
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tools for functional dissection of site-specific O-GlcNAcylation
Andrii
Gorelik†‡
 a and
Daan M. F.
van Aalten‡
*ab
a and
Daan M. F.
van Aalten‡
*ab
aCentre for Gene Regulation and Expression, School of Life Sciences, University of Dundee, Dundee, UK. E-mail: dmfvanaalten@dundee.ac.uk
bInstitute for Molecular Precision Medicine, Xiangya Hospital, Central South University, Changsha, China
First published on 12th June 2020
Abstract
Protein O-GlcNAcylation is an abundant post-translational modification of intracellular proteins with the monosaccharide N-acetylglucosamine covalently tethered to serines and threonines. Modification of proteins with O-GlcNAc is required for metazoan embryo development and maintains cellular homeostasis through effects on transcription, signalling and stress response. While disruption of O-GlcNAc homeostasis can have detrimental impact on cell physiology and cause various diseases, little is known about the functions of individual O-GlcNAc sites. Most of the sites are modified sub-stoichiometrically which is a major challenge to the dissection of O-GlcNAc function. Here, we discuss the application, advantages and limitations of the currently available tools and technologies utilised to dissect the function of O-GlcNAc on individual proteins and sites in vitro and in vivo. Additionally, we provide a perspective on future developments required to decipher the protein- and site-specific roles of this essential sugar modification.
 Andrii Gorelik and Daan van Aalten | Andrii Gorelik (left in the picture) received a Bachelor's degree in industrial biotechnology in 2014 from the Kyiv Polytechnic Institute (Ukraine). He then moved to the School of Life Sciences at the University of Dundee (UK) to complete a Wellcome Trust-funded PhD in 2018, establishing novel methods for functional dissection of site-specific protein O-GlcNAcylation under the supervision of Prof. Daan van Aalten. During his time in Dundee, he developed a particular interest in post-translational modifications (PTMs). He is presently undertaking postdoctoral research at the Francis Crick Institute and Imperial College London with Prof. Ed Tate in the field of PTM chemical biology, focusing on protein N-myristoylation. Daan van Aalten (right in the picture) is visiting professor at Xiangya Hospital, Central South University, Changsha, China and Professor of Biological Chemistry at the School of Life Sciences, Dundee. Originally graduated as a chemist (1994) followed by a biocomputing PhD (1997), Daan has been working on the interface between cell signalling and glycobiology since joining Dundee as a PI (1999) using a multidisciplinary approach covering everything from synthetic chemistry to genetics. Daan's work has been focused in the O-GlcNAc signalling field for the past 10 years where he has developed chemical biology tools, and uncovered novel molecular/biological mechanisms of this modification. |
Post(co)-translational protein O-GlcNAcylation
Post-translational modifications (PTMs) expand the function and regulation of proteins beyond the genetically encoded polypeptide. The number of known types of PTMs is close to several hundred,1 ranging from substantial alterations (proteolytic cleavage resulting in large protein fragments, attachment of ubiquitin chains, glycosylation with polysaccharides)2–4 to small tags (phosphorylation, acetylation, methylation, lipidation).5–8One of these PTMs, O-GlcNAcylation, is the attachment of a single O-linked N-acetylglucosamine onto serine and threonine side chains of intracellular proteins,9 which, in some cases, is also thought to be co-translational.10 Installation of the sugar is mediated by the O-GlcNAc transferase (OGT)11,12 on nuclear and cytoplasmic proteins, covering 10–20% of the whole human proteome (Fig. 1a).13 The O-GlcNAcase (OGA) opposes OGT by hydrolysing the O-glycosydic bond to release GlcNAc.14 Thus, a single pair of enzymes makes O-GlcNAc a reversible and highly dynamic modification that has been implicated in a myriad of processes such as signalling, metabolism, stress response and transcription.15
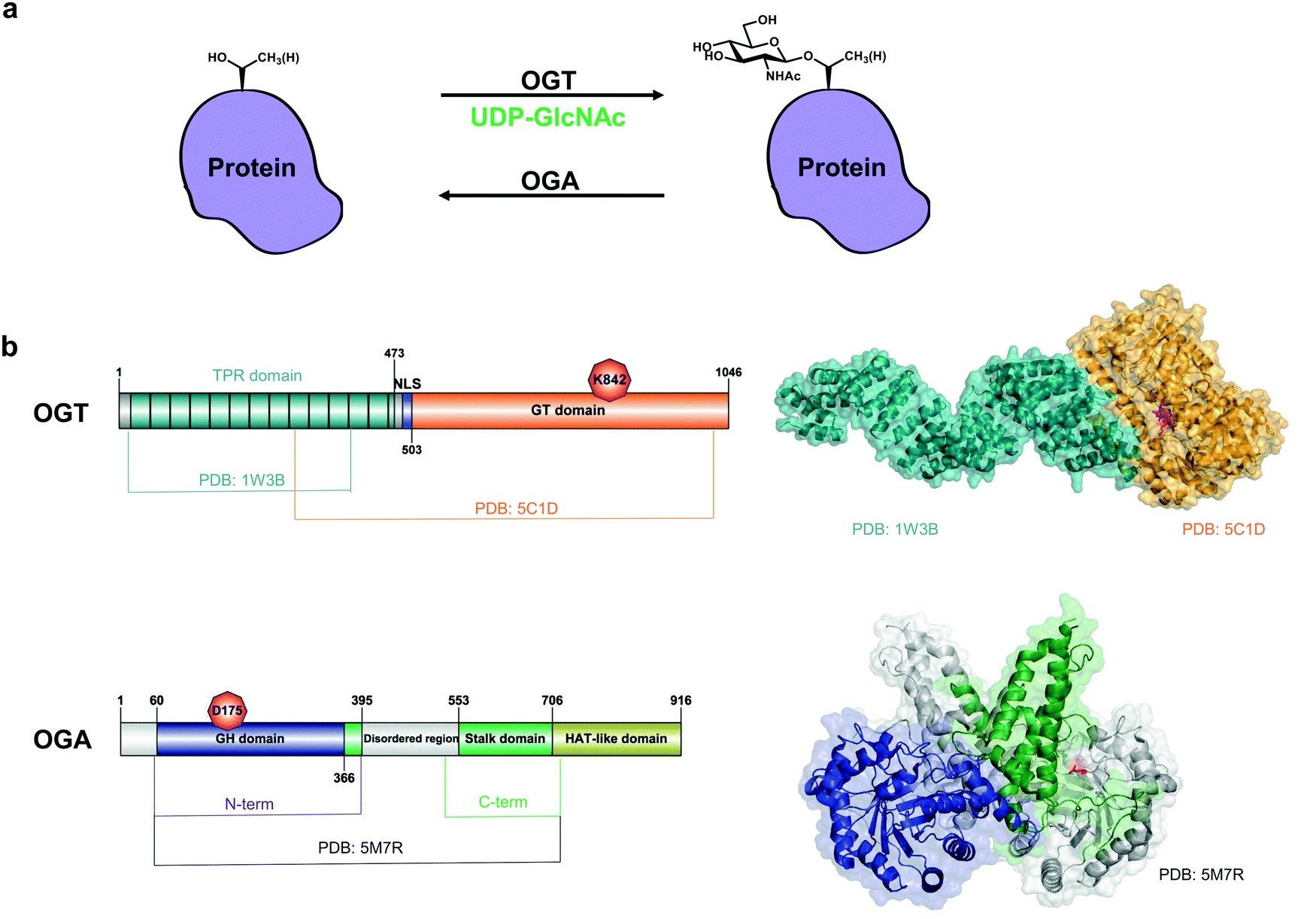 | ||
| Fig. 1 O-GlcNAc cycling on nucleocytoplasmic proteins. (a) OGT and OGA control the addition and removal of GlcNAc on serine and threonine residues. (b) Domain architecture and structures of OGT (composite of PDB: 1W3B and PDB: 5C1D) and OGA (PDB: 5M7R, lacks the disordered region). Catalytic site residues K842 (OGT) and D175 (OGA) are coloured in red. GT domain – glycosyltransferase domain. NLS – nuclear localization sequence,40 GH domain – glycoside hydrolase domain. | ||
OGT is a family GT41 GT-B glycosyltransferase16 that consists of a 13.5 tetratricopeptide repeat (TPR) domain and a Rossman-like catalytic domain (Fig. 1b).17 Although the 13.5-TPR OGT is the major isoform, two additional isoforms may arise from alternative splicing and alternative start codons: a truncated OGT with 2.5 TPRs localised in the nucleus and cytoplasm and a 9.5-TPR OGT containing a mitochondrial targeting sequence.18,19 The function of the latter two isoforms is unclear. Moreover, it appears that full-length nucleocytoplasmic OGT is sufficient for O-GlcNAcylation of mitochondrial proteins.20 O-GlcNAc transfer by OGT involves an ordered bi-bi catalytic mechanism: binding of the donor sugar nucleotide UDP-GlcNAc is followed by the target protein acceptor substrate.17,21 A second catalytic activity of OGT, host cell factor-1 (HCF-1) cleavage, is performed in the same active site and results in proteolytic maturation of this transcriptional co-regulator.22 More recently, OGT was shown to possess a third (and unexpected) activity – catalysis of S-GlcNAc transfer onto cysteines.23,24 Although the crystal structures of the TPRs and the truncated OGT have been solved (Fig. 1b),17,25 the full-length OGT structure is yet to be determined and may provide a further understanding of how OGT recognizes its substrates. The TPR domain of OGT mediates substrate recognition through a so-called asparagine ladder.17 Using protein microarrays and mutational analysis, the five asparagines in the lumen of the TPR domain have been shown to contribute to the binding of OGT protein substrates beyond its active site.26,27 Recently, additional aspartates in the TPR domain that influence recognition of some OGT substrates have been identified.28 OGT does not utilise a defined motif for O-GlcNAcylation. However, in a high-throughput screen a preferred O-GlcNAc sequon has been determined as T-P-V-gS/T-R-A12 which is in good agreement with P-X-gT-X-A and P-V-gS29 and P/V-P/V-V-gS/T-S/T,30,31 previously identified in large-scale proteomics studies.
