
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvances in catalytic routes for the production of carboxylic acids from biomass: a step forward for sustainable polymers
J.
Iglesias
 a,
I.
Martínez-Salazar
b,
P.
Maireles-Torres
c,
D.
Martin Alonso
de,
R.
Mariscal
b and
M.
López Granados
*b
a,
I.
Martínez-Salazar
b,
P.
Maireles-Torres
c,
D.
Martin Alonso
de,
R.
Mariscal
b and
M.
López Granados
*b
aChemical & Environmental Engineering Group, Universidad Rey Juan Carlos, C/Tulipan, s/n, Mostoles, Madrid 28933, Spain
bEQS Group (Sustainable Energy and Chemistry Group), Institute of Catalysis and Petrochemistry (CSIC), C/Marie Curie, 2, 28049 Madrid, Spain. E-mail: mlgranados@icp.csic.es
cUniversidad de Málaga, Departamento de Química Inorgánica, Cristalografia y Mineralogía (Unidad Asociada al ICP-CSIC), Facultad de Ciencias, Campus de Teatinos, 29071 Málaga, Spain
dGlucan Biorenewables LLC, Madison, WI 53719, USA
eDepartment of Chemical and Biological Engineering, University of Wisconsin-Madison, 1415 Engineering Drive, Madison, WI 53706, USA
First published on 13th July 2020
Abstract
Polymers are ubiquitously present in our daily life because they can meet a wide range of needs and fields of applications. This success, based on an irresponsible linear consumption of plastics and the access to cheap oil, is creating serious environmental problems. Two lines of actions are needed to cope with them: to adopt a circular consumption of plastics and to produce renewable carbon-neutral monomers. This review analyses the recent advances in the chemocatalytic processes for producing biomass-derived carboxylic acids. These renewable carboxylic acids are involved in the synthesis of relevant general purpose and specialty polyesters and polyamides; some of them are currently derived from oil, while others can become surrogates of petrochemical polymers due to their excellent performance properties. Polyesters and polyamides are very suitable to be depolymerised to other valuable chemicals or to their constituent monomers, what facilitates the circular reutilisation of these monomers. Different types of carboxylic acids have been included in this review: monocarboxylic acids (like glycolic, lactic, hydroxypropanoic, methyl vinyl glycolic, methyl-4-methoxy-2-hydroxybutanoic, 2,5-dihydroxypent-3-enoic, 2,5,6-trihydroxyhex-3-enoic acids, diphenolic, acrylic and δ-amino levulinic acids), dicarboxylic acids (2,5-furandicarboxylic, maleic, succinic, adipic and terephthalic acids) and sugar acids (like gluconic and glucaric acids). The review evaluates the technology status and the advantages and drawbacks of each route in terms of feedstock, reaction pathways, catalysts and economic and environmental evaluation. The prospects and the new research that should be undertaken to overcome the main problems threatening their economic viability or the weaknesses that prevent their commercial implementation have also been underlined.
 J. Iglesias | Jose Iglesias was born in Benavente (Spain) in 1976. He received his MEng from Universidad Complutense, Madrid (1999, UCM), and his PhD in Chemical Engineering from Universidad Rey Juan Carlos (2005, URJC) working on the development of new catalysts for olefin asymmetric oxidation. Currently, he holds a position as Associate Professor in Chemical Engineering in the Department of Chemical, Energy and Mechanics Technology at URJC. His main research interests are focused on the rational design of heterogeneous catalytic systems for green and fine chemistry, lately applied to the chemical valorisation of biomass. |
 I. Martínez-Salazar | I. Martínez-Salazar received her MS degree in Organic Chemistry in 2018 from Universidad Autónoma de Madrid (UAM). She is conducting her PhD research at Institute of Catalysis and Petrochemistry (ICP-CSIC), where she joined in February 2019 under the guidance of Dr Rafael Mariscal. As a doctoral student, she is working on the development of new solid catalysts for the synthesis of adipic acid from biomass for use as a renewable monomer. |
 P. Maireles-Torres | Prof. Pedro Maireles-Torres received his BS degree in 1987 and PhD degree in 1991 at Málaga University (Spain). After a post doc stay of four years in Montpellier (France), he came back to Málaga University, being promoted to associate professor in 1999, and full professor in 2009. Initially, the research was aimed at the synthesis of mesoporous materials from layered metal(IV) phosphates and heteroatom-doped silica, and their use in heterogeneous catalysis, ion-exchange and sorption processes. From 2004, the research interest is focused on heterogeneous catalysis for the production of biofuels and value-added chemicals from lignocellulosic biomass and vegetable oils. |
 D. Martin Alonso | Dr Martin Alonso earned his Bachelor degree in Chemical Engineering at the University of Salamanca (Spain) and his PhD at Catalysis and Petrochemistry Research Institute (Spain) working in the production of biodiesel using heterogeneous catalysis. In 2009 he moved to UW-Madison to work with Prof. James A. Dumesic at the University of Wisconsin-Madison studying new catalytic processes to convert biomass into chemicals and fuels. He is the (co)author of 52 peer-reviewed manuscripts and currently he is the Director of Research and Development at Glucan Biorenewables LLC, a start-up company focused on biomass fractionation and the production of furanic chemicals. |
 R. Mariscal | Rafael Mariscal received his BS (1987) in Chemistry from the University of Cordoba and his PhD from the Complutense University of Madrid in 1991 on catalytic oxidative coupling of methane. This experimental work was carried out at the Institute of Catalysis and Petrochemistry (ICP-CSIC). After a postdoctoral stay at the Technological University of Delft, he returned to ICP-CSIC, where he currently develops his career as a scientific researcher. His research interests are focused on the heterogeneous catalytic process applied to energy and environment. In the last decade, he has concentrated his activities to obtain biofuels, chemicals and monomers from biomass. |
 M. López Granados | Manuel López Granados is a Scientific Researcher at the Institute of Catalysis and Petrochemistry of CSIC (ICP-CSIC). He received his BSc degree from the University of Córdoba (1986) and then moved to ICP-CSIC to conduct the research needed for receiving his PhD from the University Complutense of Madrid (1991). He was a postdoctoral researcher at the University of Utrecht (The Netherlands) and Notre Dame (USA) between 1991–1993. Then he returned to Spain to ICP-CSIC where he got a permanent position at ICP. His research interest is now focused on the development of catalytic technologies for the valorisation of biomass to valuable renewable chemicals, monomers and biofuels. |
1. Introduction
Carboxylic acids are involved in the synthesis of an extensive family of polymers: polyesters and polyamides. The deployment of polyesters and polyamides in particular, and of polymers in general, is a story of success. A small number of polymers that can be prepared in a relatively simple manner are capable of covering many possible applications: packaging, piping, use in the automobile industry, furniture, household utensils, containers, toys, textile fibres, coatings, etc.1 It can be said that there is a plastic for any need demanded by consumers. This success has resulted in an average annual growth of the production of plastics in the range of 3–4%. The global production of plastics reached 348 million tons by 2017.2 In those 348 million tons, polyesters and polyamide fibres are not included; in 2014, the consumption of polyester and polyamide fibres exceeded 46 and 4 million tons, respectively.3To date, the utilisation of polymers has mainly relied on the ease of access to low-cost oil and on the adoption of an unsustainable and irresponsible linear model of utilisation with little or no consideration of either the disposal or end-use of plastics that, in the best of the cases, go to landfills or are burned, leading to considerable contamination.4–6
Sustainability, in general, is a key parameter for the future of polymers. First, this requires the replacement of the oil feedstock by other renewable resources, such as non-food biomass.7–10 Polyester and polyamide synthesis involves carboxylic molecules, and, fortunately, there are many routes for producing carboxylic acids from non-food lignocellulosic biomass suitable for polymerisation.8,11–13
Second, sustainability also requires the adoption of a circular utilisation of polymers to prevent leakage of polymers into the environment. Chemical recycling and/or repurposing to other polymers are the preferred options.7,14,15 Mechanical reutilisation is problematic due to the difficulties of effective separation technologies to prevent contamination with other plastics, metals, chemicals, pigments, etc., which eventually results in a reduced quality of the recycled product.7,8,15 Polyester and polyamides are good candidates for chemical recycling or repurposing because they can be either chemically depolymerised to their constituent monomers or chemically repurposed as other polymers.11,16–18
Biodegradability is not the best end-of-life option (microorganisms benefit only from biodegradation)19 and may even be a problem because durability is required for most of the plastic uses and affect storage times considerably. However, biodegradability of polymers can be desirable when recycling is not viable and for specific single use plastics (including tableware or food packaging) or for those intentionally designed to be released into the environment (such as agricultural films). In this context biodegradable polyesters already exist, such as poly(butylene adipate-co-terephthalate), poly(hydroxyalkanoates), poly(butylene succinate) and poly(L-lactic acid) (the latter when composted).20,21
This review will cover the main achievements accomplished so far in the chemical catalytic routes for producing carboxylic acids of interest in the production of commercialised polymers and that are amenable to manufacture from lignocellulose. More specifically, we will outline the most recent advances in the catalytic synthesis of mono- and dicarboxylic acids and hydroxycarboxylic acids. This review is also extended to carboxylic acids for other not yet commercial polymers with the potential to replace some oil-derived commercial polymers because of their suitable properties. The chemical profile of this journal precludes the description of other routes based on biotechnological processes. Fatty acids are not included in this review because they are obtained in practice from vegetable oil. Lignin-derived ferulic acid is also out of the scope of this review: enzymatic and chemical hydrolysis methods using NaOH are the more extended protocols to extract it from lignocellulose.
Excellent reviews outlining the production of monomers from biomass have already been published (see ref. 12, 13 and 22); they have been written in a wider context describing all existing possible renewable polymers and monomers, their properties and the main features of the synthesis process. Our review presents a narrower and more specialised target: a comprehensive description of the key technical aspects of the chemocatalytic routes involved in the synthesis of (only) renewable carboxylic acids from lignocellulose. Recent reviews have revised the heterogeneous catalytic processes for the synthesis of biobased polyesters11 or functionalised carboxylic acids23 from cellulose. Our review is not restricted to cellulosic sugars but also includes hemicellulosic sugars and lignin.
For a more precise discussion of the investigations, we have gathered the information in tables that summarise the reaction conditions and the most relevant catalytic properties, i.e., conversion of the substrate and yield of the acid. When the reaction was conducted under continuous mode, the space-time yield (STY) of the product is an excellent predictor for comparing different catalysts, irrespective of the reaction conditions used (under the assumption that the conditions have been optimised for the maximum yield of the product). Unfortunately, in most cases, the investigations were conducted in batch reactors using a different set of reaction conditions, especially different reaction times and concentrations of reactants and catalysts. This makes a direct comparison between the different catalysts quite complicated. In an attempt to reasonably compare the different catalysts for these cases, we have included the productivity per mass of catalyst in terms of g product gcat−1 h−1. We are aware that this cannot be used as an absolute metric for the superior performance of a catalyst because it does not convey information on the concentration of the substrate and the stability of the catalyst. In addition, its value can still be subjected to improvements by finer adjustment of the reaction conditions. However, this parameter provides relevant information: first, how productive a given transformation is in comparison to others in this review and, second, the relative amount of catalyst needed to achieve high yields. Consequently, this allows us to separate catalysts that did not perform very well from the group of catalysts that warrant further investigation.
In some cases, the literature is so scarce that all of the investigations conducted are discussed. In other cases, such as the production of adipic acid, terephthalic acid and furandicarboxylic acid, the number of studies is so overwhelming that a precise comparison of all catalytic studies is beyond the scope of this review. In these cases, we have substantially summarised the topic, depicting the key technical features of the process and the most active and robust family of catalysts identified so far for the process. In addition to reviewing the key features of the different catalytic technologies, we also underline the future prospects and new investigations that must be conducted to overcome the problems and weaknesses that prevent their commercial implementation. Although we have included a section devoted to the overall concluding remarks and future prospects of the overall topic, we have also included at the end of each section a short summary with the specific future challenges.
2. Monocarboxylic acids
2.1. Hydroxy monocarboxylic acids
Hydroxyacids constitute a highly interesting category of bifunctional chemicals, which are liable to be transformed into polyesters because the coexistence of an alcohol and an acid functionality in the same molecule theoretically results in an easy condensation process that provides linear polymers. Hydroxyacids are characterized by a notably high proportion of oxygen in their composition, especially those with short carbon chains, and their production from biomass – a typical oxygen-rich feedstock – makes deep deoxygenation reactions unnecessary. Furthermore, conditioning of the oxygen functionalities is still required to provide carboxylic acid and hydroxyl groups. In this way, different hydroxyacids have been reported to be produced from biomass feedstocks, ranging from C2 glycolic acid, which is the shortest component of this category, to hydroxyacids with chain lengths of 6–14 carbons.Hydroxyacids occur in nature in several ways. On one hand, simple, short carbon chain compounds, such as glycolic or lactic acids, are formed in metabolic routes in the vast majority of living organisms.24 On the other hand, polyesters like poly-hydroxyalkanoates (PHA) are known to occur as energy storage compounds in several types of microbial organisms.25,26 These polyesters, due to their biological origin, are biodegradable and biocompatible materials; thus, they have applications in several areas, with most being related to medicine.27,28 Despite knowing about the existence of both hydroxyacids and PHA in microorganisms since 178029 and 1920,30 respectively, the polymer industry has not focused their attention on the fabrication of these biobased monomers and polymers until a few decades ago.31,32 The main reason is the high costs associated with their production, as making biobased polyesters is an attractive option only in the context of high oil prices. Both of them are produced at an industrial scale using biological pathways, although the production of hydroxyacids (e.g., lactic acid) greatly exceeds that of PHA.32 Unlike hydroxyacids, PHAs are exclusively produced by means of fermentation routes. Thus, PHAs are outside of the topic covered by this review because this section is devoted to the main chemocatalytic routes for the synthesis of hydroxyacids for polyester production. These include the shortest hydroxyacid (glycolic acid), C3 hydroxyacids (lactic acid and 3-hydroxypropionic acid), and some examples of larger C4–C6 hydroxyacids, which have shown high potential in the preparation of functional polymers.
The synthesis of glycolic acid has been industrially approached through different procedures using several feedstocks, namely, formaldehyde and acetic acid.37 The first industrial process for GA fabrication was that developed by Du Pont and was based on a hydrative carbonylation of formaldehyde in the presence of sulfuric acid under very harsh reaction conditions (473–523 K; 300–700 bar).38 This process coexisted with a method developed by Hoechst,39 involving the chlorination of acetic acid and the subsequent hydrolysis of the resulting chloroacetic acid in the presence of sodium hydroxide. Although this last method requires milder operating conditions compared to the formaldehyde-based method, it involves extensive consumption of water. Within the above context, biotechnological processes are emerging as highly promising environmentally friendly alternatives to classical chemical processes.24 Nevertheless, the requirements for low-concentration substrates, which is a typical feature of biotechnological methods, limit the amplification of this technology to the industrial scale. However, other chemical routes from biomass-derived substrates can be considered a reasonably efficient option for the production of glycolic acid, which may be used as alternatives to both classical chemical routes and the yet-to-be-developed biotechnological processes.
A hydrothermal treatment of lignocellulosic biomass has been reported as an effective way to produce an energy-dense carbon material together with a water stream loaded with a wide variety of organic acids, including GA. This strategy can be applied to the synthesis of hydroxyacids because of its inherent simplicity; however, its selectivity is poor, thus making it a less practical application for GA production.40,41 Some authors have studied the use of niobia on silica42 and basic hydrotalcites43 in the treatment of cellulose in water at 453 K. These investigations have aimed to take advantage of the activity of these materials during the depolymerization of cellulose and the fragmentation of the evolving monosaccharide units into small molecules, which is conducted mainly through retro-aldol condensation pathways; the above mechanism makes them easier to transform into organic acids. Under these conditions, niobia materials were able to transform 60% of the converted cellulose into glycolic acid. However, this transformation required long contact times (>24 h) to achieve high product yields, resulting in impractical results for an industrial scale.
Metal oxides have been considered as an alternate method; for instance, the use of Mo-based Keggin-type polyoxometalates (POM) is an interesting option. Molybdenum heteropolyacids combine two interesting functions – Brønsted- and Lewis-type acidity. Hydrolytic transformations, such as the hydrolysis of cellulose to glucose, can be promoted by Brønsted acid sites, whereas Lewis sites allow glycolytic transformations, such as retro-aldol condensation, as well as oxidation transformations. Thus, in the presence of phosphomolybdic acid in aqueous media, cellulose has been described to undergo a hydrolysis step to yield glucose, which subsequently evolves through two consecutive retro-aldol condensations yielding three molecules of glycolaldehyde – the main intermediate; the above molecules are then oxidized to glycolic acid.44,45 This strategy allowed the conversion of complex feedstock into GA with a reasonably high selectivity. Thus, commercial cellulose powder (90.5% conversion, 49.3% yield), sugarcane bagasse (91.7% conversion, 32.2% yield) and hay (80.1% conversion, 27.9% yield) provide moderate quantities of glycolic acid, along with a minor amount of short-chain organic acids. The reaction procedure consists of treating the substrate (200 mg) in water (20 mL) at 453 K and 60 bar O2 with a substrate to catalyst molar ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for a short contact time (1 h). Moreover, heteropolyacids can be reused for 9 runs with a negligible decay in catalytic activity, though a complex procedure – consisting of the removal of the remaining substrate by filtration, drying by vacuum evaporation, esterification of the glycolic acid for its recovery by distillation, and a thermal treatment in oxygen – has to be applied to recover the catalyst.
:1 for a short contact time (1 h). Moreover, heteropolyacids can be reused for 9 runs with a negligible decay in catalytic activity, though a complex procedure – consisting of the removal of the remaining substrate by filtration, drying by vacuum evaporation, esterification of the glycolic acid for its recovery by distillation, and a thermal treatment in oxygen – has to be applied to recover the catalyst.
A different approach to the production of glycolic acid is an oxidation pathway from renewable biomass feedstock, such as glycerol or its derived chemical dihydroxyacetone (DHA) (Fig. 1). This strategy has been investigated for different catalytic systems, including supported noble and non-noble metals and heteropolyacids, among others. First proposed by the group of Sankar et al., gold and palladium nanoparticle-based catalysts have been demonstrated to be highly active in the transformation of glycerol into GA under mild temperature conditions (below 393 K).46 Under optimal conditions – 15 mL of glycerol in water (0.3 mol L−1), NaOH:glycerol = 2:1, substrate to metal molar ratio = 500, and a contact time of 4 h – Au on carbon is able to convert glycerol into sodium glycolate with an ∼50% yield, regardless of whether oxygen or hydrogen peroxide is used as the oxidant. The resultant sodium glycolate, derived from the use of an alkali reaction medium, requires a multistep purification strategy to obtain the final glycolic acid. To circumvent the complex workup to transform glycolate salts into GA, the use of hydrotalcite-supported Au NPs under base-free conditions has been proposed.47 This option allows a 78% conversion of glycerol solutions (0.1 mol L−1, 5 mL), with a 53% selectivity to GA after coming into contact with Pt on the hydrotalcite (substrate to metal molar ratio of 800) for 6 h in an oxygen atmosphere. In both cases, the Au on carbon and the Pt on hydrotalcites, the overall reaction pathway is based on the generation of tartronic acid, followed by a decarboxylation step, which can be considered an inefficient reaction pathway in terms of atom economy.
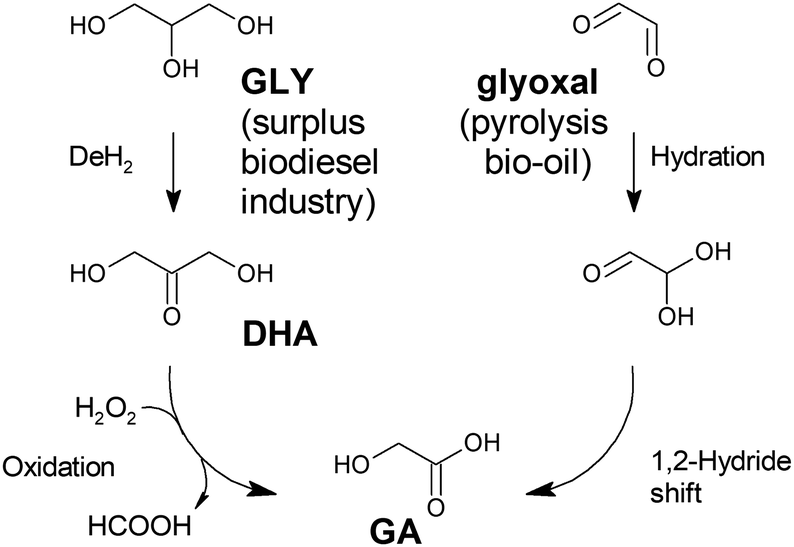 | ||
| Fig. 1 Reaction pathways for the production of glycolic acid from renewable feedstocks. GLY: glycerol; DHA: dihydroxy acetone; GA: glycolic acid. | ||
As an alternative to these previous works, Xi et al.48 formulated the use of copper-based oxidation (non-noble metal) catalysts to convert DHA to glycolic acid using hydrogen peroxide as the oxidant. Cu/Al2O3 displays a very high selectivity (100% substrate conversion, 94% yield to glycolic acid) in the conversion of DHA at room temperature after 24 h. The proposed reaction mechanism for this oxidation pathway involves the cleavage of a C–C bond in the transformation of DHA into glycolic acid, which yields formic acid, formamide or an alkyl formate if a secondary amine or an alcohol is present in the reaction media. This strategy allows the recovery of a third carbon from the DHA backbone for a different application. The above recovery overcomes the main drawback ascribed to oxidation pathways, which is their low atom economy. However, this strategy is far from being applicable in industry, as the high H2O2/DHA ratio required to obtain high GA yields (∼3) makes its application difficult on a large scale.
Another strategy used for producing GA from renewable biomass feedstock was reported by Dapsens et al.49 These authors start from glyoxal, a C2 compound present in pyrolysis bio-oil, which is converted into GA with the aid of a Lewis-acid catalyst through a 1,2-hydride shift pathway. This strategy is similar to that followed in the production of other hydroxyacids, such as lactic acid, which will be seen in the following sections. Thus, different solid Lewis acid catalysts have been tested in the transformation of glyoxal into GA, including USY zeolites with different extra-framework aluminium (EFAL) and gallium contents and with Ti- and Sn-silicalites prepared through different methods. The results are evidence of the superior performance of tin-functionalized silicalite, which provides an excellent substrate conversion (96%) and very high GA yield (over 90%) when treating 5 wt% glyoxal aqueous solutions (200 mg glyoxal, 80 mg catalyst, 3.8 g water) at 363 K for 18 h. Furthermore, the selected catalyst displays excellent reusability and stability under continuous flow operating conditions, showing high potential for being scaled up to industrial production.
In summary, the interesting properties of poly(glycolic acid) and related copolymers make this polyester a highly interesting option for future applications, namely, in the field of biomedicine. Consequently, an increasing demand is expected for this bioplastic and its monomers. A sustainable and cost-efficient process for the production of the main monomer, glycolic acid, remains a challenge. Biotechnological and chemocatalytic pathways involving routes with a high atom economy will probably play a major role in the development of industrial processes to replace the environmentally unfriendly methods currently existing for GA production. Within the field of chemistry, the transformation of glyoxal – either that present in high-temperature pyrolysis bio-oil or synthesized from sugar monosaccharides through a combination of retro-aldol and oxidation pathways – seems to be one of the most efficient routes for the production of glycolic acid. Nevertheless, there are still some challenges to overcome, such as testing the capability of Lewis acid catalysts – e.g., high EFAL USY – to drive the transformation of glyoxal into glycolic acid in real bio-oil samples, or increasing the selectivity of bifunctional acid-oxidation catalysts – e.g., polyoxometalates – in the direct transformation of sugars into GA using environmentally friendly oxidants such as air.
Lactic acid tends to polymerize if highly concentrated, leading to dimers, trimers, and low molecular weight oligomers, but the production of high molecular weight PLA is only achieved through a few condensation strategies.54 Molecular weight is one of the properties of PLA that influences its thermal and mechanical properties, and a minimum MW is necessary to make PLA suitable for commercial applications.55 In this context, a two-step process in which lactide – a cyclic dimer of lactic acid – is obtained as an intermediate is one of the most prolific strategies at the industrial scale.56 In this process, lactic acid is prepolymerized to lactide and then transformed by a ring-opening polymerization into high molecular weight PLA. Another important feature that influences the final properties of PLA is tacticity. Isotactic and stereoblock copolymers of PLA display superior thermal, mechanical and chemical properties compared to those of syndiotactic or atactic PLA.57,58 Since tacticity in PLA is linked to the chirality of the monomer, its control during the synthesis or the separation of the different enantiomers is of major importance to obtain commercial grade PLA.
With a global production of 714 kton in 2013 and a forecast of sustained growth of approximately 15% in yearly demand, a worldwide production of more than 1960 kton of lactic acid is expected in 2020, making LA the most manufactured hydroxyacid.59 LA is industrially produced by anaerobic fermentation of carbohydrate-rich feedstock,60 most of them edible raw materials (e.g., sucrose), although some other non-edible carbohydrate sources (e.g., lignocellulose biomass hydrolysate) are being explored as starting points.61 Specialized bacteria (Lactobacillus delbrueckii, Lactobacillus casei, Enterococcus faecalis, etc.) are conventionally used, although the use of yeasts has expanded in recent years. Fermentations with wild species of these microorganisms provide optically pure L-(+)-S enantiomers of LA, whereas the production of the D-(−)-R enantiomer requires bioengineered strains.62 Fermentations are conducted at mild temperatures (313–318 K) in batch or semi-batch units operating in dilute broths (∼5 g substrate per L), causing the characteristic low volumetric productivity of fermentations. The pH value is a key operational parameter that has to be adjusted to a range of 5–7 and is typically accomplished by the addition of lime (Ca(OH)2); the above pH range prevents the inhibition of bacteria at low pH values. Lactate salts are thus produced, and this influences the recovery of lactic acid, which is usually accomplished by the gypsum process. Fermentation broths are conventionally treated with H2SO4 to turn calcium lactate into lactic acid at pH < 2, while also yielding gypsum in high quantities (∼1 kg CaSO4 per kg LA), which is disposed. Finally, LA purification requires an expensive multistep process. Lactic acid has to be concentrated and purified to the required grade, which depends on the final application. In summary, biotechnological production of lactic acid is usually performed starting from edible feedstock, using a reasonably low volumetrically productive process and involving the generation of a large amount of waste (CaSO4). In this way, the main costs associated with the production of lactic acid fall upon the starting feedstock and downstream product recovery. To decrease these costs, alternative chemical routes, which usually involve low restrictions both in operation and raw material possibilities compared to that with fermentation, have been explored.
A lactonitrile process, once dominant at the industrial scale, has been the only chemical process in the industrial fabrication of this hydroxyacid.63 It uses acetaldehyde together with HCN to produce lactonitrile, which is subsequently hydrolysed to yield a racemic mixture of lactic acid. Nevertheless, environmental, energy and economic issues related to this technology have forced it to be substituted with fermentation processes.64–66 No other chemical routes have been scaled up for the industrial production of lactic acid. However, this has been an area of extraordinary research over the last decade because of the expansion in the number of commercial applications of lactic acid and the development of catalytic processes for the valorization of biomass-derived molecules, namely, glycerol and sugar monosaccharides.
2.1.2.1. Lactic acid from glycerol. Glycerol is an appropriate raw starting material for the production of lactic acid, as it already contains a C3 carbon skeleton, making C–C cleavage or coupling reactions unnecessary. In addition, glycerol is conventionally obtained as a 6-fold surplus in the biodiesel industry as it is needed for different applications; thus, it is a rather economic and plentiful feedstock. Glycerol transformation into lactic acid is conventionally assumed to occur through a cascade of reactions67 involving the oxidative dehydrogenation of glycerol into a triose–glyceraldehyde (GLA) or dihydroxy acetone (DHA) (Fig. 2). These species are in equilibrium, and both can evolve through dehydration into an enol (2-hydroxy acrolein, 2-HAC), which is tautomerized to pyruvaldehyde (PAL). PAL is then transformed into lactic acid through different pathways, depending on the used catalyst – a benzylic acid-type rearrangement if a base or a hydration and 1,2-hydride shift if a Lewis acid – or to alkyl lactate if transformed in the presence of an alcohol.
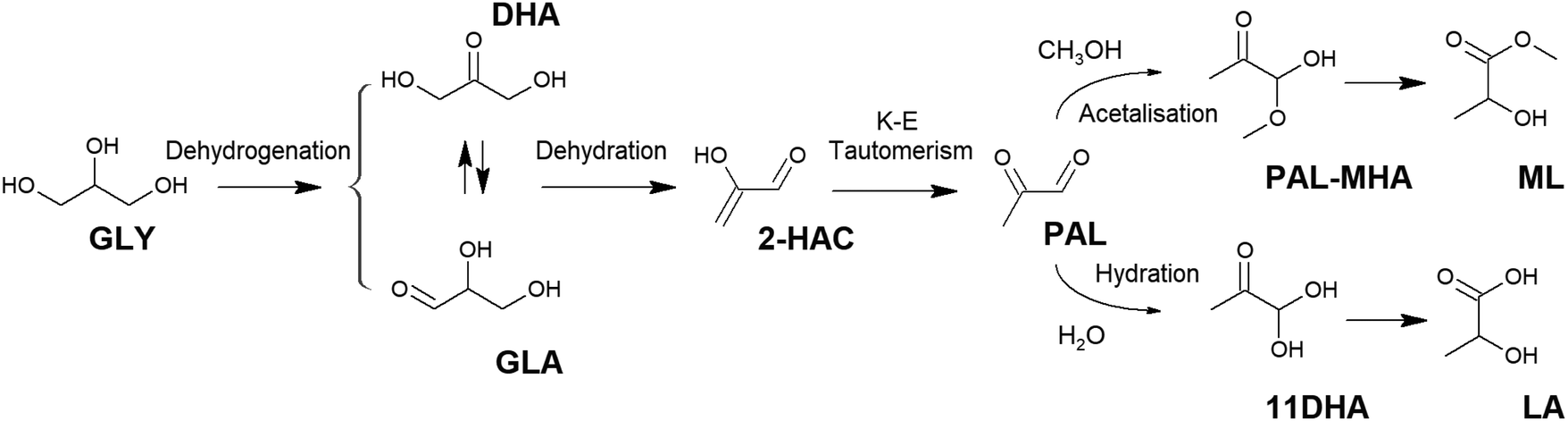 | ||
| Fig. 2 Transformation of glycerol into lactic acid and methyl lactate. GLY = glycerol; DHA = Dihydroxyacetone; GLA = glyceraldehyde; 2-HAC = 2-hydroxy acrolein; PAL = pyruvaldehyde; PAL-MHA = pyruvaldehyde methyl hemiacetal; ML = methyl lactate; 11DHA = 1,1-dihydroxy acetone; LA = lactic acid. | ||
Glycerol dehydrogenation can be performed through a hydrothermal treatment under high-temperature conditions in the presence of soluble alkali and alkaline-earth bases (NaOH, KOH, Ca(OH)2, Ba(OH)2, etc.)68–70 or by using metal species with dehydrogenation activity – Pt, Pd, Au, Ni, Cu, etc. – in the presence of soluble bases.71–73 The dehydration of trioses to PAL and its transformation into lactic acid can be performed using the same alkalis as above or in the presence of acids. Hydrothermal alkali treatments with strong soluble bases, both with and without the assistance of metal catalysts, have been extensively reported. However, these alternatives lead to the production of lactic acid as lactate salts, and thus, their recovery and purification are conditioned in the same way as in fermentation routes. On the other hand, the use of oxidation pathways in the presence of acid sites, provides lactic acid as a free acid and seems to be one of the most interesting options for the valorization of glycerol.74
Bifunctional catalysts that are able to convert glycerol into lactic acid in a single step have been prepared by supporting different metals with dehydrogenation/oxidation activity on solid acids. The influence of the solid support acidity has been assessed by several authors with alumina-supported gold nanoparticles. Alumina is a well-known amphoteric metal oxide whose acid/base properties can be tuned by mixing with other metal oxides.75,76 These studies have revealed that basic supports, such as MgO/Al2O3, enhance the dehydrogenation–oxidation activity of gold, leading to formic acid as the major product. In contrast, doping alumina with MoO3 and WO3 enhances the acidity of the material, which moderates the activity of gold and shifts its selectivity towards the formation of DHA; DHA is the key intermediate in the transformation from glycerol to lactic acid. Komanoya et al. performed studies on different combinations of active metals supported on Lewis acids77 (Table 1, entries 1–4). Platinum nanoparticles supported on titania supports is a good example of this combination. Pt nanoparticles provide oxidation capability, whereas titanium oxide contributes to water-tolerant Lewis acidity. These materials converted at 423 K – high enough temperature conditions to promote the dehydration of DHA – aqueous glycerol solutions into lactic acid with a 70% substrate conversion and a 70% selectivity towards LA after reacting for 18 h. However, the catalytic performance of these bifunctional catalysts is still far from acceptable at the industrial scale, which is evident from the low productivity offered by these catalytic systems (Table 1). The use of more sophisticated Lewis acid solid supports, such as zeolitic materials, has also been addressed (Table 1, entries 5–10). High EFAL zeolites display much higher Lewis acidity compared to metal oxides, thus requiring milder reaction conditions to complete acid-driven reactions such as DHA dehydration. These materials were first explored using nanoparticles of a Pt–Au alloy to build bifunctional catalysts.78,88 BEA, MOR, and FAU zeolites with low Si/Al ratios and high EFAL contents were tested as supports due to the extraordinarily high selectivity that these materials offer in the direct conversion of trioses into lactic acid.29,89 USY zeolites show better catalytic performance than other zeolites, probably because the large pore size of the faujasite structure partially alleviates the negative effect of pore blocking caused by the immobilization of the metal phase. In addition, the combination of highly active Pt–Au alloy nanoparticles with strong Lewis acid sites, leads to materials with higher lactic acid productivity than those thus far reported. However, Pt–Au is a highly active oxidation phase, which leads to side products due to glycerol over-oxidation. Au nanoparticles – showing moderate oxidative activity compared to Au–Pt – in combination with USY zeolites provide improved selectivity, though lower productivity, in the transformation of glycerol into methyl lactate (selectivity ∼74%) when compared to bimetallic materials; the above result is due to the selectivity enhancement of the oxidation reaction in the lactic pathway.79
| # | Catalyst | Operating conditions | Catalytic results | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Solv.a | S ccb (wt%) | C/Sc (wt%) | PO2d (bar) | T (K) | t (h) | X S (%) | YLacth (%) | Produc.i (g gcat−1 h−1) | Deact.j | |||
| a Solvent. b Substrate concentration weight ratio in %. c Catalyst to substrate weight ratio, in %. d Reactant gas pressure in bar. e Temperature conditions in Kelvin. f Reaction time. g Substrate conversion. h Yield towards product. i Productivity. j Catalyst deactivation: strong (st), moderate (m), low (l) and negligible (ne); n.s. (not studied), n.r. (not reported). k Air instead of O2. l NPs: nanoparticles. | ||||||||||||
| Metal oxides | ||||||||||||
| 1 | Pt/TiO2 | H2O | 18.4 | 54.0 | 5 | 323 | 18 | 70 | 49 | 0.05 | m (4 runs) | 77 |
| 2 | Pt/Nb2O5 | H2O | 18.4 | 54.0 | 5 | 323 | 18 | 82 | 39 | 0.04 | n.s. | 77 |
| 3 | Pt/ZrO2 | H2O | 18.4 | 54.0 | 5 | 323 | 18 | 79 | 36 | 0.04 | n.s. | 77 |
| 4 | Pt/Al2O3 | H2O | 18.4 | 54.0 | 5 | 323 | 18 | 68 | 28 | 0.03 | n.s. | 77 |
| Al-Containing zeolites | ||||||||||||
| 5 | Au–Pt/USY-712 | H2O | 27.6 | 4.7 | 3 | r.t. | 2 | 71 | 24 | 2.55 | n.s. | 78 |
| 6 | Au–Pt/USY-600 | H2O | 27.6 | 4.7 | 3 | r.t. | 2 | 70 | 24 | 2.55 | n.s. | 78 |
| 7 | Au–Pt/H-MOR | H2O | 27.6 | 4.7 | 3 | r.t. | 2 | 40 | 19 | 2.02 | n.s. | 78 |
| 8 | Au–Pt/H-β | H2O | 27.6 | 4.7 | 3 | r.t. | 2 | 59 | 13 | 1.38 | n.s. | 78 |
| 9 | Au/USY-712 | MeOH | 23.0 | 43.4 | 30k | 433 | 5 | 81 | 60 | 0.28 | n.s. | 79 |
| 10 | Au/USY-600 | MeOH | 23.0 | 43.4 | 30k | 433 | 5 | 84 | 57 | 0.26 | n.s. | 79 |
| Sn-Containing materials | ||||||||||||
| 11 | Pt/Sn-MFI | H2O | 18.4 | 21.7 | 62 | 373 | 24 | 90 | 81 | 0.16 | st (3 runs) | 80 |
| 12 | Pt/Sn-β (F) | H2O | 18.4 | 21.7 | 62 | 373 | 24 | 93 | 28 | 0.05 | n.s. | 80 |
| 13 | Au/Sn-USY | MeOH | 18.4 | 21.7 | 50 | 413 | 10 | 81 | 69 | 0.32 | n.s. | 81 |
| 14 | Au NPs + Sn-USY | MeOH | 18.4 | 21.7 | 50 | 433 | 10 | 88 | 77 | 0.35 | m (3 runs) | 81 |
| 15 | Au–Sn-Montmorillonitel | MeOH | 23.0 | 15.2 | 30 | 483 | 5 | 78 | 54 | 0.71 | st (5 runs) | 82 |
| 16 | Au/CuO + Sn-MCM-41 | MeOH | 23.0 | 86.9 | 30 | 413 | 4.5 | 79 | 51 | 0.13 | l (5 runs) | 83 |
| 17 | Au/CuO + Sn-β | MeOH | 9.2 | 86.9 | 150 | 363 | 4 | 86 | 60 | 0.17 | l (7 runs) | 84 |
| Polyoxometalates | ||||||||||||
| 18 | H3PMo12O40 | H2O | 100.0 | 10.0 | 5 | 333 | 5 | 90 | 79 | 1.58 | n.s. | 85 |
| 19 | H3PMo12O40/C-30 wt% | H2O | 100.0 | 10.0 | 5 | 333 | 5 | 98 | 92 | 1.84 | m (5 runs) | 85 |
| 20 | AlPMo12O40 | H2O | 100.0 | 10.0 | 5 | 333 | 5 | 94 | 91 | 1.81 | m (12 runs) | 86 |
| 21 | CrPMo12O40 | H2O | 100.0 | 10.0 | 5 | 333 | 5 | 88 | 85 | 1.71 | m (12 runs) | 86 |
| 22 | Ag3PMo12O40 | H2O | 100.0 | 10.0 | 10 | 333 | 5 | 99 | 93 | 1.86 | m (12 runs) | 87 |
A further step in the use of zeolites as Lewis acid supports has been taken by different groups using tin-functionalized zeolitic materials (Table 1, entries 11–17). The Lewis acidity in these zeolites is provided by the incorporation of heteroatoms in the zeolite structure. This strategy allows for fine tuning the acid properties of the support to match the reaction rates of the different steps in the reaction cascade from glycerol to lactic acid. Their combination with Pt and Au particles has been reported for MFI, BEA,80 and FAU81 structures. Pt-based catalysts prepared from Sn-MFI and Sn-β zeolites have been tested in an aqueous phase transformation of glycerol into lactic acid at 373 K. Under these conditions, Pt/Sn-MFI provides excellent catalytic performance, surpassing that obtained for the Pt/Sn-β material, which is less active in the conversion of PAL to lactic acid.80 This different behaviour is ascribed to the different hydrophilicity shown by both zeolites. Since the β zeolite is synthesized by a fluoride method, the small number of framework defects leads to a highly hydrophobic material; thus, β zeolite shows a poorer affinity to polar compounds compared to Pt/Sn-MFI. A PVA-templated Au/Sn-USY zeolite has also been tested in the transformation of glycerol in methanol media.81 It is postulated that the Au precursor interacts with extra-framework Sn species (SnO2), which helps to prevent excessive growth of the metal particles and leads to a high dispersion of this phase; thus, the activity in the oxidation of GLY to DHA/GLA is enhanced. On the other hand, Sn sites incorporated in a faujasite structure are highly active under the tested conditions, thus leading to a remarkable combined activity in the conversion of GLY to methyl lactate (77% yield). The calculated productivity values, which are more modest than those of the other systems in Table 1, suggest that Sn-USY is a better option than an MFI zeolite; this is probably because the large pores of the faujasite material cause fewer mass transfer issues. Nevertheless, these materials suffer a moderate deactivation in recycling tests, with the growth of the Au particles being the reason for the observed loss in catalytic activity. The strategy of supporting noble metals on Sn-functionalized supports has been extended to other materials. Thus, although not a zeolite, Sn-montmorillonite, a clay prepared by ion exchange with Sn species, has also been tested as a Lewis acid solid support to bear Au particles.82 Though not as selective as Sn-zeolite-based catalysts, these materials provide evidence of the high potential of this strategy in the preparation of bifunctional catalysts for the direct transformation of glycerol into methyl lactate. Nevertheless, the materials demonstrate the same low reusability as zeolites (while being even more intense) for the same reasons; the growth of Au metal particles during the catalytic tests reduces their activity in the oxidation of glycerol.
As an alternative to the previously mentioned works, other groups have proposed the use of a physical mixture of CuO-stabilized Au nanoparticles with Sn-functionalized catalysts. Au on CuO has been previously suggested to display excellent catalytic activity in glycerol oxidation towards DHA.90 Thus, Tang et al. reported the use of Sn-MCM-41 as a Lewis acid catalyst,83 whereas Zhou and co-workers employed Sn-USY.84 Though less selective towards the formation of methyl lactate, the above strategy, which is based on supporting the oxidative active metal phase (gold) and the Lewis acid sites (tin) on different catalysts, has allowed the preservation of the integrity of metal nanoparticles. Nevertheless, the catalytic results do not compensate for the complexity of the catalyst synthesis procedure.
Finally, Keggin-type heteropolyacids and salts, as explored by Tao et al.,85–87 have been revealed to be an excellent option among the various acidic catalysts for the direct conversion of glycerol into lactic acid (Table 1, entries 18–22). The redox properties of polyoxometalates (POM) can drive the dehydrogenation of GLY, whereas their Brønsted and Lewis acid sites promote the transformation of DHA/GLA into PAL, which eventually leads to LA. Treatment of GLY in the presence of phosphomolybdic acid in water provides excellent substrate conversion (∼90%) with acceptable selectivity (∼80%) towards lactic acid in reasonably concentrated aqueous glycerol solutions (∼10 wt%). The above study demonstrates the very high catalytic activity of a homogeneous POM in the oxidation of glycerol to DHA/GLA, resulting in a very high productivity of lactic acid compared to any other catalytic system. However, the high solubility of this homogeneous catalytic system makes its recovery and reutilization difficult.85 Nevertheless, the catalytic results mentioned above have made it attractive to invest efforts in immobilization to obtain easily recoverable heterogeneous catalysts. Two different strategies have been tested to obtain reusable POM catalysts. The immobilization of H3PMo12O40 on a C10-alkyl chain-functionalized carbon support enhances its activity and selectivity towards the desired product, probably because the hydrophobic n-decyl functionalities facilitate the adsorption of glycerol.85 Alternatively, an ion exchange with Lewis acid metal cations, such as Al3+ and Cr3+, provides PMO salts with a low solubility.86 Since the acidity of the catalyst in water is not affected much by these substitutions, both salts provide good catalytic activity – comparable to that of supported phosphomolybdic acid – and good stability; furthermore, the catalyst is demonstrated to be reusable for 12 consecutive runs with moderate deactivation. Finally, another salt with improved activity in oxidation reactions has been tested by exchanging acidic protons with silver cations.87 The resultant PMO salt demonstrates high catalytic activity in the oxidation of glycerol to DHA/GLA and is quite sensitive to oxygen pressure. This salt, which has a reasonably low solubility in reaction media, displays very high activity (99% substrate conversion) and selectivity to lactic acid (93.0%). It also displays good stability; for instance, it is reusable for 12 runs without noticeable loss in its production of lactic acid.
2.1.2.2. Lactic acid from carbohydrates. The production of lactic acid from carbohydrates through chemical pathways has been extensively studied over the last decade, and several excellent reviews can be found dealing with this topic.11,23,29,91 Here, we collect an update of the main findings published in recent years in the production of lactic acid and alkyl lactates by chemocatalytic processes. Thus, the alternatives proposed for the valorization of different renewable raw materials are presented, together with the conditions that the reaction pathways impose on the design of different heterogeneous catalytic systems. Finally, special attention is devoted to the productivity and reusability of the reported heterogeneous catalytic systems, both of which are crucial parameters to consider the advancement of these processes to the industrial scale.
Many types of carbohydrates have been explored as starting raw materials, including polysaccharides and monosaccharides, and among these, hexoses (glucose and fructose), pentoses (mainly xylose), and trioses (dihydroxyacetone and glyceraldehyde) have been thoroughly tested as feedstocks. Glucose and xylose are plentiful in vegetable biomass, but trioses do not naturally occur as main components of a defined raw material; however, trioses can be produced by a mild pyrolysis of lignocellulosic biomass.92
A hydrothermal treatment of poly- and monosaccharides in the presence of base catalysts allows the conversion of carbohydrates into small organic acids, including lactic acid. However, either quite low lactic acid yield or similar issues to those of the biological pathways (separation issues because of the production of lactate salts) are then faced.93–103
Chemocatalytic transformation routes mediated by Lewis acid catalysts have been revealed to be the most efficient chemical procedure for the transformation of a wide variety of carbohydrates into lactic acid or alkyl lactates.91,104,105 These transformations require converting carbohydrates into C3 skeleton compounds, namely, trioses. This requires carbohydrates larger than C3 to undergo C–C cleavage reactions. Retro-aldol condensation is one of the Lewis acid catalysed transformations able to transform monosaccharides into small sugars. However, sugar retro-aldol condensation is regioselective. Thus, if the C–C cleavage occurs between the α and β carbons, referred to the carbonyl group, different products are obtained depending on whether the substrate is an aldose or a ketose (Fig. 3). Therefore, retro-aldol glycolysis of a ketose (like fructose) produces the same two trioses that are also obtained in the oxidative dehydrogenation of glycerol – glyceraldehyde (GLA) and dihydroxyacetone (DHA) – whereas if splitting occurs on an aldose (e.g., glucose), a C2 fragment (glycolaldehyde, GAL) and a tetrose (C4 fragment, e.g., erythrose) are produced as the main products.106 In the case of pentoses, retro-aldol splitting always leads to a C2 (GAL) and a C3 fragment (GLA or DHA), regardless of whether it starts from an aldose (xylose) or a ketose (xylulose).107 Only the production of trioses provides a direct pathway for the formation of lactic acid, whereas the formation of GAL and a tetrose lead to different products.
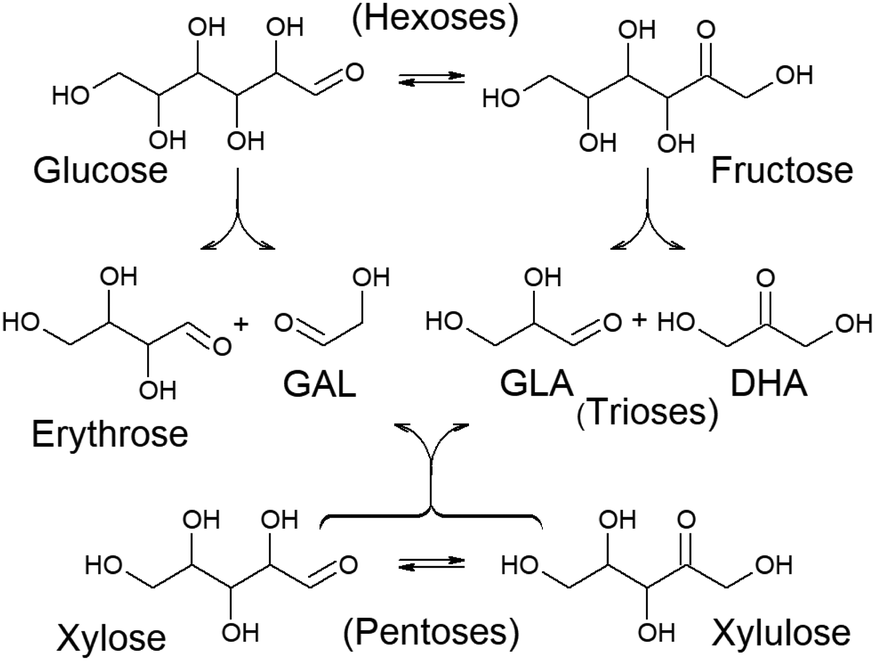 | ||
| Fig. 3 Retro-aldol condensation of hexoses and pentoses and the resultant products. GAL = glycolaldehyde; GLA = glyceraldehyde; DHA = dihydroxyacetone. | ||
Glucose, an aldose of six carbons that commonly occurs as a cyclical pyranose, is the most abundant monosaccharide present in lignocellulosic biomass. Its use as a starting point can be considered the most direct form of exploitation of carbohydrates for the production of lactic acid. The transformation begins with the opening of the sugar ring through a fast intramolecular hydride shift.108 The resultant linear aldose is then isomerized into a ketose through a Lobry de Bruyn–Alberda van Ekenstein-type rearrangement.93 This is an isomerization liable to be driven by different Lewis acids. Tin species incorporated in zeolites seem to satisfy the necessary requirements to perform this step with great efficiency.109 Fructose undergoes C–C cleavage through a retro-aldol condensation pathway – again, a Lewis acid-driven transformation – as two trioses – GLA and DHA – are produced as reaction intermediates that subsequently evolve into lactic acid or alkyl lactates.110 The last transformation, which is the isomerization of trioses into lactic acid, actually involves a cascade of transformations, which have already been mentioned for glycerol and requires the presence of acid catalysts. Converting trioses into lactic acid implies their dehydration towards an enol – 2-hydroxyacrolein (Fig. 4 shows the details of this step when performed in the presence of Brønsted acids). The above enol is in equilibrium with the corresponding ketone through a keto–enol tautomerism, thus yielding pyruvaldehyde (PAL). Hydration or an alcohol attack on the aldehyde carbonyl group of PAL forms the corresponding geminal diol or a hemiacetal, which subsequently undergoes an isomerization step that consists of a 1,2-hydride shift and is driven by Lewis acids. This last transformation yields lactic acid or alkyl lactate, depending on whether water or alcohol is used as the reaction solvent. Typically, alcohol media provide better results in terms of selectivity towards alkyl lactate and, unlike water, alcohol media prevent catalyst deactivation. Accordingly, bifunctional Brønsted and Lewis acid catalysts and the use of alcohol as the reaction solvent seem to be the preferred combination to promote the efficient conversion of trioses into alkyl lactate. For Brønsted sites, no special requirements are needed, as even mild acids, such as carboxylic acids, can promote the dehydration of trioses by increasing the operation temperature. For Lewis acid sites, the described cascade of transformations can be accomplished in the presence of a wide variety of Lewis acids.111–115 Nevertheless, Sn species offer superior catalytic performance, especially if supported in zeolite frameworks,116 because of their extraordinary catalytic activity in key steps of the cascade, such as the retro-aldol glycolysis or 1,2-hydride shift isomerization.
