
Tumor localization of oncolytic adenovirus assisted by pH-degradable microgels with JQ1-mediated boosting replication and PD-L1 suppression for enhanced cancer therapy†
Haishi
Qiao‡
a,
Xingmei
Chen‡
a,
Qiming
Wang
b,
Junmei
Zhang
a,
Dechun
Huang
a,
Enping
Chen
a,
Hongliang
Qian
a,
Yinan
Zhong
*a,
Qi
Tang
*c and
Wei
Chen
 *a
*a
aDepartment of Pharmaceutical Engineering, School of Engineering, China Pharmaceutical University, Nanjing 210009, PR China. E-mail: zhongyinan@126.com; w.chen@cpu.edu.cn
bCollege of Bioscience and Biotechnology, Hunan Agricultural University, Changsha 410128, PR China
cKey Laboratory of Antibody Technology, National Health Commission, Nanjing Medical University, Nanjing 211166, PR China. E-mail: qitang@njmu.edu.cn
First published on 3rd March 2020
Abstract
Oncolytic therapy is a fast-developing cancer treatment field based on the promising clinical performance from the selective tumor cell killing and induction of systemic antitumor immunity. The virotherapy efficacy, however, is strongly hindered by the limited virus propagation and negative immune regulation in the tumor microenvironments. To enhance the antitumor activity, we developed injectable pH-degradable PVA microgels encapsulated with oncolytic adenovirus (OA) by microfluidics for localized OA delivery and cancer treatments. PVA microgels were tailored with an OA encapsulation efficiency of 68% and exhibited a pH-dependent OA release as the microgel degradation at mildly acidic conditions. PVA microgels mediated fast viral release and increased replication in HEK293T and A549 cells at a lower pH, and the replication efficiency could be further reinforced by co-loading with one BET bromodomain inhibitor JQ1, inducing significant cytotoxicity against A549 cells. An in vivo study revealed that OA release was highly located at the tumor tissue assisted by PVA microgels, and the OA infection was also enhanced with the addition of JQ1 treatment, meanwhile greatly inhibiting the PD-L1 expression to overcome the immune suppression. OA/JQ1 co-encapsulated injectable microgels exhibited a superior in vivo antitumor activity on the A549 lung tumor-bearing mice by the combination of inhibited proliferation, amplified oncolysis, and potential immune regulation.
1 Introduction
Oncolytic viral therapy represents an emerging new class of cancer therapeutics in clinical applications based on the direct lysis of tumor cells and activated antitumor immune responses.1,2 Among all the oncolytic viruses, oncolytic adenovirus (OA) has been considered as one of the promising candidates for cancer treatments because of its biomedical advantages.3,4 Not only being gene vectors for efficient gene transfection, but OA propagation is also highly cancer-specific with significant prohibition in the normal cells because of the intrinsic defense mechanism involved with the endogenous proteins for tumor suppression, e.g. pRb and p53.5,6 Moreover, the OA-mediated oncolytic therapy can also activate a systemic immune response for immunotherapy.7,8 However, the antitumor efficiency of oncolytic viruses is often impaired by the poor retention at tumor tissues and limited spreading inside the tumor cells as well as the antiviral innate immune response.9–11Based on this, the OA delivery by nano/microsystems is acting as a potential approach to reinforce therapeutic efficiency by providing persistence of OA at the target tissue, escaping immune system recognition, and ultimately eliminating the need for repeated administrations with high doses.12–15 A local-sustained OA delivery assisted by hydrogel-based scaffolds for cancer virotherapy exhibited significant inhibition on the tumor cell growth because of their high water content and good biocompatibility and superior tumor localization.16–18 For example, Choi et al. engineered alginate-based hydrogels to encapsulate OA for prolonged local OA release at the U343 glioma tumor tissue in vivo, resulting in an enhanced antitumor activity.19 They further developed gelatin-based hydrogels encapsulated with OA-TRAIL and realized increased tumoral accumulation and infection, leading to enhanced antitumor efficacy. In addition, due to the prolonged OA-TRAIL persistence at the tumor tissue by gelatin hydrogels, the immune response was activated with higher intratumoral infiltration of CD4+/CD8+T cells for a combinational cancer immunotherapy.20 Despite the fact that the sustained local OA delivery promoted the therapeutic efficacy, clinical trials of OA have shown a limited outcome owing to the replicative ability in tumor cells and resistance to oncolytic immunotherapy such as the PD-L1 expression induced by OA.21–23
To enhance viral replication inside the tumor cells, several chemical sensitizers have been used for enhancing the propagation of the oncolytic virus in preclinical studies including histone deacetylase inhibitors, dimethyl fumarate or immune-suppressants such as cyclophosphamide and rapamycin.24–27 Li et al. reported that the BET bromodomain inhibitor (+)-JQ1 (JQ1) was able to improve the infection of the herpes simplex virus (HSV) and OA by elevating the transcription level of virus genome for enhanced viral replication.28,29 In addition, JQ1 was also reported to provide antitumor activity against several tumor models including pancreatic ductal adenocarcinoma, osteosarcoma, and neuroblastoma via BET bromodomain inhibition.30–32 More importantly, JQ1 exhibited a robust inhibition on the PD-L1 expression to overcome the immune suppression and improve the cancer immunotherapy, but it is worthy to note its short half-life in vivo.33,34
Microgels (MGs) with micro-sized hydrogels because of the high surface/volume ratio can be injected as tissue constructs with few phagocytosis or recognition by the RES system (reticuloendothelial).35–37 To simultaneously improve the virus propagation and reduce the negative immune regulation at the tumor tissue, here, we fabricated pH-degradable PVA MGs using microfluidics for the biorthogonal encapsulation and sustained localized delivery of conditionally replicating adenovirus (Scheme 1). PVA-based MGs have been used in several biomedical applications such as protein delivery and cell delivery, which are mostly due to their good water swelling behaviors and biocompatibility.37,38 JQ1 encapsulated in the MGs would prolong the half-life and achieve the controlled release to enhance the propagation of OA, inhibit tumor cell proliferation and PD-L1 expression, realizing the significantly combinational antitumor performance against the A549 tumor model.
