
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect observation of the electronic states of photoexcited hematite with ultrafast 2p3d X-ray absorption spectroscopy and resonant inelastic X-ray scattering†
Ahmed S. M.
Ismail‡
 a,
Yohei
Uemura‡
ab,
Sang Han
Park‡
c,
Soonnam
Kwon
c,
Minseok
Kim
c,
Hebatalla
Elnaggar
a,
Federica
Frati
a,
Yasuhiro
Niwa
d,
Hiroki
Wadati
ef,
Yasuyuki
Hirata
e,
Yujun
Zhang
e,
Kohei
Yamagami
e,
Susumu
Yamamoto
e,
Iwao
Matsuda
e,
Ufuk
Halisdemir
g,
Gertjan
Koster
g,
Bert M.
Weckhuysen
a and
Frank M. F.
de Groot
*a
a,
Yohei
Uemura‡
ab,
Sang Han
Park‡
c,
Soonnam
Kwon
c,
Minseok
Kim
c,
Hebatalla
Elnaggar
a,
Federica
Frati
a,
Yasuhiro
Niwa
d,
Hiroki
Wadati
ef,
Yasuyuki
Hirata
e,
Yujun
Zhang
e,
Kohei
Yamagami
e,
Susumu
Yamamoto
e,
Iwao
Matsuda
e,
Ufuk
Halisdemir
g,
Gertjan
Koster
g,
Bert M.
Weckhuysen
a and
Frank M. F.
de Groot
*a
aInorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitslaan 99, 3584 CG Utrecht, The Netherlands. E-mail: f.m.f.degroot@uu.nl
bInstitute for Catalysis, Hokkaido University, Kita 21 Nishi 10, Kita-ku, Sapporo, Hokkaido 001-0021, Japan
cPAL-XFEL, Pohang Accelerator Laboratory, 77 Cheongam-Ro, Nam-Gu, Pohang, Gyeongbuk 37673, South Korea
dPhoton Factory, Institute for Materials Structure Science, KEK, Tsukuba 305-0801, Japan
eInstitute for Solid State Physics, University of Tokyo, Kashiwa, Chiba 277-8581, Japan
fGraduate School of Material Science, University of Hyogo, Kamigori, Hyogo 678-1297, Japan
gFaculty of Science and Technology and MESA + Institute for Nanotechnology, University of Twente, P.O. Box 2171, 7500 AE Enschede, The Netherlands
First published on 16th October 2019
Abstract
Hematite, α-Fe2O3, is an important semiconductor for photoelectrochemical water splitting. Its low charge carrier mobility and the presence of midgap states provide favourable conditions for electron–hole recombination, hence affecting the semiconductor's photoelectrochemical efficiency. The nature of the excited state and charge carrier transport in hematite is strongly debated. In order to further understand the fundamental properties of the hematite photoexcited state, we conducted femtosecond 2p (L3) X-ray absorption (XAS) and 2p3d resonant inelastic scattering (RIXS) measurements on hematite thin-films at the Pohang Accelerator Laboratory X-ray Free Electron Laser (PAL-XFEL). The observed spectral changes and kinetic processes are in agreement with previous 3p XAS reports. The potential additional information that could be acquired from 2p3d RIXS experiments is also discussed.
Introduction
X-ray absorption spectroscopy (XAS) combined with the pump–probe method has become a powerful technique to investigate electronic states of photocatalytic materials.1–4 XAS provides information on the element specific electronic structure of the material under investigation. In addition, XAS can be measured under operando conditions.5,6 In combination with calculations, XAS enables the determination of the detailed local structure and electronic states of materials.7,8 There have been pioneering studies to investigate photoexcited states of metal oxides with the pump–probe XAS methodology.7,9–16 While synchrotron pump probe XAS experiments are limited by the pulse duration of X-rays (∼100 ps) or by the reachable flux for slicing experiments, the birth of X-ray free electrons lasers (XFEL) has expanded the horizon of the time resolved studies based on the pump–probe XAS methodology.1,9,11,17–20 XFELs can provide ultrashort and intense X-ray pulses down to 10 fs. The number of photons in an X-ray pulse from XFELs are at least millions of times larger than that of an X-ray pulse in a synchrotron. Such short pulse duration has enabled the study of femtosecond dynamics, which have also been achieved with femtosecond optical laser spectroscopies. In addition to the capability of tracking ultrafast dynamics, such high intense X-ray pulses enable sophisticated photon-in/photon-out spectroscopic measurements such as X-ray emission spectroscopy (XES)21–23 and resonant inelastic X-ray scattering (RIXS)24–30 with a very short temporal resolution. RIXS spectra provide additional opportunities to observe electronic excitations in materials, which would be impossible only with optical spectroscopies. At PAL-XFEL,31,32 one can measure the L3 XAS of 3d transition metal elements with a time resolution of less than 300 fs.33 In addition, 1 eV resolved 2p3d RIXS34–36 can also be measured, which extends the information provided on the electronic states of 3d metals. Here, we report the first time resolved L3 XAS and 2p3d RIXS experiments in PAL-XFEL to observe the photoexcited state of hematite (α-Fe2O3).Hematite is intensely studied as a semiconductor for the solar-assisted water splitting processes. This is mainly due to its rich abundance in the earth crust and to its optical bandgap of ∼2 eV that enables it to absorb light in the visible wavelength range of the solar spectrum.37,38 However, the intrinsic electronic properties of hematite make it difficult for the semiconductor to carry out the water splitting reaction directly, since its hole diffusion length is so small, 2–4 nm, that the electrons and holes can easily recombine in short time scales.39,40 Therefore, ultrafast spectroscopic investigations of the nature of the hematite photoexcited states have gained increasing interest due to its relevance to its water splitting efficiency.12,38,41–43 For example, Vura-Weiss et al. conducted the first femtosecond extreme-ultra violet (XUV) spectroscopic studies on hematite thin films and reported that the observed photoinduced Fe(II) states originate mainly from ligand to metal charge transfer (LMCT) i.e. the charge transfer from oxygen to iron, and not from the optical d–d excitation.44 Further theoretical and experimental XUV spectroscopic reports suggested that an additional reason for the low hematite performance is the formation of small polarons that localize the minority carriers at recombination centres.45,46
Our time resolved studies of 2p X-ray absorption spectroscopy (XAS) and 2p3d resonant inelastic X-ray scattering (RIXS) of iron elucidate the electronic structure of the photoexcited hematite. The results also demonstrate the potential of 2p3d RIXS experiments to understand the electronic nature of excited states in photocatalytic materials.