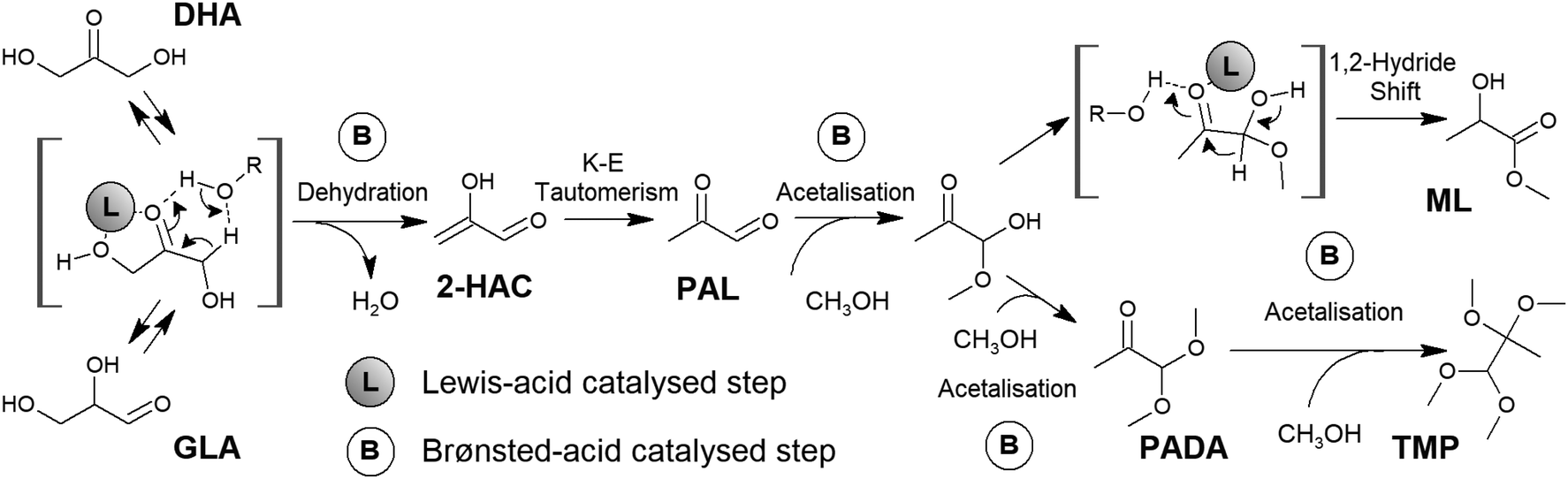 | ||
| Fig. 4 Transformation of trioses into methyl lactate in the presence of bifunctional Brønsted-Lewis acid catalysts. DHA: dihydroxyacetone; GLA: glyceraldehyde; 2-H-2PAL: 2-hydroxyacrolein; PAL: pyruvaldehyde; ML: methyl lactate; PADA: pyruvaldehyde dimethyl acetal; TMP: 1,1,2,2-tetramethoxypropane. Reaction intermediates have been highlighted in brackets. | ||
Since the discovery of a reaction pathway for the production of lactic acid from monosaccharides,110 the retro-aldol condensation of carbohydrates larger than trioses has been described as the rate-limiting stage. Accordingly, a large number of works devoted to the study of this transformation can be found in the literature. Because of their high catalytic activity, Sn-containing zeolites are the most studied group of catalysts in sugar retro-aldol condensation; among them, Sn-β has been revealed to be the most efficient catalyst because of its better catalytic performance in retro-aldol condensation compared to any other zeolite structure.117 This can be ascribed to several features of this unique material. On the one hand, the large pore size of the BEA structure allows the monosaccharides to diffuse inside the porous structure. Thus, mass transfer issues such as those occurring in medium and small pore zeolites, like MFI, are not there when using Sn-β.118–120 On the other hand, theoretical calculations have indicated that Sn sites incorporated in a BEA structure are stronger Lewis acid species than when they are incorporated in other zeolites, such as FAU;108 thus, the above observation verifies the enhanced catalytic activity of Sn-β in Lewis-acid driven transformations.
The synthesis of Sn-β through classical hydrothermal methods leads to almost defect-free materials with isolated tin species in a tetrahedral coordination; this coordination provides the Lewis acidity of the zeolite material. However, such synthesis procedures do not allow high metal loadings to be utilized and require long crystallization times.121–123 An efficient alternative to hydrothermal crystallization is post-synthetic metalation,124–127 which consists of a group of methods that allow the incorporation of high metal contents in zeolites by substituting aluminium or silicon, for metal atoms through reasonably simple and rapid procedures; additionally, the above methods can be accomplished while preserving the crystalline structure. Nevertheless, a post-synthetic stagnation of zeolites provides a different type of tin species compared to that of hydrothermal methods. Closed tin sites, corresponding to tetrahedrally coordinated (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiO)4Sn species, are obtained in zeolites prepared by hydrothermal crystallization methods, especially if crystallized through a fluoride method. In contrast, open tin sites are more abundant in materials prepared by post-synthetic metalation, and they are usually described as tri-coordinated (SiO)3Sn(OH) sites attached to the zeolite framework.128–130 Open tin sites display both stronger Lewis and Brønsted acidity compared to that of closed tin sites.131,132 Open tin sites have been described as the active catalytic sites both in 1,2-hydride shift transformations126,133,134 – which are required for the isomerization of aldoses into ketoses and the transformation of pyruvaldehyde hemiacetals into alkyl lactates – and retro-aldol condensation of sugars116,135 – which are needed in the production of trioses from large monosaccharides (Fig. 5). However, it has been described that the reversible interconversion of open and closed sites is feasible by hydration–dehydration processes,136 so that, regardless of the synthesis method, the speciation of tin is, theoretically, tunable after synthesis. Fig. 5 depicts the mechanism and reaction intermediates occurring at tin open sites in the isomerization of sugars through a 1,2-hydride shift pathway and in the retro-aldol condensation splitting of ketoses. The superior performance of tin-functionalized materials in a lactate synthesis from sugar monosaccharides has resulted in a large number of published works in scientific literature. Table 2 lists the most representative studies on the transformation of sugar monosaccharides into lactic acid/alkyl lactates in the presence of heterogeneous acid catalysts.
SiO)4Sn species, are obtained in zeolites prepared by hydrothermal crystallization methods, especially if crystallized through a fluoride method. In contrast, open tin sites are more abundant in materials prepared by post-synthetic metalation, and they are usually described as tri-coordinated (SiO)3Sn(OH) sites attached to the zeolite framework.128–130 Open tin sites display both stronger Lewis and Brønsted acidity compared to that of closed tin sites.131,132 Open tin sites have been described as the active catalytic sites both in 1,2-hydride shift transformations126,133,134 – which are required for the isomerization of aldoses into ketoses and the transformation of pyruvaldehyde hemiacetals into alkyl lactates – and retro-aldol condensation of sugars116,135 – which are needed in the production of trioses from large monosaccharides (Fig. 5). However, it has been described that the reversible interconversion of open and closed sites is feasible by hydration–dehydration processes,136 so that, regardless of the synthesis method, the speciation of tin is, theoretically, tunable after synthesis. Fig. 5 depicts the mechanism and reaction intermediates occurring at tin open sites in the isomerization of sugars through a 1,2-hydride shift pathway and in the retro-aldol condensation splitting of ketoses. The superior performance of tin-functionalized materials in a lactate synthesis from sugar monosaccharides has resulted in a large number of published works in scientific literature. Table 2 lists the most representative studies on the transformation of sugar monosaccharides into lactic acid/alkyl lactates in the presence of heterogeneous acid catalysts.
 | ||
| Fig. 5 Plausible mechanisms for (a) 1,2-hydride shift isomerisation of aldoses in ketoses, and (b) retro-aldol condensation of fructose to GLA and DHA, catalysed by open tin sites. Adapted from van der Graaff et al.126 and Elliot et al.135 | ||
| # | Catalyst | Operating conditions | Catalytic results | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Subs.a | Solv.b | S ccc (wt%) | C/Sd (wt%) | T (K) | t (h) | X S (%) | YLacth (%) | Produc.i (g gcat−1 h−1) | Deact.j | |||
| a Substrate. b Solvent. c Substrate concentration weight ratio in %. d Catalyst to substrate weight ratio in %. e Temperature conditions in Kelvin. f Reaction time. g Substrate conversion. h Yield towards product. i Productivity. j Catalyst deactivation: strong (st), moderate (m), low (l) and negligible (ne); n.s. (not studied), n.r. (not reported). | ||||||||||||
| Zeolites | ||||||||||||
| 1 | Sn-β (Si/Sn = 125) HT | Glucose | MeOH | 2.8 | 71.0 | 433 | 20 | >99 | 43 | 0.03 | n.s. | 153 |
| 2 | Sn-β (Si/Sn = 125) HT | Sucrose | MeOH | 2.8 | 71.0 | 433 | 20 | >99 | 64 | 0.05 | ne (6 runs) | 153 |
| 3 | Sn-MWW (Si/Sn = 52) PS | Glucose | MeOH | 2.8 | 71.0 | 433 | 20 | >99 | 44 | 0.03 | ne (3 runs) | 138 |
| 4 | Sn-USY (Si/Sn = 34) PS | Sucrose | MeOH | 2.4 | 42.0 | 433 | 2 | >99 | 30 | 0.38 | ne (3 runs) | 139 |
| 5 | Sn-β (Si/Sn = 200) HT | Glucose | H2O | 2.0 | 133.0 | 473 | 0.5 | >99 | 58 | 0.87 | m (5 runs) | 154 |
| 6 | Sn-β (Si/Sn = 112) PS–Hyerar | Glucose | MeOH | 3.1 | 65.0 | 433 | 10 | >99 | 58 | 0.09 | l (5 runs) | 140 |
| 7 | Sn-β (Si/Sn = 50) PS–Hyerar | Glucose | MeOH | 2.8 | 71.0 | 433 | 20 | >99 | 53 | 0.04 | st (3 runs) | 141 |
| 8 | Sn-β (Si/Sn = 50) PS-NPs | Glucose | MeOH | 3.1 | 65.0 | 433 | 10 | >99 | 47 | 0.07 | ne (8 runs) | 142 |
| 9 | Zn–Sn-β (Zn/Sn = 1/1) PS | Glucose | H2O | 2.2 | 71.0 | 463 | 2 | >99 | 48 | 0.34 | n.s. | 104 |
| 10 | Pb–Sn-β (Pb/Sn = 4/7) PS | Glucose | H2O | 2.2 | 89.0 | 463 | 2 | >99 | 52 | 0.29 | n.s. | 104 |
| 11 | Sn-β (Si/Sn = 200) PS + K2CO3 | Sucrose | MeOH | 3.0 | 33.0 | 443 | 4 | 94 | 72 | 0.54 | n.s. | 155 |
| 12 | Sn-USY (Si/Sn = 34) PS + KCl | Glucose | MeOH | 5.9 | 21.0 | 443 | 6 | >99 | 42 | 0.33 | m (4 runs) | 149 |
| Mesostructured materials | ||||||||||||
| 13 | Sn-MCM-41 (Si/Sn = 55) | Glucose | MeOH | 3.5 | 71.0 | 433 | 20 | 100 | 43 | 0.03 | l (3 runs) | 156 |
| 14 | Sn-SBA-15 (Si/Sn = 150) | Glucose | MeOH | 2.5 | 75.0 | 433 | 20 | >99 | 35 | 0.02 | ne (4 runs) | 157 |
| Metal organic frameworks | ||||||||||||
| 15 | Fe-MIL-100 | Fructose | H2O | 0.5 | 100.0 | 463 | 2 | 87 | 32 | 0.14 | st (4 runs) | 157 |
| 16 | ZIF-8 | Glucose | MeOH | 3.5 | 71.0 | 433 | 20 | 100 | 20 | 0.01 | n.s. | 158 |
| 17 | Mg-MOF-74 | Glucose | MeOH | 0.8 | 33.0 | 493 | 6 | 100 | 35 | 0.18 | l (4 runs) | 159 |
| 18 | Sn-UZAR-S10 | Glucose | MeOH | 3.5 | 71.0 | 433 | 20 | 100 | 10 | 0.01 | n.s. | 160 |
Notably, all the materials tested in the conversion of sugar monosaccharides into lactic acid demonstrated low production (Table 2) compared to those shown for the catalytic transformation of glycerol (Table 1). This is most likely due to the rate-limiting retro-aldol condensation of the sugar monosaccharides to obtain the C3 skeleton trioses in this pathway. Nevertheless, catalytic tests reveal important differences between the reported catalysts. Sn-β material, prepared through both hydrothermal crystallization and post-synthetic metalation, can convert hexoses into methyl lactate with reasonably good yields (∼40%).110,137 This material even shows enhanced catalytic activity (ML yield ∼64%) in the transformation of dimers such as sucrose (Table 2, entries 1 and 2), whose conversion requires hydrolysis into glucose and fructose; thus, the above observation suggests the existence of an increased confinement effect in the pores of the Sn-β zeolite material.110 The Sn-β catalyst has demonstrated superior catalytic activity in the conversion of sugar mono- and disaccharides compared to that of any other zeolite structure, including MFI,119 MWW,138 and FAU (Table 2, entries 3 and 4).139 In fact, it is able to provide higher lactate production from glucose than that of any other tested catalyst (Table 2, entry 5), even with short reaction times if appropriate temperature conditions are applied. However, the reusability of this material decreases as harsh reaction conditions are applied, most likely because of damage to the structure. Additionally, the small pore size associated with zeolites, even in the case of β zeolite, makes converting larger oligosaccharides, such as inulin, quite difficult139 because of mass transfer limitations.
Some authors have explored different strategies to enhance the textural properties of zeolites, which can be a plausible solution to the limitation imposed by mass transfer, by synthesizing Sn-containing hierarchical140,141 or nanosized zeolites.142 In both cases, the materials provided higher methyl lactate yields (47–58%, Table 2, entries 6–8) when treating glucose methanolic solutions compared to that of conventionally prepared materials. Nevertheless, these materials do not show improvement in the production of methyl lactate and demonstrate some deactivation, probably because structural damage during the reaction of these zeolites is more intense than in conventional ones.143 Although overcoming mass transfer issues enhance the catalytic activity of Sn-zeolites, the influence of this strategy is still far from providing quantitative lactate yields. Thus, some authors have focused their efforts on different strategies, such as the combination of different metal species to obtain tandem catalysts that are able to efficiently promote the cascade of transformations from hexoses to alkyl lactates. Combining tin salts with transition metal ions showing Lewis acidity, has been described to enhance the conversion of sugar monosaccharides to methyl lactate compared to the values obtained in the presence of individual metal species.111,144,145 This strategy has been tested in heterogeneous catalysts through the immobilization of tin species together with a second Lewis acid metal functionality – Zn,146 Pb,104 and Cr147 – incorporated in β zeolites. These materials show better catalytic performance than the monometallic systems in lactic acid production from glucose providing product yields of approximately 50% under conventional operation conditions (Table 2, entries 9 and 10). Unfortunately, the stability of these systems has not been provided.
A more efficient strategy to enhance alkyl lactate production with tin-based catalysts is the addition of alkali salts to the reaction medium (Table 2, entries 11 and 12).148 This strategy is based on inhibiting the occurrence of important hydrolytic side reactions, including the dehydration of hexoses to 5-hydroxymethylfurfural,149 the etherification of sugar monosaccharides to alkyl glycosides,150 and the acetalization of key reaction intermediates, such as pyruvaldehyde;151 these last reactions are present when using alcohol solvents. All these side reactions are promoted by Brønsted-type acid sites, such as those present in aluminium-containing materials,152 including Sn-containing zeolites prepared by post-synthetic metalation methodologies with intermediate dealumination stages. In this way, inhibiting the catalytic activity of the Brønsted acid sites allows the retro-aldol transformation pathway to advance in the conversion of sugars to alkyl lactate. Indeed, it has been postulated that alkali metal cations, such as sodium or potassium, modify the environment of the active tin sites and enhance their capacity to drive C–C cleavage reactions, such as sugar epimerization133,134 or retro-aldol condensation.161 This proximal interaction between the alkali cations and the tin sites seems to be linked to the stabilization of highly catalytically active open tin sites.162 In this way, the addition of alkali metal cations to tin-based catalysts can be considered an efficient strategy to enhance their intrinsic activity in the rate-limiting retro-aldol condensation of monosaccharides to intermediate trioses.161 This strategy has been successfully applied to different Sn-containing materials, but in the case of the Sn-β catalyst, it has been demonstrated to be highly influential; the achieved methyl lactate yield obtained from sucrose when treated with an alkali-free Sn-β zeolite prepared by post-synthetic metalation changes from 20% to 72% when K2CO3 is added to the reaction medium.155 Strong evidence of the increase in the catalytic activity of Sn sites by interaction with alkali cations can be found in the comparatively high methyl lactate production achieved with these materials (Table 2, entries 11 and 12).
Apart from tin-containing zeolites, a wide variety of alternative solid acid catalysts have been tested in the conversion of hexoses into alkyl lactates. An interesting class of those catalysts are those based on mesostructured silica as supports. These materials feature large pores, which avoid mass transfer issues such as those affecting the majority of zeolites, especially when treating large substrates. Thus, Sn incorporated into MCM-41156 and SBA-15157 materials provides quite good catalytic activity in the conversion of sugar monosaccharides (Table 2, entries 13 and 14) but also promotes the transformation of sugar oligomers directly to alkyl lactates; in the case of a Sn-SBA-15 material, it attains methyl lactate yields of above 57% when treating inulin in the presence of methanol at 433 K for 20 h.157 Nevertheless, lactate production obtained with these materials is quite low, thus, they provide negligible advantages over zeolite-based catalysts. Another interesting group of Lewis acid catalysts, metal organic frameworks (MOFs), have also been tested for lactic acid production from hexoses (Table 2, entries 15–18). Within this group, many different types of materials, such as Fe-MIL-100,163 ZIF-8,158 Mg-MOF-74,159 and Sn-UZAR-S10,160 have been tested in the conversion of hexoses to lactic acid/alkyl lactates. Nevertheless, these materials display comparatively lower catalytic performance than tin-based zeolites, as MOFs require harsh reaction conditions to achieve lactic acid/alkyl lactate yields in a range of 10–35%.
The valorization of pentoses, such as xylose, xylulose or lysose, to lactic acid/alkyl lactate has been much less studied than the case of hexoses, mainly because pentoses provide lower lactate production. In addition, treating pentoses leads to a wide variety of products coming from the glycolysis pathway. The retro-aldol condensation of these monosaccharides leads to the formation of a triose (GLA if starting from an aldose or DHA if treating a ketose) and glycolaldehyde. Trioses, as previously described, can be directly transformed into lactic acid, but it is not the same for glycolaldehyde. This C2 product can evolve through a multitude of reaction pathways, including acetalization, or through aldol condensation to yield tetroses and hexoses, which introduces more complexity to the reaction media. In this way, the conversion of the simplest sugar, C2 glycolaldehyde, can produce alkyl lactates/lactic acid, along with many other interesting products, as will be seen in the next section. For instance, Sn-β has demonstrated high catalytic activity in the conversion of pentoses – e.g., xylose, ribose, arabinose – to methyl lactate in methanol media. This material provides a methyl lactate yield of 42% after treating xylose for 16 h in methanol media.107 Additionally, the above reaction provides some other products coming from the conversion of glycolaldehyde, such as glycolaldehyde dimethyl acetal (GADMA) – produced through the acetalization of GAL – or methyl vinyl glycolate (MVG) – evolving from the tetrose produced through the aldol condensation of two molecules of GAL.181
Trioses have been extensively studied in lactic acid/alkyl lactate production because no variation in carbon chain length is required, and thus, the rate-limiting reaction stage – retro-aldol glycolysis – is avoided. Among the two isomers of trioses – GLA or DHA – the latter has been more frequently reported as a feedstock, not only because of its reduced price but also because of the minimal differences in terms of substrate conversion or product selectivity when starting from these two trioses. Aluminium, gallium, tin, lead and several other multivalent cations, which show Lewis acid properties, have demonstrated excellent catalytic activity in the conversion of trioses to lactic acid under homogeneous conditions.182–186 Thus, most of the studies dealing with heterogeneous catalysts are based on these metals, which have been immobilized on a large variety of supports. The vast majority are focused on combining the metal Lewis acid functionalities with – weak – Brønsted acid sites in the same catalyst, as both are required for efficiently converting trioses into lactic acid.
In this regard, the work by de Clippel et al.164 is one of the best examples of a rational design of bifunctional Brønsted/Lewis heterogeneous acid catalysts (Table 3, entry 1). This material, consisting of a hybrid carbon-MCM-41 support functionalized with tin species, displays weak Brønsted acidity, provided by oxygenated species in the carbon support, together with unique Lewis acid properties from the supported tin species. The result is a highly active and selective catalyst able to quantitatively convert dihydroxyacetone (100% conversion) into ethyl lactate (100% selectivity) under very mild reaction conditions. Although this material is probably the benchmark to beat in terms of catalytic activity, selectivity, and productivity, its complex synthesis method makes its application difficult at the industrial scale. Nevertheless, the literature contains many examples of heterogeneous catalysts applied to the conversion of trioses into lactic acid, whose simplicity makes their advancement to an industrial scale much easier.
| # | Catalyst | Operating conditions | Catalytic results | Ref. | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Solv.a | S cc b (wt%) | C/Sc (wt%) | T (K) | t (h) | X S (%) | YLactg (%) | P Lact (g gcat−1 h−1) | Deactiv.i | |||
| a Solvent. b Substrate concentration weight ratio in %. c Catalyst to substrate weight ratio in %. d Temperature conditions in Kelvin. e Reaction time. f Substrate conversion. g Yield towards product. h Productivity. i Catalyst deactivation: strong (st), moderate (m), low (l) and negligible (ne); n.s. (not studied), n.r. (not reported). | |||||||||||
| 1 | Sn–C-MCM-41 Ox@573K | EtOH | 2.2 | 111.0 | 363 | 6 | 100 | 100 | 0.15 | ne (3 runs) | 164 |
| Aluminum containing catalysts | |||||||||||
| 2 | H-MOR (Si/Al = 10) | MeOH | 3.5 | 71.0 | 388 | 24 | 74 | 8 | <0.01 | n.s. | 165 |
| 3 | H-ZSM5 (Si/Al = 11.5) | MeOH | 3.5 | 71.0 | 388 | 24 | 76 | 17 | 0.01 | n.s. | 165 |
| 4 | H-β (Si/Al = 12.5) | MeOH | 3.5 | 71.0 | 388 | 24 | 88 | 42 | 0.02 | n.s. | 165 |
| 5 | USY (Si/Al = 6.0) | MeOH | 3.5 | 71.0 | 388 | 24 | 99 | 96 | 0.06 | ne (4 runs) | 165 |
| 6 | USY (Si/Al = 2.5) | MeOH | 4.4 | 111.0 | 383 | 4 | n.r. | 82 | 0.18 | ne (4 runs) | 166 |
| 7 | Al-MCM-41 (Si/Al = 10) | EtOH | 4.4 | 111.0 | 363 | 6 | n.r. | 5 | 0.01 | n.s. | 167 |
| Gallium containing catalysts | |||||||||||
| 8 | Ga-MCM-41 (Si/Ga = 10) | EtOH | 4.4 | 111.0 | 363 | 6 | n.r. | 16 | 0.02 | st (5 runs) | 167 |
| 9 | Ga-USY (6 wt% Ga) | MeOH | 3.0 | 67.0 | 393 | 24 | 99 | 82 | 0.05 | n.s. | 168 |
| Tin containing catalysts | |||||||||||
| 10 | Sn-β (Si/Sn = 125) | MeOH | 3.5 | 71.0 | 353 | 24 | 100 | >99 | 0.06 | ne (3 runs) | 169 |
| 11 | Sn-MWW (Si/Sn = 52) | MeOH | 2.9 | 69.0 | 393 | 24 | >99 | >99 | 0.06 | n.s. | 138 |
| 12 | Sn-MFI (Si/Sn = 200)-Hyerar | MeOH | 3.5 | 800.0 | 343 | 24 | >99 | 85 | 0.01 | n.s. | 119 |
| 13 | Sn-USY (Si/Sn = 100) | MeOH | 2.9 | 113.0 | 313 | 5 | ∼100 | 97 | 0.17 | m (5 runs) | 170 |
| 14 | Sn-MCM-41 (Si/Sn = 9.9) | EtOH | 0.6 | 225.0 | 338 | 6 | 100 | 92 | 0.07 | ne (5 runs) | 171 |
| 15 | Sn-Mont (23 wt% Sn) | MeOH | 2.8 | 44.0 | 423 | 15 | >99 | 97 | 0.15 | n.s. | 172 |
| 16 | Sn–Al2O3 (7.9 wt% Sn) | EtOH | 4.3 | 44.0 | 373 | 7 | 99 | 68 | 0.22 | n.s. | 173–175 |
| 17 | SnO2–Nb2O5 (Sn/Nb = 6.1) | H2O | 2.2 | 71.0 | 433 | 6 | 99 | 99 | 0.23 | m (6 runs) | 176 |
| 18 | SnO2·NbOPO4·(Nb/Sn = 3) | H2O | 3.5 | 71.0 | 413 | 6 | ∼100 | 92 | 0.22 | St (6 runs) | 177 |
| 19 | Sn–Ca5(PO4)3(OH) | BuOH | 1.6 | 80.0 | 383 | 6 | n.r. | 74 | 0.15 | n.s. | 178 |
| 20 | Sn3(PO4)4·PEG2000 | H2O | 3.5 | 71.0 | 413 | 4 | ∼100 | 96 | 0.34 | m (5 runs) | 179 |
| 21 | SnO2·Si3(PO4)4 (Si/Sn = 6) | H2O | 3.5 | 71.0 | 413 | 5 | ∼100 | 94 | 0.26 | l (3 runs) | 180 |
For aluminium-based catalysts, the first steps in the development of heterogeneous catalysts for triose valorization to lactic acid are accomplished using zeolites with a high proportion of extra-framework Al3+, which provide Lewis acidity. Al3+ cations have demonstrated excellent catalytic activity in a Meerwein–Schmidt–Ponndorf–Verley-type hydrogen transfer and Oppenauer oxidation reactions,187,188 whose coupling at adjacent groups in the same molecule is another way to understand an intramolecular 1,2-hydride shift;29 the above mechanism is a key step in the isomerization of the hemiacetal, which comes from pyruvaldehyde, to alkyl lactate (Fig. 4). In this way, coupling Brønsted and Lewis acidity in Al-rich zeolites, such as USY materials, provides very high catalytic activity in the conversion of dihydroxyacetone to methyl lactate (Table 3, entries 2–6).165,166 These materials, with a faujasite structure, demonstrate superior performance compared to other zeolites with smaller pore sizes, such as HZSM-5 or mordenite,165 thus leading to higher lactate production. Other Al-containing materials with wide pores, such as amorphous silica-alumina or MCM-41 (Table 3, entry 7),167 display poor catalytic activity, suggesting that the control of the reaction space inside the zeolite pores exerts a confinement effect, which is absent in large pore-sized materials. Gallium has also been tested in zeolite structures168 and in MCM-41167 for promoting the conversion of DHA to methyl lactate (Table 3, entries 8 and 9). Ga species provide good catalytic activity in the transformation of concentrated DHA (up to 15%) to methyl lactate (∼90% yield), probably because gallium species exhibit stronger Lewis acidity compared to that of aluminium. However, similar lactate production is achieved to those shown by analogous Al-containing materials.
Tin-containing materials are, by far, the most active catalysts in the transformation of trioses into alkyl lactates, and numerous examples of this type of catalyst can be found in the literature. Sn-zeolites, comprising BEA,169 MWW,138 MFI,119 and FAU,170 have been reported to be excellent catalysts in these reactions (Table 3, entries 10–13). In this sense, large pore-sized zeolites, such as Sn-β and Sn-USY, layered zeolites, such as Sn-MWW and hierarchical Sn-MFI, provide high lactate yields from DHA, as the mass transfer does not control the reaction rate of such small substrates. Accordingly, the lactate production achieved with these materials is much higher than that provided with medium pore-sized zeolites. Thus, zeolites promote a nearly quantitative conversion of DHA into methyl lactate under very mild reaction conditions; thus, a zeolite alternative is probably one of the closest to advance to an industrial scale. Studies on the scalable production of Sn-MFI through alkaline metalation – alkali-assisted metal grafting on zeolites consisting of a simultaneous desilication and metalation process – and their use in the direct transformation of DHA into methyl lactate, demonstrate the very high stability of these catalysts and their possibility to be used in the continuous production of lactic acid.189 Techno-economic and environmental analyses of different processes devoted to the transformation of DHA into lactic acid in the presence of Sn-MFI suggest that this alternative can be more economically competitive and environmentally friendly than the conventional fermentation-based production of lactic acid. Considering the low lactate production shown by Sn-MFI, it is expected that other zeolites with better catalytic performance, such as Sn-β and Sn-USY, will provide even higher profitability.
Though theoretically less active than its zeolite counterparts, the use of tin as a Lewis acid functionality has been extended to other solids for trioses to lactate production (Table 3, entries 14–21). These catalytic systems include mesostructured materials such as MCM-41,171 high capacity ion-exchange clays such as montmorillonite,171 mixed metal oxides (with Al2O3173–175 or Nb2O5176), and different metal phosphates.177–180 The syntheses of these materials also look for the combination of Lewis and Brønsted acid-type catalysts to promote the cascade of transformations from dihydroxyacetone to methyl lactate. However, controlling the loading and strength of the Brønsted acid sites is not always easy depending on the solid support, and thus, several supports, such as alumina or hydroxyapatite, produce reasonably high yields of side products (25–30%). These include pyruvaldehyde dimethyl acetal (PADA) and tetramethoxy propane (TMP) that comes from the acetalization of PAL with methanol and is mediated by Brønsted acid sites. Nevertheless, the high lactate production values calculated for these materials suggest that when using a high loading of tin, meso-macroporous materials are some of the most interesting options available to build an efficient catalyst for a one-pot transformation of trioses into alkyl lactates, especially when using suitable catalytic supports such as niobium oxide or niobium phosphate.
2.1.2.3. Summary and prospects of lactate synthesis. The production of lactic acid has been approached through a wide variety of methodologies. Scientific literature suggests that using glycerol as the starting raw material is a more productive option compared to the conversion of sugar monosaccharides, although sugar monosaccharides are more plenty and potentially a cheaper feedstock. However, the chemocatalytic conversion of renewable feedstock into lactic acid is becoming a highly interesting option as an alternative to biological pathways. Unfortunately, there are still some difficulties in finding an efficient chemical process that is able to compete with commercial fermentation technology. Most of these issues are linked to the development of new catalytic systems. Within this context, and since the industrial production of sugar monosaccharides is easier to realize in the form of aqueous streams, it is most desirable to develop catalyst systems that are able to function under aqueous phase conditions and that are able to convert concentrated sugar solutions. Additionally, the development of robust catalysts able to preserve their integrity and catalytic activity even under harsh reaction conditions in aggressive reaction media, such as water systems, is a crucial aspect to face in the design of industrial catalytic systems for lactic acid production.
Finally, tacticity of poly(lactic acid) is an important feature that influences the final properties of PLA. Since tacticity in PLA is linked to the chirality of the monomer, its control during the synthesis or the separation of the different enantiomers is of major importance to obtain commercial-grade PLA. Chemocatalytic pathways display an advantage over fermentation in their production of the D-(R)-enantiomer of lactic acid, which is essential in stereoblock polymers. However, the enantioselective production of lactic acid/alkyl lactates through chemocatalytic pathways is still an unexplored feature that must be addressed in the future. This will allow the control of tacticity to occur rather than with an expensive enantiomer separation at the end, thus placing the chemocatalytic option as the most advantageous alternative for lactic acid production.
3-Hydroxypropanoic acid (3-HPA) is an isomer of lactic acid and was proposed by the United States Department of Energy as one of the top 12 value-added platform molecules among renewable biomass products.191 3-HPA is produced from sugars via fermentation by engineered microorganisms. Several enzymes have exhibited the potential to easily accomplish the biotransformation of glucose or glycerol into 3-HPA.194–196 However, in addition to the low productivity of the biological route, the separation and purification is an important issue to be solved. In addition, glycerol has emerged as another suitable fermentable feedstock for the production of 3-HPA; mainly raw glycerol derived from the biodiesel industry, due to its competitive price.
There are many possibilities to synthesize 3-HPA through chemocatalysis, such as the hydrolysis of propiolactone, the hydration of acrylic acid with a strong mineral acid, or the catalytic oxidation of 1,2-propanediol.197 However, these synthesis routes usually involve expensive raw materials and harsh reaction conditions. Alternatively, the synthesis of 3-HPA from renewable feedstock has also been addressed (Fig. 6). Thus, for instance, glyceric acid, a glycerol derivative obtained by oxidation, can be transformed into 3-HPA by a reduction step in the presence of hydriodic acid, which catalyses the production of 3-iodopropionic acid, subsequently hydrolysed to 3-HPA.198 The reaction pathway involves the production of acrylic acid as the key intermediate, through the substitution of the hydroxyl moieties of glyceric acid after iodination with HI but before the elimination of iodine. I2 is then reduced with H2 in the presence of a rhodium catalyst to form HI again, which attacks the double bond of acrylic acid to form 3-iodopropionic acid. This can be easily converted into 3-HPA by hydration. This methodology is quite efficient in the conversion of glyceric acid to 3-iodopropionic acid and yields a quantitative conversion in a few hours at a moderate temperature (373 K), while starting from a reasonably concentrated aqueous glyceric acid solution (20 wt%). The catalytic system has been shown to be quite stable and reusable. Nevertheless, the methodology involves extensive use of iodine and HI, and it requires testing at a larger scale.
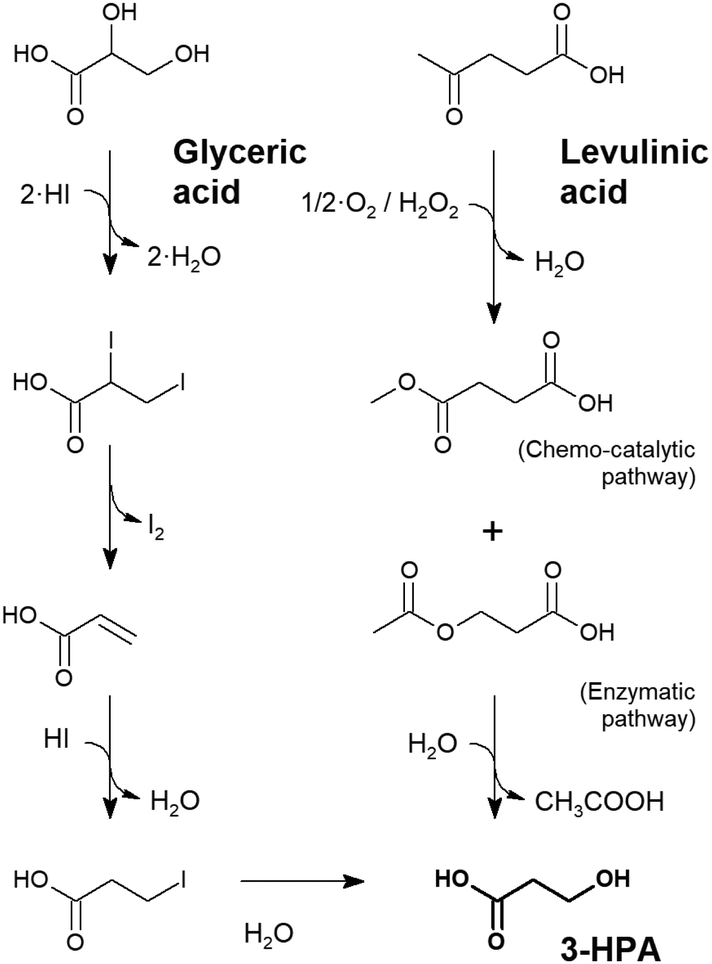 | ||
| Fig. 6 Synthesis routes for 3-hydroxypropionic acid from renewable sources. Left branch, from glyceric acid; right branch, from levulinic acid. | ||
A different approach to the preparation of 3-HPA is the use of levulinic acid, a C5 γ-keto-acid, which can be produced from glucose-containing feedstock, such as lignocellulose biomass. The production of this platform molecule has been tested at a semi-industrial scale, and different industrial initiatives and pilot plants have been built for its production from renewable biomass;199,200 thus, the processes using levulinic acid as a raw feedstock will probably become a reality in the future, as explained elsewhere in this review. The most direct pathway to transform levulinic acid into 3-hydroxypropionic acid consists of a Baeyer–Villiger oxidation to yield 3-acetoxy-propionic acid, which can be later hydrolysed to acetic and 3-hydroxypropionic acids. This strategy has been applied to the conversion of methyl levulinate using sulfonic acids, transition metal sulfonates201 and tungstic acid202 as catalysts and hydrogen peroxide as an oxidant. However, this strategy demonstrates a lack of selectivity for the acetoxy product, as the oxidative regioselectivity of the ketone group is displaced in favour of dimethyl succinate. As an alternative to these catalytic systems, the use of Baeyer–Villiger monooxygenases has been revealed to be a much more selective and productive option; the above process operates with oxygen as the oxidant and at temperatures close to ambient conditions (303 K).203 The methodology has been validated for gram-scale production of alkyl-3-acetoxy-propionates,204 while showing an almost quantitative conversion of the substrates and total selectivity towards the 3-HPA derivative.
2.1.4.1. C4 hydroxyacids. As previously seen, the retro-aldol condensation pathway does not always produce trioses from sugar monosaccharides but other interesting short-chain sugars, such as glycolaldehyde or tetroses (e.g., erythrose). Both products can be formed through the retro-aldol condensation of a C6 aldohexose (e.g., glucose). However, glycolaldehyde can also evolve from the glycolysis of a pentose, whereas tetroses can also be produced by aldol condensation of two glycolaldehyde molecules. In this way, tetroses, unlike C5 and C6 sugar monosaccharides, are scarcely present in nature, but can be produced thereof through glycolytic and aldol condensation pathways. These C4 sugars are an interesting feedstock for the preparation of α-hydroxyacids,89,164 which can be further transformed into a new generation of hyper-crosslinked functional and biodegradable polyesters. Commercial interest is thus far very limited because no application has yet been identified for these polymers.
Tin has been demonstrated to promote the transformation of tetrose sugars into methyl vinyl glycolate (MVG) and methyl-4-methoxy-2-hydroxybutanoate (MMHB) (Fig. 7). The proposed reaction mechanism for these transformations205 involves a cascade of reactions in which a tetrose undergoes a double dehydration, with an intermediate isomerization, to yield vinylglyoxal. This key intermediate can be alkylated by the attack of an alcohol molecule to the double bond and evolve by acetalization and isomerization through a 1,2-hydride shift to yield an α-hydroxyacid. Whether MVG or MMHB is formed depends on the existence of confinement effects around the Sn catalytic sites.206 Thus, if a soluble salt is used, such as tin tetrachloride, or tin-functionalized mesoporous materials – e.g., Sn-MCM-41 or Sn-SBA-15 – are employed as catalysts, then the attack of an alcohol molecule to the double bond of the key intermediate – vinylglyoxal – is free to occur and yields MMHB as the main product. However, if a microporous tin-containing catalyst is used as a catalyst – e.g., Sn-zeolites, such as β or MFI – the formation of MVG is enhanced at the expense of MMHB. This difference in behaviour between micro- and mesoporous catalysts has been ascribed to the existence of steric restrictions in the vicinity of the catalytic tin sites. Accordingly, steric hindrances around the catalytic centres hinder the alkylation of vinylglyoxal, thus yielding MVG in a higher ratio.
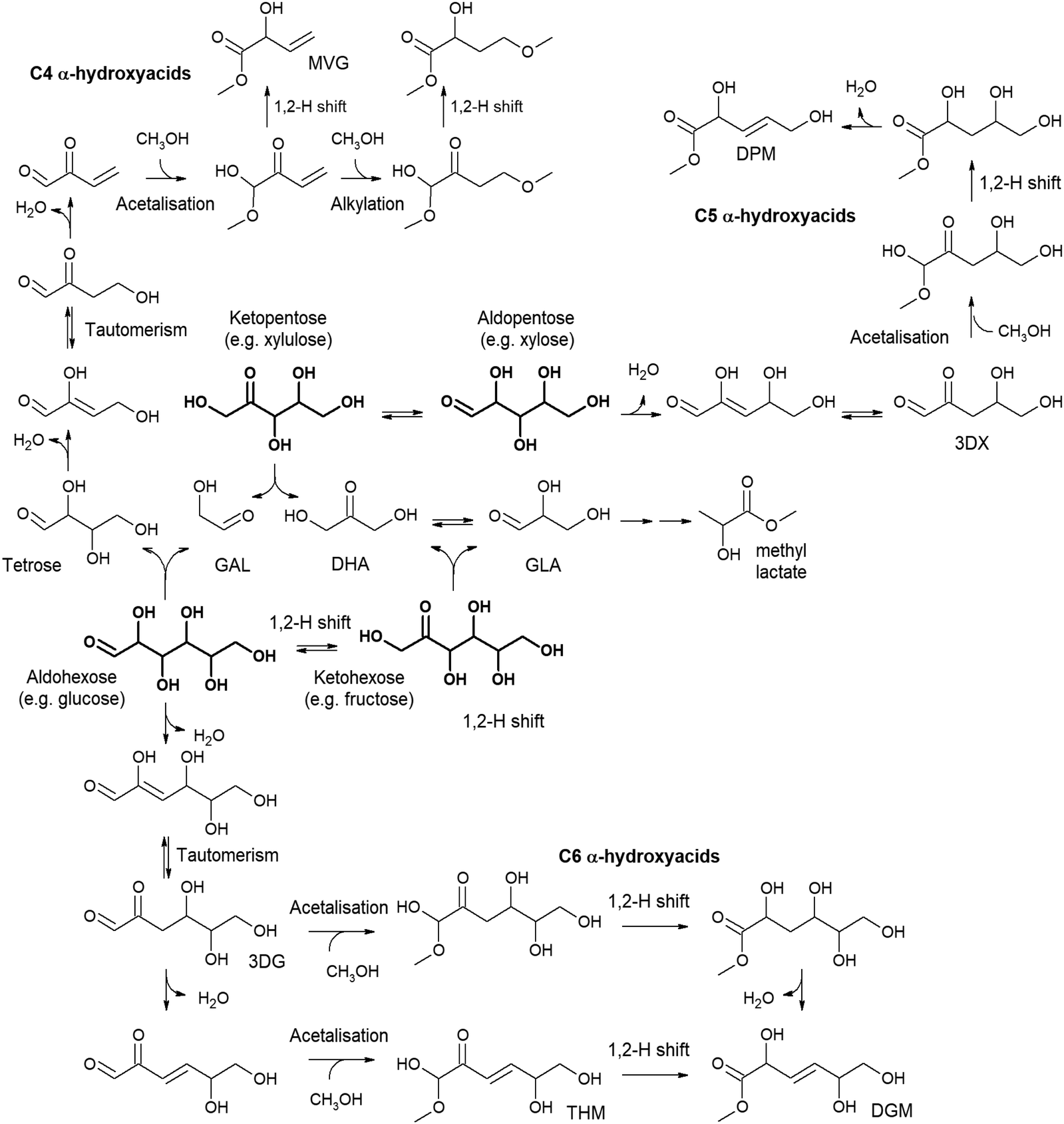 | ||
| Fig. 7 Transformation of sugar monosaccharides through plausible retro-aldol + aldol condensation routes and the related α-hydroxyacids that are produced. 3DG: 3-deoxyglucosone; MTHH: methyl-2,5,6-trihydroxyhex-3-enoate; MDG: methyl-3-deoxygluconate, DHA: dihydroxyacetone; GLA: glyceraldehyde; GAL: glycolaldehyde; ML: methyl lactate; 3DX: 3-deoxyxylosone; MDHP: methyl-2,5-dihydroxypent-3-enoate; MVG: methyl vinyl glycolate; MMHB: methyl-4-methoxy-2-hydroxybutanoate. | ||
Though highly interesting for the synthesis of functional polyesters, the preparations of MVG and MMHB are based on synthetic routes starting from tetroses, whose presence in natural sources is scarce. Thus, the synthesis of tetroses is a matter of interest, and most of the studies on their production have focused on taking advantage of the activity of several Lewis acids in sugar epimerization through the Bílik reaction.207,208 This transformation can be described as a 1,2-carbon shift combining consecutive retro-aldol and aldol condensation stages, in which an aldohexose undergoes a C–C bond cleavage occurring between the C2 and C3 carbons, followed by the creation of a new C–C bond between the C1 and C3 carbons. Thus, if the second stage does not occur, then the hexose splits into C2 and C4 sugars.207 Tungsten, molybdic acids, and MoO3, which are all strong Lewis acid solids, can convert an aldose, such as glucose or mannose, into glycolaldehyde through a double retro-aldol condensation in a reasonably efficient way.113,209 For instance, MoO3 can drive the conversion of mannose into glyceraldehyde with a 44.7% yield after a contact time of 60 s in methanol media. However, the addition of borate salts to this type of catalyst stabilizes the tetrose, preventing the second retro-aldol condensation step from occurring; thus, the selectivity of the catalytic system is tuned to the formation of erythrose and its isomers. Moreover, borate salts also interact with glycolaldehyde, leading to its aldolic condensation to produce new C4 sugars and enhancing the productivity of the system. Under optimal conditions, 25% of a methanolic solution of mannose can be converted into eythrose.192 Nevertheless, this strategy is based on the use of soluble borate salts in reasonably high amounts relative to the substrate, which makes practical applications difficult.
As an alternative to the synthesis of tetroses through the modified Bílik reaction, Tolborg et al. reported a simplified strategy based on the conversion of glycolaldehyde into tetroses using medium pore-sized shape-selective zeolites such as Sn-MFI.210 These catalytic systems can drive the aldol condensation of glycolaldehyde to tetroses in a very efficient way, as the limited space inside the pores of the MFI structure prevents a second aldol condensation to produce hexoses. Under optimal conditions, Sn-MFI is able to largely transform aqueous solutions of glycolaldehyde dimer (74%), with an outstanding selectivity towards C4 products (96%) and excellent carbon balance; the above can be accomplished in water at 353 K after a short contact time (0.5 h). The above phenomenon, namely, the transformation of glycolaldehyde into tetrose, has also been described with a Sn-β zeolite,107 but the large pore size of the BEA structure does not provide as high a shape selectivity as the MFI structure does, since the formation of hexoses is also detected.
2.1.4.2. C5–C6 hydroxyacids. Analogous to the formation of C4-hydroxyacids from tetroses, several hydroxyl-rich hydroxyacid esters showing C5 and C6 carbon backbones have been recently identified when treating pentoses and hexoses in the presence of tin-containing catalysts.181 The identified chemical routes for the production of these highly functionalized hydroxyacids are based on the initial dehydration of xylose (an aldopentose) or glucose (an aldohexose), into 3-deoxyxylosone211 or 3-deoxyglucosone,212 respectively. These transformations require the absence of retro-aldol condensation transformations to preserve the integrity of the carbon backbones of the pentoses and hexoses. These key intermediates undergo similar reaction pathways to those already described for tetroses, such as acetalization and 1–2 hydride shift pathways (Fig. 7), to yield methyl-2,5-dihydroxypent-3-enoate (MDHP), if starting from xylose, and methyl-2,5,6-trihydroxyhex-3-enoate (MTHH), if starting from glucose, as the main product. These monomers are better produced in the presence of tin-containing β zeolites, which are prepared through the fluoride method in the absence of alkali salts and bases. With the above zeolite it is presumed that the key active sites that promote this reaction stage, starting with beta dehydration, are probably the tin sites in closed environments. Nevertheless, the production of these large functional hydroxyacids is still quite low, and a deeper understanding of the reaction mechanism and the requirements for the active sites are still required to enable their production in high quantities.
2.1.4.3. Summary and prospects of the synthesis of C4–C6 hydroxyacids. A number of studies have opened new alternatives for the selective transformation of sugar monosaccharides, not only for hexoses and pentoses but also for tetroses, into C4–C6 α-hydroxyacids bearing different functionalities that are liable to be transformed into functional polymers with tunable properties. Despite the high potential of these hydroxyacids to produce functional polymers, there are still few reports dealing with the synthesis of these novel polymers. Therefore, the foreseen advantages coming from the presence of the different functionalities in the hydroxyacids need to be clarified to ascertain the potential interest of the polymer industry in these new monomers.
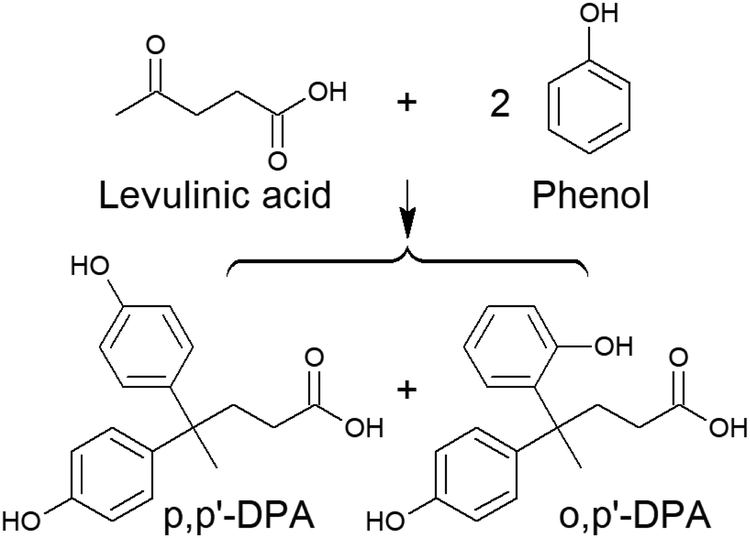 | ||
| Fig. 8 Synthesis of diphenolic acid by condensation of phenol with levulinic acid. | ||
Levulinic acid (LEVA) has been identified by the US Department of Energy as one of the top 10 platform molecules derived from biomass.191 In addition to DPA, γ-aminolevulinic acid (DALA)217–219 with potential for the production of renewable polymers, can also be obtained from LEVA and will be discussed later. LEVA can be produced from lignocellulosic biomass either by direct hydrolysis–dehydration of C6 cellulosic sugars (not commercial yet) or from hemicellulosic pentoses; the latter requires a multi-step process involving furfural formation by dehydration of pentoses, followed by its hydrogenation to furfurylic alcohol that must be subsequently transformed into LEVA.219–221 In any case, the use of LEVA as a feedstock for monomer production is still penalised by its high cost. The procurement and processing of inexpensive lignocellulosic feedstocks and advanced cost-effective and competitive technologies to produce it are still needed to decrease its final price and make it more attractive for these applications. Although still far from commercialisation, the catalytic technologies to produce DPA from LEVA will be reviewed, as once inexpensive LEVA is available, these monomers can be price competitive.
Fig. 8 shows the condensation reaction of levulinic acid with phenol via Friedel–Crafts hydroxyalkylation. The p,p′-DPA isomer is that proposed as an alternative to bisphenol. The condensation reaction is a typical Brønsted acid-catalysed reaction. Strong Brønsted mineral acids (H2SO4 and HCl) present many drawbacks (corrosiveness, difficulties in handling as well as separation, and downstream waste treatment)222 that prompted the development of solid acid catalysts. But to the best of our knowledge, few investigations have been conducted so far, that will be briefly revised below.
A mesoporous H3PW12O40/silica composite prepared by direct sol–gel condensation provided an excellent TOF over 0.015 s−1 by using a phenol/LEVA molar ratio of 4 and a weight catalyst/LEVA of 0.127, at 373 K after 8 h (LEVA conversion of 80.3% and p,p′/o,p′ = 2.9).223–225 with a maximum productivity of 1.954 g DPA gcat−1 h−1. The best catalytic performance among the different forms of silica studied was obtained for SBA-15 silica with a mesoporous texture that eased the accessibility and diffusion rate of substrates and bulky DPA. The catalyst was rapidly deactivated. No leaching was detected and the catalyst recovered its initial catalytic activity after calcination under air at 693 K. No information of other products was provided but the formation of phenol oligomers and LEVA has also been reported in other works dealing with this condensation process.226
Sulfonated hyperbranched poly(arylene oxindole) catalysed the condensation of phenol and LEVA, with a 1:1 molar ratio of thiols as a promoter.226,227 By using a phenol/LEVA molar ratio of 3 and a catalyst/LEVA weight ratio of 0.127 at 373 K, under an inert atmosphere for 16 h, a LEVA conversion of 69.5 and DPA yield of 52.9 mol% were achieved, with a TOF of 3.6 × 10−4 s−1 and a p,p′/o,p′ of 19.5 (0.644 g DPA gcat−1 h−1). Other solid acid catalysts, such as Amberlyst-15DRY, Nafion NR50 and pure H3PW12O40 heteropolyacid, were also studied in this condensation process, but they presented lower activity, DPA yield and p,p′/o,p′ ratio. This polymer, soluble in water, can be recovered by ultrafiltration,228 but the authors did not provide data about the recycling and solubility of sulfonated hyperbranched poly(arylene oxindole).
Later, Liu et al. using Brønsted acidic ionic liquids (BAILs) and a phenol/LEVA molar ratio of 4, at 333 K after 48 h229 achieved a high DPA yield (91 mol%), with a p,p′-DPA/o-p′-DPA ratio = 100. A maximum TOF of 2.34 × 10−5 s−1 and productivity of 0.0329 g DPA gcat−1 h−1 were reached. Alkyl levulinates can be used instead of LEVA (diphenolic esters are more suitable than DPA for further transformation), a mixture of DPA and diphenolic esters was produced by the hydrolysis of diphenolic esters to DPA. The highest diphenolic ester yield (83–87 mol%) was attained by reacting LEVA and phenol for 48 h and then adding alcohol for 8 h, instead of the corresponding alkyl levulinate (the DPA yield was 2–6 mol%). The BAIL catalyst suffered a mild deactivation, and the yield of ethyl diphenolic ester declined from 83 to 74 mol%, after 5 recycling runs.
In summary, the highest TOF and productivity values were obtained with a H3PW12O40/silica catalyst, much higher than those attained with homogeneous organic catalysts. However, the drawbacks associated with the low number of active sites and the high susceptibility to leaching require the development of more robust, active and inexpensive solid acid catalysts.
