
Microencapsulation improves chondrogenesis in vitro and cartilaginous matrix stability in vivo compared to bulk encapsulation†
Fanyi
Li‡§
ab,
Clara
Levinson‡
c,
Vinh X.
Truong
 a,
Lee Ann
Laurent-Applegate
d,
Katharina
Maniura-Weber
e,
Helmut
Thissen
b,
John S.
Forsythe
a,
Marcy
Zenobi-Wong
*c and
Jessica E.
Frith
*a
a,
Lee Ann
Laurent-Applegate
d,
Katharina
Maniura-Weber
e,
Helmut
Thissen
b,
John S.
Forsythe
a,
Marcy
Zenobi-Wong
*c and
Jessica E.
Frith
*a
aDepartment of Materials Science and Engineering, Monash Institute of Medical Engineering, Monash University, Wellington Road, Clayton, VIC 3800, Australia. E-mail: Jessica.frith@monash.edu
bCSIRO Manufacturing, Bayview Avenue, Clayton, VIC 3168, Australia
cTissue Engineering + Biofabrication, Department of Health Sciences and Technology, ETH Zürich, Switzerland. E-mail: Marcy.zenobi@hest.ethz.ch
dUnit of Regenerative Therapy, Lausanne University Hospital of Lausanne, Switzerland
eEmpa, Swiss Federal Laboratories for Materials Science and Technology, Laboratory for Biointerfaces, St. Gallen, Switzerland
First published on 21st January 2020
Abstract
The encapsulation of cells into microgels is attractive for applications in tissue regeneration. While cells are protected against shear stress during injection, the assembly of microgels after injection into a tissue defect also forms a macroporous scaffold that allows effective nutrient transport throughout the construct. However, in most of current strategies that form microgel-based macroporous scaffold or higher-order structures, cells are seeded during or post the assembly process and not microencapsulated in situ. The objective of this study is to investigate the chondrogenic phenotype of microencapsulated fetal chondrocytes in a biocompatible, assembled microgel system vs. bulk gels and to test the stability of the constructs in vivo. Here, we demonstrate that cell microencapsulation leads to increased expression of cartilage-specific genes in a TGF-β1-dependent manner. This correlates, as shown by histological staining, with the ability of microencapsulated cells to deposit cartilaginous matrix after migrating to the surface of the microgels, while keeping a macroscopic granular morphology. Implantation of precultured scaffolds in a subcutaneous mouse model results in vessel infiltration in bulk gels but not in assembled microgels, suggesting a higher stability of the matrix produced by the cells in the assembled microgel constructs. The cells are able to remodel the microgels as demonstrated by the gradual disappearance of the granular structure in vivo. The biocompatible microencapsulation and microgel assembly system presented in this article therefore hold great promise as an injectable system for cartilage repair.
1. Introduction
Autologous chondrocyte implantation (ACI) is the gold standard for cartilage repair and has led to relatively good clinical outcomes. However, the process is invasive, costly, and the delay before actual tissue regeneration is long as it takes at least 26 weeks for the defect to be completely filled with repair tissue.1 The current state of the art has advanced from simple injection of human articular chondrocytes (hACs) covered by a periosteal flap, to implantation of the cells within a scaffold material.1 Among these scaffolds, hydrogels have shown great promise and several reviews describe their use in tissue engineering.2–5However, bulk hydrogels have limitations due to the nanometer-sized pore size, which greatly limit cell migration, proliferation and ECM deposition in the case of cell-laden gels, or host cell infiltration in the case of acellular hydrogels.6 Macroporous scaffolds have been proposed as an alternative,7,8 as they allow greater diffusion of nutrients to cells throughout the whole scaffold, and they also provide space for cell migration into the implanted scaffold for in situ repair. Cell migration into the defect is an important part of the regeneration process,9 which may explain the improved regeneration with macroporous scaffolds.10 Microgels are polymer colloid particles, which can be fabricated using various techniques, such as mould-casting based photolithography,11 electro-spraying,12 microfluidics13 or inverse suspension polymerisation.14 They represent a novel way to produce injectable, ultimately macroporous systems, as an alternative to the use of porogens or emulsions.15,16 Microgels have found utility in various biomedical applications such as drug delivery systems17,18 and more recently tissue engineering.10,13
In the case of cartilage repair, microgels have been used for various purposes. For instance, they have been used as a platform to investigate the chondrogenic differentiation capacity of mesenchymal stem cells (MSCs) in 3D compared to 2D culture,2 or as a supporting material to create “artificial microtissues” implanted after in vitro preculture.19 In addition, microgels have been utilized as chondrogenic growth factor delivery systems.20,21 We have previously introduced the use of gelatin norbornene (GelNB) – poly(ethylene glycol)dithiol (PEGdiSH) microgels as building blocks to safely inject cells for the treatment of cartilage lesions.22,23 We demonstrated that this material is biocompatible, cell-friendly and that it promotes an enhanced chondrogenic phenotype of encapsulated human mesenchymal stem cells (hMSCs) as compared to a bulk hydrogel system as well as “gold standard” pellet culture.22 The positive effect of the microgel structure on the maintenance of chondrogenic marker expression has also been demonstrated in vivo, therefore underlining the advantages of a bottom-up rather than a top-down approach for cartilage tissue engineering.19
Microgel fabrication must allow facile encapsulation of cells with high viability to ultimately protect cells from shear stress damage during arthroscopic injection. In addition, the assembly method should provide relative stability to the final construct, to cope with the mechanically challenging environment of the joint. Several methods have been developed to assemble microgels and form scaffolds with higher porosity for tissue engineering purposes.24,25 Griffin and colleagues used an enzyme, transglutaminase factor XIII, to crosslink peptides at the surface of the microgels.10 White light with eosin Y has also been used to crosslink and stabilise HA microgels, thus reducing the processing time to 1 minute.26 Later, novel click chemistry was used to assemble PEG-based microgels, for example using SPAAC14 and thiol–ene photo click reactions.26 More recently, the thermo-responsive behaviour of gelatin was utilised to physically crosslink gelatin methacryloyl (GelMA) microgels at low temperature (4 °C) and then stabilise the structure via UV photopolymerisation.27 In another study, UV light triggered thiol-ene click chemistry was used to crosslink PEG-norbornene microgels with a PEG-dithiol linker.28 However, despite elegant chemistries, and demonstrated of biocompatibility in the above strategies, cells were incorporated during the microgel crosslinking process and not encapsulated within the microgels. Therefore, cells are grown on the surface of the assembled microgels. Besides, these approaches offer no protection to cells during the injection process. Moreover, the microgel assembly process in some studies are light-triggered or highly temperature sensitive, which make these approaches difficult for use as an injectable system for clinical applications. We have previously introduced an approach using 4-arm PEG-succinimidyl glutaramide (PEG-NHS) to bind GelNB-PEGdiSH microgels, which are fabricated via a customised pipette tip-based microfluidic device (the final PEG-NHS treated assembled-microgels is referred to as “NHSA-microgels”).23 The addition of PEG-NHS allowed crosslinking of the microgels and we demonstrated that it was possible to cast the microgel suspension into an in vitro cartilage defect model to produce a stable macroporous construct. Furthermore, the viability of the embedded cells was not affected. This makes this strategy promising for the use as an injectable therapy for cartilage lesions.
An important parameter for the successful development of a microgel-based treatment for cartilage lesions is the choice of the cell type to be embedded. ACI uses the patient's own chondrocytes after isolation and 2D expansion to reach a sufficient number of cells and in recent years, most of the cell-based approaches entering clinical trials have relied on the use of autologous chondrocytes despite the lower cell yield,29 the slower proliferation rate30 and the tendency of the cells to dedifferentiate during in vitro culture.31 To envision a more scalable translational approach, the field has turned towards other cell sources, such as human mesenchymal stem cells (hMSCs) and chondroprogenitors/chondrocytes from fetal32 and infant33 origins. MSCs are widely investigated for their immunosuppressive properties and their capacity to differentiate into chondrocytes. We have reported that hMSCs can differentiate into chondrocytes in our gelatin-PEG microgel system.23 However, there are limited amounts of MSCs in biopsies, and these cells can undergo hypertrophy, further differentiating into osteoblasts.34–36 These drawbacks call for alternative cell sources that are already advanced in the differentiation process but can still proliferate while keeping their chondrogenic phenotype. A promising alternative is the use of human fetal chondroprogenitor cells (hCCs) isolated from the fetal epiphysis. These cells can retain their chondrogenic potential up to 20 passages37 and were shown to successfully deposit cartilaginous ECM in several hydrogels in vitro and in vivo, namely alginate and hyaluronan, for cartilage engineering purposes.32,38 Due to the high proliferative potential of hCCs, sufficient cells can be generated from one donor (35 billion cells estimated) to treat hundreds of patients, which could reduce the variable clinical results among patients treated with ACI. Indeed, the chondrogenic potential of autologous chondrocytes can vary drastically among patients.39 More importantly, hCCs have a strong migratory behaviour, which is an important part of the healing process when using microgels,10 since the cells first migrate to, and proliferate on the surface of the microgels before producing ECM.23 Therefore, both human articular chondrocytes (hACs) and hCCs were assessed for their suitability as a cell source in the assembled microgel system, with hMSCs serving as the control. In addition, we found most of in situ microencapsulated MSCs migrated to the surface of microgels after one week culture and that this facilitated more cell–cell interactions and produced more cartilaginous matrix compared to bulk hydrogels. We hypothesised that the promising chondrogenic differentiation results and cartilaginous matrix production relied upon this distinction between tissue formation between microgels and within the hydrogel matrix of bulk hydrogels. The initial selection of the cell source was determined based on the criteria of (1) high cell viability following the microfluidic microencapsulation process and (2) fast migration speed of cells from the inner core of the microgels to their surface.