Experiments and calculations
Sample preparation
A 50 nm hematite (α-Fe2O3) polycrystalline thin-film was prepared on indium tin oxide (ITO)/fused silicon dioxide substrate by pulsed laser deposition (PLD). First, a 5 nm ITO thin film was deposited on a fused silicon dioxide substrate by translating a KrF excimer laser (λ = 248 nm) on a rotating ITO target with an incident angle of 45° with a fluency of 2 J cm−2 and a repetition rate of 1 Hz. The ITO target was then replaced with hematite target and hematite thin-film was deposited at the same deposition conditions. The deposition experiment was conducted at 700 °C and under 1.3 kPa oxygen pressure. The average surface roughness of the hematite thin-film was determined to be 4.7 nm using Atomic Force Microscopy. X-ray Photoelectron Spectroscopy (XPS) was employed to confirm iron and oxygen electronic properties in hematite. Further information on the sample characterization can be found in the ESI.†The X-ray setup at PAL-XFEL
All XAS and RIXS spectra were measured in the soft X-ray spectroscopy and scattering (SSS) beamline of PAL-XFEL. An illustration of the SSS beamline of PAL-XFEL is shown in Fig. 1. In the SSS beamline, soft X-rays between 250 eV and 1200 eV are available. In order to monochromatise the X-ray, a grating optics with 200 lines per mm was employed and the estimated energy resolution of the monochromatised X-ray was about 0.5 eV at 710 eV for the XAS experiments and about 1.0 eV for the RIXS experiments at 710 eV. The monochromatised X-ray was focused by a pair of Kirkpatrik–Baez mirrors to reduce the X-ray size to less than 50 μm (H) × 50 μm (V) on the sample position. Since the X-ray intensities are different in each X-ray pulse, it was necessary to measure the monochromatised X-ray intensity (I0) and the fluorescent X-ray intensity (If) shot by shot in order to obtain XAS correctly. Each I0 intensity was observed by the electrons emitted from a Si3N4 thin-film. The photoelectrons were measured with a microchannel plate (MCP) detector. The If intensity was measured with another MPC detector by obtaining fluorescent X-ray photons from the sample. Two boxcar integrators (SR250, Stanford Research Systems) were employed to collect both the signals from the MCP detectors. The boxcar integrators were synchronised with the trigger signals from the XFEL in order to gather only the signals from the X-ray pulses. The output signals from the boxcars were transferred to the digitizers (SR245, Stanford Research Systems). A Ti:sapphire laser was employed to excite the sample. Its fundamental wavelength was converted to 400 nm with a BBO crystal. The laser pulses were transferred into the measurement chamber and focused on the sample position. The spot size of the laser was approximately 210 μm (H) × 210 μm (V) for its 1/e2 value. The angle between the X-ray and the laser was set to about 1°. The excitation laser fluence was set to ∼8 mJ cm−1, which was found to be the optimum laser fluence for exciting ∼20% of the iron atoms and in the same time causing minimal to no sample damage or nonlinear effects.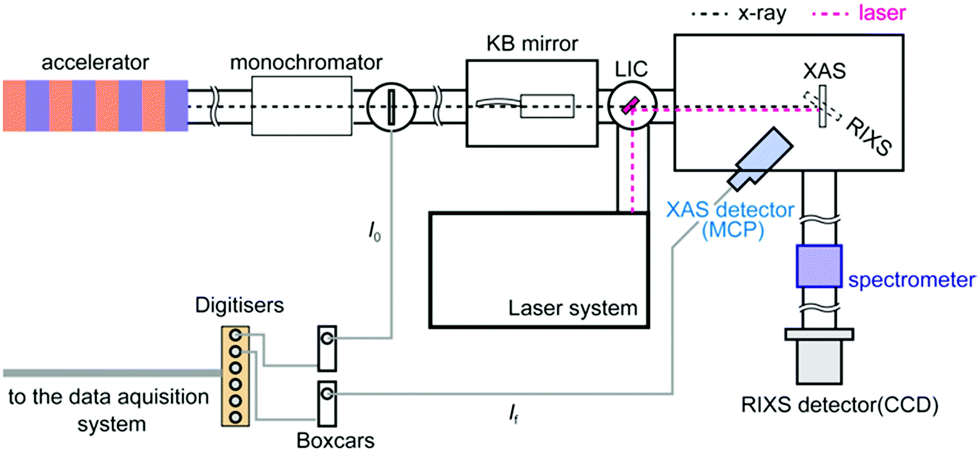 | ||
| Fig. 1 The experimental setup of the SSS beamline of PAL-XFEL. A microchannel plate (MCP) detector was employed to measure fluorescent X-ray and a CCD detector was employed to measure RIXS signals. | ||
The X-ray setup at Spring-8
Transient XAS spectra of the Fe L3 edge in the picoseconds regime were measured at the BL07LSU beamline in SPring-8.47 During the beamtime, the operation mode of SPring-8 was “H-mode”,48 where one electron bunch between bunch trains is detected. The X-rays from the isolated bunch arrives every ∼4.76 μs (∼210 kHz). In order to synchronise with the excitation laser (∼1 kHz), an X-ray chopper was employed to select X-ray pulses. XAS spectra were measured in a Partial Electron Yield (PEY) geometry. An MCP detector was employed to measure the intensity of electrons coming out from the sample surface. The fluence of the excitation laser was the same as in PAL-XFEL and the temporal resolution of the set up was estimated to be 80 ps. Further details on the setup were explained in ref. 47.Theoretical calculations
The X-ray absorption and X-ray emission processes have been modelled with crystal field multiplet theory. The use of the charge transfer multiplet (CTM) model is based on the argument that the 2p core hole state is localised on one site allowing a local model to describe the 2p3d excitation and the 3d2p X-ray emission decay. The 2p XAS spectra are simulated from transitions from the 3d5 ground state to the 2p53d6 final state, which are based on the model described by de Groot et al. and Vura-Weiss et al.44,50 The parameters we used for the calculations are identical to the parameters used in these studies, i.e. atomic values are used for the electron–electron interactions and spin–orbit couplings and a value of 0.5 eV was used for the octahedral crystal field. The multiplet-averaged Fe(II) calculation is shifted by 0.7 eV with respect to Fe(III), following the experimentally determined shift between Fe(II) and Fe(III) sites in CoFe2O4.51Results and discussion