2.2. Acrylic acid
Acrylic acid (AA) possesses two main uses in the polymer industry:230 (i) production of acrylic esters and resins, including alkyl acrylates (alkyl: methyl, ethyl, butyl and 2-ethylhexyl), used as ingredients in paint formulations, adhesives, polishes, coatings, textiles and plastics; and resins for moulding powders for signs, construction units, and decorative emblems, and (ii) production of polyacrylic acid polymers, which are cross-linked polyacrylates and super absorbents, used for nappies and feminine hygiene products. AA also finds application in the production of detergent polymers and flocculants for wastewater treatment plants.The global market for AA was approximately 5400 ktons in 2015, but its worldwide consumption rate could reach more than 8000 ktons by 2020 due to new applications, including personal care products, detergents and products that are used for adult incontinence. The market price is $1600–1800 per ton for low-grade AA and $1900–2200 per ton for glacial grade AA.231 The consumption is divided between Asia-Pacific (46%), the USA (27%) and Western Europe (21%), with BASF, Dow and Arkema being the most important producers.
AA became commercially available by the Reppe reaction processes based on the Reppe reaction of acetylene with water and carbon monoxide or an alcohol and carbon monoxide (Fig. 9). This reaction can be either conducted at atmospheric pressure in the presence of HCl and nickel tetracarbonyl at 313 K or at high pressure (Reppe process), as reported by BASF and Badische Corp., with conditions of 14 MPa and 473 K and a nickel bromide–copper(II) bromide catalyst. In addition, the reaction of ketene, produced by the pyrolysis of acetic acid or acetone, with formaldehyde was also used, although these two processes have been largely replaced by the direct oxidation of propene, which is described next.
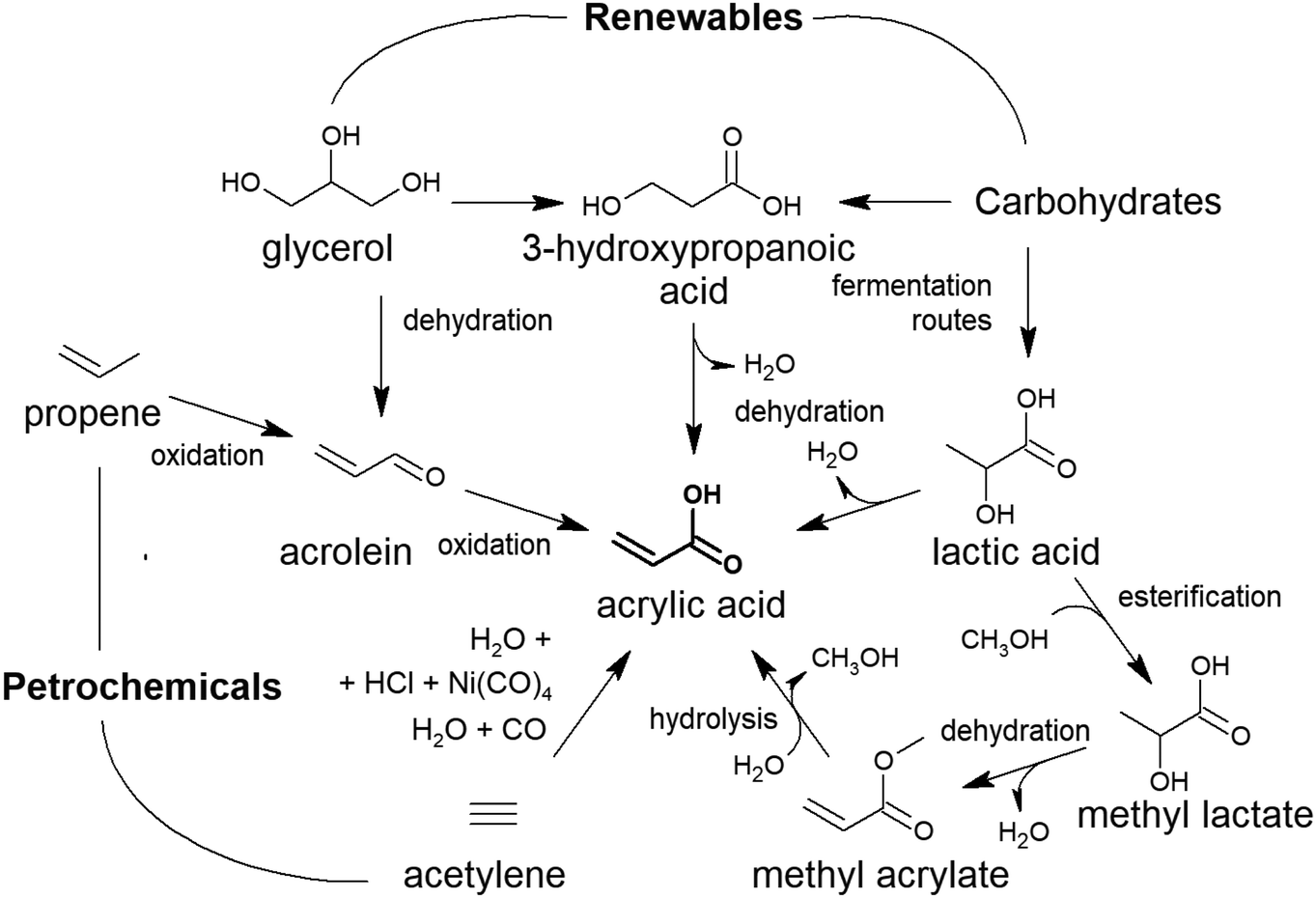 | ||
| Fig. 9 Pathways for acrylic acid synthesis: petrochemical vs. renewable route. | ||
Modern industrial plants use the vapor phase catalytic oxidation of propene with air and steam. Two types of processes exist: single- and two-step processes. The highest yield in the single-step process is 50–60%,232 at temperatures between 573 and 773 K.233 The two-step process consists in the oxidation to acrolein and its subsequent oxidation to AA. In the first reactor, propene is oxidised to acrolein at 593 K by using a Bi–Mo oxide catalyst (yield greater than 85%). In the second step, almost complete acrolein conversion and an AA yield higher than 87% have been attained at reaction temperatures between 493 and 573 K and with multicomponent metal oxide catalysts.234 AA is usually obtained as an aqueous solution of 20 to 70 wt%. Korea's LG Chem employs a new reactor design and purification technique, minimising by-products, improving catalyst life and resulting in capital and energy savings.235
On the other hand, Rohm and Haas, in collaboration with Englehard, are developing a process based on cheaper propane that is expected to decrease manufacturing costs and reduce pollution.236,237 However, the activation of propane requires high reaction temperatures, favouring total oxidation reactions.238
In recent years, much effort has been made towards the development of renewable and sustainable processes replacing petrochemical ones. They are mainly based on (i) 3-hydroxypropionic acid (obtained by fermentation of sugars), (ii) glycerol and (iii) lactic acid or methyl lactate, which are also obtained by carbohydrate fermentation.230 Currently none of these processes are commercialised because they must compete with the well-established and cost-effective petrochemical processes based on propene. Below, we describe the main key technical features of the renewable processes, stressing on the key improvements required so that they can become competitive.
Several factors play a key role in catalysts based on vanadium oxides: (i) vanadium lattice oxygen, (ii) vanadium–oxygen bond strength, (iii) host structure and oxygen atom ordering and environment, (iv) dynamic redox cycles between different oxidation states of the metal, (v) multi-functionality of the active sites, (vi) spatial isolation of the active sites and (vii) phase cooperation.240 The oxidation of glycerol seems to follow the Mars–van Krevelen mechanism.241,242 First, the adsorbed reactant is oxidised by an oxygen ion from the oxide lattice, followed by desorption of the product, and then an oxygen molecule from the gas phase restores the fully oxidised state of the catalyst. This mechanism would justify the requirement for molecular oxygen (to restore the oxidation state of multivalent metal oxide sites) to minimise coke formation, thus demonstrating the importance of O2 for the selective conversion of glycerol.
Table 4 summarises some representative studies on the oxydehydration of glycerol to AA by using heterogeneous catalysts based on metal oxides that exhibit a dynamic redox cycle (mainly V but also Mo and W). In general, reaction temperatures are higher than 500 K, and dilute aqueous solutions of glycerol are fed. AA yields lower than 60% are mostly reported and are reflected in space-time yields below 1 gAA gcat−1 h−1, with the highest corresponding to H, Fe-MCM-22, with a value of approximately 3.53 gAA gcat−1 h−1 (entry 8 in Table 4). In some cases, deactivation studies are not included, and catalytic data are given at short times on stream (a few hours). However, WV0.12Nb0.16Ox maintained a STY value of 1.11 after 37 h (entry 6 in Table 4), and H, Fe-MCM-22 and Fe-MFI (entries 7 and 8 in Table 4) maintained AA yield values similar to those observed initially after 10 and 8 h, respectively.
| # | Catalyst | Operating conditions | Catalytic results | Ref. | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| S cca (wt%) | O2/glyb (mol) | GHSVc | LHSVd | T (K) | TOSf (h) | X S (%) | Yh (%) | Produc.i (g gcat−1 h−1) | |||
| a Substrate concentration in water, weight ratio in %. b O2/glycerol molar ratio. c Gas hourly speed velocity (mL gcat−1 min−1). d Liquid hourly speed velocity (mL glycerol solution gcat−1 min−1). e Temperature conditions in Kelvin. f Time on stream. g Substrate conversion. h Yield towards product (acrylic acid). i Productivity. | |||||||||||
| 1 | V2O5 | 10.0 | 5.1 | 300 | 0.500 | 573 | 8 | 40 | 2 | 0.05 | 242 |
| 2 | V2O5/MFI | 10.0 | 5.1 | 300 | 0.500 | 623 | 8 | 100 | 17 | 0.41 | 242 |
| 3 | Mo3V oxide | 40.0 | 2.0 | 120 | 0.060 | 573 | 5 | 100 | 26 | 0.32 | 243 |
| 4 | W1V0.25Ox | 20.0 | 2.0 | 4 | 0.008 | 573 | 1 | 90 | 26 | 0.02 | 244 |
| 5 | VMoTeNbOx | 40.0 | 2.0 | 120 | 0.060 | 573 | 5 | 100 | 28 | 0.34 | 243 |
| 6 | WV0.12Nb0.16Ox | 43.0 | 2.0 | 256 | 0.100 | 538 | 37 | 100 | 50 | 1.11 | 245 |
| 7 | 2.5 wt% PO4/W2.2V0.4Nb2.4O14 | 50.6 | 2.8 | 100 | 0.145 | 558 | 2 | 100 | 59 | 2.27 | 246 |
| 8 | H, FeMCM-22 | 36.6 | 8.0 | 300 | 0.333 | 593 | 10 | 93 | 57 | 3.53 | 247 |
| 9 | Fe-MFI | 40.0 | 3.4 | 50 | 0.333 | 693 | 8 | 96 | 27 | 1.84 | 248 |
Arkema has filed a patent for the process in which the reactive vaporisation of aqueous glycerol solutions in a fluidised catalytic bed reactor is performed. The acrylic acid yield was very low (<5%).249 The substitution of vanadium for less toxic and inexpensive redox transition metal like iron catalysts has also been attempted (Table 4, entries 8 and 9).
A challenging although very attractive alternative is to conduct the reaction using a two-bed configuration; in the first bed, glycerol is oxidised to acrolein, and the latter is finally converted to AA in the second bed. Thus, Witsuthammakul and Sooknoi250 used a medium-pore zeolite (HZSM-5) in the first bed that was able to fully convert glycerol with 81 mol% selectivity to acrolein using a relatively low glycerol concentration (10–30 wt%) at 573 K and a contact time of 3440 g s mol−1. Then, in a second bed consisting of V-Mo oxides, with a contact time of 6918 g s mol−1, an AA yield close to 47 mol% was obtained at 48% acrolein conversion. Higher reaction temperatures (>573 K) and glycerol concentrations (>30 wt%) favour the degradation of adsorbed oxygenated intermediates, oligomerisation/condensation processes and coke deposition, causing catalyst deactivation. This can be minimised by the incorporation of water into the feed. Further investigation is needed to match the reaction conditions on both catalytic beds to selectively convert glycerol to acrolein and to improve its selective conversion to acrylic acid in the second bed.250
The conversion of LA into AA by using a mixture of Na2SO4 and CaSO4 as catalysts in a fixed-bed tubular reactor was first reported by Holmen in 1958.254 Since then, other more advanced catalytic technologies have been studied. The catalytic dehydration of lactic acid to AA has been performed in a continuous flow fixed-bed reactor at atmospheric pressure by using different alkali-metal modified zeolites. Thus, a lactic acid conversion of 98% with a selectivity for AA of 77% could be reached with a K-ZSM-5 zeolite at 638 K, with an LHSV of 2 h−1 for a 40 wt% lactic acid aqueous solution (Table 5, entry 1).255 LA conversion decreased from 98 to 91%, whereas the AA selectivity increased from 65 to 79% after a time-on-stream of 60 h. The high stability of ZSM-5-based catalysts, compared with other types of zeolites, was explained by their moderate acid strength and the absence of three-dimensional supercages, which minimise hydrocarbon coking and fast deactivation.
| # | Catalyst | Operating conditions | Catalytic results | Ref. | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Solv.a | S ccb (wt%) | GHSVc | LHSVd | T (K) | TOSf (h) | X S (%) | Yh (%) | Produc.i (g gcat−1 h−1) | |||
| a Solvent. b Substrate concentration in water, weight ratio in %. c Gas hourly speed velocity (mL gcat−1 min−1). d Liquid hourly speed velocity (mL LA solution gcat−1 min−1). e Temperature conditions in Kelvin. f Time on stream. g Substrate conversion. h Yield towards product (acrylic acid). i Productivity. j HAP (hydroxyapatite), n.r. (not reported). | |||||||||||
| 1 | K-ZSM-5 | H2O | 40.0 | 56 | 0.015 | 638 | 8 | 98 | 75 | 0.23 | 255 |
| 2 | Sr-HAPj | H2O | 38.0 | 40 | 0.020 | 623 | 6 | 72 | 45 | 0.18 | 256 |
| 3 | NaCa-HAP | H2O | 38.0 | 40 | 0.020 | 623 | 6 | 90 | 78 | 0.30 | 257 |
| 4 | Ba2P2O7 | H2O | 20.0 | 2 | 0.029 | 673 | 6 | 100 | 76 | 0.23 | 258 |
| 5 | KI/NaY | H2O | 29.0 | 20 | 0.050 | 598 | 2 | 98 | 66 | 0.48 | 259 |
| 6 | Na2HPO4/NaY | H2O | 34.0 | 20 | 0.125 | 613 | 2 | 78 | 57 | 1.23 | 260 |
| 7 | Ca/P = 1.3_HAP | H2O | 50.0 | 7 | 0.091 | 648 | 300 | 100 | 60 | 1.44 | 261 |
| 8 | 2.8K/NaY | H2O | 29.0 | 20 | 0.050 | 598 | 6 | 99 | 50 | 0.36 | 262 |
| 9 | 2% La/NaY | H2O | 38.0 | 20 | 0.123 | 623 | n.r. | 100 | 56 | 1.35 | 263 |
Sr-Substituted hydroxyapatite also showed a high activity in lactic acid conversion to AA (conversion of 72% and AA yield of 45%) at 623 K, with a lactic acid flow of 0.45 mL h−1 (38 wt% lactic acid aqueous solution) after 6 h of TOS (Table 5, entry 2).256 This catalytic result agrees with the relatively strong basic properties and the moderate balance of acid–base catalytic properties of Sr-hydroxyapatite, in comparison with other alkaline-earth metal cations, which might be required for AA production from lactic acid. Nevertheless, a non-stoichiometric Ca-HAP catalyst containing Na+ ions in place of deficient Ca2+ ions showed a remarkable AA yield of 78% and a lactic acid conversion of 90% under similar experimental conditions, as well as stability over 6 h of time-on-stream (Table 5, entry 3).257 It was proposed that this excellent performance may be due to the existence of fewer acid site nanodomains to produce acetaldehyde, fewer basic sites responsible for 2,3-pentanedione production, and more acid–base pair sites that might be effective in the selective dehydration of lactic acid to AA than are present in the rest of the Ca-HAP catalysts prepared via different methodologies.
A dibarium pyrophosphate (Table 5, entry 4) has shown excellent hydrothermal stability associated with its hydrolytic resistance in water, as inferred from its very low solubility, whereas the absence of strong acid sites limits coke formation, resulting in stability against deactivation.258
In addition to these examples, Table 5 gathers other families of catalysts for the dehydration of lactic acid to AA under very different experimental conditions. The best STY value (1.44 gAA gcat−1 h−1), after 300 h of TOS, was attained with hydroxyapatite with a Ca/P molar ratio of 1.3 at a high reaction temperature (648 K) with the highest aqueous lactic acid solution concentration (50 wt%), whereas the rest of the catalysts exhibited STY values lower than 0.7. Concerning the stability of other catalysts, the KI/NaY catalyst maintained an AA yield close to 40% after 260 h of TOS, with an LA conversion higher than 95%.259 However, in the case of Na2HPO4/NaY, the AA selectivity decreased continuously, and after 30 h of TOS, the AA yield was lower than 25%.260 This deactivation was due to LA polymerisation on the catalyst surface, but the initial catalytic activity was fully recovered after treatment in air at 773 K for 3 h, thus confirming the cause of activity loss.
Previously, liquid-phase conversion of LA to AA in continuous flow mode was also studied in water at high temperatures (723 K) and high pressures (100 MPa), but worse efficiency was achieved, with a maximum selectivity for AA of 44% at lactic acid conversion of 23% at a residence time of 0.8 s.264
Vapour-phase dehydration of methyl lactate is another alternative for AA production, and it has been performed, for instance, in the presence of Ca3(PO4)2-based catalysts. The best catalytic performance was achieved with Ca3(PO4)2–Ca2(P2O7) (50:50), with a conversion of 91% and a selectivity towards AA of 75% and methyl acrylate of 5% (total: 80%) at 663 K and an LHSV of 0.175 h−1 after 27 h.265,266 The moderate acid–base strength, which was regulated via the surface properties of both components, after calcination at 773 K was used to explain this efficiency. A similar AA yield has been achieved with a (K2HPO4 + Al2(SO4)3)/MCM-41 catalyst at 673 K.267
Recently, kinetic and spectroscopic studies on the role of co-fed species (water, methanol and methyl acetate) in the dehydration mechanism of methyl lactate over alkali-metal-exchanged zeolites have been published.268 The multiple competing roles of water were demonstrated, where the rate-limiting step changed from methyl lactate dehydration to sodium lactate dehydration as the concentration of water increased.
In addition to the three chemical platforms mentioned so far, other nonconventional processes have been investigated, one directly from partial oxidation of xylose, the second most abundant sugar after glucose, and another using aldol condensation of dimethoxymethane and methyl acetate. In the first case, AA was produced by using a two-fluid nozzle to atomise a xylose–water solution into a capillary fluidised bed operating at 573 K.269 The vanadyl pyrophosphate catalyst attained a maximum AA yield of 17%, in addition to the formation of acrolein (11%) and mainly maleic anhydride (25%) by using a gas residence time of 0.2 s after 2 h with a xylose conversion of 100%. Much work is still required to disfavour the caramelisation kinetics and catalyst agglomeration during the vaporisation of carbohydrates.
Regarding glycerol as a feedstock for AA production, in practice, acrolein is the key intermediate, being formed via acid-catalysed glycerol dehydration. This is an important drawback, since a two-step catalytic system is required to avoid overoxidation. Moreover, strategies to avoid coke formation, ensuring that surface catalytic sites are available for new reactant molecules and extra-framework oxygen species are continuously regenerated, are required to minimise the amount of non-condensable gases (methane and carbon oxides) to improve the carbon balance and atom efficiency. The need for more sustainable catalytic processes also must direct research to inexpensive, nontoxic, abundant and easily available transition metals as alternatives to vanadium-based catalysts.
2.3. δ-Aminolevulinic acid
δ-Aminolevulinic acid (5-aminolevulinic acid, DALA) is a non-proteinogenic five-carbon amino acid that naturally occurs in all plant and animal cells213,270,271 and for cancer treatment.272 It is an active ingredient in several environmentally benign, highly selective, broad-spectrum herbicides. Moreover, DALA can provide novel opportunities for amino acid-based polymers13 that can be modified to introduce new functionalities for drug anchoring, imaging and molecular testing,273–276 and they are expected to be non-toxic and readily excreted from the body.The complexity and low yields of the current DALA preparation justify its very high price. DALA is mainly produced by using enzymatic and metabolic engineering strategies, which have been reviewed in different works.277,278 The chemical route of DALA from levulinic acid (LEVA) is also quite complicated since it requires the selective introduction of an amino group at the C5-position of LEVA, forming a C–N bond (Fig. 10). Several amination methods are available, the most widely used being the bromination of LEVA in an alcohol solvent, generating a mixture of 5-bromo- and 3-bromoesters, which are separated by distillation.279 The 5-bromolevulinate is then aminated using a nucleophilic nitrogen species, such as sodium azide.280 Another method consists of a three-step synthesis including bromination of LEVA in methanol, reaction of methyl 5-bromolevulinic acid with potassium phthalimide and subsequent hydrolysis of the resulting phthalimide derivative, but this method is highly inefficient since a single atom from potassium phthalimide is used in the final DALA.281
 | ||
| Fig. 10 Synthesis of 5-aminolevulinic acid using sodium azide. | ||
A more sustainable route based on robust and selective heterogeneous solid catalysis that allows a decrease in the number of steps and uses less expensive starting reactants and low-toxicity intermediates is still waiting to be developed as an alternative to homogeneous catalysis, very inefficient with regard to the atom economy.
3. Dicarboxylic acids
3.1. Furandicarboxylic acid
2,5-Furandicarboxylic acid (FDCA) has been identified as one of the most promising building blocks derived from biomass.191,282 Among its many uses to produce pharmaceuticals and specialty chemicals, the production of polyethylene furoate (PEF), a substitute for polyethylene terephthalate (PET) has attracted the most interest. PET is used as a packing material, especially for beverages and foods, and to make synthetic fibres. According to a study funded in 2013 by the FP7 of European Commission, the addressable market volume for PET and PBT (polybutylene terephthalate) was 41.6 million of tons per year, while other minor products such as polycarbonates, plasticizers, polyamides and polyesters could be replaced in smaller quantities ranging from 1 to 3 million of tons per year.283 The study projected a market in 2020 for the utilization of FDCA in PEF formulations of around 322000 tons, with other uses ranging from 6000 tons to 80000 tons. The market driver for these products is the final price followed by the performance with a minor component of the renewable characteristic. Many catalytic processes have been reported in the last 10 years improving the knowledge about the reaction and claiming improvements in the production cost. The necessity to obtain more data under commercial-scale relevant conditions and to demonstrate the performance and recyclability of the material has delayed the commercial production of FDCA and today we still talk about projected production and market value, i.e. up to $850 millions by 2025.284 Avantium and Corbion are leading the production of FDCA at the demonstration scale probing that PEF could have a better performance than PET, being more than a replacement. With similar density and thermal properties (Tg around 358–368 K and Tm around 483–493 K for PEF and 518–538 K for PET),285 PEF has demonstrated 10×, 5× and 2× better barrier properties for O2, CO2 and water respectively,286,287 which could translate to a longer shelf life for food or a lower amount of plastic required.
 | ||
| Fig. 11 Production of FDCA from lignocellulosic biomass. | ||
The main challenge for the utilization of heterogeneous metal catalysts is the low solubility of FDCA (calculated solubility of FDCA in water and methanol at room temperature is 0.99 and 13.10 mg g−1, respectively).299 While HMF is highly soluble, as FDCA is formed, it precipitates over the catalyst surface causing severe catalyst deactivation. To solve that problem most of the research has been done adding a base to the solvent to form FDCA-salts which are much more soluble or working at low HMF/FDCA concentrations. Both alternatives are successful in helping to understand the reaction mechanism and kinetics; however, neither of them has commercial application because of the high cost of FDCA purification, which is the last step of the process. Very high FDCA purity >99.5% is required if it is going to be polymerized to produce PEF; however, few works address this concern. Similar problems occur with the electrocatalytic and enzymatic processes, which typically require low substrate concentrations resulting in high efficiency processes but expensive purification steps; nevertheless, electrocatalytic processes are gaining attention in the literature.
The second route is the oxidation of furfural to furoic acid followed by the disproportionation to furoate and furan300 or a cascade reaction involving esterification, chloromethylation and oxidation.301,302 Because of the complexity and low yield of these reactions, the production of FDCA from furfural is barely studied.
Ava-Biochem utilizes the simple thermal route, heating fructose in the presence of aqueous sulfuric acid to produce HMF despite the low yields.303,304 After performing a chromatographic separation, they produce high purity HMF that can be oxidized by catalytic or enzymatic methods. Ava-Biochem produces high-purity products that can be used as specialty chemicals, but the economics of the process is not clear, as plans for commercial operation (30000 tons of FDCA per year by 2019) have been delayed. The new target is a facility that can produce 5000 tons per year.305 A similar process has been reported by Petrobras working at low conversion (<30%) and high selectivity (>80%). After a chromatographic separation, they recycle the sugars to increase the overall conversion, and oxidise the 5-HMF using a heterogeneous catalyst. Before the oxidation step, they add a base to increase the pH, neutralise the solution and minimise the deactivation of the oxidation catalyst.306 The main challenge of this process is the economic production of 5-HMF and reducing the cost of 5-HMF purification, as chromatographic methods are complicated to scale up for commodity chemicals. Avoiding the use of bases during the oxidation step will also be critical. Avantium decided to use the protection route using methoxymethylfurfural (MMF) instead of 5-HMF. To produce MMF, they carried out the dehydration of fructose using methanol as the solvent in a monophasic system, and achieved MMF yields close to 60% at a 20 wt% fructose concentration.307 In addition to their stability, another advantage of MMF is its boiling point (<493 K), which is lower than that of 5-HMF. This allows for the recovery and purification of MMF by distillation before oxidation. However, heterogeneous catalysts, such as Pt/C, that normally work well with 5-HMF are not capable of oxidising MMF, probably because of the protection of the –OH group, so they moved forward with the AMOCO process and carried out the oxidation step using acetic acid as the solvent and a homogeneous Mn/Co/Br catalyst.308,309 Avantium, via its affiliated company Synvina, continues to progress on the commercialisation of FDCA and PEF technologies with plans to start a small commercial plant (5000 tons) by 2023.310 This approach has been successful in producing high-purity FDCA and PEF at the demonstration scale, attracting the interest of large companies, such as Coca-Cola, Danone, ALPLA or Mitsui; however, using fructose as a feedstock is a limitation for the economics of the process. In the future, they plan to use low-cost glucose as a feedstock, which will require new processes for producing biomass-derived glucose (project Mekong).311 Additionally, research will be necessary to improve the MMF yields obtained from glucose and to develop heterogeneous catalysts capable of oxidising MMF.
ADM and DuPont have formed a joint venture to produce furandicarboxylic methyl esters (FDMEs). ADM has expertise in FDCA/FDME production,312,313 and they collaborate with DuPont to directly produce FDCA esters, as they are claimed to have advantages in the polymerisation step. In April 2018, they opened their first pilot plant.314 In addition to the limitations mentioned above, the recovery of the alcohols used to produce FDCA esters remains a challenge.
Mascal et al. proposed an interesting alternative based on chloromethylfurfural (CMF), which can be a building block similar to 5-HMF.315,316 Using a biphasic system composed of dichloroethane and high-concentration HCl, they reported yields of CMF up to 90% from different feedstocks, including real biomass heated at 353–373 K for 3 h in aq. HCl/ClCH2CH2Cl.317 Origin Materials has scaled up this technology, but instead of producing FDCA, they convert CMF into para-xylene to make terephthalic acid.318 First, CMF is converted into dimethylfuran (DMF),319 which is converted into p-xylene via the Diels–Alder reaction320 (more details are provided in Section 3.5). The use of high concentrations of HCl in a biphasic reactor and the recovery of dichloroethane are the main challenges for this process. Origin Materials maintains the ability to produce FDCA in the future, as they have licensed technology from Eastman.321 As occurs with other processes at the pilot plant stage, they also use homogeneous catalysts for the final oxidation step (AMOCO process), a clear indication that more research is necessary in the development of heterogeneous catalysts.
Novamont decided to use ionic liquids as the solvent for the dehydration of the sugars, reporting yields up to 95% using tetraethylammonium bromide and HPWO/SiO2 as the catalyst and temperatures from 353 to 373 K for 15 min in a series of steps.322 The oxidation of 5-HMF is performed using a platinum catalyst in the presence of a base to keep the pH between 7 and 12 and using a low-pressure atmosphere of oxygen.323 Although the separation of 5-HMF from the ionic liquid is not described in those patents, Novamont announced the construction of a demonstration plant for 2021.324 Replacing fructose with glucose as the feedstock, avoiding the use of salts during the oxidation step and the ability to recover 5-HMF and the ionic liquids will be critical for the development of this technology, which is very promising because of the high yield achieved.
Other interesting lab-scale technologies for producing FDCA from sugars have been reported. Zhang et al. reported the production of 5-HMF at 87% yield from fructose in 2 h at 393 K using isopropanol as the solvent and an acid catalyst such as HCl or Amberlyst-15.325 A base was incorporated before the oxidation step that uses a metal catalyst, such as Au/HT (368 K, 7 h) or Pt/C (297 K, 4.25 h), and bubbling oxygen. They had to evaporate the isopropanol and add water to dissolve 5-HMF and separate it from the impurities, typically humins, that remain insoluble and otherwise deactivate the catalyst.326 While the overall yield of FDCA is high, 83%, the economics of the process need to be studied, as evaporating solvents is typically expensive. Liu et al. worked at higher fructose and 5-HMF concentrations than those used in the previously mentioned study using DMSO/water as the solvent.327 Using a 19% fructose solution, they obtained an 80% 5-HMF yield in the first step using Amberlyst-15 as the catalyst and DMSO as the solvent. In the second step, they used Ru/C and O2 to oxidise 5-HMF in the presence of NaHCO3, without purification of 5-HMF, at an 81% yield, which resulted in an overall FDCA yield from fructose of 65%. Unfortunately, no data about the separation of FDCA, the stability of the catalyst in the presence of fructose-derived 5-HMF or the economics of the process were reported.
Dumesic et al. reported an alternative solution to address the low solubility of FDCA and to avoid the use of a base in the reaction medium during the oxidation step by using GVL/water as the solvent.328,329 They reported that FDCA solubility in GVL/water mixtures greatly increases with temperature (>12 wt% at 403 K). GVL/water can also be used in the first step to dehydrate fructose at high concentrations (15 wt% fructose) and high yields (>70%) using FDCA as the catalyst, so no removal of the acid catalyst is needed. After a purification step to remove the humins, crude 5-HMF is oxidised to FDCA at a 91% yield using Pt/C as the catalyst and O2 for the environment. By reducing the temperature, FDCA precipitates with a purity >99%. The techno-economic analysis suggested an FDCA price of approximately $1500 per kg.329
Alternatives to these catalytic processes have been developed by Corbion and Novozymes/DuPont. Corbion developed a 2-step process combining the chemical dehydration of fructose to 5-HMF, followed by the enzymatic oxidation of 5-HMF to FDCA. The advantage of this process is that the microorganism can work in the presence of some of the impurities produced during the dehydration of fructose.286 Another interesting option is the electrocatalytic oxidation of 5-HMF, which has been reported by several authors.330–332 The main challenge of this option is the recovery and purification of FDCA from the electrolyte and the economics of the process.
Even though furfural is a commercial product and less expensive than 5-HMF, only Braskem has a process for producing FDCA using furfural as a feedstock. The first step is the oxidation of furfural using a metal catalyst, Au, Pt or Pd, in the presence of a base, followed by disproportionation to furan, which is continuously removed, and a mixture of 2,4- and 2,5-FDCA. The production of 2,4-FDCA is a problem, as it reduces the yield of 2,5-FDCA (used to produce PEF) and requires additional purification steps to separate 2,4-FDCA from 2,5-FDCA.333 The low yields and complicated purification steps are the main challenges of this process. There has been no indication that this process is being moved to demonstration or commercial scale.
 | ||
| Fig. 12 Reaction mechanism for the oxidation of HMF to FDCA. | ||
| # | Catalyst | Operating conditions | Catalytic results | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Solv.a | S ccb (wt%) | C/Sc (wt%) | PO2d (bar) | T (K) | t (h) | X S (%) | YFDCAh (%) | Produc.i (g gcat−1 h−1) | Deact.j | |||
| a Solvent. b Substrate concentration weight ratio in %. c Catalyst to substrate weight ratio in %. d Reactant gas pressure in bar. e Temperature conditions in Kelvin. f Reaction time. g Substrate conversion. h Yield towards product. i Productivity. j Catalyst deactivation: strong (st), moderate (m), low (l) and negligible (ne); n.s. (not studied), n.r. (not reported). k tert-Butyl hydroperoxide (TBHP) was used as the oxidant. l Reaction conducted in a continuous mode in a fixed bed at the WHSV (weight hourly space velocity) indicated in the table. m Requires regeneration by treating with H2. n Can be regenerated. | ||||||||||||
| 1 | Au/Al2O3 | H2O (NaOH) | 1.0 | 80.0 | 10 | 343 | 4 | 100 | 99 | 0.38 | l | 337 |
| 2 | Au/GC | H2O (NaOH) | 0.2 | 100.0 | 7 | 383 | 6 | 100 | >97 | 0.20 | l | 338 |
| 3 | Au/CeO2 | H2O (NaOH) | 0.3 | 67.0 | 5 | 403 | 2.5 | 100 | 88 | 0.65 | n.s. | 339 |
| 4 | Pt-Nanocrystals | H2O (NaHCO3) | 0.3 | 4.0 | 1 | 373 | 10 | 100 | 95 | 2.94 | n.s. | 340 |
| 5 | Pd–Au/HT (1:4) |
H2O/NaOH | 0.4 | 50.0 | 1 | 333 | 6 | 100 | 90 | 0.37 | l | 341 |
| 6 | Ni–MnOx | H2O (NaHCO3) | 1.3 | 79.0 | 8 | 373 | 28 | 100 | 94 | 0.05 | n.s. | 342 |
| 7 | MnOx–CeO2 | H2O (KHCO3) | 0.6 | 167.0 | 20 | 383 | 12 | 100 | 91 | 0.06 | l | 343 |
| 8 | Pt/NC-CeO2 | H2O | 0.2 | 167.0 | 4 | 423 | 4 | 100 | 99 | 0.18 | ne | 344 |
| 9 | Pt/ZrO2 | H2O | 0.5 | 60.0 | 4 | 373 | 12 | 100 | 97 | 0.17 | n.s. | 345 |
| 10 | Ru/MgO | H2O | 1.3 | 160.0 | 6 | 413 | 4 | 100 | 90 | 0.17 | ne | 346 |
| 11 | Ru/MgAlO | H2O | 1.3 | 80.0 | 6 | 413 | 4 | 100 | 99 | 0.38 | st | 346 |
| 12 | Co–Mn | H2O | 0.6 | 167.0 | 10 | 393 | 5 | 99 | 95 | 0.14 | ne | 347 |
| 13 | 5% Ru/C | DMSO/H2O (NaHCO3) | 10.0 | 41.0 | 40 | 383 | 12 | 100 | 93 | 0.23 | lm | 327 |
| 14 | MnFe2O4 | CH3CN | 0.8 | 187.5 | TBHPk | 373 | 5 | 100 | 80 | 0.11 | ne | 348 |
| 15 | Pt/C | GVL/H2O | 7.5 | 103.0 | 40 | 383 | 20 | 100 | 94 | 0.06 | n.s. | 329 |
| 16 | H3PMo12O40 | [Bmim]Cl | 1.3 | 400.0 | 10 | 413 | 6 | 100 | 89 | 0.05 | l | 349 |
| 17 | Ru/Al2O3 | H2O (Na2CO3) | 1.3 | 30 | 413 | 1 h−1l |
100 | 62 | 0.80 | ln | 350 | |
| 18 | Pt@Dowex-Na | H2O | 0.3 | 8 | 393 | 0.08 h−1l |
100 | >99 | 0.27 | ne | 351 | |
| 19 | Pt/C | GVL/H2O | 1.0 | 40 | 373 | 0.01 h−1l |
100 | 94 | 0.01 | ne | 329 | |
Pt, Pd and Ru are also active and selective catalysts that have been demonstrated to be successful in the production of FDCA in the presence and absence of a base. The morphology of these metals has shown that the oxidation reaction is a structure-sensitive reaction. Liu et al. studied the conversion of 5-HMF in the presence of Pt nanocrystals and observed important differences in the generated oxygen species. They reported that on the Pt(100) surface, the O2 molecules dissociated into O atoms and formed activated OH molecules with H2O (entry 4, Table 6). However, on the Pt(111) surface, the O2 molecules were reduced instead of being dissociated, resulting in the formation of ˙O2− and showing a lower FDCA yield than that of a reaction utilising a catalyst rich in Pt(100).340 Pt and Ru (entries 8–11, 13) have shown good performance in water and continuous operation (entries 17 and 18), and they are promising catalysts that deserve further study.
To reduce the cost of the catalyst, some authors are trying to replace noble metals with less expensive options. Non-noble metals, such as Ni–Mn and MnO (entries 6 and 7), are active, but the productivity of the reaction is almost an order of magnitude lower than that utilising Au catalysts. The utilisation of Mn oxides is an option that has proven successful.342,343,347,348 Rao et al. used a Co–Mn catalyst in aqueous NaHCO3 and obtained FDCA yields >95% with the catalyst showing good stability (entry 12, Table 6). The FDCA yield decreased to 73% when they used crude 5-HMF produced from sugars.347 They report an economic study starting from starch to produce glucose and then 5-HMF in a biphasic reactor. With the limited data reported (no information about mass and energy balances), it seems that just the cost of the raw materials was close to $2000 per ton of FDCA.
One of the most important limitations is the use of dilute solutions of 5-HMF, which typically results in greater catalytic results but penalises the economics of future processes because of the purification cost. Liu et al. studied the oxidation of highly concentrated 5-HMF using Ru/C as the catalyst (entry 13, Table 6). They observed that the highest yields of FDCA were achieved in water or DMSO/water mixtures in the presence of a weak base (NaHCO3). Even though a high concentration of 5-HMF was used, there was no mention about the solubility of FDCA, which may be limited in the absence of the base and be the reason for the poor carbon balance in some of the experiments. The catalyst suffered deactivation but could be regenerated by a reducing treatment. The FDCA yield decreased from 93 to 81% when 5-HMF produced from fructose was used in the oxidation without intermediate purification.327
Another limitation is that most of the studies focus on batch reactions, and there are few studies in continuous mode. Da Fonseca Ferreira et al. used Ru/Al2O3 and a concentration of 5-HMF of 1.25 wt%, but they required the presence of Na2CO3, and they only achieved a 62% FDCA yield350 (entry 17, Table 6). Liguori et al.351 studied the continuous oxidation of 5-HMF using a Pt/resin catalyst and oxygen (entry 18, Table 6). They were able to achieve an FDCA yield >99% using pure water as the solvent and demonstrated the stability of the catalyst for 5 days. The main limitations are the high Pt loading of 15.6 wt% and the low concentration of the 5-HMF used during the experiments (0.3 wt%). The role of the resin in the reaction was not discussed, but relatively low yields (38%) were reported when using a simple Pt/C catalyst. Dumesic et al. reported an FDCA yield of 93.7% using Pt/C, GVL/water as the solvent and 1 wt% 5-HMF329 (entry 19, Table 6).
In the future, new processes investigated at a laboratory scale must include the integration of 5-HMF production and 5-HMF oxidation. Ideally, both reactions should be carried out in the same solvent to avoid the separation of 5-HMF. Consequently, it will be important to better understand the formation of humins and other degradation products during 5-HMF production, as they deactivate the solid catalyst used for the oxidation. Studies should be performed at high 5-HMF/FDCA concentrations, at least at 10 wt%, to minimise capital cost, reduce energy requirements and facilitate solvent recovery and reutilisation. At those concentrations, avoiding the FDCA precipitation will be necessary to utilise heterogeneous catalysts. Using mixtures of solvents to increase the solubility of FDCA could be an option to solve the solubility problem. New technologies should go together with techno-economic and life cycle analysis. To date, some of these analyses have been published using data from the literature,354,355 but the lack of first-hand data makes it complicated to elaborate accurate mass and energy balances that combine all the steps involved in the production of FDCA.
Another field of research that needs to be improved is separation and purification. Hallet et al.356 recently studied the separation of furanic compounds from ionic liquids, which will be useful in the design of new technologies based on the utilisation of ionic liquids as solvents. In this sense, not all impurities have the same effect; for example, FCA is a chain terminator that will be greatly detrimental to the polymerisation of FDCA, while DFF could be less problematic. Avantium is thoroughly studying the removal of some of the impurities produced during the oxidation of 5-HMF. For example, they hydrogenated by-products to produce 2,5-bishydroxymethylfuran (BHMF), which can be removed by evaporation.357
3.2. Maleic acid
Maleic anhydride (MA) and its hydrated form, maleic acid (MAc), are used for the production of a number of polymers with a wide variety of applications: personal care products, fibre reinforced plastics, surface coating applications, improved polymer compatibilisers in packaging films and coating applications, textile fibres and paper sizing, pigment dispersion, varnishes, engineering plastics, paper sizing, carpet and floor care products, food packaging, etc.358 The global production of maleic anhydride in 2012 was 1.78 million tons, with a market value of over 314 million USD.359MA is currently produced by the well-proven and established partial oxidation of benzene and mostly butane358,360,361 (both petrochemical feedstocks). However, other renewable routes based on the oxidation of biomass-derived platforms have been demonstrated at the laboratory scale. They produce either MA or MAc depending on the reaction conditions (one can be interconverted into the other by simple hydration or dehydration steps). These renewable platforms are butanol, levulinic acid (LEVA), 5-hydroxymethylfurfural (5-HMF), and furfural. Only furfural is currently industrially available, which may explain the most abundant scientific literature for this platform. In general, O2 is used as an oxidant in both gas phase and liquid phase processes, but aqueous H2O2 and electrochemical oxidation have also been utilised.
This process is proposed to be an oxy-dehydration process first involving the dehydration of butanol to 1-butene that is then isomerised to 2-butene, which is, subsequently, oxidised to crotonaldehyde. The latter can be cyclised to furan that is further oxidised to an endoperoxide, and, finally, MA is formed via 5-hydroxyfuranone. Another possibility is that crotonaldehyde is oxidised to formyl crotonic acid (via a crotonic acid intermediate), which is, subsequently, oxidised to maleic acid and cyclised to MA. Remarkably, no noticeable deactivation was observed for 6 h when using high-grade biobutanol.
Interestingly, the reaction was also conducted with biobutanol produced by fermentation, but the presence of impurities results in a reduced MA yield. The removal of impurities after a purification treatment with bleaching earth and activated charcoal resulted in similar behaviour to that of high-grade butanol. The main challenge for this process remains in the development of a catalyst with a higher yield of anhydride and robustness against deactivation in the long term when using biobutanol obtained from fermentation.
The selective route involves the cleavage of the C4–C5 bond,10 releasing formaldehyde and succinic acid that is cyclised to the anhydride and, subsequently, dehydrogenated to MA. The C–C scission between the C3–C4 bond results in the unselective formation of acetaldehyde and malonic acid. LEVA can also be dehydrated to angelica lactones and, eventually, to cyclopentanedione and cyclopentenedione or decarboxylation to methyl vinyl ketone. The unselective overoxidation of all the above-mentioned compounds to CO2 and H2O is also possible. The intrinsic activity of supported vanadates in the oxidative C–C cleavage of bonds in ketones is very sensitive to both the vanadium oxide structure and support nature, as demonstrated by the same authors, with studies conducted on vanadate species deposited on different supports (SiO2, γ-Al2O3, TiO2, and CeO2) using 2-pentanone.368
These results should foster more research to identify better catalysts, especially using lower grade, biomass-derived LEVA than that so far considered. The experiments must be conducted under well-specified reaction conditions; otherwise, a comparison between different catalyst systems is impractical.
The liquid phase oxidation of high-grade 5-HMF to MA using O2 has been studied using V- or V-Mo-based catalysts,369–373 with the exception of one amino-functionalised silica catalyst.374Table 7 summarises the results. The reaction was conducted at relatively mild temperatures (T ≤ 373 K) and O2 pressure (10–20 atm). Acetic acid or acetonitrile were the preferred solvents, and a mixture of MA and MAc was then obtained. When the reaction was conducted in water, the hydrated form (MAc) was also obtained with a higher selectivity, at the expense of MA.372
| # | Catalyst | Operating conditions | Catalytic results | Ref. | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Solv.a | S ccb (wt%) | C/Sc (wt%) | PO2d (bar) | O2/FURe (mol) | T (K) | t (h) | X S (%) | Yi (%) | Produc.j (g gcat−1 h−1) | Deact.k | |||
| a Solvent. b Substrate concentration weight ratio in %. c Catalyst to substrate weight ratio in %. d Reactant gas pressure in bar. e O2/furfural molar ratio. f Temperature conditions in Kelvin. g Reaction time. h Substrate conversion. i Yield towards product (MA + MAc). j Productivity to MAc. k Catalyst deactivation: strong (st), moderate (m), low (l) and negligible (ne); n.s. (not studied) n.r. (not reported). | |||||||||||||
| 1 | V2O5 | AcOH | 2.4 | 3.6 | 5 | 7.5 | 373 | 4 | 99 | 79 | 5.05 | l (5 runs) | 369 |
| 2 | H5PV2Mo10O40 | CH3CN/AcOH | 9.2 | 100.0 | 10 | 6.3 | 363 | 8 | 100 | 64 | 0.07 | n.s | 370 |
| 3 | VON–GO | AcOH | 2.4 | 4.0 | 20 | 30.0 | 363 | 4 | 100 | 91 | 5.23 | l (6 runs) | 371 |
| 4 | VO–NH2–GO | AcOH | 2.4 | 4.0 | 20 | n.r. | 363 | 4 | 100 | 95 | 5.48 | l (5 runs) | 372 |
| 5 | VO(acac)2 | CH3CN | 7.0 | 10.6 | 10 | n.r. | 363 | 4 | 99 | 52 | 1.13 | n.s. | 373 |
| 6 | SiO2–NH2 | H2O | 1.0 | 50.0 | 10 | n.r. | 363 | 8 | 36 | 30 | 0.07 | n.s. | 374 |
A radical mechanism has been proposed that is initiated by the cleavage of the C–C bond between the furan ring and the hydroxymethyl group, releasing formic acid.371 More details of the mechanism are given elsewhere371 that, although it explains essential features of the reaction (such as that the reaction is deeply restrained by the addition of free radical inhibitors and CO, CO2 and HCOOH are released in the course of the reaction), is not supported by any other experimental evidence.371
In any case, the highest yield reported so far (95%) was obtained using a 2.4 wt% FUR solution in acetic acid after 4 h, at 363 K and 20 atm using oxo-vanadium species deposited onto an amino-functionalised graphene oxide (Table 7, entry 4).372 Similar results were obtained with a non-functionalised graphene oxide.371 A quite small amount of catalyst was used; consequently, both systems show remarkably high productivity: above 5 g of MAc gcat−1 h−1. These results are more notable considering the low loading of vanadium deposited onto the graphene oxide. It would be very interesting to study the behaviour of this type of catalyst using higher furfural concentrations than that mentioned above to determine whether these high yields and productivities remain. Unfortunately, both catalysts were slightly but continuously deactivated after 5 runs.371,372 A minor loss of V species by leaching was observed in these two cases. This complicates a possible industrial application. However, it must be considered that the slight deactivation could have also been a consequence of the unavoidable losses of catalysts occurring while handling the catalysts (catalysts must be removed from the solution before conducting another reuse run). The amount of catalyst used in these experiments is quite small, less than 100 mg. The accumulated losses while reusing may be comparable to the observed slight decrease in the activity. Long-term experiments in flow reactors are needed to assess the stability of these catalysts.
A more durable although less selective (yield of 79%) silica-supported V2O5 catalyst has also been investigated working under quite similar reaction conditions. Other V catalysts, H5PV2Mo10O40 (Table 7, entry 2) and VO(acetylacetone)2 (Table 7, entry 5), are less productive than the silica-supported V2O5 catalyst.370,373 The amino-functionalised silica was also less productive (Table 7, entry 6).374 In summary, the challenge of developing a highly productive and durable catalyst for this reaction is yet to be resolved.
Four different oxidation processes of furfural to maleic acid or anhydride are possible, namely, liquid-phase oxidation with O2, gas-phase oxidation with O2, liquid-phase oxidation with H2O2 and electrochemical oxidation. These processes are described in the following paragraphs.
3.2.4.1. Liquid-phase oxidation of furfural to MA/MAc using O2. Liquid phase oxidation with O2 is conducted at similar relatively mild temperatures (T < 383 K) and O2 pressures (10–20 atm) and using similar vanadium-based catalysts as previously mentioned for the liquid-phase oxidation of 5-HMF, except for soluble heteropolyacids and notable Fe-porphyrin (Fe-PP)-based systems (see Table 8).
| # | Catalyst | Operating conditions | Catalytic results | Ref. | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Solv.a | S ccb (wt%) | C/Sc (wt%) | PO2d (bar) | O2/FURe (mol) | T (K) | t (h) | X S (%) | Yi (%) | Produc.j (g gcat−1 h−1) | Deact.k | |||
| a Solvent. b Substrate concentration weight ratio in %. c Catalyst to substrate weight ratio in %. d Reactant gas pressure in bar. e O2/furfural molar ratio. f Temperature conditions in Kelvin. g Reaction time. h Substrate conversion. i Yield towards product (MA + MAc). j Productivity. k Catalyst deactivation: strong (st), moderate (m), low (l) and negligible (ne); n.s. (not studied), n.r. (not reported). | |||||||||||||
| Oxidation of furfural with homogeneous catalysts | |||||||||||||
| 1 | H3PMo12O40 | H2O-tetrachloroethane | 7.0 | 190.0 | 20 | 12 | 383 | 14 | 50 | 35 | 0.02 | n.s. | 378 |
| 2 | H3PMo12O40 + Cu(NO3)2 | H2O | 17.4 | 210.0 | 20 | 5 | 371 | 14 | 95 | 49 | 0.02 | n.s. | 379 |
| 3 | H5PV2Mo10O40 + Cu(CF3SO3)2 | CH3CN-AcOH | 7.8 | 15.0 | 20 | 10 | 383 | 14 | 99 | 54 | 0.31 | n.s. | 380 |
| Oxidation of furfural with heterogeneous catalysts | |||||||||||||
| 4 | Mo4VO14 | AcOH | 1.8 | 15.6 | 20 | n.r. | 393 | 16 | 100 | 65 | 0.31 | st (5 runs) | 381 |
| 5 | VO-NH2-GO | AcOH | 1.8 | 5.2 | 20 | n.r. | 363 | 8 | 82 | 62 | 1.81 | st (5 runs) | 372 |
| 6 | VON-GO | AcOH | 1.8 | 5.2 | 20 | 30 | 363 | 10 | 86 | 60 | 1.39 | n.s. | 371 |
| 7 | CaCu2P2O7 | H2O | 5.0 | 20.0 | 8 | 5 | 388 | 18 | 68 | 37 | 0.13 | m (4 runs) | 382 |
| 8 | FeT(p-Cl)PPCl | H2O | 6.3 | 16.7 | 12 | 14 | 363 | 10 | 96 | 44 | 0.32 | l (5 runs) | 383 |
| 9 | FeT(p-Br) PPCl/SBA15 | H2O | 4.8 | 20.8 | 10 | 10 | 373 | 6 | 76 | 56 | 0.54 | l (5 runs) | 384 |
The yield of MA + MAc obtained with soluble heteropolyacid soluble catalysts (Table 8, entries 1–3)378–380 (homogeneous catalysts) is similar to that of reactions utilising heterogeneous catalysts. However, their reutilisation for several runs is not straightforward because it requires the separation of the soluble catalyst from the liquid phase. This prevents their practical application. The authors mentioned that it is possible to separate soluble heteropolyacids by the use of water-organic solvent biphasic systems378–380 but they did not demonstrate this protocol. This problem could also be circumvented using heteropolyacids supported on different solid supports, but no information has been disclosed.
Regarding the heterogeneous catalyst based on vanadium, it must be mentioned that, as for 5-HMF oxidation, acetic acid or acetonitrile were needed to obtain a high yield; water resulted in a lower yield. In the case of vanadium species anchored on graphene oxide, the productivity was well above 1 g of MA/MAc per g of catalyst per h (in the case of VO/GO, the productivity was 1.81 g gcat−1 h−1).371,372 Unfortunately, these vanadium-based catalysts are deactivated. However, as also explained for 5-HMF, this can be an experimental flaw related to the accumulated loss of catalyst while handling the catalyst in the reuse runs. Long-term experiments under continuous flow are needed to assess deactivation.