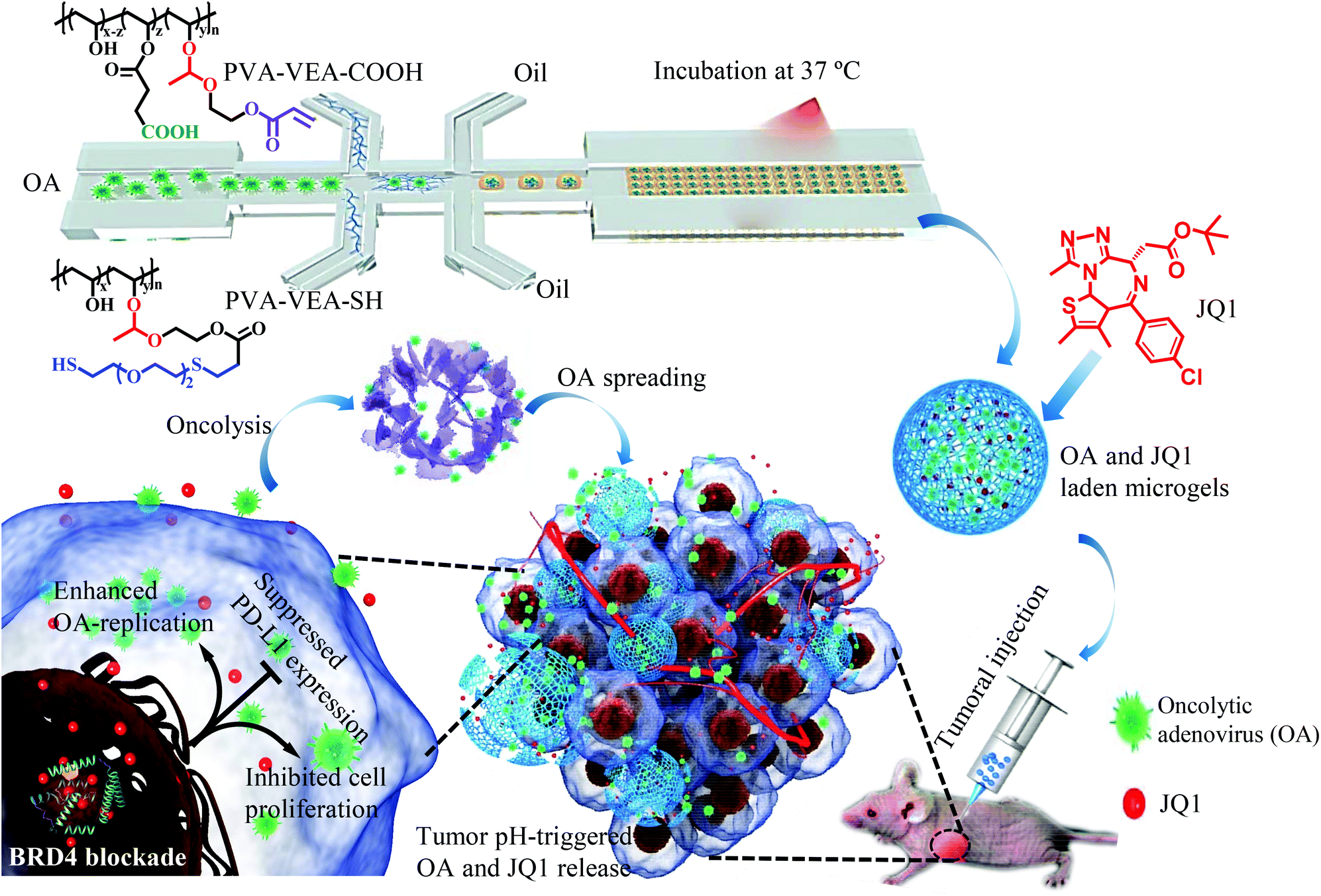 | ||
| Scheme 1 Illustration of pH-degradable microgels (MGs) for the simultaneous encapsulation of OA and JQ1 for an enhanced oncolytic viral treatment with JQ1-meidated boosting viral replication and PD-L1 suppression. | ||
2 Experimental section
2.1 Microgel fabrication
The microgel-encapsulated OA (AMGs) were prepared by microfluidics technology using poly(dimethylsiloxane) (PDMS) chips according to our previous report.37 Briefly, PVA-VEA-COOH and PVA-VEA-SH were dissolved separately in PBS (100 mg mL−1) with N2 perfusion to remove oxygen and used as two dispersed phases. OA was suspended in PBS (5.0 × 1010 mL−1) and served as the virus phase. Paraffin oil containing 1.0 wt% of EM 90 (modified polyether-polysiloxane) and 2.0 wt% of MGPR 90 (polyglycerol polyricinoleate) was used as the continuous oil phase. The flow rates of the four phases were adjusted by syringe pumps to control the size of the microgels approximately at 150–200 μm. After incubation at 37 °C for 1 h, the collected microgels were washed with PBS to remove the paraffin and surfactant. For the preparation of OA and JQ1 co-laden microgels (JAMGs), JQ1 in ethanol (50 mg mL−1) was mixed into the microgels at a feeding weight ratio of 10% by vortex for 2 min, and purified by suspending in the PBS and centrifugation three times.2.2 pH-Dependent release of OA and JQ1
For the in vitro release study, 5.0 mL of JAMGs were placed into PB (pH 7.4 and 6.5) at 37 °C under slow shaking. At desired time points, 0.5 mL of the supernatant was collected after centrifugation (1000 rpm) and replaced with a fresh medium. The amount of OA in the release medium was quantified by a multiscan spectrum (Molecular devices spectramax@i3x, USA) according to a standard curve. The amount of JQ1 was determined by HPLC (Agilent1260, USA) with UV detection at 254 nm. Release experiments were performed three times, and the results are presented as the mean ± standard deviation (SD).2.3 In vitro infection test
Human lung adenocarcinoma A549 cells and human embryonic kidney cell line expressing the adenovirus E1 region (HEK293T) cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 1% nonessential amino acids, and 1 mM sodium pyruvate. All the medium and supplements were obtained from Biological Industries. The cells were cultured at 37 °C in a humidified atmosphere with 5% CO2. HEK293T cells were seeded in 12-well plates (5.0 × 105 cells per well) for 8 h, followed by the addition of AMGs at a MOI (multiplicity of infect) of 3 at pH 7.4 and 6.5 for different time periods. In another study, HEK293T cells were treated with AMGs at a MOI of 1 containing different concentrations of JQ1 at pH 7.4 and pH 6.5 conditions for 16 h incubation. In both cases, the cells were observed via fluorescence microscopy (Leica DMI3000 B, Germany). The infection efficiency of OA in the HEK293T cells was also determined via flow cytometry (BD Accuri C6). Data from 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 events were collected for further analysis and represented the average values of relative fluorescence intensities in three independent experiments.
000 events were collected for further analysis and represented the average values of relative fluorescence intensities in three independent experiments.
2.4 In vitro cytotoxicity against A549 cells
A549 cells were seeded at a density of 5.0 × 105 cells per well in 12-well plates for 8 h, followed by the addition of AMGs at a MOI of 3 containing different concentrations of JQ1 for 24 h incubation. The cells infected by OA were observed via fluorescence microscopy (Leica DMI3000 B, Germany).A549 cells (1.0 × 105) were seeded in 12-well plates and infected with AMGs at a MOI of 1 containing different concentrations of JQ1 at pH 6.5 and 7.4. After 120 h OA infection, cells were fixed with 4% paraformaldehyde for 10 min and stained with a 0.5% crystal violet solution at room temperature for 10 min. The plates were rinsed with water and dried at room temperature overnight for photographing.
A549 cells were seeded at the density of 4.0 × 103 cells per well in a 96-well plate for 12 h and subsequently treated with naked OA or AMGs at different MOIs for 72 h. In another study, A549 cells were seeded at the density of 4 × 103 cells and treated with naked OA or AMGs (MOI = 3) containing different concentrations of JQ1. In both cases, after 72 h cell incubation, 10 μL of MTT (5.0 mg mL−1) was added to each well for another 4 h incubation. After that, the medium was removed and added with 100 μL of DMSO. The absorbance at 570 nm was measured in a microplate reader (MD@i3x, USA). The experiments were conducted in triplicate, and the results were presented as the mean ± SD.
