
The combination of halogen and hydrogen bonding: a versatile tool in coordination chemistry†
Lucia
González
a,
Sara
Graus
a,
Rosa M.
Tejedor
 ab,
Anjana
Chanthapally
c,
José Luis
Serrano
a and
Santiago
Uriel
*a
ab,
Anjana
Chanthapally
c,
José Luis
Serrano
a and
Santiago
Uriel
*a
aDepartamento de Química Orgánica, Instituto de Nanociencia de Aragón (INA)-Instituto de Ciencia de Materiales de Aragón (ICMA), Universidad de Zaragoza-CSIC, Zaragoza, Spain
bCentro Universitario de la Defensa, Academia General Militar, 50090 Zaragoza, Spain
cDepartment Chemistry, National University of Singapore, 43, Singapore
First published on 21st July 2020
Abstract
4-Iodo-N-(4-pyridyl)benzamide (INPBA) and four derived coordination complexes were synthesized in order to explore the combination of halogen and hydrogen bonding interactions in coordination chemistry. An electron-withdrawing carbonyl group attached to the aromatic ring bearing an iodine atom has been introduced to increase its halogen bonding ability. Single crystal X-ray diffraction analyses of INPBA, [Ag(INPBA)2]NO3 (1), [ZnBr2(INPBA)2] (2), [Zn(OCOPh)2(INPBA)2] (3) and [HgI2(INPBA)]n (4) show the versatility of the INPBA building block yielding a variety of different interesting structures. Iodine atom arrangement plays a key role by reinforcing and extending crystalline structures into diverse 3D supramolecular networks via I⋯O, I⋯N, and I⋯I halogen bonding interactions. Besides this, the overall supramolecular architecture of the coordination complexes is modulated by the N–H⋯O, N–H⋯Br, and C–H⋯O hydrogen bonds formed by the carboxamide group. The combination of both, halogen and hydrogen bonds, allows very different coordination networks to be designed.
Introduction
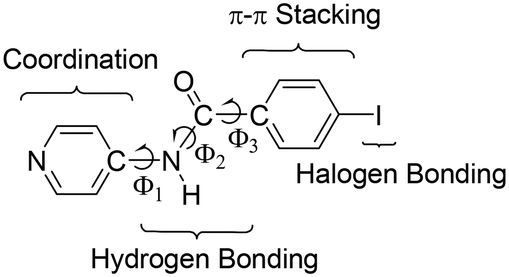
The physical and chemical properties of molecular materials in the solid state are controlled by the arrangement of molecular building blocks. The adequate design of these supramolecular organizations affords control over the properties of these materials.1–4 One of the most powerful strategies to control the solid-state structure of metal-containing compounds is the assembly of metallotectons through non-covalent interactions. Therefore, organic ligand characteristics such as rigidity, flexibility, binding and potential interaction sites5 can largely determine the supramolecular organization. In the solid state, non-covalent interactions such as hydrogen bonding,6,7 π–π stacking,8 CH⋯π interaction,8 and, the most recent and therefore least explored, halogen bonding interactions9–13 direct the structure of molecular materials. Moreover, synergy between non-covalent interactions can determine the supramolecular assembly.14To study the ability to direct the structure of the combination of hydrogen and halogen bonds in coordination chemistry, we have prepared 4-iodo-N-(4-pyridyl)benzamide (INPBA) (Chart 1) and four metal complexes of this ligand with metal ions Ag+, Zn2+ and Hg2+, which give rise to complexes with a low coordination number. The cooperation between the halogen bonding interactions and other non-covalent interactions has been previously studied15–20 to develop metal-free liquid crystals,21 two-dimensional structures22 and porous materials.23
 | ||
| Chart 1 4-Iodo-N-(4-pyridyl)benzamide (INPBA). | ||
INPBA has three active functional groups (Chart 1): iodine, pyridine and carboxamide. The iodo-substituent is partially activated24,25 by the electron-withdrawing carbonyl of the amide group attached to the aromatic ring26 and is able to take part in C–I⋯oxygen, C–I⋯nitrogen, C–I⋯π, C–I⋯I–C and C–I⋯X–M halogen bondings.27–30 As in previous studies, halogen bonding is expected to generate different supramolecular architectures, including helical chains.31–37 Furthermore, INPBA contains a carboxamide group that plays an important role in the supramolecular organizations through its participation in both hydrogen and halogen bonds.
Taking into account this previous experience, we have designed in this paper three new coordination complexes namely [Ag(INPBA)2]NO3 (1), [ZnBr2(INPBA)2] (2), and [Zn(OCOPh)2(INPBA)2] (3), and one extended coordination polymer, [HgI2(INPBA)]n (4). The synthesis and the crystal structure of INPBA and the metal complexes are described. The different supramolecular structures obtained in the solid state have been analysed based on non-covalent interactions observed by Hirshfeld surface calculations. Also, the ability of INPBA as a halogen bonding donor is evaluated by theoretical calculations.