Fe-porphyrins (Table 8, entries 8 and 9) displayed lower productivity values than those of the vanadium-based catalysts (in the range of 0.3–0.5 g of MA gcat−1 h−1) but, interestingly, presented better reutilisation properties that make these systems worthy of further study. When discussing productivity, we must bear in mind that the experiments were conducted in water, a solvent much greener than acetic acid and acetonitrile, but less selective. A fairer comparison is that conducting the reaction in these solvents (so far not conducted). In addition, the molecular weight of porphyrin molecules is quite significant (higher than 300 g mol−1), and the supported systems contained less than 1 wt% metalloporphyrins. In practice, the reaction rate per site is quite high, and yield and productivity may be more favourable if larger loadings are deposited on the support. Despite using water as a solvent, these systems presented similar yields to those achieved with the use of vanadium-based catalysts and quite good reusability properties. The challenge is to make these Fe-porphyrins more stable and more productive to increase their chances of a practical application.
3.2.4.2. Gas-phase oxidation of furfural to MA using O2. The scientific literature covering this reaction is quite scarce, and only a limited number of catalytic systems have been studied (see Table 9). The reaction is conducted between 543 and 633 K and at GHSV values between 600 and 36
000 h−1. In some cases, the studies dated back the first half of the last century, and relevant information, such as exact reaction conditions, the yield of MA and the Space Time Yield (STY) of MA in terms of gMA gcat−1 h−1, cannot be inferred. Most of the catalysts were based on vanadium, although some promoters, such as Ni, Na, Ag, Fe, Sn, Bi and, especially, Mo and P, have also been incorporated.28–31,385–393 In most cases, unsupported systems were used as catalysts, but alumina and pumice have also been utilised as supports.
| # | Catalyst | Operating conditions | Catalytic results | Ref. | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gas phasea | S ccb (vol/vol) | O2/FURc (mol) | T (K) | GHSVe (h−1) | X S (%) | Yg (%) | STYh (g gcat−1 h−1) | Deact.i | |||
| a Gas phase. b Substrate concentration in vol/vol (%). c O2/furfural molar ratio. d Temperature conditions in Kelvin. e Gas hourly space velocity in h−1. f Substrate conversion. g Yield towards product (MA). h STY (space-time yield). i Catalyst deactivation: strong (st), moderate (m), low (l) and negligible (ne); n.s. (not studied) n.r. (not reported). j MA selectivity. k Reactivation in weeks. | |||||||||||
| 1 | V2O5/pumice | O2–N2 | n.r. | 44 | 593 | 600–1000 | 100 | 25 | n.s. | Stable | 385 |
| 2 | Fe molybdate | O2–N2 | 0.02 | 1200 | 543 | 2461 | n.r. | 85 | 0.0016 | 386 | |
| 3 | V–Mo–P–O | O2–N2 | 0.3–0.4 | n.r. | 573 | 15700–18000 |
n.r. | 60j | 0.47 | n.s. | 387 |
| 4 | V–Mo–P/α-Al2O3 | O2–N2 | 0.8 | 3–117 | 575–623 | 8000–36000 |
n.r. | n.r. | n.s. | n.s. | 388 |
| 5 | VOx/Al2O3 | O2–N2 | 0.3–1.0 | 10–40 | 583 | 1720 | 100 | 86 | 0.11–0.33 | m (20 h) | 389 and 390 |
| 6 | VPO | O2–N2 | 6.0–10.0 | 2 | 633 | 2400–7200 | 100 | 90 | 1.31–2.26 | ne (25 h) | 391 |
Fig. 13 summarises the pathway of the reaction that has been proposed based on the products detected. Furfural is initially oxidised to furoic acid, which is rapidly decarboxylated to furan, and furan is oxidised to furan-5(2H)-one, which is, finally, oxidised to MA.389–391 Any of these products can also be fully oxidised to carbon oxides and water, which eventually results in a loss of MA yield. Molar yields between 25 and 90% and STY from 0.1 to 2.3 g gcat−1 h−1 have been reported (see Table 9).
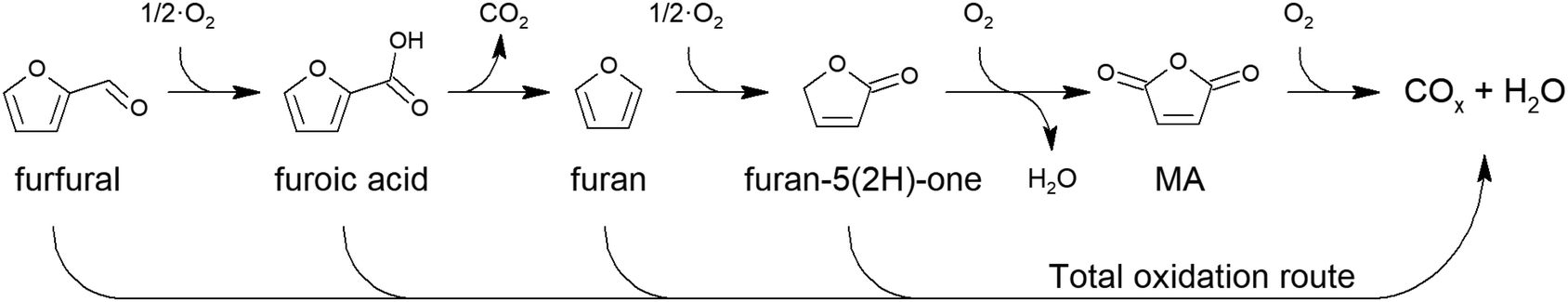 | ||
| Fig. 13 Oxidation of furfural to maleic anhydride with gas phase O2 (adapted from ref. 387). | ||
A non-selective reaction that must be seriously considered is the formation of heavy furanic resins by oligomerisation of furfural. Furfural is very prone to oligomerisation, and O2 triggers this reaction;385,394 therefore, resin formation has to be minimised to obtain a high MA yield. The formation of resins can go unnoticed because resins are heavy compounds with high boiling points and cannot be transported in the gas phase, remaining either in the catalyst bed or in the lines downstream of the catalyst bed.385 Long-term operation may result in plugging. Detection by conventional methods, such as GC or HPLC, is difficult; but its formation can indirectly be noticed and quantified by the lack of a carbon balance.390 VPO systems have recently been reported to achieve a yield of MA as high as 90% (Table 9, entry 6).391 The overall selectivity of the different products was reported to be exactly 100%, consequently, resins may have been unaccounted for. These remarkable results require confirmation.
The deposits of maleates and resins are involved in the deactivation of catalysts.390 Fortunately, the activity is fully restored by burning these deposits off at high temperatures (773 K) under air.390 The deposition of these species can also be prevented by conducting the reaction under reaction mixtures with a high oxidising potential, namely, a high O2/furfural mol ratio and high temperatures (to achieve full furfural conversion). Within this context, it is worth mentioning that a Fe molybdate catalyst has been reported to be not deactivated but, on the contrary, activated with time on-stream and takes weeks (10–20 weeks) to reach its full potential.386
The conflicting results regarding resin formation and deactivation issues require further research to unveil important fundamental aspects, such as the surface species involved in the reaction mechanism, the deactivation phenomena and the effect of contact time, reaction temperature, and the concentrations of FUR and O2 on the kinetics of the reaction. The limited number of articles and, in some cases, the lack of detailed information about the reaction conditions and the preparation of the catalyst prevent any conclusion about the nature of the active and stable species and, consequently, the design of more active and robust catalysts. Further investigation is required to shed light on these issues.
Although gas-phase O2-oxidation requires relatively high temperatures (above 543 K), energy integration allows for a minimum selling price of maleic anhydride that is attractively close to the current commercial price of petrochemically produced MA.395 For the gas-phase oxidation of furfural to become more competitive, it still requires further improvements in the space-time yield and productivity, the yield of MA and the long-term stability of the catalyst.395 The economic viability is very sensitive to the price of furfural;395 consequently, it is also essential that the catalyst can process low-grade furfural that is less expensive than high-grade furfural used in all of the studies conducted so far.
3.2.4.3. Liquid-phase oxidation of furfural to MA/MAc using aqueous H2O2. A third route to produce MAc from furfural is liquid-phase oxidation using H2O2 as the oxidant. MAc is formed rather than MA because the reaction takes place in aqueous solutions. A practical application seems very unlikely. The techno-economic evaluation mentioned in the previous section for the gas-phase oxidation with O2 of furfural was also extended to liquid phase H2O2-oxidation.395 Notwithstanding the mild conditions (343–363 K and at atmospheric pressure), this reaction is estimated to be far from economic feasibility (the minimum selling prices are almost ten-fold higher than the current price of petrochemical MA). The reasons are the high H2O2 cost, the low productivity, the diluted aqueous solution used in the process and the complicated purification protocol needed to separate MAc from the other by-products.395 This route can only become attractive in a scenario of substantially low-cost H2O2 (several times less expensive than the current price). In the hope that the cost of H2O2 can be reduced in the future, a very brief review of the studies conducted so far will be presented below. Further details concerning the wide range of catalysts and the mechanism of reaction can be consulted in ref. 221 and 396.
Generally, there are two families of catalysts for this reaction. The first family of catalysts is those possessing either acidic or basic Brønsted sites. The reaction proceeds according to the following well-accepted mechanism: the initial step is the Baeyer–Villiger oxidation of the aldehyde group of furfural through furyl hydroxyhydroperoxide and a formylfuroate ester (see Fig. 14). The latter is hydrolysed, releasing formic acid (actually the most concentrated product of the reaction) and 2-hydroxyfuran, which is in keto–enol tautomeric equilibrium with furan-2(3H)-one and furan-2(5H)-one. These two furanones are the starting point for further hydrolysis and oxidation steps, the former (route 1 in Fig. 14) ending in succinic acid (SAc) and the latter (route 2) in 5-hydroxy-furan-2(5H)-one (HFONE) and, finally, in MAc. Badovskaya et al.397 have proposed that SAc can also be derived from HFONE via another keto–enol tautomeric equilibrium. This reaction presents a quite complicated mechanism for only obtaining MAc. However, the MAc yield can be maximised by tuning the reaction conditions.
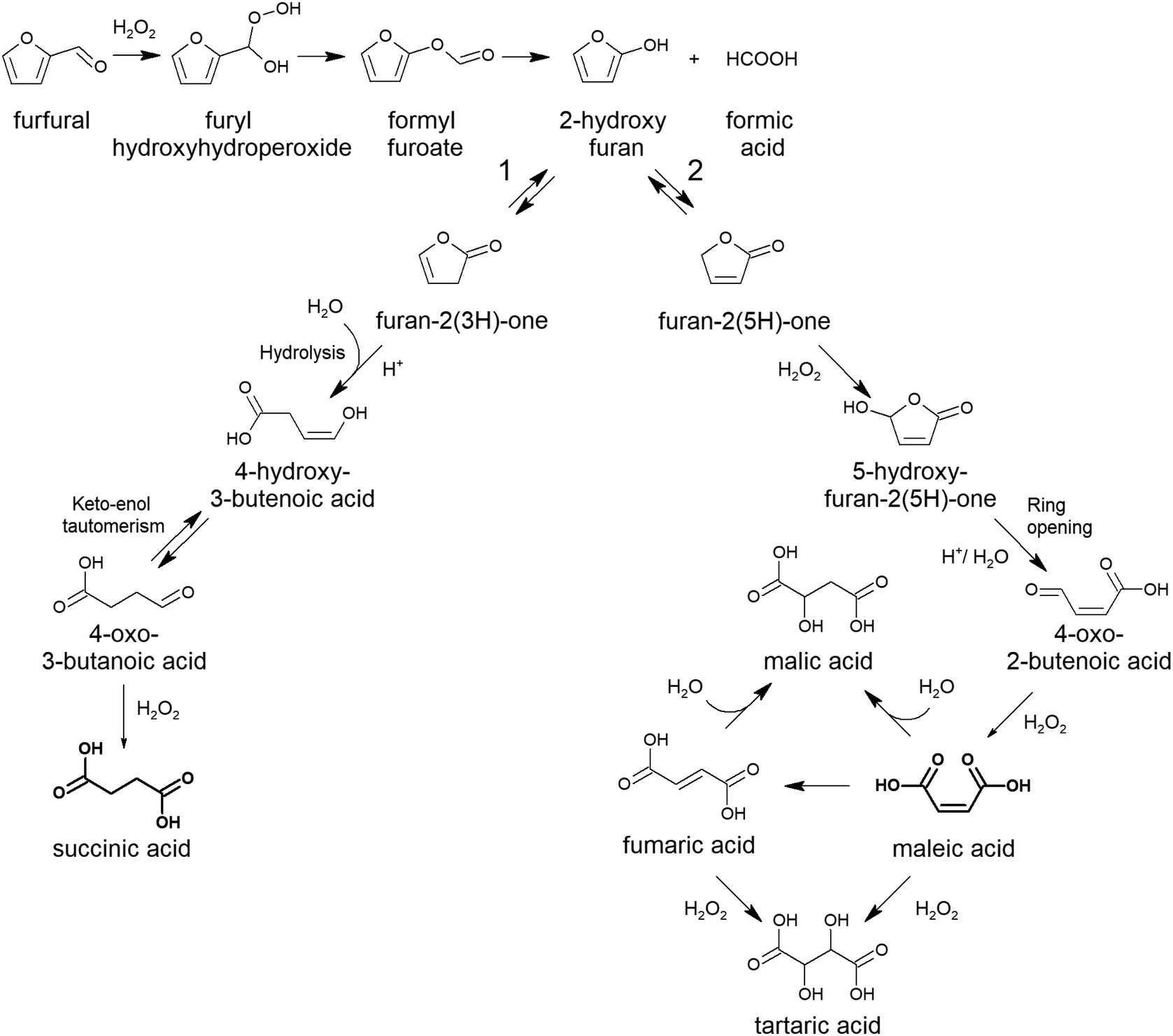 | ||
| Fig. 14 Oxidation of furfural with H2O2 in the liquid phase using Brønsted acid or basic catalysts. | ||
Typical reaction conditions that favour MAc formation are temperatures between 303 and 343 K, reaction times much longer than 3–4 h and a H2O2/FUR mol ratio much higher than 3 (the stoichiometric ratio). Shorter reaction time (less than 1 h) directs the production to furan-2(5H)-one and HFONE whereas a H2O2/FUR mol ratio lower than 3 maximises the formation of SAc. Relatively severe conditions give rise to overoxidation to formic acid or even to CO2 and H2O. As discussed thoroughly in the next section, this type of catalyst is more appropriate for oxidation to SAc than for MAc production.
It is worth noting that, in practice, the reaction proceeds without the incorporation of a catalyst but at a much lower rate. Faster rates require the presence of acid catalysts (basic catalysts are consumed during the reaction because of the neutralisation by the acids formed in the reaction). Among the variety of soluble and solid catalysts studied (see ref. 221), the best yield reported thus far has been obtained using formic acid as a catalyst (which is actually one of the products of the reaction). A yield as high as 95% of MAc was obtained at 353 K after 8 h of reaction using a 1.6 wt% of furfural, a H2O2/FUR mol ratio of ca. 10, in a solution containing ca. 80 wt% formic acid (FAc).398 In addition to the high concentration of formic acid required, which is translated into very low productivity (lower than 0.003 g MAc gcat−1 h−1), the problem of this homogeneous process is that separation from FAc and further purification of MAc is required. A similar separation problem is faced when using other homogeneous catalysts, such as H2SO4, a much stronger acid than formic acid. In this case, a yield as high as 85% was obtained with a much lower catalyst loading (0.01%);398 consequently, a much higher equivalent productivity (more than 1 kg MAc gcat−1 h−1) was obtained. Betaine chloride has also been used as a homogeneous catalyst;399 a yield of 61% MA and 31% fumaric acid, an isomer of MA (equivalent to a productivity of ca. 0.4 g MAc gcat−1 h−1) was achieved using a relatively high concentration of furfural (10 wt%) and betaine (40 wt%) after 30 min at 373 K. Acetone is needed for separation of the catalysts from the reaction medium, and acetone is later distilled to remove it from the products.
In principle, solid catalysts, such as sulphonic acid resins or sulphonated silica or carbon, are better options than liquid catalysts because they can easily be separated from the reaction by filtration. A yield of ca. 80% was obtained using Amberlyst-15,398 a sulphonic acid resin, a 1.6 wt% solution of furfural, a H2O2/FUR mol ratio of ca. 10, and a catalyst loading of 0.7 wt% at 298 K and 7 h of reaction. Deactivation is caused either by leaching of the acid sites or fouling due to deposits. The latter cannot be removed by calcination because sulphonic acid sites will also be gasified. Amberlyst-15 has been reported to be stable for three runs.400,401 However, stability must be studied for a larger number of runs.402
Another family of catalysts is that including titanium-silicalite (TS-1), a pure silica zeolite with an MFI structure in which Ti4+ cations are incorporated within the framework, replacing Si4+ cations.402–405 This reaction does not proceed through the mechanism mentioned in Fig. 14 but through a different mechanism that does not involve the formation of SAc (explained elsewhere404). This catalyst is more selective for the formation of MAc than the other catalysts mentioned above, and SAc is a minor product that comes from unavoidable B–V oxidation (the acids formed during the reaction autocatalyse the B–V reaction). The formation of HFONE takes place rapidly, its oxidation to MAc is much slower, and high reaction temperatures (343 K) and a high H2O2/FUR mol ratio (>7) are required to achieve the maximum yield of MAc (70%) in 4–6 h. The TS-1 catalyst is deactivated, and the principal cause of deactivation is the deposition of heavy products on the surface of the zeolite.403,405 The catalyst can be fully regenerated by burning off these deposits by calcination at 823 K.
Interestingly, deactivation of the TS-1 catalyst was suppressed by using GVL/H2O mixtures instead of only water; in this case, the deposition of heavy products was prevented. Further improvement of the yield of MAc can be accomplished (83%) by the creation of mesopores within the primary zeolite particles and the increase of the hydrophobicity of TS-1 channels using hydrothermal treatment with tetrapropylammonium hydroxide.47
Irrespective of the type of catalyst used for this reaction, the critical barrier for the practical application is the price of H2O2 itself, which must be substantially reduced for the process to become competitive.395
3.2.4.4. Electrochemical oxidation of furfural to MAc. The electrochemical oxidation of aqueous furfural to MAc is very attractive because the oxidation of biomass-derived furfural is coupled with the H2 evolution reaction at the cathode. In addition, renewable electricity (wind, photovoltaic, hydroelectric, etc.) can be used. Early investigations used a carbon-based electrode.406,407 Recent studies have been conducted using a PbO2 anode408 and 0.1 wt% FUR in a 0.1 M H2SO4 solution (in basic medium, furfural is oxidised to furoic acid) and at 2.0 V vs. RHE. A maximum yield of 65% MAc was achieved. Interestingly, H2 was also produced. It was proposed that the overall reaction in the cell was:
| Furfural (C5H4O2) + 4H2O → MAc (C4H4O4) + CO2 + 4H2 |
The faradaic efficiency was only 46% because H2O is also oxidised at the electrode. The formation of humins or furanic resins was also proposed, which results in lower yields of MAc.408 Pt or MnO2 anodes gave even worse MAc yields and faradaic efficiencies than the PbO2 anode. It was proposed that FUR is initially oxidised to 2-furanol, which is in tautomeric keto–enol equilibrium with furan-2(3H)-one and furan-2(5H)-one, that is subsequently oxidised to MAc via HFONE.409 Furoic acid was also proposed to be involved as an intermediate between FUR and 2-furanol.408
Further investigations must be directed at improving the faradaic efficiency and the chemical selectivity at relatively higher furfural concentrations to make the process more attractive for practical applications. Techno-economic evaluation studies of this process, including the purification steps to separate the MAc from the solution, are needed to assess its economic viability.
A possibility to decrease the cost in the liquid phase oxidation is processing low-grade furfural solutions directly obtained from the biomass instead of using high-grade furfural. High-grade furfural requires two energy-demanding distillation steps, and, consequently, using low-grade furfural allows for skipping or simplification of the distillation process. Low-grade furfural is particularly interesting if the oxidation is conducted with relatively high-concentration furfural solutions (10–20 wt%) in the same solvent used for processing the lignocellulosic biomass. The challenge then is to find robust catalysts resistant to the impurities present in low-grade furfural solutions and to develop an affordable separation and purification process for MA/MAc. Precipitation is a good first, simple option; distillation or evaporation of the solvent seem prohibitive.
The same can be said about the oxidation of butanol, LEVA, or 5-HMF. Their market price discourages their use, but using low-grade, concentrated 5-HMF and LEVA solutions, directly obtained from biomass, could be an alternative route.
The use of low-grade furfural solutions is more problematic for gas-phase oxidation because evaporation is then needed. In addition, gas-phase processes have been revealed to be less productive than those in the liquid phase. However, gas-phase oxidation presents the advantage of easy downstream separation by deposition of MA (the melting point of MA is ca. 326 K), which may be critical in an economic evaluation of the global process.
3.3. Succinic acid
In the context of polymers, the importance of succinic acid (SAc) is related to the production of polybutylene succinate (PBS) and succinic-based polyurethanes.410 In the case of PBS, a polyester formed by the condensation of succinic acid and 1,4-butanediol (BDO), the latter can also be produced from SAc.411 In addition, SAc also has other industrial applications (pharmaceutical, food and agrochemical industries and in the production of γ-butyrolactone and tetrahydrofuran411).The market size of SAc in the last decade was approximately 40–50 kton of SAc per year.411,412 The production of SAc has been dominated by hydrogenation of petrochemical maleic anhydride. A renewable process based on the fermentation of sugars is becoming increasingly competitive. In 2011, the proportion of biomass-derived SAc was quite low (3%);413 in 2016, the total capacity of the fermentation route was reported to be in the range of petrochemical production (55 kton per year412). Annual growth rates of 25–30% were projected, and several companies announced the opening of new fermentation plants with a production capacity totalling 400 kton per year.412
The price of SAc fermentation ($2000–3000 per ton) is in the range of the petrochemical SAc.412–414 Its competitiveness is very sensitive to the price of oil. Fermentation presents several drawbacks, including the precise control of operation parameters for the handling of microorganisms, low productivity, problems associated with the separation of succinic acid from the broth and downstream purification that penalises its competitiveness, especially in a scenario where oil is less expensive than biomass.
Other routes of producing succinic acid from different biomass-derived platforms have been demonstrated. They are still at a very low readiness technological level (only studied at a laboratory scale), but they are attractive because they are based on chemo-catalysis and do not present the problems associated with the fermentation route. These routes are reviewed below. Three different renewable SAc precursors have been studied: furfural, 5-hydroxymethylfurfural and levulinic acid. Liquid phase oxidation with H2O2 (liquid phase) and with O2 (liquid and gas phases) has been carried out. Finally, a recently proposed renewable modification of the petrochemical route is also summarised.
Table 10 summarises the most active solid Brønsted acid catalysts studied in this reaction. Soluble catalysts are not easily separable from the reaction medium, so the table only deals with solid catalysts. A wide variety of catalysts (such as Amberlyst resins, Nafion, silica-supported Nafion, KIT-6 silica-supported V-Zr pyrophosphates and carbon or graphene oxide functionalised with sulphonic groups) have been studied.400,415–420 Tuning the reaction conditions can increase the yield of SAc. Relatively low H2O2/FUR ratios (between 3 and 6) facilitate SAc formation via furan-2(3H)-one, preventing the formation of MAc. Larger ratios (>6) accelerate the formation of MAc, and then lower SAc yields are obtained. Mild temperatures (between 333 and 353 K), a H2O2/FUR mol ratio between 3 and 6 and a reaction time between 1 and 24 h were used. Quite high productivity, between 1 and 3 g SAc gcat−1 h−1, has been obtained with carbon and graphene oxide functionalised with sulphonic groups418,419 (Table 10, entries 5 and 6), largely because low loadings of catalysts and short reaction times were used. Deactivation of the catalyst is one problem that must be addressed. In the best-case scenario, the deactivation is slight but continuous, which threatens their use in practical applications. The graphene oxide-based catalyst (Table 10, entry 6) showed a very low deactivation rate (which, as explained in the previous Section 3.2, can also be compatible with the accumulated loss of the catalyst during reuse studies). Long-term experiments in fixed bed continuous flow reactors are needed to really evaluate the deactivation. Supported V-Zr pyrophosphate (Table 1, entry 7) was also very productive, but its SAc yield was quite low (worse atom efficiency); besides, acetonitrile was used as a solvent.
| # | Catalyst | Operating conditions | Catalytic results | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Solv.a | S ccb (wt%) | C/Sc (wt%) | H2O2/FURd mol | T (K) | t (h) | X S (%) | YSAch (%) | Produc.i (g gcat−1 h−1) | Deact.j | |||
| a Solvent. b Substrate concentration weight ratio in %. c Catalyst to substrate weight ratio in %. d H2O2/furfural molar ratio. e Temperature conditions in Kelvin. f Reaction time. g Substrate conversion. h Yield towards product. i Productivity. j Catalyst deactivation: strong (st), moderate (m), low (l) and negligible (ne); n.s. (not studied), n.r. (not reported). | ||||||||||||
| 1 | Amberlyst-15 | H2O | 2.7 | 52.0 | 4 | 353 | 24 | 100 | 74 | 0.07 | l (3 runs) | 400, 415 and 416 |
| 2 | Nafion NR50 | H2O | 32.7 | 52.0 | 4 | 353 | 24 | 100 | 41 | 0.04 | n.s. | 400, 415 and 416 |
| 3 | Nafion SAC13 | H2O | 2.7 | 52.0 | 4 | 353 | 24 | 100 | 28 | 0.03 | n.s. | 400, 415 and 416 |
| 4 | Smopex 101 | H2O | 10.6 | n.r. | 4 | 353 | 24 | 100 | 65 | n.r. | n.s. | 417 |
| 5 | C-SO3H | H2O | 5.8 | 37.5 | 5 | 333 | 1 | 100 | 81 | 2.66 | m (13 runs) | 418 |
| 6 | Graphene oxide-SO3H | H2O | 2.5 | 10.4 | 6 | 343 | 6 | 100 | 88 | 1.73 | l (6 runs) | 419 |
| 7 | V-Zr-P oxide on KTI-6 silica | CH3CN | 19.1 | 6.3 | 3 | 343 | 1 | 81 | 29 | 4.23 | ne (4 runs) | 420 |
Regarding the economic viability, the techno-economic evaluation conducted for the oxidation of furfural to MAc with H2O2 concluded that the process was uneconomical because of the high price of H2O2.395 A specific techno-economic study for this reaction is required to rigorously compare the price of SAc obtained with H2O2 with that of petrochemically produced SAc. Less H2O2 is needed for succinic acid production than for MAc production, so the price may be competitive.
Other alternative substrates for SAc production at quite interesting productivities are 5-HMF or levulinic acid. Thus, from 5-HMF, a yield of 92% succinic anhydride was obtained using sulphonic ionic liquids as catalysts at 333 K and 14 h of reaction (with a productivity of less than 0.2 g SAc gcat−1 h−1).421 From levulinic acid, a maximum yield of succinic acid of 36% (at a conversion of 48%) was obtained using tungstic acid as a catalyst after 3 h at 363 K from a 15 wt% levulinic acid solution (equivalent to ca. 2.8 g SAc gcat−1 h−1).202 Finally, a 61% yield was achieved from a 5 wt% LEVA solution using H2SO4 as the catalyst after ca. 4 h at 363 K (0.4 g SAc gcat−1 h−1).422 When considering these alternative platforms, it must be noted that furfural is thus far the only substrate that is currently commercially available as a commodity. Consequently, until 5-HMF or levulinic acid are commercially available at a low price, furfural seems to be the best option for oxidation to SAc using H2O2 as the oxidant.
Regarding LEVA, different homogeneous processes have been reported, such as using a mixture of Co(AcO)4·4H2O, Mn(AcO)4·4H2O and HBr in acetic acid solution (between 393–473 K and 30 bar O2)423 or using nitric acid (3–11 M) along with the addition of small amounts of sodium nitrite as oxidants and V2O5 as a catalyst (between 313–373 K).424,425 Their yields of SAc were quite modest, and these processes seem quite impracticable at a large scale due to either the use of harsh, unsustainable reactants (second case) or difficulties in reusing the catalysts (first case). Oxidative demethylation of levulinic acid with I2 dissolved in t-BuOK and t-butanol–H2O solutions yielded a much higher SAc yield (above 83%) in approximately 1 h than that reported above.424 This process was even studied using cellulose to provide levulinic acid with an overall yield of 72% based on the initial amount of cellulose. However, this homogeneous I2-driven demethylation process used unsustainable and unaffordable reactants.
Interestingly, the use of solid catalysts based on RuCl3 anchored on different amino-functionalised supports (SiO2 or carbon) has also been investigated426,427 with quite interesting results. Remarkably, full conversion of LEVA to SAc (a yield of 100%, equivalent to a productivity of 0.29 g SAc gcat−1 h−1) was obtained at 453 K and 10 bar O2 after 4 h of reaction using RuCl3 anchored on multiwalled carbon nanotubes functionalised with 2-aminophenol.426 The productivity was quite modest in comparison to those presented in previous paragraphs because the studies were conducted with a very low concentration of LEVA (ca. 0.6%) and a high wt% catalyst/LEVA ratio (close to 85%). However, this system also showed excellent reusability properties: after 3 runs, the SAc yield was still above 96% (by-products were 3-hydroxypropanoic, acetic and formic acids).426 These very promising results must be confirmed for long-term experiments using fixed-bed continuous flow reactors, with more concentrated LEVA solutions and with low catalytic loading.
The route through which the reaction proceeds is not disclosed, but the presence of basic sites provided by the amino groups seems to be instrumental. Within this context, it is worth mentioning that these systems based on RuCl3 anchored on N-doped carbon also showed high yields of SAc but directly from the oxidation of glucose.426,428–430 Interestingly, a yield of SAc of 87% was obtained with a N-doped graphene support at 433 K and 16 bar O2 and after 20 h. These results are also very encouraging because, although studies were conducted with very dilute glucose solutions (0.9 wt% of glucose), they represent the production of SAc in a one-pot, single-step reaction from glucose, and the intermediate transformation of glucose to LEVA is not required. Actually, the pathway of the reaction proposed for the conversion of glucose to SAc does not involve the formation of LEVA but rather the formation of tartaric acid as a primary intermediate.426,428–430
Concerning the oxidation of 5-HMF with O2, extensive research has been devoted to the oxidation of the side chains of 5-HMF to other chemicals, like diformylfuran, 5-hydroxymethyl-2-furan carboxylic acid, 5-formyl-2-furan carboxylic acid and furan dicarboxylic acid (see ref. 329, 431 and 432 and references therein), not to SAc. The investigation of the oxidation involving the furan ring of 5-HMF to yield SAc is, to the best of our knowledge, limited to the investigation conducted by Parvulescu et al. with a Fe3O4@SiO2–CoOx catalyst.374 In this work, a yield of 73% at a conversion of 5-HMF of 79% was obtained at 383 K and 10 bar O2 and 8 h of reaction (Fe3O4 was incorporated to separate the catalyst magnetically from the solution for the reuse studies). The co-addition of NaOH was needed for the oxidation to proceed to SAc; otherwise, MAc was the major oxidation product. Interestingly, according to the authors the catalyst was stable for 9 runs (but no data were presented). This interesting result requires more studies, especially to confirm the stability of the catalyst under long-term, continuous flow experiments and when employing a more concentrated 5-HMF solution (0.5 wt% 5-HMF was used in this study374). This information is essential for assessing the practical implementation of this process.
Surprisingly, as far as we know, the only study on this topic is the patent of A. P. Dunlop of the Quaker Oats Company filed in 1954 that utilised vanadium oxide supported on pumice as a catalyst, reporting a yield of 83% at 648 K. This 1954 patent is not very detailed, and, unfortunately, no information was disclosed about the specific LEVA and O2 molar concentrations and the contact time used for achieving this yield.433 Consequently, despite the relevance, this is a very unexplored topic.
000 g SAc gPd−1. An LCA of this CTH process indicates that even when using oil-derived FAc, the CTH process is greener than the conventional hydrogenation process with H2, and the benefits in some of the significant environmental parameters studied in this LCA are even larger if biomass-derived FAc is involved.437
3.4. Adipic acid
Adipic acid (AdA) is mainly used in the production of nylon-6,6, polyurethanes and adipic esters, but it is also employed in the food, pharmaceutical and cosmetic industries.438–440 The main global producers of adipic acid are Invista, Ascend, Honeywell, BASF, Radici, China Shenma and PetroChina. Its market price is in the range of $1500–1700 per metric ton, and the worldwide production of AdA is approximately 2.6 million tons with a growing demand of 3–5% per year.438,441 High-purity fibre-grade AdA is used to produce nylon 6,6, while lower grade AdA is employed in the synthesis of polyurethanes.Currently, its production is based on petrochemical routes. Benzene is reduced to cyclohexane, which is oxidised in air with a cobalt-based catalyst to obtain the so-called KA-oil (ketone-alcohol oil). This is a mixture of cyclohexanone and cyclohexanol, which is treated with nitric acid to produce AdA.456 This process is not sustainable due to the low conversion of cyclohexane, which requires a large amount of energy for recycling and limits the adipic acid yield. Furthermore, this process produces nitrous oxide emissions, a gas with notable greenhouse effects.
The need to find alternative sustainable routes to produce adipic acid has stimulated investigations in the last decade.438,457 In this review, the most promising routes starting from the following biomass chemical platforms will be outlined and updated: biobased KA-oil, γ-valerolactone, cyclopentanone, 5-hydroxymethyl furfural, succinic acid, glucaric acid, glucarates and muconic acid (Fig. 15). In many of these platforms, several steps are required, and some of them involve fermentation processes. A meticulous description of all of these processes would require a review for each process; thus, we concentrate on the most significant technical aspects of the chemical steps in the context of the whole process. Table 11 summarises the most relevant information of the last catalytic step of all discussed processes except that involving muconic acids, which is included in Table 12. More thorough and detailed information can be found in the cited references.
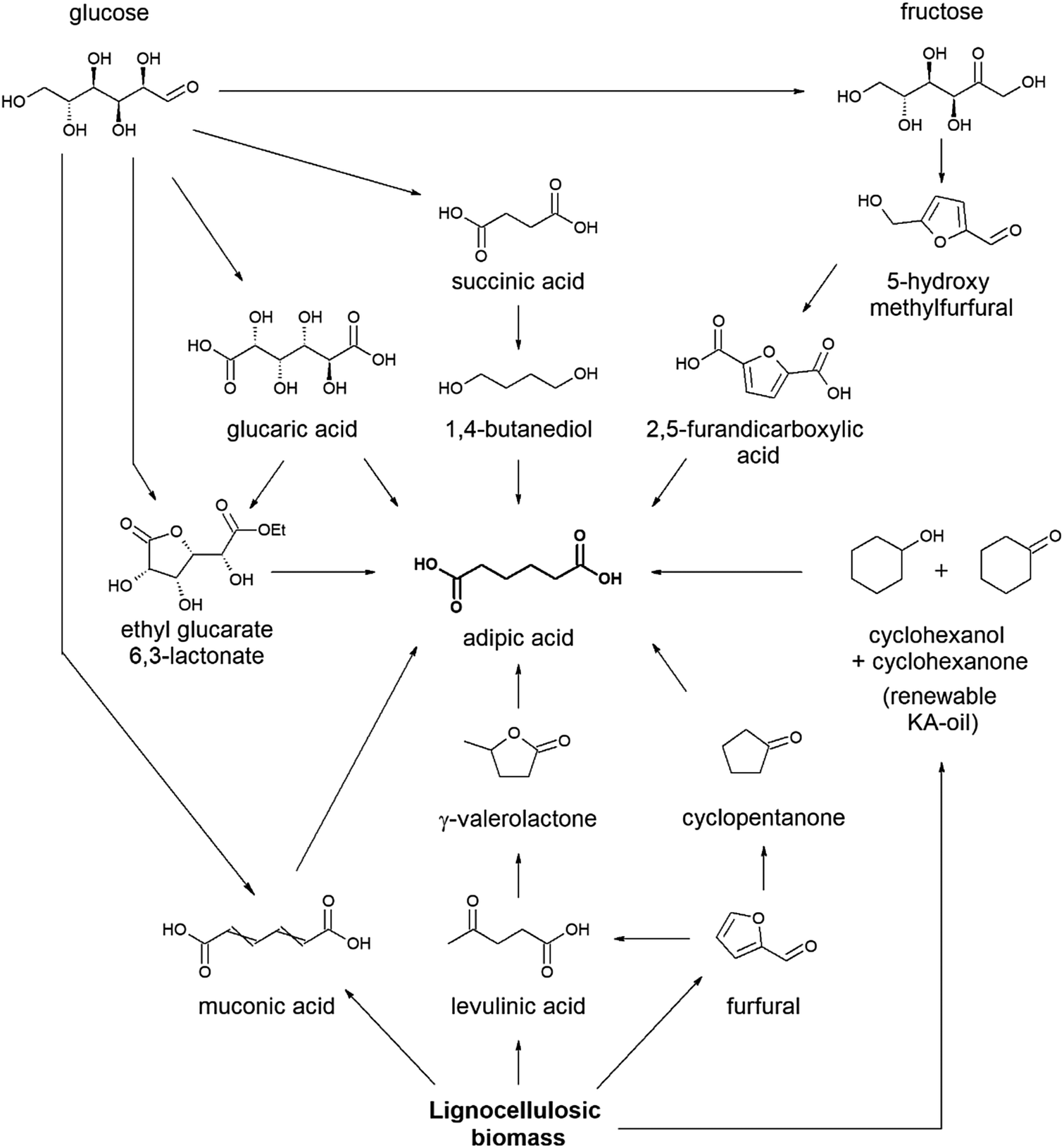 | ||
| Fig. 15 Routes for the synthesis of adipic acid from biomass. | ||
| # | Catalyst | Operating conditions | Catalytic properties | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Solv.a | S ccb (wt%) | C/Sc (wt%) | P (bar) | T (K) | t (h) | X S (%) | YAdAh (%) | Produc.i (g gcat−1 h−1) | Deac.j | |||
| a Solvent. b Substrate concentration weight ratio in %. c Catalyst to substrate weight ratio in %. d Reactant gas pressure in bar. e Temperature conditions in Kelvin. f Reaction time. g Substrate conversion. h Yield towards product. i Productivity. j Catalyst deactivation: strong (st), moderate (m), low (l) and negligible (ne); n.s. (not studied), n.r. (not reported). k Used catalyst obtained by calcination at 773 K for 5 h, under a nitrogen atmosphere. | ||||||||||||
| KA-oil | ||||||||||||
| 1 | K6P2Mo6W12O62 | H2O2 30% | n.r. | 8.3 | n.r. | 363 | 20 | n.r. | 69 | 0.61 | n.s. | 442 |
| 2 | [NH4]β-{[H3NC(CH2O)3]2MnMo6O18} | H2O2 30%, 2% DMSO | 87.0 | 0.2 | n.r. | 293 | 20 | n.r. | 93 | 34.27 | l | 443 |
| γ-Valerolactone | ||||||||||||
| 3 | RhCl3·3H2O | H2O–HBr–AcOH | 93.0 | 5.3 | 28 CO | 493 | 4 | 64 | 40 | 2.75 | n.s. | 444 |
| 4 | Pd(OAc)2, DTBPX, MSA | Diglyme–H2O | 21.0 | 2.0 | 60 CO | 378 | 2 | 65 | 63 | 11.49 | n.s. | 445 |
| 5 | Pd(OAc)2, DTBPX, MSA | Diglyme–H2O | 15.0 | 2.8 | 60 CO | 378 | 5 | n.r. | 48 | 5.00 | n.s. | 446 |
| Cyclopentanone | ||||||||||||
| 6 | PdCl2 | MeOH–CuCl2 | 3.3 | 25.0 | 2 CO | 293 | 96 | 85 | 73 | 0.05 | n.s. | 447 |
| 7 | MgO | DMC | 14.0 | 14.0 | n.r. | 533 | 5 | 86 | 44 | 1.09 | lk | 448 |
| 8 | MgO | DMC | 2.2 | 42.0 | 5 N2 | 533 | 3 | 77 | 47 | 0.65 | n.s. | 449 |
| 9 | CeO2-nanorod | DMC | 2.2 | 42.0 | 5 N2 | 533 | 5 | 97 | 84 | 0.69 | n.s. | 449 |
| Tetrahydrofuran-2,5-dicarboxylic acid | ||||||||||||
| 10 | 5% Pd(NO3)2/silica | AcOH -HI | 3.0 | 100.0 | 49 H2 | 433 | 3 | n.r. | 99 | 0.30 | n.s | 450 |
| 2,5-Furandicarboxylic acid | ||||||||||||
| 11 | Pt–MoOx/TiO2 | H2O | 4.0 | 6.0 | 15 H2 | 473 | 4 | 99 | 21 | 0.82 | st | 451 |
| 12 | Pt/Nb2O5·xH2O | H2O | 0.3 | 128.0 | 30 H2 | 473 | 8 | 99 | 38 | 0.03 | ne | 452 |
| Glucaric acid | ||||||||||||
| 13 | 1.65% Rh/4.7% Pt/silica Davisil 635 | AcOH–HBr | 5.0 | 65.0 | 49 H2 | 433 | 3 | n.r. | 89 | 0.32 | n.s | 453 |
| 14 | 0.4% Rh/0.61% Pt/ZrO2 | AcOH–HBr | 19.0 | 10.0 | 62 H2 | 433 | 1.5 | n.r. | 98 | 4.54 | n.s. | 454 |
| Ethyl glucarate-6,6-lactone | ||||||||||||
| 15 | 1% KReO4–0.75% Pd/C | MeOH–H3PO4 | 28.0 | 12.0 | 5 H2 | 423 | 18 | n.r. | 88 | 0.27 | n.s. | 455 |
| # | Catalyst | Operating conditions | Catalytic properties | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Solv.a | S ccb (wt%) | C/Sc (wt%) | PH2d (bar) | T (K) | t (h) | X S (%) | YAdAh (%) | Produc.i (g gcat−1 h−1) | Deac.j | |||
| a Solvent. b Substrate concentration weight ratio in %. c Catalyst to substrate weight ratio in %. d Reactant gas pressure in bar. e Temperature conditions in Kelvin. f Reaction time. g Substrate conversion. h Yield towards product. i Productivity. j Catalyst deactivation: strong (st), moderate (m), low (l) and negligible (ne); n.s. (not studied), n.r. (not reported). k c,c-NaM as substrates instead of c,c-MA. | ||||||||||||
| c,c-MA | ||||||||||||
| 1 | 1% Rh/AC | EtOH | n.r. | n.r. | 24 | 297 | n.r. | n.r. | 49 | n.r. | ne | 477 |
| 2 | 1% Pd/C | EtOH | 1.0 | 7.5 | 24 | 297 | n.r. | 97 | 94 | n.r. | n.s. | 476 |
| 3k | 5% Pd/C | H2O | 0.2 | 7.0 | 1 | 343 | 1.5 | n.r. | 95 | 9.30 | ne | 478 and 479 |
| 4 | Ni/Al2O3 | H2O | 1.0 | 60.0 | 10 | 333 | 5 | 100 | 98 | 0.34 | ne | 480 |
| t,t-MA | ||||||||||||
| 5 | 5% Pd/C | H2O | 0.2 | 8.0 | 1 | 343 | 1 | n.r. | 100 | 12.85 | ne | 478 and 479 |
| 6 | 5% Pt/C | H2O | 1.0 | 10.0 | 7 | 297 | 8 | n.r. | 92 | 1.18 | n.s. | 474 |
| 7 | Ru10Pt2/SiO2 | EtOH | 6.0 | 1.0 | 30 | 353 | 5 | n.r. | 96 | 19.74 | n.s. | 481 |
| 8 | Re/TiO2 | MeOH | 18–25 | n.r. | 69 | 483 | 5 | 100 | 88 | n.r. | n.s. | 482 |
A way to transform lignin-derived phenol into cyclohexanone is by single-step hydrogenation using palladium-supported catalysts and a Lewis acid459 (Fig. 16).
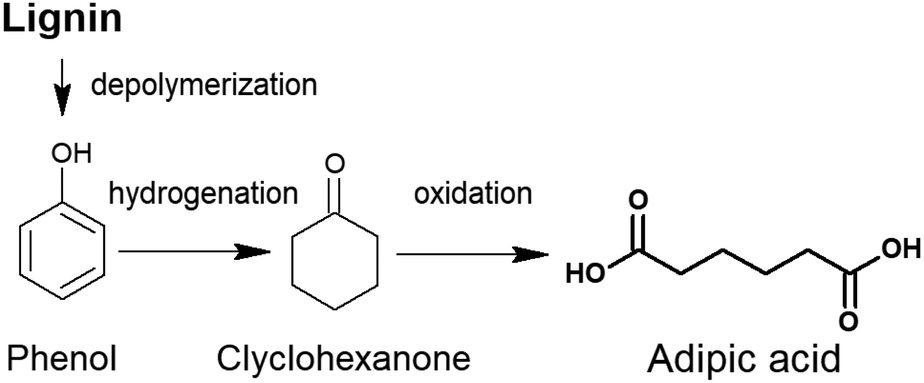 | ||
| Fig. 16 Lignin conversion to adipic acid. | ||
The most studied green route for converting cyclohexanone and KA-oil to adipic acid is oxidation with hydrogen peroxide using polyoxometalate (POM)-based catalysts. Thus, adipic acid yields from cyclohexanone and KA-oil of 59% and 69%, respectively, were obtained using a Dawson-type POM (α-K6P2Mo6W12O62) catalyst at 363 K (entry 1, Table 11).442 More recently, an Anderson POM ([NH4]β-{[H3NC(CH2O)3]2MnMo6O18}) catalyst was reported to obtain a 93% yield of adipic acid from KA-oil at room temperature (ca. 34 g of AdA gcat−1 h−1, entry 2, Table 11).443 These developments avoid the use of nitric acid, which makes them environmentally friendly approaches. However, even though hydrogen peroxide is a green oxidant (water is formed during its reduction), it is also expensive. Consequently, given the difficult transformation of lignin into phenol and the price of H2O2, this route may not be able to compete with the existing petrochemical route of adipic acid production. Currently, lignin conversion should be optimised, and other green oxidants should probably be studied if an economic analysis of the use of H2O2 proves unfavourable.
The conversion of GVL to adipic acid involves the hydrolysis of GVL with acid catalysts, giving a mixture of pentenoic acids, followed by its hydrocarbonylation to adipic acid (more details are given below). The ring-opening of GVL can also be achieved at high temperatures, and if it is performed in the presence of methanol and in the gas phase, it can lead to a mixture of isomeric methyl pentenoates instead of pentenoic acids.445 These pentenoates can be transformed into adipic acid using a palladium diphosphine catalyst in homogeneous media via tandem isomerisation–hydroxycarbonylation, obtaining a 64% yield (entry 4, Table 11).445
A patent also reported the conversion of GVL to adipic acid in a homogeneous mixture of water and acetic acid, using HBr as the promoter and RhCl3 as the catalyst, giving a 40% yield (entry 3, Table 11).444
A techno-economic analysis of the production of AdA from lignocellulose (via GVL) was conducted by Han.439 The analysis was based on results previously reported in the scientific literature and concluded that such a process can economically compete with the current petro-based process. This proposal combines the catalytic conversion of cellulose and hemicellulose to GVL462 and its subsequent conversion to adipic acid446 (Fig. 17). First, it is proposed that cellulose and hemicellulose are transformed into levulinic acid and furfural using sulfuric acid as the catalyst; furfural is converted to furfuryl alcohol that is next transformed into levulinic acid; then, levulinic acid is converted to GVL with high yields using RuSn/C as the catalyst.462 The biomass residues (lignin and humins) are separated and conveniently used for heat and power generation. GVL is then converted to pentenoic acid isomers with SiO2/Al2O3, and, afterward, the production of adipic acid takes place with a palladium acetate catalyst.446 The overall molar yield of adipic acid was 25.9%. An economic analysis of the process was performed, and it proved that the minimum selling price of biobased AdA would be $633–1316 per metric ton. The market price of petro-based adipic acid is $1500–1700,438 which means that, according to Han,439 this renewable process is economically competitive with petro-based adipic acid. Nonetheless, in addition to a practical evaluation of economic viability with real data from a demonstration plant, a complete life cycle analysis of the environmental impacts of the whole process is still needed before this can be considered a green and renewable process for industrialisation.
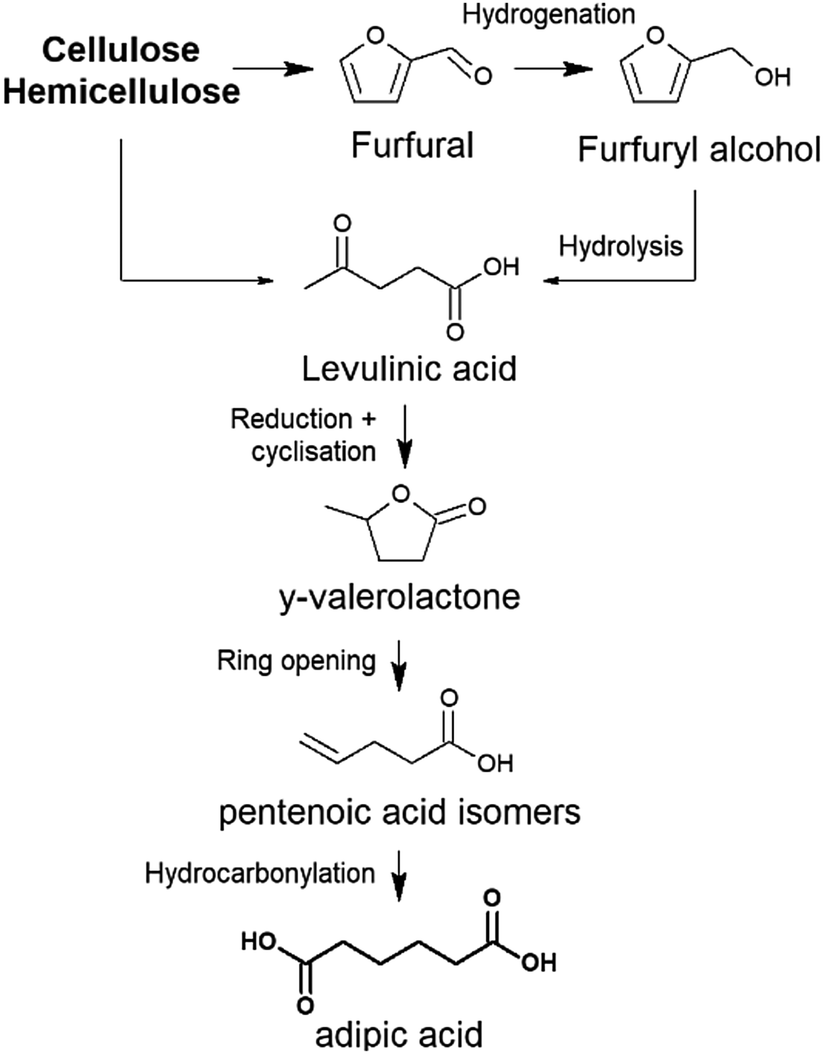 | ||
| Fig. 17 Catalytic route for the production of adipic acid from hemicellulosic and cellulosic sugars via GVL proposed by Han et al.439 | ||
A basic catalyst is needed so CP can react with DMC to yield DAP as shown in Fig. 18. The main challenges of this conversion are the decomposition of the product in the reaction media and the selectivity to DAP due to several other side reactions that can take place, such as methylations and aldol condensation of CP.448
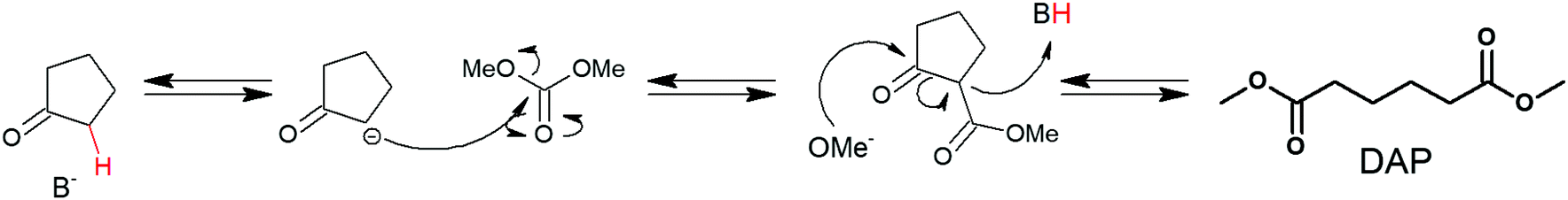 | ||
| Fig. 18 Mechanism of the synthesis of dimethyl adipate from cyclopentanone in the presence of a basic catalyst. | ||
In practice, the conversion of CP to DAP with DMC has been performed at 533 K using MgO448,449 (44–47% yield, entry 7 and 8, Table 11) as a catalyst and a CeO2 nanorod. The last one gave a 84% DAP yield (entry 9, Table 11).449 However, this last approach used a 42% wt cat/CP ratio and a diluted mixture (2.2% wt CP/DMC), which are not ideal conditions because separation and purification from diluted solutions are energy demanding processes.
This reaction is attractive because it combines CO2 and biomass, making it a renewable and green process. However, as mentioned before, this conversion presents several challenges that have not been thoroughly studied yet.