Finally, a limitation of highly porous scaffolds for tissue engineering is their lower structural stability. Before proceeding to further development of a given system, it is important to consider the stability of the microgels and of the ECM deposited by the cells in this system. In this regard, subcutaneous implantation in mice represents a first step towards the understanding of such constructs in a more challenging environment than in vitro culture.
The aim of this study is to investigate the cells from different sources with the GelNB-PEGdiSH microgels system. We hypothesize that using the microfluidic technique to microencapsulate cells in microgels and to assemble these cell-laden microgels into a macroporous structure will lead to better chondrogenesis than in conventional bulk gels in vitro, and to greater stability in vivo.
2. Results and discussion
2.1. Human cell type screening
In order to assess whether the alternative human cell types survive during the microgel fabrication process and are compatible with the gelatin-PEG material, hACs and hCCs, as well as hMSCs (the cell type used in our previous studies) as a control, were in situ encapsulated in the 600 μm-diameter microgels with high monodispersity as our previous report22 (Fig. 1A). Live/dead staining at 24 hours post-encapsulation showed a high viability of over 80% for all three cell types (Fig. 1B and C), with no differences between the different cell sources. This demonstrates that both the in situ pipette tip-based microfluidic encapsulation process and the visible light-induced thiol–ene photo click polymerisation are compatible with all three cell types. Further live/dead staining was conducted to monitor cell viability in microgels cultured in basal medium for up to one week (Fig. 1B and C). This showed a significant increase in the proportion of live hACs from day 4 to day 7 (P = 0.0119), which we attribute to the high proliferation rate of hACs in the microgels. After 7 days in culture, this increase in viable hACs resulted in a slightly but significantly higher hAC viability (91 ± 2%) as compared to hMSCs (87 ± 2%, P = 0.0023). hCC viability (89 ± 1%) was not statistically different from that of hMSCs. This is consistent with our previous research showing that gelatin-PEG microgels provide a suitable microenvironment for long-term cell maintenance.22 The excellent cytocompatibility may be attributed to the gelatin, which is a major component of the microgels and has an intrinsic property for cell adhesion. This is particularly favourable for anchorage-dependent cells, such as the hACs, hCCs and hMSCs that are investigated here. Furthermore, the small dimensions and spherical geometry of the microgels may facilitate rapid nutrient and waste exchange.13,19,40,41 This highly efficient mass transfer property is therefore considered an additional benefit to enhance the biocompatibility of the microgels.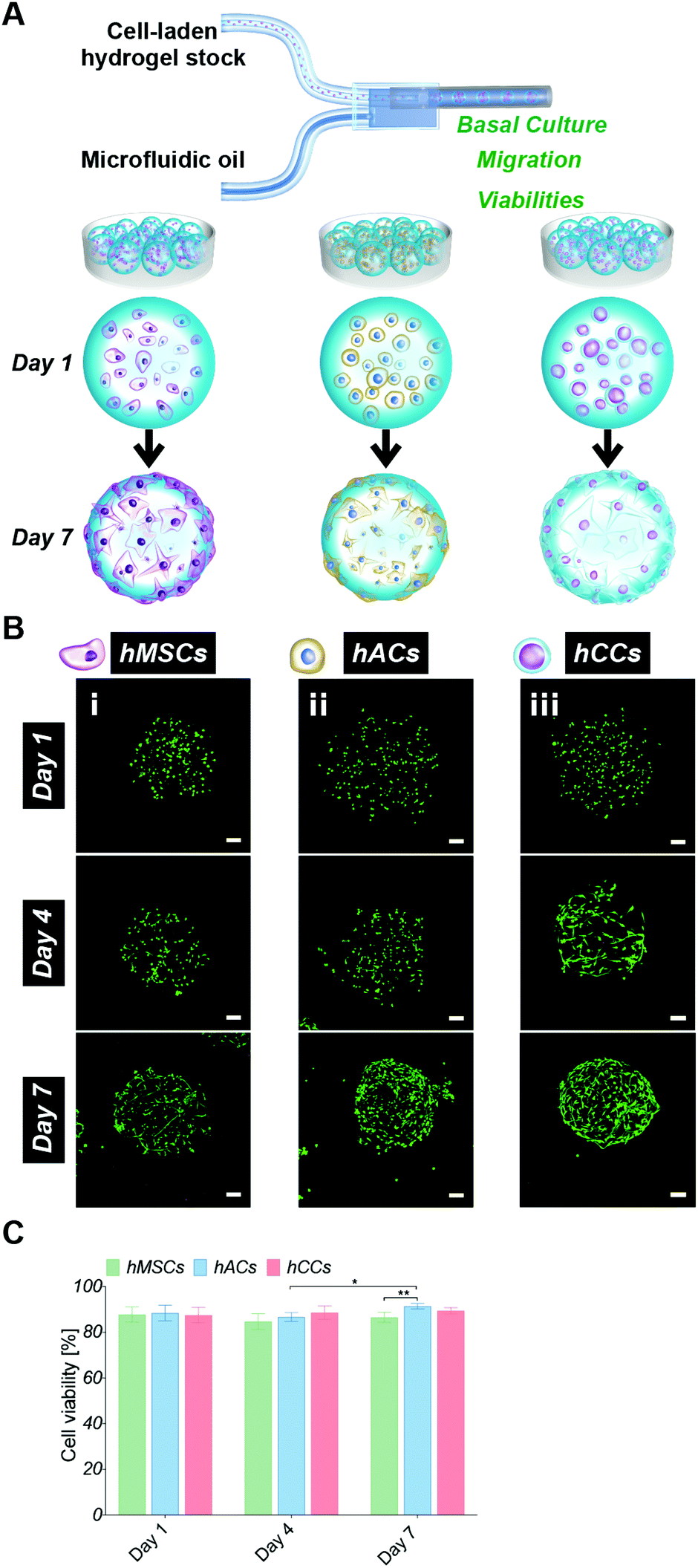 | ||
| Fig. 1 (A) Schematic representation of microfluidic in situ microencapsulation of human cells (hMSCs, hACs and hCCs) for cell screening analysis. (B) Live/dead staining of cell-laden microgels on day 1, 4, and 7 (i: hMSCs, ii: hACs and iii: hCCs, z-stack fluorescent images showing live cells in green and dead cells in red, scale bar: 100 μm. (C) Quantitative measurements of hMSC, hAC and hCC viabilities in microgels (*P < 0.05 and **P < 0.01). | ||
Cell migration behaviour in the microgels was monitored over 7 days by analysing the fluorescent intensity profiles of 3D reconstruction images (Fig. 2). At day 1, cells were homogenously distributed in the microgels, confirming that the droplet pipette-tip based microfluidic technique allowed uniform cell microencapsulation. Additionally, analysis of the cell morphologies indicated very few cell protrusions at day 1. This is likely due to the cells requiring more time to fully sense, adapt to and interact with the surrounding matrix. Surprisingly, after four days in culture, obvious differences in cell distribution were already observed between the three different cell types. There were several hMSCs relocated close to the microgel surface but still some cells residing near the core (Fig. 2A). However, there was no noticeable change to the hAC intensity profile compared to day 1, which indicated that the uniform distribution of hACs was maintained (Fig. 2B). Conversely, all the hCCs were found at the microgel periphery with no cells in the centre (Fig. 2C). This observation suggests that hCCs migrate faster than hMSCs and both migrate to a greater extent than hACs. Contrary to the cellular morphologies at day 1, protrusions were evident in all three cell types at day 4, suggesting that the cells had started to spread and interact with the surrounding matrix.