2p X-ray absorption spectroscopy of photoexcited hematite
The Fe L3 edge XAS spectrum and transient (difference) spectra of hematite are shown in Fig. 2. Each difference spectrum was calculated by subtracting the reference spectrum (the spectrum at a delay time of −10 ps) from the photoexcited XAS spectrum at the corresponding delay time. A distinctive change was observed at 709 eV in the difference XAS spectra at positive delay times. The maximum of the peaks around 709 eV in each difference spectrum at a positive delay time are comparable to each other, while the peak width at a delay time of 10 ps is larger. It is expected that the laser excites electrons in the Fe 3d orbitals, effectively reducing Fe(III) to Fe(II). Vura-Weis et al. have observed the excited state of hematite with transient 3p XAS experiments and they calculated the XAS spectra of the excited states with multiplet calculations.44 Similar calculations of the 2p XAS spectra of Fe(II) were performed by de Groot et al. and Haverkort et al.49,50 The difference spectra between the calculated Fe L3 XAS of Fe(II) and the calculated the XAS of Fe(III) are shown in Fig. 2(c). The XAS spectra of Fe(II) were scaled in order to reproduce the spectral shapes of the difference XAS (Δμtcalc = μtFe(II)(E + ΔE) − αμtFe(III)(E)). We estimated the values of α and ΔE value based on the knowledge of XAS of the mixed-valence iron compounds such as Fe3O4, where a detailed fitting procedure determined a shift between octahedral Fe(II) and octahedral Fe(III) to be 0.7 eV.51 Different α values were explored in order to reproduce the spectral features of the difference XAS. The combination of α = 0.5 and ΔE = 0.7 eV were found to be the best values to reproduce the experimental spectra. The main peak position of the difference spectra in Fig. 2(b) was reproduced by the calculated spectra. The photoexcited state at a delay time of 0.2 ps is the initial excited state and is equal to the time resolution of our experiment as shown below in Fig. 3(b). We expect that no structural change occurs at this time scale, implying that the crystal field value 10Δq can be approximated as being equal to the ground state value of Fe(III).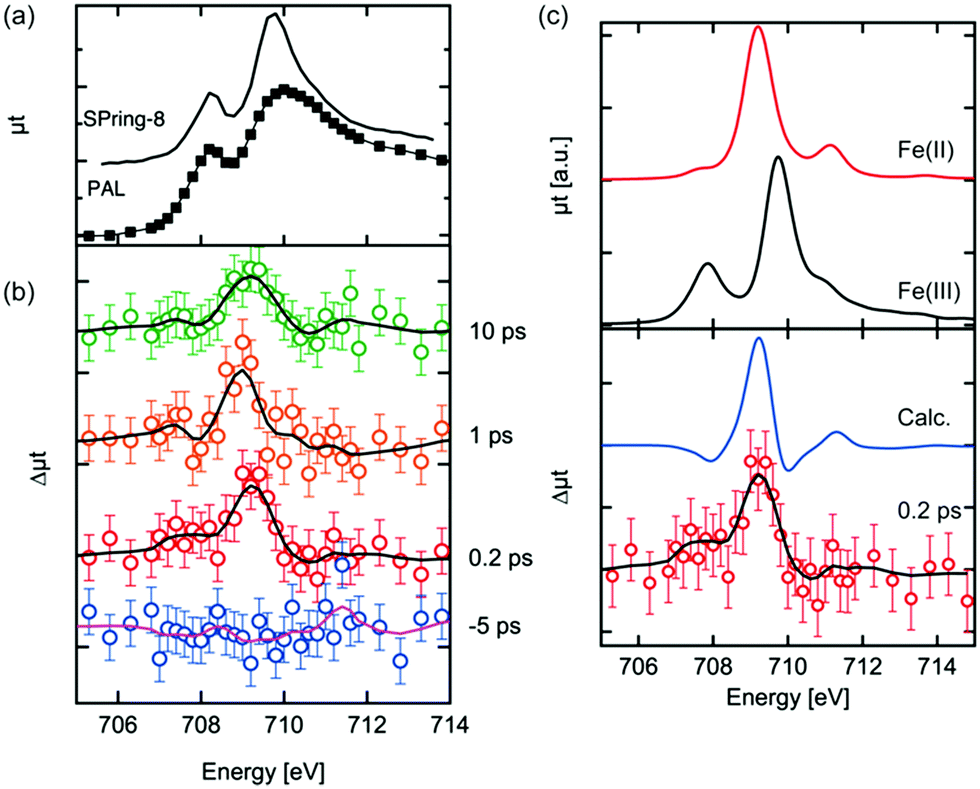 | ||
| Fig. 2 Fe L3 edge XAS spectrum of the hematite thin film (a), the transient Fe L3 edge XAS spectra of photoexcited hematite at several delay times (b) and the calculated spectra of Fe L3 edge XAS by the multiplet theory. The solid black line in panel (c) is the smoothed representation of the experimental data points that helps to clarify the spectral shape of the transient spectra. | ||
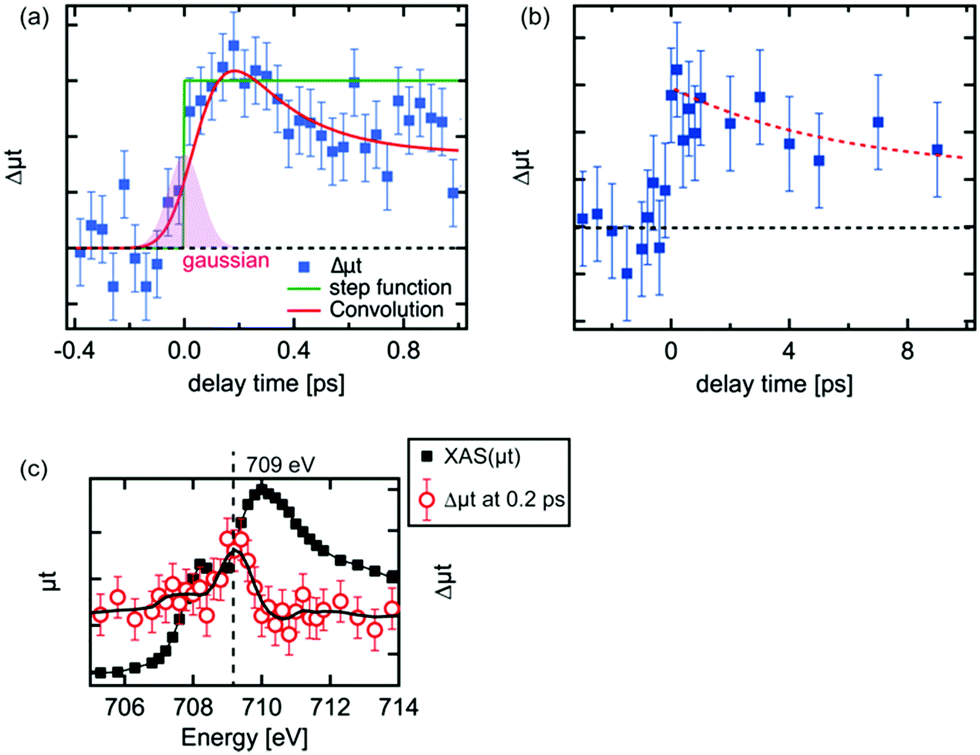 | ||
| Fig. 3 The kinetic traces of the transient XAS at 709 eV. (a) From a delay time of −0.4 ps to that of 1.0 ps, (b) from a delay time of −3 ps to that of 10 ps, (c) Fe L3 XAS and the transient XAS spectrum at 0.2 ps. | ||
Kinetic traces of 2p3d X-ray absorption spectroscopy of photoexcited hematite
The kinetic traces of the XAS at 709 eV are shown in Fig. 3(a) and (b). To avoid possible artificial effects resulting from the long-term fluctuations at the XFEL, we sequentially recorded the photoexcited signal and the reference signal (at a delay time of −10 ps) for each delay time. For shorter delay times, the kinetic trace was observed between −0.4 ps and 1.0 ps. The transient signal increased upon irradiation by the laser pulses. In order to estimate the time resolution of the experimental setup, the rise of the transient signal was fitted with a convolution of a step function and a Gaussian function. The time resolution for the experiments was estimated to be 180 fs by the full width of half maximum (FWHM) of the Gaussian function. The estimated value in the experiment seems to be reasonable considering the laser pulse duration (∼50 fs), the pulse duration of X-ray (<100 fs) and the jitter between the optical laser and the XFEL, although this value is slightly shorter from the time resolution estimated by the change of the reflectance of the laser pulse from a YAG single crystal.33 It should be noted that an arrival timing tool or a timing compensation tool is yet to be installed in the beamline.52,53 As a result, a precise measurement of the delay time between each X-ray pulse and optical laser pulse or the compensation of the timing drift between them could not be performed. Therefore, we measured the position of the delay stage for a delay time of 0 ps at the beginning and the end of each 12 h shift. Based on these measurements, we have established that the position for a delay time 0 did not shift by more than 100 