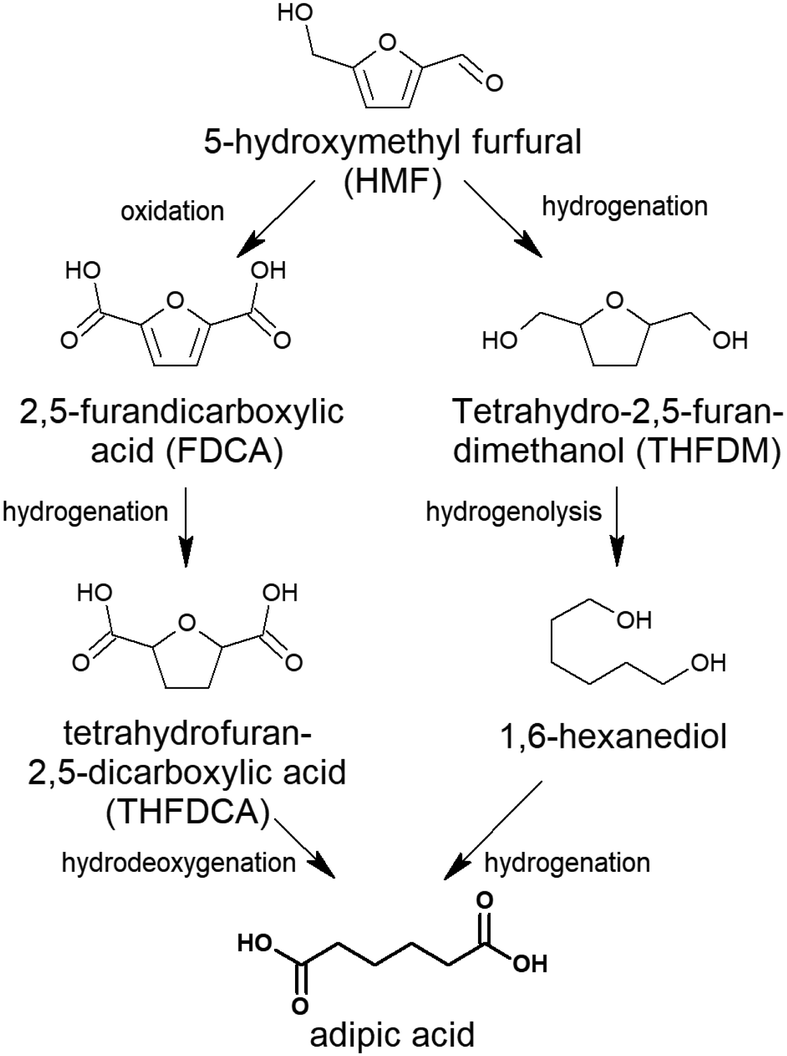 | ||
| Fig. 19 Routes for adipic acid synthesis from 5-HMF. | ||
A multi-step process has been reported showing a route for the conversion of lignocellulose to adipic acid via THFDM: (1) acid-catalysed hydrolysis/isomerisation/dehydration of lignocellulose to HMF in the presence of H2SO4; (2) hydrogenation of HMF to THFDM by a RANEY® nickel catalyst; (3) hydrogenolysis with copper chromite to 1,6-hexanediol; and (4) synthesis of adipic acid using biocatalysis (microorganisms). Each of these processes takes place in a batch reactor.464 The optimisation of these individual reactions has also been studied. The conversion of HMF to THFDM has been investigated using supported Ru, Ni, Pt, Pd and their combinations.465,466 The subsequent reaction to 1,6-hexanediol is described to be catalysed by a bifunctional system composed of Rh-Re/SiO2 and a solid acid (Nafion SAC-13, commercially available).467 The final step, synthesis of adipic acid from 1,6-hexanediol, requires the use of microorganisms (fermentation), as mentioned before.
A second route has been reported via FDCA. FDCA can be synthesised directly from fructose468 or via HMF (see Section 3.1). Rennovia Inc. patented the hydrogenation of FDCA to tetrahydrofuran-2,5-dicarboxylic acid (THFDCA) using Pd/SiO2 as a catalyst, giving an 88% yield. Subsequently, THFDCA is transformed into adipic acid in the presence of HBr or HI in acetic acid, with Pd/SiO2 or Rh/SiO2 (99% yield, entry 10, Table 11).450 More recently, THFDCA was converted into adipic acid in the absence of metal catalysts. This system uses H2 and HI in an organic acid solvent. HI combines the activation of hydrogen and Brønsted acidity, working as a bifunctional catalyst and giving an 89% yield.469 This route involves several steps that require more separation and purification processes, which leads to a loss in the overall yield. A way to reduce the number of steps would be the direct transformation of FDCA into adipic acid, which has been reported using Pt–MoOx/TiO2451 and Pt/Nb2O5·xH2O452 as catalysts at 473 K; however, the adipic acid yields are still low (21% and 38%, respectively, entries 11 and 12, Table 11). Finally, it must also be mentioned that, as stated before, fructose can be transformed into HMF or directly into FDCA; however, fructose is not an abundant source, so the transformation of other sugars still needs to be optimised.
Succinic acid can be directly transformed into adipic acid using a mutated enzyme (NRPS-PKS, non-ribosomal peptide synthetase-polyketide synthase).470 However, it can also be transformed into 1,4-butanediol (BDO) and later into adipic acid via chemo-catalysis. The only practical route to synthesise renewable BDO is from renewable succinic acid by the use of bimetallic catalysts. Thus, a yield of 89% BDO can be obtained via hydrogenation of succinic acid using 1% Pd–4% Re/TiO2 as a catalyst.471 Next, BDO undergoes homogeneous catalytic carboxylation using a Rh(PPh3)COCl catalyst, resulting in a 74% yield of adipic acid. This is called the Monsanto process438 and was patented in 1977, but it is not commercially used.
Finally, it is worth noting that the current price of succinic acid is higher than that of adipic acid, which impacts the economic viability of the succinic acid route of AdA production.
Rennovia Inc. worked on this transformation with a process run at 50% single-pass conversion to glucaric acid to achieve higher yields. This was followed by separation and reaction recycling of intermediates. The long-term catalyst stability of both processes was proven. In fact, Rennovia Inc. had a miniplant with Johnson Matthey Davy Technologies where the adipic acid process was being operated, including incorporation of all major recycle streams. Economic analysis showed that the production costs were expected to be 20–25% below those of conventional petro-based adipic acid production. Furthermore, a life cycle analysis revealed a lower carbon footprint and an 80% reduction in greenhouse gas emissions.472 However, Rennovia Inc. ceased operations in 2018 due to insufficient financing and could not progress from pilot-scale to commercial production.
The disadvantage of this process is the transformation of calcium glucarate into ethyl glucarate 6,3-lactone when glucaric acid can be directly transformed into adipic acid, as mentioned before. Furthermore, this is the sole example found utilising this technology, and there is still much investigation to be done to implement this process industrially.
cis,cis-Muconic acid synthesis was reported via fermentation of glucose with strains of E. coli475 and, more recently, from lignin using an engineered bacteria (P. putida KT2440).476 The highest yield of AdA from biobased c,c-MA was obtained by a 1% Pd/C catalysed hydrogenation in ethanol (97% yield, entry 2, Table 12) (Fig. 20). Even though this catalyst gives high yields, it is deactivated by significant leaching, and it has not been tested for long-term stability. 1% Rh/AC can also catalyse this reaction, although the yield is notably lower (49%). Nonetheless, this catalyst was tested in a continuous trickle bed reactor and, interestingly, showed a stable performance after 24 h.476,477 Unfortunately, its productivity could not be estimated because relevant information for the calculation was not disclosed (entry 1, Table 12).
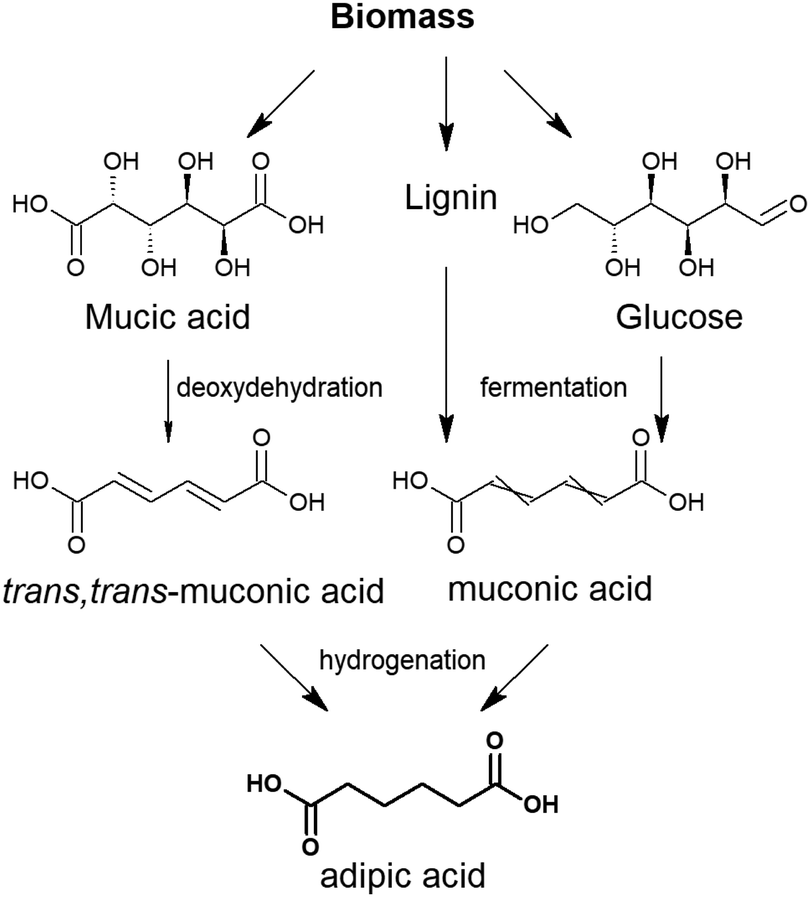 | ||
| Fig. 20 Routes to obtain renewable adipic acid from muconic acids. | ||
The production of adipic acid from c,c-NaM and t,t-MA has also been compared. The hydrogenation of sodium muconate leads to sodium adipate, which, with further purification, leads to adipic acid. The aim of the study was to optimise the reaction conditions and to discover whether the purification step was necessary at the beginning or at the end of this transformation. The results revealed that the hydrogenation of t,t-MA is faster than the reduction of c,c-NaM (entries 3 and 5, Table 12) and that temperature is the main parameter that affects the reaction, with 343 K being the optimal condition.478 Very high AdA productivity was obtained with a 5% Pd/C catalyst (ca. 9 g of AdA gcat−1 h−1, ca. 13 g of AdA gcat−1 h−1, entries 3 and 5 Table 12), but long-term deactivation studies using more concentrated solutions are needed to assess the suitability of this catalyst for the reaction.
A different approach is the transformation of agroresidues to mucid acid, which is subsequently hydrogenated to t,t-MA and then to adipic acid, as mentioned before (Fig. 20). This route presents an overall yield of 8.4% AdA combining biological and chemical catalysis.474 For this transformation, sugar beet residue goes through a fermentation step for the synthesis of mucid acid; this step is not very productive and penalises the overall yield. Mucic acid is then deoxyhydrogenated to t,t-MA in 3-pentanol using NH4ReO4 as a catalyst. This acid is subsequently hydrogenated with Pt/C at room temperature for 8 h, reaching a 92% yield of adipic acid with a net productivity of ca. 1 g AdA gcat−1 h−1 (entry 6, Table 12).
The largest productivities in t,t-MA hydrogenation were obtained with Pt/C (entry 5, Table 12) and Ru10Pt2/SiO2 (entry 7, Table 12) catalysts. Interestingly, in the former case, the stability of the catalyst was studied and found to be negligible.
It should be mentioned that muconic acid has been the most studied route of AdA production. Interestingly, it only requires two or three steps for the overall process of conversion from biomass. In general, the conversion of muconic acid to adipic acid gives high yields, yet deactivation studies have not been conducted for all of the catalysts (Table 12). Furthermore, this route requires previous fermentation steps and, as any other production process involving fermentation, presents the challenge of the separation of biologically produced acids and their further purification, which could account for more than 50% production costs. Nonetheless, it is interesting to outline that a high overall yield can be obtained by combining biological conversion and chemical catalysis for the transformation of lignin into adipic acid. A life cycle assessment of adipic acid production from lignin predicted and improved the environmental impact compared to that of the conventional petrochemical route; even so, some steps of this process are being optimised to further reduce the environmental impacts.483 Before any comparison of the economics of this process with those of petrochemical AdA production is conducted, the long-term stability of the other catalysts has to either be assessed or improved.
A common challenge for the processes mentioned above is that most of them involve several steps, and each requires separation and purification stages, which affect the overall yield and economic viability. To mitigate these problems, an investigation into the integration of all or some steps (in only one, if possible) is required. However, not all transformations may allow for this simplification due to the differences in reaction conditions and in problems associated with the presence of by-products that may poison the catalyst and prevent the next reaction from proceeding. Within this context, there is also a lack of investigations in continuous mode operations using fixed bed reactors. This configuration allows for the use of different catalytic beds with different reaction conditions (temperature, pressure, contact time, etc.) that may favour the simplification and intensification of the overall process.
Catalyst stability is an important factor for technical and economic viability. Many studies of the catalytic steps mentioned above have overlooked the investigation of stability.
Another aspect that requires improvements in many of the processes described above is the concentration of the solutions: an excessively diluted product involves larger equipment and increased difficulty in product recovery, which results in higher processing costs.484 Most of the processes mentioned above use quite dilute feeds (less than 10 wt%), and further investigations are required using more concentrated samples.
3.5. Terephthalic acid
Terephthalic acid (TPA) is predominantly used in the production of polyester fibres, films, and polyethylene terephthalate (PET), the latter used in bottles and food product containers, and a number of consumer goods (water, beverages, detergents, etc.). The worldwide production of purified TPA was estimated at 50 million tons per year, with an annual increase of 6%.441The conventional production process is based on oxidation of p-xylene (pX) which is oxidised to TPA by application of the Amoco process (catalytic aerobic partial oxidation of pX with air).298,485 Advantageously, inexpensive pX is derived from crude oil and a high TPA yield of 93% is achieved. However, this process presents separation/purification and environmental problems. The leading manufacturers are Yisheng, Heng Li, BP, Reliance, and Xianglu, among others. The price of purified TPA was reported to be 850 US$ per ton.486 Ethylene glycol monomer is made via the dehydration of commercial bioethanol; consequently, the synthesis of biobased TPA is a required step for producing 100% biobased PET.
The catalytic processes proposed for producing biobased purified TPA are (i) those aiming at obtaining renewable pX as an intermediate, which is oxidised into TPA using the existing infrastructure for TPA production from fossil origins (Amoco process), and (ii) those through intermediates other than pX not using the facilities of the conventional method. Fig. 21 summarises all these processes. A description of each of the catalytic routes will be made next.
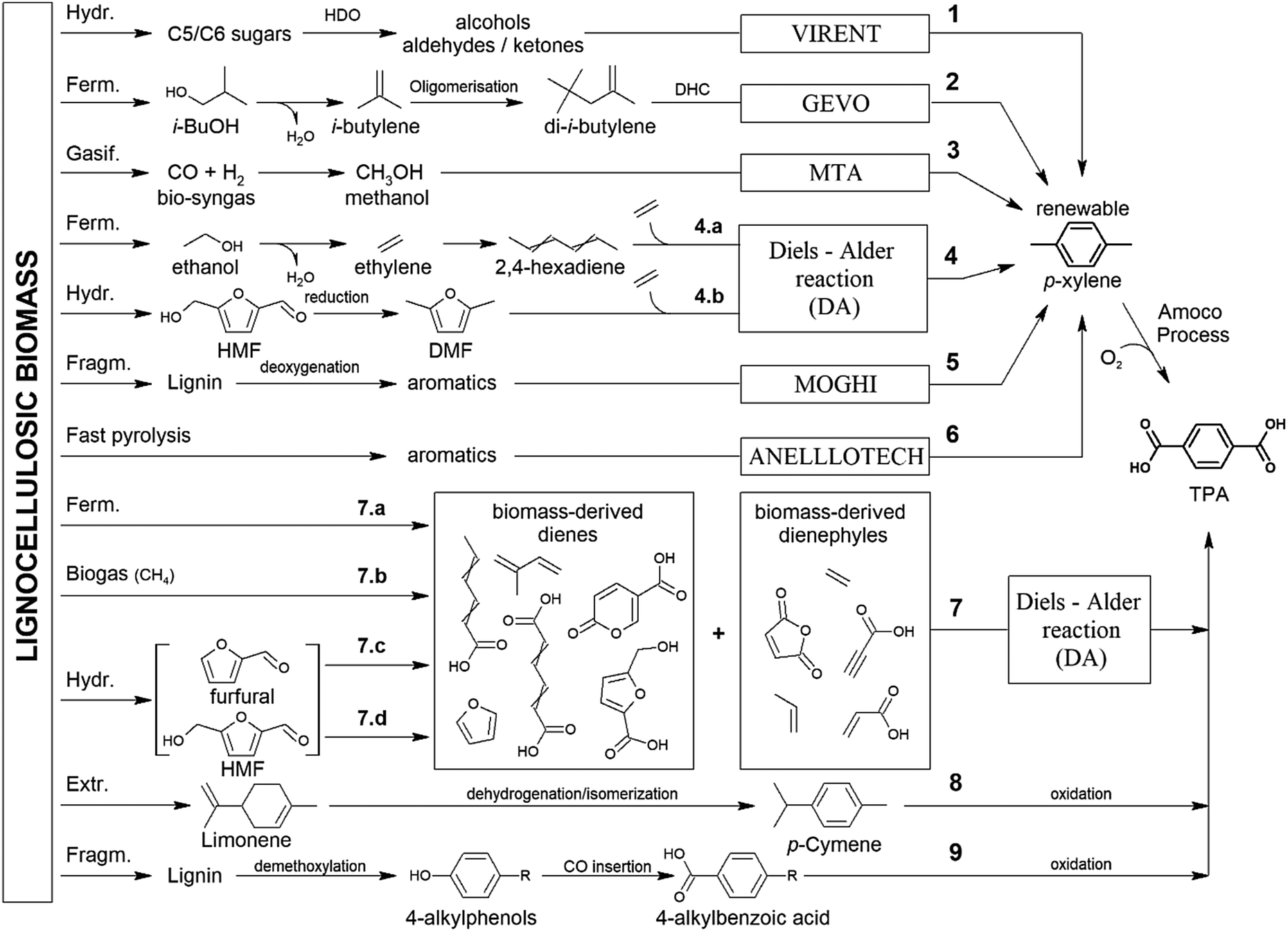 | ||
| Fig. 21 Summary scheme of the biobased TPA synthesis routes. Acronyms: hydrolysis (Hydr.), hydrodeoxygenation (HDO), fermentation (Ferm.), dehydrocyclisation (DHC), gasification (Gasif.), fragmentation (Fragm.) and extraction (Extr.). | ||
The Virent process. This technology deals with the transformation of soluble C6/C5 carbohydrates, obtained by hydrolysis of lignocellulose, into a BTX stream rich in pX (pathway 1, in Fig. 21). This approach consists of two essential catalytic stages. First, the hydrodeoxygenation of carbohydrates gives oxygenated intermediates, such as alcohols, aldehydes, and ketones, that are produced with supported metal catalysts between 473–553 K and at an H2 pressure of up to 90 bar. The second step is the catalytic condensation of these oxygenated intermediates to produce streams with 64 vol% aromatic products (benzene, toluene, xylenes, and ethylbenzene, among others) by using acid catalysts, such as ZSM-5-supported Cu, Ni, Ga or Zn at 398–723 K and up to 50 bar.487,488 This technology was developed by Virent Inc. and commercialised as BioForming®.
The Gevo process. The Gevo company process consists of three steps (route 2, Fig. 21). Isobutanol, obtained mainly by fermentation of sugars, is dehydrated to isobutylene in the presence of an acid catalyst with yields higher than 95%. Isobutylene is oligomerised (ZSM-5, 433 K) to give a mixture of oligomers containing 69% iso-octenes (mainly 2,4,4-trimethyl-1-pentene, also known as diisobutylene). The iso-octene fraction is purified by distillation and subjected to a dehydrocyclisation reaction catalysed by a chromium-doped alumina catalyst to give 75% selectivity and an overall p-xylene yield of 18.7 wt% from isobutanol.489
The MTA (methanol-to-aromatics) process. The bio-methanol conversion to gasoline, and, more specifically, to aromatics, has been known for years and has been patented by Mobil.490–492 Although the focus has been on the production of gasoline, several studies have also been reported in which catalyst modification aims to obtain high aromatic yields (Fig. 21, pathway 3). Thus doping H-ZSM-5 with Zn, P and SiO2 can simultaneously give a high methanol conversion (>95%) and high yield of aromatics (61.7%) at 748 K.493 The catalyst exhibited good stability in continuous reaction/regeneration cycles. Biomass-derived methanol can be obtained by different routes,494 among which the oxidation of methane derived from fermentation of biogas and hydrogenation of CO2 and CO is highlighted. The key question is whether sufficient and inexpensive biomethanol is currently feasible.
Processes based on the Diels–Alder (DA) reaction. This approach is based on aromatisation through the DA reaction of biobased dienes and dienephiles (route 4 in Fig. 21). Two σ bonds are generated between the diene molecule and the dienophile, forming a cycloadduct that is subsequently dehydrated to afford the aromatic compound (see Fig. 22). DA reactions can occur spontaneously depending on the nature of the diene and dienophile used and operation conditions. However, to accelerate the reaction at moderate conditions, the presence of an acid catalyst is required. An exhaustive review on this topic has recently been reported;495,496 we will briefly mention the key aspects.
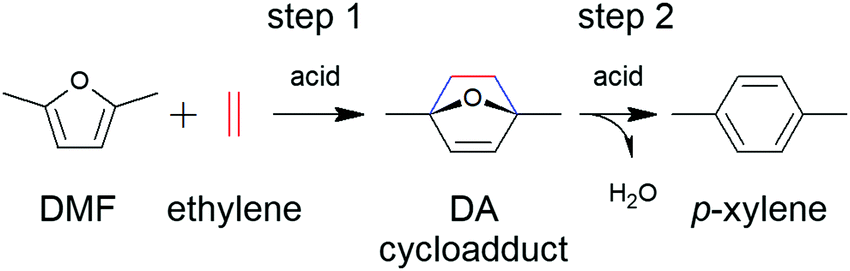 | ||
| Fig. 22 Mechanism of the Diels–Alder cycloaddition for DMF and ethylene. | ||
In general, 2,5-dimethylfuran (DMF) is mainly selected as the starting diene. DMF can be obtained from lignocellulose via hydrolysis of cellulose to sugars that are subsequently dehydrated to HMF, and further hydrogenolysed to DMF.497,498 Bioethylene derived from the dehydration of bioethanol is generally preferred as a dienophile. Other dienophiles, such as acrolein499 and acrylic acid,500,501 have also been used.
A particular case using ethylene as the unique starting olefin molecule has been developed by researchers from the University of North Carolina (UNC)502,503 (route 4.a in Fig. 21). In practice, bioethylene, obtained by dehydration of bioethanol, is trimerised to obtain 1-hexene, that is disproportionated via dehydrogenation to 2,4-hexadiene and hexane. The DA reaction of 2,4 hexadiene with ethylene gives 3,6-dimethylcyclohexene, which undergoes dehydrogenation to produce pX and hydrogen, reaching 93% yield using Pt/Al2O3 as a catalyst at 673 K.
Table 13 summarises the main catalytic system results using the most common molecules, such as DMF and ethylene, to produce pX by the DA reaction. Regarding the nature of the acid centres of the catalysts, it should be mentioned that Lewis acidity is typically desired, although good catalytic behaviour has also been reported for catalysts with Brønsted-type centres or with the presence of both types of acid centres. Dauenhauer et al.504–507 (entry 1, Table 13) reported that HY could efficiently catalyse the cycloaddition of DMF and ethylene and subsequent dehydration to pX in n-heptane solvent. The yield of pX reached 72% at 573 K and 62 bar ethylene pressure. The use of n-heptane was beneficial for improving the selectivity of pX by enhancing the hydrophobic environment in the zeolite, thus inhibiting several side reactions, in particular, the hydrolysis of DMF. A detailed DFT calculation suggests that the cycloaddition of DMF and ethylene is relatively favourable with Lewis sites, whereas Brønsted acids are less active. In contrast, Brønsted catalysts are more efficient for the dehydration of the cycloadduct intermediate to pX than Lewis acids. These analyses suggest that the rational design of a bifunctional catalyst combining Brønsted and Lewis acid sites would have the potential to achieve both a high DA reaction rate (by Lewis acid catalysis) and a high dehydration rate (by Brønsted acid catalysis). In this way, Wang et al.508 (entry 2) demonstrated that the WOx–ZrO2 catalyst, with both kinds of acid sites, is more active than the catalysts that predominantly have only Lewis acid sites. In their study, a hexadecane-like solvent was used. Several solid Brønsted and/or Lewis acids have been tested using different solvents, such as THF, n-heptane, hexane and 1,4 dioxane. Details regarding the operating conditions and catalytic properties can be seen in the mentioned table.
| # | Catalyst | Operating conditions | Catalytic results | Ref. | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Solv.a | S cc b (wt%) | C/Sc (wt%) | P (bar) | C2H4/DMFe (mol) | T (K) | t (h) | X S (%) | YpXi (%) | Produc.j (g gcat−1 h−1) | Deact.k | |||
| a Solvent. b Substrate concentration weight ratio in %. c Catalyst to substrate weight ratio in %. d Reactant C2H4 gas pressure in bar. e C2H4/DMF molar ratio. f Temperature conditions in Kelvin. g Reaction time. h Substrate conversion. i Yield towards product. j Productivity. k Catalyst deactivation: strong (st), moderate (m), low (l) and negligible (ne); n.s. (not studied), n.r. (not reported), regenerable (reg). l Ethanol solvent and reagent. | |||||||||||||
| 1 | H-Y | n-Heptane | 30.0 | 2.3 | 62 | 0.6 | 573 | 24 | 95 | 72 | 1.44 | n.s. | 504–507 |
| 2 | WOx–ZrO2 | Hexadecane | 10.0 | 7.5 | 50 | 0.6 | 523 | 6 | 60 | 46 | 1.13 | m reg | 508 |
| 3 | H-[B]-BEA | THF | 11.0 | 7.3 | 28 | 1.0 | 503 | 24 | 97 | 69 | 0.44 | n.s. | 509 |
| 4 | Nanosponge-BEA | n-Heptane | 30.0 | 0.9 | 50 | 0.8 | 523 | 24 | 99 | 80 | 4.10 | n.s. | 510 |
| 5 | Zr-BEA | n-Heptane | 18.0 | n.r. | 62 | 0.6 | 523 | 24 | 99 | 90 | n.r. | n.s. | 511 |
| 6 | Nanosheets-ZSM5 | n-Heptane | 30.0 | 0.9 | 50 | 0.8 | 523 | 24 | 89 | 76 | 3.89 | st reg | 512 |
| 7 | WO3/SBA-15 | n-Heptane | 18.0 | 3.9 | 54 | 0.6 | 523 | 24 | 64 | 57 | 0.68 | w reg | 513 |
| 8 | SiO2–SO3H | n-Heptane | 5.0 | 25.0 | 45 | 5.0 | 523 | 6 | 77 | 68 | 0.50 | st | 514 |
| 9 | P-BEA | n-Heptane | 18.0 | 4.6 | 62 | 1.1 | 523 | 24 | 99 | 97 | 0.97 | w | 515 |
| 10 | Si–Al aerogel | Dioxane | 10.0 | 4.3 | 30 | 2.2 | 523 | 6 | 90 | 60 | 2.56 | n.s. | 516 |
| 11 | HSiW/SiO2 | Dioxane | 10.0 | 4.3 | 30 | 2.2 | 523 | 6 | 94 | 80 | 3.41 | st | 517 |
| 12 | NbOx based | n-Heptane | 14.0 | 12.0 | 54 | 0.7 | 523 | 6 | 87 | 81 | 1.24 | w reg | 518 |
| 13 | KY | n-Heptane | 14.0 | 10.4 | 20 | 0.5 | 523 | 15 | 50 | 42 | 0.15 | ne | 519 |
| 14 | Desilicated-ZSM5 | n-Heptane | 31.0 | 2.3 | 40 | 0.4 | 523 | 20 | 51 | 30 | 0.74 | w reg | 520 |
| 15 | Al-HBEA | n-Heptane | 21.0 | 6.7 | 40 | 0.4 | 573 | 20 | 100 | 97 | 0.80 | w reg | 521 |
| 16 | HUSY12 | EtOH | 67.0 | 3.0 | 1l | 1.0 | 573 | 12 | 95 | 64 | 1.97 | n.s. | 522 |
Concerning the deactivation of the catalysts used in the DA reaction of DMF and ethylene (reuse column), it can be said that, in general, the zeolitic catalysts, in those cases where deactivation had been studied, are regenerable and can be used again. Zhao et al.521 studied the deactivation of H-beta zeolite catalysts with Al modifications by coke deposition (entry 15). H-beta zeolite, after further doping with alumina, shows excellent and durable performances with a pX yield of 97%. The coke content decreases from 12 to 7 wt% in the spent catalyst, with mainly soft carbonaceous species remaining, due to its appropriate Brønsted/Lewis acid ratio. Thus, H-beta zeolite has good stability and regeneration capability.
Dehydration of the cycloadduct requires high temperatures to proceed at high rates. Since the retro-DA reaction is also accelerated at high temperatures, a high ethylene pressure (usually higher than 2 MPa) is needed to shift the equilibrium to the adduct and to restrain the retro-DA reaction. To cope with the problem of using a high ethylene gas pressure, Teixeira et al.522 proposed using ethanol as a solvent that is dehydrated in situ to ethylene in the presence of an acid zeolite catalyst (entry 16). Among the different investigated zeolites, HUSY-12 zeolite, with a medium acid density (Si/Al ratio = 6), showed the highest yield (64% at 573 K) of pX production for the reaction of DMF/ethanol (molar ratio 1:1).
A more detailed description of the different catalytic systems used for this reaction can be found elsewhere.457,523,524 Cycloaddition of DMF–ethylene is thermally feasible, and the kinetic limitation of the DA and dehydration reactions is dependent on the Lewis acids or Brønsted acids in catalysts. However, it is still a challenge to obtain low-cost HMF from cellulosic biomass, which is the precursor of DMF.
The Moghi process. A more direct route for producing aromatic compounds, in particular p-xylene, is the depolymerisation and deoxygenation of lignin (pathway 5 in Fig. 21). Lignin is frequently viewed as a by-product that can be burned to produce energy rather than as an attractive and promising feedstock for providing low-cost and renewable aromatic monomers. Therefore, the production of aromatic hydrocarbons in large volumes from lignin is a desirable long-term goal since it is a sustainable and economically attractive solution. Lignin is hydrophobic and highly resistant to biological or chemical degradation; however, once it is dissolved, its resistance to chemical reactions is markedly reduced.525 The process developed by the Biochemtex company, called Moghi technology,526 involves the deoxygenation of lignin in a continuous, three-phase bubble reactor in the presence of free, floating catalyst particles. The lignin conversion reaction occurs in the temperature range of 463–643 K. To reduce char formation, it is crucial to control the pressure during the reaction in the range of 70–300 bar. By combining a metal catalyst, such as RANEY® nickel, with lignin, water and hydrogen at these specified temperatures and pressures, a range of useful chemicals, including BTX, can be produced. The reaction step is then followed by the separation of the organic molecules produced and the recovery of the catalyst particles. The assumed pX yield is approximately 10 wt% due to the low selectivity of the process.
The Anellotech process. A new approach to producing a BTX aromatic stream directly from lignocellulosic biomass in a single fluidised bed reactor was developed by Anellotech (route 6 in Fig. 21). This process uses a catalytic fast pyrolysis approach for rapid heating of the biomass under anaerobic conditions at very short residence times and immediate conversion of the resulting gases to aromatics using ZSM5-based catalysts.527,528 An 11 wt% pX yield was achieved. This route is attractive because of its simplicity (one fluidised bed reactor) and the availability and low cost of the feedstock and zeolite catalysts. However, high reaction temperatures ≥823 K are required.
Processes based on DA cycloaddition. A significant effort has focused on the development of synthetic pathways for biobased TPA production without the formation of p-xylene as an intermediate. Most of the approaches reported in the literature are based on the Diels–Alder reaction (route 7 in Fig. 21), and subsequent dehydration or dehydrogenation to produce the aromatic compound. The ease of formation of this intermediate will depend significantly on the different dienes and dienophiles selected and, more specifically, on the electron-donating and electron-withdrawing nature of the functional groups on the diene and dienophile compounds, respectively. Nonetheless, the proposed routes use substrates that already contain one or two carboxylic acids present in the structure of TPA and thus avoid an Amoco oxidation step.
A wide number of biomass-derived dienes (muconic acid/ester, isoprene, sorbic acid/ester, coumalic acid/ester, furan, and hydroxymethyl furoic acid/ester) and dienophiles (ethylene, propylene, acrylic acid, propiolic acid, and maleic anhydride) have been proposed for conducting DA reactions. All of these reagents can be classified according to the main process by which they are obtained from biomass, such as sugar fermentation to different dienes and dienophiles, fermentation to biomethane and sugar hydrolysis and subsequent dehydration to furanic compounds (furfural and hydroxymethylfurfural). A more in-depth description of each of these routes is provided below.
Production from sugar fermentation derivatives (pathway 7.a). The different options described in the literature for sugar fermentation routes to produce dienes are shown in Fig. 23. A first short route is that using muconic acid/ester as the substrate (route I in Fig. 23). A mixture of cis,cis- and cis,trans-muconic acid isomers is obtained from the fermentation of glucose and lignin-derived compounds.529,530 These isomers have to be converted to trans,trans-muconic acid (t,t-MA) isomer and then reacted with ethylene. Isomerisation to t,t-MA reached a 90% yield after refluxing with aqueous iodine/THF solutions. The esterification of t,t-MA improves its solubility and the DA reaction and subsequent TPA selectivity. Then, the DA cycloaddition reaction gives cyclohex-2-ene-1,4-dicarboxylic acid/ester in the presence of silicotungstic acid (Lewis acid) at 16 bar ethylene and 423 K. The dehydrogenation reaction is catalysed by 1 mol% Pt/C in cyclohexane under 17 bar air and 423 K, reaching a total final diester terephthalate yield of 81%.531,532
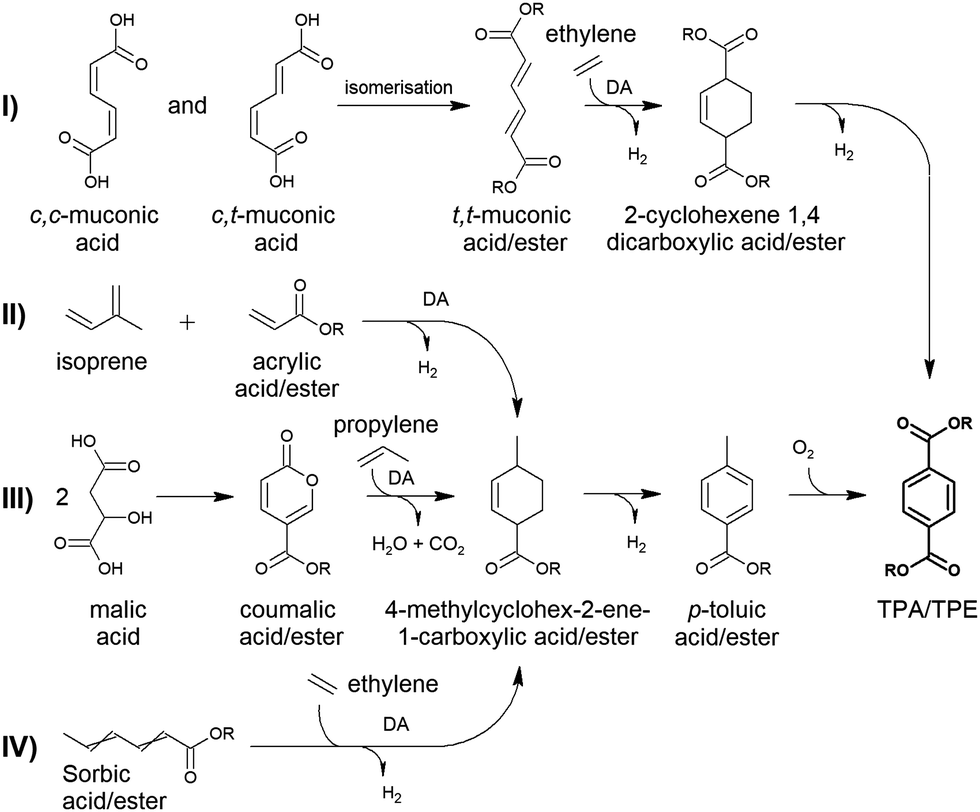 | ||
| Fig. 23 Different routes of TPA production by Diels Alder reaction of biomass derived dienes obtained via fermentation. | ||
The pathway II represented in Fig. 23 involves the cycloaddition of isoprene with acrylic acid. Both compounds can be obtained by the microbial synthesis of glucose.533 The main challenge is preventing isoprene polymerisation catalysed by acrylic acid and accelerated by the same Lewis acid catalysts required to improve the selectivity in the DA cycloaddition step. In the absence of solvent, cycloaddition was observed using TiCl4 as a homogeneous catalyst (94% yield and para/meta ratio 23). Subsequently, aromatisation of the 4-methylcyclohex-2-ene-1-carboxylic acid by the Pd catalyst provides p-toluic acid with a 77% yield, which must be converted subsequently into TPA acid with Co2+/Mn2+-catalysed O2 oxidation (94% yield).
The use of biobased 2-pyrones to produce biobased aromatics has also been described (pathway III in Fig. 23).534,535 Malic acid, obtained by microbial synthesis from glucose, self-condenses to produce coumalic acid (ca. 70% yield) by fuming sulfuric acid, which is esterified to methyl coumalate. Methyl coumalate is reacted with propylene using 1,4 dioxane as the solvent and a Pd/C catalyst at 453 K for 4 h to produce p-toluic acid (98% yield).535 TPA was obtained by oxidation and hydrolysis. One of the principal drawbacks of this process is the 70% yield of coumalic acid from malic acid condensation. Without a high yield (>90%) in this step, the overall yield of TPA from malic acid is significantly reduced.
The fourth route implies sorbic acid/ester as a substrate that can be produced either microbially (at quite limited quantities) or from bioethanol oxidation to acetaldehyde, trimerisation to 2,4-hexadienal (sorbaldehyde), and final oxidation to sorbic acid. Bérard et al.536 reported that ethyl sorbate reacted with ethylene to form ethyl 4-methylcyclohex-2-enoate at 463 K and 40 bar without a catalyst after 18 h and reached a 64% yield. Subsequent dehydrogenation of the cycloadduct in the presence of a 5 wt% Pt/C catalyst at 423 K and under an air pressure of 16 bar for 24 h yielded ethyl p-toluate with a 30% isolated yield. An alternative route (not represented in Fig. 23) is the DA reaction between sorbic and acrylic acids under mild conditions, followed by a combined dehydrogenation/decarboxylation process, providing p-toluic acid in high yields.537
In the case of biomass-derived di- or monoacids as substrates, such as muconic, malic (or coumalic) and sorbic acids, the previous esterification produces an improvement in TPA yields. However, it is supposed that there are two extra stages in the overall process to obtain TPA, including esterification and, finally, terephthalate hydrolysis. These stages may make the process not commercially attractive.
Production from biomethane obtained by biogas (route 7.b). Zhang et al.538 described the DA reaction of isoprene and propiolic acid; both compounds can be obtained from renewable methane derived from low cost biogas539 (see Fig. 24). The DA reaction gives 4-methyl-1,4-cyclohexadiene-1-carboxylic acid with a 67% yield. Subsequent reaction of this cycloadduct with O2 catalysed by Co(OAc)2/Mn(OAc)2 affords TPA, reaching an 85% yield under the cascade reaction process.
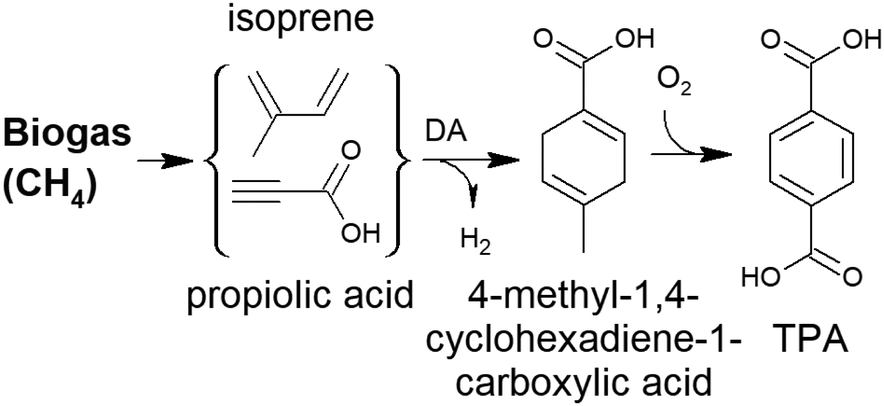 | ||
| Fig. 24 TPA production from biomethane. | ||
Production from furfural obtained by hydrolysis of pentoses (route 7.c). Furfural, a commercial renewable commodity already mentioned in Sections 3.2 and 3.3,221 is the starting molecule of furan and maleic anhydride required to produce TPA (see Fig. 25). Renewable furan is industrially produced from furfural via decarboxylation,540 whereas it has been reported that maleic anhydride could be obtained from furfural.389,390 Tachibana et al.541 recently reported the synthesis of TPA from furfural. The DA cycloadduct formed between furan and maleic anhydride needs to be dehydrated to phthalic anhydride, which was converted via phthalic acid and dipotassium phthalate to TPA. This route comprises multistep synthesis, and more details of the reaction conditions and the catalysts proposed for each of the stages can be found in this reference. Some homogeneous and corrosive catalysts have been employed in these steps, and an overall TPA yield from furfural lower than 20% was obtained.
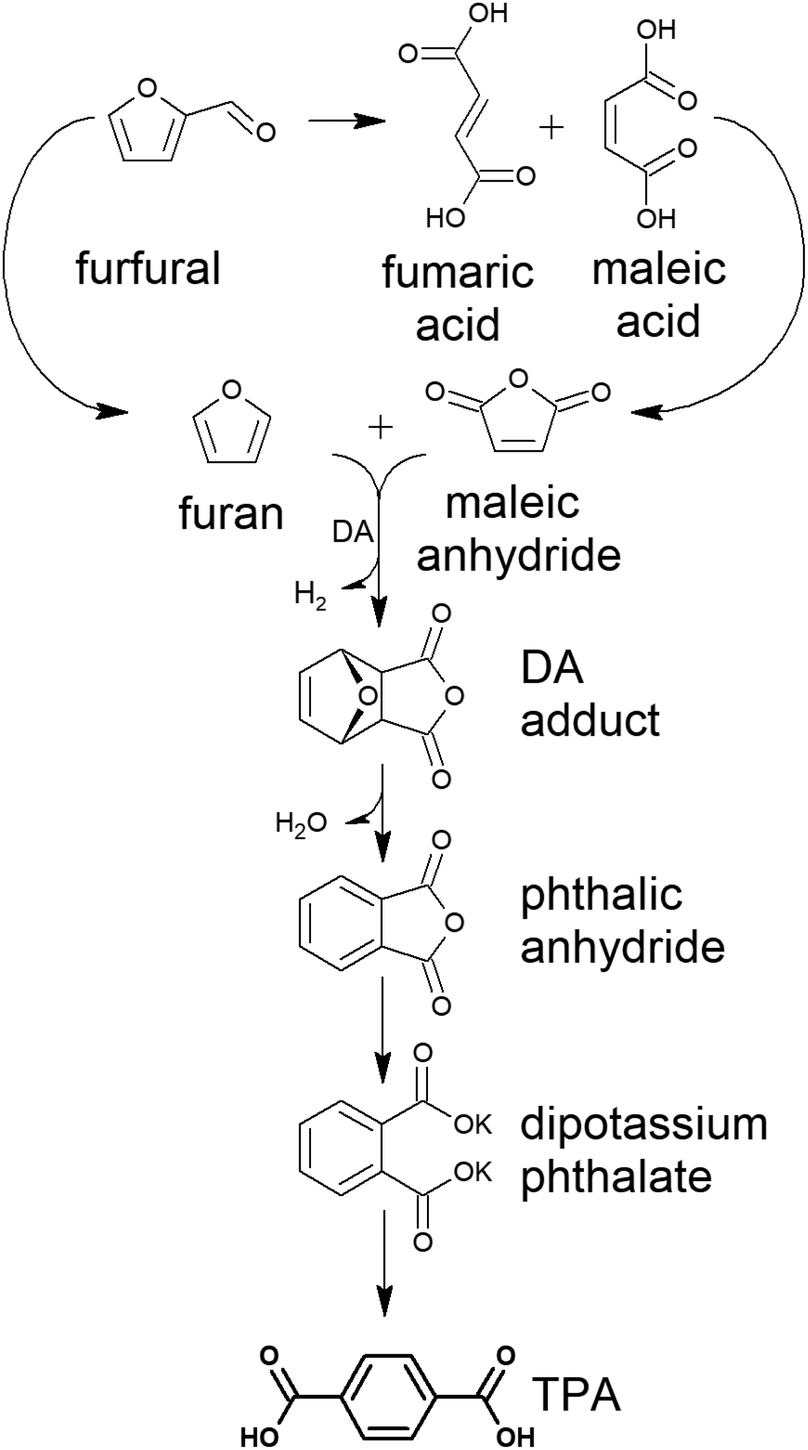 | ||
| Fig. 25 TPA production from biomass-derived furfural (adapted from ref. 541). | ||
Production from HMF obtained by hydrolysis of hexoses (route 7.d). In the previous section, the formation of p-xylene using DMF (a derivative of HMF) as the diene was described (route 4.b). However, this strategy is not atom efficient because DMF preparation from HMF requires an additional hydrogenation step at high hydrogen pressures. The removal of this hydrogenation step could be economically advantageous. HMF has three oxygen atoms in its structure, whereas DMF only contains one oxygen atom; the oxygen atom on the latter is lost after dehydration of the DA adduct to give p-xylene, and the goal product, TPA, contains four oxygen atoms. It does not make sense to remove the oxygen atoms from the initial molecule (HMF) to later incorporate them back into the final product.
Consequently, Pacheco et al.542,543 introduced an alternative approach in which, initially, HMF would be partially oxidised to generate 5-(hydroxymethyl)furoic acid (HFCA) to reach a more atomically efficient process in which less oxygen is required (see Fig. 26 and route 7.d in Fig. 21). To protect the hydroxyl and acid groups of HFCA, methanol is used to generate the corresponding ethers and esters. These compounds readily react with ethylene to produce, after dehydration of the DA adduct, the aromatic product 4-hydroxymethyl benzoic acid (HMBA), which could be further oxidised to afford TPA. The partial oxidation of HMF makes the reactant more active in the Diels–Alder reaction and avoids side reactions. Unfortunately, a maximum yield of aromatics of only 24% at 463 K and 70 bar ethylene is obtained using Zr-beta and Sn-beta catalysts and dioxane as the solvent. The authors claim that Brønsted acid sites should be prevented due to their ability to promote undesired side reactions, including coke formation, leading to low selectivity of the cycloaddition products. Both DA and dehydration steps are proposed to be catalysed by Lewis acid sites.
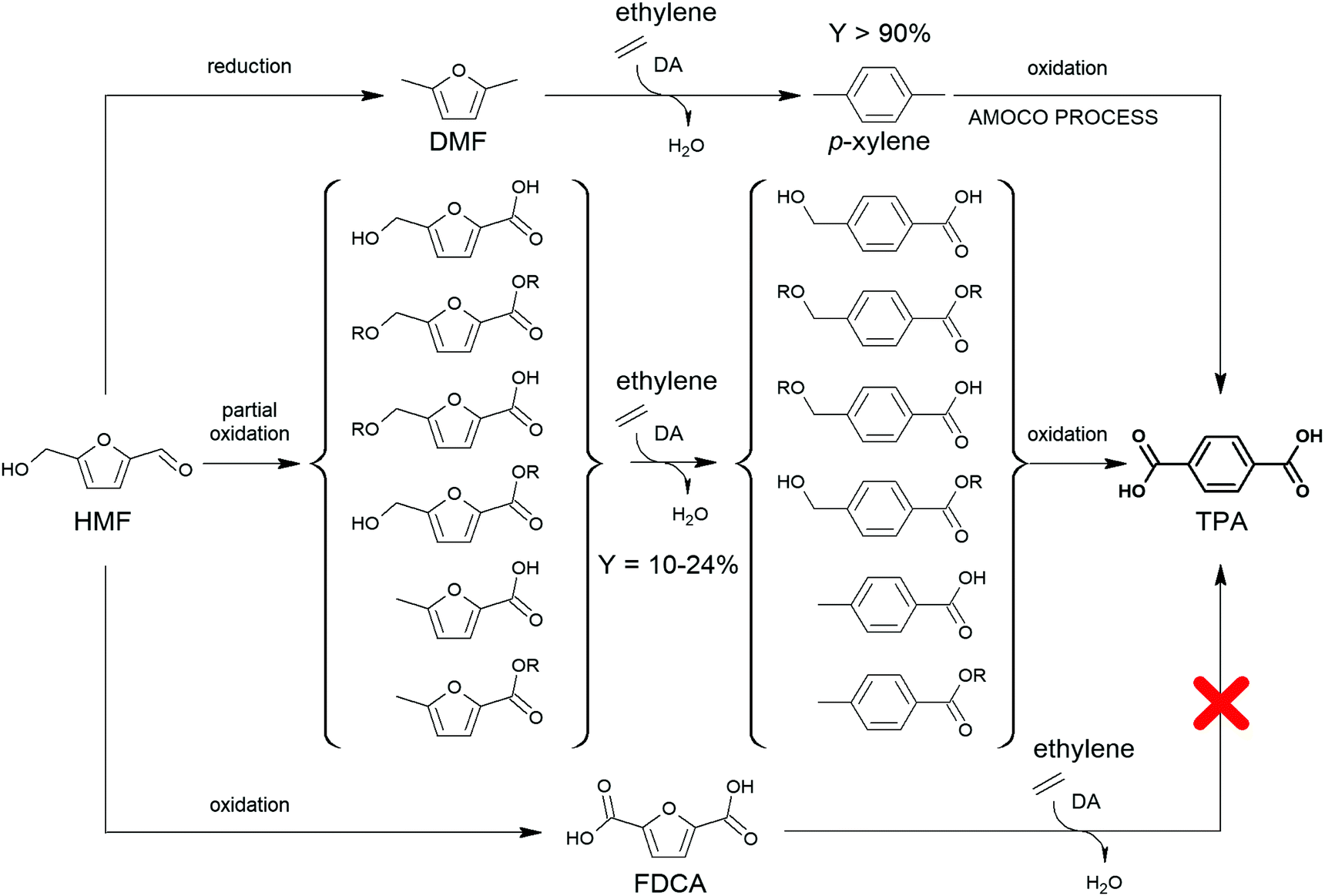 | ||
| Fig. 26 Diels–Alder routes to TPA production from biomass-derived HMF (adapted from ref. 542). | ||
Biobased TPA from HMF can also be proposed starting from 2,5-furan dicarboxylic acid (FDCA). The chemical oxidation of HMF to FDCA is a well-known, very selective reaction.544 The DA reaction of FDCA with ethylene forms the adduct intermediate 7-oxa-bicyclo[2.2.1]-hept-2-ene-1,4-dicarboxylic acid (or its esters), which contains 5 oxygen atoms. The subsequent dehydration of the ether to TPA is the final step. However, the FDCA derivative is practically inactive in DA cycloaddition.
Techno-economic and environmental aspects will be essential for choosing between the two alternative routes for TPA production from HMF summarised in Fig. 26: hydrogenation or decarbonylation to DMF (route 4b) or partial oxidation to HFCA (route 7d). DMF route can achieve high yields (>90%), but this approach presents unfavourable atom efficiency. The HFCA route is more atom efficient, but progression of the DA reaction is limited, and low yields (<25%) are obtained.
The limonene route. A new approach based on limonene, which can be extracted at a low cost from orange peels, has also been developed (pathway 8 in Fig. 21). Limonene is selectively transformed into para-cymene by its incorporation into a solution of ethylenediamine, anhydrous FeCl3 and sodium at 373 K for 8 h. The oxidation of p-cymene to TPA was conducted in a two-step process under heated refluxing conditions, the first step of which included the use of a mineral acid, and the second of which incorporated KMnO4 as the catalyst. In one example, the overall yield of TPA from limonene was approximately 85%. Since limonene is oxidised to TPA, the overall reaction from limonene to TPA has a theoretical mass yield of 122%.545 Unprecedented synthesis of biobased TPA in the presence of O2 has been reported.546 In this case, the oxidation of p-cymene has also been reported in the presence of a Mn–Fe mixed-oxide heterogeneous catalyst reaching a TPA yield of only 51% at 413 K, 20 bar O2, and 24 h of reaction.