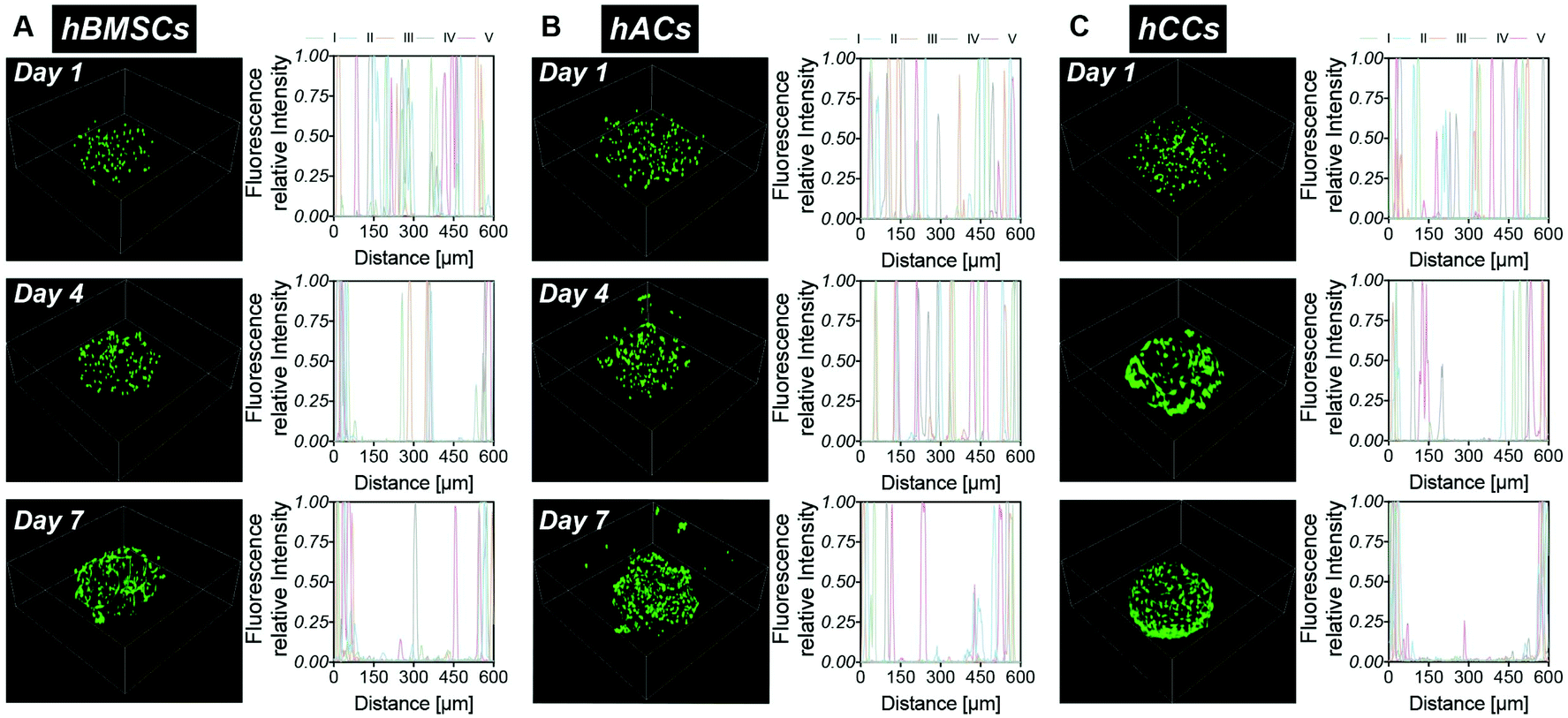 | ||
| Fig. 2 hMSC, hAC and hCC migration in the microgels – 3D reconstruction images of live/dead staining and intensity profile analysis for 5 samples (I, II, III, IV and V represent different samples) of the middle slice of the microgels on day 1, 4 and 7. (A) hMSCs. (B) hACs. (C) hCCs. | ||
After one week in culture, in line with our previous findings,22 the majority of hMSCs were located near the microgel surface with only a few at the core (Fig. 2A). Almost all hCCs had migrated to the microgel surface and the hACs had also now migrated away from the interior of the microgels with less in the core. In terms of morphology, both hCCs and hMSCs presented longer membrane protrusions than hACs. This difference in filopodial extension observed in Fig. 1Bi–iii could be due to the presence of more hCCs and hMSCs located at the surface of the microgels, leading to fewer constraints from the surrounding gel matrix and more cell–cell contacts compared to hACs within the gel. Another possible reason is the different expression levels of migratory factors such as matrix metalloproteases and integrins.42
Overall, the gelatin microgels provided both a cytocompatible and dynamic microenvironment, enabling long-term survival and migration of all three cell types. Here, hCCs demonstrated the most rapid migration to the microgel surface. Based on our previous findings, which show that tissue formation is driven by hMSCs that migrate into the spaces between the microgels, it was hypothesised that the high migratory behaviour of hCCs would lead to rapid cartilage tissue regeneration in the gelatin microgel system. This fast migration capacity of hCCs appears to be well supported by their original isolation process.37 Briefly, hCCs are derived from cells which migrated out of a cultured piece of proximal ulnar epiphysis tissue explant from a 14-week gestation donor. Hence, it seems likely that the cell selection process favours a cell type that possesses a rapid migration ability. In addition to their fast migration, hCCs are considered to be an emerging cell source for cartilage regeneration which avoids donor-to-donor variations and makes them particularly attractive for clinical applications.32,37 hCCs were consequently selected over hMSCs and hACs for the subsequent in vitro and in vivo chondrogenic studies.
2.2. hCC-laden bulk hydrogel and microgel chondrogenic differentiation in vitro
hCC-laden microgels were assembled by reacting microgels with 4-arm PEG-NHS (“NHSA-microgels”) and the in vitro chondrogenic potential was assessed. The bulk hydrogels and NHSA-microgels were cultured for three weeks in chondrogenic media ((+)TGF-β1) or in (+)TGF-β1 media for the first week before being switched to chondrogenic control media ((−)TGF-β1) for the following two weeks (Fig. 3A).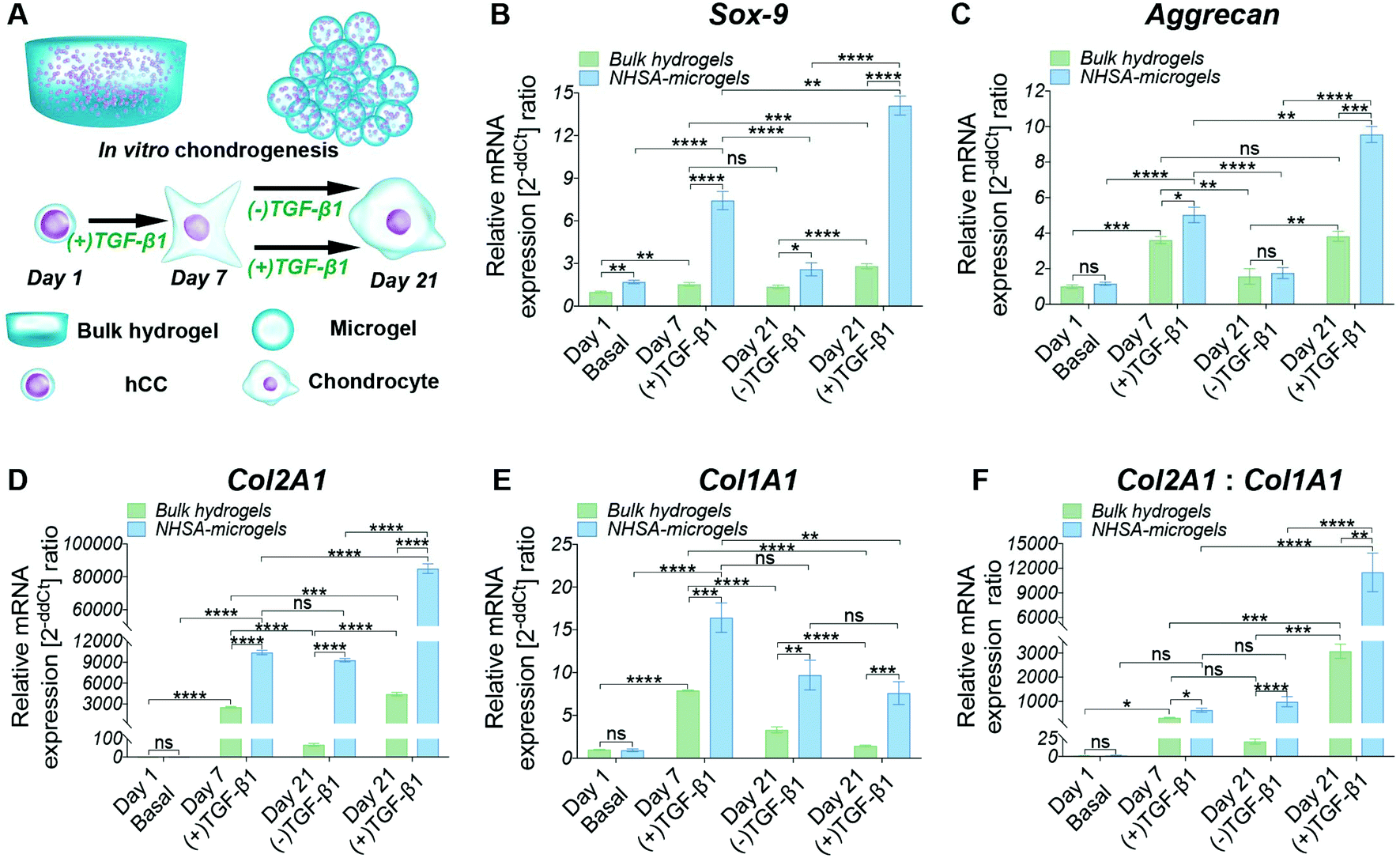 | ||
| Fig. 3 (A) Schematic representation of hCC-laden bulk hydrogel and NHSA-microgel in vitro chondrogenesis: either cultured in chondrogenic media ((+)TGF-β1) for three weeks or in chondrogenic media ((+)TGF-β1) for the first week and control media ((−)TGF-β1) for the following two weeks. RT-qPCR analysis of hCC chondrogenesis in bulk hydrogels and NHSA-microgels was done after 1, 7 and 21 days of culture. (B–E) The fold changes of Sox-9, Aggrecan, Col2A1 and Col1A1 are expressed using RPL13a as the reference gene. (F) The chondrogenic differentiation index (gene expression ratio of Col2A1 and Col1A1). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. | ||
Continuous supplementation of TGF-β1 led to a steady increase in chondrogenic gene expression in microgels and, to a lower extent, in bulk hydrogels. However, Aggrecan gene expression in hCCs encapsulated in bulk hydrogels and cultured in (+)TGF-β1 media for three weeks did not show significant increase compared to one-week culture (Fig. 3C, P = 0.9869). This may indicate that hCCs had reached a peak expression level of Aggrecan after one week culture in the bulk hydrogel, which could only then be maintained with prolonged TGF-β1 supplementation. Although a decrease of chondrogenic gene expression could be expected upon stopping growth factor supplementation, no significant reduction of Col2A1 expression level was observed in the NHSA-microgels after TGF-β1 withdrawal (P = 0.6497). This indicates that following one week preculture in (+)TGF-β1 media, hCCs are able to maintain a high level of Col2A1 expression for at least 2 weeks in vitro without additional TGF-β1 supplementation. This was particularly encouraging for the following in vivo studies that hCC preculture in NHSA-microgels in (+)TGF-β1 media would be expected to promote sustained chondrogenic differentiation.
In addition to the differences in (+)TGF-β1 vs. (−)TGF-β1 treatment regimes, significant differences were also identified between the bulk hydrogels and NHSA-microgels. Sox-9, Aggrecan and Col2A1 expression in NHSA-microgels were substantially higher than in the bulk hydrogels from the same media, at each time point. This demonstrates the superiority of NHSA-microgels compared to the bulk hydrogels for hCC chondrogenesis. The most striking result was the vastly increased expression of the Col2A1 gene in NHSA-microgels (Fig. 3D). Compared to day 1 samples, hCCs cultured in NHSA-microgels for 7 days exhibited more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold elevation of Col2A1 expression, which further increased with continuous TGF-β1 supplementation to yield an approximately 85000-fold upregulation at day 21. This was 20 times higher than the 4000-fold upregulation in bulk hydrogels. Type-II collagen has been identified as the most predominant component in articular cartilage43 and the exceptionally elevated expression of Col2A1 indicates promising cartilaginous matrix deposition in the NHSA-microgels. Given that this remarkable Col2A1 expression upregulation was also observed in our previous studies with hMSCs microencapsulation,22,23 we conclude that the effects are due to the spherical geometry, small dimensions of the microgels and therefore increased mass transport in the microgel system, rather than the source of microencapsulated cells.
000-fold elevation of Col2A1 expression, which further increased with continuous TGF-β1 supplementation to yield an approximately 85000-fold upregulation at day 21. This was 20 times higher than the 4000-fold upregulation in bulk hydrogels. Type-II collagen has been identified as the most predominant component in articular cartilage43 and the exceptionally elevated expression of Col2A1 indicates promising cartilaginous matrix deposition in the NHSA-microgels. Given that this remarkable Col2A1 expression upregulation was also observed in our previous studies with hMSCs microencapsulation,22,23 we conclude that the effects are due to the spherical geometry, small dimensions of the microgels and therefore increased mass transport in the microgel system, rather than the source of microencapsulated cells.
In addition to the typical chondrogenic markers, Col1A1 was monitored to examine the quality of the chondrogenic differentiation (Fig. 3E). Interestingly, in both bulk hydrogel and NHSA-microgels, the highest upregulation of Col1A1 was obtained at day 7, with levels subsequently decreasing in both (+)TGF-β1 and (−)TGF-β1 media. This indicates that the hCCs might be in the early chondrogenic condensation process during the first week, a period in which cell–cell interactions are formed similarly to the process of fetal chondrogenesis, associated with type-I collagen production.44 The decrease of Col1A1 expression from day 7 to 21 may indicate the initiation of hyaline-like cartilage tissue development, in which the ratio of type-I: type-II collagen is reduced. Col1A1 was also compared with Col2A1 to determine the chondrogenic differentiation index (Col2A1:Col1A1) (Fig. 3F). Higher type-II collagen gene expression level was observed in all samples compared to the type-I collagen regardless of time point or geometry, which is suggestive of hyaline type cartilage formation in these systems. However, the differentiation index in the NHSA-microgels was significantly higher than bulk hydrogels (P = 0.0004), which aligns well with previous chondrogenic genes results, and further substantiates that the NHSA-microgels were better than the bulk hydrogel for hCC chondrogenesis.
Generally, the Sox-9, Aggrecan and Col2A1 gene expression levels were higher in NHSA-microgels compared to the bulk hydrogel, confirming that NHSA-microgels provide a better microenvironment for hCC chondrogenesis compared to bulk hydrogels. Even though the chondrogenic gene expression levels were attenuated without continuous supplementation of TGF-β1 in the culture media, the absolute expression value remained significantly higher than the normalised conditions, which implied that chondrogenic differentiation of hCCs was not terminated or reversed.
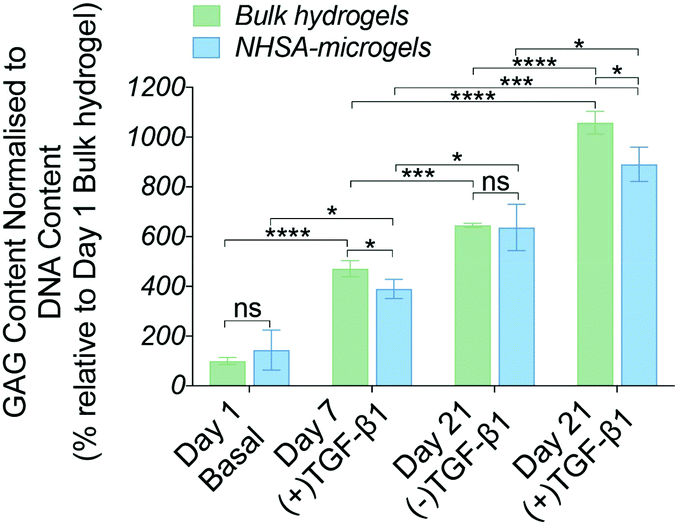 | ||
| Fig. 4 Biochemical analysis in bulk hydrogels and NHSA-microgels over 21 days in in vitro culture. Values represent DMMB assay measurements of GAG accumulation normalised to DNA content (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001). | ||
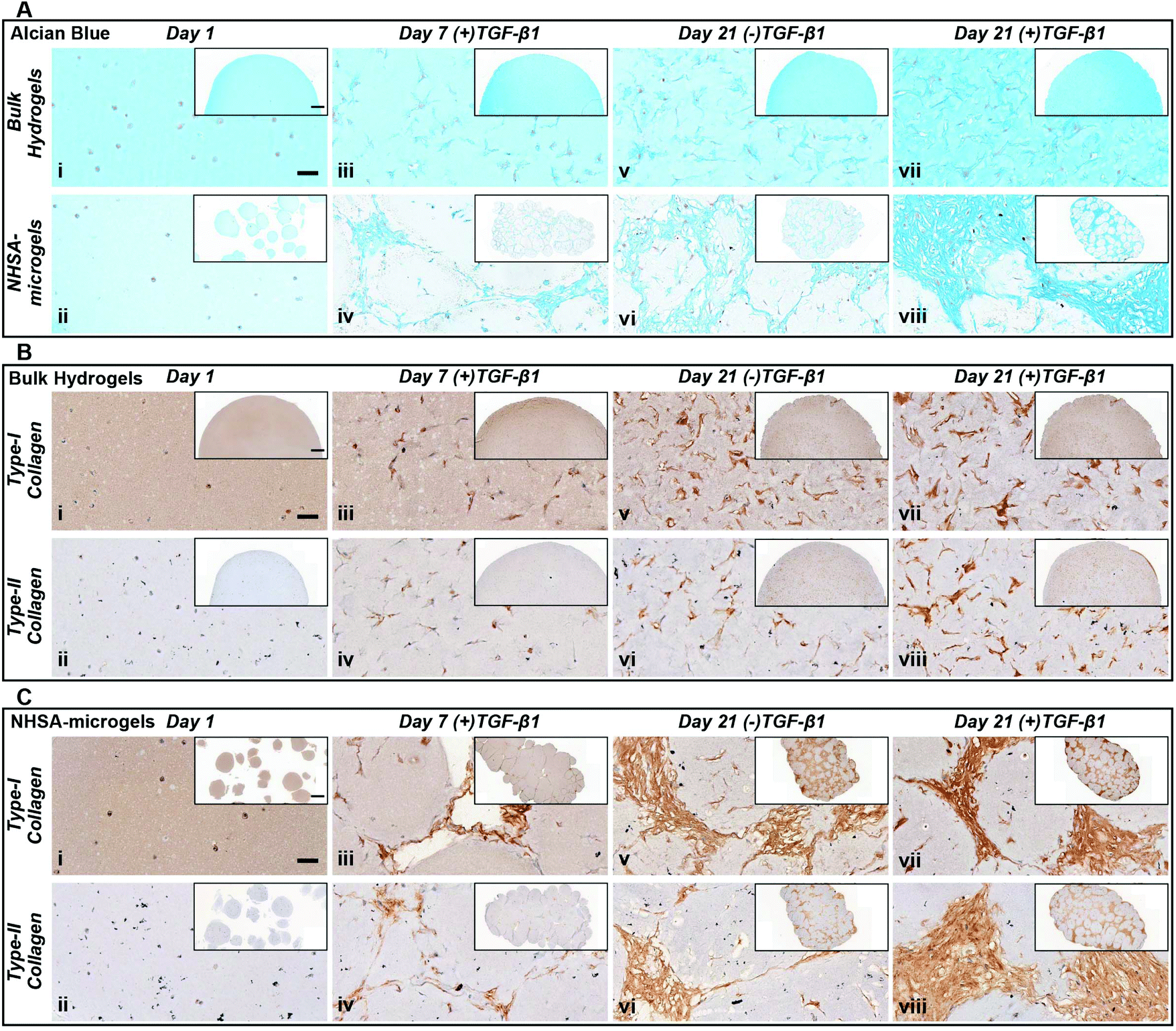 | ||
| Fig. 5 Histological characterisation of bulk hydrogels and NHSA-microgels at day 1, 7 & 21 of in vitro culture. (A) Alcian blue (pH 1) staining – bulk hydrogel (top row) & NHSA-microgels (bottom row). (B) Type-I and type-II collagen immunohistochemical staining on bulk hydrogels. (C) Type-I and type-II collagen immunohistochemical staining on NHSA-microgels (scale bar: 50 μm/inset: 500 μm). | ||
In NHSA-microgels, the most intense Alcian blue staining was located in the cavities between the microgels (Fig. 5Avi & viii), showing that most GAGs were deposited in these spaces rather than within the hydrogel matrix. This was expected since the cells had already migrated out of the gelatin microgels and proliferated in the macropores, as evidenced by the few nuclei (nuclear fast red staining) inside the microgels. Similar to bulk gels, the most intense staining was observed in the (+)TGF-β1 media-cultured samples as compared to those without continuous TGF-β1 supplementation (Fig. 5Aviii). The (+)TGF-β1 media-cultured samples formed a highly condensed tissue structure, particularly in the gaps between the microgels, while the gaps in between the (−)TGF-β1 media-cultured microgels were filled with relatively loose matrix. This further supports the observation that hCCs require continuous exposure to TGF-β1 to more effectively undergo chondrogenesis. Nevertheless, in both (+)TGF-β1 and (−)TGF-β1 media, NHSA-microgels displayed a lack of positive staining close to the hCC pericellular region, which demonstrated a pocket morphology formation within the newly formed matrix. This feature is similar to the mature chondrocytes living in lacunae within a typical articular cartilage tissue.47 Although hCCs on the surface of microgels were highly stretched and in close contacted with each other, the cells in the newly formed tissue between the microgels adopted a more rounded morphology and were sparsely distributed. Due to the close resemblance of this structure to native articular cartilage, these phenotypes were believed to be the additional evidence for hyaline type cartilage formation in the NHSA-microgels, upon further remodelling and gelatin microgel resorption.48
Type-I and type-II collagen levels and distribution were also monitored throughout the culture period (Fig. 5B & C). After one week of culture, there was positive staining for both type-I and type-II collagen in all samples, with a distribution at the intracellular and pericellular territories. In the bulk hydrogel, type-II collagen remained located at the pericellular environment at day 21 in either (+)TGF-β1 or (−)TGF-β1 media. This does not follow the same trend as for the GAGs that were distributed throughout the whole bulk hydrogel (Fig. 5Bvi & viii). The morphology and distribution of type-II collagen in NHSA-microgels at day 21 was similar to the GAG deposition as defined by Alcian blue staining (Fig. 5A & C). Generally, type-II collagen was deposited in the cavities between the microgels. Samples cultured in (+)TGF-β1 media exhibited a more condensed collagen matrix structure compared to (−)TGF-β1 media. This result further confirmed that continuous supplementation of TGF-β1 stimulates hCCs to produce more cartilaginous matrix in the construct. Interestingly, the distribution and morphology of type-I collagen were identical to type-II collagen in both bulk hydrogels and NHSA-microgels. This is in contrast to our previous hMSC studies, in which significantly more type-II collagen was synthesised compared to type-I collagen.23 Strong type-I collagen staining has also been reported in several other studies using hCCs for cartilage regeneration.32,38 Hence, the unexpectedly high type-I collagen production may be specific to the hCC phenotype. Although the reasons for this result are not yet completely understood, one possible explanation is that the hCCs may still be in an early chondrogenic developmental stage, similar to fetal cartilage tissue which shows an extensive amount of type-I collagen.49,50 Furthermore, Sox-9 has been recognised as an important cartilage marker to regulate early chondrogenic development. The gene expression results demonstrated sustained Sox-9 upregulation over three weeks of culture, which further supports our hypothesis that hCCs might still be undergoing early chondrogenesis in both bulk hydrogel and NHSA-microgels. Prolonged culture with further analysis of Sox9/Scleraxis expression ratio and type-I/II collagen staining,51 may allow this hypothesis to be examined in the future and determine whether the newly-formed ECM would turn into a more like hyaline- or fibro-cartilage in terms of collagen distribution.
Taken together, the histological analyses demonstrated in vitro cartilaginous matrix formation in both bulk hydrogel and NHSA-microgels. Although bulk hydrogels had a higher overall GAG content, histological analysis suggests a better quality of the newly deposited ECM in the interstitial spaces within the assembled NHSA-microgels. In fact, it exhibited a structure similar to native articular cartilage tissue, in which hCCs showed a rounded morphology and were sparsely distributed, also residing in lacunae within the structure. Additionally, even though continuous supplementation of TGF-β1 to the hCCs lead to a more condensed matrix deposition, one week of pre-chondrogenic induction could already initiate chondrogenesis with continuous cartilaginous matrix production over time.
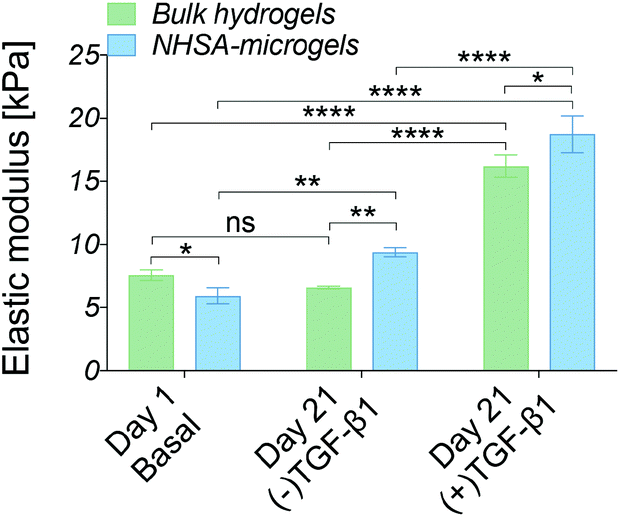 | ||
| Fig. 6 Biomechanical characterisation of bulk hydrogels and NHSA-microgels using nanoindentation at day 1 & 21 of in vitro culture (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001). | ||
The cell-laden bulk hydrogel cultured in the (−)TGF-β1 media for three weeks maintained an elastic modulus of 6.5 ± 0.5 kPa with no significant change compared to day 1. On the contrary, the modulus of the cell-laden NHSA-microgels cultured in (−)TGF-β1 media from day 7 to 21 was higher at the three week timepoint, reaching an elastic modulus of 9.5 ± 0.3 kPa, which was significantly higher than that of the bulk hydrogel (P = 0.0088). This indicated that the cartilaginous matrix in NHSA-microgels after three weeks culture (one week (+)TGF-β1 media and two weeks (−)TGF-β1 media) was not only histologically better, with a hyaline-like cartilaginous matrix phenotype (rounded cell morphology within lacunae, cells sparsely distributed in the dense ECM, Fig. 5A and C), but also led to a higher modulus of the NHSA-microgels samples compared to bulk hydrogels. Moreover, an increase in elastic modulus was detected in the samples that were continuously cultured in the (+)TGF-β media for 3 weeks, achieving a modulus of 16.2 ± 0.7 kPa for the bulk hydrogel and 18.8 kPa ± 1.2 kPa for the NHSA-microgels. This again suggested that continuous supplementation of TGF-β1 considerably improves chondrogenic outcomes when relying on hCCs for ECM synthesis. Taken as a whole, general maintenance or enhancement of elastic modulus was achieved in all samples after three weeks culture. Promisingly, although NHSA-microgels demonstrated a lower modulus compared to bulk hydrogels at day 1, they reached a slightly, but significantly (P = 0.0181) higher elastic modulus compared to bulk hydrogels after three weeks chondrogenesis.
Overall, based on in vitro gene expression, biochemical, histological and biomechanical analysis, chondrogenic differentiation of hCCs was achieved within all different samples, but showing the best results for NHSA-microgels cultured in the presence of TGF-β1 for 3 weeks.
2.3 hCC-laden bulk hydrogel and microgel implantation in vivo
The in vitro studies demonstrated the ability to initiate and maintain hCC chondrogenesis in both bulk hydrogels and NHSA-microgels by pre-culture in chondrogenic media. Therefore, in order to evaluate the scaffold stability and whether this preculture strategy could be effective in a clinical scenario, the pre-cultured samples (Fig. 7A, two weeks in (+)TGF-β1 media) were implanted subcutaneously in a nude mouse model. Histological analysis was conducted two weeks and five weeks after in vivo implantation. Biomechanical analysis was also performed on the samples implanted for two weeks in vivo. Gelatin can be degraded by matrix metalloproteinases (MMPs),52 therefore the first question was whether the hydrogels would be stable in vivo. Both bulk hydrogels and NHSA-microgels were visible under the skin throughout the in vivo maturation period. Additionally, no apparent signs of toxicity (i.e. irritability and necrosis) were observed near the implanted scaffold. A typical thin fibrous capsule around the scaffold was evident macroscopically and via histology staining and it could easily be removed for the mechanical testing after 2 weeks (Fig. 7). There was no obvious cell infiltration from the host within the bulk hydrogel or NHSA-microgels (Fig. 7Bi & Ci).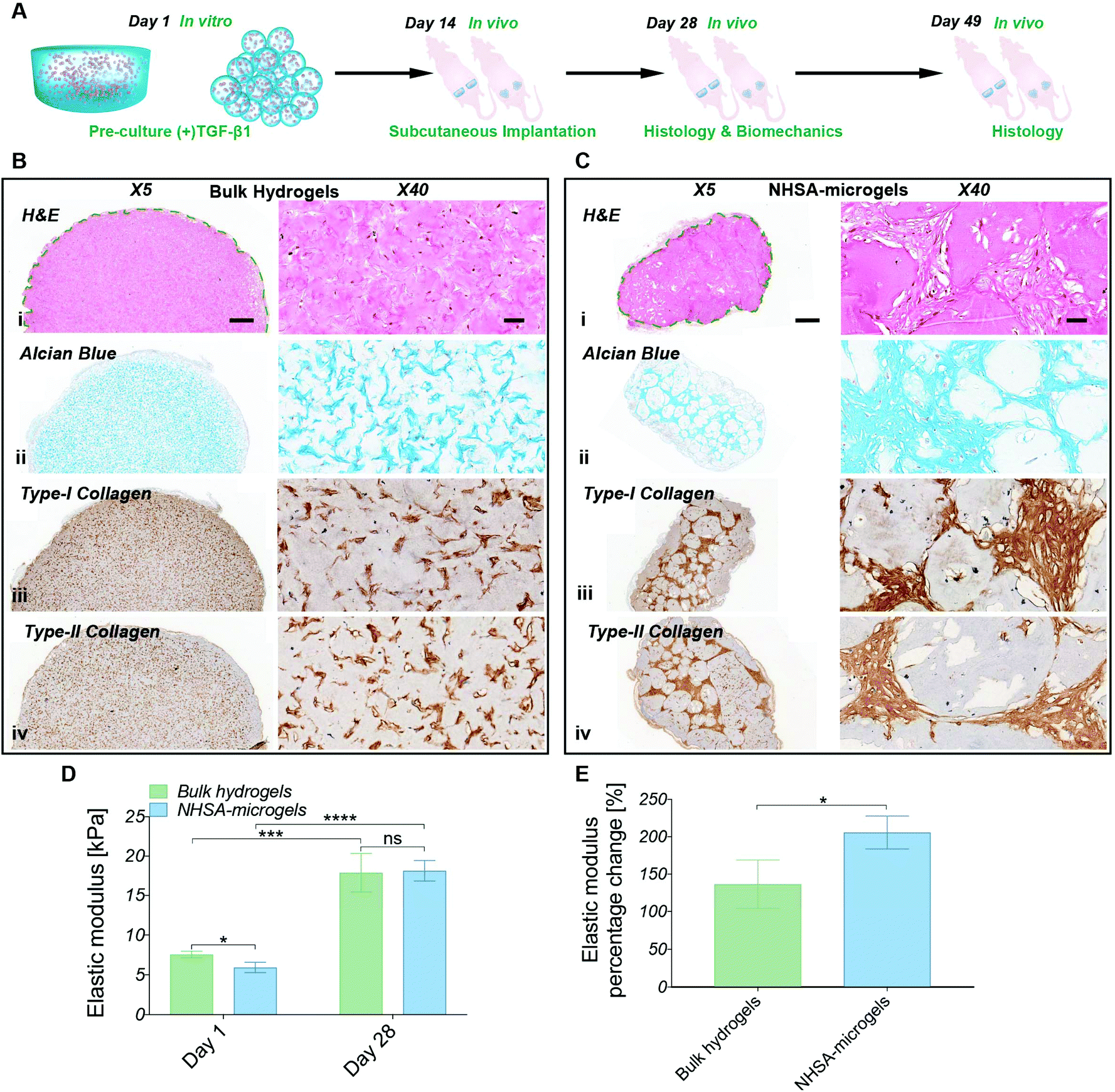 | ||
| Fig. 7 (A) Schematic representation of hCC-laden bulk hydrogels and NHSA-microgel in vivo chondrogenesis: after in vitro pre-culture in chondrogenic media ((+)TGF-β1) for two weeks, samples were subcutaneously implanted into nude mice (left mice – bulk hydrogel; right mice – NHSA-microgels) and maintained in vivo for an additional two weeks (day 28) or five weeks (day 49). (B) Histological analysis of bulk hydrogel at day 28 (i: H&E, ii: Alcian blue, iii: type-I collagen, iv: type-II collagen). (C) Histological analysis of NHSA-microgel at day 28 (i: H&E, ii: Alcian blue, iii: type-I collagen, iv: type-II collagen) (scale bar: X5/500 μm; X40/50 μm). (D) Nanoindentation tests at day 1 and day 28. (E) Percentage elastic modulus increment profile showing hCC-laden bulk hydrogel and NHSA-microgel mechanical properties enhancement after 28 days maintenance in vivo (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001). | ||
Consistent with the in vitro study, after two weeks in vivo, the hCCs were elongated and uniformly distributed in the bulk hydrogels, whilst for NHSA-microgels most of the hCCs had a round morphology and were located in the space between the NHSA-microgels (Fig. 7Bi & Ci). In addition to cell morphology and distribution, the cartilaginous matrix distribution was also consistent with in vitro results. Specifically, GAGs, type-I and type-II collagens were concentrated in the intracellular and pericellular environment in the bulk hydrogels, thus leading to patterned, rather than homogeneous, histological staining (Fig. 7Bii–iv). In NHSA-microgels, the newly formed tissue occupied the inter-gel spaces and had a relatively dense structure with a morphology analogous to native articular cartilage (Fig. 7Cii–iv). Therefore, the overall morphology and distribution of regenerated tissue in vivo were consistent with the in vitro studies.
Numerous cavities were present in the microgels in vivo, which were not observed in the bulk hydrogel or in vitro studies. These cavities were generally located around the surface of the microgels, and are likely due to accelerated degradation of the microgels in vivo. Although the exact underlying mechanism is still not entirely clear, several possible factors may be responsible. For example, the in vivo environment may stimulate hCCs in the NHSA-microgels to secrete more MMPs, such as MMP-2 and MMP-9 resulting in faster degradation. There may also be a high concentration of MMPs produced by endogenous cells under the skin of the mice, which more efficiently infiltrate the NHSA-microgels due to their highly porous structure. Although the accelerated in vivo degradation may shorten the survival time of the scaffold, this may enhance the tissue development and remodelling process to achieve a more uniform cartilaginous matrix distribution because the microgels would be eventually replaced by the regenerated ECM.
Similar to the histological analyses, the in vivo biomechanical analysis was consistent with previous in vitro studies showing that the production of the cartilaginous matrix resulted in an increase in elastic modulus of the scaffolds (Fig. 7D). After two weeks in vitro preculture and another two weeks in vivo, NHSA-microgels had an elastic modulus of 18.1 ± 1.1 kPa compared to 17.9 ± 2.0 kPa for the bulk hydrogel, which was comparable to previous in vitro results with a maximum of 18.8 kPa ± 1.2 kPa. Although, a similar final modulus was achieved in bulk hydrogels and NHSA-microgels, NHSA-microgels achieved a 2-fold increase in elastic modulus compared to the starting point, which was significantly higher than the change in bulk hydrogels with 1.3-fold upregulation (Fig. 7E, P = 0.0375).
After five weeks in vivo, the implanted bulk hydrogel and NHSA-microgels were still visible under the skin with no macroscopic signs of inflammation or toxicity (Fig. 8A & B). In bulk hydrogels, in contrast to the elongated shape of the cells in vitro and after 2 weeks in vivo, hCCs adopted a more rounded shape (Fig. 8Ci). In the NHSA-microgels, the individual microgels were almost fully degraded and replaced by ECM, explaining the disappearance of the overall granular structure of the samples. This resulted in a more uniform cell distribution throughout the newly formed tissue (Fig. 8Di). Of note, this remodelling of the gelatin material by the cells led to a shrinkage of the samples, which would have to be compensated by injection of more material in the defect than the volume of the lesion, in the clinical context.
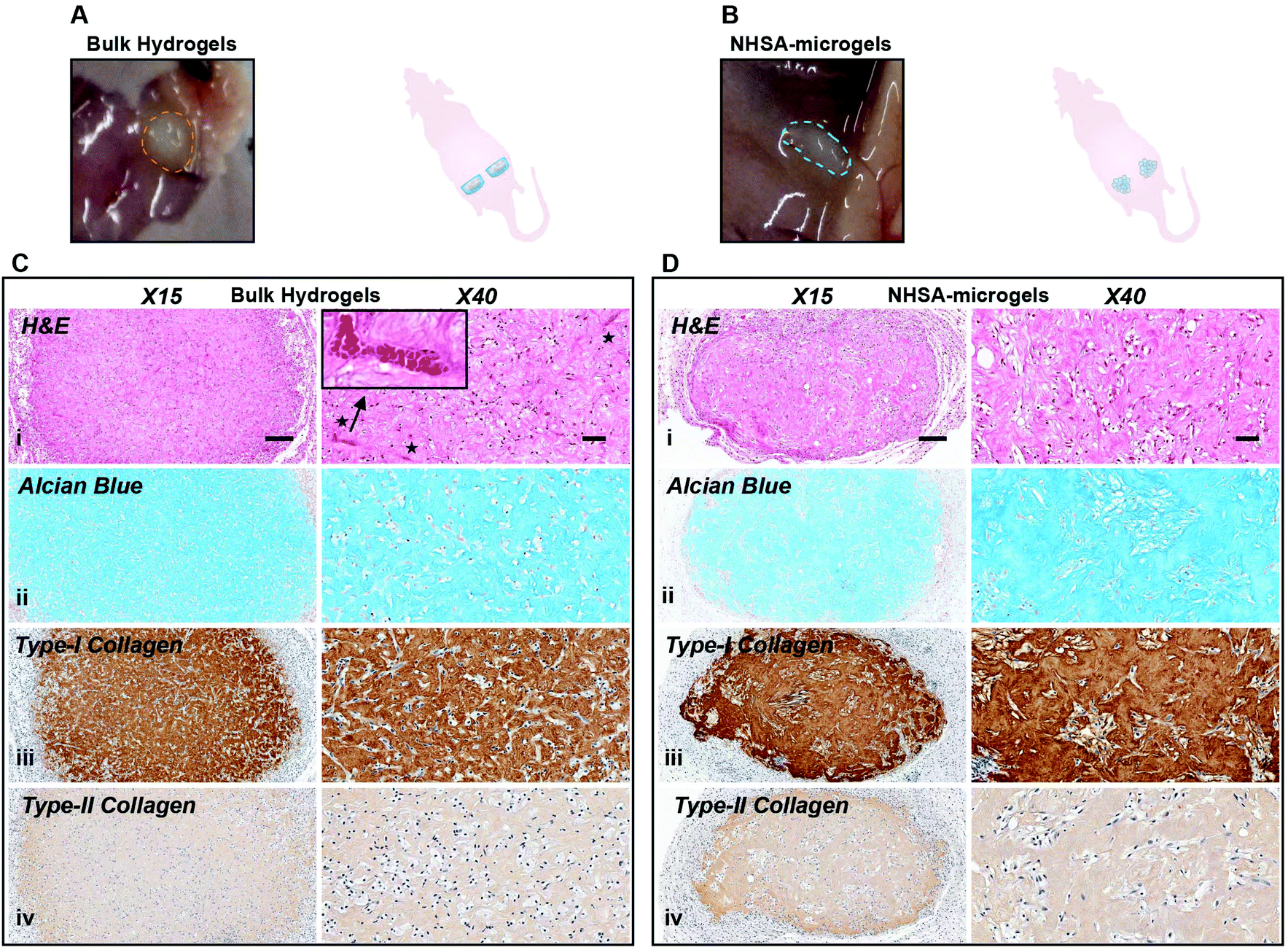 | ||
| Fig. 8 hCC-laden bulk hydrogels and NHSA-microgels after in vivo chondrogenesis at day 49. (A and B) Appearance of bulk hydrogel & NHSA-microgel explants. (C and D) Histological analysis of bulk hydrogels & NHSA-microgels (i: H&E, ii: Alcian blue, iii: type-I collagen, iv: type-II collagen; scale bar: X5/500 μm; X40/50 μm). | ||
Alcian blue and collagen staining demonstrated homogenous distribution of both GAGs and collagens within the entire bulk hydrogels, but none in the intracellular/pericellular regions (Fig. 8Cii–iv). This was also observed in the NHSA-microgels with cartilaginous matrix uniformly distributed across the whole construct (Fig. 8Dii–iv), which was assumed to be a result of the active cell–material interactions. This reinforced our previous observation that a more uniform cartilaginous matrix could be formed with prolonged culture. Although a native articular cartilage morphology was regenerated in NHSA-microgels, the levels of type-I collagen were still high in comparison to type-II collagen levels in both bulk hydrogels and NHSA-microgels (Fig. 8Ciii, iv & 8Diii, iv). This might be due to a lack of continuous chondrogenic induction in the in vivo environment, meaning that more than two weeks preculture might be necessary to fully support differentiation. More encouragingly, even though both bulk hydrogels and NHSA-microgels experienced extensive cellular remodelling and degradation, no vascularisation was evident in the NHSA-microgels after five weeks in vivo (Fig. 8Ci & Fig. S3†), which conforms to the characteristic of native articular cartilage tissue, in which blood vessels are absent. However, vascularisation was observed in the bulk hydrogels (Fig. 8Di & Fig. S3†). This suggested that the deposited cartilaginous matrix in NHSA-microgels was phenotypically more stable than the matrix in bulk hydrogel.
In summary, there were differences of distribution and morphology in newly formed ECM between the bulk hydrogels and NHSA-microgels after two weeks in vivo with NHSA-microgels being better. Although this had evened out by five weeks in vivo, showing similar matrix phenotype in bulk hydrogels and NHSA-microgels, the regenerated tissue in NHSA-microgels demonstrates superior quality compared to the bulk hydrogel with the ability to resist vascularisation standing out.
Overall the data are encouraging and it is likely that further optimisation of pre-transplantation culture conditions will achieve a more favourable in vivo outcome. For instance, a more detailed investigation of the preculture times to ensure a prolonged chondrogenesis tendency and improved cartilaginous matrix retention in vivo is warranted. Moreover, physical entrapment or covalent conjugation of chondrogenic growth factors within the microgel network may provide an alternative approach to deliver sustained chondrogenic signals to encapsulated hCCs within the in vivo environment. Apart from chondrogenic supplementation, both the bulk hydrogel and NHSA-microgels experienced shrinkage after three weeks in vitro culture or five weeks in vivo maturation, which is crucial consideration for implementation into clinical practice. This is most likely due to the progressive degradation of the hydrogel materials, remodelling and formation of condensed tissue during the chondrogenesis process. In short, these preliminary in vivo findings follow the major outcomes in the in vitro studies. The encouraging preliminary in vivo discoveries have demonstrated the great potential of using hCC-laden NHSA-microgels for further clinical translation in articular cartilage regeneration.