fs. Hence, we concluded that there is no significant timing drift on a time scale of 12 h.The decay process shown in Fig. 3(a) is a fast decay process, whose kinetic constant (1/k) is ∼0.2 ± 0.1 ps based on the fit on the decrease of the intensity of the transient signal. A fast process within 1 ps after photoexcitation was observed by several research groups43,44 and similar values for the process were proposed (0.24 ps,44 0.25 ps43). The kinetic trace in Fig. 3(a) in the longer delay times (>0.2 ps) was fitted well with a convolution of a Gaussian function (FWHM: 0.18 ps) and an exponential function (f(t) = A × exp(−t/k) + B, where A and B are arbitrary constants). Considering our experimental conditions, the fast process we observed should correspond to the fast process observed in the previous studies.42–44 This process is ascribed to be a relaxation process of the excited electrons in higher energy states to the bottom of the conduction band.42,44
For delay times over 1 ps, the peak intensity of the difference signal gradually decreased as in Fig. 3(b). The kinetic trace in Fig. 3(b) was fitted as a single exponential function and its kinetic constant of the decay process (1) was estimated to be ∼6 ps. A similar time constant was observed in optical spectroscopy,43 which was claimed to be a recombination process between the relaxed electrons and the holes, while such process was not found in the XUV spectroscopies.41,44
The electronic structure of the iron sites in transition metal oxides such as α-Fe2O3 is described by charge transfer multiplet theory, where the ground state of Fe in α-Fe2O3 is described as |3d5 + 3d6![[L with combining low line]](https://www.rsc.org/images/entities/char_004c_0332.gif) 〉. The 3d5 configuration represents the ground state of Fe(III) and “3d6” represents the state where one electron is excited from O2p to Fe3d with a charge transfer energy (Δ) that is different from the band gap due to strong correlation effects. The ground state is the bonding combination of both configurations. Upon optical laser excitation, a valence electron is excited into the conduction band, which in the CTM model refers to an anti-bonding state |3d6 + 3d5〉. For simplicity we approximate the ground state and laser excited state to be |3d5〉 and |3d6〉 respectively, keeping in mind that both states are mixed. In addition, the |3d6〉R and |3d6〉R′ states represent the relaxed states from the initial excitation. The overall process can then be described as:
〉. The 3d5 configuration represents the ground state of Fe(III) and “3d6” represents the state where one electron is excited from O2p to Fe3d with a charge transfer energy (Δ) that is different from the band gap due to strong correlation effects. The ground state is the bonding combination of both configurations. Upon optical laser excitation, a valence electron is excited into the conduction band, which in the CTM model refers to an anti-bonding state |3d6 + 3d5〉. For simplicity we approximate the ground state and laser excited state to be |3d5〉 and |3d6〉 respectively, keeping in mind that both states are mixed. In addition, the |3d6〉R and |3d6〉R′ states represent the relaxed states from the initial excitation. The overall process can then be described as:
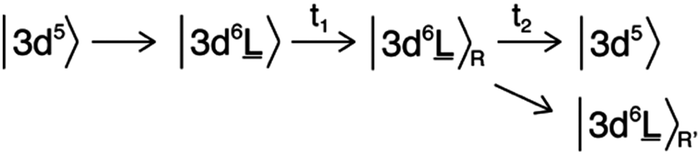 〉 state will have a |2p53d7〉 final state similar to a Fe(II) ground state. This justifies the first-order approximation of its spectral shape that is related to a Fe(II) species in the local electronic structure of α-Fe2O3.
〉 state will have a |2p53d7〉 final state similar to a Fe(II) ground state. This justifies the first-order approximation of its spectral shape that is related to a Fe(II) species in the local electronic structure of α-Fe2O3.
2p3d resonant inelastic scattering of photoexcited hematite
2p3d RIXS measures the 2p XAS excitation of the 3d5 ground state to 2p53d6, followed by the decay back to the 3d5 manifold. A recent static 2p3d RIXS study of α-Fe2O3 has been published by Miyawaki et al.54 We note that the lifetime of the 2p core hole state is ∼10 fs based on its lifetime broadening. We assume that the 2p core hole decay is faster than any other reorganisation process and that 2p3d RIXS is effectively an instantaneous probe.The 2p3d RIXS spectra were measured at 710 eV, i.e. at the maximum of the static α-Fe2O3 2p XAS spectrum. The delay times of each measurement were 1 ps and 10 ps as shown in Fig. 4. The RIXS spectra were normalised to the intensity of elastic X-ray peak (energy loss (Eloss) ∼ 0 eV). Each RIXS spectrum was recorded in two hours. Due to the increased possibility for time drift in the delay times around 0.2 ps, we could not record the RIXS spectrum for such short delay times. In addition, in order to guarantee that RIXS from photoexcited states are observed, the kinetic traces at 709 eV were observed before each RIXS measurement (Fig. 4).