Lignin demethoxylation route. Recently, Song et al.547 reported a new chemocatalytic synthesis route via the transformation of corn-stover-derived lignin oil into TPA (route 9 in Fig. 21). This process follows three steps: (i) selective demethoxylation keeping the phenolic group and benzene ring untouched by the hydrotreatment of lignin with a supported molybdenum catalyst (MoOx) at 593 K under 30 bar H2 after 2 h; (ii) insertion of CO into the C–OH bond in 4-alkylphenols to obtain the corresponding alkylphenol in the presence of a Pd-based catalyst; and (iii) finally, 4-alkylbenzoic acid alkylchain oxidation into carboxyl groups in the presence of a homogeneous Co–Mn–Br catalyst, giving the single product, TPA (purity greater than 99%). Unfortunately, the overall yield of this route was reported to be 15.5 wt%.
Table 14 gives an overview of the different technologies described in this section, highlighting their main advantages and drawbacks. The data in this table indicate the biomass-derived substrate used in each of the routes discussed in this section. Two more columns are also included: one displays the number of steps required to obtain the final product from the starting substrate, and another column displays the overall yield (% carbon-based) of each process. The values of both columns refer to the transformation of the starting precursor into the final product (pX or TPA). Therefore, they do not include the steps or yields of the conversion from the biomass to the starting precursor. Although the steps necessary to achieve the renewable starting platform are not taken into account and although some authors provided yield on a weight basis and others on a molar basis, the table is still valid for providing an estimation of the most promising processes and for qualitatively addressing which aspects of the processes require subsequent research.
| # | Route | Substrate | Steps | Ya (%C) | Product | Advantages | Drawbacks |
|---|---|---|---|---|---|---|---|
| a Yield towards product based on carbon %. b Yield towards product in weight %. c Does not include the transformations of methane into DA reagents (isoprene and propiolic acid). | |||||||
| 1 | 1 | C5, C6 sugars | 2 | 26b | pX | Cheap and available substrate | Low yield |
| 2 | 2 | Isobutanol | 3 | 19b | Available substrate | Low yield, economic evaluation | |
| 3 | 3 | Methanol | 1 | 28 | To achieve high amounts of substrate | High cost substrate, low yield | |
| 4 | 4a | Ethanol | 5 | 49 | Moderate yield | Low atom efficiency, multiple steps | |
| 5 | 4b | DMF (via HMF red) | 2 | 90 | High yield | Low atom efficiency, high cost of HMF | |
| 6 | 5 | Lignin | 1 | 10b | Cheap and available substrate | Low selectivity | |
| 7 | 6 | Biomass | 1 | 11b | Cheap and available substrate | Low yield, harsh conditions | |
| 8 | 7a | Muconic acid | 3 | 81 | TPA | High yield, short pathway | Limited feedstock resource |
| Isoprene & acrylic ac | 3 | 72 | |||||
| Malic acid | 4 | 65 | |||||
| Sorbic acid | 3 | — | |||||
| 9 | 7b | Methane | 4 | 57c | Abundant substrate | Multi-step to achieve DA reactants | |
| 10 | 7c | Furfural | 6 | 19 | Commercial substrate | Multi steps, harsh reaction conditions | |
| 11 | 7d | HMF | 3 | 24 | High atom efficiency | High cost of HMF, low yield | |
| 12 | 8 | Limonene | 2 | 85 | Short pathway, high yield | Limited feedstock resource | |
| 13 | 9 | Lignin | 3 | 16b | Cheap and available substrate | Low yield | |
The synthesis of TPA from isobutanol (route 2) and the DA cycloaddition of DMF and bioethylene (path 4 b) appear as the more advanced and attractive routes in the short to medium term. In both routes, the chemistry has been well established, but there are still some challenges to be addressed. For instance, the cycloaddition of DMF and ethylene typically requires high temperatures and pressures, whereas the maximum theoretical yield of p-xylene from isobutanol is quite low.
The direct route to biobased TPA from limonene (route 8) produces the highest yield of TPA (85%) but suffers from the use of stoichiometric oxidation catalysts. Methyl and isopropyl groups of p-cymene are, unlike the commercial Amoco cobalt/manganese/bromine oxidation system, only effectively oxidised by KMnO4. High costs will be associated with the use and recovery of KMnO4, and this route would be substantially more attractive if a robust oxidation catalyst working under mild conditions could be used at 1 mol% loading or below.
Interestingly, Volanti et al.548 recently carried out a life cycle assessment (LCA) comparing the environmental performance of these three alternative routes (the isobutanol Gevo process, the DMF-ethylene DA reaction, and the limonene route) with that of the conventional process of production from crude oil. Generally, the use of biobased feedstocks should produce a drastic reduction of the carbon footprint in the manufacture of chemical products, by decreasing both the energy required to manufacture the product and the use of (biogenic) carbon, particularly if organic waste streams are converted into raw materials for the production of starting materials. However, the utilisation of other contaminating effluents and energy-demanding steps can unbalance the final environmental impact. Surprisingly, the results of the LCA conducted by Volanti et al. proved that the limonene route is, thus far, the most favourable from an environmental point of view, despite the fact that KMnO4 is used. These surprising results emphasise the importance of LCA for assessing the environmental impact of a process.
Concerning other routes not analysed in the above-mentioned LCA, route 7.a (using diacids, such as malic acid and muconic acid, obtained via fermentation) is an attractive candidate. These diacids already contain the two carboxylic acid groups present in the structure of TPA, which eliminates the Amoco oxidation step required for those p-xylene routes. However, these syntheses involve one or more esterification steps and, ultimately, produce dimethyl terephthalate. This ester must be hydrolysed back to the free acid to afford TPA. These additional steps to the synthetic route should be removed for these syntheses to become commercially attractive.
Route 7.b for TPA production involves three steps: cycloaddition, aromatisation, and aryl methyl oxidation. After aromatisation of the para- and meta-cycloadducts followed by methyl group oxidation, TPA and isophthalic acid (IPA) are obtained. Both acrylic acid and isoprene are very interesting feedstocks because they can be produced from different biomass feedstocks. Another advantage is its ability to produce both TPA and IPA. The PET polymer obtained from TPA contains 3–5 wt% IPA to provide transparency and lower the melting point, making the polymer easier to process. An additional advantage is the development of a Lewis acid-catalysed solvent-free cycloaddition of an unprotected carboxylic acid. Catalysing reactions of unprotected carboxylic acids has been a long-standing problem in synthetic chemistry, often necessitating the use of an ester derivative instead of the free acid.
In conclusion, there are basically two groups of processes to obtain biobased TPA: (i) those that have a high yield and a short reaction path but have a starting substrate derived from biomass that is scarce and expensive and for which the reaction conditions should be relatively mild; and (ii) those that are multi-stage with low yields/selectivities and sometimes requiring severe reaction conditions despite starting from inexpensive and available substrates. The improvements of these deficiencies, together with techno-economic analysis and LCA, will be critical for selecting the most successful processes that will eventually be imposed.
4. Sugar acids
4.1. Gluconic acid
D-Gluconic acid ((2R,3S,4R,5R)-2,3,4,5,6-pentahydroxyhexanoic acid) is produced by the oxidation of glucose with O2, either by hypochlorite solution or by electrolytic oxidation of glucose solution in the presence of bromide, and by a fermentation process with microorganisms (Acetobacter aceti, Aspergillus niger fungus549 and Gluconobacter suboxidans bacteria550). The worldwide annual production of gluconic acid is approximately 60 ktons,551 with an average price oscillating between $1.5 and 8.5 kg−1.552 Gluconic acid and its derivatives have important applications as food additives, as supplements to prevent calcium deficiency and anaemia, in the pharmaceutical industry, in cosmetics, as additives for cement, and in metallurgy.553 In the polymer field, it is an intermediate in the preparation of novel degradable polyesters, polyurethanes, poly(D-glucaroamidoamine)s and hydroxylated polyamides.13 Hlasiwetz and Habermann were the first to prepare gluconic acid by oxidation of glucose with chlorine but in the form of its barium and calcium salts. Other by-products, such as formic acid, glycolic acid, oxalic acid, and carbon dioxide, can be generated depending on the sugar and type of oxidant.553Although the production of gluconic acid is mainly accomplished by the oxidation of glucose (Fig. 27), other carbohydrates, such as cellobiose554,555 and cellulose,554,556 can also be used as feedstocks. Current research pursues the development of new greener chemical routes for the selective oxidation of carbohydrates to gluconic acid in the presence of heterogeneous catalysts, with oxygen or air as an oxidant, under mild experimental conditions.
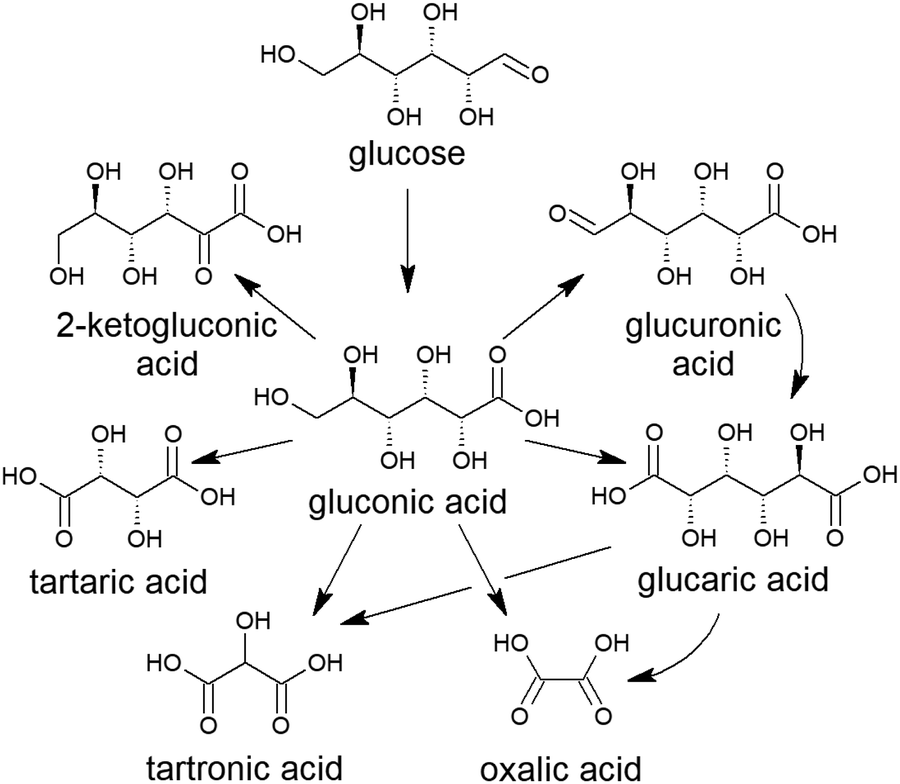 | ||
| Fig. 27 Different carboxylic acids that can be formed by oxidation of glucose to glucaric acid. | ||
The catalytic liquid phase aerobic oxidation of glucose in gluconic acid has been extensively studied using catalysts based on noble metals, mainly Pt, Pd and Au (Table 15). Batch reactors were employed, where the system was pressurised or the oxidant (air or pure oxygen) was bubbled in the sugar solution. Strict pH control was required for attaining a high glucose conversion, usually by addition of a sacrificial strong base; pH > 11 can favour undesired reactions, such as Cannizzaro, retro-Claisen, condensation and isomerisation.557 However, at low pH values, the strong adsorption of reaction products on the catalyst surface deactivates the catalyst, as observed with a Pt/C catalyst.558 In this sense, to avoid the use of a basic medium, Pt was supported on a basic hydrotalcite (HT) (Table 15, entry 1), attaining a glucose conversion and gluconic acid yield similar to those of Pt/Al2O3 in the presence of Na2CO3.559 A pH close to 9 was measured in the presence of Pt/HT, which seems to be the optimum reaction pH.
| # | Catalyst | Operating conditions | Catalytic results | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S cca (wt%) | C/Sb (wt%) | O2c (mL min−1) | pH | T (K) | t (h) | X S (%) | Yh (%) | Produc.i (g gcat−1 h−1) | Deact.j | |||
| a Substrate concentration in water, weight ratio in %. b Catalyst to substrate weight ratio, in %. c Oxygen flow in mL min−1. d n.c. (without pH control). e Temperature conditions in Kelvin. f Reaction time. g Substrate conversion. h Yield towards product (glucaric acid). i Productivity. j Catalyst deactivation: strong (st), moderate (m), low (l) and negligible (ne); n.s. (not studied), n.r. (not reported). k MPa. l Air. m Gluconate. | ||||||||||||
| 1 | 3.5 wt% Pt/HT | 2.3 | 43.0 | 0.1k | n.c.d | 323 | 12 | 99 | 83 | 0.18 | ne (1 run) | 559 |
| 2 | 5 wt% Pt/C | 10.8 | 2.9 | 10.0 | 9 | 333 | 7 | 85 | 66 | 3.54 | ne (1 run) | 561 |
| 3 | 4 wt% Pd/C | 2.7 | 15.0 | 30.0 | n.r. | 298 | 2 | 100 | 98 | 3.56 | st (4 runs) | 560 |
| 4 | 2 wt% Pd/C | 10.8 | 3.9 | 10.0 | 9 | 333 | 7 | 95 | 91 | 3.63 | ne (1 run) | 561 |
| 5 | 2.5 wt% Pd/C | 0.2 | 11.0 | 0.5 | 9 | 323 | 4 | n.r. | 50 | 1.24 | ne (1 run) | 562 |
| 6 | PdBi/C | 29.9 | 1.6 | 1500.0l | 9 | 313 | 2.6 | 100 | 99m | 25.91 | ne (5 runs) | 568 |
| 7 | Au38/AC-120 | 5.0 | 9.3 | 60.0 | 9.5 | 333 | 0.6 | 100 | 100 | 19.51 | ne (7 runs) | 576 and 577 |
| 8 | Au/CMK-3 | 1.8 | 11.0 | 0.3k | n.c. | 383 | 2 | 92 | 81 | 4.01 | m (4 runs) | 580 |
| 9 | 0.45 wt% Au/TiO2 | 1.8 | 5.6 | 500.0 | 11 | 313 | 2 | 100 | 98 | 9.53 | ne (17 runs) | 581 |
| 10 | 1.8 wt% Au/CeO2 | 2.0 | 13.0 | 100.0 | 9.5 | 333 | 1 | 90 | 90 | 7.54 | n.s. | 582 |
| 11 | Au/Al2O3 | 3.6 | 67.0 | 0.1k | n.c. | 393 | 2 | 60 | 60 | 0.49 | m (4 runs) | 583 |
| 12 | Au–Pd/MgO | 9.0 | 13.0 | 0.1k,l | n.c. | 333 | 24 | 62 | 62 | 0.22 | st (2 runs) | 584 |
In some cases, the leaching of active phases, such as Pd, was observed due to the excellent complexing properties of gluconic acid.560 This explained the reduction of glucose conversion (nearly 15%) observed with Pd/C after 4 catalytic runs, although gluconic acid selectivity was maintained at 98%.
The catalytic activity of Pd- and Pt-based catalysts has been compared, finding that 2 wt% Pd supported on carbon allowed attaining a gluconic acid productivity similar to that of 5 wt% Pt, thus demonstrating the superior activity of Pd (Table 15, entries 2 and 4).561 Moreover, a similar catalytic performance of both pre-reduced (by H2) and non-reduced Pt catalysts and a twofold increase in the Pd/C activity after pre-reduction were observed, which was justified by the ability of glucose to reduce surface Pt2+ sites to catalytically Pt(0) active sites, whereas surface palladium oxides were less prone to be reduced to Pd(0) due to the lower reduction potential of Pd in comparison with that of Pt. These authors also observed quite similar TOFs irrespective of the Pt particle size, contrary to that observed with Pd catalysts, where particles smaller than 3 nm were deactivated faster than those larger than 6 nm. This fact was corroborated by Haynes et al. (Table 15, entry 5), who found the best catalytic results for an average Pd particle size of 7 nm, while larger particles were quickly deactivated.562 However, catalysts showed progressive deactivation, regardless of the particle size, by the formation of oxygen layers on Pd particles, but no product adsorption or Pd leaching was observed. The formation of Pd–O species as a cause of deactivation was also proposed by Nikov et al. with Pd (0.5 wt%) supported on Al2O3, although they observed the adsorption of reaction products.563
The catalytic performance of Pt- and Pd-based catalysts can be improved by the addition of promoters, such as Bi.564–570 The formation of intermetallic BiPd phases was detected,564,566,568 identifying a Bi2Pd phase as the most active, whereas BiPd3 was inactive. However, gluconic acid and/or gluconates seem to favour the leaching of Bi species during the catalytic test,566 which are also involved in the catalytic process.569 However, Besson et al. did not observe Bi species in solution.568 Other studies have concluded that Bi could stabilise Pd particles as small as 1 nm, avoiding the Pd over-oxidation previously noted, since oxygen was adsorbed on Bi atoms instead of Pd atoms. During a reutilisation study, gluconic acid yields remained almost constant at 40% after normalisation with respect to the catalyst mass during 13 consecutive catalytic cycles.569
Au NPs are also suitable catalysts, due to their greater inertness to the oxidation of secondary alcohol groups,571 compared to secondary ones.557 In addition, Au NPs exhibit lower activity in C–C bond scission or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond isomerisation, which should significantly disfavour undesired catalytic processes. In this sense, Au/C catalysts, prepared by supporting Au nanoparticles (NPs) by the immobilisation of colloidal gold particles on carbon,557,572,573 were very selective, with a catalytic performance similar to that of commercial Pd and Pt catalysts but a higher selectivity,557 under mild experimental conditions (pH = 7–9.5, T = 323–373 K, P(O2) = 0.1–0.3 MPa). The selectivity towards gluconate was greater than 99%, which was explained by both the chemoselective inertness of Au for secondary alcohols and pH selected that avoid the isomerisation of glucose to fructose. Au-Based catalysts were less prone to deactivation by product adsorption, but Au leaching (up to 70 wt% after 6 runs) was observed in reutilisation tests, an important drawback for industrial implementation.
C bond isomerisation, which should significantly disfavour undesired catalytic processes. In this sense, Au/C catalysts, prepared by supporting Au nanoparticles (NPs) by the immobilisation of colloidal gold particles on carbon,557,572,573 were very selective, with a catalytic performance similar to that of commercial Pd and Pt catalysts but a higher selectivity,557 under mild experimental conditions (pH = 7–9.5, T = 323–373 K, P(O2) = 0.1–0.3 MPa). The selectivity towards gluconate was greater than 99%, which was explained by both the chemoselective inertness of Au for secondary alcohols and pH selected that avoid the isomerisation of glucose to fructose. Au-Based catalysts were less prone to deactivation by product adsorption, but Au leaching (up to 70 wt% after 6 runs) was observed in reutilisation tests, an important drawback for industrial implementation.
Regarding the structure sensitivity, Comotti et al. observed that the catalytic activity was inversely proportional to the size in the range of 2.5 to 4.5 nm, but nanoparticles larger than 10 nm were inactive.572 However, the catalytic activity per Au atom was independent of the nanoparticle size. Furthermore, supported (0.45 wt% Au/C) and unsupported Au nanoparticles with a size of 3.6 nm exhibit a similar catalytic behaviour, indicating that the metal–support interaction did not affect the intrinsic metal activity.
The preparation of supported Au catalysts by using conventional methodologies, mainly impregnation and deposition–precipitation, failed for supports with a low point of zero charge, such as silica, carbon and organic polymers. Haruta et al. carried out important work by depositing Au NPs onto many different supports (cellulose,574 inorganic oxides,575 carbon,576–578 and ion exchange resins579) by deposition–reduction (DR) and solid grinding (SG) methods with volatile organo-gold complexes. In particular, SG was very effective in depositing Au NPs on carbon, with a mean diameter as small as 1.9 nm.578 Nevertheless, Au NPs smaller than 10 nm showed a high catalytic activity in glucose oxidation, irrespective of the deposition method and carbon support. The TOF values attained with carbon-supported NPs smaller than 3 nm were approximately one-third those of Au NPs supported on metal oxides.575
An excellent catalytic performance was achieved with a catalyst based on Aux(PET)y clusters (PET = phenylethanethiolate, @SCH2CH2Ph) immobilised on activated carbon and annealed at 393 K in air, with gluconic acid yields close to 100% (Table 15, entry 7).576,577 The strong interaction between the S–Au–S motifs of “Au(PET)” and the aldehyde group of glucose seems to favour sugar oxidation by reactive oxygen species (e.g., peroxides) on the surface of activated carbon. It was recyclable due to its thermal stability, without appreciable loss of catalytic activity after 7 cycles, without sintering of Au NPs.
In some cases, although the catalytic activity decreased during reuse, mainly due to the adsorption of carboxylic acids on the catalyst surface, as observed for Au NPs confined in the channels of a mesoporous CMK-3 carbon (Table 15, entry 8),580 the spent catalyst could recover its initial catalytic performance after treatment with an aqueous sodium hydroxide solution at 363 K.
Remarkably, the Au/TiO2 catalyst exhibited high selectivity and noticeable robustness, maintaining complete glucose conversion and 99.5% selectivity to gluconic acid after 17 reuses (Table 15, entry 9).581
Using CeO2 as a support for Au nanoparticles results in the formation of small Au NPs (average size of 3.5 nm) (Table 15, entry 10).582 In addition, it is well known that ceria has the ability to accumulate oxygen, and this catalyst is very active and completely recyclable after simple washing with water.
On the other hand, Megías-Sagayo et al. demonstrated that the selectivity pattern depended on the type of support.583 Among all the supports studied (Al2O3, CeO2 and mixed Al2O3–CeO2, ZrO2–CeO2 and ZrO2–CeO2), Au/Al2O3 was the most selective catalyst (Table 15, entry 11) due to its low Lewis acidity (with high Lewis acidity favouring selectivity towards lactic acid to the detriment of gluconic acid formation). Unfortunately, a clear drop in glucose conversion was detected after 4 catalytic runs, and leaching of gold species was observed, which accounted for 10% of the overall glucose conversion.
A basic metal oxide (MgO) was also used to support Au–Pd NPs (Table 15, entry 12) to avoid the use of a sacrificial base or pH control.584 However, although the catalyst was active and selective, its reutilisation was not feasible due to support leaching in the reaction medium.
Au-Containing bi- and trimetallic colloidal nanoparticles, including Au/Pt,585,586 Au/Pd587 and Au/Pt/Ag,588 have also been evaluated. Their catalytic activity was always higher than that of monometallic Au NPs and up to 3.8 times higher in the case of Au/Pt/Ag nanoparticles.588 However, the main drawback of these colloidal nanoparticles was their isolation for reutilisation in consecutive runs. Thus, the long-term activity study was performed by continuously withdrawing and adding new glucose solution after each 2 h of reaction, and 60% conversion was preserved for at least 8 h. Anionic Au atoms (and Pt when present) on the catalyst surface act as catalytic sites, activating the oxygen molecule by donating an excess of electronic charge to its antibonding molecular orbital, forming peroxo- or superoxo-like oxygen.
A gluconic acid selectivity of 100% was attained with gold NPs supported on Al2O3 at 313 K and pH 9 under oxygen flow.589,590 It was observed that variations in the specific activity were negligible for Au loadings higher than 4 wt%. Nevertheless, the catalysts with the lowest Au loading were robust against Au lixiviation, being reused for 20 runs without loss of activity. The 0.25 wt% Au/Al2O3 catalyst was also tested in a continuous stirred tank reactor for 70 days at pH 9 and 313 K without loss of activity or selectivity.591
To the best of our knowledge, there are few works dealing with the oxidation of cellulose to gluconic acid, although cellulose is the most abundant glucose biopolymer. However, cellobiose, a disaccharide consisting of two β-glucose molecules, has been used as a feedstock for gluconic acid production. Since a two-step process is needed, including hydrolysis of cellobiose to glucose and its subsequent oxidation to gluconic acid, a bifunctional catalyst is required, consisting of an acid support to promote the hydrolysis process and supported metal NPs to accomplish the oxidation reaction. The support, in addition to being involved in the hydrolysis of cellobiose thanks to its acid sites, can also tune the oxidation capacity of metal NPs by modifying their electronic density and, consequently, affecting the occurrence of undesirable side reactions.592,593 The cellobiose oxidation proceeds under base-free conditions, although a higher reaction temperature (373–433 K) and oxygen pressure (0.3–0.5 MPa), as well as a longer reaction time (up to 24 h), are needed to attain high gluconic acid yields. However, the formation of by-products, such as glycolic, acetic, oxalic, glyceric, glucaric, succinic and formic acids, fructose, sorbitol and ethylene glycol, was also detected.592–594 Different acidic supports, such as Al2O3, HZSM-5 and HY zeolites, heteropolyacids, TiO2, and several types of carbon, have been used to incorporate gold nanoparticles.
Heteropolyacids have also been studied by An et al. as acid supports, and an Au/Cs1.2H1.8PW12O40 system with Au nanoparticles with a mean diameter of 2.7 nm displayed a gluconic acid yield of 97% from cellobiose at 418 K for 3 h.554 These authors concluded that a higher Cs loading led to lower gluconic acid yields (47–60%), in contrast to previous results that reported a yield (96.4%) similar to that of Au/Cs2HPW12O40.592 In both cases, the reutilisation studies demonstrated the stability of these catalysts after 5 and 6 runs, maintaining gluconic acid yields higher than 90 and 71%, respectively.
Similar to that observed from glucose, doping with a second transition metal to form bimetallic phases, such as Au–Cu, Au–Ru, Au–Co and Au–Pd,555 improves the catalytic performance of an Au/TiO2 catalyst, increasing the gluconic acid yield from cellobiose from 72 to 88.5% (at 418 K after 2 h) with Au–Cu/TiO2,593,595 whereas Au–Ru/TiO2 required a longer reaction time (9 h) to attain a yield of 85.4%.
In summary, different heterogeneous catalytic processes for the oxidation of glucose to gluconic acid have already been reported, showing their potential to replace the enzymatic route currently implemented in industry. However, additional research effort is still required to overcome the main drawbacks identified in these investigations, such as those associated with catalyst reutilisation for minimising the leaching of the active phase. The exhaustive pH control (basic) of the reaction medium later complicates the separation and purification of gluconic acid because it makes necessary a post-neutralisation step of gluconate to form gluconic acid. The use of inexpensive sources of glucose as a feedstock, such as the cellulose contained in lignocellulose, requires the development of new catalytic technologies. At the heart of these technologies, there must be robust, novel bifunctional catalysts exhibiting acid and metallic sites capable of processing raw or barely treated lignocellulose at once. The latter must have a very positive impact on the economy of the process. In this respect, there is also a lack of techno-economic studies of the most promising catalytic routes to assess their potential as alternatives to enzymatic processes.
4.2. Glucaric acid
Glucaric acid is the oxidation product of gluconic acid and possesses four hydroxyl and two terminal carboxylic groups. This renewable dicarboxylic acid has been included as a “top value-added chemical from biomass” in the US DOE report due to its large spectrum of applications.191 The global glucaric acid market size, estimated at $550.4 million in 2016, is increasing due to the demand for detergents and soaps, food ingredients, corrosion inhibitors, and de-icing applications.596 Moreover, glucaric acid can contribute to the development of novel sugar-containing polymers, with an estimated annual market of 60 ktons for linear monosaccharide derivatives.218 It is also a source of co-monomers for the manufacture of functionalised polyesters for biomedical and pharmaceutical applications. Recently, much attention has been paid to poly(D-glucaramidoamine) (PGAA), a polymer derived from glucaric acid and consisting of carbohydrate groups, secondary amines and amide groups as the main functionalities. It finds applications as an efficient and degradable gene delivery vehicle. Moreover, there are several other types of polymers prepared from glucaric acid. For instance, glucaric acid is a potential feedstock for the production of adipic acid used in the manufacture of nylon-66. Glucaric acid acetate and aromatic amines can polymerise to produce poly(m-xylylene-acetyl glucaramide) and poly(p-xylylene-acetyl glucaramide).597Although glucaric acid occurs naturally in fruits and vegetables, in industry, glucaric acid is produced by glucose oxidation with nitric acid at low temperatures (298–313 K) and pressures (0.1–0.2 MPa).598 It is isolated as its potassium or calcium glucarate salts after base neutralisation. The formation of by-products, such as gluconic, oxalic, tartaric and 5-ketogluconic acids, decreases the glucaric acid yield to 45%. In addition to nitric acid oxidation, other greener and consequently more sustainable alternatives from glucose exist: electrochemical, chemical, biochemical and chemocatalytic oxidation.
Rivertop Renewables has started up the commercial manufacturing of glucarate-based products in a joint venture of Rivertop Renewables and DTI, using an improved process for the production of glucarates, which reduces the amount of chemical inputs and consumption, thus minimising the generation of wastes in comparison with the commercial process based on the use of concentrated nitric acid or bleaching agents.605 However, with the pursuit of developing green chemistry processes, heterogeneous catalysts in the presence of air, molecular oxygen or hydrogen peroxide are needed for the production of glucaric acid and glucarates from glucose. Interestingly, these processes can also be applied to other polysaccharides.
Glucaric acid is obtained from glucose in a two-step process: (i) oxidation of glucose to gluconic acid in the presence of mono- and bimetallic catalysts with various promoters such as Bi to minimise deactivation (see Section 4.1), and (ii) oxidation of gluconic acid to glucaric acid, which is more difficult, and consequently, there are fewer papers dealing with this process. Most catalysts reported for the oxidation of glucose, gluconic acid, or gluconate to glucaric acid are based on noble metals such as Pt, Pd and Au incorporated into several supports. The addition of promoters seems to have a positive effect. The catalytic results of representative systems in glucaric acid production are shown in Table 16. Moreover, most papers dealing with this oxidation process have used oxygen as an oxidant.
| # | Catalyst | Operating conditions | Catalytic results | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S cca (wt%) | C/Sb (wt%) | PO2c (bar) | pH | T (K) | t (h) | X S (%) | Yg (%) | Produc.h (g gcat−1 h−1) | Deact.i | |||
| a Substrate concentration in water, weight ratio in %. b Catalyst to substrate weight ratio in %. c Pressure of O2 in bar. d Temperature conditions in Kelvin. e Reaction time. f Substrate conversion. g Yield towards product (glucaric acid). h Productivity. i Catalyst deactivation: strong (st), moderate (m), low (l) and negligible (ne); n.s. (not studied), n.r. (not reported). j mL min−1. | ||||||||||||
| 1 | 5 wt% Pt/C | 3.6 | 111.0 | 1 | 10 | 328 | 2 | 100 | 50 | 0.26 | n.s. | 599 |
| 2 | 5 wt% Pt/C | 10.0 | 40.0 | 14 | 8.5 | 353 | 10 | 99 | 74 | 0.22 | l (5 runs) | 600 |
| 3 | Au/titanate_NT | 4.5 | 68.0 | 5 | 9.5 | 353 | 6 | 73 | 14 | 0.04 | ne (5 runs) | 601 |
| 4 | Pt1Cu3/TiO2 | 9.0 | 4.0 | 1 | >13 | 318 | 6 | 100 | 9 | 0.44 | ne (3 run) | 602 |
| 5 | PtPd/TiO2 | 5.0 | 4.0 | 60j | 11.5 | 318 | 6 | 100 | 25 | 1.22 | ne (3 runs) | 603 |
| 6 | Au3Bi/AC | 5.0 | 25.0 | 10 | n.r. | 333 | 3 | 100 | 29 | 0.45 | ne (24 h) | 604 |
The oxidation of glucose to glucaric acid requires more drastic conditions because glucaric acid adsorbs more strongly, blocking active sites for new gluconic acid molecules to be oxidised.600 However, from both feedstocks, the highest glucaric acid yield attained was similar, 50–55%, by using Pt/C, although different experimental conditions were used. Productivity values of 0.116 and 0.100 gglucaric acid gcat−1 h−1 were reached from gluconic acid and glucose aqueous solutions, respectively.599
The influence of the support on glucose oxidation was initially evaluated by using TiO2, ZrO2, SiO2 and SiO2–TiO2 by Rennovia Inc. A yield of 71% was attained with a 4 wt% Au–4 wt% Pt–TiO2 catalyst at 392 K after 3 h with a high oxygen pressure (2.7 MPa).606 It was also found that the catalytic performance decreased in the order Pt/C > Pt/SiO2 > Pt/Al2O3. A TOF of 0.244 s−1 at 20% glucose conversion was achieved with Pt/C, and the high catalytic activity was well correlated with the high surface area.600
Oxidation of carbohydrates requires a basic pH, and for this reason, Dirkx et al. evaluated the catalytic performance of Pt/C at pH values between 8 and 11.599 An increase in the pH favoured the formation of (the corresponding salts of) tartronic and tartaric acids (Fig. 27). Later, Lee et al. used a similar catalyst,600 obtaining a glucaric acid yield of 74% under base-free and mild basic conditions, with a TOF of 0.244 s−1. Under acidic conditions, gluconic acid was the major product, oligomerisation products explained the low carbon balance. However, in a highly basic solution, C–C bond cleavage, mainly from glucaric acid, is favoured, decreasing the selectivity to glucaric acid and producing low-carbon-chain carboxylic acids, such as tartronic and oxalic acids (Fig. 27). This was confirmed by Solmi et al. by using Au/activated carbon catalysts, as the use of a glucose/NaOH molar ratio of 3 resulted in a large spectrum of products with traces of gluconic acid and no formation of glucaric acid.604 These authors did not observe glucose oxidation in the absence of a base.
Usually, low reaction temperatures are employed for glucose oxidation (318–338 K), since high reaction temperatures cause degradation of glucaric acid,600 as observed by using oxygen as an oxidant at 318–338 K and pH = 8–11.599 A detrimental effect of a high concentration of oxygen on the catalytic activity was also observed. In addition, regarding the side reactions, the selectivity towards glucaric acid was affected by C–C cleavage of gluconic acid, yielding mono- and dicarboxylic acids, irrespective of the reactant.
The O2/glucose ratio in the liquid phase must be optimised to minimise the formation of by-products. A high O2 pressure enhances glucose conversion and facilitates oxidation of gluconic acid to glucaric acid, although the carbon balance is worst due to the formation of undetected products.600
An increase of the glucose/active metal phase ratio improves the gluconic acid selectivity, but glucose conversion and glucaric acid selectivity were barely modified.600 Regarding the glucose concentration in water, it must be optimised to obtain a suitable balance between glucaric acid and by-product formation, despite achieving total glucose conversion, as observed with AuBi/activated carbon with glucose concentrations between 2.5 and 20 wt%.604
Au nanoparticle size has no role in glucose conversion, with all catalysts being very active, but a key role was observed for the prevalence of one of the two parallel reaction pathways of gluconic acid transformation into either glucaric acid or by-products.604 The catalyst with the smallest nanoparticles (4.1 nm) was the least selective for glucaric acid since side reactions leading to degradation products of the gluconic acid intermediate were enhanced. Regarding the stability of catalysts, a Pt/C catalyst was stable after five catalytic cycles, and no Pt leaching was detected.600
The effect of Bi and Au addition on the activity and stability of Pt/C catalysts has been studied at pH 7 in air at 333 K.607 Gluconate oxidation to glucoronate requires previous complete glucose conversion. The incorporation of Bi as a promoter (PtBi/C) improves the initial specific reaction rate compared to that of the parent Pt/C catalyst. However, for larger Bi concentrations, the 2-ketogluconate yield increases to the detriment of glucarate production due to the formation of a surface complex where the gluconate molecules chelate the Bi promoter atoms, which are dehydrogenated to 2-ketogluconate (Fig. 27). On the other hand, the addition of Au on Pt was intended to decrease the catalyst deactivation. The initial reaction rate on PtAu/C was barely higher than that on the parent Pt/C, but after 24 h of reaction, the conversion was maintained at 91% compared with 67% in the absence of Au. This positive effect could be associated with a decrease in the poisoning of the metal surface, since oxygen or acidic by-product adsorption is disfavoured because the surface Pt-Au alloy has less affinity for oxygen than pure Pt.
On the other hand, Au and AuBi nanoparticles supported on activated carbon were compared without pH control.604 The best catalytic results were found for a bimetallic AuBi/C catalyst, with Au/Bi = 3:1, which attained a glucaric acid yield of 31% after 3 h of reaction at 333 K and 1 MPa O2 pressure. The reutilisation study (reaction time of 30 min, AuBi/C catalyst) demonstrated its reuse in successive runs, although deactivation was important. Glucaric acid was only observed in the first catalytic cycle with a selectivity lower than 5%. In all cases, the gluconic acid selectivity was maintained at approximately 83%. No leaching of Bi and Au was detected. TG analysis of the spent catalysts revealed the existence of strongly adsorbed heavy compounds on the catalyst surface, whereas agglomeration and sintering of NPs was inferred by STEM. This latter finding could be explained by the weak support–NP interaction and local overheating spots.
Bimetallic PtCu catalysts have also been prepared with TiO2 as a support.602 A Pt1Cu3 alloy attained a TOF of 0.984 s−1, as determined for a substrate conversion of 4–22% and a glucaric acid selectivity of 46% (at 318 K and 0.1 MPa of O2 pressure from a basic solution of sodium gluconate). The corresponding monometallic catalysts gave rise to TOF values lower than 0.153 s−1. Under these experimental conditions, after 5 h of reaction time, complete gluconate conversion was reached, with a selectivity towards glucaric acid of 32.3%, thus giving a productivity of 0.44 gglucaricacid gcat−1 h−1 for the bimetallic catalyst. The catalytic study demonstrated that the oxidation process was sensitive to the surface morphology of Pt NPs. The PtCu alloy seems to more significantly promote the C–C cleavage reactions by retro-aldolisation than the monometallic phases. Moreover, Cu species on Pt improved the oxidation rates of C–O to CO, increasing the oxidation rate of sodium gluconate. By using glucose as a feedstock, a lower glucaric acid yield (25.4%) at complete glucose conversion after 24 h of reaction was attained. No remarkable loss of catalytic activity was observed after three consecutive runs.
This effect of bimetallic morphology on the catalytic performance was evaluated with PtPd nanoparticles with alloy, core–shell, and cluster-in-cluster structures immobilised on TiO2.603 It was found, corroborating previous studies, that the bimetallic phase exhibits a synergistic effect, enhancing the catalytic activity and glucaric acid selectivity compared to those of their monometallic counterparts. Thus, a complete conversion of glucose was achieved at 318 K after 10 h, with a TOF of 0.68 s−1. Monometallic catalysts showed a high gluconic acid selectivity (S = 57–76%) with negligible glucaric acid formation (S ∼ 4%) after 12 h. In contrast, the glucaric acid selectivity reached 31% on the bimetallic catalyst. Other reaction products, such as tartronic, oxalic, glyceric, glycolic and lactic acids, were also detected on the bimetallic catalyst, while the selectivity towards these products was low on monometallic catalysts. A glucose inhibition effect in the secondary oxidation reaction, as previously noted, was also suggested, such that glucose might be strongly adsorbed on the bimetallic PtPd surface, thus preventing side reactions such as C–C cleavage of glucose. This could be more favourable from gluconic acid. The catalyst was stable after three runs, without a significant loss of catalytic activity.
The catalytic performance of bimetallic Au–Pd nanoparticles (NPs) prepared by colloidal synthesis and immobilised on titanate nanotubes (Ti-NTs) in glucose oxidation has also been studied under initially basic and relatively mild conditions but with lower glucaric acid selectivity (maximum of 18.5%).601
In addition to the previous conventional O2-driven transformation, iron tetra(2,3-bis(butylthio)maleonitrile)porphyrazine (FePz(SBu)8) was recently supported on an H-ZSM-5 zeolite to obtain a supported biomimetic photocatalyst.608 The photocatalytic oxidation of glucose in water under visible light (λ > 420 nm) irradiation was studied by using H2O2 as the oxidant. Under such conditions, glucose can be efficiently converted into value-added chemicals such as glucaric acid, gluconic acid, arabinose, glycerol and formic acid. However, glucose conversion is less than 50%, and selectivity towards glucaric acid does not exceed 15%, making this catalyst more selective to gluconic acid.
Electrolytic processes have also been proposed as alternatives to enzymatic and chemocatalytic methods for the oxidation of glucose to glucaric acid.609–611 A self-supported MOF nanorod array, [Ni3(BTC)2/NiF], has been demonstrated to be a highly energy-efficient and stable bifunctional electrocatalyst for integrating HER and replacing OER by electro-oxidation of sodium gluconate to glucaric acid, which is more favourable.610 The results show a nearly 100% faradaic efficiency for the HER at the cathode, a glucaric acid yield of 97.9% at the anode, and long-term durability. Nanostructured NiFe oxide (NiFeOx) and nitride (NiFeNx) catalysts synthesised from NiFe layered double hydroxide nanosheet arrays on three-dimensional Ni foams have been demonstrated to be highly active and selective in anodic glucose oxidation.609 A faradaic efficiency of 87% and glucaric acid yield of 83% were achieved by glucose electrolysis, which takes place via a guluronic acid pathway, as evidenced by in situ infrared spectroscopy. A techno-economic analysis has revealed that the electrochemical oxidation of glucose produces glucaric acid at a 54% lower cost than the current chemical approach, and the concomitant production of H2 is also very interesting. This electrocatalytic glucose oxidation process, in comparison with conventional chemical oxidation and microbial fermentation, provides higher glucaric acid yield at a shorter reaction time and with a lower E-factor (the mass ratio of the generated waste to target products). However, an important drawback is the large amount of KOH (370 tons per 1000 tons of glucaric acid), thus requiring specific equipment. Finally, the oxidation of glucose was investigated by cyclic voltammetry in alkaline medium for Cu, Pt and Au electrodes.611 However, although a selectivity towards glucaric acid of 38.4% was achieved at a low potential, the catalytic activity was too low to attain acceptable glucose conversion. In all cases, gluconic acid was the major oxidation product.
On the other hand, by using low-cost feedstocks, such as hydrolysates acquired from the hydrolysis of lignocellulosic biomass for the production of glucaric acid, Derrien et al. evaluated the possible inhibitory effect of impurities present in such hydrolysates on glucose oxidation in the presence of AuPt/ZrO2.612,613 At 373 K, 4 MPa air and a glucose/metal molar ratio of 80, a glucaric acid yield of 50% was attained after 10 h from the oxidation of commercial glucose (free of impurities), and the catalyst was completely reusable. Regarding impurities, in individual studies dealing with only one type of impurity, it was demonstrated that acids (mainly sulfuric or acetic acid) did not influence gluconic acid production, but the glucaric acid yield was slightly lower. This could indicate that these acids favour side reactions from glucaric acid. The inhibitory effect of furanic compounds was more evident, with a 3 h delay in the maximum production of gluconic acid and a final glucaric acid yield of 40%. In the presence of cellobiose as a disaccharide residue, the oxidation of gluconic acid to glucaric acid was not complete after 24 h, and the glucaric acid yield was slightly lower after 8 h of reaction.
A techno-economic and lifecycle assessment of the production of glucaric acid from corn stover biomass has demonstrated that both routes are economically viable, leading to minimum selling prices of glucaric acid of ∼$2.53 kg−1 and ∼$2.91 kg−1 for the heterogeneous catalytic process and the homogeneous glucose oxidation route, respectively.614 The heterogeneous process is accomplished in the presence of a 5 wt% Pt/C catalyst, using air at pressures of 3.5–4.0 MPa and temperatures of 353–363 K, with glucaric acid yields of 50–60%. Concerning the homogeneous process, nitric acid was used as the catalyst at a relatively low temperature of 298–313 K in a reactor pressurised at 0.1–0.2 MPa with air. It was concluded that the heterogeneous catalytic oxidation process achieves a 22% lower environmental impact than the homogeneous process.
The economic viability of energy self-sufficient biorefineries for processing sugarcane lignocelluloses into sorbitol or glucaric acid and electricity has been evaluated by Kapanji et al.615 However, they faced the challenge of limited information available in the public domain concerning glucaric acid production due to proprietary rights.
In summary, the production of glucaric acid through green routes such as heterogeneous catalysis is still in its infancy, and considerable efforts must be devoted to substitute the current commercial technology based on the use of nitric acid. The main drawback to overcome is related to the actual selectivity of the proposed catalysts and their stability during the reutilisation tests, since glucaric acid is produced from gluconic acid oxidation as an oxidised intermediate product from glucose. However, there are several mechanistic pathways, favouring the degradation of gluconic and glucaric acids into low-molecular-weight acids (Fig. 27). Moreover, the direct transformation of lignocellulosic glucose into glucaric acid using catalytic technologies requires further development, especially in the case of much cheaper agroresidues that would reduce the cost of the glucaric acid.
5. Concluding remarks and future general prospects
In this review, the most relevant technical aspects of the chemocatalytic technologies needed for the synthesis of carboxylic acid monomers have been comprehensively described. The levels of comprehensiveness of the different technologies described in this review are very dissimilar; some of the processes have very high technological readiness levels (TRLs) and are close to commercialisation like adipic acid production from glucose (Rennovia's process) or FDCA synthesis from glucose (Avantium's process). Some other processes displayed very low TRLs, and only promising catalytic technologies have been identified so far; this is the case for maleic acid, acrylic acid and lactic acid, just to mention a few. In other cases, catalysts with enough selectivity and robustness (mostly in the presence of a real feedstock and recycling impurities) have not yet been developed, and there is still a great deal of investigation to be done.Nonetheless, a wide variety of catalysts have already been studied for all processes described in this review, and the main features of the active sites have been revealed. Further investigation must be supported by these previous results, and any new catalysts proposed must be benchmarked with these technologies that have already demonstrated good performance. Specific guidelines for future investigations to cope with the weaknesses already identified have been briefly discussed in each section of this manuscript. Next, the main guidelines for future studies, common to all the technologies, are discussed.
Considering the high risk of new technologies, reducing the overall number of steps should be a goal. Moreover, a holistic and more integral approach is needed. Thus, the catalytic process for the synthesis of a monomer must be compatible with the preceding processes required for the chemical platform on which the monomer is synthesised (monomer precursor) and with the final polymerisation reaction. And all the catalytic steps must be suitably integrated with the purification and separation steps, which usually contribute greatly to the final cost of the products.
One of the first options for improving productivity is to utilise more concentrated solutions. A rapid look at the tables and the column of tables giving the wt% of the substrate used in the reactions tells us that many studies used very dilute feeds, often below 5 wt%. Obtaining similar results at higher concentrations (above 10 wt%) is needed. An additional positive outcome is that separation and purification processes needed to obtain high-grade monomers are more productive and cost-effective with high-concentration solutions, as capital and energy costs are typically reduced. However, working with concentrated solutions increases the concentration of impurities which maximises the side reactions associated with the higher concentration of reactants and intermediates and makes the deactivation phenomena more intense. In addition, mass and heat transfer may become more problematic. Besides, in many cases, the surface of the catalyst can get saturated with reactants, and then the reaction rate does not depend on their concentration; in these cases, to reach similar conversion, the temperature and pressure of the reaction may have to be increased to compensate for the increase in the concentration of reactant, and this may also come with undesirable side reactions, lower selectivity and faster deactivation of the catalyst. The other possibilities, i.e., maintaining the catalyst/substrate ratio or increasing the contact time, both adversely impact the cost and the productivity of the process. Consequently, the catalysts must be redesigned to mitigate these problems.
Productivity in laboratory investigations can also be improved by using fixed bed reactors under continuous flow conditions instead of the widespread utilisation of batch reactors. Using fixed bed reactors is a straightforward method of increasing the substrate/catalyst mass ratio and, consequently, the productivity (space-time yield).616 Moreover, the scaling of a process to an industrial level of operation likely requires flow operation. Another advantage of fixed reactors is that different reaction conditions can be set along the reactor, which facilitates the integration of the process.462,617,618 Studies with dilute solutions using batch reactors are suitable for rapid screening of families of catalysts and improving the understanding of the reaction (reaction orders, kinetic parameters, reaction pathways, etc.), but when one needs to improve the productivity and to truly assess the performance and robustness of a catalytic technology, one has to use continuous flows of concentrated solutions (well above 10 wt%, if possible) for long reaction times (weeks rather than days). In summary, closing the gap between using dilute solutions in batch reactors and concentrated solutions in flow operation is essential for improving productivity.
In the best-case scenario, the production of a monomer entails only two reaction steps: first, the production of the monomer precursor from the biomass; and, second, the transformation of the precursor into the monomer. However, in most cases, the process requires more complex reaction schemes involving multiple steps that complicate the practical implementation and economic viability of the process. Research must be directed toward integrating, or at least simplifying, the reaction scheme for producing the precursor and the monomer.
Thus, for example, correct selection of the solvent(s) used in the synthesis of the monomer precursor can be one of the drivers for simplification. The solvents must be renewable or, at least, green, but a good option is to conduct the reaction for the synthesis of the monomer in the same solvent(s) used for the synthesis of its precursor. It would be especially interesting to conduct research with solvents that have shown the capacity to fractionate lignocellulose into its main components: sugars (from cellulose and hemicellulose) and lignin.619 If the reaction conditions for the processes are similar, the investigation must be completed by integrating both steps in a cascade reaction scheme, designing bifunctional catalysts, or by using fixed bed reactors with different reaction conditions and/or catalyst beds in different zones of the reactor.617
The investigation must also move from high-grade precursors, easily available on the market, to low-grade precursors. Most of the catalytic technologies discussed in this review use high grade precursors. Removing problematic impurities implies extensive purification steps (distillation, precipitation, recrystallisation, extractions with solvent, etc.) that are frequently complex, energy-demanding and expensive. Besides, the transformation of the precursor into the monomer is frequently conducted in the liquid phase; therefore, it does not make much sense to remove a solvent if the precursor will later be dissolved in another solvent (furthermore, once the carboxylic acid is produced, another purification is required to remove impurities incompatible with the polymerisation reaction and/or detrimental to the performance of the polymer). Using the same solvent in both processes allows for the integration and intensification of the synthesis of the precursor and its transformation into the carboxylic acid. Consequently, productivity will increase and purification steps will be simplified. This entails a search for catalysts that are compatible with the solvent and the higher levels of impurities associated with relatively low-grade monomer precursors.
A last comment about the nature of the catalysts: this review has been directed at reusable catalysts. The easiest approach is to use solid catalysts that can be easily separated from the batch reaction medium or effortlessly loaded into a fixed bed reactor. However, in some cases, the performance of heterogeneous catalysts has not reached that of homogeneous ones. A solution is to heterogenise the homogeneous catalysts by supporting them on stable supports; this research is yet to be done.
Some of the monomers described in this review are drop-in monomers, that is, they are currently derived from oil by means of efficient and mature petrochemical processes. Processes from biomass conversion still need to improve their carbon efficiency (high selectivity), productivity (high volumetric-space velocity, high reagent concentration), and separation and/or purification protocols to minimise costs. In this context, it is also worth mentioning that with biomass, the starting C/O atomic ratio is 1 (carbohydrates) or slightly lower (lignin), but at the end, the ratio of the product is often larger than 1. Consequently, the weight-to-weight conversion is negative, unlike that for oil-derived processes which is positive. All of these drawbacks must be confronted and mitigated for a biomass pathway to be economically competitive.
Techno-economic evaluations provide information about the economic viability of biomass-derived processes in comparison with that of petrochemical processes. These evaluations not only give the final cost of the product but also identify bottlenecks in its economic viability and, consequently, are indispensable tools to direct future investigations for improving the competitiveness of a process.376,395,484 The catalyst alone, very often, is not the critical economic aspect but it is always at the heart of the technical viability. Any modification of the process must be compatible with the catalytic technology or prompt the development of a new technology congruent with the modifications. In the case of having different renewable technologies for the synthesis of renewable carboxylic acids, techno-economic evaluation is also vital for selecting the most viable technology. Surprisingly, there is a lack of techno-economic evaluations of biomass-derived monomer production processes, which must be corrected without delay. The studies should be done by multidisciplinary teams combining direct lab results with techno-economic calculations. While the utilisation of the literature is suitable, it has to be done carefully to avoid oversimplifications; for example, by extrapolating economic evaluations and applying them to different catalytic processes or assuming that the catalysts performance will not change upon modification of a process, i.e., changing the solvent or concentration. In addition, these studies should always provide a list of assumptions, including the most relevant information used in the calculations, so the readers are able to judge the study and easily select the assumptions that must be modified according to their own experience and results.
Finally, it must be clearly stated that a renewable origin of carboxylic acids does not imply their environmental sustainability. Comparative life cycle analysis (LCA) between the biomass and petrochemical processes inventories the environmental impacts of both routes. In the case of an unfavourable impact, whichever the approach is (from cradle to grave, gate to gate, Eco-LCA, etc.), LCA identifies the parts of a process with the highest impact that, consequently, must be first confronted and mitigated.614,620–622 Therefore, LCA may also have an effect on the modification of catalytic technology or even call for the development of new technologies so that environmental viability is accomplished. Again, as for the economic evaluation, the LCAs of the processes used for the synthesis of carboxylic acids are unexpectedly scarce, and this must be corrected. Future investigations must concentrate on developing new catalysts that cope with the environmental drawbacks detected in LCAs.