OGA is a GH84 family glycoside hydrolase16 consisting of an N-terminal catalytic domain and a histone acetyltransferase (HAT)-like domain separated by a disordered region and a stalk domain (Fig. 1b). The HAT-like domain lacks the ability to bind acetyl coenzyme A (the substrate for acetylation) and its role remains enigmatic.32 The putative short human OGA isoform lacks the HAT-like domain and a portion of the stalk domain, resulting in reduced activity due to the inability to form a functional dimer.33 Overexpression of this isoform with a GFP tag results in its localisation to lipid droplets.34 However, the short OGA isoform has not been detected at an endogenous protein level. The first crystal structures of a monomeric bacterial orthologue of human OGA from Clostridium perfringens (CpOGA) in complex with its substrate peptides exhibited a V-shaped peptide conformation in the active site.35,36 Later it was shown that human OGA forms a dimer with a substrate-binding cleft where O-GlcNAc peptides assume the same V-shaped conformation upon binding in the active site, confirming the findings obtained with CpOGA.33,37,38 While the interactions of OGA active site residues with GlcNAc are conserved, exactly how (and whether) protein substrates bind beyond the active site is unknown.39 OGA itself contains a single O-GlcNAcylation site at Ser405, which may regulate OGA stability.24
The roles of O-GlcNAc on proteins have been studied in animal models (such as mice and fruit flies), where OGT deletion is lethal and causes severe developmental abnormalities.41–43 Excess of O-GlcNAc can also have negative impact on normal cell physiology as OGA knockout is lethal in mice,44 whereas increased O-GlcNAc levels have been linked to oncogenic reprogramming.45 Since its discovery, the relationship between O-GlcNAcylation and phosphorylation has been closely studied due to the frequent occurrence on the same or adjacent Ser/Thr residues which results in mutual regulation.46 The role of O-GlcNAc in stress response is exemplified by its participation in stress granule formation and oxidative stress.47 At individual protein level, lack of O-GlcNAc on tau and α-synuclein has been associated with pathological brain conditions such as Alzheimer's and Parkinson's disease.48,49 Moreover, O-GlcNAc can mediate transcription through modification of RNA Polymerase II50 and regulate autophagosome maturation by modifying SNAP29 depending on the cellular metabolic state.51
Transcriptionally, OGT expression can be decreased through nuclear degradation of an intron-retained OGT transcript in response to high overall O-GlcNAcylation levels to maintain O-GlcNAc homeostasis.52 Similar to OGT, OGA expression is also responsive to the modulation of overall O-GlcNAc modification. Pharmacological inhibition of OGA in cells increases total O-GlcNAcylation levels, while also increasing OGA expression and decreasing expression of OGT as a compensatory mechanism.53
Recently, ogt missense mutations associated with X-linked intellectual disability have been identified.54 These occur in the TPR domain of OGT54–56 as well as in the catalytic domain.57,58 Some of these mutations cause a decrease in endogenous OGT activity.55,57 Therefore, it is possible that abolished O-GlcNAcylation status on a subset of OGT substrates or even a single substrate/site could contribute to the intellectual disability phenotype, an avenue that is currently being explored in our laboratory using several methods discussed in this review.
Currently, the O-GlcNAc field is experiencing rapid growth with researchers from related disciplines discovering that O-GlcNAc modulates the function of their proteins of interest. Examples include discoveries of the role of O-GlcNAcylation in preventing the aggregation of a Polycomb member, polyhomeotic, in Drosophila,59 as well as in controlling the anti-inflammatory function on receptor-interacting serine/threonine-protein kinase 3 (RIPK3)60 and in activating phosphoglycerate kinase 1 (PGK1) to promote glycolysis,61 to name a few. As the roles of O-GlcNAc are becoming recognized, there is an emerging need for tools to dissect site-specific O-GlcNAcylation.
Visualisation of protein- and site-specific O-GlcNAcylation
A crucial step in studying any PTM is its detection. Although mass spectrometry (MS) provides a high-throughput method for O-GlcNAc identification and site-mapping (reviewed elsewhere62), it is complicated by time-consuming sample preparation, expensive equipment and (often difficult) analysis. Therefore, the use of gel and western blot methods is generally required which will be discussed below.In the early days of the field, O-GlcNAc was detected by galactosyltransferase labelling with an [3H]-galactose followed by autoradiography and by lectin binding (for instance, with fluorescently labelled wheat germ agglutinin, which binds terminal GlcNAc residues).63,64 This was later surpassed by a superior chemoenzymatic method based on the mutant β-1,4-galactosyltransferase (GalTY289L) that transfers an azide derivative of GalNAc (GalNAz) onto O-GlcNAcylated proteins.65 The azide provides a reactive handle for the attachment of tags using copper-catalysed azide–alkyne cycloaddition (CuAAC) or copper-free strain-promoted azide–alkyne cycloaddition (SPAAC),66,67 allowing qualitative and quantitative detection of O-GlcNAc.66,68 The use of mass tags (such as polyethylene glycols PEG5000- or PEG2000-alkyne) allows resolution of GlcNAc-modified proteins on a gel and determination of absolute stoichiometry using a western blot with an antibody against the target protein (eliminating the need for pulldowns and O-GlcNAc-specific antibodies).66 Additionally, the number of bands shifted indicates the number of O-GlcNAc sites. Unfortunately, the detection limit of the PEGylation method is approximately 5% (depending on the quality of the antibody) and the labelling efficiency relies on full completion of the galactosyltransferase reaction.66 The efficiency of the subsequent chemical addition of a PEG mass tag may also vary depending on the reagent concentration. To address this issue, the SPAAC reaction has been optimised using a synthetic protein standard (bearing stoichiometric O-GlcNAc modification) and commercial reagents.69 As an alternative to mass tagging, a native polyacrylamide gel electrophoresis (native PAGE) method based on co-polymerised CpOGA has been developed in our laboratory (C. Fu and D. M. F. van Aalten, Analyst, in press). The retardation of an O-GlcNAc-modified band happens as a result of high-affinity binding by the catalytically inactive CpOGA. Conveniently, this approach does not require any chemical modification of the sample.
Sometimes the stoichiometry of O-GlcNAc on a given protein is below the detection limit of antibodies. In order to amplify the O-GlcNAc signal in such cases, a proximity-ligation based method has been developed. First, O-GlcNAc is chemoenzymatically labelled with GalNAz, followed by a reaction with an alkyne-biotin. The next step is the coupling of antibody-DNA conjugates targeted to either biotin or the protein of interest. If the protein is O-GlcNAcylated, the antibodies are brought into proximity, leading to ligation of DNA tags. qPCR is subsequently performed to quantify the signal.70 While this method allows detection of O-GlcNAcylated proteins in small amounts of sample, the specificity of O-GlcNAc signal heavily relies on the specificity of an antibody against a protein of interest which could otherwise detect off-targets.