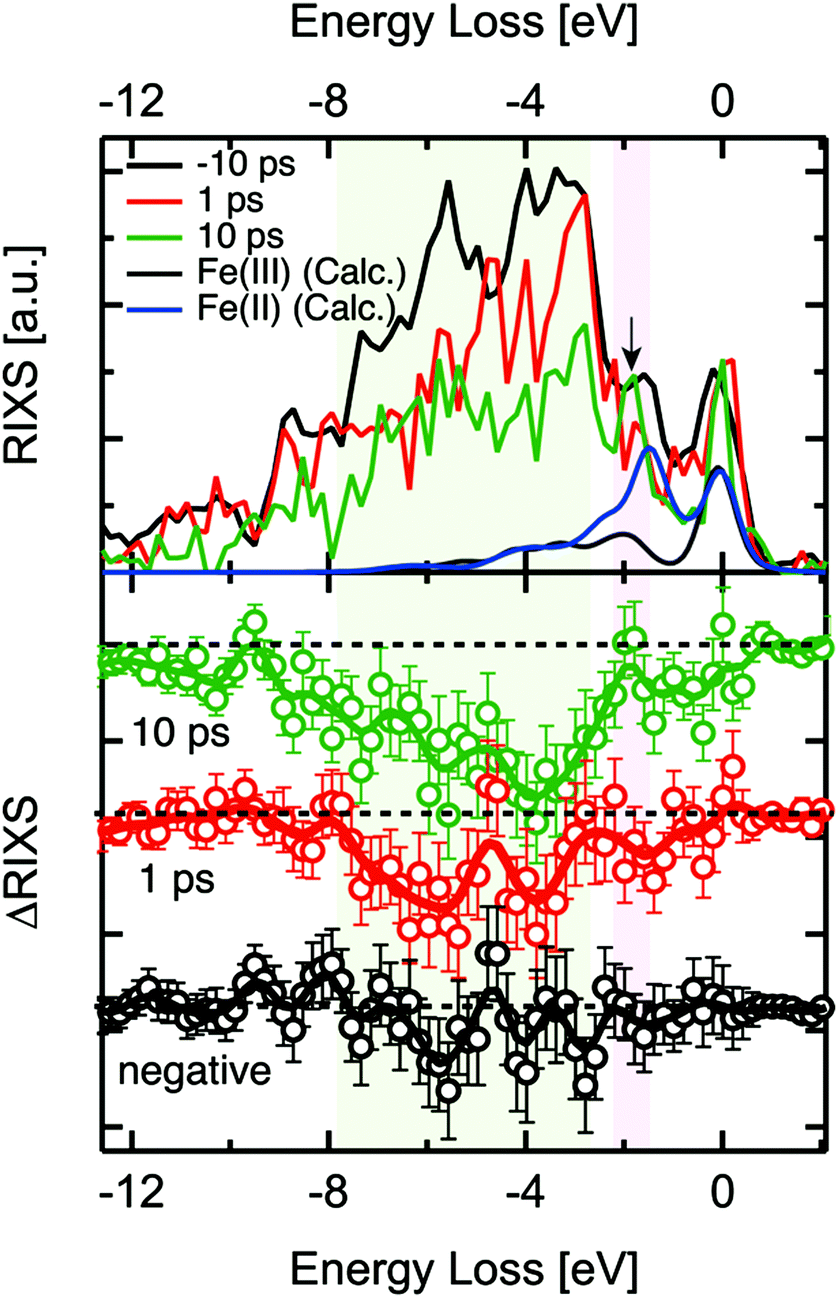 | ||
| Fig. 4 2p3d RIXS spectra for −10 ps, 1 ps, 10 ps (top) and the difference RIXS spectrum for each delay time (bottom). The incident X-ray energy was 710 eV. The calculated spectra were normalised so that their elastic scattering intensities were half of the experimental spectra for clarity. | ||
When we compared the transient RIXS at a delay time of 1 ps to that of 10 ps, we noticed that the overall difference signal seemed to increase in the later delay time. We are not certain about the interpretation of this effect due to the low energy resolution of the spectra and the interference between Fe(II) and Fe(III) features. One possible explanation could be the long-life time of the Fe(II) excited states that are generated at hematite defect sites. We observed in another related time-resolved XUV study that these states could have a life time of up to ∼350 ps. It is possible that the concentration of these states increases over time after subsequent optical excitations, which could be reflected in the increase of the difference signal in RIXS at 10 ps.
Examining the time evolution of the RIXS difference signals showed spectral changes in the coloured areas at between −3 eV and −8 eV. The theoretical 2p3d RIXS spectrum of Fe(III) contains an elastic peak and a range of d–d excitations between −2 and −7 eV (Fig. 4). The ratio between both the RIXS spectra is given by the excitation energy, essentially as given by the XAS calculations. This implies that Fe(III) has ∼10 times more intensity than Fe(II). The spectrum of Fe(II) has a strong peak at ∼1.8 eV related to the 5T2 → 5T1 d–d transition, which will compensate the intensity loss of Fe(III) at ∼1.8 eV, implying that only between −3 and −8 eV intensity is lost. We note that an excitation energy of 710 eV, more than ∼90% of the signal is related to Fe(III) based on the theoretical 2p XAS spectral shapes, which implies that we mainly see Fe(III) in the 2p3d RIXS. One could also look specifically at the Fe(II) species, but this would require comparing the RIXS spectra recorded at the excitation energy of 710 eV as well as that of 709 eV, which was not measured due to the time constraints. We essentially observe a decrease of the region between −2 and −7 eV indicating that the amount of Fe(III) species is reduced. The data quality unfortunately does not allow us to make more specific statements regarding the 2p3d RIXS spectral shapes.
We note that by measuring the 2p3d RIXS at 709 eV we would specifically target the transient Fe(II) species. Its 2p3d RIXS spectrum would reveal its d–d excitations and as such the nature of its state in much more details compared to 2p XAS. In addition there would be much less signal related to the Fe(III) species.
Photoexcited states of hematite
It has been proposed that several different excited states exist for the photo-absorption process in hematite by optical spectroscopy.43 From our XAS and RIXS results we expect three different excited states in the first 10 ps, in line with the observed states proposed by optical spectroscopic measurements. The initial photoexcited state was observed at a delay time of 0.2 ps. After photo-absorption, the 3d5 ground state of Fe(III) is excited to the 3d6 charge transfer state. The charge transfer state symmetry is similar to that of a Fe(II) ground state and results in transient peak at 709 eV. In a single particle picture, the 3d6 state can be interpreted as an electron in the conduction band coupled to a hole in the valence band (Fig. 5). Since the wavelength of the optical laser is larger than the band gap of hematite, the excited electrons and holes have excess energy (often known as hot carriers). The hot electron–hole excitations relate to hot electrons and holes which are distributed in the conduction and valence bands, respectively. The transient signal intensity at 709 eV decreases after 1 ps, which corresponds to the relaxation process of the hot carriers. During the relaxation of the hot carriers, the hot electrons relax to the bottom of the conduction band while the holes relax to the top of the valence band releasing extra energy. The fast decay in Fig. 2 and the simulation of the difference spectra in Fig. 3 agree with this hypothesis. During the relaxation the local structure of the Fe atoms is affected, which modifies the Fe–O and Fe–Fe bond distances. These structural changes affect the electronic structure (10Δq, etc.) of the excited Fe atoms, thereby also changing the Fe L3 XAS spectral shape.
 | ||
| Fig. 5 Schematic diagram of the possible pathways of the excited states of hematite upon visible light excitation. Before photoexcitation (|3d5〉), the valence of Fe is 3+, which has 5 electrons in the 3d orbitals. The 3d orbitals of Fe are split into 3 tg orbitals and 2 eg orbitals because of its octahedral symmetry. After photoexcitation (|3d6〉, ∼200 fs), an excited electron occupies tg or eg orbitals. Within 1 ps (|3d6〉R′ ∼ 1 ps), the excited electron relaxes to occupy the tg orbital. In the later delay time (∼10 ps), some of the excited Fe species change to another excited state (|3d6〉R′) while others relax to the ground state. It is noted that the excited electron is spin-down because the spin-up states are all occupied. At a neighboring site, the spin-up and spin-down signature is reversed as α-Fe2O3 is an antiferromagnet. | ||
The transient signal intensity decreased further after 10 ps, which agrees with the charge recombination process observed in optical spectroscopy. The population of the excited electrons is decreased by the recombination events, i.e. by the decay of anti-bonding 3d6 to bonding 3d5 states. The intensity of the difference spectra at 709 eV is attributed to the number of Fe(II) atoms and the decrease of Fe(II) atoms leads to the reduction of the difference signals around 10 ps. Interestingly, we observe that the Fe(II) species remains present for several hundreds of picoseconds.