Acronyms
| 11-DHA | 1,1-Dihydroxy acetone |
| 2-HAC | 2-Hydroxy acrolein |
| 3-HPA | 3-Hydroxypropionic acid |
| 3DG | 3-Deoxyglucosone |
| 3DX | 3-Deoxyxylosone |
| 5-HMF | 5-Hydroxymethyl furfural |
| AdA | Adipic acid |
| AcOH | Acetic acid |
| AcryA | Acrylic acid |
| BDO | 1,4-Butanediol |
| BHMF | 2,5-Bishydroxymethylfuran |
| BTX | Benzene, toluene and xylene |
| B-V | Baeyer–Villiger |
| c,c-MA | cis,cis-Muconic acid |
| CMF | Chloromethylfurfural |
| CP | Cyclopentanone |
| DA | Diels–Alder reaction |
| DALA | δ-Aminolevulinic acid |
| DAP | Dimethyl adipate |
| DFF | 2,5-Diformylfuran |
| DHA | Dihydroxy acetone |
| DMC | Dimethyl carbonate |
| DMF | 2,5-Dimethylfuran |
| DMSO | Dimethyl sulfoxide |
| DPA | Diphenolic acid |
| DTBPX | Di-tert-butylphosphino-o-xylene |
| EFAL | Extra-framework aluminium |
| EGL | Ethyl glucarate-6,6-lactonate |
| EtOH | Ethanol |
| FAc | Formic acid |
| FCA | 5-Formyl-furandicarboxylic acid |
| FDCA | 2,5-Furandicarboxylic acid |
| FDME | Furandicarboxylic methyl esters |
| FUR | Furfural |
| GA | Glycolic acid |
| GADMA | Glycolaldehyde dimethyl acetal |
| GAL | Glycoladehyde |
| GLA | Glyceraldehyde |
| GLU | Glucose |
| GLY | Glycerol |
| GVL | γ-Valerolactone |
| HFCA | 5-Hydroxymethyl-2-furancarboxylic acid, also known as 5-hydroxymethyl furoic acid |
| HFONE | 5-Hydroxy-furan-2(5H)-one, also known as 5-hydroxyfuranone |
| IPA | Isophthalic acid |
| KA-oil | Ketone-alcohol oil |
| LA | Lactic acid |
| LCA | Life cycle analysis |
| LEVA | Levulinic acid |
| MA | Maleic anhydride |
| MAc | Maleic acid |
| MeOH | Methanol |
| MDG | Methyl-3-deoxygluconate |
| MDHP | Methyl-2,5-dihydroxypent-3-enoate |
| ML | Methyl lactate |
| MMF | Methoxymethylfurfural |
| MMHB | Methyl-4-methoxy-2-hydroxybutanoate |
| MTA | Methanol to aromatics |
| MTHH | Methyl-2,5,6-trihydroxyhex-3-enoate |
| MOF | Metal organic frameworks |
| MSA | Methanesulfonic acid |
| MVG | Methyl vinyl glycolate |
| ODCB | o-Dicholorobenzene |
| PAL | Pyruvaldehyde |
| PAL-MHA | Pyruvaldehyde methyl hemiacetal |
| PADA | Pyruvaldehyde dimethyl acetal |
| PBT | Polybutylene terephthalate |
| PEF | Polyethylene furoate |
| PET | Polyethylene terephthalate |
| PHA | Poly(hydroxy alkanoates) |
| PLA | Poly(lactic acid) |
| POM | Polyoxymetalates |
| PVA | Poly(vinyl alcohol) |
| pX | para-Xylene |
| STY | Space-time yield |
| THFDCA | Tetrahydrofuran-2,5-dicarboxylic acid |
| THFDM | Tetrahydro-2,5-furandimethanol |
| TMP | 1,1,2,2-Tetramethoxypropane |
| TOF | Turnover frequency |
| TPA | Terephthalic acid |
| t,t-MA | trans,trans-Muconic acid |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministry of Research, Innovation (MICINN) (RTI2018-094918-B-C41, RTI2018-094918-B-C42 and RTI2018-094918-B-C44), is gratefully acknowledged. This research was funded by Comunidad de Madrid (Spain) and ERDF (European Regional Development Fund), grant number S2018/EMT-4344 (BIOTRES-CM).Notes and references
- The Basics: Polymer Definition and Properties, https://plastics.americanchemistry.com/plastics/The-Basics/, accessed 11 February 2020.
- Plastics – the Facts. https://www.plasticseurope.org/application/files/6315/4510/9658/Plastics_the_facts_2018_AF_web.pdf, 2018.
- https://www.textileworld.com/textile-world/fiber-world/2015/02/man-made-fibers-continue-to-grow/, https://www.textileworld.com/textile-world/fiber-world/2015/02/man-made-fibers-continue-to-grow/.
- Is burning plastic waste a good idea? https://www.nationalgeographic.com/environment/2019/03/should-we-burn-plastic-waste/#close, accessed 11 February 2020.
- R. Verma, K. S. Vinoda, M. Papireddy and A. N. S. Gowda, Procedia Environ. Sci., 2016, 35, 701–708 CrossRef CAS.
- C. J. Rhodes, Sci. Prog., 2019, 102, 218–248 CrossRef PubMed.
- S. Lambert and M. Wagner, Chem. Soc. Rev., 2017, 46, 6855–6871 RSC.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS PubMed.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- G. Q. Chen and M. K. Patel, Chem. Rev., 2012, 112, 2082–2099 CrossRef CAS PubMed.
- R. De Clercq, M. Dusselier and B. F. Sels, Green Chem., 2017, 19, 5012–5040 RSC.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- C. Ablasser, A. Hilger and O. Hinrichsen, Chem. Ing. Tech., 2014, 86, 1948–1953 CrossRef CAS.
- M. Hong and E. Y. X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- C. Alberti, R. Figueira, M. Hofmann, S. Koschke and S. Enthaler, ChemistrySelect, 2019, 4, 12638–12642 CrossRef CAS.
- A. R. Rahimi and J. M. Garciá, Nat. Rev. Chem., 2017, 1, 1–11 CrossRef.
- C. Jehanno, M. M. Pérez-Madrigal, J. Demarteau, H. Sardon and A. P. Dove, Polym. Chem., 2019, 10, 172–186 RSC.
- https://www.biobasedconsultancy.com/uploads/files/InnProBio_Factsheet_n3.pdf, accessed 11 February 2020.
- European Bioplastics, Accountabilit y is key, Environmental Communication Guide for Bioplasics, https://docs.european-bioplastics.org/publications/EUBP_Environmental_communications_guide.pdf.
- 2016 Bio-based Building Blocks and Polymers, http://www.bio-based.eu/market_study/media/16-12-16-Bio-based-Building-Blocks-and-Polymers-short-version.pdf, 2017.
- B. M. Stadler, C. Wulf, T. Werner, S. Tin and J. G. de Vries, ACS Catal., 2019, 9, 8012–8067 CrossRef CAS.
- S. Li, W. Deng, S. Wang, P. Wang, D. An, Y. Li, Q. Zhang and Y. Wang, ChemSusChem, 2018, 11, 1995–2028 CrossRef CAS PubMed.
- L. Salusjärvi, S. Havukainen, O. Koivistoinen and M. Toivari, Appl. Microbiol. Biotechnol., 2019, 103, 2525–2535 CrossRef PubMed.
- C. Du, J. Sabirova, W. Soetaert and S. Ki Carol Lin, Curr. Chem. Biol., 2012, 6, 14–25 CAS.
- M. Winnacker, Eur. J. Lipid Sci. Technol., 2019, 121, 1900101 CrossRef CAS.
- Y. Chen, S.-T. Hung, E. Chou and H.-S. Wu, Mini. Rev. Org. Chem., 2017, 15, 105–121 CrossRef.
- Z. Luo, Y. Wu, Z. Li and X. J. Loh, Biotechnol. J., 2019, 1900283, 1900283 CrossRef PubMed.
- M. Dusselier, P. Van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 RSC.
- J. M. Luengo, B. García, A. Sandoval, G. Naharro and E. R. Olivera, Curr. Opin. Microbiol., 2003, 6, 251–260 CrossRef CAS PubMed.
- P. R. Gruber, E. S. Hall, J. J. Kolstad, M. L. Iwen, R. D. Benson and R. L. Borchordt, US5247059A, 1993.
- G. Q. Chen, Chem. Soc. Rev., 2009, 38, 2434–2446 RSC.
- R. G. Hill, Biomaterials, Artificial Organs and Tissue Engineering, Elsevier Inc., 2005, pp. 97–106 Search PubMed.
- Z. Zhang, O. Ortiz, R. Goyal and J. Kohn, Principles of Tissue Engineering, Elsevier Inc., 4th edn, 2013, pp. 441–473 Search PubMed.
- C. C. Chu, Biotextiles As Medical Implants, Elsevier Inc., 2013, pp. 275–334 Search PubMed.
- X. Hua, R. Cao, X. Zhou and Y. Xu, Bioresour. Technol., 2018, 268, 402–407 CrossRef CAS PubMed.
- K. Miltenberger, Ullmann's Encycl. Ind. Chem., 2000, 481–492 Search PubMed.
- M. Shattuck, US2443482A, 1948.
- H. Klug, DE2810906A1, 1979.
- H. S. Kambo, J. Minaret and A. Dutta, Waste Biomass Valorization, 2018, 9, 1181–1189 CrossRef CAS.
- G. Nazeri, S. B. Liaw, Y. Yu and H. Wu, Fuel, 2018, 218, 174–178 CrossRef CAS.
- N. Candu, F. Anita, I. Podolean, B. Cojocaru, V. I. Parvulescu and S. M. Coman, Green Process. Synth., 2017, 6, 255–264 CAS.
- C. Guarín, L. Gavilà, M. Constantí and F. Medina, Chem. Eng. Sci., 2018, 179, 83–91 CrossRef.
- J. Zhang, X. Liu, M. Sun, X. Ma and Y. Han, ACS Catal., 2012, 2, 1698–1702 CrossRef CAS.
- A. Bayu, S. Karnjanakom, A. Yoshida, K. Kusakabe, A. Abudula and G. Guan, Catal. Today, 2019, 332, 28–34 CrossRef CAS.
- M. Sankar, N. Dimitratos, D. W. Knight, A. F. Carley, R. Tiruvalam, C. J. Kiely, D. Thomas and G. J. Hutchings, ChemSusChem, 2009, 2, 1145–1151 CrossRef CAS PubMed.
- A. Takagaki, A. Tsuji, S. Nishimura and K. Ebitani, Chem. Lett., 2011, 40, 150–152 CrossRef CAS.
- X. Dai, S. Adomeit, J. Rabeah, C. Kreyenschulte, A. Brückner, H. Wang and F. Shi, Angew. Chem., Int. Ed., 2019, 58, 5251–5255 CrossRef CAS PubMed.
- P. Y. Dapsens, C. Mondelli, B. T. Kusema, R. Verel and J. Pérez-Ramírez, Green Chem., 2014, 16, 1176–1186 RSC.
- G. Rychen, G. Aquilina, G. Azimonti, V. Bampidis, M. de L. Bastos, G. Bories, A. Chesson, P. S. Cocconcelli, G. Flachowsky, B. Kolar, M. Kouba, M. López-Alonso, S. López Puente, A. Mantovani, B. Mayo, F. Ramos, M. Saarela, R. E. Villa, R. J. Wallace, P. Wester, A. Lundebye, C. Nebbia, D. Renshaw, M. L. Innocenti and J. Gropp, Safety of lactic acid and calcium lactate when used as technological additives for all animal species, John Wiley & Sons, Ltd, 2017, vol. 15 Search PubMed.
- C. S. Pomelli, C. Chiappe, F. Barontini, M. Puccini, M. Seggiani and S. Vitolo, Clean Technol. Environ. Policy, 2017, 19, 259–267 CrossRef CAS.
- K. Butterwick and N. J. Lowe, J. Am. Acad. Dermatol., 2009, 61, 281–293 CrossRef CAS PubMed.
- R. A. A. Alsaheb, A. Aladdin, N. Z. Othman, R. A. Malek, O. M. Leng, R. Aziz, H. A. El Enshasy and H. A. El, J. Chem. Pharm. Res., 2015, 7, 729–735 Search PubMed.
- M. S. Lopes, A. L. Jardini and R. M. Filho, Chem. Eng. Trans., 2014, 38, 331–336 Search PubMed.
- L. T. Sin, A. R. Rahmat and W. A. W. A. Rahman, Applications of Poly(lactic Acid), Elsevier, 2012 Search PubMed.
- Y. Hu, W. A. Daoud, K. K. L. Cheuk and C. S. K. Lin, Materials, 2016, 9, 133 CrossRef PubMed.
- P. VanWouwe, M. Dusselier, E. Vanleeuw and B. Sels, ChemSusChem, 2016, 9, 907–921 CrossRef CAS PubMed.
- H. Tsuji, Adv. Drug Delivery Rev., 2016, 107, 97–135 CrossRef CAS PubMed.
- A. Komesu, J. A. R. de Oliveira, L. H. da Silva Martins, M. R. Wolf-Maciel and R. Maciel-Filho, Lactic Acid Production to Purification: A Review, 2017, vol. 12 Search PubMed.
- C. Miller, A. Fosmer, B. Rush, T. McMullin, D. Beacom and P. Suominen, Comprehensive Biotechnology, Elsevier Inc., 2nd edn, 2011, vol. 3, pp. 179–188 Search PubMed.
- E. Cubas-Cano, C. González-Fernández, M. Ballesteros and E. Tomás-Pejó, Biofuels, Bioprod. Biorefin., 2018, 12, 290–303 CrossRef CAS.
- K. Okano, T. Tanaka, C. Ogino, H. Fukuda and A. Kondo, Appl. Microbiol. Biotechnol., 2010, 85, 413–423 CrossRef CAS PubMed.
- G. Gómez Millán, S. Hellsten, J. Llorca, R. Luque, H. Sixta and A. M. Balu, ChemCatChem, 2019, 11, 2022–2042 CrossRef.
- G. Juodeikiene, D. Vidmantiene, L. Basinskiene, D. Cernauskas, E. Bartkiene and D. Cizeikiene, Catal. Today, 2015, 239, 11–16 CrossRef CAS.
- A. G. Daful, K. Haigh, P. Vaskan and J. F. Görgens, Food Bioprod. Process., 2016, 99, 58–70 CrossRef CAS.
- A. Gezae Daful and J. F. Görgens, Chem. Eng. Sci., 2017, 162, 53–65 CrossRef CAS.
- Y. Zhang, Z. Shen, X. Zhou, M. Zhang and F. Jin, Green Chem., 2012, 14, 3285–3288 RSC.
- H. Kishida, F. Jin, Z. Zhou, T. Moriya and H. Enomoto, Chem. Lett., 2005, 34, 1560–1561 CrossRef CAS.
- C. A. Ramírez-López, J. R. Ochoa-Gómez, M. Fernández-Santos, O. Gómez-Jiménez-Aberasturi, A. Alonso-Vicario and J. Torrecilla-Soria, Ind. Eng. Chem. Res., 2010, 49, 6270–6278 CrossRef.
- T. Shimanouchi, S. Ueno, K. Shidahara and Y. Kimura, Chem. Lett., 2014, 43, 535–537 CrossRef CAS.
- X. Jin, D. Roy, P. S. Thapa, B. Subramaniam and R. V. Chaudhari, ACS Sustainable Chem. Eng., 2013, 1, 1453–1462 CrossRef CAS.
- H. Yin, C. Zhang, H. Yin, D. Gao, L. Shen and A. Wang, Chem. Eng. J., 2016, 288, 332–343 CrossRef CAS.
- Z. Jiang, Z. Zhang, T. Wu, P. Zhang, J. Song, C. Xie and B. Han, Chem. – Asian J., 2017, 12, 1598–1604 CrossRef CAS PubMed.
- R. Abdullah, S. N. Mohamed Saleh, K. Embong and A. Z. Abdullah, Chem. Eng. Commun., 2019, 1–31 Search PubMed.
- T. Ntho, J. Aluha, P. Gqogqa, M. Raphulu and G. Pattrick, React. Kinet. Mech. Catal., 2013, 109, 133–148 CrossRef CAS.
- Z. Yuan, Z. Gao and B. Q. Xu, Cuihua Xuebao, 2015, 36, 1543–1551 CAS.
- T. Komanoya, H. Kobayashi, K. Hara, W. J. Chun and A. Fukuoka, J. Energy Chem., 2013, 22, 290–295 CrossRef.
- R. K. P. Purushothaman, J. Van Haveren, A. Mayoral, I. Melián-Cabrera and H. J. Heeres, Top. Catal., 2014, 57, 1445–1453 CrossRef CAS.
- R. K. Pazhavelikkakath Purushothaman, J. Van Haveren, I. Melián-Cabrera, E. R. H. Van Eck and H. J. Heeres, ChemSusChem, 2014, 7, 1140–1147 CrossRef CAS PubMed.
- H. J. Cho, C. C. Chang and W. Fan, Green Chem., 2014, 16, 3428–3433 RSC.
- T. Lu, X. Fu, L. Zhou, Y. Su, X. Yang, L. Han, J. Wang and C. Song, ACS Catal., 2017, 7, 7274–7284 CrossRef CAS.
- J. Li, D. Li, C. L. Liu, C. Xu and W. S. Dong, J. Chem. Technol. Biotechnol., 2019, 94, 1958–1967 CrossRef CAS.
- Z. Tang, S. L. Fiorilli, H. J. Heeres and P. P. Pescarmona, ACS Sustainable Chem. Eng., 2018, 6, 10923–10933 CrossRef CAS PubMed.
- L. Zhou, Y. Xu, X. Yang, T. Lu and L. Han, Energy Convers. Manage., 2019, 196, 277–285 CrossRef CAS.
- M. Tao, X. Yi, I. Delidovich, R. Palkovits, J. Shi and X. Wang, ChemSusChem, 2015, 8, 4195–4201 CrossRef CAS PubMed.
- M. Tao, D. Zhang, X. Deng, X. Li, J. Shi and X. Wang, Chem. Commun., 2016, 52, 3332–3335 RSC.
- M. Tao, D. Zhang, H. Guan, G. Huang and X. Wang, Designation of highly efficient catalysts for one pot conversion of glycerol to lactic acid, Sci. Rep., 2016, 6, 29840 CrossRef CAS PubMed.
- A. Villa, G. M. Veith and L. Prati, Angew. Chem., Int. Ed., 2010, 49, 4499–4502 CrossRef CAS PubMed.
- R. De Clercq, M. Dusselier and B. F. Sels, Reaction Pathways and Mechanisms in Thermocatalytic Biomass Conversion I: Cellulose Structure, Depolymerization and Conversion by Heterogeneous Catalysts, Springer, Singapore, 2015, pp. 27–55 Search PubMed.
- S. Sen Liu, K. Q. Sun and B. Q. Xu, ACS Catal., 2014, 4, 2226–2230 CrossRef.
- A. Bayu, A. Abudula and G. Guan, Fuel Process. Technol., 2019, 196, 106162 CrossRef CAS.
- T. R. Carlson, J. Jae, Y. C. Lin, G. A. Tompsett and G. W. Huber, J. Catal., 2010, 270, 110–124 CrossRef CAS.
- F. Jin and H. Enomoto, Energy Environ. Sci., 2011, 4, 382–397 RSC.
- X. Yan, F. Jin, K. Tohji and H. Enomoto, AIP Conf. Proc., 2007, 898, 182–185 CrossRef CAS.
- L. Li, F. Shen, R. L. Smith and X. Qi, Green Chem., 2017, 19, 76–81 RSC.
- X. Yan, F. Jini, A. Kishita, H. Enomoto and K. Tohji, AIP Conf. Proc., 2008, 987, 50–53 CrossRef CAS.
- X. Yan, F. Jin, K. Tohji, T. Moriya and H. Enomoto, J. Mater. Sci., 2007, 42, 9995–9999 CrossRef CAS.
- C. A. Ramírez-López, J. R. Ochoa-Gómez, S. Gil-Río, O. Gómez-Jiménez-Aberasturi and J. Torrecilla-Soria, J. Chem. Technol. Biotechnol., 2011, 86, 867–874 CrossRef.
- Z. Huo, Y. Fang, D. Ren, S. Zhang, G. Yao, X. Zeng and F. Jin, ACS Sustainable Chem. Eng., 2014, 2, 2765–2771 CrossRef CAS.
- C. Sánchez, I. Egüés, A. García, R. Llano-Ponte and J. Labidi, Chem. Eng. J., 2012, 181–182, 655–660 CrossRef.
- F. Wang, Y. Wang, F. Jin, G. Yao, Z. Huo, X. Zeng and Z. Jing, Ind. Eng. Chem. Res., 2014, 53, 7939–7946 CrossRef CAS.
- H. Choudhary, S. Nishimura and K. Ebitani, Appl. Catal., B, 2015, 162, 1–10 CrossRef CAS.
- D. Esposito and M. Antonietti, ChemSusChem, 2013, 6, 989–992 CrossRef CAS PubMed.
- M. Xia, W. Dong, M. Gu, C. Chang, Z. Shen and Y. Zhang, RSC Adv., 2018, 8, 8965–8975 RSC.
- S. Li, W. Deng, Y. Li, Q. Zhang and Y. Wang, J. Energy Chem., 2019, 32, 138–151 CrossRef.
- R. S. Assary and L. A. Curtiss, Energy Fuels, 2012, 26, 1344–1352 CrossRef CAS.
- M. S. Holm, Y. J. Pagán-Torres, S. Saravanamurugan, A. Riisager, J. A. Dumesic and E. Taarning, Green Chem., 2012, 14, 702–706 RSC.
- B. D. Montejo-Valencia and M. C. Curet-Arana, J. Phys. Chem. C, 2019, 123, 3532–3540 CrossRef CAS.
- M. Moliner, Y. Román-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS PubMed.
- M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602–605 CrossRef CAS PubMed.
- Y. Wang, W. Deng, B. Wang, Q. Zhang, X. Wan, Z. Tang, Y. Wang, C. Zhu, Z. Cao, G. Wang and H. Wan, Nat. Commun., 2013, 4, 2141, DOI:10.1038/ncomms3141.
- X. Lei, F. F. Wang, C. L. Liu, R. Z. Yang and W. S. Dong, Appl. Catal., A, 2014, 482, 78–83 CrossRef CAS.
- Y. Yan, L. Feng, G. Li, S. Lin, Z. Sun, Y. Zhang and Y. Tang, Borate-stabilized transformation of C6 aldose to C4 aldose, 2017 Search PubMed.
- J. Wang, G. Yao and F. Jin, Mol. Catal., 2017, 435, 82–90 CrossRef CAS.
- M. Dusselier, R. De Clercq, R. Cornelis and B. F. Sels, Catal. Today, 2017, 279, 339–344 CrossRef CAS.
- P. Ferrini, J. Dijkmans, R. De Clercq, S. Van de Vyver, M. Dusselier, P. A. Jacobs and B. F. Sels, Coord. Chem. Rev., 2017, 343, 220–255 CrossRef CAS.
- W. N. P. Van Der Graaff, C. H. L. Tempelman, E. A. Pidko and E. J. M. Hensen, Catal. Sci. Technol., 2017, 7, 3151–3162 RSC.
- C. M. Lew, N. Rajabbeigi and M. Tsapatsis, Microporous Mesoporous Mater., 2012, 153, 55–58 CrossRef CAS.
- H. J. Cho, P. Dornath and W. Fan, ACS Catal, 2014, 4, 2029–2037 CrossRef CAS.
- P. Y. Dapsens, C. Mondelli, J. Jagielski, R. Hauert and J. Pérez-Ramírez, Catal. Sci. Technol., 2014, 4, 2302–2311 RSC.
- A. Corma, L. T. Nemeth, M. Renz and S. Valencia, Nature, 2001, 412, 423–425 CrossRef CAS PubMed.
- M. Renz, T. Blasco, A. Corma, V. Fornés, R. Jensen and L. Nemeth, Chem. – Eur. J., 2002, 8, 4708–4717 CrossRef CAS.
- S. Tolborg, A. Katerinopoulou, D. D. Falcone, I. Sádaba, C. M. Osmundsen, R. J. Davis, E. Taarning, P. Fristrup and M. S. Holm, J. Mater. Chem. A, 2014, 2, 20252–20262 RSC.
- C. Hammond, S. Conrad and I. Hermans, Angew. Chem., Int. Ed., 2012, 51, 11736–11739 CrossRef CAS PubMed.
- J. Dijkmans, J. Demol, K. Houthoofd, S. Huang, Y. Pontikes and B. Sels, J. Catal., 2015, 330, 545–557 CrossRef CAS.
- W. N. P. Van Der Graaff, G. Li, B. Mezari, E. A. Pidko and E. J. M. Hensen, ChemCatChem, 2015, 7, 1152–1160 CrossRef CAS.
- P. Y. Dapsens, C. Mondelli and J. Pérez-Ramírez, New J. Chem., 2016, 40, 4136–4139 RSC.
- P. Wolf, M. Valla, A. J. Rossini, A. Comas-Vives, F. Núñez-Zarur, B. Malaman, A. Lesage, L. Emsley, C. Copéret and I. Hermans, Angew. Chem., Int. Ed., 2014, 53, 10179–10183 CrossRef CAS PubMed.
- J. C. Vega-Vila, J. W. Harris and R. Gounder, J. Catal., 2016, 344, 108–120 CrossRef CAS.
- J. W. Harris, M. J. Cordon, J. R. Di Iorio, J. C. Vega-Vila, F. H. Ribeiro and R. Gounder, J. Catal., 2016, 335, 141–154 CrossRef CAS.
- J. Dijkmans, M. Dusselier, W. Janssens, M. Trekels, A. Vantomme, E. Breynaert, C. Kirschhock and B. F. Sels, ACS Catal., 2016, 6, 31–46 CrossRef CAS.
- B. D. Montejo-Valencia, J. L. Salcedo-Pérez and M. C. Curet-Arana, J. Phys. Chem. C, 2016, 120, 2176–2186 CrossRef CAS.
- R. Bermejo-Deval, M. Orazov, R. Gounder, S. J. Hwang and M. E. Davis, ACS Catal., 2014, 4, 2288–2297 CrossRef CAS.
- S. Li, T. Josephson, D. G. Vlachos and S. Caratzoulas, J. Catal., 2017, 355, 11–16 CrossRef CAS.
- S. G. Elliot, E. Taarning, R. Madsen and S. Meier, ChemCatChem, 2018, 10, 1414–1419 CrossRef CAS.
- G. Qi, Q. Wang, J. Xu, Q. Wu, C. Wang, X. Zhao, X. Meng, F. Xiao and F. Deng, Commun. Chem., 2018, 1 Search PubMed.
- C. M. Osmundsen, M. Spangsberg Holm, S. Dahl and E. Taarning, Proc. R. Soc. London, Ser. A, 2012, 468, 2000–2016 CrossRef CAS.
- Q. Guo, F. Fan, E. A. Pidko, W. N. P. Van Der Graaff, Z. Feng, C. Li and E. J. M. Hensen, ChemSusChem, 2013, 6, 1352–1356 CrossRef CAS PubMed.
- I. Tosi, A. Sacchetti, J. S. Martinez-Espin, S. Meier and A. Riisager, Top. Catal., 2019, 62, 628–638 CrossRef CAS.
- X. Yang, J. Bian, J. Huang, W. Xin, T. Lu, C. Chen, Y. Su, L. Zhou, F. Wang and J. Xu, Green Chem., 2017, 19, 692–701 RSC.
- J. Zhang, L. Wang, G. Wang, F. Chen, J. Zhu, C. Wang, C. Bian, S. Pan and F. S. Xiao, ACS Sustainable Chem. Eng., 2017, 5, 3123–3131 CrossRef CAS.
- X. Yang, Y. Liu, X. Li, J. Ren, L. Zhou, T. Lu and Y. Su, ACS Sustainable Chem. Eng., 2018, 6, 8256–8265 CrossRef CAS.
- W. N. P. van der Graaff, C. H. L. Tempelman, F. C. Hendriks, J. Ruiz-Martinez, S. Bals, B. M. Weckhuysen, E. A. Pidko and E. J. M. Hensen, Appl. Catal., A, 2018, 564, 113–122 CrossRef CAS.
- K. Nemoto, Y. Hirano, K. I. Hirata, T. Takahashi, H. Tsuneki, K. I. Tominaga and K. Sato, Appl. Catal., B, 2016, 183, 8–17 CrossRef CAS.
- W. Deng, P. Wang, B. Wang, Y. Wang, L. Yan, Y. Li, Q. Zhang, Z. Cao and Y. Wang, Green Chem., 2018, 20, 735–744 RSC.
- W. Dong, Z. Shen, B. Peng, M. Gu, X. Zhou, B. Xiang and Y. Zhang, Sci. Rep., 2016, 6, 26713, DOI:10.1038/srep26713.
- M. Xia, Z. Shen, S. Xiao, B. yu Peng, M. Gu, W. Dong and Y. Zhang, Appl. Catal., A, 2019, 583, 117126 CrossRef CAS.
- L. Zhou, L. Wu, H. Li, X. Yang, Y. Su, T. Lu and J. Xu, J. Mol. Catal. A: Chem., 2014, 388–389, 74–80 CrossRef CAS.
- J. Iglesias, J. Moreno, G. Morales, J. A. Melero, P. Juárez, M. López-Granados, R. Mariscal and I. Martínez-Salazar, Green Chem., 2019, 21, 5876–5885 RSC.
- I. Tosi, A. Riisager, E. Taarning, P. R. Jensen and S. Meier, Catal. Sci. Technol., 2018, 8, 2137–2145 RSC.
- F. Wang, Y. Wen, Y. Fang and H. Ji, ChemCatChem, 2018, 10, 4154–4161 CrossRef CAS.
- A. A. Marianou, C. M. Michailof, A. Pineda, E. F. Iliopoulou, K. S. Triantafyllidis and A. A. Lappas, Appl. Catal., A, 2018, 555, 75–87 CrossRef CAS.
- M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602–605 CrossRef CAS PubMed.
- Y. Y. Sun, L. Shi, H. Wang, G. Miao, L. Kong, S. Li and Y. Y. Sun, Sustain, Energy Fuels, 2019, 3, 1163–1171 CAS.
- S. Tolborg, I. Sádaba, C. M. Osmundsen, P. Fristrup, M. S. Holm and E. Taarning, ChemSusChem, 2015, 8, 613–617 CrossRef CAS PubMed.
- B. Murillo, A. Sánchez, V. Sebastián, C. Casado-Coterillo, O. de la Iglesia, M. P. López-Ram-de-Viu, C. Téllez and J. Coronas, J. Chem. Technol. Biotechnol., 2014, 89, 1344–1350 CrossRef CAS.
- J. Pang, M. Zheng, X. Li, L. Song, R. Sun, J. Sebastian, A. Wang, J. Wang, X. Wang and T. Zhang, ChemistrySelect, 2017, 2, 309–314 CrossRef CAS.
- B. Murillo, B. Zornoza, O. De La Iglesia, C. Téllez and J. Coronas, J. Catal., 2016, 334, 60–67 CrossRef CAS.
- X. Lu, L. Wang and X. Lu, Catal. Commun., 2018, 110, 23–27 CrossRef CAS.
- B. Murillo, B. Zornoza, O. de la Iglesia, S. Wang, C. Serre, C. Téllez and J. Coronas, Eur, J. Inorg. Chem., 2019, 2624–2629 CAS.
- S. G. Elliot, S. Tolborg, R. Madsen, E. Taarning and S. Meier, ChemSusChem, 2018, 11, 1198–1203 CrossRef CAS PubMed.
- R. Otomo, R. Kosugi, Y. Kamiya, T. Tatsumi and T. Yokoi, Catal. Sci. Technol., 2016, 6, 2787–2795 RSC.
- S. Huang, K. L. Yang, X. F. Liu, H. Pan, H. Zhang and S. Yang, RSC Adv., 2017, 7, 5621–5627 RSC.
- F. De Clippel, M. Dusselier, R. Van Rompaey, P. Vanelderen, J. Dijkmans, E. Makshina, L. Giebeler, S. Oswald, G. V. Baron, J. F. M. Denayer, P. P. Pescarmona, P. A. Jacobs and B. F. Sels, J. Am. Chem. Soc., 2012, 134, 10089–10101 CrossRef CAS PubMed.
- R. M. West, M. S. Holm, S. Saravanamurugan, J. Xiong, Z. Beversdorf, E. Taarning and C. H. Christensen, J. Catal., 2010, 269, 122–130 CrossRef CAS.
- P. P. Pescarmona, K. P. F. Janssen, C. Delaet, C. Stroobants, K. Houthoofd, A. Philippaerts, C. De Jonghe, J. S. Paul, P. A. Jacobs and B. F. Sels, Green Chem., 2010, 12, 1083–1089 RSC.
- L. Li, C. Stroobants, K. Lin, P. A. Jacobs, B. F. Sels and P. P. Pescarmona, Green Chem., 2011, 13, 1175–1181 RSC.
- P. Y. Dapsens, B. T. Kusema, C. Mondelli and J. Pérez-Ramírez, J. Mol. Catal. A: Chem., 2014, 388–389, 141–147 CrossRef CAS.
- E. Taarning, S. Saravanamurugan, M. S. Holm, J. Xiong, R. M. West and C. H. Christensen, ChemSusChem, 2009, 2, 625–627 CrossRef CAS PubMed.
- X. Yang, L. Wu, Z. Wang, J. Bian, T. Lu, L. Zhou, C. Chen and J. Xu, Catal. Sci. Technol., 2016, 6, 1757–1763 RSC.
- K. D. Kim, Z. Wang, Y. Jiang, M. Hunger and J. Huang, Green Chem., 2019, 21, 3383–3393 RSC.
- J. Wang, Y. Masui and M. Onaka, Appl. Catal., B, 2011, 107, 135–139 CrossRef CAS.
- E. Pighin, V. K. Díez and J. I. Di Cosimo, Appl. Catal., A, 2016, 517, 151–160 CrossRef CAS.
- E. Pighin, V. K. Díez and J. I. Di Cosimo, Catal. Today, 2017, 289, 29–37 CrossRef CAS.
- E. A. Pighin, J. I. Di Cosimo and V. K. Díez, Mol. Catal., 2018, 458, 189–197 CrossRef CAS.
- X. Wang, Y. Song, L. Huang, H. Wang, C. Huang and C. Li, Catal. Sci. Technol., 2019, 9, 1669–1679 RSC.
- X. Wang, Y. Song, C. Huang and B. Wang, Sustainable Energy Fuels, 2018, 2, 1530–1541 RSC.
- Z. Zhang and Z. Zhao, Cuihua Xuebao, 2011, 32, 70–73 Search PubMed.
- X. Wang, F. Liang, C. Huang, Y. Li and B. Chen, Catal. Sci. Technol., 2015, 5, 4410–4421 RSC.
- X. Wang, F. Liang, C. Huang, Y. Li and B. Chen, Catal. Sci. Technol., 2016, 6, 6551–6560 RSC.
- H. S. Chen, A. Wang, H. Sorek, J. D. Lewis, Y. Román-Leshkov and A. T. Bell, ChemistrySelect, 2016, 1, 4167–4172 CrossRef CAS.
- A. Bayu, A. Yoshida, S. Karnjanakom, K. Kusakabe, X. Hao, T. Prakoso, A. Abudula and G. Guan, Green Chem., 2018, 20, 4112–4119 RSC.
- E. Jolimaitre, D. Delcroix, N. Essayem, C. Pinel and M. Besson, Catal. Sci. Technol., 2018, 8, 1349–1356 RSC.
- Y. Hayashi and Y. Sasaki, Chem. Commun., 2005, 2716–2718 RSC.
- C. B. Rasrendra, B. A. Fachri, I. G. B. N. Makertihartha, S. Adisasmito and H. J. Heeres, ChemSusChem, 2011, 4, 768–777 CrossRef CAS PubMed.
- S. Lux and M. Siebenhofer, Chem. Biochem. Eng. Q., 2015, 29, 575–585 CrossRef CAS.
- P. N. Paulino, R. F. Perez, N. G. Figueiredo and M. A. Fraga, Green Chem., 2017, 19, 3759–3763 RSC.
- R. Cohen, C. R. Graves, S. B. T. Nguyen, J. M. L. Martin and M. A. Ratner, J. Am. Chem. Soc., 2004, 126, 14796–14803 CrossRef CAS PubMed.
- M. Morales, P. Y. Dapsens, I. Giovinazzo, J. Witte, C. Mondelli, S. Papadokonstantakis, K. Hungerbühler and J. Pérez-Ramírez, Energy Environ. Sci., 2015, 8, 558–567 RSC.
- L. T. Mika, E. Cséfalvay and Á. Németh, Chem. Rev., 2018, 118, 505–613 CrossRef CAS PubMed.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass Volume I-Results of Screening for Potential Candidates from Sugars and Synthesis Gas, US Department of Energy, Energy Efficiency and Renewable Energy, 2004 Search PubMed.
- S. Choi, C. W. Song, J. H. Shin and S. Y. Lee, Metab. Eng., 2015, 28, 223–239 CrossRef CAS PubMed.
- F. de Fouchecour, A.-K. Sanchez-Castañeda, C. Saulou-Berion and H. E. Spinnler, Biotechnol. Adv., 2018, 36, 1207–1222 CrossRef CAS PubMed.
- X. Jiang, X. Meng and M. Xian, Appl. Microbiol. Biotechnol., 2009, 82, 995–1003 CrossRef CAS PubMed.
- B. 2014, BASF, Cargill and Novozymes achieved another milestone in bio-based acrylic acid, https://www.cargill.com/news/releases/2014/NA31686855.jsp, accessed 3 January 2020.
- C. Li, Q. Zhu, Z. Cui, B. Wang, Y. Fang and T. Tan, Chem. Eng. Sci., 2018, 183, 288–294 CrossRef CAS.
- C. Della Pina, E. Falletta and M. Rossi, Green Chem., 2011, 13, 1624–1632 RSC.
- T. Li, S. Liu, B. Wang, J. Long, J. Jiang, P. Jin, Y. Fu, H. Yu and W. Yang, Green Chem., 2019, 21, 4434–4442 RSC.
- D. J. Hayes, S. Fitzpatrick, M. H. B. Hayes and J. R. H. Ross, in Biorefineries – Industrial Processes and Products, ed. B. Kamm, P. R. Gruber and M. Kamm, Wiley-VCH Verlag GmbH & Co., 2006, vol. 1, pp. 139–164 Search PubMed.
- D. Hayes, Optimal Use of DIBANET Feedstocks and Technologies, 2013.
- Y. Wang, F. Vogelgsang and Y. Román-Leshkov, ChemCatChem, 2015, 7, 916–920 CrossRef CAS.
- D. Carnevali, M. G. Rigamonti, T. Tabanelli, G. S. Patience and F. Cavani, Appl. Catal., A, 2018, 563, 98–104 CrossRef CAS.
- M. J. Fink and M. D. Mihovilovic, Chem. Commun., 2015, 51, 2874–2877 RSC.
- Y.-Y. Liu, C.-X. Li, J.-H. Xu and G.-W. Zheng, Efficient Synthesis of Methyl 3-Acetoxypropionate by a Newly Identified Baeyer-Villiger Monooxygenase, 2019 Search PubMed.
- M. Dusselier, P. VanWouwe, F. deClippel, J. Dijkmans, D. W. Gammon and B. F. Sels, ChemCatChem, 2013, 5, 569–575 CrossRef CAS.
- R. De Clercq, M. Dusselier, J. Dijkmans, R. I. Iacobescu, Y. Pontikes and B. F. Sels, ACS Catal., 2015, 5, 5803–5811 CrossRef CAS.
- W. R. Gunther, Y. Wang, Y. Ji, V. K. Michaelis, S. T. Hunt, R. G. Griffin and Y. Román-Leshkov, Nat. Commun., 2012, 3, 1109, DOI:10.1038/ncomms2122.
- A. Takagaki, S. Furusato, R. Kikuchi and S. T. Oyama, ChemSusChem, 2015, 8, 3769–3772 CrossRef CAS PubMed.
- S. Lin, X. Guo, K. Qin, L. Feng, Y. Zhang and Y. Tang, ChemCatChem, 2017, 9, 4179–4184 CrossRef CAS.
- S. Tolborg, S. Meier, S. Saravanamurugan, P. Fristrup, E. Taarning and I. Sádaba, ChemSusChem, 2016, 9, 3054–3061 CrossRef CAS PubMed.
- S. G. Elliot, C. Andersen, S. Tolborg, S. Meier, I. Sádaba, A. E. Daugaard and E. Taarning, RSC Adv., 2017, 7, 985–996 RSC.
- S. Tolborg, S. Meier, I. Sádaba, S. G. Elliot, S. K. Kristensen, S. Saravanamurugan, A. Riisager, P. Fristrup, T. Skrydstrup and E. Taarning, Green Chem., 2016, 18, 3360–3369 RSC.
- J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. G. Neuenscwander, S. W. Fitzpatrick, R. J. Bilski and J. L. Jarnefeld, Resour. Conserv. Recycl., 2000, 28, 227–239 CrossRef.
- C. Zúñiga, G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, Polymer, 2012, 53, 1617–1623 CrossRef.
- J. Ertl, E. Cerri, M. Rizzuto and D. Caretti, AIP Conf. Proc., 2014, 1599, 326–329 CrossRef CAS.
- H. R. Kricheldorf, R. Hobzova and G. Schwarz, Polymer, 2003, 44, 7361–7368 CrossRef CAS.
- K. Yan, C. Jarvis, J. Gu and Y. Yan, Renewable Sustainable Energy Rev., 2015, 51, 986–997 CrossRef CAS.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- F. D. Pileidis and M.-M. Titirici, ChemSusChem, 2016, 9, 562–582 CrossRef CAS.
- D. W. Rackemann and W. O. Doherty, Biofuels, Bioprod. Biorefin., 2011, 5, 198–214 CrossRef CAS.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. López Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2011, 13, 520 RSC.
- Y. Guo, K. Li and J. H. Clark, Green Chem., 2007, 9, 839–841 RSC.
- Y. Guo, K. Li, X. Yu and J. H. Clark, Appl. Catal., B, 2008, 81, 182–191 CrossRef CAS.
- K. Li, J. Hu, W. Li, F. Ma, L. Xu and Y. Guo, J. Mater. Chem., 2009, 19, 8628–8638 RSC.
- S. Van De Vyver, J. Geboers, S. Helsen, F. Yu, J. Thomas, M. Smet, W. Dehaen and B. F. Sels, Chem. Commun., 2012, 48, 3497–3499 RSC.
- K. Shimizu, S. Kontani, S. Yamada, G. Takahashi, T. Nishiyama and A. Satsuma, Appl. Catal., A, 2010, 380, 33–39 CrossRef CAS.
- S. Van De Vyver, J. Thomas, J. Geboers, S. Keyzer, M. Smet, W. Dehaen, P. A. Jacobs and B. F. Sels, Energy Environ. Sci., 2011, 4, 3601–3610 RSC.
- H. F. Liu, F. X. Zeng, L. Deng, B. Liao, H. Pang and Q. X. Guo, Green Chem., 2013, 15, 81–84 RSC.
- X. Xu, J. Lin and P. Cen, Chin. J. Chem. Eng., 2006, 14, 419–427 CrossRef CAS.
- Acrylic acidfrom gluycerin, https://ihsmarkit.com/products/chemical-technology-pep-acrylic-acid-from-glycerin-2011.html.
- F. T. Maler and W. Bayer, Encycl. Chem. Process. Des., 1976, 1, 401 Search PubMed.
- N. Kurata, T. Ohara and K. Oda, US3475488, 1969.
- T. Ohara, T. Sato, N. Shimizu, G. Prescher, H. Schwind, O. Welberg, K. Marten and H. Greim, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH, 2003 Search PubMed.
- Acrylic Acid Production and Manufacturing Process, https://www.icis.com/explore/resources/news/2007/11/01/9074872/acrylic-acid-production-and-manufacturing-process/, accessed 28 December 2019.
- J. C. Védrine, E. K. Novakova and E. G. Derouane, Catal. Today, 2003, 81, 247–262 CrossRef.
- M. M. Lin, Appl. Catal., A, 2001, 207, 1–16 CrossRef CAS.
- E. K. Novakova, J. C. Védrine and E. G. Derouane, J. Catal., 2002, 211, 226–234 CAS.
- T. Dishisha, S. H. Pyo and R. Hatti-Kaul, Microb. Cell Fact., 2015, 14, 1–11 CrossRef PubMed.
- A. S. Paula, L. G. Possato, D. R. Ratero, J. Contro, K. Keinan-Adamsky, R. R. Soares, G. Goobes, L. Martins and J. G. Nery, Microporous Mesoporous Mater., 2016, 232, 151–160 CrossRef CAS.
- P. Mars and D. W. van Krevelen, Chem. Eng. Sci., 1954, 3, 41–59 CrossRef CAS.
- L. G. Possato, W. H. Cassinelli, T. Garetto, S. H. Pulcinelli, C. V. Santilli and L. Martins, Appl. Catal., A, 2015, 492, 243–251 CrossRef CAS.
- J. Deleplanque, J. L. Dubois, J. F. Devaux and W. Ueda, Catal. Today, 2010, 157, 351–358 CrossRef CAS.
- L. Shen, H. Yin, A. Wang, X. Lu and C. Zhang, Chem. Eng. J., 2014, 244, 168–177 CrossRef CAS.
- A. Chieregato, M. D. Soriano, F. Basile, G. Liosi, S. Zamora, P. Concepción, F. Cavani and J. M. López Nieto, Appl. Catal., B, 2014, 150–151, 37–46 CrossRef CAS.
- K. Omata, K. Matsumoto, T. Murayama and W. Ueda, Catal. Today, 2016, 259, 205–212 CrossRef CAS.
- M. B. dos Santos, H. M. C. Andrade and A. J. S. Mascarenhas, Microporous Mesoporous Mater., 2019, 278, 366–377 CrossRef CAS.
- M. M. Diallo, S. Laforge, Y. Pouilloux and J. Mijoin, Catal. Commun., 2019, 126, 21–25 CrossRef CAS.
- J. L. Dubois and G. Patience, US8530697B2, 2013.
- A. Witsuthammakul and T. Sooknoi, Appl. Catal., A, 2012, 413–414, 109–116 CrossRef CAS.
- P. Mäki-Arvela, I. L. Simakova, T. Salmi and D. Y. Murzin, Chem. Rev., 2014, 114, 1909–1971 CrossRef.
- R. K. Grasselli and F. Trifirò, Rend. Lincei, 2017, 28, 59–67 CrossRef.
- D. Sun, Y. Yamada, S. Sato and W. Ueda, Green Chem., 2017, 19, 3186–3213 RSC.
- G. Holmen, US2859240, 1958.
- C. Yuan, H. Liu, Z. Zhang, H. Lu, Q. Zhu and Y. Chen, Cuihua Xuebao, 2015, 36, 1861–1866 CAS.
- Y. Matsuura, A. Onda, S. Ogo and K. Yanagisawa, Catal. Today, 2014, 226, 192–197 CrossRef CAS.
- Y. Matsuura, A. Onda and K. Yanagisawa, Catal. Commun., 2014, 48, 5–10 CrossRef CAS.
- C. Tang, J. Peng, G. Fan, X. Li, X. Pu and W. Bai, Catal. Commun., 2014, 43, 231–234 CrossRef CAS.
- P. Sun, D. Yu, Z. Tang, H. Li and H. Huang, Ind. Eng. Chem. Res., 2010, 49, 9082–9087 CrossRef CAS.
- J. Zhang, Y. Zhao, M. Pan, X. Feng, W. Ji and C. T. Au, ACS Catal., 2011, 1, 32–41 CrossRef CAS.
- V. C. Ghantani, S. T. Lomate, M. K. Dongare and S. B. Umbarkar, Green Chem., 2013, 15, 1211–1217 RSC.
- P. Sun, D. Yu, K. Fu, M. Gu, Y. Wang, H. Huang and H. Ying, Catal. Commun., 2009, 10, 1345–1349 CrossRef CAS.
- H. Wang, D. Yu, P. Sun, J. Yan, Y. Wang and H. Huang, Catal. Commun., 2008, 9, 1799–1803 CrossRef CAS.
- T. M. Aida, A. Ikarashi, Y. Saito, M. Watanabe, R. L. Smith and K. Arai, J. Supercrit. Fluids, 2009, 50, 257–264 CrossRef CAS.
- J. M. Lee, D. W. Hwang, Y. K. Hwang, S. B. Halligudi, J. S. Chang and Y. H. Han, Catal. Commun., 2010, 11, 1176–1180 CrossRef CAS.
- J. H. Hong, J. M. Lee, H. Kim, Y. K. Hwang, J. S. Chang, S. B. Halligudi and Y. H. Han, Appl. Catal., A, 2011, 396, 194–200 CrossRef CAS.
- C. Li, B. Wang, Q. Zhu and T. Tan, Appl. Catal., A, 2014, 487, 219–225 CrossRef CAS.
- B. M. Murphy, T. Mou, B. Wang and B. Xu, ACS Catal., 2018, 8, 9066–9078 CrossRef CAS.
- T. Ghaznavi, C. Neagoe and G. S. Patience, Biomass Bioenergy, 2014, 71, 285–293 CrossRef CAS.
- S. I. Beale and P. A. Castelfranco, Plant Physiol., 1974, 53, 297–303 CrossRef CAS PubMed.
- C. Sasikala, C. V. Ramana and P. R. Rao, Biotechnol. Prog., 1994, 10, 451–459 CrossRef CAS.
- N. Fotinos, M. A. Campo, F. Popowycz, R. Gurny and N. Lange, Photochem. Photobiol., 2006, 82, 994–1015 CrossRef CAS PubMed.
- N. Khemthongcharoen, R. Jolivot, S. Rattanavarin and W. Piyawattanametha, Adv. Drug Delivery Rev., 2014, 74, 53–74 CrossRef CAS.
- M. Hiorth, S. Nilsen and I. Tho, Pharmaceutics, 2014, 6, 494–511 CrossRef CAS PubMed.
- J. Rak, V. Vladimír and R. Smucler, EP3085358A1, 2016.
- S. F. Chang, Y. T. Yang, W. L. Li, C. T. Lin and T. Tsai, J. Dent. Sci., 2010, 5, 30–35 CrossRef.
- Z. Kang, W. Ding, X. Gong, Q. Liu, G. Du and J. Chen, World J. Microbiol. Biotechnol., 2017, 33, 1–7 CrossRef CAS PubMed.
- S. Liu, G. Zhang, X. Li and J. Zhang, Appl. Microbiol. Biotechnol., 2014, 98, 7349–7357 CrossRef CAS.
- S. F. MacDonald, Can. J. Chem., 1974, 52, 3257–3258 CrossRef CAS.
- H.-J. Ha, S.-K. Lee, Y.-J. Ha and J.-W. Park, Synth. Commun., 1994, 24, 2557–2562 CrossRef CAS.
- E. Benedikt and H.-P. Köst, Z. Naturforsch., B, 1986, 41, 1593–1594 Search PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- WP 8.1. Determination of market potential for selected platform chemicals - PDF, https://docplayer.net/95007975-Wp-8-1-determination-of-market-potential-for-selected-platform-chemicals.html, accessed 28 October 2019.
- 2,5-Furandicarboxylic Acid (FDCA) Market Size Worth Around $850 Million By 2025: Acumen Research and Consulting, https://www.globenewswire.com/news-release/2019/04/12/1803565/0/en/2-5-Furandicarboxylic-Acid-FDCA-Market-Size-Worth-Around-850-Million-By-2025-Acumen-Research-and-Consulting.html, accessed 28 October 2019.
- R. J. I. Knoop, W. Vogelzang, J. Van Haveren and D. S. Van Es, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4191–4199 CrossRef CAS.
- Breaking boundaries in biotechnology, https://www.corbion.com/bioplastics, accessed 30 October 2019.
- E. De Jong, M. A. Dam, L. Sipos and G. J. M. Gruter, ACS Symp. Ser., 2012, 1105, 1–13 CrossRef CAS.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- X. Tong, Y. Ma and Y. Li, Appl. Catal., A, 2010, 385, 1–13 CrossRef CAS.
- T. Wang, M. W. Nolte and B. H. Shanks, Green Chem., 2014, 16, 548–572 RSC.
- B. Saha and M. M. Abu-Omar, Green Chem., 2014, 16, 24–38 RSC.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G. J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC.
- J. Zhang, J. Li, Y. Tang, L. Lin and M. Long, Carbohydr. Polym., 2015, 130, 420–428 CrossRef CAS PubMed.
- H. Li, Z. Fang, R. L. Smith and S. Yang, Prog. Energy Combust. Sci., 2016, 55, 98–194 CrossRef.