The donor substrate promiscuity of OGT has been extensively exploited to generate various metabolic reporters for global O-GlcNAc profiling by mass-spectrometry and in-gel visualisation. These modified sugars can be fed to cells where they enter specific metabolic pathways and can be utilised by OGT as donor substrates. These include GlcNAc derivatives with alkyne and azide functionalities such as GlcNAz,71 GlcNAlk,72 6AzGlcNAc,73 4-deoxy-GlcNAz,74 2AzGlc,75 6AlkGlcNAc,76 6AzGlc77 and GlcNDAz.78
Antibodies remain the most popular tool to detect O-GlcNAc. The two most widely used pan-specific O-GlcNAc antibodies are RL2 (originally developed for detecting nuclear pore proteins)64,79 and CTD110.6 (developed based on the O-GlcNAcylated RNA polymerase II C-terminal domain).80 Although both of these commercially available antibodies are monoclonal, RL2 exclusively binds to O-GlcNAc, while CTD110.6 may also recognise N-GlcNAc, GlcNAcylated O-mannose, cross-reacts with terminal β-GlcNAc on complex N-glycans of cell surface glycoproteins and more recently has been shown to efficiently detect S-GlcNAc on cysteines.24,81–84 Therefore, care must be taken when using CTD110.6 antibody whose specificity must be tested in each case with an OGA-treated negative control. Although RL2 binds O-GlcNAc with greater specificity, it may not recognise O-GlcNAcylation in certain contexts such as demonstrated with α-synuclein stoichiometrically O-GlcNAc-modified at several reported O-GlcNAcylation sites.85
Unlike in the field of phosphorylation, very few site-specific O-GlcNAc antibodies have been generated. Nevertheless, these have greatly aided the study of site-specific O-GlcNAc modification and are summarised in Table 1. For instance, reciprocal interplay between O-GlcNAcylation and proximal phosphorylation could be examined using antibodies against O-GlcNAcylated Thr58 on c-Myc (first site-specific O-GlcNAc antibody ever produced)86 and O-GlcNAcylated Ser400 on tau.46 Our laboratory had generated a Ser395-O-GlcNAc-specific TGF-β activated kinase-1 (TAK1) binding protein-1 (TAB1) antibody that revealed increased O-GlcNAcylation as a modulator of TAK1 signalling upon IL-1 stimulation and osmotic stress.87 More recently, we produced a Ser517-O-GlcNAc-specific CRMP2 antibody to show an age-dependent increase in CRMP2 O-GlcNAcylation associated with short-term memory impairment.88
| O-GlcNAc antibody | Site (human) | Ref. |
|---|---|---|
| c-Myc | Thr58 | 86 |
| Collapsin response mediator protein-2 (CRMP2) | Ser517 | 88 |
| Histone H2A | Ser40 | 89 |
| Histone H2A | Thr101 | http://www.glycoscientific.com/ |
| Histone H2B | Ser112 | 90 |
| Histone H3 | Thr32 | http://www.glycoscientific.com/ |
| Histone H4 | Ser47 | http://www.glycoscientific.com/ |
| Insulin receptor substrate-1 (IRS1) | Ser1011 | 91 |
| Insulin receptor substrate-2 (IRS2) | Thr1155 | http://www.glycoscientific.com/ |
| NAD-dependent protein deacetylase Sirtuin-1 (SIRT1) | Ser549 | 92 |
| Tau | Ser400 | 93 and 94 |
| TGF-beta-activated kinase 1-binding protein 1 (TAB1) | Ser395 | 87 |
| TGF-beta-activated kinase 1-binding protein 3 (TAB3) | Ser408 | 95 |
Enzymatic methods for generating protein-specific O-GlcNAcylation
Access to stoichiometrically modified proteins is often required to dissect the mechanistic consequences of PTMs in vitro and in vivo. However, producing highly O-GlcNAc-modified proteins is challenging. Traditionally, the simplest approach to obtain an O-GlcNAcylated protein of interest has been in vitro reaction with OGT (Fig. 2a).96 Despite the ease of implementation, to achieve high O-GlcNAc stoichiometry, long incubation times are generally needed which in itself may have undesired effects on the stability of the protein under investigation. Alternatively, to preserve protein stability, OGT can be co-expressed with its substrates in E. coli or insect cells (Fig. 2b). Application of this system in a bacterial setting has been demonstrated for the RNA-Polymerase II C-terminal domain, TAB1, calcium/calmodulin-dependent protein kinase type IV (CaMKIV), Tau, Coactivator Associated Arginine Methyltransferase 1 (CARM1) and nuclear pore glycoprotein p62 (nup62).96–98 However, an endogenous glycosidase in E. coli may compromise the yield of the O-GlcNAc-modified proteins produced with this approach.99 In insect (Sf9) cells simultaneous expression of CREB and OGT led to a three-fold increase of CREB glycosylation to almost 90%.66 Although this co-expression approach may not be compatible with all OGT substrates, the main advantage is the ability to produce co-translationally O-GlcNAc-modified proteins which is impossible via OGT reaction in vitro.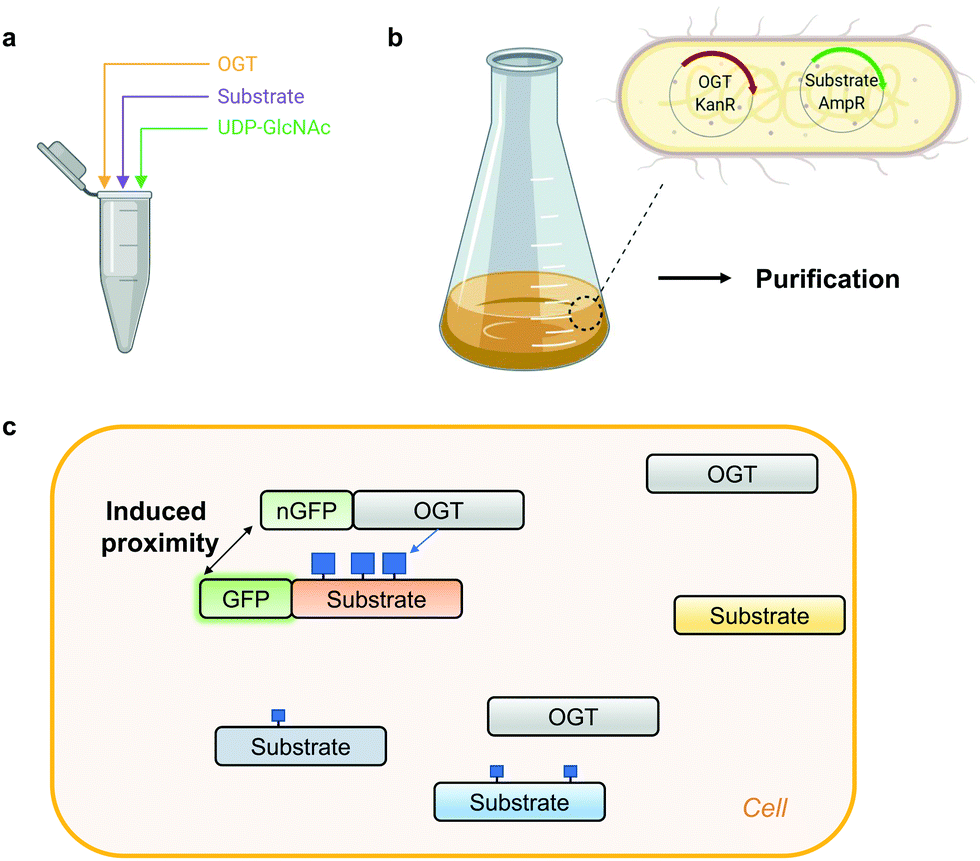 | ||
| Fig. 2 Methods for generating protein-specific O-GlcNAcylation. (a) In vitro OGT reaction. (b) Co-expression of OGT with its substrate in E. coli, followed by purification. KanR – kanamycin resistance, AmpR – ampicillin resistance. (c) Proximity OGT approach for protein-selective O-GlcNAcylation in cells.100 nGFP – anti-GFP nanobody. Blue squares denote O-GlcNAc. | ||
Recombinant O-GlcNAc-modified protein on its own is usually not sufficient to dissect the function of this modification. To acquire control over a single-protein O-GlcNAcylation status in a complex cellular environment, a creative way to induce proximity of OGT to its substrates in cells has been proposed recently (Fig. 2c).100 Exploiting the fusion of OGT to a nanobody against a tag (GFP or RFP) on a protein of interest or a nanobody against endogenous protein target, Woo and colleagues managed to increase O-GlcNAcylation stoichiometry on a subset of OGT substrates. In its current form this proximity induction relies on the overexpression of OGT which concomitantly elevates global O-GlcNAc levels. This strategy may have potential to increase O-GlcNAcylation on a protein of interest, if the impact of OGT overexpression on the global O-GlcNAcome can be addressed. Alternatively, other proximity approaches could be explored, such as the use of aptamers or small molecules (PROTAC principle). For example, the use of a heterobifunctional small molecule has already been utilised to bring protein phosphatase 1 (PP1) in proximity to its targets resulting in a protein-specific decrease in phosphorylation of AKT kinase and EGFR.101 One of the most important considerations for the future development of such probes is that the interaction of the proximity-inducing molecules does not affect the catalytic or scaffolding function of OGT or its substrate.
Current methods to increase protein-specific O-GlcNAcylation still result in heterogeneous mixtures of modified and unmodified proteins and make functional analysis particularly cumbersome in case of multiple glycosylation sites. Thus, knowledge of specific O-GlcNAc sites and application of approaches to generate site-specifically O-GlcNAcylated proteins could circumvent this issue.
Chemical biology methods for dissecting site-specific O-GlcNAcylation in vitro
One approach that has been explored to incorporate site-specific O-GlcNAcylation is expressed protein ligation (EPL), a semi-synthetic strategy to obtain homogenous and stoichiometric site-specific PTMs of a target protein (Fig. 3a).102,103 EPL involves synthesis of a peptide bearing a desired functionality (e.g. post-translational modification), which is then ligated via S-to-N acyl-transfer to a thioester generated from a recombinant intein-fusion protein.102 To date, tau,104 α-synuclein,49,85,105 HSP27106 and an unnaturally modified ubiquitin69 are the only four reported O-GlcNAcylated proteins produced using EPL. This method has been used to dissect the mechanistic consequences of O-GlcNAcylation on α-synuclein (Thr72, Thr75, Thr81, Ser87),49,85,105 which is a small (<15 kDa) protein amenable to EPL. This approach revealed that O-GlcNAc on α-synuclein affects aggregation and toxicity in vitro.85 Apart from a handful of O-GlcNAcylated proteins, S-GlcNAcylated casein kinase 2 (CK2)107 and α-synuclein108 have been produced by EPL in order to increase the stability of the modification against hydrolysis. Through structural and biophysical studies it was shown that S-GlcNAc serves as a good non-hydrolysable analogue for O-GlcNAc that can be recognized by some O-GlcNAc-specific antibodies (Ser395-O-GlcNAc-TAB1 and CTD110.6).24,108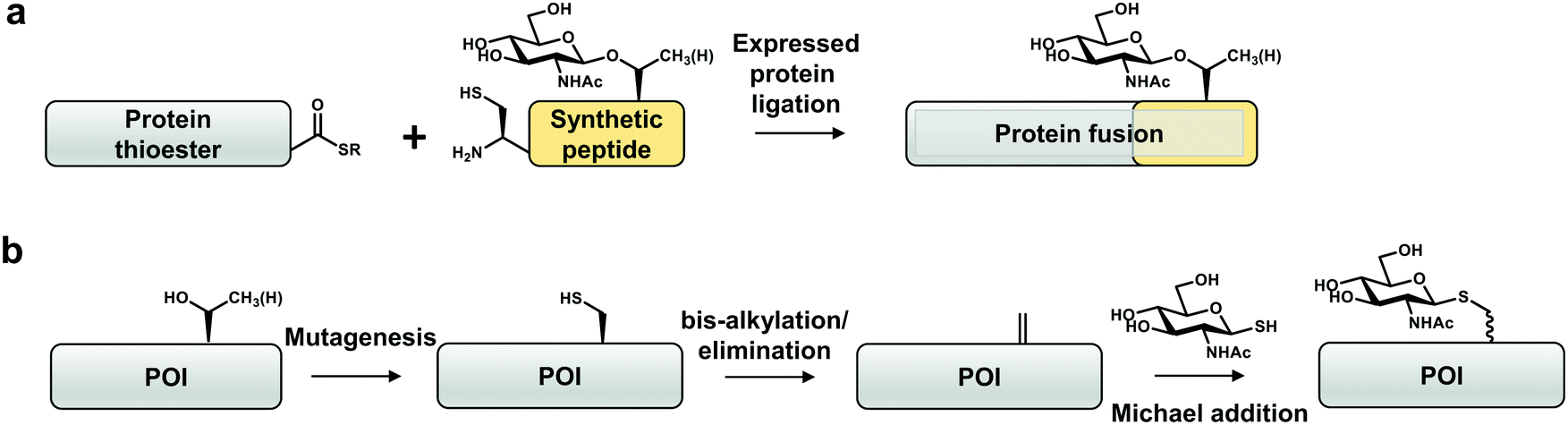 | ||
| Fig. 3 Site-specific chemical biology approaches to study O-GlcNAcylation in vitro. (a) Expressed protein ligation. (b) “Tag-and-modify” dehydroalanine approach. POI – protein of interest. | ||
In future the utility of CHF or CF2 functionalized GlcNAc as potentially nonhydrolyzable analogues for EPL could be investigated as these have been shown to be good steric and electronic representations in the context of O-phosphorylation.109 The main disadvantage of EPL is that it must be optimised for every protein and the maximum length of the peptide for ligation is currently limited. Importantly, this method requires good expression of a soluble intein-fusion protein, which frequently poses a problem. Insertion of modified peptides into the central portions of large, globular proteins involves iterative ligation steps and is particularly challenging. A cysteine residue that EPL introduces at the site of ligation cannot always be converted to a native amino acid (e.g. to alanine via desulphurisation). Therefore, the site of ligation must be carefully considered.
In another approach, taking advantage of the potent nucleophilicity and low abundance of cysteine in proteins, cysteine-conversion chemical methods have been developed by Davis and co-workers to obtain proteins with site-specific O-GlcNAc mimics in vitro (Fig. 3b).110–112 These rely on the chemical transformation of a genetically installed cysteine into dehydroalanine by an alkylating reagent (for example, O-mesitylenesulfonylhydroxylamine (MSH) or 2,5-dibromohexanediamide (DBHDA)) under denaturing conditions. The dehydroalanine of the unfolded protein is then incubated with a reactive GlcNAc derivative (e.g. GlcNAc-thiol) and the modified protein is refolded.110–112 Notably, a thiol-linked GlcNAc prepared in such a way is resistant to reduction by DTT.113 Moreover, endoglycosidase-A exhibits trans-glycosylation activity for a synthetic S-linked GlcNAc (S-GlcNAc)-modified protein with a high modification efficiency.112 Cys-S-GlcNAcylation at position 101 (Thr in the native protein) of histone H2A was shown to destabilise H2A/H2B dimers, promoting an open chromatin state. Installation of S-GlcNAc at position 112 of histone H2B (Ser in the native protein) allowed identification of interactors, among which are subunits of the FACT chromatin remodelling complex.110,111 As evidence for physiological mimicry, O-GlcNAc-homohomo-Ser prepared by this approach can be hydrolysed by human OGA.114 This method is limited to recombinant proteins with no or few native cysteine residues, that otherwise must first be substituted by other amino acids (Ser or Ala). Furthermore, this approach does not allow control of stereo-specificity of the modification rendering it difficult to interpret the results of the experiment.
Another method for installing S-GlcNAc employs a thio-glycoligase capable of transferring GlcNAc to cysteines, engineered from a Streptomyces plicatus hexosaminidase via mutation of the catalytic glutamate (E314A).115 Withers and co-workers managed to produce S-GlcNAcylated synuclein peptides and tau protein using this new method.115 As with the dehydroalanine approach, this technique requires mutation of all native cysteines that are not destined for S-GlcNAc modification and its utility is limited to in vitro reactions.
Genetic methods for dissecting site-specific O-GlcNAcylation in vivo
Loss-of-function mutations are the most common approach to probe site-specific O-GlcNAcylation in cultured cells (e.g. Ser/Thr mutation to Ala). For example, Ser529Ala mutation on phosphofructokinase 1 was shown to prevent O-GlcNAc-dependent increase in its activity,116 while Thr228Ala mutation of Oct4 reduces stem cell self-renewal and reprogramming.117 The caveat of this approach lies in the loss of the side chain which in itself could have an impact on protein folding and stability. Therefore, Ala mutagenesis is often used in combination with other methods to down- or upregulate OGT or OGA levels and their enzymatic activity. OGA knockout, knockdown and inhibition is an effective way to elevate total O-GlcNAc modification levels in vivo and in cell culture, since O-GlcNAcylation is often sub-stoichiometric in cells (<10%) due to the high OGA activity.118–120 However, with this approach, functional dissection of the roles of individual O-GlcNAc sites is impeded by potential ambiguous phenotypes through effects on many other OGT substrates.Genetic code expansion (GCE) technology allows site-specific incorporation of unnatural amino acids (including PTMs) in vitro and in vivo by utilising evolved orthogonal amber suppressor tRNA synthetases (Fig. 4a).121–125 The GCE method often requires evolution of an archaeal pyrrolysine-tRNA synthetase (PylRS) to aminoacylate a corresponding amber suppressor tRNA, which in turn allows decoding of the unnatural amino acid (UAA) of interest at an amber stop (or quadruplet121) codon in a genetically predetermined fashion (Fig. 4a). Since the amber codon is the least abundant of the three stop codons, its use reduces the off-target incorporation of the amino acid where endogenous amber stop codons occur.
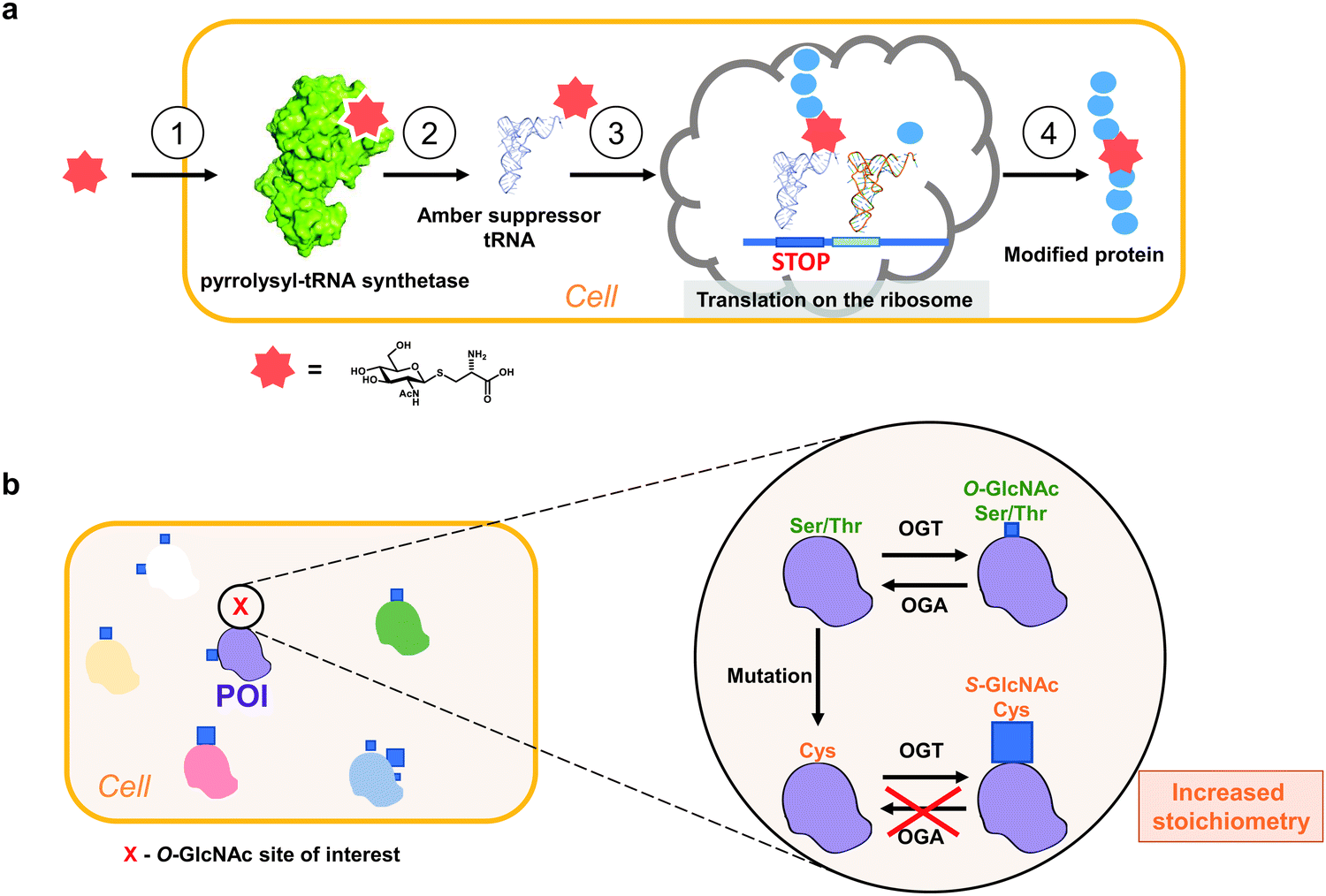 | ||
| Fig. 4 Approaches to study site-specific O-GlcNAcylation in vivo. (a) Principle of a genetic code expansion approach with a metabolically stable Cys-S-GlcNAc amino acid: (1). UAA uptake, (2). tRNA aminoacylation, (3). UAA translation in response to a stop codon, (4). Production of a modified protein. (b) Genetic recoding approach to introduce site-specific S-GlcNAc mimicry of O-GlcNAcylation.24 Blue squares denote GlcNAc. POI – protein of interest. | ||
Co-translational incorporation of a glycosylated amino acid by the GCE technology could become a promising tool to site-specifically examine the gain-of-function O-GlcNAc modification in vivo.122–124 With over 200 UAAs incorporated to date,126 the expanded genetic code includes several PTMs such as phosphorylated amino acids (serine, threonine and tyrosine), acetylated lysine and a scaffold for ubiquitin- and SUMO-modified lysine.127–132 Most of the UAAs incorporated by the PylRS-based GCE are lysine derivatives that resemble the natural substrate of PylRS, pyrrolysine.126 Amino acids as polar as GlcNAcylated serine or cysteine have not yet been added to the expanded genetic code. Apart from the specificity and activity of the evolved PylRS, incorporation of UAAs largely depends on the tolerance of the corresponding aminoacylated tRNA by the translation machinery, such as the ribosome, elongation factor Tu and release factors. These problems were previously encountered with phosphoserine, for which some components of the translational machinery had to be additionally engineered.128
A major obstacle in applying GCE to O-GlcNAc is the uptake and stability of the synthetic O-GlcNAcylated amino acid. E. coli, which is used as a host system for evolving PylRS, metabolises Ser-O-GlcNAc as a carbon source,133 precluding the use of this UAA for GCE.134 De-acetylation of a per-O-acetylated Ser-O-GlcNAc variant (which would hypothetically increase the cellular uptake) does not occur in E. coli as it does not express the requisite deacetylases.133 The latter strategy would be more suited to mammalian cells, where pan-specific de-acetylating enzymes do exist. Importantly, however, we showed that a synthetic amino acid Cys-S-GlcNAc, a Ser-O-GlcNAc analogue, is efficiently internalised by E. coli where it remains metabolically stable, satisfying the first essential step and prerequisite for directed evolution of PylRS.135
Recombinant proteins with a glycosylation mimic have been obtained indirectly through PylRS amber suppression, by introducing an amino acid with the alkene functionality followed by a click chemistry reaction.136 Recently, cellular incorporation of dehydroalanine was demonstrated.137 This allowed the authors to install thio-linked GlcNAc at a defined position on GFP which was recognised by a pan-specific O-GlcNAc antibody.137
Although glycosylated amino acids have not been incorporated by GCE, there are some clues as to the feasibility of this approach reported in several studies.138,139 In these reports glycosylated recombinant proteins were produced in a cell-free system using chemically aminoacylated amber suppressor tRNAs.138,139 While the efficiencies of amber suppression varied between different glycosylated amino acids reaching up to 30%,138 in one of the studies, it was shown that Thr-O-GalNAc could be incorporated only at the protein N-terminus, suggesting that glycosylated polypeptides are not well tolerated by the ribosome.139
The major advantage of GCE is the ability to produce stoichiometrically-modified proteins in living systems. On the other hand, the level of incorporation strongly depends on the suppression efficiency of the stop codon. This does not pose significant issues for purified recombinant proteins (where the expression can be scaled up). However, when interpreting the effects of a post-translational modification in vivo, it is challenging to provide appropriate controls (i.e. achieving the same expression as for the unmodified protein). Therefore, any observed effects resulting from incorporation of a UAA could also be the result of altered expression levels. Undesired off-target effects could also be observed in case of mis-incorporation of the amino acid in unintended proteins in place of endogenous stop codons, resulting in skewed phenotypes. Future developments of GCE are required to address these issues.
To gain insight into site-specific O-GlcNAcylation in cells, our laboratory has explored a surprisingly simple approach to genetically introduce a non-hydrolysable S-GlcNAc in vitro and in vivo using Ser/Thr to Cys mutagenesis and relying on the fortuitous promiscuity of OGT that happens to possess efficient S-GlcNAc transferase activity (Fig. 4b).24 Notably, this method does not require any exogenous biomolecular machinery, overexpression of (mutant) genes or chemical synthesis and can be combined with the CRISPR-Cas9 genome editing technology. With this approach, the stoichiometry of cysteine GlcNAcylation can replicate that of OGA inhibition in cells due to the hydrolytic stability of S-GlcNAc. However, unlike OGA inhibition, S-GlcNAc mutagenesis does not affect global O-GlcNAc levels, minimizing undesired indirect effects. By applying this method to a single Ser O-GlcNAcylation site on OGA and converting it to a Cys S-GlcNAcylation site, we managed to increase the stoichiometry almost five-fold (from 15% to over 70%), similar to that achieved with the OGA inhibitor treatment.24 We showed that the high GlcNAcylation stoichiometry decreased OGA stability in cells, while the overall thermal stability remained unchanged relative to the wild type protein.24 Interestingly, in the same sequence contexts S-GlcNAc appears to be more stable to CID MS/MS fragmentation23 and may assist in mapping and detection of O-GlcNAc sites. Additionally, homogenous site-specifically S-GlcNAcylated recombinant proteins can be produced through GlcNAcylation of a Cys mutant protein and subsequent treatment with OGA (for example, the highly-active CpOGA) to remove unwanted O-GlcNAcylation (if multiple modification sites are present). It must be noted that in cases where OGA inhibition does not elevate O-GlcNAcylation levels on a protein of interest in cells, this approach may not be applicable. While this S-GlcNAc genetic recoding approach has been applied in cultured human and mouse cells, it has yet to be demonstrated that this leads to site-specific S-GlcNAc incorporation in animal models.
Conclusions
The functional consequences of protein O-GlcNAcylation still remain poorly understood due to the limited number of tools to study its site-specific effects. As can be appreciated from this review, no single method to study protein- and site-specific O-GlcNAcylation can be universally applied and all of the discussed techniques have their advantages and disadvantages. Thus, a combination of these approaches can help us achieve a comprehensive understanding of the functions of the O-GlcNAc PTM on specific proteins and sites. The future developments in the field require optimization of contemporary methods and invention of new strategies to gain control of protein- and site-specific modification. Expanding the genetic code with glycosylated amino acids would represent just one of these future advances. Ingenious methods are required to achieve protein- and site-specific O-GlcNAcylation with minimal side effects and to avoid unnecessary non-physiological perturbation in living systems. The use of non-hydrolysable analogues of O-GlcNAc (such as S-, CHF- and CF2-linked GlcNAc or other analogues24,74,140) in an intracellular setting must be considered to withstand the high activity of OGA that could otherwise abolish the efforts of site-specific installation of O-GlcNAc. Spatiotemporal control of site-specific O-GlcNAcylation in cells is also important, since O-GlcNAc is a signalling molecule that can lead to rapid activation of signalling cascades. Last but not least, low O-GlcNAcylation stoichiometry at a given site can be the result of protein–protein interactions that “mask” such modification sites and prevent subsequent post-translational modification by OGT, thus requiring implementation of techniques for co-translational site-specific GlcNAc incorporation. Bridging the gap between chemistry and biology is required to invent approaches that fine-tune O-GlcNAc stoichiometry with a single protein and amino acid residue precision. Such interdisciplinary amalgamation will be instrumental in investigating the diverse functions of protein O-GlcNAcylation.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was funded by a Wellcome Trust Investigator Award (110061) to DvA and a Wellcome Trust 4-year PhD studentship (105310/Z/14/Z) to AG. Fig. 2a and b were created with biorender.com.References
- R. Aebersold, et al., How many human proteoforms are there?, Nat. Chem. Biol., 2018, 14, 206–214 CrossRef CAS PubMed.
- T. Klein, U. Eckhard, A. Dufour, N. Solis and C. M. Overall, Proteolytic Cleavage - Mechanisms, Function, and ‘omic’ Approaches for a Near-Ubiquitous Posttranslational Modification, Chem. Rev., 2018, 118, 1137–1168 CrossRef CAS PubMed.
- R. Yau and M. Rape, The increasing complexity of the ubiquitin code, Nat. Cell Biol., 2016, 18, 579–586 CrossRef CAS PubMed.
- K. W. Moremen, M. Tiemeyer and A. V. Nairn, Vertebrate protein glycosylation: Diversity, synthesis and function, Nat. Rev. Mol. Cell Biol., 2012, 13, 448–462 CrossRef CAS PubMed.
- F. Ardito, M. Giuliani, D. Perrone, G. Troiano and L. L. Muzio, The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review), Int. J. Mol. Med., 2017, 40, 271–280 CrossRef CAS PubMed.
- E. Verdin and M. Ott, 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond, Nat. Rev. Mol. Cell Biol., 2015, 16, 258–264 CrossRef CAS.
- A. Jambhekar, A. Dhall and Y. Shi, Roles and regulation of histone methylation in animal development, Nat. Rev. Mol. Cell Biol., 2019, 20, 625–641 CrossRef CAS PubMed.
- B. Chen, Y. Sun, J. Niu, G. K. Jarugumilli and X. Wu, Protein Lipidation in Cell Signaling and Diseases: Function, Regulation, and Therapeutic Opportunities, Cell Chem. Biol., 2018, 25, 817–831 CrossRef CAS PubMed.
- G. W. Hart, Nutrient regulation of signaling and transcription, J. Biol. Chem., 2019, 294, 2211–2231 CrossRef CAS PubMed.
- Y. Zhu, et al., O-GlcNAc occurs cotranslationally to stabilize nascent polypeptide chains, Nat. Chem. Biol., 2015, 11, 319–325 CrossRef CAS PubMed.
- R. S. Haltiwanger, M. A. Blomberg and G. W. Hart, Glycosylation of nuclear and cytoplasmic proteins: Purification and characterization of a uridine diphospho-N-acetylglucosamine:polypeptide beta-N-acetylglucosaminyltransferase, J. Biol. Chem., 1992, 267, 9005–9013 CAS.
- S. Pathak, et al., The active site of O-GlcNAc transferase imposes constraints on substrate sequence, Nat. Struct. Mol. Biol., 2015, 22, 744–750 CrossRef CAS PubMed.
- J. Ma and G. W. Hart, Protein O-GlcNAcylation in diabetes and diabetic complications, Expert Rev. Proteomics, 2013, 10, 365–380 CrossRef CAS PubMed.
- Y. Gao, L. Wells, F. I. Comer, G. J. Parker and G. W. Hart, Dynamic O-glycosylation of nuclear and cytosolic proteins: Cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain, J. Biol. Chem., 2001, 276, 9838–9845 CrossRef CAS PubMed.
- X. Yang and K. Qian, Protein O-GlcNAcylation: emerging mechanisms and functions, Nat. Rev. Mol. Cell Biol., 2017, 18, 452–465 CrossRef CAS PubMed.
- B. I. Cantarel, et al., The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics, Nucleic Acids Res., 2009, 37, D233–D238 CrossRef CAS PubMed.
- M. B. Lazarus, Y. Nam, J. Jiang, P. Sliz and S. Walker, Structure of human O-GlcNAc transferase and its complex with a peptide substrate, Nature, 2011, 469, 564–567 CrossRef CAS PubMed.
- D. Nolte and U. Müller, Human O-GlcNAc transferase (OGT): Genomic structure, analysis of splice variants, fine mapping in Xq13.1, Mamm. Genome, 2002, 13, 62–64 CrossRef CAS PubMed.
- J. A. Hanover, et al., Mitochondrial and nucleocytoplasmic isoforms of O-linked GlcNAc transferase encoded by a single mammalian gene, Arch. Biochem. Biophys., 2003, 409, 287–297 CrossRef CAS PubMed.
- R. Trapannone, D. Mariappa, A. T. Ferenbach and D. M. F. van Aalten, Nucleocytoplasmic human O-GlcNAc transferase is sufficient for O-GlcNAcylation of mitochondrial proteins, Biochem. J., 2016, 473, 1693–1702 CrossRef CAS PubMed.
- M. Schimpl, et al., O-GlcNAc transferase invokes nucleotide sugar pyrophosphate participation in catalysis, Nat. Chem. Biol., 2012, 8, 969–974 CrossRef CAS PubMed.
- M. B. Lazarus, et al., HCF-1 is cleaved in the active site of O-GlcNAc transferase, Science, 2013, 342, 1235–1239 CrossRef CAS PubMed.
- J. C. Maynard, A. L. Burlingame and K. F. Medzihradszky, Cysteine S-linked N-acetylglucosamine (S-GlcNAcylation), A New Post-translational Modification in Mammals, Mol. Cell. Proteomics, 2016, 15, 3405–3411 CrossRef CAS PubMed.
- A. Gorelik, et al., Genetic recoding to dissect the roles of site-specific protein O-GlcNAcylation, Nat. Struct. Mol. Biol., 2019, 26, 1071–1077 CrossRef CAS PubMed.
- M. Jínek, et al., The superhelical TPR-repeat domain of O-linked GlcNAc transferase exhibits structural similarities to importin α, Nat. Struct. Mol. Biol., 2004, 11, 1001–1007 CrossRef PubMed.
- K. Rafie, et al., Recognition of a glycosylation substrate by the O-GlcNAc transferase TPR repeats, Open Biol., 2017, 7, 170078 CrossRef PubMed.
- Z. G. Levine, et al., O-GlcNAc Transferase Recognizes Protein Substrates Using an Asparagine Ladder in the Tetratricopeptide Repeat (TPR) Superhelix, J. Am. Chem. Soc., 2018, 140, 3510–3513 CrossRef CAS PubMed.
- C. M. Joiner, Z. G. Levine, C. Aonbangkhen, C. M. Woo and S. Walker, Aspartate Residues Far from the Active Site Drive O-GlcNAc Transferase Substrate Selection, J. Am. Chem. Soc., 2019, 141, 12974–12978 CrossRef CAS PubMed.
- J. F. Alfaro, et al., Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7280–7285 CrossRef CAS PubMed.
- Z. Wang, et al., Enrichment and site mapping of O-linked N-acetylglucosamine by a combination of chemical/enzymatic tagging, photochemical cleavage, and electron transfer dissociation mass spectrometry, Mol. Cell. Proteomics, 2010, 9, 153–160 CrossRef CAS PubMed.
- R. J. Chalkley, A. Thalhammer, R. Schoepfer and A. L. Burlingame, Identification of protein O-GlcNAcylation sites using electron transfer dissociation mass spectrometry on native peptides, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8894–8899 CrossRef CAS PubMed.
- F. V. Rao, et al., Structure of a bacterial putative acetyltransferase defines the fold of the human O-GlcNAcase C-terminal domain, Open Biol., 2013, 3, 130021 CrossRef PubMed.
- N. L. Elsen, et al., Insights into activity and inhibition from the crystal structure of human O-GlcNAcase, Nat. Chem. Biol., 2017, 13, 613–615 CrossRef CAS PubMed.
- C. N. Keembiyehetty, A. Krzeslak, D. C. Love and J. A. Hanover, A lipid-droplet-targeted O-GlcNAcase isoform is a key regulator of the proteasome, J. Cell Sci., 2011, 124, 2851–2860 CrossRef CAS PubMed.
- F. V. Rao, et al., Structural insights into the mechanism and inhibition of eukaryotic O-GlcNAc hydrolysis, EMBO J., 2006, 25, 1569–1578 CrossRef CAS PubMed.
- M. Schimpl, V. S. Borodkin, L. J. Gray and D. M. van Aalten, Synergy of peptide and sugar in O-GlcNAcase substrate recognition, Chem. Biol., 2012, 19, 173–178 CrossRef CAS PubMed.
- B. Li, H. Li, L. Lu and J. Jiang, Structures of human O-GlcNAcase and its complexes reveal a new substrate recognition mode, Nat. Struct. Mol. Biol., 2017, 24, 362–369 CrossRef CAS PubMed.
- C. Roth, et al., Structural and functional insight into human O-GlcNAcase, Nat. Chem. Biol., 2017, 13, 610–612 CrossRef CAS PubMed.
- B. Li, H. Li, C. W. Hu and J. Jiang, Structural insights into the substrate binding adaptability and specificity of human O-GlcNAcase, Nat. Commun., 2017, 8, 666 CrossRef PubMed.
- H. G. Seo, et al., Identification of the nuclear localisation signal of O-GlcNAc transferase and its nuclear import regulation, Sci. Rep., 2016, 6, 34614 CrossRef CAS PubMed.
- R. Shafi, et al., The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5735–5739 CrossRef CAS PubMed.
- M. C. Gambetta, K. Oktaba and J. Muller, Essential role of the glycosyltransferase sxc/Ogt in polycomb repression, Science, 2009, 325, 93–96 CrossRef CAS PubMed.
- D. Mariappa, A. T. Ferenbach and D. M. F. Van Aalten, Effects of hypo O-GlcNAcylation on Drosophila development, J. Biol. Chem., 2018, 293, 7209–7221 CrossRef CAS PubMed.
- Y. R. Yang, et al., O-GlcNAcase is essential for embryonic development and maintenance of genomic stability, Aging Cell, 2012, 11, 439–448 CrossRef CAS PubMed.
- C. Slawson and G. W. Hart, O-GlcNAc signalling: implications for cancer cell biology, Nat. Rev. Cancer, 2011, 11, 678–684 CrossRef CAS PubMed.
- G. W. Hart, C. Slawson, G. Ramirez-Correa and O. Lagerlof, Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease, Annu. Rev. Biochem., 2011, 80, 825–858 CrossRef CAS PubMed.
- M. R. Martinez, T. B. Dias, P. S. Natov and N. E. Zachara, Stress-induced O-GlcNAcylation: An adaptive process of injured cells, Biochem. Soc. Trans., 2017, 45, 237–249 CrossRef CAS PubMed.
- F. Liu, K. Iqbal, I. Grundke-Iqbal, G. W. Hart and C.-X. Gong, O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer’s disease, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10804–10809 CrossRef CAS PubMed.
- N. P. Marotta, et al., O-GlcNAc modification blocks the aggregation and toxicity of the protein alpha-synuclein associated with Parkinson's disease, Nat. Chem., 2015, 7, 913–920 CrossRef CAS PubMed.
- S. M. Ranuncolo, S. Ghosh, J. A. Hanover, G. W. Hart and B. A. Lewis, Evidence of the involvement of O-GlcNAc-modified human RNA polymerase II CTD in transcription in vitro and in vivo, J. Biol. Chem., 2012, 287, 23549–23561 CrossRef CAS PubMed.
- B. Guo, et al., O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation, Nat. Cell Biol., 2014, 16, 1215–1226 CrossRef CAS PubMed.
- S. K. Park, et al., A Conserved Splicing Silencer Dynamically Regulates O-GlcNAc Transferase Intron Retention and O-GlcNAc Homeostasis, Cell Rep., 2017, 20, 1088–1099 CrossRef CAS PubMed.
- Z. Zhang, E. P. Tan, N. J. VandenHull, K. R. Peterson and C. Slawson, O-GlcNAcase Expression is Sensitive to Changes in O-GlcNAc Homeostasis, Front. Endocrinol., 2014, 5, 206 Search PubMed.
- K. Vaidyanathan, et al., Identification and characterization of a missense mutation in the O-linked β-N-acetylglucosamine (O-GlcNAc) transferase gene that segregates with X-linked intellectual disability, J. Biol. Chem., 2017, 292, 8948–8963 CrossRef CAS PubMed.
- A. P. Willems, et al., Mutations in N-acetylglucosamine (O-GlcNAc) transferase in patients with X-linked intellectual disability, J. Biol. Chem., 2017, 292, 12621–12631 CrossRef CAS PubMed.
- N. Selvan, et al., O-GlcNAc transferase missense mutations linked to X-linked intellectual disability deregulate genes involved in cell fate determination and signaling, J. Biol. Chem., 2018, 293, 10810–10824 CrossRef CAS PubMed.
- V. M. Pravata, et al., A missense mutation in the catalytic domain of O-GlcNAc transferase links perturbations in protein O-GlcNAcylation to X-linked intellectual disability, FEBS Lett., 2019, 594, 717–727 CrossRef PubMed.
- V. M. Pravata, et al., Catalytic deficiency of O-GlcNAc transferase leads to X-linked intellectual disability, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 14961–14970 CrossRef CAS PubMed.
- M. C. Gambetta and J. Müller, O-GlcNAcylation Prevents Aggregation of the Polycomb Group Repressor Polyhomeotic, Dev. Cell, 2014, 31, 629–639 CrossRef CAS PubMed.
- X. Li, et al., O-GlcNAc Transferase Suppresses Inflammation and Necroptosis by Targeting Receptor-Interacting Serine/Threonine-Protein Kinase 3, Immunity, 2019, 50, 576–590 CrossRef CAS PubMed.
- H. Nie, et al., O-GlcNAcylation of PGK1 coordinates glycolysis and TCA cycle to promote tumor growth, Nat. Commun., 2020, 11, 36 CrossRef CAS PubMed.
- J. W. Thompson, A. W. Sorum and L. C. Hsieh-Wilson, Deciphering the Functions of O-GlcNAc Glycosylation in the Brain: The Role of Site-Specific Quantitative O-GlcNAcomics, Biochemistry, 2018, 57, 4010–4018 CrossRef CAS PubMed.
- W. G. Kelly and G. W. Hart, Glycosylation of chromosomal proteins: Localization of O-linked N-acetylglucosamine in Drosophila chromatin, Cell, 1989, 57, 243–251 CrossRef CAS.
- G. D. Holt, et al., Nuclear pore complex glycoproteins contain cytoplasmically disposed O-linked N-acetylglucosamine, J. Cell Biol., 1987, 104, 1157–1164 CrossRef CAS PubMed.
- N. Khidekel, et al., A chemoenzymatic approach toward the rapid and sensitive detection of O-GlcNAc posttranslational modifications, J. Am. Chem. Soc., 2003, 125, 16162–16163 CrossRef CAS PubMed.
- J. E. Rexach, et al., Quantification of O-glycosylation stoichiometry and dynamics using resolvable mass tags, Nat. Chem. Biol., 2010, 6, 645–651 CrossRef CAS PubMed.
- C. F. Teo and L. Wells, Monitoring protein O-linked β-N-acetylglucosamine status via metabolic labeling and copper-free click chemistry, Anal. Biochem., 2014, 464, 70–72 CrossRef CAS PubMed.
- P. M. Clark, et al., Direct in-gel fluorescence detection and cellular imaging of O-GlcNAc-modified proteins, J. Am. Chem. Soc., 2008, 130, 11576–11577 CrossRef CAS PubMed.
- N. Darabedian, J. W. Thompson, K. N. Chuh, L. C. Hsieh-Wilson and M. R. Pratt, Optimization of Chemoenzymatic Mass Tagging by Strain-Promoted Cycloaddition (SPAAC) for the Determination of O-GlcNAc Stoichiometry by Western Blotting, Biochemistry, 2018, 57, 5769–5774 CrossRef CAS PubMed.
- P. V. Robinson, C. T. Tsai, A. E. De Groot, J. L. McKechnie and C. R. Bertozzi, Glyco-seek: Ultrasensitive Detection of Protein-Specific Glycosylation by Proximity Ligation Polymerase Chain Reaction, J. Am. Chem. Soc., 2016, 138, 10722–10725 CrossRef CAS PubMed.
- D. J. Vocadlo, H. C. Hang, E.-J. Kim, J. A. Hanover and C. R. Bertozzi, A chemical approach for identifying O-GlcNAc-modified proteins in cells, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9116–9121 CrossRef CAS PubMed.
- B. W. Zaro, Y.-Y. Yang, H. C. Hang and M. R. Pratt, Chemical reporters for fluorescent detection and identification of O-GlcNAc-modified proteins reveal glycosylation of the ubiquitin ligase NEDD4-1, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8146–8151 CrossRef CAS PubMed.
- K. N. Chuh, B. W. Zaro, F. Piller, V. Piller and M. R. Pratt, Changes in metabolic chemical reporter structure yield a selective probe of O-GlcNAc modification, J. Am. Chem. Soc., 2014, 136, 12283–12295 CrossRef CAS PubMed.
- J. Li, et al., An OGA-resistant probe allows specific visualization and accurate identification of O-GlcNAc-modified proteins in cells, ACS Chem. Biol., 2016, 11, 3002–3006 CrossRef CAS PubMed.
- B. W. Zaro, A. R. Batt, K. N. Chuh, M. X. Navarro and M. R. Pratt, The Small Molecule 2-Azido-2-deoxy-glucose Is a Metabolic Chemical Reporter of O-GlcNAc Modifications in Mammalian Cells, Revealing an Unexpected Promiscuity of O-GlcNAc Transferase, ACS Chem. Biol., 2017, 12, 787–794 CrossRef CAS PubMed.
- K. N. Chuh, et al., The New Chemical Reporter 6-Alkynyl-6-deoxy-GlcNAc Reveals O-GlcNAc Modification of the Apoptotic Caspases That Can Block the Cleavage/Activation of Caspase-8, J. Am. Chem. Soc., 2017, 139, 7872–7885 CrossRef CAS PubMed.
- N. Darabedian, J. Gao, K. N. Chuh, C. M. Woo and M. R. Pratt, The metabolic chemical reporter 6-azido-6-deoxy-glucose further reveals the substrate promiscuity of O-GlcNAc transferase and catalyzes the discovery of intracellular protein modification by O-glucose, J. Am. Chem. Soc., 2017, 140, 7092–7100 CrossRef PubMed.
- S.-H. Yu, et al., Metabolic labeling enables selective photocrosslinking of O-GlcNAc-modified proteins to their binding partners, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4834–4839 CrossRef CAS PubMed.
- C. M. Snow, A. Senior and L. Gerace, Monoclonal antibodies identify a group of nuclear pore complex glycoproteins, J. Cell Biol., 1987, 104, 1143–1156 CrossRef CAS PubMed.
- F. I. Comer, K. Vosseller, L. Wells, M. A. Accavitti and G. W. Hart, Characterization of a mouse monoclonal antibody specific for O-linked N-acetylglucosamine, Anal. Biochem., 2001, 293, 169–177 CrossRef CAS PubMed.
- T. Isono, O-GlcNAc-specific antibody CTD110.6 cross-reacts with N-GlcNAc2-modified proteins induced under glucose deprivation, PLoS One, 2011, 6, e18959 CrossRef CAS PubMed.
- M. Ogawa, et al., GTDC2 modifies O-mannosylated α-dystroglycan in the endoplasmic reticulum to generate N-acetyl glucosamine epitopes reactive with CTD110.6 antibody, Biochem. Biophys. Res. Commun., 2013, 440, 88–93 CrossRef CAS PubMed.
- Y. Tashima and P. Stanley, Antibodies that detect O-linked beta-D-N-acetylglucosamine on the extracellular domain of cell surface glycoproteins, J. Biol. Chem., 2014, 289, 11132–11142 CrossRef CAS PubMed.
- R. A. Reeves, A. Lee, R. Henry and N. E. Zachara, Characterization of the specificity of O-GlcNAc reactive antibodies under conditions of starvation and stress, Anal. Biochem., 2014, 457, 8–18 CrossRef CAS PubMed.
- P. M. Levine, et al., α-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson’s disease, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 1511–1519 CrossRef CAS PubMed.
- K. Kamemura, B. K. Hayes, F. I. Comer and G. W. Hart, Dynamic interplay between O-glycosylation and O-phosphorylation of nucleocytoplasmic proteins: Alternative glycosylation/phosphorylation of Thr-58, a known mutational hot spot of c-Myc in lymphomas, is regulated by mitogens, J. Biol. Chem., 2002, 277, 19229–19235 CrossRef CAS PubMed.
- S. Pathak, et al., O-GlcNAcylation of TAB1 modulates TAK1-mediated cytokine release, EMBO J., 2012, 31, 1394–1404 CrossRef CAS PubMed.
- V. Muha, et al., Loss of CRMP2 O-GlcNAcylation leads to reduced novel object recognition performance in mice, Open Biol., 2019, 9, 190192 CrossRef PubMed.
- M. Hirosawa, et al., Novel O-GlcNAcylation on Ser40 of canonical H2A isoforms specific to viviparity, Sci. Rep., 2016, 6, 31785 CrossRef CAS PubMed.
- R. Fujiki, et al., GlcNAcylation of histone H2B facilitates its monoubiquitination, Nature, 2011, 480, 557–560 CrossRef CAS PubMed.
- A. L. Klein, M. N. Berkaw, M. G. Buse and L. E. Ball, O-linked N-acetylglucosamine modification of insulin receptor substrate-1 occurs in close proximity to multiple SH2 domain binding motifs, Mol. Cell. Proteomics, 2009, 8, 2733–2745 CrossRef CAS PubMed.
- H. Shan, J. Sun, M. Shi, X. Liu, Z. Shi, W. Yu and G. Yuchao, Generation and characterization of a site-specific antibody for SIRT1 O-GlcNAcylated at serine 549, Glycobiology, 2018, 28, 482–487 CrossRef CAS PubMed.
- S. A. Yuzwa, et al., Mapping O-GlcNAc modification sites on tau and generation of a site-specific O-GlcNAc tau antibody, Amino Acids, 2011, 40, 857–868 CrossRef CAS PubMed.
- A. Cameron, et al., Generation and characterization of a rabbit monoclonal antibody site-specific for tau O-GlcNAcylated at serine 400, FEBS Lett., 2013, 587, 3722–3728 CrossRef CAS PubMed.
- T. Tao, Z. He, Z. Shao and H. Lu, TAB3 O-GlcNAcylation promotes metastasis of triple negative breast cancer, Oncotarget, 2016, 7, 22807–22818 CrossRef PubMed.
- D. L. Shen, T. M. Gloster, S. A. Yuzwa and D. J. Vocadlo, Insights into O-linked N-acetylglucosamine (O-GlcNAc) processing and dynamics through kinetic analysis of O-GlcNAc transferase and O-GlcNAcase activity on protein substrates, J. Biol. Chem., 2012, 287, 15395–15408 CrossRef CAS PubMed.
- K. H. Lim, C. H. Ha and H. I. Chang, Production of O-GlcNAc modified recombinant proteins in Escherichia coli, J. Microbiol. Biotechnol., 2002, 12, 306–311 CAS.
- K. C. Sohn and S. I. Do, Transcriptional regulation and O-GlcNAcylation activity of zebrafish OGT during embryogenesis, Biochem. Biophys. Res. Commun., 2005, 337, 256–263 CrossRef CAS PubMed.
- O. Y. Goodwin, M. S. Thomasson, A. J. Lin, M. M. Sweeney and M. A. E. Macnaughtan, E. coli sabotages the in vivo production of O-linked β-N-acetylglucosamine-modified proteins, J. Biotechnol., 2013, 168, 315–323 CrossRef CAS PubMed.
- D. H. Ramirez, et al., Engineering a Proximity-Directed O-GlcNAc Transferase for Selective Protein O-GlcNAcylation in Cells, ACS Chem. Biol., 2020, 15, 1059–1066 CrossRef CAS PubMed.
- S. Yamazoe, et al., Heterobifunctional Molecules Induce Dephosphorylation of Kinases–A Proof of Concept Study, J. Med. Chem., 2019, 63, 2807–2813 CrossRef PubMed.
- T. W. Muir, D. Sondhi and P. A. Cole, Expressed protein ligation: a general method for protein engineering, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6705–6710 CrossRef CAS PubMed.
- R. E. Thompson and T. W. Muir, Chemoenzymatic Semisynthesis of Proteins, Chem. Rev., 2020, 120, 3051–3126 CrossRef CAS PubMed.
- S. Schwagerus, O. Reimann, C. Despres, C. Smet-Nocca and C. P. Hackenberger, Semi-synthesis of a tag-free O-GlcNAcylated tau protein by sequential chemoselective ligation, J. Pept. Sci., 2016, 22, 327–333 CrossRef CAS PubMed.
- Y. E. Lewis, et al., O-GlcNAcylation of α-Synuclein at Serine 87 Reduces Aggregation without Affecting Membrane Binding, ACS Chem. Biol., 2017, 12, 1020–1027 CrossRef CAS PubMed.
- A. T. Balana, et al., O-GlcNAcylation of small heat shock proteins enhances their anti-amyloid chaperone activity, bioRxiv, 2019, 869909, DOI:10.1101/869909.
- M. K. Tarrant, et al., Regulation of CK2 by phosphorylation and O-GlcNAcylation revealed by semisynthesis, Nat. Chem. Biol., 2012, 8, 262–269 CrossRef CAS PubMed.
- C. A. De Leon, P. M. Levine, T. W. Craven and M. R. Pratt, The Sulfur-Linked Analogue of O-GlcNAc (S-GlcNAc) Is an Enzymatically Stable and Reasonable Structural Surrogate for O-GlcNAc at the Peptide and Protein Levels, Biochemistry, 2017, 56, 3507–3517 CrossRef CAS PubMed.
- V. D. Romanenko and V. P. Kukhar, Fluorinated phosphonates: Synthesis and biomedical application, Chem. Rev., 2006, 106, 3868–3935 CrossRef CAS PubMed.
- L. Lercher, et al., Generation of a synthetic GlcNAcylated nucleosome reveals regulation of stability by H2A-Thr101 GlcNAcylation, Nat. Commun., 2015, 6, 7978 CrossRef CAS PubMed.
- R. Raj, L. Lercher, S. Mohammed and B. G. Davis, Synthetic Nucleosomes Reveal that GlcNAcylation Modulates Direct Interaction with the FACT Complex, Angew. Chem., Int. Ed., 2016, 55, 8918–8922 CrossRef CAS PubMed.
- M. Fernández-González, et al., Site-selective chemoenzymatic construction of synthetic glycoproteins using endoglycosidases, Chem. Sci., 2010, 1, 709–715 RSC.
- G. J. L. Bernardes, J. M. Chalker, J. C. Errey and B. G. Davis, Facile conversion of cysteine and alkyl cysteines to dehydroalanine on protein surfaces: Versatile and switchable access to functionalized proteins, J. Am. Chem. Soc., 2008, 130, 5052–5053 CrossRef CAS PubMed.
- T. H. Wright, et al., Posttranslational mutagenesis: A chemical strategy for exploring protein side-chain diversity, Science, 2016, 354, aag1465 CrossRef PubMed.
- G. Tegl, et al., Facile Formation of β-thioGlcNAc Linkages to Thiol-Containing Sugars, Peptides, and Proteins using a Mutant GH20 Hexosaminidase, Angew. Chem., Int. Ed., 2019, 58, 1632–1637 CrossRef CAS PubMed.
- W. Yi, et al., Phosphofructokinase 1 glycosylation regulates cell growth and metabolism, Science, 2012, 337, 975–980 CrossRef CAS PubMed.
- H. Jang, et al., O-GlcNAc regulates pluripotency and reprogramming by directly acting on core components of the pluripotency network, Cell Stem Cell, 2012, 11, 62–74 CrossRef CAS PubMed.
- N. Khidekel, et al., Probing the dynamics of O-GlcNAc glycosylation in the brain using quantitative proteomics, Nat. Chem. Biol., 2007, 3, 339–348 CrossRef CAS PubMed.
- H. C. Dorfmueller, V. S. Borodkin, M. Schimpl and D. M. F. van Aalten, GlcNAcstatins are nanomolar inhibitors of human O-GlcNAcase inducing cellular hyper-O-GlcNAcylation, Biochem. J., 2009, 420, 221–227 CrossRef CAS PubMed.
- T. M. Gloster, et al., Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells, Nat. Chem. Biol., 2011, 7, 174–181 CrossRef CAS PubMed.
- J. W. Chin, Expanding and reprogramming the genetic code, Nature, 2017, 550, 53–60 CrossRef CAS PubMed.
- R. J. Ernst, et al., Genetic code expansion in the mouse brain, Nat. Chem. Biol., 2016, 12, 776–778 CrossRef CAS PubMed.
- A. Bianco, F. M. Townsley, S. Greiss, K. Lang and J. W. Chin, Expanding the genetic code of Drosophila melanogaster, Nat. Chem. Biol., 2012, 8, 748–750 CrossRef CAS PubMed.
- S. Greiss and J. W. Chin, Expanding the genetic code of an animal, J. Am. Chem. Soc., 2011, 133, 14196–14199 CrossRef CAS PubMed.
- K. Sakamoto, et al., Site-specific incorporation of an unnatural amino acid into proteins in mammalian cells, Nucleic Acids Res., 2002, 30, 4692–4699 CrossRef CAS PubMed.
- A. Dumas, L. Lercher, C. D. Spicer and B. G. Davis, Designing logical codon reassignment – Expanding the chemistry in biology, Chem. Sci., 2015, 6, 50–69 RSC.
- H. Neumann, S. Y. Peak-Chew and J. W. Chin, Genetically encoding Nε-acetyllysine in recombinant proteins, Nat. Chem. Biol., 2008, 4, 232–234 CrossRef CAS PubMed.
- D. T. Rogerson, et al., Efficient genetic encoding of phosphoserine and its nonhydrolyzable analog, Nat. Chem. Biol., 2015, 11, 496–503 CrossRef CAS PubMed.
- S. Virdee, et al., Traceless and site-specific ubiquitination of recombinant proteins, J. Am. Chem. Soc., 2011, 133, 10708–10711 CrossRef CAS PubMed.
- M. Fottner, et al., Site-specific ubiquitylation and SUMOylation using genetic-code expansion and sortase, Nat. Chem. Biol., 2019, 15, 276–284 CrossRef CAS PubMed.
- M. S. Zhang, et al., Biosynthesis and genetic encoding of phosphothreonine through parallel selection and deep sequencing, Nat. Methods, 2017, 14, 729–736 CrossRef CAS PubMed.
- X. Luo, et al., Genetically encoding phosphotyrosine and its nonhydrolyzable analog in bacteria, Nat. Chem. Biol., 2017, 13, 845–849 CrossRef CAS PubMed.
- A. K. Antonczak, Z. Simova and E. M. Tippmann, A critical examination of Escherichia coli esterase activity, J. Biol. Chem., 2009, 284, 28795–28800 CrossRef CAS PubMed.
- Z. Zhang, et al., Retraction, Science, 2009, 326, 1187 CAS.
- A. Gorelik, Genetic encoding of a stable O-GlcNAc analogue, PhD thesis, University of Dundee, 2018.
- E. Kaya, et al., Synthesis of threefold glycosylated proteins using click chemistry and genetically encoded unnatural amino acids, ChemBioChem, 2009, 10, 2858–2861 CrossRef CAS PubMed.
- B. Yang, et al., Genetically Introducing Biochemically Reactive Amino Acids Dehydroalanine and Dehydrobutyrine in Proteins, J. Am. Chem. Soc., 2019, 141, 7698–7703 CrossRef CAS PubMed.
- N. E. Fahmi, L. Dedkova, B. Wang, S. Golovine and S. M. Hecht, Site-specific incorporation of glycosylated serine and tyrosine derivatives into proteins, J. Am. Chem. Soc., 2007, 129, 3586–3597 CrossRef CAS PubMed.
- T. Matsubara, K. Iijima, T. Watanabe, T. Hohsaka and T. Sato, Incorporation of glycosylated amino acid into protein by an in vitro translation system, Bioorg. Med. Chem. Lett., 2013, 23, 5634–5636 CrossRef CAS PubMed.
- H. Wang, et al., Ac4GlcNAcF3, an OGT-tolerated but OGA-resistant regulator for O-GlcNAcylation, Bioorg. Med. Chem. Lett., 2019, 29, 802–805 CrossRef CAS PubMed.
Footnotes |
| † Current address: Department of Chemistry, Imperial College London, London, UK; The Francis Crick Institute, London, UK. |
| ‡ AG and DvA wrote the manuscript. |
| This journal is © The Royal Society of Chemistry 2020 |