Conclusions
In this study, we have reported the first femtosecond Fe L3 XAS and femtosecond 2p3d RIXS of hematite using the PAL-XFEL. Upon photoexcitation, electrons are transferred from the hematite valence band (mainly O 2p) to the conduction band (mainly Fe 3d). We detected three relaxation events after photoexcitation: the first process is the relaxation of hot electrons to a lower energy state. This relaxation process of the hot carriers with a kinetic constant of 0.2 ps was observed by Fe L3 XAS with a time resolution of 180 fs. The second process is a fast carrier recombination, which was observed as the decrease of the transient XAS signal at 709 eV with a kinetic constant of 6 ps. In this process, part of the Fe(II)-like species relaxed back to 3d5 Fe(III). However, our observations also show that some Fe(II) species remain at a longer time scale (in Fig. S2 in ESI†). This long-living photoexcited state could be a trapping state of the excited electrons with a local structural change around photoexcited iron atoms in hematite to form small polarons.To the best of our knowledge, this is the first report on femtosecond transient RIXS of a solid-state material. Our result demonstrates exciting possibilities for studying the photoexcited states of solid-state materials using femtosecond RIXS. In future experiments, where RIXS spectra will be measured at different excitation energies and better energy resolution, more detailed information will be revealed about specific electronic states of the photoexcited materials. It could also be possible to describe charge carrier dynamics in ultrafast time scales with electronic configurations at each distinctive delay time.
Although there have been several pioneering works of time-resolved 2p3d RIXS,24–30 the opportunity to perform time-resolved 2p3d RIXS experiments was limited since only the Linac Coherent Light Source (LCLS, Menlo Park, CA, USA) was able to provide the soft X-rays needed.55,56 In addition to the Pohang XFEL, new soft X-ray beamlines are being implemented in novel XFEL facilities such as in LCLS-II,57 European XFEL33 and Swiss FEL.36 Such powerful FEL facilities, providing ultrashort soft X-ray pulses, will make it possible to address structural and electronic configurations of excited states of semiconductors consisting of 3d transition metals thereby opening possibilities to understand the mechanism of e.g. a solar-assisting water splitting reaction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No 340279), Netherlands Center for Multiscale Catalytic Energy Conversion (MCEC), a Gravitation Program from the Netherlands Organisation for Scientific Research (NWO), Grant for collaborative research in Institute for Catalysis, Hokkaido University (18A1005) and a basic science research program funded by the Ministry of Education, Government of the Netherlands (No. NRF-2017R1D1A1A02018484). This work was carried out by the joint research in the Synchrotron Radiation Research Organization and the Institute for Solid State Physics, the University of Tokyo (Proposal No. 2019A7592). The experiments were performed at SSS end-station of PAL-XFEL (proposal no. 2018-2nd-SSS-001 or 2019-1st-SSS-020) funded by the Ministry of Science and ICT of Korea.References
- M. Chergui and E. Collet, Chem. Rev., 2017, 117, 11025–11065 CrossRef CAS PubMed.
- C. J. Milne, T. J. Penfold and M. Chergui, Coord. Chem. Rev., 2014, 277–278, 44–68 CrossRef CAS.
- C. Bressler and M. Chergui, Annu. Rev. Phys. Chem., 2010, 61, 263–282 CrossRef CAS PubMed.
- C. Bressler and M. Chergui, Chem. Rev., 2004, 104, 1781–1812 CrossRef CAS PubMed.
- Y. Iwasawa, K. Asakura and M. Tada, XAFS Techniques for Catalysts, Nanomaterials, and Surfaces, Springer International Publishing, 2017 Search PubMed.
- J. A. Van Bokhoven and C. Lamberti, X-Ray Absorption and X-Ray Emission Spectroscopy: Theory and Applications, John Wiley & Sons, UK, 2016 Search PubMed.
- T. J. Penfold, J. Szlachetko, F. G. Santomauro, A. Britz, W. Gawelda, G. Doumy, A. M. March, S. H. Southworth, J. Rittmann, R. Abela, M. Chergui and C. J. Milne, Nat. Commun., 2018, 9, 478 CrossRef PubMed.
- T. J. Penfold, S. Karlsson, G. Capano, F. A. Lima, J. Rittmann, M. Reinhard, M. H. Rittmann-Frank, O. Braem, E. Baranoff, R. Abela, I. Tavernelli, U. Rothlisberger, C. J. Milne and M. Chergui, J. Phys. Chem. A, 2013, 117, 4591–4601 CrossRef CAS.
- Y. Uemura, D. Kido, A. Koide, Y. Wakisaka, Y. Niwa, S. Nozawa, K. Ichiyanagi, R. Fukaya, S. Adachi, T. Katayama, T. Togashi, S. Owada, M. Yabashi, K. Hatada, A. Iwase, A. Kudo, S. Takakusagi, T. Yokoyamaa and K. Asakura, Chem. Commun., 2017, 53, 7314–7317 RSC.
- Y. Obara, H. Ito, T. Ito, N. Kurahashi, S. Thürmer, H. Tanaka, T. Katayama, T. Togashi, S. Owada, Y.-I. Yamamoto, S. Karashima, J. Nishitani, M. Yabashi, T. Suzuki and K. Misawa, Struct. Dyn., 2017, 4, 044033 CrossRef.
- Y. Uemura, D. Kido, Y. Wakisaka, H. Uehara, T. Ohba, Y. Niwa, S. Nozawa, T. Sato, K. Ichiyanagi, R. Fukaya, S.-I. Adachi, T. Katayama, T. Togashi, S. Owada, K. Ogawa, M. Yabashi, K. Hatada, S. Takakusagi, T. Yokoyama, B. Ohtani and K. Asakura, Angew. Chem., Int. Ed., 2016, 55, 1364–1367 CrossRef CAS.
- D. Hayes, R. G. Hadt, J. D. Emery, A. A. Cordones, A. B. F. Martinson, M. L. Shelby, K. A. Fransted, P. D. Dahlberg, J. Hong, X. Zhang, Q. Kong, R. W. Schoenlein and L. X. Chen, Energy Environ. Sci., 2016, 9, 3754–3769 RSC.
- H. Wen, M. Sassi, Z. Luo, C. Adamo, D. G. Schlom, K. M. Rosso and X. Zhang, Sci. Rep., 2015, 5, 15098 CrossRef CAS.