2.5 In vivo imaging of OA infection and flow cytometry analysis
In vivo imaging of the OA infection was performed in A549-Luc lung tumor-bearing nude mice. The experimental mice were purchased from the Model Animal Research Centre at Nanjing University (Nanjing, China). All animal procedures were performed in accordance with the guidelines for Care and Use of Laboratory Animals of China Pharmaceutical University (Nanjing, China) and approved by the Animal Ethics Committee of the Ministry of Health in China. Naked OA or AMGs (1.0 × 109 VPs per tumor) were intratumorally injected into the tumor tissues. In order to monitor the live fluorescence imaging by OA infection, the mice were anesthetized with isoflurane and then, the fluorescent images were acquired by a fluorescence imaging system (IVIS® Lumina™, USA) at the interval time points at the excitation of 480 nm and emission of 750 nm. After 24 h, the tumor tissues were separated to examine the frozen section, and digital images were obtained under a confocal laser scanning microscope (C2, Nikon, JAPAN). In another study, naked OA or AMGs (1.0 × 109 VPs per tumor) at JQ1 concentrations of 10 mg kg−1 were intratumorally injected into the tumor tissues. After 48 h, the separated tumor tissues were sheared into pieces and digested into cell suspension. The OA infection was investigated using a fluorescence microscope (Leica DMI3000 B, Germany) and analyzed by FACS Calibur (BD Biosciences).2.6 In vitro and In vivo detection of PD-L1 expression
A549 cells (1.0 × 105) were seeded in 12-well plates for 8 h, and subsequently pre-incubated with different concentrations of JQ1 for 30 min, followed by treating with IFN-γ (30 ng mL−1) stimulation for 24 h. Moreover, after cell seeding, A549 cells were co-cultured with OA at different MOIs for 24 h. In another study, A549 cells were co-cultured with OA at a MOI of 1 containing different concentrations of JQ1 for 24 h. For all the cases, the cells were harvested and stained with the anti-PD-L1 antibody (Biolegend, LOT: 3209706, clone: 29E.2A3) and analyzed via flow cytometry. The cells without any treatment were stained with the anti-PD-L1 antibody and used as a control. For the in vivo evaluation of the PD-L1 expression, naked OA or AMGs (1.0 × 109 VPs) were intratumorally injected into the tumor tissues with or without JQ1 (10 mg kg−1). After 72 h, the separated tumor tissues were fixed and performed via immunohistochemistry (IHC) for IFN-γ and PD-L1 analysis.2.7 In vivo antitumor activity
In vivo antitumor activity was examined on a A549-Luc lung tumor xenograft mouse model. As the tumor size in volume reached to 50–100 mm3, the mice were randomly divided into seven groups with five mice in each group: PBS, naked OA, free JQ1, combination of naked OA and JQ1 (OA + JQ1), JQ1-loaded microgels (JMGs), OA-encapsulated microgels (AMGs) and OA/JQ1 co-laden microgels (JAMGs). Treatment with different formulations was administrated intra-tumorally at a JQ1 dosage of 10 mg kg−1 and 1.0 × 109 VPs per tumor, respectively, and the time with the first injection was designated as day 0. The injection performance was carried out on days 0, 5 and 10. Then, the tumor size was recorded every 3 d and tumor volume was calculated according to the common formula: V = 0.5 × L × W2, wherein L and W were the tumor dimensions at the longest and widest sides, respectively.V/V0 was recorded as the relative tumor volume (V0: the initial tumor size at day 0). W/W0 was recorded as the relative body weight of the mice (W0: the body weight on day 0). Finally, after the intravenous injection of D-luciferin potassium salt, the luminescence intensity of tumor xenograft mice was observed by an IVIS device. Then, the tumor tissues were isolated from the euthanatized mice and photographed, HE staining and IHC (immunohistochemistry). The normal tissues of the excised liver, heart, lung, spleen, and kidney were fixed with 4% paraformaldehyde and embedded in paraffin. The sliced organ tissues on the glass slides were stained by HE and imaged using a microscope (Nikon, Japan).
3 Results and discussion
3.1 Preparation of microgels loaded with OA
The poor retention and the limited replication of the oncolytic virus in the tumor microenvironments as well as the immune suppression induced by oncolytic viral therapy have imposed major obstacles to cancer treatments.39 In order to enhance the infection efficiency towards tumor tissues, we have developed pH-degradable PVA-derivatives of PVA-VEA-COOH and PVA-VEA-SH (Fig. S1†) and combined microfluidics with a Michael-type addition crosslinking reaction to form microgels for the tumor localization of OA. BET bromodomain inhibitor JQ1 was simultaneously encapsulated into microgels for boosting OA replication and PD-L1 repression to achieve efficient oncolytic virotherapy. PVA-VEA-COOH and PVA-VEA-SH separately dissolved in water and mixed with OA suspension within microfluidic chips were disintegrated by the continuous oil phase to form microdroplets with a monodispersed diameter of 150 μm, followed by incubation to form the crosslinked OA-encapsulated microgels (AMGs) with a small swollen size of ∼200 μm (Fig. 1A). The OAs engineered with EGFP were obviously detected in the AMGs via fluorescence microscopy, and the SEM image shows the nanoporous structures of microgels as sophisticated scaffolds for OA delivery (Fig. 1A).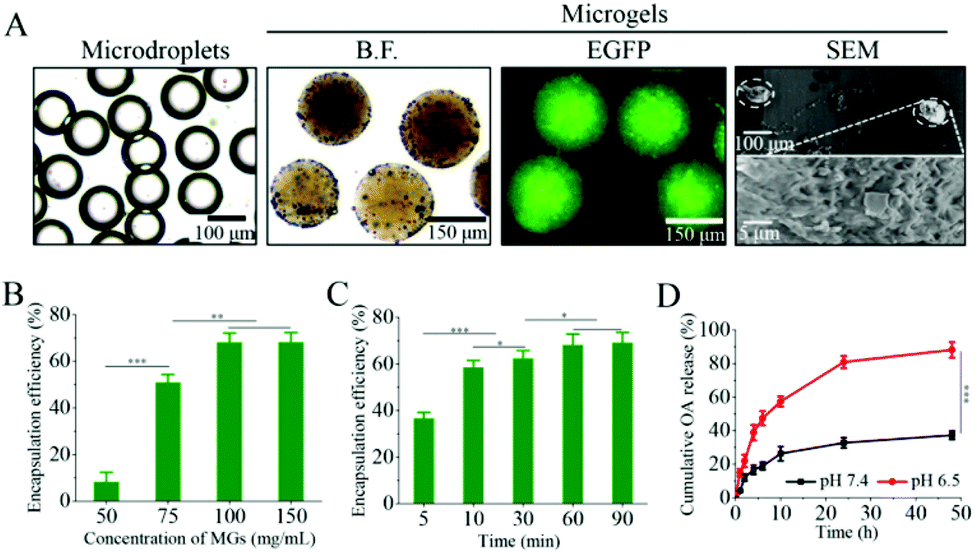 | ||
| Fig. 1 (A) Images of OA-encapsulated microdroplets in paraffin oil and microgels (MGs) in PBS prepared by microfluidics technology. Conditions: VOil = 20 μL min−1, and VOA = 2.0 μL min−1 and VW = 2.0 μL min−1. From left to right: bright field (B.F.) images of microdroplets in oil, MGs in PBS, fluorescence image and SEM image (the inset showed the image with higher magnification). (B) Relative OA encapsulation efficiency of MGs prepared with different polymer concentrations and crosslinking for 60 min. (C) Relative OA encapsulation efficiency of MGs prepared with different crosslinking times at a polymer concentration of 100 mg mL−1. (D) The cumulative release profile of OA from MGs in response to different pH conditions at 37 °C. The data are presented as the average ± SD (n = 3, ***p < 0.001). | ||
To demonstrate whether the concentration of the polymer and crosslinking time affected the loading efficiency (LE) of OA, AMGs were prepared with different polymer concentrations ranging from 50 to 150 mg mL−1 and the crosslinking time various from 5 to 90 min. As shown in Fig. 1B, the LE of OA was improved as the polymer concentration increased and reached to the highest LE value of 68% at the polymer concentration over 100 mg mL−1 (Fig. 1B). The results indicated that the higher polymer concentration induced microgels with the increasing crosslinking density, leading to the more sophisticated networks to prevent the premature OA release.37 The incubation time for microgel crosslinking also determined the LE of OA, and the results showed that the LE level remained stable at 68% when the microgels were prepared at the polymer concentration of 100 mg mL−1 with the incubation time of 60 min for crosslinking (Fig. 1C).
3.2 pH-Dependent OA release
VEA-functionalized PVA-based nano/microgels have been developed for a pH-triggered drug and cell delivery,37,40 in which VEA-involved acetal groups could be cleaved at mildly acidic conditions. The in vitro release profile of OA was performed under different pH 7.4 and pH 6.5 (mimicking tumor microenvironmental pH condition) at 37 °C. As displayed in Fig. 1D, OA release from AMGs was restricted at physiological pH with a minimal amount of 25% in 10 h, and the release amount was detected with 38% even after 48 h. However, the OA release was accelerated significantly at pH 6.5, in which approximately 60% of OA was released in 10 h, and the release amount reached 88% in 48 h due to the microgel degradation at an acidic pH condition.Despite the fact that the tumor-immune microenvironment can be activated in response to the oncolytic virus, there was a simultaneous immune suppression effect through PD-L1 upregulation due to the infection of the oncolytic virus, resulting in the resistance to oncolytic immunotherapy.21,41 JQ1, as one of the bromodomain and extra-terminal (BET) inhibitors, can not only directly inhibit the proliferation of tumor cells, but also can overcome immune tolerance by suppressing the intratumoral PD-L1 expression.30,33 It is also reported that JQ1 can enhance the infection of the oncolytic virus by increased replication in host cells.28,29 Therefore, OA microgel systems involved with JQ1 would possibly perform a combination of direct tumor inhibition and enhanced OA infection, accompanying with the intratumoral PD-L1 suppression for cancer therapy. JQ1-loaded microgels (JMGs) showed a LE of 85% at a loading content of 7.8%, which was mainly due to the strong hydrophobic interaction. The in vitro release also displayed visibly accelerated JQ1 release from microgels at pH 6.5 compared with that at physiological pH (Fig. S2†), by which JQ1 and OA would be simultaneously released as the pH-dependent microgel degradation.