Results and discussion
4-Iodo-N-(4-pyridyl)benzamide (INPBA) was prepared by treating 4-iodobenzoyl chloride with 4−aminopyridine in the presence of triethylamine. INPBA crystallizes in the monoclinic P21/c space group, with one independent molecule in the asymmetric unit. The crystallographic data of INPBA and complexes 1 to 4 are listed in Table 1. The bond distances and angles of INPBA were as expected. This ligand consists of three structural parts: a pyridyl unit, a central carboxamide group and a p-iodophenyl unit (Chart 1). These structural moieties form three regions with conformational degrees of freedom defined by the torsion angles Φ1 (Φ1 = C(9)–C(8)–N(1)–C(7)), Φ2 (Φ2 = C(8)–N(1)–C(7)–C(4)) and Φ3 (Φ3 = N(1)–C(7)–C(4)–C(3)), which allow it to fit the local environment (Fig. 1 and S1 in the ESI†). The INPBA central C–C–N–C spacer unit is almost planar, as shown by the torsion angle (Φ2 = −177.3°). However, the aromatic rings are markedly twisted out of this plane (Φ1 = 33.7° and Φ3 = 28.4°) (Table 2). This conformation is similar to that of N-(4-pyridyl)benzamide38 (NPBA) (Φ1 = −16.8°, Φ2 = 178.4° and Φ3 = −34.3°) and 4-fluoro-N-(pyridin-4-yl)benzamide39 (FNPBA) (Φ1 = 17.6°, Φ2 = −177.8° and Φ3 = 35.4°) (Fig. S2, ESI†).| Compound | INPBA | 1 | 2 | 3 | 4 |
|---|---|---|---|---|---|
| a Cu-Kα radiation. | |||||
| Empirical formula | C12H9IN2O | C24H18N5O5I2Ag | C24H18N4O2Br2I2Zn | C38H28I2N4O6Zn | C12H9HgI3N2O |
| Formula weight | 324.11 | 818.10 | 873.41 | 955.81 | 778.50 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Orthorhombic | Monoclinic |
| a, Å | 5.3119(3) | 9.3872(5) | 9.7184(2) | 14.2615(6) | 4.303(12) |
| b, Å | 14.3087(6) | 11.4744(4) | 27.7576(6) | 8.9996(3) | 14.824(5) |
| c, Å | 15.0594(9) | 13.2529(6) | 9.8707(2) | 29.6078(13) | 25.840(8) |
| α, deg | 107.591(4) | ||||
| β, deg | 90.017(5) | 110.075(5) | 91.040(2) | ||
| γ, deg | 94.437(4) | ||||
| V, Å3 | 1144.61(11) | 1251.65(10) | 2662.27(10) | 3800.1(3) | 1648.3(9) |
| T, K | 248(2) | 292(2) | 100(2) | 100(2) | 293(2) |
| Space group | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c | Pbcn | P21/c |
| Z | 4 | 2 | 4 | 4 | 4 |
| μ(Mo Kα), mm−1 | 2.777 | 3.318 | 23.252 | 2.321 | 14.950 |
| Θ range, deg | 1.96 to 28.32 | 1.75 to 26.37 | 3.18 to 74.29 | 2.68 to 26.37 | 3.15 to 25.00 |
| Refl. collected | 8308 | 8687 | 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 071 071 |
40876 |
5264 |
| Uniq reflect/Rint | 2604/0.0384 | 5123/0.0239 | 5330/0.0612 | 3879/0.0326 | 2884/0.0340 |
| R 1/wR2 (I > 2σ) | 0.0376/0.0655 | 0.0295/0.0638 | 0.0500/0.1287 | 0.0315/0.0726 | 0.0420/0.0778 |
| R 1/wR2 (all data) | 0.0529/0.0703 | 0.0336/0.0677 | 0.0521/0.1304 | 0.0382/0.0764 | 0.0666/0.0872 |
| Max. shift/esd | 0.001 | 0.009 | 0.001 | 0.001 | 0.004 |
| Residual ρ/e Å−3 | 0.669 and −0.633 | 1.181 and −0.975 | 1.895 and −2.123 | 2.016 and −1.927 | 1.161 and −1.361 |
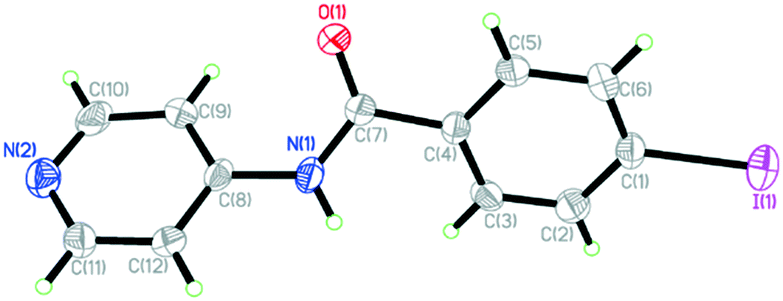 | ||
| Fig. 1 ORTEP diagram showing thermal ellipsoids at the 50% probability level and numbering scheme for the INPBA crystal structure. | ||
INPBA optimized torsional angles Φ2 and Φ3 (B3LYP-D3/6-311G++(d,p) and DGDZVP, see the Experimental section) are −179° and 25.8°, respectively, similar to the experimental conformation. However, the calculated angle Φ1 is 1.2°, indicating a planar arrangement of the pyridyl and amide groups. The electrostatic potential on the 0.002 electron per Å molecular surface of optimized INPBA (Fig. 2) exhibits the characteristic positive electrostatic potential at the end region of the iodine atom along the C–I bond that corresponds to the σ-hole. The maximum value for the relative electrostatic potential on the iodine atom is 33.7 kcal mol−1 for INPBA. This value is intermediate between the electrostatic potential calculated for weak halogen-bonding donors such as 2,8-diiodo-6H,12H-5,11-methanodibenzodiazocine (31.0 kcal mol−1)37 and the powerful halogen bond donor 1,4-bis(iodoethynyl)benzene (46.2 kcal mol−1).21
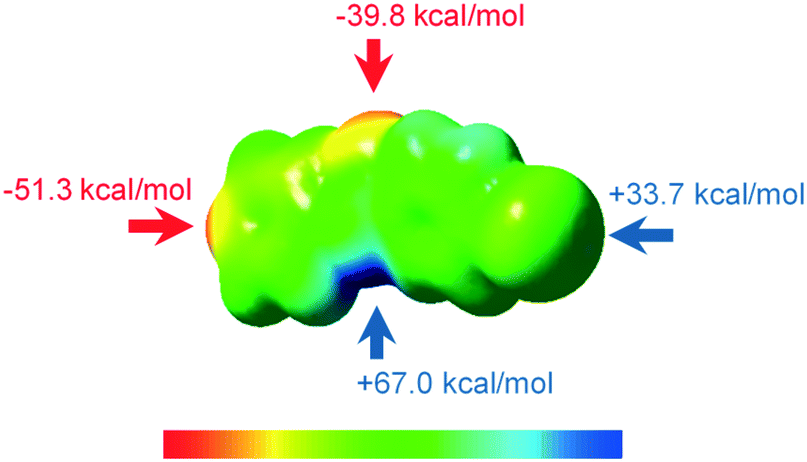 | ||
| Fig. 2 Computed electrostatic potentials of the optimized INPBA obtained by the molecular electron density of 0.002 electron per Å. Electrostatic values range from −51.4 (red) to 67.1 (blue) kcal mol−1. | ||
The lower ESP value for the iodine atom of N-(4-iodophenyl)isonicotinamide (27.0 kcal mol) (Fig. S3, ESI†) compared with 33.7 kcal mol−1 for INPBA proves the effect on the binding of the electron-withdrawing carbonyl group to the halogenated aromatic ring and this suggests that INPBA has a stronger halogen-bonding ability. The hydrogen atom of the INPBA amide group also shows a high positive electrostatic potential. As expected, two minima of electrostatic potential are located on the pyridine nitrogen atom and on the oxygen atom of the amide group. These results show that INPBA can act as a halogen bonding donor or acceptor and a hydrogen bonding acceptor or donor as evidenced by its crystalline structure described below.
The supramolecular arrangement of INPBA in the crystal structure shows chains of molecules, along the b axis, aligned head-to-tail by halogen bonding C(1)–I(1)⋯N(2) (2.989 Å, 175.3°) (Table 3).
| C–I⋯A | C–I⋯A (Å) | C–I (Å) | <(C–I⋯A) (deg) | |
|---|---|---|---|---|
| Symmetry codes: (i) x, 1 + y, z; (ii) −x, −y, 1 − z; (iii) −x, 1/2 + y, 1/2 − z; (iv) −1/2 + x, 1.5 − y, −z; (v) 1 + x, 1.5 − y, −1/2 + z. | ||||
| INPBA | C(1)–I(1)⋯N(2)i | 2.989(3) | 2.111(4) | 175.3(1) |
| 1 | C(22)–I(2)⋯O(5)ii | 3.498(4) | 2.104(4) | 152.5(1) |
| 2 | C(22)–I(2)⋯I(1)iii | 3.938(1) | 2.092(6) | 166.2(2) |
| 3 | C(1)–I(1)⋯O(1)iv | 3.257(2) | 2.098(3) | 148.8(1) |
| 4 | C(1)–I(1)⋯I(3)v | 3.858(1) | 2.10(1) | 175.5(3) |
The C–I⋯N interaction distance (green in Fig. 3) corresponds to a reduction of approximately 15% of the sum of the van der Waals radii of N and I (3.53 Å).The chains of INPBA are joined by hydrogen bonds (in blue in Fig. 3a) between carboxamide groups, thus resulting in layers in the ab plane that pack in an anti-parallel fashion to afford a centric crystal. Hydrogen bond distances and angles are gathered in Table S1 in the ESI.† These layers are practically isolated from each other due to the twisted conformation of the phenyl moieties, preventing the π–π stacking interactions (Fig. 3(b)).
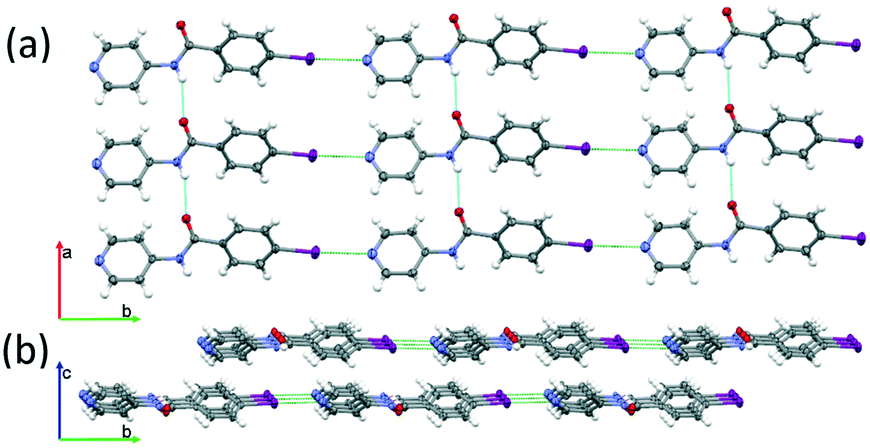 | ||
Fig. 3
INPBA structure: (a) 2D sheets formed by Npyr⋯I halogen bonding in green and perpendicular N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C carboxamide hydrogen bonds in blue; (b) isolated layers. Thermal ellipsoids are drawn at the 50% probability level. C carboxamide hydrogen bonds in blue; (b) isolated layers. Thermal ellipsoids are drawn at the 50% probability level. | ||
Dimers XB-exp and HB-exp extracted from the crystalline structure (Fig. S4 in the ESI†) were optimized with B3LYP-D3 using DGDZVP for the iodine atom and 6-311G++(d,p) for the rest of the atoms. The optimized dimers were designated as HB-opt and XB-opt (Fig. S4 in the ESI†). The interaction energies and the key geometric parameters of the optimized dimers are gathered Table S2 in the ESI.† The C–I⋯N contact of the optimized dimers exhibits structural parameters close to the experimental data. The negative interaction energy (−6.6 kcal mol−1) proves the non-covalent interaction between the σ-hole of the iodine atom and the nitrogen atom of the pyridyl group. The dimer based on the hydrogen bond converged into HB-opt that exhibits a H⋯O distance considerably shorter than the sum of the van der Waals radii and the experimental distance. The calculated interaction energy of HB-opt was −16.6 kcal mol−1. The interaction energies and the geometric parameters of the optimized dimers confirm that both hydrogen and halogen bonds collaborate to support the crystalline structure of INPBA.
Solid network of metal complexes
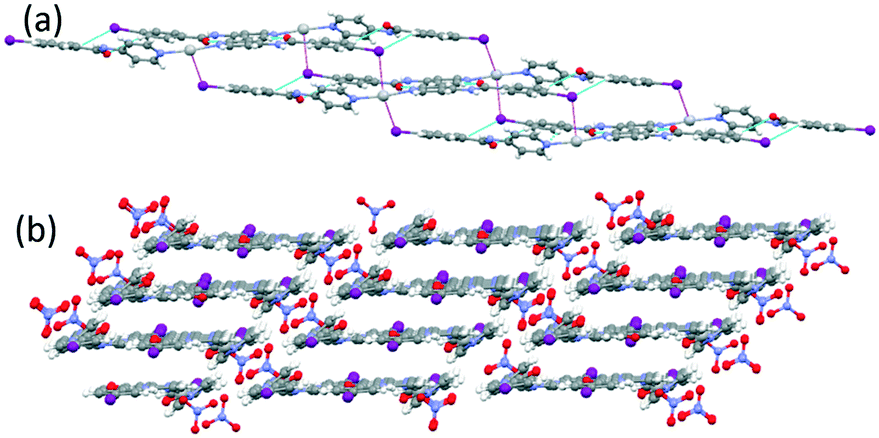
The reaction of INPBA with AgNO3, ZnBr2 and Zn(NO3)2, in the presence of sodium benzoate, produced three discrete metal complexes, namely [Ag(INPBA)2]NO3 (1), [ZnBr2(INPBA)2] (2), and [Zn(OCOPh)2(INPBA)2] (3). The reaction with HgI2 yielded the extended coordination polymer [HgI2(INPBA)]n (4). The solid state network of the complexes depends on the coordination number and the geometry of the metal ions and it is determined by the concomitant (or not) formation of hydrogen and halogen bonding and π–π stacking interactions. triclinic space group and its asymmetric unit contains two INPBA ligands coordinated in a linear fashion to a silver atom and a nitrate anion. The pyridine rings of these two ligands form an angle of 36.5° and the carbonyl groups CO(1) and CO(2) as shown in Fig. 4a are on the same side (cis conformation). This arrangement allows the formation of dimers by hydrogen bonding (C(5)–H⋯O(1) = 2.594 Å, 172.6°; C(11)–H⋯O(2) = 2.719 Å, 143.2° and C(20)–H⋯I(1) = 3.336 Å, 152.0°) as shown in Fig. 4a. These dimers are bound by hydrogen bonds that involve the iodine atom and these bonds give rise to ribbons (C(9)–H⋯I(1) = 3.3421 Å, 139.1°), as shown in Fig. 4b. The edge-to-edge hydrogen bonding interactions (CAr–H⋯I) promote a planar arrangement, in a similar way to the 1,4 and 1,3-bis(iodoethynyl)benzene derivatives.22
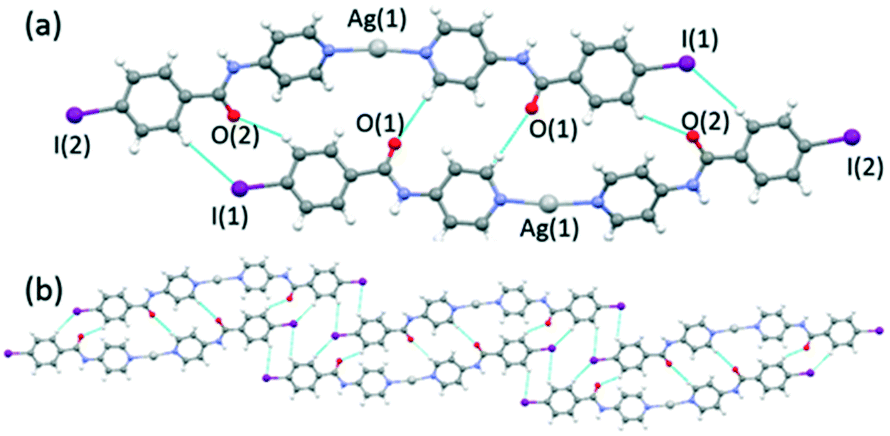 | ||
| Fig. 4 (a) A view showing Ag(I) coordination in 1 and hydrogen bonded dimers and (b) ribbons formed by edge-to-edge hydrogen bonding interactions (CAr–H⋯I). | ||
The central region of the iodine atom shows a negative electrostatic potential, which interacts with the silver cation (in violet in Fig. 5a). These results are consistent with the charge polarization model at the heavy halogen.27 The C–I⋯Ag distances (3.472 and 3.585 Å) are significantly shorter than the van der Waals contact, 3.70 Å (van der Waals radius for Ag = 1.72 and I = 1.98 Å), and the C–I⋯Ag angles are close to 90° (97.9 and 108.6°). These interactions link up the ribbons described above forming planes (Fig. 5a).
 | ||
| Fig. 5 A view showing C–I⋯Ag interactions in violet (a) and nitrate anion linking planes by C–I⋯O halogen bonding and Ag⋯O interactions (b) in 1. | ||
Finally, the nitrate anions connect the planes by hydrogen bonds (N(2)–H⋯O(5) = 3.024 Å, 158.8° and N(4)–H⋯O(4) = 2.958 Å, 161.3°), an Ag⋯O bifurcated interaction (Ag(1)⋯O(3) = 2.856 Å; Ag(1)⋯O(4) = 2.934 Å) and halogen bonding C–I(2)⋯O(5) (3.498 Å, 152.5°), which gives rise to a three-dimensional network (Fig. 5b).
 | ||
| Fig. 6 Coordination environment of zinc(II) in 2 (a) and 3 (b). | ||
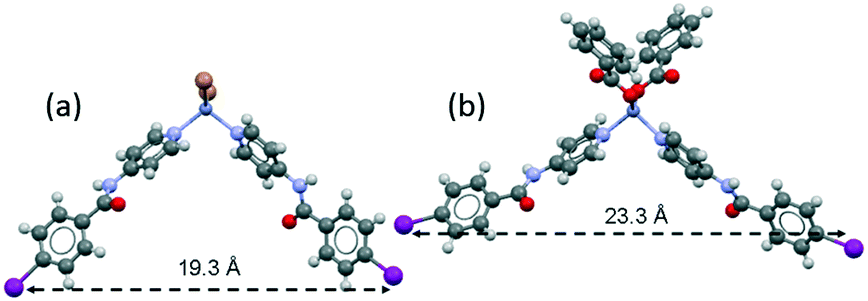
The supramolecular arrangement of 2 is formed due to C–I⋯I halogen bonding interactions and hydrogen bonds formed by each of the two amide groups. A summary of the data for the halogen and hydrogen bonding interactions is provided in Table 3 and Table S1 (ESI†), respectively. Each iodine atom of 2 has a different role; I(2) is a halogen bond donor while the I(1) atom acts as a halogen bond acceptor (C–I(2)⋯I(1) = 3.938 Å, 166.2°). The I(2) iodine atom is oriented towards the equatorial belt of the I(1) iodine atom, thus forming an angle close to 90° (Idonor⋯Iaceptor–C = 75.1°), which promotes the formation of a helical chain that propagates along the 21 screw axis in the b-direction. The length of the repeating unit of the helical chain is 27.8 Å and the distance between contiguous zinc cations is 24.2 Å, as shown in Fig. 7a and b. Each of these chains is linked to two enantiomeric helices (Fig. 7c) through hydrogen bonds between the amide group along the c-axis (N(4)–H⋯O(2) = 2.795 Å, 149.2°) of the INPBA ligand with the more tilted conformation with respect to the carboxamide group (Φ1 = −18.4°; Φ3 = −26.8°) (Table 2).
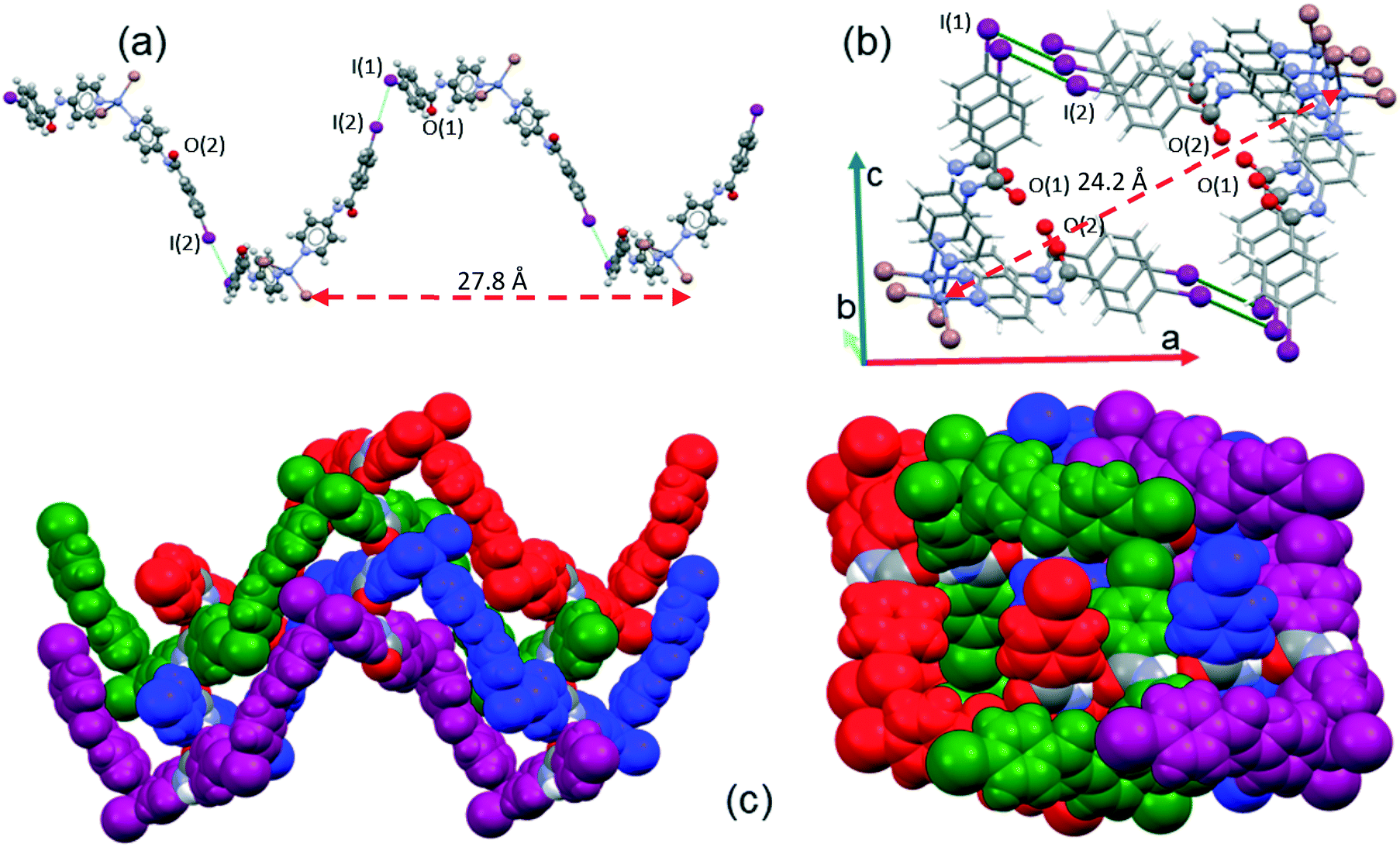 | ||
| Fig. 7 (a) A view along the b-axis of a helical strand of 2 formed by I⋯I halogen bonding; (b) view along the c-axis, and (c) an enantiomeric helical strand hydrogen bonded along the b- and c-axes. | ||
The second amide group forms hydrogen bonds with one of the bromine atoms (N(2)–H⋯Br(1) = 3.743 Å, 150.9°)40 and this leads to a ladder arrangement due to the formation of weak hydrogen bonds (C(5)–H⋯O(1)C = 3.577 Å, 158.1°) (Fig. S5 in the ESI†). Evidence for the different environments of the carbonyl groups is provided by two bands at 1695 and 1670 cm−1 in the ATR-IR spectra.
The supramolecular organization of 3 is determined by the halogen bonding between the iodine atom and carbonyl oxygen atom of the carboxamide group (C–I(1)⋯O(1)C = 3.257 Å, 148.8°) and hydrogen bond N(1)–H⋯O(3)C (2.822 Å, 167°) in which the hydrogen bond acceptor is the carbonyl oxygen of the ester group. Halogen bonding provides corrugated layers that extend along the ac plane while hydrogen bonding connects these planes along the b-axis to give a three-dimensional arrangement (Fig. 8).
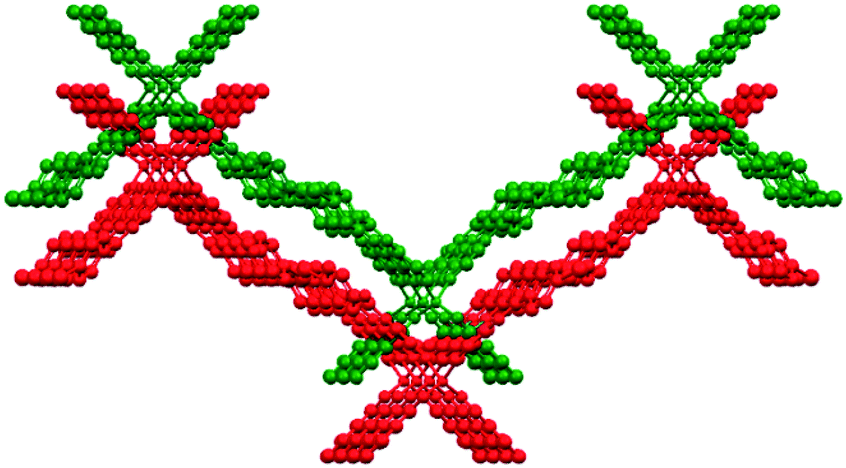 | ||
| Fig. 8 Corrugated layers stacking along the b-axis in 3. | ||
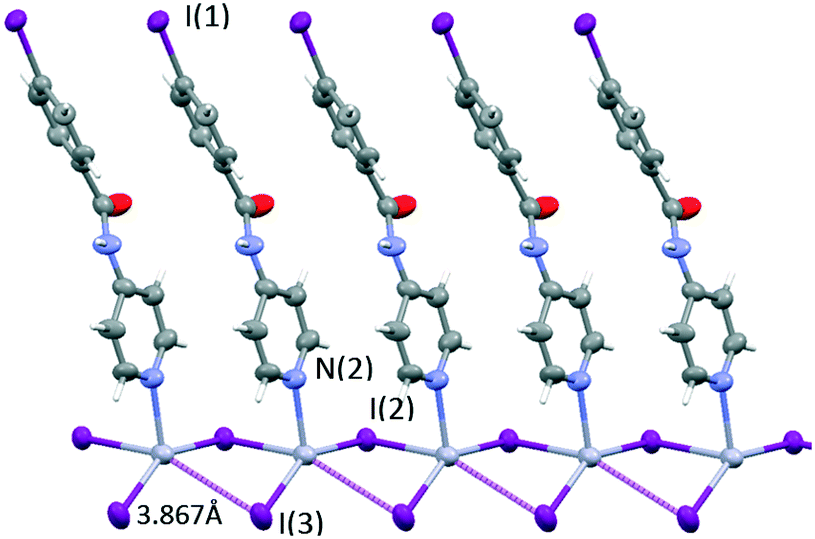 | ||
| Fig. 9 A view showing the rhomboidal base pyramid coordination sphere of mercury(II) in 4. Thermal ellipsoids are drawn at the 50% probability level. | ||
The rhomboidal base has two short Hg–I bonds (2.651 and 2.666 Å) and two long ones (3.238 and 3.867 Å). The very long Hg–I interatomic distance (3.867 Å) (violet dashed lines in Fig. 9) is too long to be considered a bond (values for the van der Waals radii range from 1.8241 to 2.24 Å42,43 for mercury and 2.1 Å for iodine), but short enough to point it out. The mercury coordination geometry can also be described as a see-saw structure with a geometrical index τ444,45 of 0.74 and a dihedral angle between the I–Hg–I and I–Hg–N planes of 89.7°.
Mercury rhomboid–pyramidal coordination units share the edges of their base and all INPBA ligands point to the same side of the polymer chain along the c-axis, as shown in Fig. 9. This organization promotes π–π stacking, since the adjacent molecules of INPBA are related by a translation along the a-direction and the distance between planes containing pyridyl moieties is 3.559 Å and 3.587 Å between the planes containing phenyl rings. In addition, 1D chains are linked by halogen bonding C(1)–I(1)⋯I(3) (3.86 Å and 175°). This XB in green in Fig. 10 gives rise to corrugated planes that are connected by weak hydrogen bonds involving the oxygen of the carbonyl group (C(5)–H⋯O(1)C(7) = 2.616 Å, 168.9°), metal iodide (N(1)–H⋯I(3)–Hg(1) = 3.340 Å, 157.9°) and the carbon-bound iodine atom of the INPBA ligand (C(2)–H⋯I(1)–C(1) = 3.314 Å, 140.6°). This gives rise to a three-dimensional organization, as shown in Fig. 10.
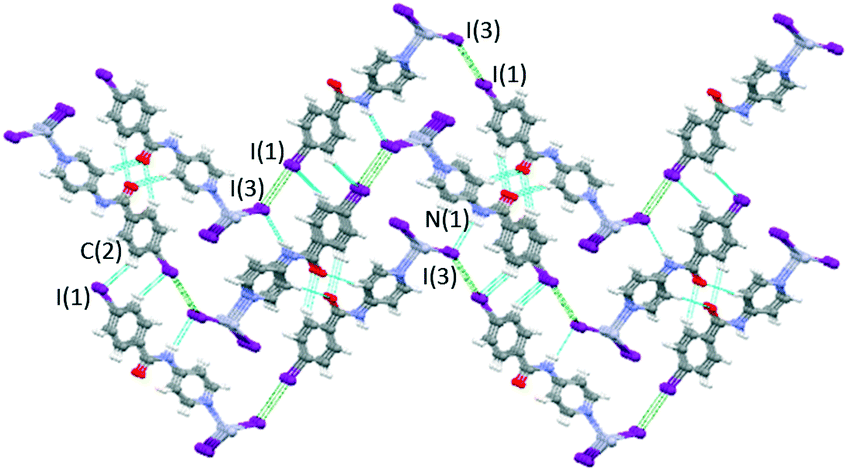 | ||
| Fig. 10 Three dimensional organization in the crystal structure of 4. Corrugated planes formed by halogen bonding, in green, connected by hydrogen bonds, in blue. | ||
The comparison of the structure of 4 with others in which the mercury atom has a similar coordination45–50 shows that the establishment of halogen bonding causes an elongation of the Hg–I bond, shortening of the Hg–N bond and a deviation of the mercury atom from the plane defined by the iodine atoms. Thus, the structure of 4 has the longest Hg–I distance (2.651 Å), the shortest Hg–N bond distance (2.368 Å) and the longest distance between the mercury atom and the plane defined by the iodine atoms (0.66 Å). These effects are consistent with halogen bonding wherein the iodine atom attached to the phenyl moiety behaves as a halogen bond donor and the iodine atom attached to mercury as a halogen bond acceptor.
Hirshfeld surface analysis
The intermolecular contacts in the crystal structures of INPBA and 1 to 4 were analysed by considering the Hirshfeld surface using the CrystalExplorer programme.51 Hirshfeld surfaces can encode different properties in a three-dimensional picture and all of this information can be summarized in a bidimensional fingerprint (di, de) graph.52 As expected, the Hirshfeld surfaces of each crystallographically independent INPBA ligand in the crystal structures of INPBA and 1 to 4 revealed the close contacts of halogen and hydrogen bonding, in addition to other weaker interactions and contacts. The main contributors to the Hirshfeld surface are weak dispersion forces such as H⋯H, C⋯H, and I⋯H. The fingerprint plots (Fig. 11) of each INPBA ligand are different, including those belonging to the same crystal structure, and they are characterized by spikes that correspond to O⋯H, C⋯H and I⋯H interactions denoted as a, b and c, respectively. The major differences between the graphs are the presence or absence of a clear area near to the centre of the plot, i.e., in the vicinity of (di, de) ≈ 1.8–2.0 Å (named d). These are related to the planar π–π stacking (C⋯C and C⋯N interactions).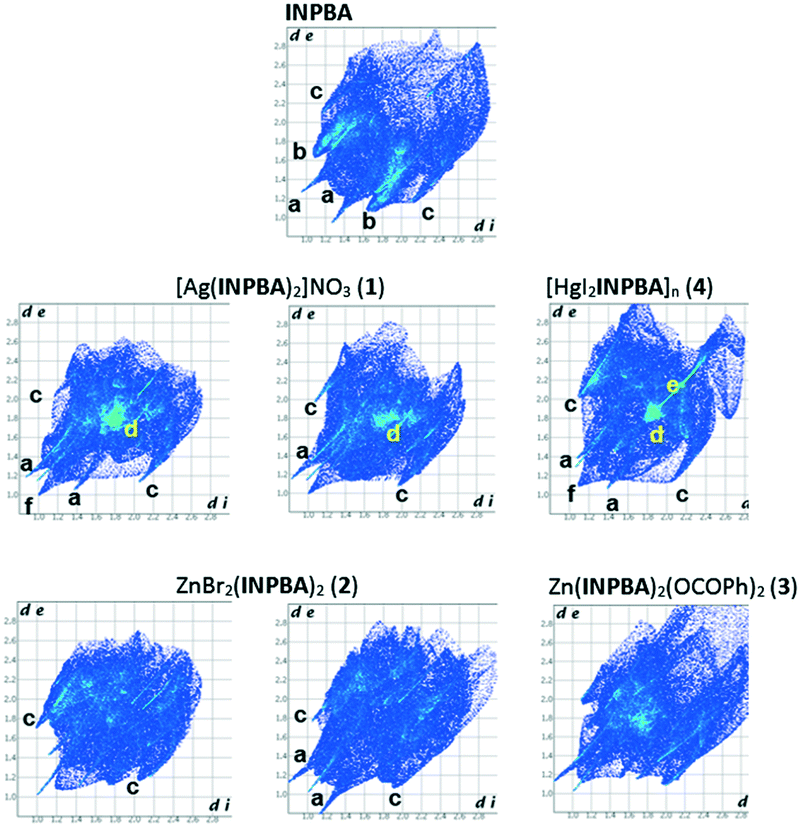 | ||
| Fig. 11 Fingerprint plots of the INPBA ligands, crystallographically different in the crystal structures of INPBA and 1 to 4. | ||
The π–π stacking contribution to the Hirshfeld surfaces for INPBA and 1 to 4 structures ranged from 0 to almost 14% in INPBA and 1, respectively. However, in the silver complex, the ligand with the more planar conformation has a greater π–π interaction but there is no correlation between the conformation and π–π stacking in the studied structures.
Conclusions
The preparation and crystal structures of 4-iodo-N-(4-pyridyl)benzamide (INPBA), three metal complexes, [Ag(INPBA)2]NO3, [ZnBr2(INPBA)2], and [Zn(OCOPh)2(INPBA)2], and a coordination extended coordination polymer [HgI2(INPBA)]n are reported. The INPBA ligand has been proven to be very versatile and gives rise to different types of interactions such as coordination with metals, π–π stacking, halogen bonding and hydrogen bonding. This ability of INPBA promotes an increase in dimensionality and improves the stability of the structures of 1–4. Halogen bonding of the types C–I⋯A− (A− = NO3−), C–I⋯OC and C–I⋯I–Hg gives rise to planes in compounds 1, 3 and 4, respectively. Helical chains with a repeating unit length of 27.76 Å are formed in 2 by C–I⋯I–C halogen bonding. Furthermore, the simultaneous action of these interactions highlights the complexity and challenge in programming the supramolecular assembly of metal organic networks, since small variations can cause very significant structural changes.
Experimental
4-Iodobenzoyl chloride, 4-aminopyridine, triethylamine, silver nitrate, zinc bromide and mercury(II) iodide were used as received from commercial sources (Sigma-Aldrich or Acros). 1H-NMR and 13C-NMR spectra were recorded on a Bruker Avance 300 spectrometer. Infrared (IR) spectra were recorded on an ATR unit-upgraded (Golden Gate) Bruker FT-IR Vertex 70 spectrophotometer. Mass spectra were recorded on a VG Auto-spec system with the ESI technique. C, H, and N analyses were carried out on a PerkinElmer 2400 microanalyzer.Synthesis of 4-iodo-N-(4−pyridyl)benzamide (INPBA)
A solution of 4-iodobenzoyl chloride (1.32 g, 5.0 mmol) and triethylamine (0.50 g, 5.0 mmol) in 50 mL of chloroform was cooled to 0 °C in an ice bath for 10 min. 4-Aminopyridine (0.470 g, 5.0 mmol) was added slowly to the cold solution over a period of 10 min. The reaction mixture was stirred at room temperature for 16 h and the concentration afforded a white precipitate. The solid was filtered off and washed several times with cold chloroform. Yield 1.33 g (82%). 1H-NMR spectrum (d6-DMSO, 300 MHz) δ from TMS: 7.63 (m, 3H, Bz), 7.78 (d, 2H, Py), 7.95 (d, 2H, o-Bz), 8.47 (d, 2H, Py), 10.60 (s, NH). 13C-NMR spectrum (d6-DMSO, 62.5 MHz): 114.70, 128.56, 129.15, 132.76, 134.96, 146.66, 150.97, 167.19. IR (ATR) νmax (neat)/cm−1: 3332, 1660, 1587, 1502, 1477, 1413, 1330, 1282, 1210, 822, 748. High resolution positive ESI-MS (a.m.u.): calcd. for M + H: 324.9834; found: 324.9827. Anal. calcd. for C12H9IN2O: C 44.47, H 2.80, N 8.64; found: C 44.22, H 2.67, N 8.61.Preparation of INPBA metal complexes 1, 2 and 4
A solution of INPBA (32 mg, 0.10 mmol) in THF (10 mL) was added to a solution of inorganic salt (0.1 mmol) in methanol (5 mL). The solution was allowed to slowly evaporate with orbital stirring over a period of 48 h, during which it generated colourless crystals.Preparation of [Zn(OCOPh)2(INPBA)2] (3)
X-ray quality single crystals of 3 were obtained by three-layer diffusion, INPBA in methanol, sodium benzoate in ethanol and zinc nitrate in water.X-ray crystallographic analysis
X-ray diffraction experiments were carried out on Oxford-diffraction Xcalibur S and Bruker AXS D8 Venture diffractometers. Mo-Kα radiation was used for the data collection for INPBA, 1, 3 and 4 and Cu-Kα for 2. XSCANS and CrysAlis software packages were used to process the data. Final cell parameters were obtained by global refinement of reflections obtained by integration of all the frame data. The structures were solved by direct methods and refined by the full-matrix method based on F2 using the SHELXTL programme. The non-hydrogen atoms of INPBA and 1 to 4 were refined anisotropically, and the hydrogen atoms of 1, 3, 4 were observed in difference electron density maps and refined isotropically. Hydrogen atoms of INPBA and 2 were included at idealized positions by using a riding model and refined isotropically. The crystal parameters and basic information related to data collection and structure refinement are summarized in Table 1.Hirshfeld surface analysis
Hirshfeld surfaces and the associated fingerprint plots were calculated using CrystalExplorer,51 which accepts a structure input file in the CIF format. Bond lengths of hydrogen atoms were set to typical neutron values (C–H = 1.083 Å, N–H = 1.009 Å, O–H = 0.983 Å). The distances from the Hirshfeld surface to the nearest atoms outside and inside the surface were characterized by the quantities de and di, respectively, as well as the normalized contact distance based on these, dnorm = (di − rivdW)/ rivdW + (de − revdW)/revdW), with rivdW and revdW being the van der Waals radii of the atoms. The 2D-fingerprint of the Hirshfeld surface summarise the complex information contained in a molecular crystal structure into a single, unique full colour plot, showing the frequency of each combination of de and di across the surface of a molecule.Computational methods
Quantum chemical calculations were performed using the Gaussian 09 package.53 Optimization of the dimers extracted from the experimental crystal structures (see Fig. S4†) was carried out using DFT in conjunction with the B3LYP exchange-correlation functional54–56 and Grimme D3BJ van der Waals dispersion correction.57 The 6-311G++(d,p) basis set58 was used for all atoms except the iodine atom, for which the DGDZVP one was employed.59 The optimization was conducted without geometry restrictions. Vibrational frequency analysis was used to characterize the stationary points as minima, by checking that they did not present any imaginary frequency. The interaction energies between the different monomers that form part of the complexes were calculated at the same level of theory as the difference between the total complex energy and the sum of the total energies of the monomers. The interaction energies (ΔE) were corrected (ΔEBSSE) using the counterpoise method of Boys–Bernardi so as to eliminate the basis set superposition error (BSSE).60 Moreover, the interaction energies of the optimized complexes were corrected for scaled (0.9887)61 zero-point energy differences (obtained from the frequency calculations described above).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors give thanks to the Crystallography Service of the University of Zaragoza-CSIC (Spain), the University of Pais Vasco (Spain), the National University of Singapore and the Servicio General de Apoyo a la Investigación SAI, Universidad de Zaragoza. This work was supported by the Ministerio de Economía, Industria y Competitividad under the projects PGC2018-093761-B-C31, PGC2018-097583-B-100 and MAT2017-84838-P. The authors acknowledge support from the Gobierno de Aragón-FEDER for funding the Crystal and Polymers Group (E47_20R, FEDER 2014-2020 Construyendo Europa desde Aragón).Notes and references
- E. R. T. Tiekink and J. J. Vittal, Frontiers in crystal engineering, Wiley, 2006 Search PubMed.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC.
- P. A. Gale and J. W. Steed, Supramolecular chemistry : from molecules to nanomaterials, Wiley, 2012 Search PubMed.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed.
- B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- L. Brammer, Chem. Soc. Rev., 2004, 33, 476–489 RSC.
- K. S. Kim, P. Tarakeshwar and J. Y. Lee, Chem. Rev., 2000, 100, 4145–4185 CrossRef CAS PubMed.
- R. Bertani, P. Sgarbossa, A. Venzo, F. Lelj, M. Amati, G. Resnati, T. Pilati, P. Metrangolo and G. Terraneo, Coord. Chem. Rev., 2010, 254, 677–695 CrossRef CAS.
- L. Brammer, G. Mínguez Espallargas and S. Libri, CrystEngComm, 2008, 10, 1712–1727 RSC.
- M. Borovina, I. Kodrin and M. Đaković, CrystEngComm, 2018, 20, 539–549 RSC.
- B. Li, S. Q. Zang, L. Y. Wang and T. C. W. Mak, Coord. Chem. Rev., 2016, 308, 1–21 CrossRef CAS.
- Y. V. Torubaev, I. V. Skabitskiy, P. Rusina, A. A. Pasynskii, D. K. Rai and A. Singh, CrystEngComm, 2018, 20, 2258–2266 RSC.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775–2825 CrossRef CAS PubMed.
- C.-Y. Lin, Z.-K. Chan, C.-W. Yeh, C.-J. Wu, J.-D. Chen and J.-C. Wang, CrystEngComm, 2006, 8, 841–846 RSC.
- S. K. Chandran, R. Thakuria and A. Nangia, CrystEngComm, 2008, 10, 1891–1898 RSC.
- H. R. Khavasi and A. A. Tahrani, CrystEngComm, 2013, 15, 5799–5812 RSC.
- H. R. Khavasi and A. Azhdari Tehrani, Inorg. Chem., 2013, 52, 2891–2905 CrossRef CAS PubMed.
- C. J. Wu, C. Y. Lin, P. C. Cheng, C. W. Yeh, J. Der Chen and J. C. Wang, Polyhedron, 2011, 30, 2260–2267 CrossRef CAS.
- J. Gamekkanda, A. Sinha, J. Desper, M. Đaković and C. Aakeröy, Crystals, 2017, 7, 226 CrossRef.
- L. González, N. Gimeno, R. M. Tejedor, V. Polo, M. B. Ros, S. Uriel and J. L. Serrano, Chem. Mater., 2013, 25, 4503–4510 CrossRef.
- L. González, R. M. Tejedor, E. Royo, B. Gaspar, J. Munárriz, A. Chanthapally, J. L. Serrano, J. J. Vittal and S. Uriel, Cryst. Growth Des., 2017, 17, 6212–6223 CrossRef.
- K. Raatikainen and K. Rissanen, Chem. Sci., 2012, 3, 1235–1239 RSC.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- H. R. Khavasi, M. Hosseini, A. A. Tehrani and S. Naderi, CrystEngComm, 2014, 16, 4546–4553 RSC.
- S. Saha and G. R. Desiraju, J. Am. Chem. Soc., 2017, 139, 1975–1983 CrossRef CAS PubMed.
- P. Politzer, P. Lane, M. C. Concha, Y. Ma and J. S. Murray, J. Mol. Model., 2007, 13, 305–311 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- C. B. Aakeröy, N. Schultheiss, J. Desper and C. Moore, CrystEngComm, 2007, 9, 421–426 RSC.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS.
- A. Farina, S. V. Meille, M. T. Messina, P. Metrangolo, G. Resnati and G. Vecchio, Angew. Chem., Int. Ed., 1999, 38, 2433–2436 CrossRef CAS.
- J. Cao, X. Yan, W. He, X. Li, Z. Li, Y. Mo, M. Liu and Y.-B. Jiang, J. Am. Chem. Soc., 2017, 139, 6605–6610 CrossRef CAS PubMed.
- T. A. Logothetis, F. Meyer, P. Metrangolo, T. Pilati and G. Resnati, New J. Chem., 2004, 28, 760–763 RSC.
- V. R. Hathwar, R. G. Gonnade, P. Munshi, M. M. Bhadbhade and T. N. G. Row, Cryst. Growth Des., 2011, 11, 1855–1862 CrossRef CAS.
- J. Lieffrig, A. G. Niassy, O. Jeannin and M. Fourmigué, CrystEngComm, 2015, 17, 50–57 RSC.
- C. J. Massena, N. B. Wageling, D. A. Decato, E. Martin Rodriguez, A. M. Rose and O. B. Berryman, Angew. Chem., Int. Ed., 2016, 55, 12398–12402 CrossRef CAS PubMed.
- L. González, S. Graus, R. M. Tejedor, P. López, J. Elguero, J. L. Serrano and S. Uriel, CrystEngComm, 2018, 20, 3167–3170 RSC.
- J. C. Noveron, M. S. Lah, R. E. Del Sesto, A. M. Arif, J. S. Miller and P. J. Stang, J. Am. Chem. Soc., 2002, 124, 6613–6625 CrossRef CAS PubMed.
- K. Donnelly, J. F. Gallagher, A. J. Lough and IUCr, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2008, 64, o335–o340 CrossRef CAS PubMed.
- K. Biradha, C.-Y. Su and J. J. Vittal, Cryst. Growth Des., 2011, 11, 875–886 CrossRef CAS.
- H. J. Bohórquez and R. J. Boyd, Chem. Phys. Lett., 2009, 480, 127–131 CrossRef.
- S. Nag, K. Banerjee and D. Datta, New J. Chem., 2007, 31, 832–834 RSC.
- S.-Z. Hu, Z.-H. Zhou and B. E. Robertson, Z. Kristallogr., 2009, 224, 375–383 CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- A. A. Tehrani, A. Morsali and M. Kubicki, Dalton Trans., 2015, 44, 5703–5712 RSC.
- Y.-H. Shen, J.-G. Liu and D.-J. Xu, Acta Crystallogr., Sect. A: Found. Crystallogr., 2005, 61, m1880–m1882 CAS.
- W. T. Chen, M. S. Wang, X. Liu, G. C. Guo and J. S. Huang, Cryst. Growth Des., 2006, 6, 2289–2300 CrossRef CAS.
- C. Hu, I. Kalf and U. Englert, CrystEngComm, 2007, 9, 603–610 RSC.
- Q.-Y. Du, J.-H. Qin and H.-X. Wu, Chin. J. Struct. Chem., 2007, 26, 817–821 CAS.
- H. P. Zhou, J. H. Yin, L. X. Zheng, P. Wang, F. Y. Hao, W. Q. Geng, X. P. Gan, G. Y. Xu, J. Y. Wu, Y. P. Tian, X. T. Tao, M. H. Jiang and Y. H. Kan, Cryst. Growth Des., 2009, 9, 3789–3798 CrossRef CAS.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, CrystalExplorer (Version 3.1), Univ. West. Aust, 2012 Search PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- N. Godbout, D. R. Salahub, J. Andzelm and E. Wimmer, Can. J. Chem., 1992, 70, 560–571 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- J. P. Merrick, D. Moran and L. Radom, J. Phys. Chem. A, 2007, 111, 11683–11700 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic files (CIF) CCDC 1423795–1423799. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce00832j |
| This journal is © The Royal Society of Chemistry 2020 |