- F. G. Santomauro, A. Lübcke, J. Rittmann, E. Baldini, A. Ferrer, M. Silatani, P. Zimmermann, S. Grübel, J. A. Johnson, S. O. Mariager, P. Beaud, D. Grolimund, C. Borca, G. Ingold, S. L. Johnson and M. Chergui, Sci. Rep., 2015, 5, 14834 CrossRef CAS.
- Y. Uemura, H. Uehara, Y. Niwa, S. Nozawa, T. Sato, S. Adachi, B. Ohtani, S. Takakusagi and K. Asakura, Chem. Lett., 2014, 43, 977–979 CrossRef CAS.
- M. H. Rittmann-Frank, C. J. Milne, J. Rittmann, M. Reinhard, T. J. Penfold and M. Chergui, Angew. Chem., Int. Ed., 2014, 53, 5858–5862 CrossRef CAS.
- N. A. Miller, A. Deb, R. Alonso-Mori, B. D. Garabato, J. M. Glownia, L. M. Kiefer, J. Koralek, M. Sikorski, K. G. Spears, T. E. Wiley, D. Zhu, P. M. Kozlowski, K. J. Kubarych, J. E. Penner-Hahn and R. J. Sension, J. Am. Chem. Soc., 2017, 139, 1894–1899 CrossRef CAS.
- H. T. Lemke, K. S. Kjaer, R. Hartsock, T. B. van Driel, M. Chollet, J. M. Glownia, S. Song, D. Zhu, E. Pace, S. F. Matar, M. M. Nielsen, M. Benfatto, K. J. Gaffney, E. Collet and M. Cammarata, Nat. Commun., 2017, 8, 15342 CrossRef CAS PubMed.
- M. L. Shelby, P. J. Lestrange, N. E. Jackson, K. Haldrup, M. W. Mara, A. B. Stickrath, D. Zhu, H. T. Lemke, M. Chollet, B. M. Hoffman, X. Li and L. X. Chen, J. Am. Chem. Soc., 2016, 138, 8752–8764 CrossRef CAS.
- H. T. Lemke, C. Bressler, L. X. Chen, D. M. Fritz, K. J. Gaffney, A. Galler, W. Gawelda, K. Haldrup, R. W. Hartsock, H. Ihee, J. Kim, K. H. Kim, J. H. Lee, M. M. Nielsen, A. B. Stickrath, W. Zhang, D. Zhu and M. Cammarata, J. Phys. Chem. A, 2013, 117, 735–740 CrossRef CAS.
- S. E. Canton, K. S. Kjær, G. Vankó, T. B. van Driel, S.-I. Adachi, A. E. Bordage, C. Bressler, P. Chabera, M. Christensen, A. O. Dohn, A. Galler, W. Gawelda, D. Gosztola, K. Haldrup, T. Harlang, Y. Liu, K. B. Móller, Z. Németh, S. Nozawa, M. Pápai, T. Sato, T. Sato, K. Suarez-Alcantara, T. Togashi, K. Tono, J. Uhlig, D. A. Vithanage, K. Wärnmark, M. Yabashi, J. Zhang, V. Sundström and M. M. Nielsen, Nat. Commun., 2015, 6, 6359 CrossRef CAS.
- K. Haldrup, W. Gawelda, R. Abela, R. Alonso-Mori, U. Bergmann, A. Bordage, M. Cammarata, S. E. Canton, A. O. Dohn, T. B. van Driel, D. M. Fritz, A. Galler, P. Glatzel, T. Harlang, K. S. Kjær, H. T. Lemke, K. B. Móller, Z. Németh, M. Pápai, N. Sas, J. Uhlig, D. Zhu, G. Vankó, V. Sundström, M. M. Nielsen and C. Bressler, J. Phys. Chem. B, 2016, 120, 1158–1168 CrossRef CAS.
- J. Kern, R. Tran, R. Alonso-Mori, S. Koroidov, N. Echols, J. Hattne, M. Ibrahim, S. Gul, H. Laksmono, R. G. Sierra, R. J. Gildea, G. Han, J. Hellmich, B. Lassalle-Kaiser, R. Chatterjee, A. S. Brewster, C. A. Stan, C. Glöckner, A. Lampe, D. DiFiore, D. Milathianaki, A. R. Fry, M. M. Seibert, J. E. Koglin, E. Gallo, J. Uhlig, D. Sokaras, T.-C. Weng, P. H. Zwart, D. E. Skinner, M. J. Bogan, M. Messerschmidt, P. Glatzel, G. J. Williams, S. Boutet, P. D. Adams, A. Zouni, J. Messinger, N. K. Sauter, U. Bergmann, J. Yano and V. K. Yachandra, Nat. Commun., 2014, 5, 4371 CrossRef CAS PubMed.
- M. Beye, F. Sorgenfrei, W. F. Schlotter, W. Wurth and A. Föhlisch, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16772–16776 CrossRef CAS.
- R. M. Jay, S. Eckert, M. Fondell, P. S. Miedema, J. Norell, A. Pietzsch, W. Quevedo, J. Niskanen, K. Kunnus and A. Föhlisch, Phys. Chem. Chem. Phys., 2018, 20, 27745–27751 RSC.
- R. M. Jay, J. Norell, S. Eckert, M. Hantschmann, M. Beye, B. Kennedy, W. Quevedo, W. F. Schlotter, G. L. Dakovski, M. P. Minitti, M. C. Hoffmann, A. Mitra, S. P. Moeller, D. Nordlund, W. Zhang, H. W. Liang, K. Kunnus, K. Kubicek, S. A. Techert, M. Lundberg, P. Wernet, K. Gaffney, M. Odelius and A. Föhlisch, J. Phys. Chem. Lett., 2018, 9, 3538–3543 CrossRef CAS PubMed.
- K. Kunnus, I. Josefsson, I. Rajkovic, S. Schreck, W. Quevedo, M. Beye, C. Weniger, S. Grübel, M. Scholz, D. Nordlund, W. Zhang, R. W. Hartsock, K. J. Gaffney, W. F. Schlotter, J. J. Turner, B. Kennedy, F. Hennies, F. M. F. de Groot, S. Techert, M. Odelius, P. Wernet and A. Föhlisch, Struct. Dyn., 2016, 3, 043204 CrossRef CAS.
- J. Norell, R. M. Jay, M. Hantschmann, S. Eckert, M. Guo, K. J. Gaffney, P. Wernet, M. Lundberg, A. Föhlisch and M. Odelius, Phys. Chem. Chem. Phys., 2018, 20, 7243–7253 RSC.
- V. Vaz da Cruz, F. Gel'mukhanov, S. Eckert, M. Iannuzzi, E. Ertan, A. Pietzsch, R. C. Couto, J. Niskanen, M. Fondell, M. Dantz, T. Schmitt, X. Lu, D. McNally, R. M. Jay, V. Kimberg, A. Föhlisch and M. Odelius, Nat. Commun., 2019, 10, 1013 CrossRef.