3.3 Intracellular JQ1-enhanced OA infection
In order to confirm that OA released from MGs could further replicate and infect the host cells, HEK293T cells were incubated with AMGs at different MOIs for 24 h, and it showed that the fluorescence intensity of OA, replicating in the host cells, became much stronger as the MOI increased (Fig. S3†). Then, the HEK293T cells were further incubated with AMGs at a MOI of 3 at pH 7.4 and 6.5 for different time periods to look into the effect of pH-dependent release on the infection. As shown in Fig. 2A, OA began to diffuse out of the microgels and infect HEK293T cells as early as 2 h incubation under an acidic condition, but little fluorescence was visible after 2 h incubation at physiological pH. It should be noteworthy to mention that the fluorescence intensity significantly increased at pH 6.5 as the incubation time extended to 16 h compared with that at pH 7.4, which was mostly due to the accelerated OA release at an acidic condition, followed by more OA replication in the HEK293T cells. The fluorescence intensity of OA, replicating in HEK293T cells, was further quantified via flow cytometry, in which the relative intensity incubated at pH 6.5 was nearly 2.2-fold higher than at pH 7.4 after 16 h, and it was quite in line with the fluorescence observation (Fig. 2B).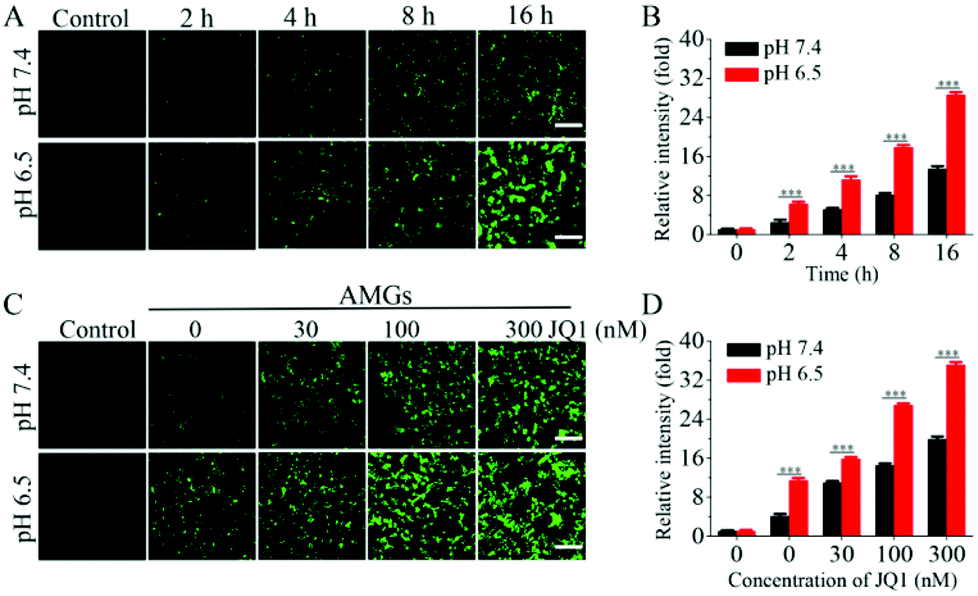 | ||
| Fig. 2 (A) Fluorescence images of the pH-dependent OA release to infect the HEK293T cells and (B) relative fluorescence intensity of EGFP by the flow cytometry analysis to quantify the infection efficiency of the HEK293T cells co-incubated with AMGs (MOI = 3) at different time periods. (C) Fluorescence images of the OA release to infect the HEK293T cells and (D) relative fluorescence intensity of EGFP by the flow cytometry analysis to quantify the infection efficiency of the HEK293T cells by 16 h co-incubation with AMGs (MOI = 1) containing different concentrations of JQ1. The data are presented as the average ± SD performed with three independent experiments (scale bars: 200 μm, ***p < 0.001). | ||
JQ1 is capable of switching BRD4 to transcription regulation of viral gene expression, which promotes the OA infection in the host cells.28 The HEK293T cells were incubated with AMGs (MOI = 1) containing different concentrations of JQ1 at pH 7.4 and 6.5 for 24 h incubation. As shown in Fig. 2C, based on the OA released from the microgels to infect the HEK293T cells, the addition of JQ1 could significantly dose-dependently increase the yield of OA production in host cells, as indicated by the enhanced fluorescence intensity. As compared to the physiological pH, the mildly acid environments caused notable OA replication due to more OA release from AMGs during the early treatment at pH 6.5 following with the stimulus of JQ1. The fluorescence intensity of the OA replication was also validated quantitatively via flow cytometry, further demonstrating that the combination of the OA release under an acidic condition and JQ1 significantly elevated the OA infection in the HEK293T cells (Fig. 2D).
3.4 In vitro tumor cell killing effects of AMGs combined with JQ1
OAs with E1-deleted were taken as models for the oncolytic virus to study the anticancer activity using the human lung carcinoma A549 cells since A549 cells with high expression of coxsackievirus-adenovirus receptors are susceptible to adenovirus infection.42,43 The results of the fluorescence microscope revealed that OA was able to infect the A549 cells, and the infection efficiency could be dose-dependently improved by the addition of JQ1-treatment (Fig. 3A), which was in accord with the results of infection in the HEK293T cells. Plaque assays showed that AMGs infected A549 cells and induced cytopathic effect (CPE) at both pH values of 7.4 and 6.5. The treatment with the addition of JQ1 could greatly inhibit the growth of A549 cells, as indicated by the enhanced CPE (Fig. 3B). MTT assay was also performed to investigate the inhibition effects of OA combined with JQ1 on cell viability. Naked OA and AMGs were incubated with the A549 cells at pH 6.5, and the results exhibited that naked OA induced more cytotoxicity as compared to AMGs, which might be because of the hysteretic OA release from the microgels compared with naked OA during the incubation period (Fig. 3C). JQ1 has the ability of directly inhibiting the proliferation of tumor cells, and here, it induced the viability of the A549 cells declining from 92% to 43% as the JQ1 concentration increased from 10 nM to 300 nM (Fig. 3D). It should be noted that the combination of OA and JQ1 greatly elevated the antitumor activity against the A549 cells. As shown in Fig. 3C, naked OA and AMGs at a MOI of 3 without JQ1 only decreased the cell viability to 54% and 78%, respectively, while the addition of JQ1 treatment significantly improved the killing activity against the A549 cells; for example, the cell viability decreased less than 20% at a JQ1 concentration of 300 nM for both treatments of OA and AMGs (Fig. 3D).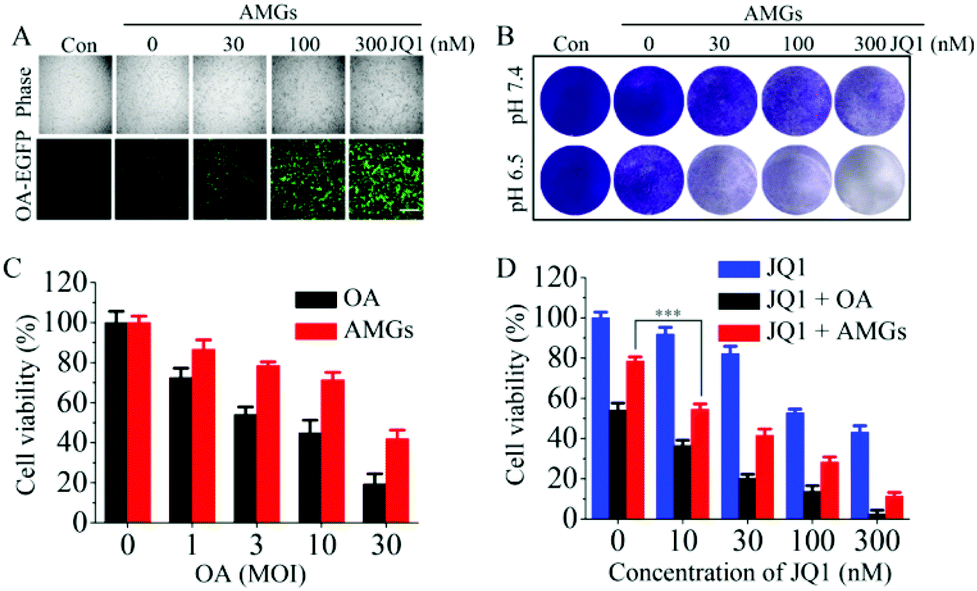 | ||
| Fig. 3 In vitro antitumor activity by OA infection on the A549 cells (A) Fluorescence images of OA release to infect the A549 cells by 24 h co-incubation with AMGs (MOI = 3) containing different concentrations of JQ1 (scale bars: 200 μm); (B) The cytopathic effect (CPE) of AMGs (MOI = 1) containing different concentrations of JQ1 on the A549 cells after 120 h incubation analyzed by crystal violet staining. Cell viability determined by the MTT assay using the A549 cells treated with different MOIs of naked OA and AMGs for 72 h (C) and treated with naked OA and AMGs (MOI = 3) containing different concentrations of JQ1 for 72 h (D). The data are presented as the average ± SD (n = 3, ***p < 0.001). | ||
3.5 In vivo biodistribution and tumor infection
In vivo fluorescence imaging was performed to investigate the tumor retention of OA in A549 tumor-bearing nude mice, in which OA with EGFP expression facilitated the biodistribution observation. The mice were subcutaneously injected with naked OA and AMGs, and the EGFP fluorescence was detected at the tumor sites at the beginning. However, the fluorescence of naked OA rapidly reduced in 2 h and was almost invisible after 4 h, while AMGs maintained the fluorescence intensity until 16 h, indicating the long tumoral retention of OA, which could play an important role in improving the OA infection of tumor tissues (Fig. 4A). In order to investigate whether the MG-mediated delivery of OA was able to enhance the infection of tumor tissues, MGs labelled with Cy5 were used to assess the in vivo infected ability of AMGs. Cy5-labelled AMGs or naked OA were intratumorally injected into the A549 tumor-bearing mice. After 48 h treatment, frozen section analysis was performed on tumor tissues collected from euthanatized mice to evaluate the infection of OA. The results showed that the AMG treatment significantly increases OA infection and spreading in tumor tissues, as indicated by the much stronger intensity and broader distribution of EGFP as compared to naked OA (Fig. 4B).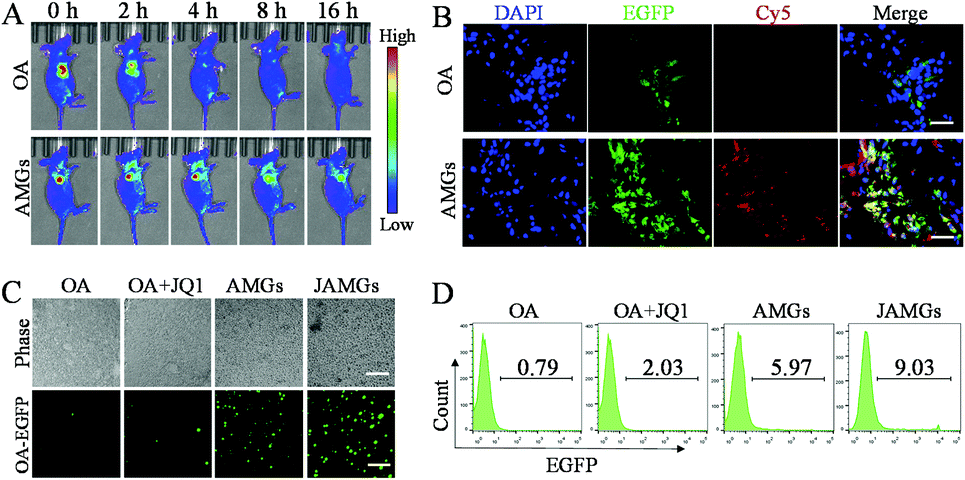 | ||
| Fig. 4 (A) In vivo living images of the A549 tumor-bearing nude mice intratumorally injected with AMGs or naked OA containing the OA number of 1.0 × 109 photographed at the indicated time. (B) CLSM images of tumor tissue slices from A549 tumor-bearing nude mice intratumorally injected with AMGs or naked OA (OA number: 1.0 × 109) after 24 h (blue: nucleus of tumor cells stained with DAPI, green: EGFP expression in tumor cells infected by OA, red: MGs labelled with Cy5, scale bar: 40 μm). (C) Fluorescence images of tumor cells and (D) flow cytometer analysis of tumor cells from the digested tumor tissues of A549 tumor-bearing nude mice intratumorally treated with AMGs or naked OA (OA number: 1.0 × 109) with or without JQ1 (10 mg kg−1) for 48 h (scale bar: 200 μm). | ||
3.6 JQ1-mediated enhanced in vivo OA infection
JQ1 exhibits antitumor activity against several types of cancers including lung adenocarcinoma, breast cancer, and thyroid tumor,44–46 but the clinical application of JQ1 is highly limited partly due to the relatively short half-life of approximately 1 h.47 Here, JQ1 was co-encapsulated into AMGs and locally injected into the tumor tissues to realize the controlled release and prevent fast metabolism. AMGs and naked OA with or without JQ1 were separately injected into tumor tissues for 48 h, and then the mice were euthanatized to separate the tumor tissues, followed by digesting into single tumor cell suspension with type IV collagenase. The cell suspension was observed under a fluorescence microscope, and it was found that the mice injected with naked OA only showed very weak fluorescence regardless of the addition of JQ1 (Fig. 4C). However, OA replicated very fast with obviously increased fluorescence with the treatment of AMGs, and the JAMG treatment induced the highest EGFP expression in the cell suspension, indicating that the MG-mediated OA delivery could significantly extend OA retention at the tumor tissue, and the OA-infected efficiency could be further elevated by the presence of JQ1. Flow cytometry results revealed that AMGs increased virus replication in tumor tissues by 7.6-fold compared with naked OA (Fig. 4D). Furthermore, the infection by the treatment of naked OA with the addition of JQ1 was improved increased above 2.6-fold than that without JQ1. In line with the fluorescence observation, JAMGs caused the most efficient OA infection, which was over 10-fold higher than that of naked OA, and even 1.5-fold higher compared with AMGs, demonstrating the dually important roles of long tumor retention and JQ1-stimulation for the boosted OA replication.3.7 Inhibition of PD-L1 expression
It is reported that oncolytic therapy by virus infection could induce the negative immune regulation by the PD-L1 expression on host cells, which might diminish the antitumor efficacy.48 IFN-γ stimulation enhanced the PD-L1 expression on the A549 cells, while this elevated PD-L1 expression was significantly inhibited by the JQ1 treatment at both concentrations of 100 nM and 300 nM (Fig. 5A). Zhu et al. also demonstrated that JQ1 significantly suppressed the PD-L1 expression on ovarian tumor cells and the tumor-associated macrophages and dendritic cells, inhibiting the ovarian tumor growth on a mouse model.34 To evaluate whether OA infection could also induce the overexpressed PD-L1 on A549 cells, viral particles at different MOIs were added to co-culture with tumor cells, and the protein level of PD-L1 was elevated dependently on the increasing MOIs of OA and quantitively determined via flow cytometry (Fig. 5B). Interestingly, the JQ1 treatment was able to reduce the expression of PD-L1 induced by OA infection (Fig. 5C), indicating the dual function of JQ1 for enhanced virus propagation and reduced negative immune regulation.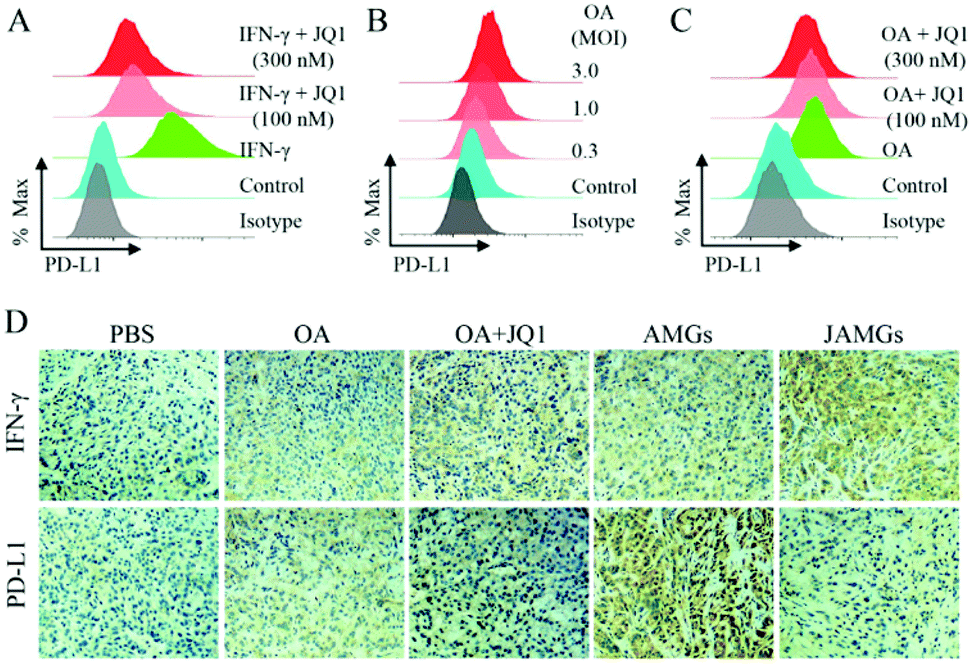 | ||
| Fig. 5 (A) Flow cytometry analysis of the expression of PD-L1 on tumor cells pretreated with or without JQ1 and then stimulated by IFN-γ (30 ng mL−1) for 24 h; (B) flow cytometry analysis of the PD-L1 expression in tumor cells induced by different MOIs of OA (0.3, 1.0 and 3.0) for 24 h; (C) flow cytometry analysis of the PD-L1 expression in tumor cells pretreated with or without JQ1 and then infected by OA (MOI = 3) for 24 h; (D) IHC staining images of IFN-γ and PD-L1 of tumors isolated from A549 lung tumor-bearing nude mice after different treatment for 72 h. | ||
Furthermore, the in vivo inhibited effect of JAMGs on PD-L1 was investigated on the A549 tumor xenograft mouse model. As shown in Fig. 5D, IFN-γ expression, which is an important indicator of the immune response, was activated by the tumoral injection of OA, and the expression was significantly enhanced by the treatment with the addition of JQ1 because of the boosted OA replication at the tumor tissue. The PD-L1 expression was accordingly increased as the OA infected the tumor tissues; however, it should be noted that the treatment involved with JQ1 could simultaneously inhibit the PD-L1 expression. The statistical analysis of the expression levels of IFN-γ and PD-L1 according to the IHC staining images consisted of the in vitro observation (Fig. S4†). Therefore, JAMGs would possibly improve the OA replication for viral therapy and meanwhile overcome the immune suppression for overall cancer therapy.
3.8 In vivo antitumor activity
The A549 lung tumor-bearing nude mice were treated by local injection with PBS, free JQ1, naked OA, combination of naked OA with JQ1 (OA + JQ1), MGs loaded with JQ1 (JMGs), MGs loaded with OA (AMGs) and MGs loaded with OA and JQ1 (JAMGs), and the administrations were performed every 5 days for three times. The results showed that naked OA treatment exhibited negligible inhibition on the tumor growth during the treatment period as a similar performance as PBS did (Fig. 6A). It is very interesting to notice that JMGs exhibit superior inhibition on the tumor growth as compared to free JQ1with the relative volume (V/V0) of 5.4 and 3.9, respectively, which might be explained from that the half-life of JQ1 was prolonged by MGs with a controlled release behavior. The combination treatment of OA/JQ1 was able to suppress the tumor growth with higher tumor inhibition than single formulation. AMGs exhibited high efficiency on the encapsulation of OA and significantly enhanced the localized infection of OA in vivo experiments, and the additional JQ1 treatment further promoted the replication of the virus in tumor cells. AMGs showed a strong inhibition on the tumor growth, which was further enhanced by the addition of JQ1 with a relative volume of 1.8, and much more efficient than other treatment groups. Mice with different treatments all presented little weight loss (Fig. 6B), and the pathologic changes in major organs by hematoxylin and eosin (HE) staining indicated that the local injection of OA/JQ1 microgel systems exhibited little systemic side effects during the treatment period (Fig. S5†).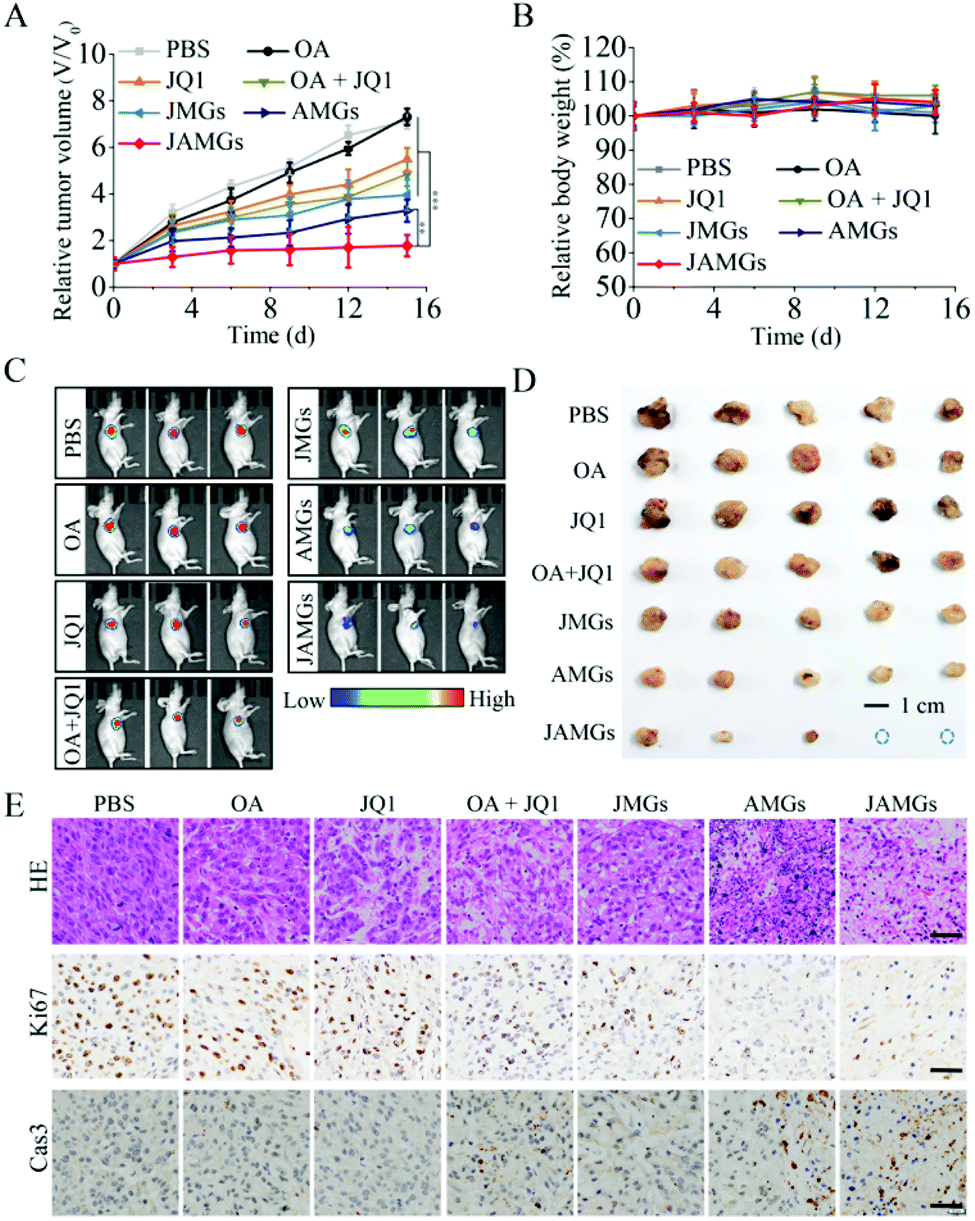 | ||
| Fig. 6 In vivo antitumor performance in the A549 tumor-bearing nude mice (n = 5) by intratumor injection with different treatments: PBS, naked OA, free JQ1, a combination of OA and JQ1, JQ1 loaded MGs (JMGs), OA-loaded MGs (AMGs), and JQ1/OA co-loaded MGs (JAMGs). The mice were administrated with different formulations at a JQ1 dose of 10 mg kg−1 and with an OA number of 1.0 × 109 per mouse, and the injections were performed on days 0, 5, and 10 for 3 times. (A) Tumor volume changes and (B) body weight changes of mice with different treatments in 15 days. (C) In vivo living images of mice performed on day 14 and (D) tumor images from each group after euthanizing the mice on day 15. (E) Histological examination of the A549 tumor xenografts with different treatment for 15 days: typical HE staining images, IHC staining images of Ki-67 and cleaved caspase-3 of tumors retrieved after treatment (scale bar: 10 μm). The data are presented as the average ± SD (n = 5, **p < 0.01, ***p < 0.001). | ||
In vivo imaging was also performed to monitor the intensity of tumor luminescence, indicating the tumor size of A549 tumor xenograft mice at 15 d. As shown in Fig. 6C, JAMG treatment on the mice showed a higher ratio of tumor shrinkage compared with other groups. Finally, the tumor tissues were isolated from the euthanatized mice for further analysis. The tumor blocks in the photograph clearly demonstrate that the JAMG treatment realized the significant therapy efficiency with smallest tumor sizes on the mice as compared to other groups (Fig. 6D), which was quite in line with the results of the tumor volume inhibition and observation of the living luminescence imaging. Tumor tissues by HE staining revealed that the necrosis of tumor cells was greatly induced by the treatment of JAMGs, which also caused remarkable proliferation inhibition and apoptosis of the tumor cell, as indicated by Ki67 reduction and the increased cleaved caspase-3 protein, respectively (Fig. 6E).
4 Conclusions
We have developed injectable PVA microgels by the combination of microfluidics technology and a Michael-type addition crosslinking reaction for an efficient oncolytic adenovirus loading and cancer viral therapy. These pH-degradable microgels provided extended persistence of OA at tumor tissues and increased tumoral OA accumulation. The OA replication and infection in the tumor cells were further strengthened by the addition of the BET bromodomain inhibitor JQ1, leading to enhanced antitumor activity. The simultaneous JQ1 delivery mediated by PVA microgels also greatly inhibited the PD-L1 expression to overcome the immune suppression during the viral therapy. These injectable pH-degradable PVA microgels provide a promising platform to the clinical application of OA and JQ1 for the treatment of malignant tumors by the multi-combination of viral, chemo and immunotherapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (NSFC 51973233, 51803238, 21878337), the National Key Research and Development Program of China (No. 2017YFD0401301), the Natural Science Foundation of Jiangsu Province (BK20170730), Jiangsu Agriculture Science and Technology Innovation Fund (CX(18)3039), and the Jiangsu Specially-Appointed Professor Program to W.C.Notes and references
- A. Ribas, R. Dummer, I. Puzanov, A. VanderWalde, R. H. I. Andtbacka, O. Michielin, A. J. Olszanski, J. Malvehy, J. Cebon, E. Fernandez, J. M. Kirkwood, T. F. Gajewski, L. Chen, K. S. Gorski, A. A. Anderson, S. J. Diede, M. E. Lassman, J. Gansert, F. S. Hodi and G. V. Long, Cell, 2017, 170, 1109 CrossRef CAS.
- S. K. Totsch, C. Schlappi, K. D. Kang, A. S. Ishizuka, G. M. Lynn, B. Fox, E. A. Beierle, R. J. Whitley, J. M. Markert, G. Y. Gillespie, J. D. Bernstock and G. K. Friedman, Oncogene, 2019, 38, 6159 CrossRef CAS PubMed.
- J. W. Choi, Y. S. Lee, C. O. Yun and S. W. Kim, J. Controlled Release, 2015, 219, 181 CrossRef CAS.
- N. Martinez-Velez, M. Garcia-Moure, M. Marigil, M. Gonzalez-Huarriz, M. Puigdelloses, J. G. Perez-Larraya, M. Zalacain, L. Marrodan, M. Varela-Guruceaga, V. Barchino, E. Raabe, M. Monje, O. J. Becher, M. P. Junier, E. A. El-Habr, H. Chneiweiss, G. Aldave, H. Jiang, J. FueyoLaspidea, J. J. Aristu, L. I. Ramos, S. Tejada-Solis, R. Diez-Valle, C. Jones, A. Mackay, J. A. Martinez-Climent, M. J. Garcia-Barchino, A. Patino-Garcia, C. Gomez-Manzano and M. M. Alonso, Nat. Commun., 2019, 10, 2235 CrossRef.
- S. J. Russell, K. W. Peng and J. C. Bell, Nat. Biotechnol., 2012, 30, 658 CrossRef CAS PubMed.
- I. K. Choi and C. O. Yun, Cancer Gene Ther., 2013, 20, 70 CrossRef CAS.
- X. Li, P. Wang, H. Li, X. Du, M. Liu, Q. Huang, Y. Wang and S. Wang, Clin. Cancer Res., 2017, 23, 239 CrossRef CAS.
- J. Martinez-Quintanilla, I. Seah, M. Chua and K. Shah, J. Clin. Invest., 2019, 130, 1407 CrossRef.
- R. Alemany, Adv. Cancer Res., 2012, 115, 93 CrossRef CAS.
- H. Jiang, C. Gomez-Manzano, Y. Rivera-Molina, F. F. Lang, C. A. Conrad and J. Fueyo, Curr. Opin. Virol., 2015, 13, 33 CrossRef CAS PubMed.
- J. J. Cody and J. T. Douglas, Cancer Gene Ther., 2009, 16, 473 CrossRef CAS PubMed.
- J. W. Choi, H. A. Kim, K. Nam, Y. Na, C. O. Yun and S. Kim, J. Controlled Release, 2015, 220, 691 CrossRef CAS PubMed.
- C. Y. Moon, J. W. Choi, D. Kasala, S. J. Jung, S. W. Kim and C. O. Yun, Biomaterials, 2015, 41, 53 CrossRef CAS.
- J. Kim, Y. Li, S. W. Kim, D. S. Lee and C. O. Yun, Biomaterials, 2013, 34, 4622 CrossRef CAS.
- D. D. Draganov, A. F. Santidrian, I. Minev, D. Nguyen, M. O. Kilinc, I. Petrov, A. Vyalkova, E. Lander, M. Berman, B. Minev and A. A. Szalay, J. Transl. Med., 2019, 17, 100 CrossRef PubMed.