- L. Hu, A. He, X. Liu, J. Xia, J. Xu, S. Zhou and J. Xu, ACS Sustainable Chem. Eng., 2018, 6, 15915–15935 CrossRef CAS.
- P. Pal and S. Saravanamurugan, ChemSusChem, 2019, 12, 145–163 CrossRef CAS PubMed.
- USDA ERS – Data Products, https://www.ers.usda.gov/data-products/, accessed 9 January 2020.
- R. A. F. Tomás, J. C. M. Bordado and J. F. P. Gomes, Chem. Rev., 2013, 113, 7421–7469 CrossRef PubMed.
- Y. Zhang, X. Guo, P. Tang and J. Xu, J. Chem. Eng. Data, 2018, 63, 1316–1324 CrossRef CAS.
- T. Pan, J. Deng, Q. Xu, Y. Zuo, Q. X. Guo and Y. Fu, ChemSusChem, 2013, 6, 47–50 CrossRef CAS PubMed.
- Y.-T. Huang, J.-J. Wong, C.-J. Huang, C.-L. Li and G.-W. B. Jang, Chemicals and Fuels from Bio-Based Building Blocks, Wiley-VCH Verlag GmbH & Co. KGaA, 2016, pp. 191–216 Search PubMed.
- R. D. Sanderson, D. F. Schneider and I. Schreuder, J. Appl. Polym. Sci., 1994, 53, 1785–1793 CrossRef CAS.
- K. M. Rapp, US4740605A, 1988.
- P. Ackermann and J. Vyscocil, DE102011001108A1, 2011, https://patents.google.com/patent/DE102011001108A1/en.
- Home - 5-HMF the key to green chemistry, https://5-hmf.com/.
- B. G. Siqueira, R. B. de Menezes, C. R. Klotz Rabello and M. Gomes, US9199957B2, 2013.
- A. S. Vagueiro de Sousa Dias, G. J. M. Gruter and R. J. van Putten, US9090581B2, 2015.
- J. C. van der Waals, E. Mazoyer, H. J. Baars and G. J. M. Gruter, Liquid Phase Aerobic Oxidation Catalysis: Industrial Applications and Academic Perspectives, Wiley-VCH Verlag, 2016, pp. 311–329 Search PubMed.
- FDCA Manufacture Technology – Free Library|Intratec.us, https://www.intratec.us/free-tools/how-to-make/fdca-manufacture-technology.
- Avantium Technology & Markets Day: “Entering the Commercialization Phase for PEF” – Avantium, https://www.avantium.com/investor-relations/avantium-technology-markets-day-entering-the-commercialization-phase-for-pef/, accessed 30 October 2019.
- Mekong – Avantium, https://www.avantium.com/renewable-chemistries/mekong/, accessed 2 November 2019.
- A. Sanborn, EP2784069B2, 2014.
- A. Sanborn, US9562028B2, 2017.
- News: DuPont Industrial Biosciences & Archer Daniels Midland Company Open Groundbreaking Biobased Pilot Facility in Illinois - DuPont Industrial Biosciences, http://biosciences.dupont.com/news/dupont-industrial-biosciences-archer-daniels-midland-company-open-groundbreaking-biobased-pilot-fa/, accessed 2 November 2019.
- M. Mascal, ChemSusChem, 2015, 8, 3391–3395 CrossRef CAS.
- S. Dutta, L. Wu and M. Mascal, Green Chem., 2015, 17, 3737–3739 RSC.
- M. Mascal and E. B. Nikitin, Green Chem., 2010, 12, 370–373 RSC.
- Origin Materials - Technology, https://www.originmaterials.com/technology.
- S. Dutta and M. Mascal, ChemSusChem, 2014, 7, 3028–3030 CrossRef CAS.
- N. Nikbin, S. Feng, S. Caratzoulas and D. G. Vlachos, J. Phys. Chem. C, 2014, 118, 24415–24424 CrossRef CAS.
- Eastman licenses proprietary FDCA technology to Origin Materials, https://www.bioplasticsmagazine.com/en/news/meldungen/20170926-Eastman-liceses-FDCA-technology-to-Origin-Materials.php.
- L. Capuzzi, F. Digioia and G. Carotenuto, WO2014180979A9, 2014.
- G. Borsoti and F. Digioia, WO2012017052A1, 2012.
- Novamont Starts Construction of a Demo Plant for the Production of Furandicarboxylic Acid – Novamont – Press, https://www.novamont.com/eng/read-press-release/novamont-starts-construction-of-a-demo-plant-for-the-production-of-furandicarboxylic-acid/, accessed 3 November 2019.
- L. Lai and Y. Zhang, ChemSusChem, 2011, 4, 1745–1748 CrossRef CAS.
- G. Yi, S. P. Teong, X. Li and Y. Zhang, ChemSusChem, 2014, 7, 2131–2135 CrossRef CAS.
- H. Liu, X. Cao, T. Wang, J. Wei, X. Tang, X. Zeng, Y. Sun, T. Lei, S. Liu and L. Lin, J. Ind. Eng. Chem., 2019, 77, 209–214 CrossRef CAS.
- J. M. R. Gallo, D. M. Alonso, M. A. Mellmer and J. A. Dumesic, Green Chem., 2013, 15, 85–90 RSC.
- A. H. Motagamwala, W. Won, C. Sener, D. M. Alonso, C. T. Maravelias and J. A. Dumesic, Sci. Adv., 2018, 4, eaap9722 CrossRef PubMed.
- S. R. Kubota and K. S. Choi, ChemSusChem, 2018, 11, 2138–2145 CrossRef CAS PubMed.
- W.-J. Liu, L. Dang, Z. Xu, H.-Q. Yu, S. Jin and G. W. Huber, ACS Catal., 2018, 8, 5533–5541 CrossRef CAS.
- R. Latsuzbaia, R. Bisselink, A. Anastasopol, H. van der Meer, R. van Heck, M. S. Yagüe, M. Zijlstra, M. Roelands, M. Crockatt, E. Goetheer and E. Giling, J. Appl. Electrochem., 2018, 48, 611–626 CrossRef CAS.
- J. van Haveren, S. Thiyagarajan and A. Teruo, US9284290B2, 2016.
- S. E. Davis, B. N. Zope and R. J. Davis, Green Chem., 2012, 14, 143–147 RSC.
- O. Casanova, S. Iborra and A. Corma, ChemSusChem, 2009, 2, 1138–1144 CrossRef CAS PubMed.
- S. E. Davis, A. D. Benavidez, R. W. Gosselink, J. H. Bitter, K. P. De Jong, A. K. Datye and R. J. Davis, J. Mol. Catal. A: Chem., 2014, 388–389, 123–132 CrossRef CAS.
- C. Megías-Sayago, A. Lolli, S. Ivanova, S. Albonetti, F. Cavani and J. A. Odriozola, Catal. Today, 2019, 333, 169–175 CrossRef.
- B. Sang, J. Li, X. Tian, F. Yuan and Y. Zhu, Mol. Catal., 2019, 470, 67–74 CrossRef.
- Q. Li, H. Wang, Z. Tian, Y. Weng, C. Wang, J. Ma, C. Zhu, W. Li, Q. Liu and L. Ma, Catal. Sci. Technol., 2019, 9, 1570–1580 RSC.
- Y. Liu, H.-Y. Ma, D. Lei, L.-L. Lou, S. Liu, W. Zhou, G.-C. Wang and K. Yu, ACS Catal., 2019, 9, 8306–8315 CrossRef CAS.
- H. Xia, J. An, M. Hong, S. Xu, L. Zhang and S. Zuo, Catal. Today, 2019, 319, 113–120 CrossRef CAS.
- K. Yu, Y. Liu, D. Lei, Y. Jiang, Y. Wang, Y. Feng, L. L. Lou, S. Liu and W. Zhou, Catal. Sci. Technol., 2018, 8, 2299–2303 RSC.
- X. Han, C. Li, X. Liu, Q. Xia and Y. Wang, Green Chem., 2017, 19, 996–1004 RSC.
- C. Ke, M. Li, G. Fan, L. Yang and F. Li, Chem. – Asian J., 2018, 13, 2714–2722 CrossRef CAS PubMed.
- H. Chen, J. Shen, K. Chen, Y. Qin, X. Lu, P. Ouyang and J. Fu, Appl. Catal., A, 2018, 555, 98–107 CrossRef CAS.
- C. A. Antonyraj, N. T. T. Huynh, K. W. Lee, Y. J. Kim, S. Shin, J. S. Shin and J. K. Cho, J. Chem. Sci., 2018, 130, 156 CrossRef.
- K. T. V. Rao, J. L. Rogers, S. Souzanchi, L. Dessbesell, M. B. Ray and C. C. Xu, ChemSusChem, 2018, 11, 3323–3334 CrossRef CAS.
- A. B. Gawade, A. V. Nakhate and G. D. Yadav, Catal. Today, 2018, 309, 119–125 CrossRef CAS.
- R. Chen, J. Xin, D. Yan, H. Dong, X. Lu and S. Zhang, ChemSusChem, 2019, 12, 2715–2724 CrossRef CAS PubMed.
- A. Danielli da Fonseca Ferreira, M. Dorneles de Mello and M. A. P. da Silva, Ind. Eng. Chem. Res., 2019, 58, 128–137 CrossRef CAS.
- F. Liguori, P. Barbaro and N. Calisi, ChemSusChem, 2019, 12, 2558–2563 CrossRef CAS PubMed.
- X. Wan, C. Zhou, J. Chen, W. Deng, Q. Zhang, Y. Yang and Y. Wang, ACS Catal., 2014, 4, 2175–2185 CrossRef CAS.
- N. K. Gupta, S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2011, 13, 824–827 RSC.
- L. Dessbesell, S. Souzanchi, K. T. Venkateswara Rao, A. A. Carrillo, D. Bekker, K. A. Hall, K. M. Lawrence, C. L. J. Tait and C. Xu, Biofuels, Bioprod. Biorefin., 2019, 13, 1234–1245 CrossRef CAS.
- S. Bello, P. Méndez-Trelles, E. Rodil, G. Feijoo and M. T. Moreira, Sep. Purif. Technol., 2020, 233, 116056 CrossRef CAS.
- A. Al Ghatta, J. D. E. T. Wilton-Ely and J. P. Hallett, ACS Sustainable Chem. Eng., 2019, 7, 16483–16492 CrossRef.
- B. Blank, A. Gordillo, M. A. Bohn, A. Kindler, S. A. Schunk, L. Zhang, C. Futter, R. Dehn, H. Werhan, J. H. Teles, M. Piepenbrink and R. Backes, WO2015197699A1, 2015.
- O. M. Musa, Handbook of Maleic Anhydride Based Materials, Springer International Publishing, Switzerland, 2016 Search PubMed.
- https://mcgroup.co.uk/news/20131213/global-maleic-anhydride-consumption-registered-35-yoy-increase.html .
- T. R. Felthouse, J. Burnett, B. Horrell, M. J. Mummey and Y.-J. Kuo, Kirk-Othmer Encyclopedia of Chemical Technology Online, 2001, pp. 1–58 Search PubMed.
- K. Lohbeck, H. Haferkorn, W. Fuhrmann and N. Fedtke, Ullmann's Encyclopedia of Industrial Chemistry, Weinheim, Germany, 2000, pp. 463–473 Search PubMed.
- A. Chieregato, J. V. Ochoa, C. Bandinelli, G. Fornasari, F. Cavani and M. Mella, ChemSusChem, 2015, 8, 377–388 CrossRef CAS PubMed.
- Q. Zhang, J. Dong, Y. Liu, Y. Wang and Y. Cao, J. Energy Chem., 2016, 25, 907–910 CrossRef.
- G. Pavarelli, J. Velasquez Ochoa, A. Caldarelli, F. Puzzo, F. Cavani and J. L. Dubois, ChemSusChem, 2015, 8, 2250–2259 CrossRef CAS PubMed.
- J. Q. Bond and A. Chatzidimitriou, US Pat., 2015/0191411A12015, 2015 Search PubMed.
- A. Chatzidimitriou and J. Q. Bond, Green Chem., 2015, 17, 4367–4376 RSC.
- J. Q. Bond and A. Chatzidimitriou, US Pat., 2018/0354883A1, 2018 Search PubMed.
- R. Zhu, A. Chatzidimitriou and J. Q. Bond, J. Catal., 2018, 359, 171–183 CrossRef CAS.
- X. Li and Y. Zhang, Green Chem., 2016, 18, 643–647 RSC.
- J. Lan, J. Lin, Z. Chen and G. Yin, ACS Catal., 2015, 5, 2035–2041 CrossRef CAS.
- G. Lv, S. Chen, H. Zhu, M. Li and Y. Yang, J. Clean. Prod., 2018, 196, 32–41 CrossRef CAS.
- G. Lv, C. Chen, B. Lu, J. Li, Y. Yang, C. Chen, T. Deng, Y. Zhu and X. Hou, RSC Adv., 2016, 6, 101277 RSC.
- Z. Du, J. Ma, F. Wang, J. Liu and J. Xu, Green Chem., 2011, 13, 554–557 RSC.
- A. Tirsoaga, M. El Fergani, V. I. Parvulescu and S. M. Coman, ACS Sustainable Chem. Eng., 2018, 6, 14292–14301 CrossRef CAS.
- S. Kumar, Furfural market size, growth to 2020, https://www.researchgate.net/publication/275800434_Furfural_Market_Size_Growth_To_2020.
- D. M. Alonso, S. H. Hakim, S. Zhou, W. Won, O. Hosseinaei, J. Tao, V. Garcia-Negron, A. H. Motagamwala, M. A. Mellmer, K. Huang, C. J. Houtman, N. Labbé, D. P. Harper, C. T. Maravelias, T. Runge and J. A. Dumesic, Sci. Adv., 2017, 3, e1603301 CrossRef PubMed.
- H. E. Hoydonckx, W. M. van Rhijn, W. van Rhijn, D. E. de Vos and P. A. Jacobs, Ullmann's Enycopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012, vol. 16, pp. 285–313 Search PubMed.
- H. Guo and G. Yin, J. Phys. Chem. C, 2011, 115, 17516–17522 CrossRef CAS.
- S. Shi, H. Guo and G. Yin, Catal. Commun., 2011, 12, 731–733 CrossRef CAS.
- J. Lan, Z. Chen, J. Lin and G. Yin, Green Chem., 2014, 16, 4351–4358 RSC.
- X. Li, B. Ho and Y. Zhang, Green Chem., 2016, 18, 2976–2980 RSC.
- M. Gál, T. Soták, J. Škriniarová, M. Hronec and E. Dobročka, Catal. Lett., 2017, 147, 2714–2723 CrossRef.
- Y. Huang, C. Wu, W. Yuan, Y. Xia, X. Liu, H. Yang and H. Wang, J. Chin. Chem. Soc., 2017, 64, 786–794 CrossRef CAS.
- Y. Xie, Y. Huang, C. Wu, W. Yuan, Y. Xia, X. Liu and H. Wang, Mol. Catal., 2018, 452, 20–27 CrossRef CAS.
- N. A. Milas and W. L. Walsh, J. Am. Chem. Soc., 1935, 57, 1389–1393 CrossRef CAS.
- W. V. Nielsen, Ind. Eng. Chem., 1949, 41, 365–368 CrossRef.
- D. R. Kreile, V. A. Slavinskaya, M. V. Shimanskaya and E. Y. Lukevits, Chem. Heterocycl. Compd., 1969, 5, 429–430 CrossRef.
- V. A. Slavinskaya, D. R. Kreile, E. E. Dzilyuma and D. E. Sile, Chem. Heterocycl. Compd, 1977, 7, 881–894 Search PubMed.
- N. Alonso-Fagúndez, M. L. Granados, R. Mariscal and M. Ojeda, ChemSusChem, 2012, 5, 1984–1990 CrossRef PubMed.
- N. Alonso-Fagúndez, M. Ojeda, R. Mariscal, J. L. G. Fierro and M. López Granados, J. Catal., 2017, 348, 265–275 CrossRef.
- X. Li, J. Ko and Y. Zhang, ChemSusChem, 2018, 11, 612–618 CrossRef CAS.
- K. Rajamani, P. Subramanian and M. S. Murthy, Ind. Eng. Chem. Process Des. Dev., 1976, 15, 232–234 CrossRef CAS.
- M. S. Murthy and K. Rajamani, Chem. Eng. Sci., 1974, 29, 601–609 CrossRef CAS.
- K. J. Zeitsch, The Chemistry and Technology of Furfural and its Many By-Products (Sugar Series vol. 13), Elsevier Science B.V., 2000 Search PubMed.
- I. Agirre, I. Gandarias, P. L. Arias and M. López Granados, Biomass Convers. Biorefin., 2019 DOI:10.1007/s13399-019-00462-w.
- M. L. Granados, Furfural: An Entry Point of Lignocellulose in Biorefineries to Produce Renewable Chemicals, Polymers, and Biofuels, 2018, pp. 239–261 Search PubMed.
- V. V. Poskonin and L. A. Badovskaya, Chem. Heterocycl. Compd., 2003, 39, 594–597 CrossRef CAS.
- X. Li, B. Ho, D. S. W. Lim and Y. Zhang, Green Chem., 2017, 19, 914–918 RSC.
- N. Araji, D. D. Madjinza, G. Chatel, A. Moores, F. Jérôme and K. De Oliveira Vigier, Green Chem., 2017, 19, 98–101 RSC.
- H. Choudhary, S. Nishimura and K. Ebitani, Chem. Lett., 2012, 41, 409–411 CrossRef CAS.
- A. Takagaki, S. Nishimura and K. Ebitani, Catal. Surv. Asia, 2012, 16, 164–182 CrossRef CAS.
- Y. Rodenas, J. L. G. Fierro, R. Mariscal, M. Retuerto and M. López Granados, Top. Catal., 2019, 62, 560–569 CrossRef CAS.
- A. C. Alba-Rubio, J. L. G. Fierro, L. León-Reina, R. Mariscal, J. A. Dumesic and M. López Granados, Appl. Catal., B, 2017, 202, 269–280 CrossRef CAS.
- N. Alonso-Fagúndez, I. Agirrezabal-Telleria, P. L. Arias, J. L. G. Fierro, R. Mariscal and M. L. Granados, RSC Adv., 2014, 4, 54960–54972 RSC.
- Y. Rodenas, R. Mariscal, J. L. G. Fierro, D. Martín-Alonso, J. A. Dumesic and M. López Granados, Green Chem., 2018, 20, 2845–2856 RSC.
- V. L. Mil’man and G. M. Sycheva, Sov. Electrochem., 1987, 1539–1540 Search PubMed.
- V. L. Mil’man and G. M. Sycheva, Sov. Electrochem., 1986, 21, 1133–1134 Search PubMed.
- S. R. Kubota and K. S. Choi, ACS Sustainable Chem. Eng., 2018, 6, 9596–9600 CrossRef CAS.
- A. M. Román, J. C. Hasse, J. W. Medlin and A. Holewinski, ACS Catal., 2019, 9, 10305–10316 CrossRef.
- A. Carlson, B. Coggio, K. Lau, C. Mercogliano and J. Millis, in Chemicals and Fuels from Bio-Based Building Blocks, ed. F. Cavani, S. Albonetti, F. Basile and A. Gandini, Wiley-VCH Verlag, 2016, pp. 173–190 Search PubMed.
- A. Cukalovic and C. V. Stevens, Biofuels, Bioprod. Biorefin., 2008, 2, 505–529 CrossRef CAS.
- M. J. Biddy, C. Scarlata and C. Kinchin, Chemicals from biomass: A market assessment of bioproducts with near-term potential. Technical Report NREL/TP-5100-65509, 2014, vol. 20.
- BioConSepT Market potential-for selected platform-chemicals report1, https://www.igb.fraunhofer.de/content/dam/igb/en/documents/publications/BioConSepT_Market-potential-for-selected-platform-chemicals_ppt1.pdf, 2011.
- From the sugar platform to biofuels and biochemicals. Final report for the European Commission Directorate-General Energy (ENER/C2/423-2012/SI2.673791), 2015.
- A. Takagaki, S. Nishimura and K. Ebitani, Catal. Surv. Asia, 2012, 16, 164–182 CrossRef CAS.
- H. Choudhary, S. Nishimura and K. Ebitani, Appl. Catal., A, 2013, 458, 55–62 CrossRef CAS.
- F. Saleem, P. Müller, K. Eränen, J. Warnå, D. Yu Murzin and T. Salmi, J. Chem. Technol. Biotechnol., 2017, 92, 2206–2220 CrossRef CAS.
- P. Maneechakr and S. Karnjanakom, Energy Convers. Manage., 2017, 154, 299–310 CrossRef CAS.
- W. Zhu, F. Tao, S. Chen, M. Li, Y. Yang and G. Lv, ACS Sustainable Chem. Eng., 2019, 7, 296–305 CrossRef CAS.
- M. Rezaei, A. Najafi Chermahini, H. A. Dabbagh, M. Saraji and A. Shahvar, J. Environ. Chem. Eng., 2019, 7, 102855 CrossRef.
- A. S. Amarasekara and N. C. Okorie, Catal. Commun., 2018, 108, 108–112 CrossRef CAS.
- S. Dutta, L. Wu and M. Mascal, Green Chem., 2015, 17, 2335–2338 RSC.
- B. Subramaniam, X. Zuo, D. H. Busch and P. Venkitasubramaniam, US2014/0343305A1, 2014.
- D. van Es, F. van der Klis and J. van Haveren, WO2012/044168A1, 2014.
- E. J. Molitor and B. D. Mullen, US Pat., 2016/0031788A1, 2016 Search PubMed.
- I. Podolean, B. Cojocaru, H. Garcia, C. Teodorescu, V. I. Parvulescu and S. M. Coman, Top. Catal., 2018, 61, 1866–1876 CrossRef CAS.
- I. Podolean, V. Kuncser, N. Gheorghe, D. MacOvei, V. I. Parvulescu and S. M. Coman, Green Chem., 2013, 15, 3077–3082 RSC.
- C. Rizescu, I. Podolean, B. Cojocaru, V. I. Parvulescu, S. M. Coman, J. Albero and H. Garcia, ChemCatChem, 2017, 9, 3314–3321 CrossRef CAS.
- C. Rizescu, I. Podolean, J. Albero, V. I. Parvulescu, S. M. Coman, C. Bucur, M. Puche and H. Garcia, Green Chem., 2017, 19, 1999–2005 RSC.
- I. Podolean, C. Rizescu, C. Bala, L. Rotariu, V. I. Parvulescu, S. M. Coman and H. Garcia, ChemSusChem, 2016, 9, 2307–2311 CrossRef CAS.
- Z. Zhang and G. W. Huber, Chem. Soc. Rev., 2018, 47, 1351–1390 RSC.
- Z. Zhang and K. Deng, ACS Catal., 2015, 5, 6529–6544 CrossRef CAS.
- A. P. Dunlop and S. Smith, US Pat., 2676186, 1954 Search PubMed.
- M. J. Gilkey and B. Xu, ACS Catal., 2016, 6, 1420–1436 CrossRef CAS.
- Z. Zhang and G. W. Huber, Chem. Soc. Rev., 2018, 47, 1351–1390 RSC.
- J. Albert, Faraday Discuss., 2017, 202, 99–109 RSC.
- M. López-Granados, J. Moreno, A. C. Alba-Rubio, J. Iglesias, D. Martin Alonso and R. Mariscal, Green Chem., 2020, 22, 1859–1872 RSC.
- R. Beerthuis, G. Rothenberg and N. R. Shiju, Green Chem., 2015, 17, 1341–1361 RSC.
- J. Han, Energy Convers. Manage., 2016, 129, 75–80 CrossRef CAS.
- A. Castellan, J. C. J. Bart and S. Cavallaro, Catal. Today, 1991, 9, 237–254 CrossRef CAS.
- A. Pellis, E. Herrero Acero, L. Gardossi, V. Ferrario and G. M. Guebitz, Polym. Int., 2016, 65, 861–871 CrossRef CAS.
- M. Moudjahed, L. Dermeche, S. Benadji, T. Mazari and C. Rabia, J. Mol. Catal. A: Chem., 2016, 414, 72–77 CrossRef CAS.
- J. Luo, Y. Huang, B. Ding, P. Wang, X. Geng, J. Zhang and Y. Wei, Catalysts, 2018, 8, 5–7 CrossRef.
- P. M. Burcke, EP0395038, 1990.
- J. D. Nobbs, N. Z. B. Zainal, J. Tan, E. Drent, L. P. Stubbs, C. Li, S. C. Y. Lim, D. G. A. Kumbang and M. van Meurs, ChemistrySelect, 2016, 1, 539–544 CrossRef CAS.
- P. K. Wong, C. Li, L. Stubbs, M. van Meurs, A. G. A. Kumbang, S. C. Y. Lim and E. Drent, US20150183703, 2015.
- O. Hamed, A. El-Qisairi and P. M. Henry, Tetrahedron Lett., 2000, 41, 3021–3024 CrossRef CAS.
- D. Wu, Z. Chen, Z. Jia and L. Shuai, Sci. China Chem., 2012, 55, 380–385 CrossRef CAS.
- G. S. Zhang, M. M. Zhu, Q. Zhang, Y. M. Liu, H. Y. He and Y. Cao, Green Chem., 2016, 18, 2155–2164 RSC.
- T. R. Boussie, E. L. Dias, Z. M. Fresco and V. J. Murphy, US20100317822, 2010.
- T. Asano, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustainable Chem. Eng., 2016, 4, 6253–6257 CrossRef CAS.
- L. Wei, J. Zhang, W. Deng, S. Xie, Q. Zhang and Y. Wang, Chem. Commun., 2019, 55, 8013–8016 RSC.
- T. R. Boussie, E. L. Dias, Z. M. Fresco, V. J. Murphy, J. Shoemaker, R. Archer and H. Jiang, WO2011109051, 2011.
- G. F. Salem, G. Zhu, A. G. Hagmeyer, A. G. Dias and L. E. Dias, WO2013173372, 2013.
- R. T. Larson, A. Samant, J. Chen, W. Lee, M. A. Bohn, D. M. Ohlmann, S. J. Zuend and F. D. Toste, J. Am. Chem. Soc., 2017, 139, 14001–14004 CrossRef CAS.
- A. Shimizu, K. Tanaka and M. Fujimori, Chemosphere: Global Change Sci., 2000, 2, 425–434 CrossRef CAS.
- S. Li, W. Deng, S. Wang, P. Wang, D. An, Y. Li, Q. Zhang and Y. Wang, ChemSusChem, 2018, 11, 1995–2028 CrossRef CAS PubMed.
- Z. Strassberger, P. Prinsen, F. Van Der Klis, D. S. Van Es, S. Tanase and G. Rothenberg, Green Chem., 2015, 17, 325–334 RSC.
- H. Liu, T. Jiang, B. Han, S. Liang and Y. Zhou, Science, 2009, 326, 1250–1252 CrossRef CAS PubMed.
- S. De, B. Saha and R. Luque, Bioresour. Technol., 2015, 178, 108–118 CrossRef CAS PubMed.
- S. Kang, J. Fu and G. Zhang, Renewable Sustainable Energy Rev., 2018, 94, 340–362 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein, M. A. Mellmer, E. I. Gurbuz and J. A. Dumesic, Energy Environ. Sci., 2013, 6, 76–80 RSC.
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023–11117 CrossRef CAS PubMed.
- M. Faber, US4400468A, 1983.
- Y. Nakagawa and K. Tomishige, Catal. Commun., 2010, 12, 154–156 CrossRef CAS.
- R. Alamillo, M. Tucker, M. Chia, Y. Pagán-Torres and J. Dumesic, Green Chem., 2012, 14, 1413–1419 RSC.
- T. Buntara, S. Noel, P. H. Phua, I. Melián-Cabrera, J. G. De Vries and H. J. Heeres, Angew. Chem., Int. Ed., 2011, 50, 7083–7087 CrossRef CAS.
- M. L. Ribeiro and U. Schuchardt, Catal. Commun., 2003, 4, 83–86 CrossRef CAS.
- M. J. Gilkey, A. V. Mironenko, D. G. Vlachos and B. Xu, ACS Catal., 2017, 7, 6619–6634 CrossRef CAS.
- J. L. Fortman, A. Hagen, L. Katz, J. Kealsing, S. Poust, J. Zhang and S. Zotchev, WO2012058686, 2012.
- C. Delhomme, D. Weuster-Botz and F. E. Kühn, Green Chem., 2009, 11, 13–26 RSC.
- A. G. F. Cavani, S. Albonetti and F. Basile, Chemicals and Fuels from Bio-Based Building Blocks, Wiley-VCH, Weinheim, 2016, vol. 1 and 2 Search PubMed.
- D. E. Kiely, L. Chen and T. H. Lin, J. Am. Chem. Soc., 1994, 116, 571–578 CrossRef CAS.
- H. Zhang, X. Li, X. Su, E. L. Ang, Y. Zhang and H. Zhao, ChemCatChem, 2016, 8, 1500–1506 CrossRef CAS.
- W. Niu, K. M. Draths and J. W. Frost, Biotechnol. Prog., 2002, 18, 201–211 CrossRef CAS.
- D. R. Vardon, M. A. Franden, C. W. Johnson, E. M. Karp, M. T. Guarnieri, J. G. Linger, M. J. Salm, T. J. Strathmann and G. T. Beckham, Energy Environ. Sci., 2015, 8, 617–628 RSC.
- D. R. Vardon, N. A. Rorrer, D. Salvachúa, A. E. Settle, C. W. Johnson, M. J. Menart, N. S. Cleveland, P. N. Ciesielski, K. X. Steirer, J. R. Dorgan and G. T. Beckham, Green Chem., 2016, 18, 3397–3413 RSC.
- S. Capelli, D. Motta, C. Evangelisti, N. Dimitratos, L. Prati, C. Pirola and A. Villa, ChemCatChem, 2019, 11, 3075–3084 CrossRef CAS.
- S. Capelli, A. Rosengart, A. Villa, A. Citterio, A. Di Michele, C. L. Bianchi, L. Prati and C. Pirola, Appl. Catal., B, 2017, 218, 220–229 CrossRef CAS.
- S. Scelfo, R. Pirone and N. Russo, Catal. Commun., 2016, 84, 98–102 CrossRef CAS.
- J. M. Thomas, R. Raja, B. F. G. Johnson, T. J. O’Connell, G. Sankar and T. Khimyak, Chem. Commun., 2003, 1126–1127 RSC.
- X. She, H. M. Brown, X. Zhang, B. K. Ahring and Y. Wang, ChemSusChem, 2011, 4, 1071–1073 CrossRef CAS PubMed.
- A. Corona, M. J. Biddy, D. R. Vardon, M. Birkved, M. Z. Hauschild and G. T. Beckham, Green Chem., 2018, 20, 3857–3866 RSC.
- J. P. Lange, Catal. Sci. Technol., 2016, 6, 4759–4767 RSC.
- W. Partenheimer, Catal. Today, 1995, 23, 69–158 CrossRef CAS.
- Purified Terephthalic Acid Price Trend, https://www.fibre2fashion.com/market-intelligence/textile-market-watch/purified-terephthalic-acid-pta-price-trends-industry-reports/1/?gcode=1, accessed 7 February 2020.
- R. D. Cortright and J. A. Dumesic, US5736478, 1998.
- R. D. Cortright and P. G. Blommel, US8362307, 2013.
- M. W. Peters, J. D. Taylor, M. M. Jenni, L. E. Manzer and D. E. Henton, US20110087000, 2011.
- A. Maneffa, P. Priecel and J. A. Lopez-Sanchez, ChemSusChem, 2016, 9, 2736–2748 CrossRef CAS PubMed.
- C. D. Chang, A. J. Silvestri and R. L. Smith, US3928483, 1975.
- W. W. Kaeding, US4088706, 1978.
- J. Zhang, W. Qian, C. Kong and F. Wei, ACS Catal., 2015, 5, 2982–2988 CrossRef CAS.
- N. S. Shamsul, S. K. Kamarudin, N. A. Rahman and N. T. Kofli, Renewable Sustainable Energy Rev., 2014, 33, 578–588 CrossRef CAS.
- A. E. Settle, L. Berstis, N. A. Rorrer, Y. Roman-Leshkóv, G. T. Beckham, R. M. Richards and D. R. Vardon, Green Chem., 2017, 19, 3468–3492 RSC.
- A. G. F. Cavani, S. Albonetti and F. Basile, Chemicals and Fuels from Bio-Based Building Blocks, 2016, vol. 2 Search PubMed.
- M. Braun and M. Antonietti, Green Chem., 2017, 19, 3813–3819 RSC.
- P. P. Upare, D. W. Hwang, Y. K. Hwang, U. H. Lee, D. Y. Hong and J. S. Chang, Green Chem., 2015, 17, 3310–3313 RSC.
- M. Shiramizu and F. D. Toste, Chem. – Eur. J., 2011, 17, 12452–12457 CrossRef CAS PubMed.
- L. Ni, J. Xin, H. Dong, X. Lu, X. Liu and S. Zhang, ChemSusChem, 2017, 10, 2394–2401 CrossRef CAS PubMed.
- L. Ni, J. Xin, K. Jiang, L. Chen, D. Yan, X. Lu and S. Zhang, ACS Sustainable Chem. Eng., 2018, 6, 2541–2551 CrossRef CAS.
- T. W. Lyons, D. Guironnet, M. Findlater and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 15708–15711 CrossRef CAS PubMed.
- M. Brookhart, M. Findlater, D. Guironnet and T. W. Lyons, US20130237732, 2013.
- C. L. Williams, C. C. Chang, P. Do, N. Nikbin, S. Caratzoulas, D. G. Vlachos, R. F. Lobo, W. Fan and P. J. Dauenhauer, ACS Catal., 2012, 2, 935–939 CrossRef CAS.
- R. Xiong, S. I. Sandler, D. G. Vlachos and P. J. Dauenhauer, Green Chem., 2014, 16, 4086–4091 RSC.
- R. E. Patet, N. Nikbin, C. L. Williams, S. K. Green, C. C. Chang, W. Fan, S. Caratzoulas, P. J. Dauenhauer and D. G. Vlachos, ACS Catal., 2015, 5, 2367–2375 CrossRef CAS.
- C. L. Williams, K. P. Vinter, R. E. Patet, C. C. Chang, N. Nikbin, S. Feng, M. R. Wiatrowski, S. Caratzoulas, W. Fan, D. G. Vlachos and P. J. Dauenhauer, ACS Catal., 2016, 6, 2076–2088 CrossRef CAS.
- D. Wang, C. M. Osmundsen, E. Taarning and J. A. Dumesic, ChemCatChem, 2013, 5, 2044–2050 CrossRef CAS.
- R. E. Patet, M. Koehle, R. F. Lobo, S. Caratzoulas and D. G. Vlachos, J. Phys. Chem. C, 2017, 121, 13666–13679 CrossRef CAS.
- T. W. Kim, S. Y. Kim, J. C. Kim, Y. Kim, R. Ryoo and C. U. Kim, Appl. Catal., B, 2016, 185, 100–109 CrossRef CAS.
- C. C. Chang, H. Je Cho, J. Yu, R. J. Gorte, J. Gulbinski, P. Dauenhauer and W. Fan, Green Chem., 2016, 18, 1368–1376 RSC.
- J. C. Kim, T. W. Kim, Y. Kim, R. Ryoo, S. Y. Jeong and C. U. Kim, Appl. Catal., B, 2017, 206, 490–500 CrossRef CAS.
- X. Feng, C. Shen, K. Ji, J. Yin and T. Tan, Catal. Sci. Technol., 2017, 7, 5540–5549 RSC.
- X. Feng, C. Shen, C. Tian and T. Tan, Ind. Eng. Chem. Res., 2017, 56, 5852–5859 CrossRef CAS.
- H. J. Cho, L. Ren, V. Vattipalli, Y. H. Yeh, N. Gould, B. Xu, R. J. Gorte, R. Lobo, P. J. Dauenhauer, M. Tsapatsis and W. Fan, ChemCatChem, 2017, 9, 398–402 CrossRef CAS.
- Y. P. Wijaya, D. J. Suh and J. Jae, Catal. Commun., 2015, 70, 12–16 CrossRef CAS.
- Y. P. Wijaya, H. P. Winoto, Y. K. Park, D. J. Suh, H. Lee, J. M. Ha and J. Jae, Catal. Today, 2017, 293–294, 167–175 CrossRef CAS.
- J. Yin, C. Shen, X. Feng, K. Ji and L. Du, ACS Sustainable Chem. Eng., 2018, 6, 1891–1899 CrossRef CAS.
- R. Y. Rohling, E. Uslamin, B. Zijlstra, I. C. Tranca, I. A. W. Filot, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2018, 8, 760–769 CrossRef CAS PubMed.
- J. McGlone, P. Priecel, L. Da Vià, L. Majdal and J. A. Lopez-Sanchez, Catalysts, 2018, 8, 253 CrossRef.
- R. Zhao, L. Xu, S. Huang and W. Zhang, Catal. Sci. Technol., 2019, 9, 5676–5685 RSC.
- I. F. Teixeira, B. T. W. Lo, P. Kostetskyy, M. Stamatakis, L. Ye, C. C. Tang, G. Mpourmpakis and S. C. E. Tsang, Angew. Chem., Int. Ed., 2016, 55, 13061–13066 CrossRef CAS PubMed.
- J. Pang, M. Zheng, R. Sun, A. Wang, X. Wang and T. Zhang, Green Chem., 2016, 18, 342–359 RSC.
- B. M. Stadler, C. Wulf, T. Werner, S. Tin and J. G. de Vries, ACS Catal., 2019, 9, 8012–8067 CrossRef CAS.
- D. I. Collias, A. M. Harris, V. Nagpal, I. W. Cottrell and M. W. Schultheis, Ind. Biotechnol., 2014, 10, 91–105 CrossRef CAS.
- A. Murray and S. Ryba, US20150337214, 2015.
- G. W. Huber, Y.-T. Cheng, Z. Wang and W. Fan, US20130324772, 2013.
- G. W. Huber, Y.-T. Cheng, T. Carlson, T. Vispute, J. Jae and G. Tompsett, US8864984, 2014.
- C. W. Johnson, D. Salvachúa, P. Khanna, H. Smith, D. J. Peterson and G. T. Beckham, Metab. Eng. Commun., 2016, 3, 111–119 CrossRef PubMed.
- J. M. Carraher, T. Pfennig, R. G. Rao, B. H. Shanks and J. P. Tessonnier, Green Chem., 2017, 19, 3042–3050 RSC.
- J. W. Frost, A. Miermont, D. Schweitzer, V. Bui, E. Paschke and D. A. Wicks, US20110288263, 2011.
- R. Lu, F. Lu, J. Chen, W. Yu, Q. Huang, J. Zhang and J. Xu, Angew. Chem., Int. Ed., 2016, 55, 249–253 CrossRef CAS PubMed.
- K. K. Miller, P. Zhang, Y. Nishizawa-Brennen and J. W. Frost, ACS Sustainable Chem. Eng., 2014, 2, 2053–2056 CrossRef CAS.
- G. A. Kraus, S. Riley and T. Cordes, Green Chem., 2011, 13, 2734–2736 RSC.
- T. Pfennig, A. Chemburkar, S. Cakolli, M. Neurock and B. H. Shanks, ACS Sustainable Chem. Eng., 2018, 6, 12855–12864 CrossRef CAS.
- S. Bérard, C. Vallée and D. Delcroix, Ind. Eng. Chem. Res., 2015, 54, 7164–7168 CrossRef.
- M. B. Banella, C. Gioia, M. Vannini, M. Colonna, A. Celli and A. Gandini, ChemSusChem, 2016, 9, 942–945 CrossRef CAS PubMed.
- P. Zhang, V. Nguyen and J. W. Frost, ACS Sustainable Chem. Eng., 2016, 4, 5998–6001 CrossRef CAS.
- M. Prussi, M. Padella, M. Conton, E. D. Postma and L. Lonza, J. Clean. Prod., 2019, 222, 565–572 CrossRef CAS PubMed.
- P. Lejemble, A. Gaset and P. Kalck, Biomass, 1984, 4, 263–274 CrossRef CAS.
- Y. Tachibana, S. Kimura and K. Kasuya, Sci. Rep., 2015, 5, 8249 CrossRef PubMed.
- J. J. Pacheco and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8363–8367 CrossRef CAS PubMed.
- J. J. Pacheco, J. A. Labinger, A. L. Sessions and M. E. Davis, ACS Catal., 2015, 5, 5904–5913 CrossRef CAS.
- M. Sajid, X. Zhao and D. Liu, Green Chem., 2018, 20, 5427–5453 RSC.
- M. Colonna, C. Berti, M. Fiorini, E. Binassi, M. Mazzacurati, M. Vannini and S. Karanam, Green Chem., 2011, 13, 2543–2548 RSC.
- F. Neaţu, G. Culică, M. Florea, V. I. Parvulescu and F. Cavani, ChemSusChem, 2016, 9, 3102–3112 CrossRef PubMed.
- S. Song, J. Zhang, G. Gözaydın and N. Yan, Angew. Chem., Int. Ed., 2019, 58, 4934–4937 CrossRef CAS PubMed.
- M. Volanti, D. Cespi, F. Passarini, E. Neri, F. Cavani, P. Mizsey and D. Fozer, Green Chem., 2019, 21, 885–896 RSC.
- B. P. Lawson, E. Golikova, A. M. Sulman, B. D. Stein, D. G. Morgan, N. V. Lakina, A. Y. Karpenkov, E. M. Sulman, V. G. Matveeva and L. M. Bronstein, ACS Sustainable Chem. Eng., 2018, 6, 9845–9853 CrossRef CAS.
- S. Ramachandran, S. Nair, C. Larroche and A. Pandey, Current Developments in Biotechnology and Bioengineering: Production, Isolation and Purification of Industrial Products, 2016 Search PubMed.
- F. W. Lichtenthaler, Acc. Chem. Res., 2002, 35, 728–737 CrossRef CAS.
- S. Anastassiadis and I. Morgunov, Recent Pat. Biotechnol., 2007, 1, 167–180 CrossRef CAS.
- A. L. Harreus, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2002 Search PubMed.
- D. An, A. Ye, W. Deng, Q. Zhang and Y. Wang, Chem. – Eur. J., 2012, 18, 2938–2947 CrossRef CAS.
- P. N. Amaniampong, X. Jia, B. Wang, S. H. Mushrif, A. Borgna and Y. Yang, Catal. Sci. Technol., 2015, 5, 2393–2405 RSC.
- M. Hernández, E. Lima, A. Guzmán, M. Vera, O. Novelo and V. Lara, Appl. Catal., B, 2014, 144, 528–537 CrossRef.
- S. Biella, L. Prati and M. Rossi, J. Catal., 2002, 206, 242–247 CrossRef CAS.
- A. Abbadi and H. van Bekkum, J. Mol. Catal., 1995, 97, 111–118 CrossRef CAS.
- A. Tathod, T. Kane, E. S. Sanil and P. L. Dhepe, J. Mol. Catal. A: Chem., 2014, 388–389, 90–99 CrossRef CAS.
- A. Liu, Z. Huang and X. Wang, Catal. Lett., 2018, 148, 2019–2029 CrossRef CAS.
- I. V. Delidovich, O. P. Taran, L. G. Matvienko, A. N. Simonov, I. L. Simakova, A. N. Bobrovskaya and V. N. Parmon, Catal. Lett., 2010, 140, 14–21 CrossRef CAS.
- T. Haynes, V. Dubois and S. Hermans, Appl. Catal., A, 2017, 542, 47–54 CrossRef CAS.
- I. Nikov and K. Paev, Catal. Today, 1995, 24, 41–47 CrossRef CAS.
- S. Karski, J. Mol. Catal. A: Chem., 2003, 191, 87–92 CrossRef CAS.
- M. Wenkin, M. Devillers, P. Ruiz and B. Delmon, Stud. Surf. Sci. Catal., 2001, 139, 295–302 CrossRef CAS.
- M. Wenkin, R. Touillaux, P. Ruiz, B. Delmon and M. Devillers, Appl. Catal., A, 1996, 148, 181–199 CrossRef CAS.
- S. Karski, J. Mol. Catal. A: Chem., 2006, 253, 147–154 CrossRef CAS.
- M. Besson, F. Lahmer, P. Gallezot, P. Fuertes and G. Flèche, J. Catal., 1995, 152, 116–121 CrossRef CAS.
- M. Wenkin, C. Renard, P. Ruiz, B. Delmon and M. Devillers, Stud. Surf. Sci. Catal., 1997, 108, 391–398 CrossRef CAS.
- M. Wenkin, P. Ruiz, B. Delmon and M. Devillers, J. Mol. Catal. A: Chem., 2002, 180, 141–159 CrossRef CAS.
- L. Prati and M. Rossi, Stud. Surf. Sci. Catal., 1997, 110, 509–516 CrossRef CAS.
- M. Comotti, C. Della Pina, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812–5815 CrossRef CAS PubMed.
- P. Beltrame, M. Comotti, C. Della Pina and M. Rossi, Appl. Catal., A, 2006, 297, 1–7 CrossRef CAS.
- T. Ishida, H. Watanabe, T. Bebeko, T. Akita and M. Haruta, Appl. Catal., A, 2010, 377, 42–46 CrossRef CAS.
- T. Ishida, N. Kinoshita, H. Okatsu, T. Akita, T. Takei and M. Haruta, Angew. Chem., Int. Ed., 2008, 47, 9265–9268 CrossRef CAS PubMed.
- C. Liu, J. Zhang, J. Huang, C. Zhang, F. Hong, Y. Zhou, G. Li and M. Haruta, ChemSusChem, 2017, 10, 1976–1980 CrossRef CAS PubMed.
- J. Zhang, Z. Li, J. Huang, C. Liu, F. Hong, K. Zheng and G. Li, Nanoscale, 2017, 9, 16879–16886 RSC.
- H. Okatsu, N. Kinoshita, T. Akita, T. Ishida and M. Haruta, Appl. Catal., A, 2009, 369, 8–14 CrossRef CAS.
- T. Ishida, S. Okamoto, R. Makiyama and M. Haruta, Appl. Catal., A, 2009, 353, 243–248 CrossRef CAS.
- P. Qi, S. Chen, J. Chen, J. Zheng, X. Zheng and Y. Yuan, ACS Catal., 2015, 5, 2659–2670 CrossRef CAS.
- A. Mirescu, H. Berndt, A. Martin and U. Prüße, Appl. Catal., A, 2007, 317, 204–209 CrossRef CAS.
- M. Signoretto, F. Menegazzo, A. Di Michele and E. Fioriniello, Catalysts, 2016, 6, 87 CrossRef.
- C. Megías-Sayago, S. Ivanova, C. López-Cartes, M. A. Centeno and J. A. Odriozola, Catal. Today, 2017, 279, 148–154 CrossRef.
- P. J. Miedziak, H. Alshammari, S. A. Kondrat, T. J. Clarke, T. E. Davies, M. Morad, D. J. Morgan, D. J. Willock, D. W. Knight, S. H. Taylor and G. J. Hutchings, Green Chem., 2014, 16, 3132–3141 RSC.
- N. Toshima and H. Zhang, Macromol. Symp., 2012, 317–318, 149–159 CrossRef CAS.
- H. Zhang and N. Toshima, J. Colloid Interface Sci., 2013, 394, 166–176 CrossRef CAS PubMed.
- H. Zhang, K. Kawashima, M. Okumura and N. Toshima, J. Mater. Chem. A, 2014, 2, 13498–13508 RSC.
- H. Zhang and N. Toshima, Appl. Catal., A, 2011, 400, 9–13 CrossRef CAS.
- C. Baatz and U. Prüße, J. Catal., 2007, 249, 34–40 CrossRef CAS.
- C. Baatz, N. Thielecke and U. Prüße, Appl. Catal., B, 2007, 70, 653–660 CrossRef CAS.
- N. Thielecke, M. Aytemir and U. Prüsse, Catal. Today, 2007, 121, 115–120 CrossRef CAS.
- J. Zhang, X. Liu, M. N. Hedhili, Y. Zhu and Y. Han, ChemCatChem, 2011, 3, 1294–1298 CrossRef CAS.
- P. N. Amaniampong, K. Li, X. Jia, B. Wang, A. Borgna and Y. Yang, ChemCatChem, 2014, 6, 2105–2114 CrossRef CAS.
- K. Morawa Eblagon, M. F. R. Pereira and J. L. Figueiredo, Appl. Catal., B, 2016, 184, 381–396 CrossRef CAS.
- P. N. Amaniampong, A. Y. Booshehri, X. Jia, Y. Dai, B. Wang, S. H. Mushrif, A. Borgna and Y. Yang, Appl. Catal., A, 2015, 505, 16–27 CrossRef CAS.
- Glucaric Acid Market – Global Industry Analysis, Size, Share, Growth, Trends, and Forecast 2017–2025, https://www.transparencymarketresearch.com/glucaric-acid-market.html, accessed 26 January 2020.
- Y. Wu, Y. Enomoto, H. Masaki and T. Iwata, J. Appl. Polym. Sci., 2019, 136, 47255 CrossRef.
- T. N. Smith, K. Hash, C.-L. Davey, H. Mills, H. Williams and D. E. Kiely, Carbohydr. Res., 2012, 350, 6–13 CrossRef CAS PubMed.
- J. M. H. Dirkx, H. S. Van der Baan and J. M. A. J. J. Van den Broek, Carbohydr. Res., 1977, 59, 63–72 CrossRef CAS.
- J. Lee, B. Saha and D. G. Vlachos, Green Chem., 2016, 18, 3815–3822 RSC.
- M. Khawaji, Y. Zhang, M. Loh, I. Graça, E. Ware and D. Chadwick, Appl. Catal., B, 2019, 256, 117799 CrossRef CAS.
- X. Jin, M. Zhao, J. Shen, W. Yan, L. He, P. S. Thapa, S. Ren, B. Subramaniam and R. V. Chaudhari, J. Catal., 2015, 330, 323–329 CrossRef CAS.
- X. Jin, M. Zhao, M. Vora, J. Shen, C. Zeng, W. Yan, P. S. Thapa, B. Subramaniam and R. V. Chaudhari, Ind. Eng. Chem. Res., 2016, 55, 2932–2945 CrossRef CAS.
- S. Solmi, C. Morreale, F. Ospitali, S. Agnoli and F. Cavani, ChemCatChem, 2017, 9, 2797–2806 CrossRef CAS.
- S. Donen and K. Jensen, US0275622A1, 2014.
- V. J. Murphy, J. Shoemaker, G. Zhu, G. F. Salem and E. L. Dias, US0306790A1, 2011.
- M. Besson, G. Flèche, P. Fuertes, P. Gallezot and F. Lahmer, Recl. des Trav. Chim. des Pays-Bas, 1996, 115, 217 CrossRef CAS.
- R. Chen, C. Yang, Q. Zhang, B. Zhang and K. Deng, J. Catal., 2019, 374, 297–305 CrossRef CAS.
- W. J. Liu, Z. Xu, D. Zhao, X. Q. Pan, H. C. Li, X. Hu, Z. Y. Fan, W. K. Wang, G. H. Zhao, S. Jin, G. W. Huber and H. Q. Yu, Nat. Commun., 2020, 11, 265 CrossRef CAS PubMed.
- L. Zhao, X. Kuang, X. Sun, Y. Zhang and Q. Weiz, J. Electrochem. Soc., 2019, 166, H534–H540 CrossRef CAS.
- G. Moggia, T. Kenis, N. Daems and T. Breugelmans, ChemElectroChem, 2020, 7, 86–95 CrossRef CAS.
- E. Derrien, P. Marion, C. Pinel and M. Besson, Org. Process Res. Dev., 2016, 20, 1265–1275 CrossRef CAS.
- E. Derrien, M. Mounguengui-Diallo, N. Perret, P. Marion, C. Pinel and M. Besson, Ind. Eng. Chem. Res., 2017, 56, 13175–13189 CrossRef CAS.
- V. B. Thaore, R. D. Armstrong, G. J. Hutchings, D. W. Knight, D. Chadwick and N. Shah, Chem. Eng. Res. Des., 2020, 153, 337–349 CrossRef CAS.
- K. K. Kapanji, K. F. Haigh and J. F. Görgens, Bioresour. Technol., 2019, 289, 121635 CrossRef CAS PubMed.
- R. Gérardy, R. Morodo, J. Estager, P. Luis, D. P. Debecker and J. C. M. Monbaliu, Top. Curr. Chem., 2019, 377, 1–35 CrossRef PubMed.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS PubMed.
- E. I. Gürbüz, D. M. Alonso, J. Q. Bond and J. A. Dumesic, ChemSusChem, 2011, 4, 357–361 CrossRef PubMed.
- W. Schutyser, T. Renders, S. Van Den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- C. M. Lam, I. K. M. Yu, S. C. Hsu and D. C. W. Tsang, J. Clean. Prod., 2018, 199, 840–848 CrossRef CAS.
- R. Aryapratama and M. Janssen, J. Clean. Prod., 2017, 164, 434–443 CrossRef CAS.
- J. Moreno, J. Iglesias, J. Blanco, M. Montero, G. Morales and J. A. Melero, J. Clean. Prod., 2020, 250, 119568 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |