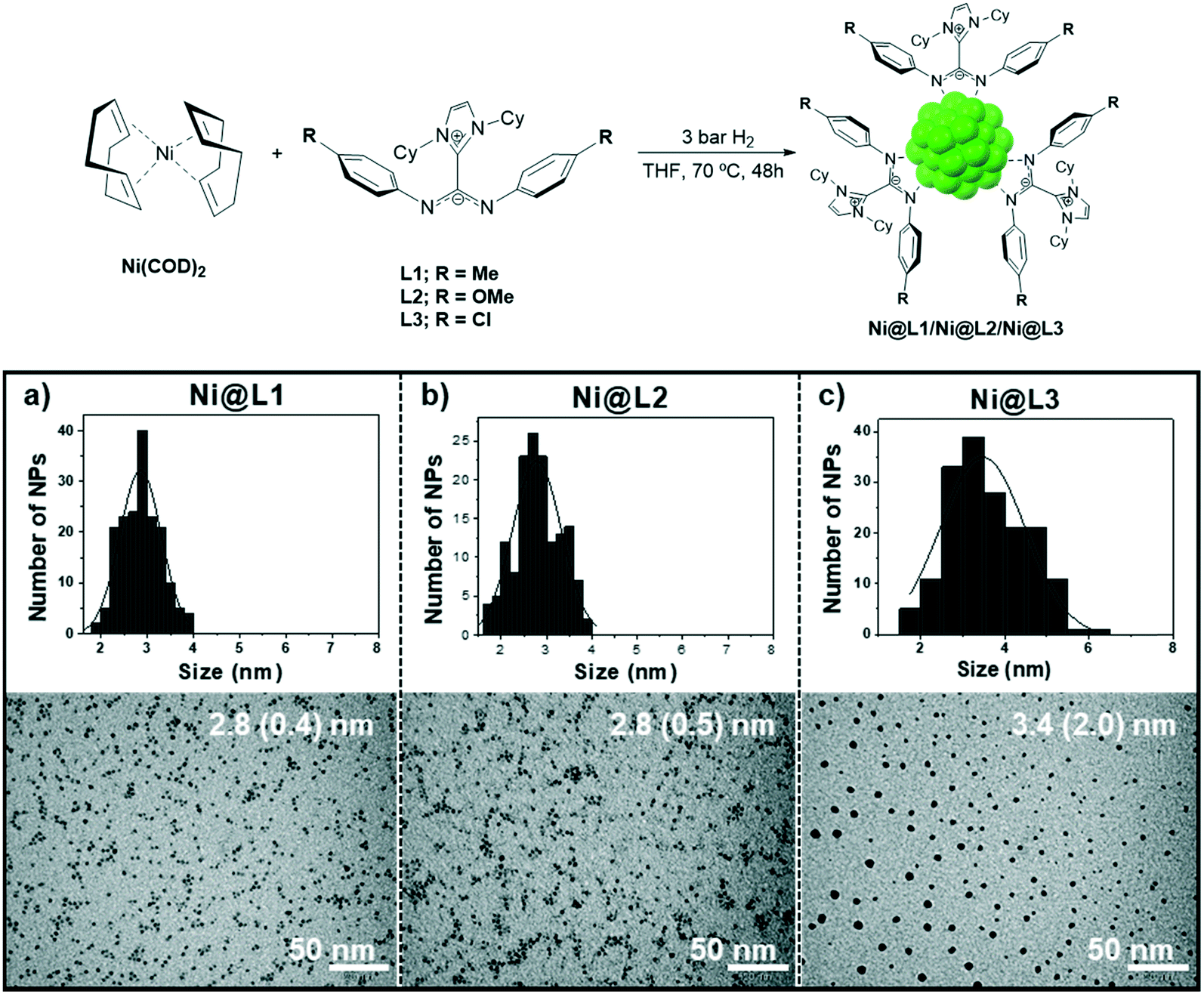 | ||
| Fig. 1 Top: Synthesis of the nickel nanoparticles stabilized by ICy·(Ar)NCN (Ni@L1, Ni@L2 and Ni@L3). Bottom: TEM images and size distribution histograms of (a) Ni@L1, (b) Ni@L2 and (c) Ni@L3. | ||
X-ray powder diffraction (XRD) of Ni@L1, Ni@L2 and Ni@L3 also revealed that the nanoparticles are well crystallized according to the fcc metal structure. In addition, very small peaks at 2θ values of ca. 42°, a characteristic of NiO, were observed in all cases (Fig. 2). As the Ni NPs were handled under argon and the XRD analyses were performed under inert atmosphere, it must be assumed that the oxide is formed during the synthesis of the NPs in THF. Wide-angle X-ray scattering (WAXS) analyses of the Ni@L NPs (Fig. S3 of ESI†) revealed mostly metallic nanoparticles with a compact structure (fcc) and a coherence length of ca. 3 nm, confirming the crystalline structure and size observed by XRD and HRTEM. Again, some signs of oxidation for the three nickel systems were observed. Atomic absorption spectroscopy (AAS) of the Ni@L NPs gave metal contents of ca. 75.9, 71.2 and 45.5 wt% for Ni@L1, Ni@L2 and Ni@L3 respectively. For Ni@L1 and Ni@L2, the Ni/L ratio is large enough to allow the coordination of all ICy·NCN molecules, since the estimated number of Ni surface atoms [Ni(s)] is up to 7–9 times larger than the calculated number of ligands per particle (Table S1 of ESI†). However, in the case of Ni@L3, the Ni(s)/L ratio has a value of 2.2. As each ligand coordinates to two Ni atoms due to steric reasons, it is not possible to accommodate all ligands onto the Ni NPs surface. Therefore, as was previously observed for Ru and Pt NPs ligated with the same amidinate ligands, the excess of L3 may be organized in a second sphere of non-coordinated ICy·(p-ClC6H4)NCN, probably bonded by π–π stacking and ionic interactions of the zwitterions.
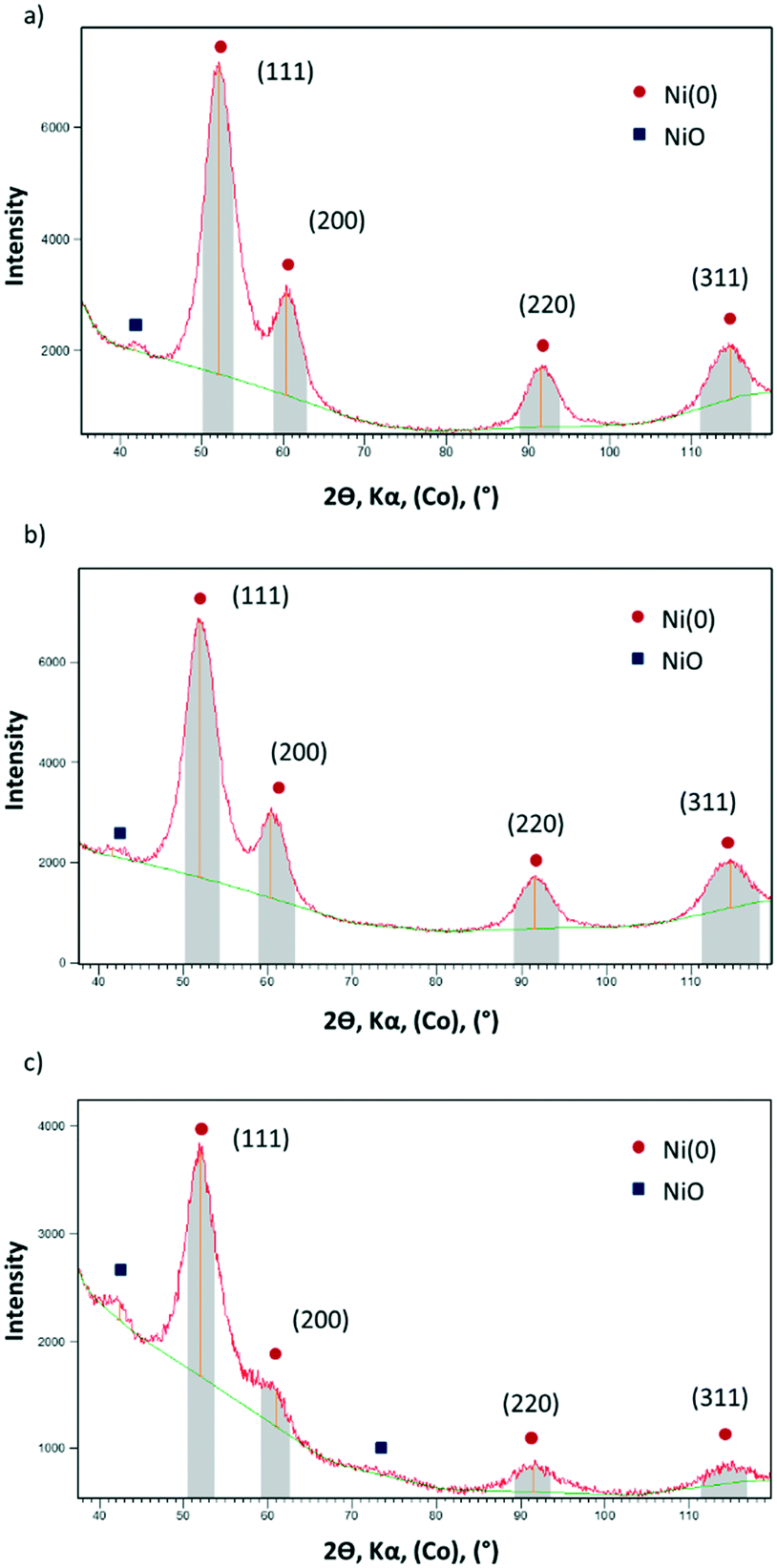 | ||
| Fig. 2 XRD diffractograms of (a) Ni@L1 (b) Ni@L2 and (c) Ni@L3. The peaks labelled in red correspond to fcc Ni(0) and the peaks labelled in blue correspond to NiO. | ||
The chemical state and coordination mode of the imidazolium-amidinate ligands (L1, L2 and L3) to the Ni surface were further investigated by X-ray photoelectron spectroscopy (XPS). The N 1s signals of the ligands present two binding energies (BEs) at ∼401.5 and ∼397.5 eV (Fig. 3a, blue). The first peak can be assigned to the nitrogen atoms of the imidazolium fragment, which bear a partial positive charge. The second one at lower BE corresponds to the N atoms of the amidinate fragment with a partial negative charge. The coordination of ligand L1 through the amidinate moiety is reflected in the loss of electron density on these N atoms, as their BE increases from 397.5 to 399.9 eV. The overlapping of this signal with the one corresponding to the imidazolium N atoms, gave a new broad peak centred at 399.9 eV (Fig. 3a, red). The latter could be deconvoluted in three contributions at 401.3, 399.8 and 398.5 eV, which correspond to the δ−, neutral and δ+ N atoms, respectively (Fig. 3b). The N 1s signal of Ni@L2 presents a peak similar to that in Ni@L1, but here the contribution corresponding to Nδ− (398.7 eV) increases at the expense of the Nδ+ contribution (401.3 eV), due to the presence of an electron donor group in the ligand (–OMe). On the other hand, in the N 1s signal of Ni@L3, which contains the electron-withdrawing chloro substituent, the δ+ is the most important contribution (401.4 eV). This can be explained by the presence of the second sphere of non-coordinated ligands, as proposed above.
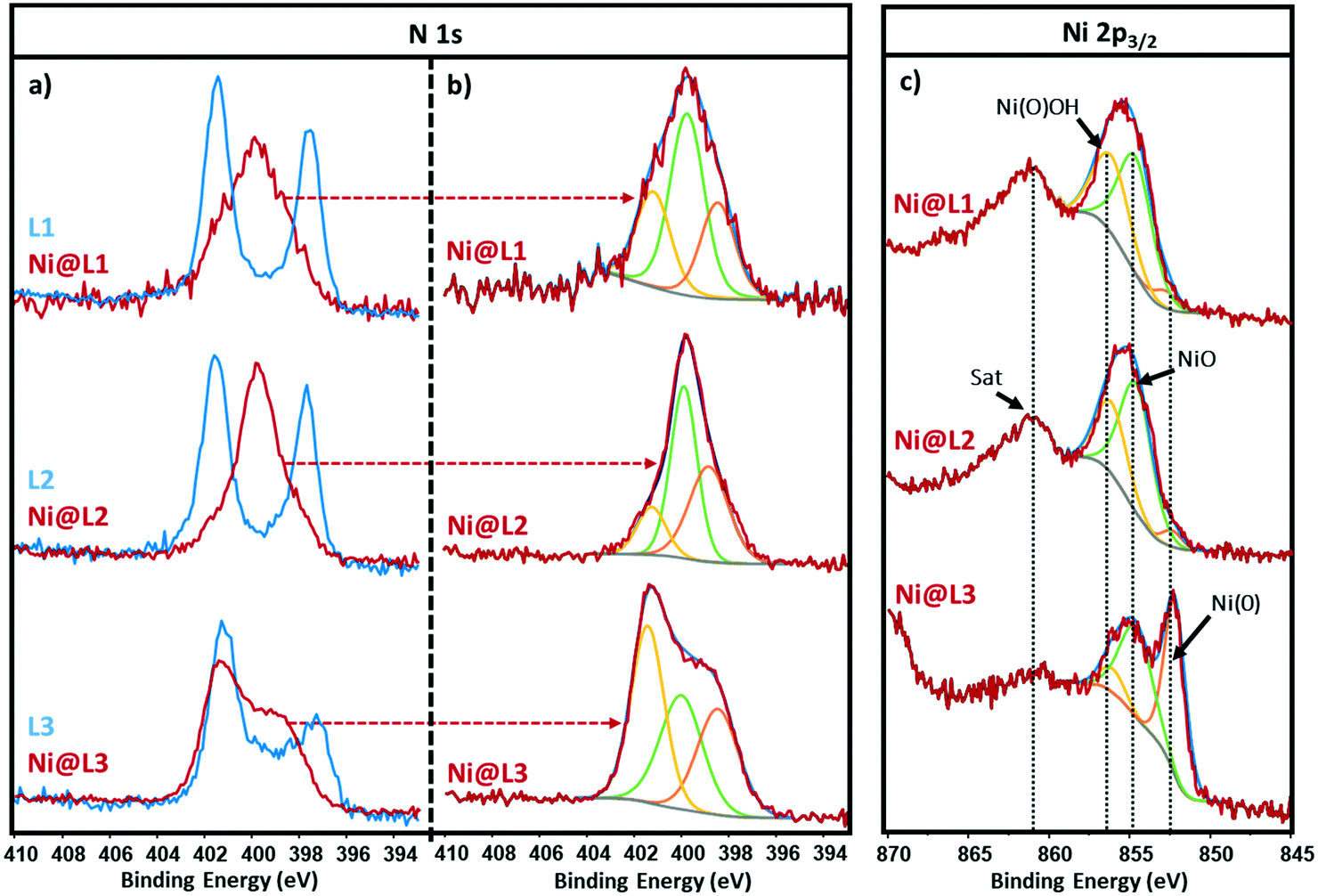 | ||
| Fig. 3 XPS spectra of Ni@L1, Ni@L2 and Ni@L3 (experimental spectra plotted in red line). Left inset, N 1s signals; (a) comparison with the spectra of the free ligands L1, L2 and L3 (blue lines); (b) same spectra, showing their deconvolution in three components: δ+ (yellow), δ0 (green), and δ− (orange). Right inset, (c) The corresponding Ni 2p3/2 signals, showing the deconvolution of the main signal in three components (yellow, green and orange), assigned to Ni(III) oxyhydroxide species [Ni(O)OH], Ni(II) oxide (NiO) and elemental Ni [Ni(0)]. | ||
The Ni 2p3/2 spectra of Ni@L1 and Ni@L2 showed a broad signal centred at ca. 855.4 eV accompanied by a satellite peak (Sat) at 861.4 eV, also known as shake-up satellite.12 The peak at 855.4 eV could be deconvoluted in three contributions at ∼852.6, ∼854.6 and ∼856.1 eV that correspond to Ni(0), nickel oxide (NiO) and nickel oxyhydroxide [Ni(O)OH], respectively (Fig. 3c, top and centre).13 As the main contributions are the ones corresponding to NiO and nickel oxyhydroxide species,12c we can assume that practically all the surface atoms of Ni@L1 and Ni@L2 were oxidized during the XPS analyses as the samples were exposed to air. Interestingly, the surface of Ni@L3 was only partially oxidized during the XPS analysis, presenting a clear peak at 852.6 eV that belongs to Ni(0) (Fig. 3c, bottom). We ascribe these differences to the higher surface coverage of Ni@L3 and the lower electron density of the Ni surface atoms of these Ni NPs because of the comparatively lower electron-donor capacity of L3.
The as-prepared Ni NPs were further characterized by vibrating sample magnetometry (VSM). Magnetization versus applied field curves evidenced a superparamagnetic behaviour at room temperature and an absence of saturation, with magnetization (MS) values at 3 T of 28, 23 and 35 A2 m2 kg−1 for Ni@L1, Ni@L2 and Ni@L3 (Fig. 4a), respectively. At low temperature (5 K), after cooling down in the presence of μ0H of 3 T (field-cooling), the three samples were close to saturation at 3 T with magnetization values of 44, 38 and 44 A2 m2 kg−1 for Ni@L1, Ni@L2 and Ni@L3, respectively (Fig. 4a). In all three cases, the MS values were below that of bulk Ni (55 A m2 kg−1).14 This can result from the surface/magnetic-core ratio which increases for small Ni NPs, increasing the amount of nonmagnetic layers at their surface.15 A further electronic role of the ligand acting as a π-acceptor cannot be discarded.16 Furthermore, the coercive field (HC) values of 10, 5 and 20 mT were observed for Ni@L1, Ni@L2 and Ni@L3, respectively (Fig. 4b). The small discrepancies observed between the three samples can result from the polydispersity of the NPs, since larger HC values are found in the most polydisperse system, Ni@L3, which contains the larger NPs. The absence in the hysteresis loops of any exchange bias, characteristic of the coupling between ferromagnetic and antiferromagnetic layers,17 demonstrates the absence of significant oxidation of the particles and suggests that the presence of NiO that was observed in the XRD and WAXS analyses should be almost negligible.
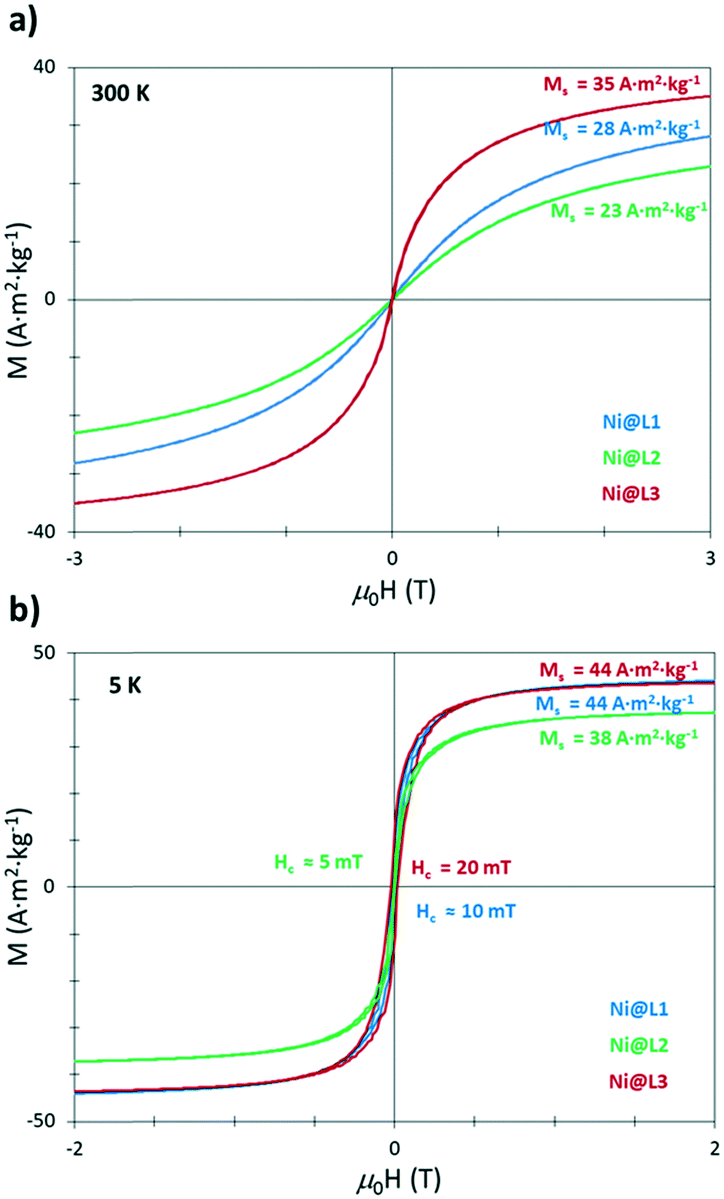 | ||
| Fig. 4 Magnetization against applied field curves for the Ni@L NPs at (a) room temperature (300 K) and (b) 5 K. | ||
Catalytic evaluation
In order to learn about the catalytic properties of the novel Ni NPs, Ni@L1 was tested in the hydrogenation of various substrates containing different functional groups including alkenes, alkynes and carbonyl groups. The catalytic results show that the nickel systems are capable of hydrogenating double and triple bonds, but the carbonyl groups were hardly affected. Table 1 presents the scope of substrates in hydrogenation reactions. The best results were obtained in the hydrogenation of 3-hexyne (1) and diphenylacetylene (2) at 1 bar H2 and room temperature for 5 hours. The selective hydrogenation of 1 and 2 gave a high selectivity towards the (Z)-alkene, with some traces of (E)-alkene and the corresponding alkane (Table 1, entries 1 and 2). The conversion values for 1 and 2 were of 92% (selec. 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:3) and 47% (selec. 92:8:0), respectively, showing that the steric effects between substrate and nanoparticles influence the catalytic activity of Ni@L1. Substrates 3–5 that contain double bonds and carbonyl groups are hydrogenated with a high preference for the double bond (selec. 92%) and they show a low conversion of the carbonyl group (Table 1, entries 3–5). Substrate 6 (furfural) reached a 17% conversion to the corresponding alcohol (2-furfuryl alcohol) with no hydrogenation of the conjugated aromatic ring (Table 1, entry 6). The conversion of 7 was of 10% containing an electron-withdrawing CF3 group, whereas acetophenone (8) conversion was negligible.
:4:3) and 47% (selec. 92:8:0), respectively, showing that the steric effects between substrate and nanoparticles influence the catalytic activity of Ni@L1. Substrates 3–5 that contain double bonds and carbonyl groups are hydrogenated with a high preference for the double bond (selec. 92%) and they show a low conversion of the carbonyl group (Table 1, entries 3–5). Substrate 6 (furfural) reached a 17% conversion to the corresponding alcohol (2-furfuryl alcohol) with no hydrogenation of the conjugated aromatic ring (Table 1, entry 6). The conversion of 7 was of 10% containing an electron-withdrawing CF3 group, whereas acetophenone (8) conversion was negligible.
| Entry | Substrate | Product | Conv.b (%) select. (%) |
|---|---|---|---|
| a Reaction conditions: substrate (0.5 mmol), Ni@L1 (0.015 mmol Ni assuming % of Ni from AAS. 3 mol% Ni loading), toluene (0.75 mL), H2 (40 bar), overnight and r.t. b Conversions and selectivities were determined by GC/MS (average of two runs). c H2 (1 bar), 5 h and r.t. | |||
| 1 |
 1
1
|
 1a
1a
|
>92%, 1a:1b:1c = 93:4:3 |
 1b
1b
|
|||
 1c
1c
|
|||
| 2 |
 2
2
|
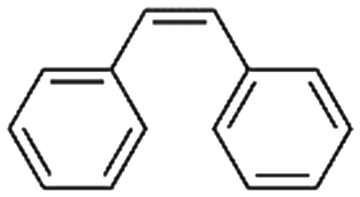 2a
2a
|
47%, 2a:2b:2c = 92:8:0 |
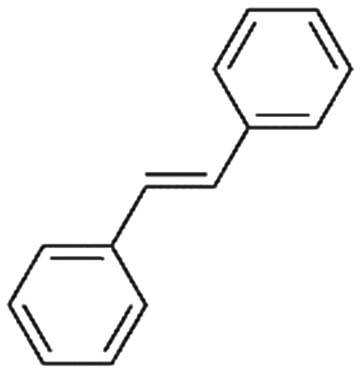 2b
2b
|
|||
 2c
2c
|
|||
| 3 |
 3
3
|
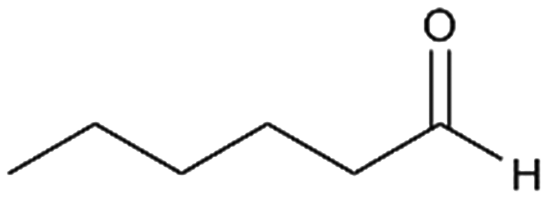 3a
3a
|
>99%, 3a:3b = 94:6 |
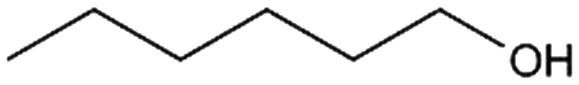 3b
3b
|
|||
| 4 |
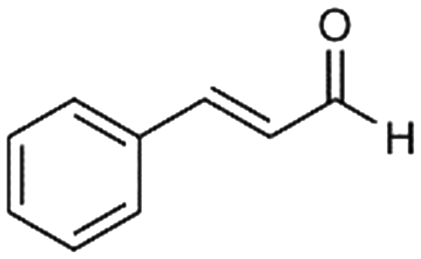 4
4
|
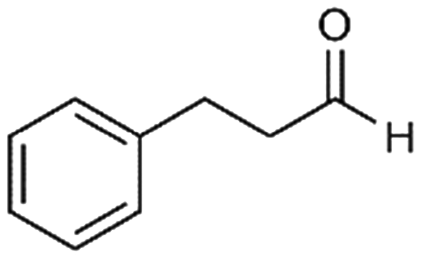 4a
4a
|
66%, 4a:4b = 97:3 |
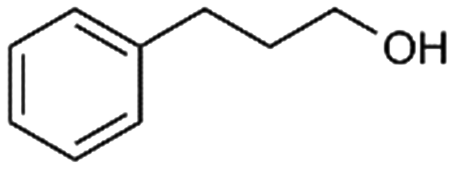 4b
4b
|
|||
| 5 |
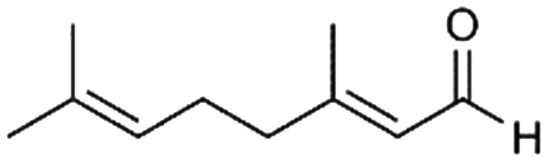 5
5
|
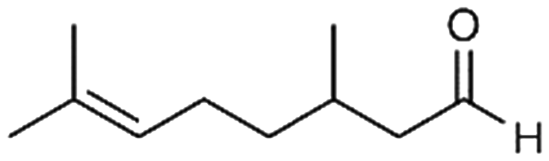 5a
5a
|
>99%, 5a:5b = 92:8 |
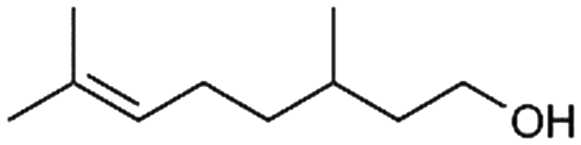 5b
5b
|
|||
| 6 |
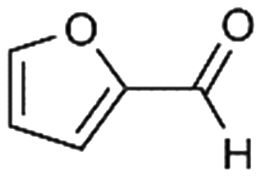 6
6
|
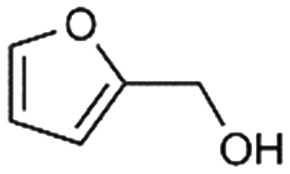 6a
6a
|
17% |
| 7 |
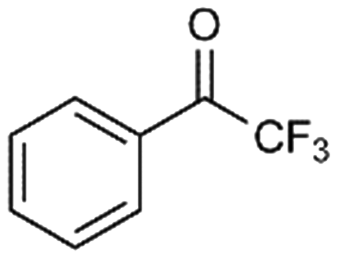 7
7
|
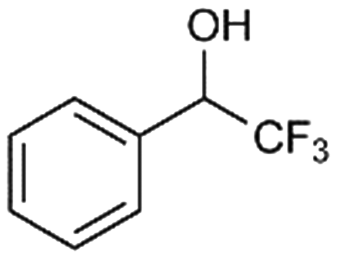 7a
7a
|
10% |
| 8 |
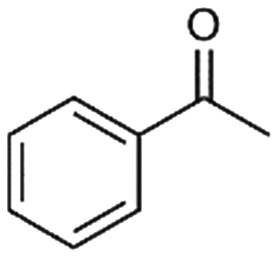 8
8
|
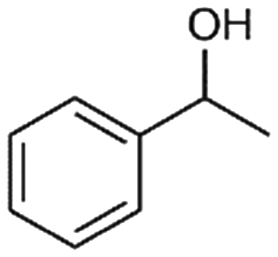 8a
8a
|
2% |
Encouraged by the result obtained with 3-hexyne (Table 1, entry 1), we decided to test the as-prepared Ni NPs in the selective hydrogenation of alkynes into alkenes. Only a few examples have been reported up to date that employ colloidal Ni NPs for the selective semi-hydrogenation of alkynes using molecular hydrogen.7 In order to investigate the influence of the electron donor/acceptor group of each ligand (R = –Me, –OMe, –Cl) on their activity and selectivity of the Ni NPs herein prepared, we chose the semi-hydrogenation of 3-hexyne as a model reaction. Table 2 summarizes the results obtained in the semi-hydrogenation reaction of 3-hexyne catalysed by Ni@L1, Ni@L2 and Ni@L3. Toluene was chosen as a reaction solvent because of the good dispersibility of the NPs in it. Interestingly, although the NPs were slightly oxidized, they still showed good and comparable activities at long reaction times (8 h, entries 10–12). At short reaction times (2 h, entries 1–3), we observed an effect of the ligand on the conversion; the stronger the electron donor on the N-aryl group, the more active is the Ni@L catalyst. After 2 h of reaction, a conversion of 45% was obtained with the catalyst that bears the –OMe group (Ni@L2), whereas the conversion for Ni@L1 and Ni@L3 were 32 and 27%, respectively. The same trend could be observed for reaction times of 5 and 6 hours (entries 4–9), which also showed a lower activity for the NPs that contain the electron withdrawing Cl-substituent (Ni@L3). A similar catalytic behaviour was previously observed with Pt NPs ligated by the same ligands in the hydrogenation of ketones.11 After 8 h, the conversion was complete for all three catalytic systems.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ni@L | Time | Conv.b (%) | Selectivityb (%) | ||
| 1a | 1b | 1c | ||||
| a Reaction conditions: 3-hexyne (0.5 mmol), Ni@L (0.015 mmol Ni assuming % of Ni from AAS. 3 mol% Ni loading), toluene (0.75 mL), H2 (1 bar), and r.t. b Conversions and selectivities were determined by 1H NMR (average of two runs). | ||||||
| 1 | Ni@L1 | 2 | 32 | 92 | 7 | 1 |
| 2 | Ni@L2 | 2 | 45 | 88 | 8 | 3 |
| 3 | Ni@L3 | 2 | 27 | 83 | 9 | 8 |
| 4 | Ni@L1 | 5 | 92 | 93 | 4 | 3 |
| 5 | Ni@L2 | 5 | 97 | 91 | 5 | 4 |
| 6 | Ni@L3 | 5 | 73 | 83 | 12 | 5 |
| 7 | Ni@L1 | 6 | 98 | 82 | 11 | 7 |
| 8 | Ni@L2 | 6 | 98 | 86 | 8 | 6 |
| 9 | Ni@L3 | 6 | 89 | 89 | 7 | 4 |
| 10 | Ni@L1 | 8 | >99 | 78 | 11 | 11 |
| 11 | Ni@L2 | 8 | >99 | 83 | 9 | 8 |
| 12 | Ni@L3 | 8 | >99 | 90 | 6 | 4 |
Fig. 5 shows the evolution of the products in the semi-hydrogenation of 3-hexyne catalysed by Ni@L1 with respect to time. The formation of the (Z)-alkene runs parallel with the consumption of the alkyne until a maximum is reached after 5 hours for Ni@L1 and Ni@L2, and 8 h for Ni@L3 (Fig. 5 and S4†). When comparing the 3-hexyne consumption slopes during the initial stages of the reactions (between 0–5 hours), it can be seen that Ni@L2 is the most active catalyst (slope: 19.25 mol s−1 Fig. S4†), followed by Ni@L1 (slope: 18.42 mol s−1) and Ni@L3 (slope: 14.86 mol s−1 Fig. S4†). Two combined phenomena, an effect of the ligand and polydispersity, may explain these differences. In terms of selectivity, when comparing the three systems at the end of the reaction, Ni@L3 is slightly more selective towards the formation of (Z)-3-hexene, which might be due to the lower activity. Though somewhat less active, Ni@L3 exhibits a remarkable chemo- and stereocontrol, achieving a 90% selectivity in the semi-hydrogenation to (Z)-3-hexene at full conversion of the alkyne.
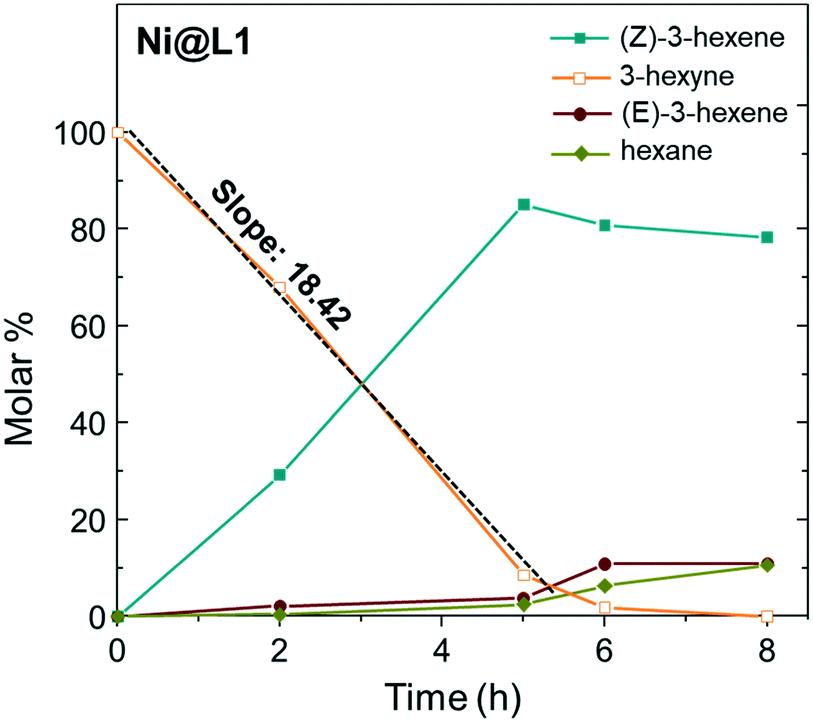 | ||
| Fig. 5 Time course of the product yield in the semi-hydrogenation of 3-hexyne using Ni@L1 as the catalyst. Reaction conditions: 0.5 mmol of 3-hexyne, 3 mmol% catalyst, 0.75 mL toluene, and 1 bar H2. | ||
The catalytic activity of the Ni@L systems herein prepared is higher than those of the Ni-based systems previously reported in the literature.7a,b,d For example, Godard and co-workers prepared Ni NPs stabilized by NHC ligands that were supported on multi-walled carbon nanotubes, which were used in the hydrogenation of internal alkynes under 5 bar H2 and at 50 °C.7b The catalytic systems prepared in the present work are three times faster under milder reaction conditions (1 bar H2 and room temperature), likely due to the non-supported nature of the catalysts. Similarly, recoverable magnetic Fe3O4@Ni nanoparticles have been employed in the selective hydrogenation of terminal alkynes to alkenes under 7 bar H2 and at room temperature, but higher catalyst loadings (7%) and longer reaction times (48 h) were required in that case to reach an 80% conversion.7g
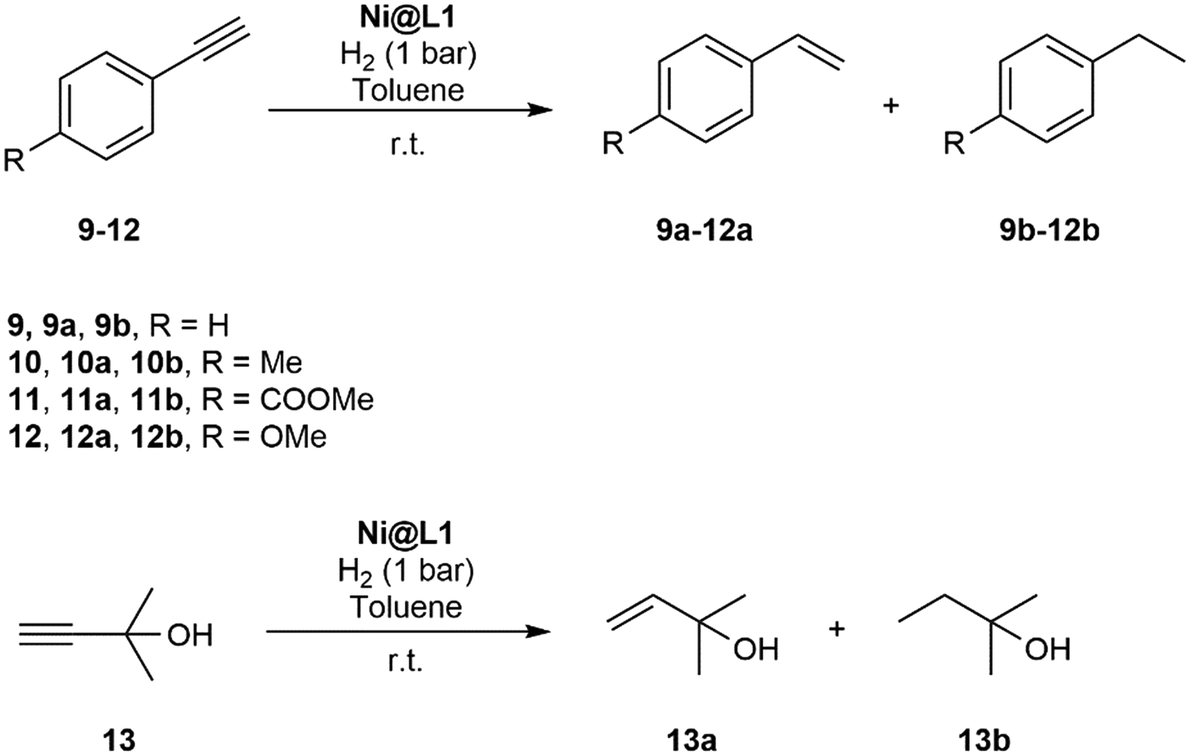
The hydrogenation of terminal alkynes catalysed by Ni@L1 under mild conditions (1 bar H2 and r.t.) gave a good selectivity to the corresponding alkenes showing mixtures of a/b ca. 90/10 (Table 3), except for 13 which showed a mixture of 39/61. In addition, there was no full conversion of the substrates after 8 h and only for 13 the conversion reached 96%. Comparing these results with the hydrogenation of internal alkynes 1 and 2, a 92% conversion was observed for 1 (Table 1, entry 1) and 47% for 2 (Table 1, entry 2), which indicates that steric factors affect both selectivity and conversion. In general, the hydrogenation of the substrates 2 and 9–12 (containing aromatic rings) is slow compared to 1 and 13 (the aliphatic ones), but the hydrogenation of aryl terminal alkynes 9–12 is faster than diarylalkyne 2. This can also be attributed to the steric factors caused by hindered coordination to the nanoparticle surface. To evaluate the robustness of the Ni NPs prepared in this work, recycling experiments were performed with Ni@L1 (Fig. 6). Taking advantage of the magnetic character of Ni@L, the nanoparticles were effortlessly recovered from the reaction medium after precipitation in the presence of an external magnet (adhesive force: 4 kg). Fig. 6 shows the recycling results during five catalytic runs (1 bar H2; 5 h). Both the conversion and selectivity towards (Z)-3-hexene were practically maintained during the first 3 runs, but after the 4th run the activity of Ni@L1 considerably decreased. This effect can be attributed to an agglomeration of the nanoparticles after 3 catalytic cycles, as evidenced by TEM (Fig. S5 and S6 of ESI†), but oxidation of the NPs during the recycling cycles cannot be discarded.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Conv.b (%) | Selectivityb (%) | |
| a | b | |||
| a Reaction conditions: substrate (0.5 mmol), Ni@L (0.015 mmol Ni assuming % of Ni from AAS. 3 mol% Ni loading), toluene (0.75 mL), H2 (1 bar), 8 h and r.t. b Conversions and selectivities were determined by GC/MS (average of two runs). | ||||
| 1 | 9 | 63 | 89 | 11 |
| 2 | 10 | 86 | 89 | 11 |
| 3 | 11 | 85 | 91 | 9 |
| 4 | 12 | 60 | 90 | 10 |
| 5 | 13 | 96 | 39 | 61 |
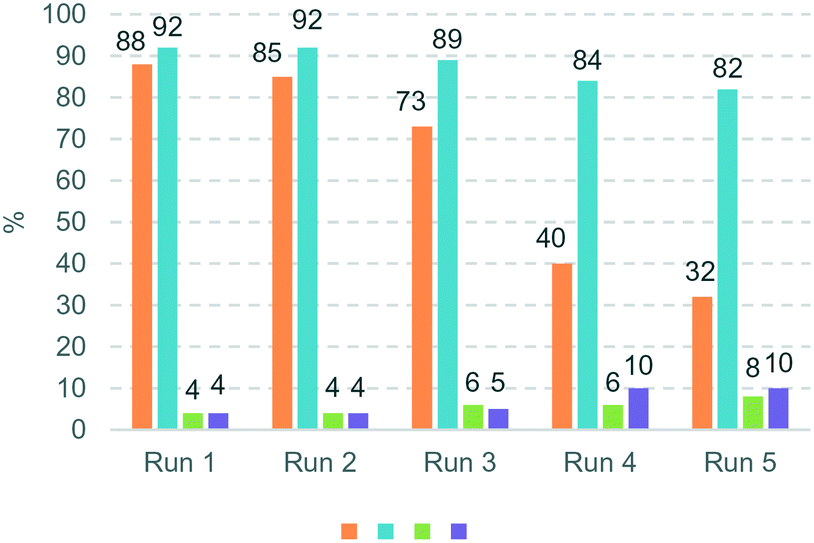 | ||
| Fig. 6 Recycling experiments using Ni@L1 as the catalyst. Reaction conditions: 0.5 mmol of 3-hexyne, 3 mmol% catalyst, 0.75 mL toluene, 1 bar H2, and 5 h. Conversions and selectivities were determined by 1H NMR (average of two runs). | ||
Experimental
All manipulations were carried out using standard Schlenk tubes, Fisher–Porter bottle techniques or in a glove-box under argon atmosphere. The organic solvents were purified before their use by a solvent purification system (MBraun). The commercial products were obtained from commercial providers and used as received. [Ni(COD)2] and 3-hexyne were bought from Sigma Aldrich. Zwitterionic imidazolium-amidinate ligands ICy·(Ar)NCN (L1, L2 and L3) were synthesized according to the literature.11 The AAS analyses of Ni@L1, Ni@L2 and Ni@L3 were performed by Kolbe (Mülheim, Germany), using a Perkin Elmer Analyst 200 atomic absorption spectrometer. The samples were prepared with an acid digestion procedure. Ni@L were analysed by TEM after the deposition of a drop of a colloidal dispersion in THF on a copper grid covered with a carbon layer. The TEM pictures were taken at the UMS-Castaing by using a JEOL JEM 1011 CX-T electron microscope (100 kV; point resolution of 4.5 Å). The particles mean size was obtained through a manual analysis of the magnified micrographs by measuring at least 200 nanoparticles on a given grid. XRD measurements were performed on a PANalytical Empyrean diffractometer [Co-Kα radiation (λ = 0.1789 nm); 45 kV; 40 mA]. The XRD samples were prepared and sealed under argon atmosphere. Magnetic studies were executed on a vibrating sample magnetometer (VSM, Quantum Device PPMS Evercool II). X-Ray photoelectron spectroscopy (XPS) analyses were carried out at CIRIMAT Laboratory (Toulouse) with the help of a Thermoelectron K-Alpha machine. The photoelectron emission spectra were obtained using a monochromatized source of Al-Kα radiation (hν = 1486.6 eV). Analyzed area = 0.15 mm2. Pass energy = 40 eV. The spectrometer was calibrated using the Au 4f7/2 (83.9 ± 0.1 eV) and Cu2p3/2 (932.8 ± 0.1 eV) photoelectron lines. XPS spectra were recorded in direct mode N(Ec). The XPS spectra were analysed with CasaXPS processing software.Synthesis of the Ni NPs
[Ni(COD)2] (150 mg, 0.55 mmol) and 30 mL of dried and deoxygenated THF, were placed in a 180 mL Fisher–Porter bottle. The resulting solution was cooled at −40 °C and a THF solution of 30 mL containing 0.2 equiv. (0.11 mmol) of the appropriate ligand was added (L1, L2 or L3). The Fisher–Porter was pressurized with 3 bar H2, and when the solution reached room temperature it was stirred at 70 °C during 48 h, leading to a black suspension. After that period, the H2 pressure was released and the suspension was concentrated to 2–3 mL under reduced pressure. Next, 50 mL of pentane were added resulting in a black precipitate, which was washed with pentane (25 mL × 2) and dried overnight under dynamic vacuum. NP diameters determined by TEM, Ni@L1: 2.8 (0.4) nm, Ni@L2: 2.8 (0.5) nm, Ni@L3: 3.4 (2.0) nm. Ni content determined by AAS, Ni@L1: 75.9%, Ni@L2: 71.1%, Ni@L3: 45.5%.Catalytic hydrogenation reactions
A HEL 24-multireactor (Vvial = 1.5 mL) was used to perform the catalytic experiments. In a typical experiment, Ni@L (0.015 mmol of Ni) in 0.75 mL of toluene (from a standard solution previously prepared) was added to a vial with 0.5 mmol of substrate. The reactor was then sealed under argon, purged with H2 three times, and finally pressurized with 1 bar of hydrogen. Then, the reaction mixture was stirred using a magnetic stir bar at room temperature during the time indicated. After that, the samples from each reaction were analysed by 1H NMR and/or GC-MS. More specifically, after the catalytic reactions, Ni NPs were easily separated from the organic solution with the help of an external magnet. For the NMR analyses, the samples were evaporated, and redissolved in CDCl3. For the GC-MS analysis, the samples were directly injected in the equipment. For the recycling experiments, the NPs were decanted with the help of a magnet, the supernatant was removed, and the NPs were washed three times with toluene (2 mL). Then, the substrate and the solvent were introduced into the HEL 24-multireactor and the reaction was loaded with H2.Conclusions
Novel Ni NPs were obtained by reaction of [Ni(COD)2] with zwitterionic imidazolium-amidinate ligands. Specifically, three amidinate ligands with different electronic properties were employed as stabilizers (L1, L2 and L3). The Ni NPs ligated with the weakest electron donor ligand (L3) presumably have a densely covered surface that protects them towards oxidation, as has been observed by XPS, probably thanks to the electron-withdrawing character of the ligand. The three different catalytic systems herein prepared were active in the hydrogenation of various substrates and especially in the semi-hydrogenation of 3-hexyne to (Z)-3-hexene under very mild reaction conditions. An influence on the activity and selectivity was observed depending on the ligand used as a stabilizer. Ni@L2 stabilized with a strongest donor ligand (R = –OMe) was the most active catalyst. The slowest catalyst was the system containing an electron-withdrawing group Ni@L3 (R = –Cl). The higher surface coverage can induce a higher selectivity in this system, as Ni@L3 showed the best selectivity towards the (Z)-3-hexene formation at full conversions. It should be highlighted that ligands do not only play a role in the stabilization of the NPs, but also in the capacity to modulate the activity and selectivity of the NPs in catalysis. The magnetic character of Ni@L permits the recovery of the Ni NPs from the reaction medium after the catalysis by an external magnet. The NPs were recyclable up to 3 times maintaining good activity values. This fact, together with the good selectivity of these catalytic systems without the need of toxic salts as poison, makes them potentially useful catalysts for semi-hydrogenation reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank CNRS, UPS-Toulouse, INSA, “IDEX/Chaires d'attractivité l'Université Fédérale Toulouse Midi-Pyrénées”, “Instituto de Tecnología Química” (ITQ; UPV-CSIC), “Juan de la Cierva” programme (IJCI-2016-27966), “Primero Proyectos de Investigación” (PAID-06-18), “Instituto de Investigaciones Químicas” (IIQ; CSIC-US), “Ministerio de Ciencia, Innovación y Universidades” (MCIU/AEI), FEDER funds of the European Union (PGC2018-095768-B-I00) and ERC Advanced Grant (MONACAT 2015-694159) for financial support. We also thank L. Datas for the TEM facilities (UMS Castaing) and S. Cayez for the HRTEM measurements.Notes and references
- (a) K. C. K. Swamy, A. S. Reddy, K. Sandeep and A. Kalyani, Tetrahedron Lett., 2018, 59, 419 CrossRef CAS; (b) J. Lei, L. Su, K. Zeng, T. Chen, R. Qiu, Y. Zhou, C.-T. Au and S.-F. Yin, Chem. Eng. Sci., 2017, 171, 404 CrossRef CAS; (c) C. Oger, L. Balas, T. Durand and J.-M. Galano, Chem. Rev., 2013, 113, 1313–1350 CrossRef CAS PubMed; (d) J. G. de Vries and C. J. Elsevier, The Handbook of Homogeneous Hydrogenation, Wiley-VCH, 2008 Search PubMed.
- (a) D. Albani, M. Shahrokhi, Z. Chen, S. Mitchell, R. Hauert, N. López and J. Pérez-Ramírez, Nat. Commun., 2018, 9, 2634 CrossRef PubMed; (b) R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783 CrossRef CAS PubMed; (c) N. López and C. Vargas-Fuentes, Chem. Commun., 2012, 48, 1379 RSC.
- (a) M. Crespo-Quesada, F. Cárdenas-Lizana, A.-L. Dessimoz and L. Kiwi-Minsker, ACS Catal., 2012, 2, 1773 CrossRef CAS; (b) A. Molnar, A. Sarkany and M. Varga, J. Mol. Catal. A: Chem., 2001, 173, 185 CrossRef CAS; (c) H. Lindlar, Helv. Chim. Acta, 1952, 35, 446 CrossRef CAS.
- J. A. Delgado, O. Benkirane, C. Claver, D. Curulla-Ferre and C. Godard, Dalton Trans., 2017, 46, 12381 RSC.
- (a) K. Philippot and P. Serp, Nanomaterials in Catalysis, Wiley-VCH, Weinheim, 2013 Search PubMed; (b) D. Astruc, Nanoparticles and Catalysis, Wiley-VCH, Weinheim, 2008 Search PubMed; (c) U. Heiz and U. Landman, Nanocatalysis, Springer, Berlin, 2007 CrossRef.
- (a) G. Vilé, N. Almora-Barrios, S. Mitchell, N. López and J. Pérez-Ramírez, Chem. – Eur. J., 2014, 20, 5926 CrossRef PubMed; (b) P. T. Witte, S. Boland, F. Kirby, R. van Maanen, B. F. Bleeker, D. A. M. de Winter, J. A. Post, J. W. Geus and P. H. Berben, ChemCatChem, 2013, 5, 582 CrossRef CAS; (c) G. La Sorella, L. Sperni, P. Canton, L. Coletti, F. Fabris, G. Strukul and A. Scarso, J. Org. Chem., 2018, 83, 7438 CrossRef CAS PubMed; (d) L. Z. Nikoshvili, A. V. Bykov, T. E. Khudyakova, T. LaGrange, F. Héroguel, J. S. Luterbacher, V. G. Matveeva, E. M. Sulman, P. J. Dyson and L. Kiwi-Minsker, Ind. Eng. Chem. Res., 2017, 56, 13219 CrossRef CAS.
- (a) A. Reina, I. Favier, C. Pradel and M. Gómez, Adv. Synth. Catal., 2018, 360, 1 CrossRef; (b) M. Díaz de los Bernardos, S. Pérez-Rodríguez, A. Gual, C. Claver and C. Godard, Chem. Commun., 2017, 53, 7894 RSC; (c) X. Wen, X. Shi, X. Qiao, Z. Wu and G. Bai, Chem. Commun., 2017, 53, 5372 RSC; (d) H. Konnerth and M. H. G. Prechtl, Chem. Commun., 2016, 52, 9129 RSC; (e) S. Carenco, A. Leyva-Pérez, P. Concepción, C. Boissière, N. Mézailles, C. Sanchez and A. Corma, Nano Today, 2012, 7, 21 CrossRef CAS; (f) V. Polshettiwar, B. Baruwati and R. S. Varma, Green Chem., 2009, 11, 127 RSC; (g) F. Alonso, I. Osante and M. Yus, Tetrahedron, 2007, 63, 93 CrossRef CAS.
- L. M. Rossi, N. J. S. Costa, F. P. Silva and R. Wojcieszak, Green Chem., 2014, 16, 2906 RSC.
- (a) L. M. Martínez-Prieto and B. Chaudret, Acc. Chem. Res., 2018, 51, 376 CrossRef PubMed; (b) L. M. Martinez-Prieto, E. A. Baquero, G. Pieters, J. C. Flores, E. de Jesus, C. Nayral, F. Delpech, P. W. N. M. van Leeuwen, G. Lippens and B. Chaudret, Chem. Commun., 2017, 53, 5850 RSC; (c) L. M. Martínez-Prieto, L. Rakers, A. M. López-Vinasco, I. Cano, Y. Coppel, K. Philippot, F. Glorius, B. Chaudret and P. W. N. M. van Leeuwen, Chem. – Eur. J., 2017, 23, 12779 CrossRef PubMed; (d) J. M. Asensio, S. Tricard, Y. Coppel, R. Andrés, B. Chaudret and E. de Jesús, Angew. Chem., Int. Ed., 2017, 56, 865 CrossRef CAS PubMed.
- L. M. Martínez-Prieto, C. Urbaneja, P. Palma, J. Campora, K. Philippot and B. Chaudret, Chem. Commun., 2015, 51, 4647 RSC.
- L. M. Martínez-Prieto, I. Cano, A. Marquez, E. A. Baquero, S. Tricard, L. Cusinato, I. del Rosal, R. Poteau, Y. Coppel, K. Philippot, B. Chaudret, J. Campora and P. W. N. M. van Leeuwen, Chem. Sci., 2017, 8, 2931 RSC.
- (a) V. Biju and M. Abdul Khadar, J. Nanopart. Res., 2002, 4, 247 CrossRef CAS; (b) V. M. A. van Sawatzky, Phys. Rev. Lett., 1993, 70, 2459 CrossRef PubMed; (c) J. C. Vedrine, G. Hollinger and M. D. Tran, J. Phys. Chem., 1978, 82, 1515 CrossRef CAS.
- D. Delgado, R. Sanchís, J. A. Cecilia, E. Rodríguez-Castellón, A. Caballero, B. Solsona and J. M. L. Nieto, Catal. Today, 2019, 333, 10 CrossRef CAS.
- D. Jiles, Introduction to Magnetism and Magnetic Materials, Chapman and Hall, London, 1991 Search PubMed.
- (a) R. Kaiser and G. Miskolczy, J. Appl. Phys., 1970, 41, 1064 CrossRef CAS; (b) I. M. L. Billas, A. Chatelain and W. A. de Heer, Science, 1994, 265, 1682 CrossRef CAS PubMed.
- N. Cordente, C. Amiens, B. Chaudret, M. Respaud, F. Senocq and M.-J. Casanove, J. Appl. Phys., 2003, 94, 6358 CrossRef CAS.
- J. Nogués and I. K. Schuller, J. Magn. Magn. Mater., 1999, 192, 203 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy02172h |
| This journal is © The Royal Society of Chemistry 2020 |