- I. C. Liao, S. Chen, J. B. Liu and K. W. Leong, J. Controlled Release, 2009, 139, 48 CrossRef CAS PubMed.
- C. M. Curtin, G. M. Cunniffe, F. G. Lyons, K. Bessho, G. R. Dickson, G. P. Duffy and F. J. O'Brien, Adv. Mater., 2012, 24, 749 CrossRef CAS PubMed.
- T. Fuhrmann, R. Y. Tam, B. Ballarin, B. Coles, I. Elliott Donaghue, D. van der Kooy, A. Nagy, C. H. Tator, C. M. Morshead and M. S. Shoichet, Biomaterials, 2016, 83, 23 CrossRef CAS PubMed.
- J. W. Choi, E. Kang, O. J. Kwon, T. J. Yun, H. K. Park, P. H. Kim, S. W. Kim, J. H. Kim and C. O. Yun, Gene Ther., 2013, 20, 880 CrossRef CAS PubMed.
- B. K. Jung, E. Oh, J. Hong, Y. Lee, K. D. Park and C. O. Yun, Biomaterials, 2017, 147, 26 CrossRef CAS PubMed.
- D. Zamarin, J. M. Ricca, S. Sadekova, A. Oseledchyk, Y. Yu, W. M. Blumenschein, J. Wong, M. Gigoux, T. Merghoub and J. D. Wolchok, J. Clin. Invest., 2018, 128, 1413 CrossRef PubMed.
- S. J. Russell and G. N. Barber, Cancer Cell, 2018, 33, 599 CrossRef CAS PubMed.
- E. Lam and E. Falck-Pedersen, J. Virol., 2014, 88, 14426 CrossRef.
- J. Qiao, H. Wang, T. Kottke, C. White, K. Twigger, R. M. Diaz, J. Thompson, P. Selby, J. de Bono, A. Melcher, H. Pandha, M. Coffey, R. Vile and K. Harrington, Clin. Cancer Res., 2008, 14, 259 CrossRef CAS PubMed.
- T. Alain, X. Lun, Y. Martineau, P. Sean, B. Pulendran, E. Petroulakis, F. J. Zemp, C. G. Lemay, D. Roy, J. C. Bell, G. Thomas, S. C. Kozma, P. A. Forsyth, M. Costa-Mattioli and N. Sonenberg, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1576 CrossRef CAS PubMed.
- X. Q. Lun, H. Zhou, T. Alain, B. Sun, L. Wang, J. W. Barrett, M. M. Stanford, G. McFadden, J. Bell, D. L. Senger and P. A. Forsyth, Cancer Res., 2007, 67, 8818 CrossRef CAS PubMed.
- A. Dyer, B. Schoeps, S. Frost, P. Jakeman, E. M. Scott, J. Freedman, E. J. Jacobus and L. W. Seymour, Cancer Res., 2019, 79, 331 CrossRef CAS PubMed.
- K. Ren, W. Zhang, X. Chen, Y. Ma, Y. Dai, Y. Fan, Y. Hou, R. X. Tan and E. Li, PLoS Pathog., 2016, 12, e1005950 CrossRef PubMed.
- B. Lv, J. Li, M. Li, Y. Zhuo, K. Ren, E. Li and G. Yang, Sci. Rep., 2018, 8, 11554 CrossRef.
- P. L. Garcia, A. L. Miller, K. M. Kreitzburg, L. N. Council, T. L. Gamblin, J. D. Christein, M. J. Heslin, J. P. Arnoletti, J. H. Richardson, D. Chen, C. A. Hanna, S. L. Cramer, E. S. Yang, J. Qi, J. E. Bradner and K. J. Yoon, Oncogene, 2016, 35, 833 CrossRef CAS.
- V. M. Wu, J. Mickens and V. Uskokovic, ACS Appl. Mater. Interfaces, 2017, 9, 25887 CrossRef CAS.
- J. Shahbazi, P. Y. Liu, B. Atmadibrata, J. E. Bradner, G. M. Marshall, R. B. Lock and T. Liu, Clin. Cancer Res., 2016, 22, 2534 CrossRef CAS PubMed.
- O. Melaiu, M. Mina, M. Chierici, R. Boldrini, G. Jurman, P. Romania, V. D'Alicandro, M. C. Benedetti, A. Castellano, T. Liu, C. Furlanello, F. Locatelli and D. Fruci, Clin. Cancer Res., 2017, 23, 4462 CrossRef CAS PubMed.
- H. Zhu, F. Bengsch, N. Svoronos, M. R. Rutkowski, B. G. Bitler, M. J. Allegrezza, Y. Yokoyama, A. V. Kossenkov, J. E. Bradner, J. R. Conejo-Garcia and R. Zhang, Cell Rep., 2016, 16, 2829 CrossRef CAS PubMed.
- C. H. Choi, H. Wang, H. Lee, J. H. Kim, L. Zhang, A. Mao, D. J. Mooney and D. A. Weitz, Lab Chip, 2016, 16, 1549 RSC.
- D. Sivakumaran, E. Mueller and T. Hoare, Soft Matter, 2017, 13, 9060 RSC.
- Y. Hou, W. Xie, K. Achazi, J. L. Cuellar-Camacho, M. F. Melzig, W. Chen and R. Haag, Acta Biomater., 2018, 77, 28 CrossRef CAS.
- X. Huang, Y. Hou, L. Zhong, D. Huang, H. Qian, M. Karperien and W. Chen, Biomacromolecules, 2018, 19, 94 CrossRef CAS PubMed.
- W. Chen, Y. Hou, Z. Tu, L. Gao and R. Haag, J. Controlled Release, 2017, 259, 160 CrossRef CAS PubMed.
- J. Wojton and B. Kaur, Cytokine Growth Factor Rev., 2010, 21, 127 CrossRef CAS.
- P. K. Bommareddy, S. Aspromonte, A. Zloza, S. D. Rabkin and H. L. Kaufman, Sci. Transl. Med., 2018, 10, eaau0417 CrossRef CAS PubMed.
- K. J. Excoffon, N. Gansemer, G. Traver and J. Zabner, J. Virol., 2007, 81, 5573 CrossRef CAS PubMed.
- P. Sharma, A. O. Kolawole, S. M. Wiltshire, K. Frondorf and K. J. D. A. Excoffon, J. Gen. Virol., 2012, 93, 155 CrossRef CAS PubMed.
- L. L. da Motta, I. Ledaki, K. Purshouse, S. Haider, M. A. De Bastiani, D. Baban, M. Morotti, G. Steers, S. Wigfield, E. Bridges, J. L. Li, S. Knapp, D. Ebner, F. Klamt, A. L. Harris and A. McIntyre, Oncogene, 2017, 36, 122 CrossRef CAS PubMed.
- X. Zhu, K. Enomoto, L. Zhao, Y. J. Zhu, M. C. Willingham, P. Meltzer, J. Qi and S. Y. Cheng, Clin. Cancer Res., 2017, 23, 430 CrossRef CAS PubMed.
- W. W. Lockwood, K. Zejnullahu, J. E. Bradner and H. Varmus, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19408 CrossRef CAS PubMed.
- P. Filippakopoulos, J. Qi, S. Picaud, Y. Shen, W. B. Smith, O. Fedorov, E. M. Morse, T. Keates, T. T. Hickman, I. Felletar, M. Philpott, S. Munro, M. R. McKeown, Y. Wang, A. L. Christie, N. West, M. J. Cameron, B. Schwartz, T. D. Heightman, N. La Thangue, C. A. French, O. Wiest, A. L. Kung, S. Knapp and J. E. Bradner, Nature, 2010, 468, 1067 CrossRef CAS.
- Z. Q. Liu, R. Ravindranathan, P. Kalinski, Z. S. Guo and D. L. Bartlett, Nat. Commun., 2017, 8, 14754 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm00172d |
| ‡ The authors Haishi Qiao and Xingmei Chen made equal contributions to this work. |
| This journal is © The Royal Society of Chemistry 2020 |