- P. Wernet, K. Kunnus, I. Josefsson, I. Rajkovic, W. Quevedo, M. Beye, S. Schreck, S. Grubel, M. Scholz, D. Nordlund, W. Zhang, R. W. Hartsock, W. F. Schlotter, J. J. Turner, B. Kennedy, F. Hennies, F. M. de Groot, K. J. Gaffney, S. Techert, M. Odelius and A. Fohlisch, Nature, 2015, 520, 78–81 CrossRef CAS.
- H.-S. Kang, C.-K. Min, H. Heo, C. Kim, H. Yang, G. Kim, I. Nam, S. Y. Baek, H.-J. Choi, G. Mun, B. R. Park, Y. J. Suh, D. C. Shin, J. Hu, J. Hong, S. Jung, S.-H. Kim, K. Kim, D. Na, S. S. Park, Y. J. Park, J.-H. Han, Y. G. Jung, S. H. Jeong, H. G. Lee, S. Lee, S. Lee, W.-W. Lee, B. Oh, H. S. Suh, Y. W. Parc, S.-J. Park, M. H. Kim, N.-S. Jung, Y.-C. Kim, M.-S. Lee, B.-H. Lee, C.-W. Sung, I.-S. Mok, J.-M. Yang, C.-S. Lee, H. Shin, J. H. Kim, Y. Kim, J. H. Lee, S.-Y. Park, J. Kim, J. Park, I. Eom, S. Rah, S. Kim, K. H. Nam, J. Park, J. Park, S. Kim, S. Kwon, S. H. Park, K. S. Kim, H. Hyun, S. N. Kim, S. Kim, S.-M. Hwang, M. J. Kim, C.-y. Lim, C.-J. Yu, B.-S. Kim, T.-H. Kang, K.-W. Kim, S.-H. Kim, H.-S. Lee, H.-S. Lee, K.-H. Park, T.-Y. Koo, D.-E. Kim and I. S. Ko, Nat. Photonics, 2017, 11, 708–713 CrossRef CAS.
- PAL-XFEL, http://pal.postech.ac.kr/paleng/.
- S. H. Park, M. Kim, C.-K. Min, I. Eom, I. Nam, H.-S. Lee, H.-S. Kang, H.-D. Kim, H. Y. Jang, S. Kim, S.-M. Hwang, G.-S. Park, J. Park, T.-Y. Koo and S. Kwon, Rev. Sci. Instrum., 2018, 89, 055105 CrossRef.
- L. J. P. Ament, M. van Veenendaal, T. P. Devereaux, J. P. Hill and J. van den Brink, Rev. Mod. Phys., 2011, 83, 705–767 CrossRef CAS.
- F. de Groot and A. Kotani, Core Level Spectroscopy of Solids, CRC Press, 2008 Search PubMed.
- A. Kotani and S. Shin, Rev. Mod. Phys., 2001, 73, 203–246 CrossRef CAS.
- K. Sivula, F. L. Formal and M. Grätzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS.
- A. G. Tamirat, J. Rick, A. A. Dubale, W.-N. Su and B.-J. Hwang, Nanoscale Horiz., 2016, 1, 243–267 RSC.
- M. Barroso, S. R. Pendlebury, A. J. Cowan and J. R. Durrant, Chem. Sci., 2013, 4, 2724–2734 RSC.
- A. Kay, I. Cesar and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 15714–15721 CrossRef CAS.
- J. Husek, A. Cirri, S. Biswas and L. R. Baker, Chem. Sci., 2017, 8, 8170–8178 RSC.
- S. R. Pendlebury, X. Wang, F. L. Formal, M. Cornuz, A. Kafizas, S. D. Tilley, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2014, 136, 9854–9857 CrossRef CAS.
- S. Sorenson, E. Driscoll, S. Haghighat and J. M. Dawlaty, J. Phys. Chem. C, 2014, 118, 23621–23626 CrossRef CAS.
- J. Vura-Weis, C.-M. Jiang, C. Liu, H. Gao, J. M. Lucas, F. M. F. de Groot, P. Yang, A. P. Alivisatos and S. R. Leone, J. Phys. Chem. Lett., 2013, 4, 3667–3671 CrossRef CAS.
- L. M. Carneiro, S. K. Cushing, C. Liu, Y. Su, P. Yang, A. P. Alivisatos and S. R. Leone, Nat. Mater., 2017, 16, 819–825 CrossRef CAS.
- A. J. E. Rettie, W. D. Chemelewski, D. Emin and C. B. Mullins, J. Phys. Chem. Lett., 2016, 7, 471–479 CrossRef CAS.
- K. Takubo, K. Yamamoto, Y. Hirata, Y. Yokoyama, Y. Kubota, S. Yamamoto, S. Yamamoto, I. Matsuda, S. Shin, T. Seki, K. Takanashi and H. Wadati, Appl. Phys. Lett., 2017, 110, 162401 CrossRef.
- SPring-8 several bunch modes, http://www.spring8.or.jp/en/users/operation_status/schedule/bunch_mode_b19/.
- M. U. Delgado-Jaime, K. Zhang, J. Vura-Weis and F. M. F. de Groot, J. Synchrotron Radiat., 2016, 23, 1264–1271 CrossRef CAS.
- M. W. Haverkort, M. Zwierzycki and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 165113 CrossRef.
- B. Liu, C. Piamonteze, M. U. Delgado-Jaime, R.-P. Wang, J. Heidler, J. Dreiser, R. Chopdekar, F. Nolting and F. M. F. de Groot, Phys. Rev. B, 2017, 96, 054446 CrossRef.
- M. R. Bionta, N. Hartmann, M. Weaver, D. French, D. J. Nicholson, J. P. Cryan, J. M. Glownia, K. Baker, C. Bostedt, M. Chollet, Y. Ding, D. M. Fritz, A. R. Fry, D. J. Kane, J. Krzywinski, H. T. Lemke, M. Messerschmidt, S. Schorb, D. Zhu, W. E. White and R. N. Coffee, Rev. Sci. Instrum., 2014, 85, 083116 CrossRef CAS.
- T. Katayama, S. Owada, T. Togashi, K. Ogawa, P. Karvinen, I. Vartiainen, A. Eronen, C. David, T. Sato, K. Nakajima, Y. Joti, H. Yumoto, H. Ohashi and M. Yabashi, Struct. Dyn., 2016, 3, 034301 CrossRef PubMed.
- J. Miyawaki, S. Suga, H. Fujiwara, M. Urasaki, H. Ikeno, H. Niwa, H. Kiuchi and Y. Harada, Phys. Rev. B, 2017, 96, 214420 CrossRef.
- T. Funk, A. Deb, S. J. George, H. Wang and S. P. Cramer, Coord. Chem. Rev., 2005, 249, 3–30 CrossRef CAS.
- X. Liu, W. Yang and Z. Liu, Adv. Mater., 2014, 26, 7710–7729 CrossRef CAS.
- LCLS-II, https://lcls.slac.stanford.edu/lcls-ii.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp03374b |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2020 |