3. Conclusions
In this work, the successful long-term culture of hACs, hCCs and hMSCs demonstrated the versatility of gelatin-based microgels for cell encapsulation. Based on their high viability and fast migration rate, hCCs were further investigated as an alternative emerging candidate for cartilage regeneration in combination with the microgel assembly approach. These cells were successfully microencapsulated and their ability to support chondrogenesis determined via the comparison to bulk hydrogels with different TGF-β1 regimes during in vitro culture and maintenance in vivo. All samples incubated either in vitro or in vivo demonstrated the ability to support cartilage formation to varying extents. In vitro culture experiments suggested hCCs have considerably better chondrogenic outcomes in NHSA-microgels compared to bulk hydrogels as evidenced by the significantly higher chondrogenic gene expression, a more native cartilage tissue-like structure and superior mechanical properties. Another important finding from the in vitro study was that the sustained supplementation of TGF-β1 during culture would greatly enhance the chondrogenesis of hCC outcomes in both bulk hydrogels and NHSA-microgels. Furthermore, there were marked differences after two weeks in vivo with NHSA-microgels showing better tissue morphology and a higher degree of mechanical improvement compared to the bulk hydrogels. The newly formed tissue was more similar between bulk hydrogels and NHSA-microgels after five weeks maintenance in vivo but the NHSA-microgels demonstrated the ability to resist vascularisation. Overall, both in vitro and in vivo results support the utility of NHSA-microgel for hCC chondrogenesis. Therefore, it is expected that this hCC-laden NHSA-microgel system will be a promising candidate for articular cartilage repair or regeneration in the future.4. Experimental
4.1. Hydrogel materials preparation
Gelatin was functionalised with norbornene groups with a defined substitution degree of 48% through a two-step process as described in the ESI (Fig. S1†). Norbornene carboxylic acid was first reacted with N-hydroxysuccinimide to produce a ester intermediate. The intermediate was subsequently grafted onto gelatin backbone to form gelatin-norbornene conjugate with further purification and lyophilised to achieve the final product. PEGdiSH crosslinker was synthesised via thiol functionalisation on both ends of the linear PEG with details being provided in the ESI (Fig. S2†).4.2. Human cell types screening
hCCs were isolated from a 14 weeks-gestation donor's proximal ulnar epiphysis (the same donor reported by Darwiche et al.,37 Centre Hospitalier Universitaire Vaudois, Ethic Committee Protocol no. 62/07 and registered under the Federal Transplantation Programme complying with the law and Biobank procedures). Experiments in which hCCs were used were performed in accordance with the Guidelines of “Hospital Department Biobank Regulations (DAL Biobank)”, and Experiments were approved by the local ethics committee (protocol #62/07: “Development of fetal cell banks for tissue engineering”, August 2007). hCCs were cultured in DMEM (41966) (+4.5 g L−1D-glucose, L-glutamine, +pyruvate) supplemented with 10% (v/v) FBS, 2 mM L-glutamine and 100 U mL−1 penicillin–streptomycin.
All MSC experiments were performed in accordance to the Guidelines of the Canton of St Gallen, Switzerland and experiments were approved by the ethics committee Kantonsspital St Gallen, Switzerland (Ethical Approval no. EKSG08/014/1B). hMSCs from 3 independent donors were isolated from femur-derived bone marrow according to the protocol in our previous report.53 The donors were aged 42, 46, 74 years old and samples were taken during hip replacement surgery. bhMSCs were cultured in alphaMEM (+L-glutamine, – ribonucleosides, – deoxyribonucleosides) supplemented with 10% FBS, 10 μg mL−1 gentamicin and 1 ng mL−1 fibroblast growth factor 2.
All cells were expanded in a tissue culture flask and maintained at 37 °C in 5% CO2 until passage 2 (hACs and hMSCs) or 4 (hCCs).
Fiji (https://imagej.net/Fiji) was used to create the z-stack images for the viability analysis. Viability was presented as a cell survival percentage, which was measured based on the counting of viable and dead cells. Icy software (Institut Pasteur, Open Source, http://icy.bioimageanalysis.org/) was used to reconstruct the 3D images for the cell migration analysis. For each cell type, five microgels were selected and the intensity profiles were plotted on the middle slice of each image to assess the migration in the microgels.
4.3. Characterisation of hCCs in vitro chondrogenesis
Both bulk hydrogel and microgel samples were cultured in the hCC expansion media for the first 24 h. On day 1, dispersed cell-laden microgels were assembled into aggregates of microgels, referred to as “NHSA-microgels”. Briefly, the culture media was first removed to reach 200 μL concentrated microgels. To this microgel suspension, 200 μL 4-arm PEG-NHS (4% (w/v)) was added and incubated for 90 min to achieve stable covalent bonding. After 1 day, NHSA-microgels and bulk hydrogels culture media was changed to chondrogenic media, which consisted of DMEM (31966) + GlutaMAXTM-I (+4.5 g L−1D-glucose, +pyruvate) supplemented with 100 U mL−1 penicillin–streptomycin, 1% (v/v) ITS + Premix, 40 μg mL−1L-proline, 50 μg mL−1L-ascorbic acid-2-phosphate and 10 ng mL−1 TGF-β1. The samples were cultured in the chondrogenic media for seven days with media change every three days. After one week in culture, half of the samples continued to be cultured in chondrogenic media (referred as “(+)TGF-β1” media), while the other half of the samples were changed to chondrogenic control media (“(−)TGF-β1” media) with TGF-β1 withdrawn for the further two weeks.
Fast SYBR Green Master Mix (Life technologies) was used for the final PCR amplification with chondrogenesis target genes (Table S1†) and GAPDH and RPL13a were selected as housekeeping gene. Then, 10 μL SYBR, 1 μL of forward and reverse primers (150 nM each), 2 μL cDNA (5 ng μL−1) and additional 7 μL DNase/RNase-free water were mixed to achieve the final 20 μL reaction mixture. All samples were prepared in triplicate and processed via a thermal cycler (StepOnePlus™ Real-Time PCR System). They underwent the cycling conditions of 95 °C for 20 s, 95 °C for 3 s, and 60 °C for 16 s for 40 cycles to obtain a standard melting curve. The data were analysed using the 2−ΔΔCT method normalized to day 1 bulk hydrogel samples with RPL13a as the reference gene (RPL13a was found to be more stable than GAPDH in different samples across different time points in this study, Fig. S4†).
Alcian blue (pH 1) staining was performed on the rehydrated sections following standard procedure. Briefly, slides were acidified for 3 min in 3% (v/v) acetic acid in deionized water, then incubated for 30 min in 1% (v/v) Alcian blue in 3% (v/v) acetic acid brought to pH 1 (Sigma-Aldrich, B8438). After rinsing in tap water and counterstaining with nuclear fast red (Sigma-Aldrich N3020) for 5 min, the slides were rinsed in tap water, dehydrated and mounted with Eukritt mounting media.
Type-I and type-II collagen colorimetric staining started with antigen retrieval by digesting the section with 1200 U ml−1 hyaluronidase (Sigma-Aldrich, H3506) 30 min at 37 °C, followed by blocking with 5% (v/v) NGS (normal goat serum) in PBS at room temperature for 1 h. The sections were then incubated with rabbit anti collagen I (Abcam, ab138492, 1:1500 dilution) and rabbit anti-collagen II (Rockland, 600-401-104, 1:200 dilution) at 4 °C overnight. Primary antibodies were diluted in 1% (v/v) NGS in PBS. After 2 washings in PBS, slides were treated with 1% (v/v) H2O2 in deionized water for 20 min and goat anti-rabbit IgG-HPR (Abcam, ab6721, 1:1500 dilution in 1% NGS) was applied to the slides for 1 h at room temperature. After rinsing 3 times with PBS, a chromogen solution was added to the slides at room temperature using the DAB substrate kit (Abcam, ab64238), for 4 min (type-I and type-II collagens). The slides were counterstained with hematoxylin (Sigma-Aldrich, MHS1) for 3 min, washed, dehydrated and mounted with Eukritt mounting media (Sigma-Aldrich, 03989). The images of histological slides were acquired via a slide scanner (Pannoramic 250, 3D Histech).
4.4. Characterisation of hCCs in vivo chondrogenesis
4.5. Statistical analysis
Statistical analysis was performed using Prism 7 software. The cell viabilities were presented as mean ± standard deviation and two-way ANOVA with Tukey's post hoc test (equal variance) was used to determine the differences between cell types and time points. The in vitro qPCR and biomechanical data were shown as mean ± standard deviation with two-way ANOVA using Tukey's multiple comparison post hoc test. The in vivo nanoindentation results were demonstrated as mean ± standard deviation with unpaired t test with Welch's corrections. The data differences were considered significant when p < 0.05.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. L. acknowledges support from the New Horizons Monash-CSIRO Council [Joint PhD scholarship] and Faculty of Engineering at Monash University [Graduate research international travel grant]. M. Z. W. acknowledges the support of the Swiss National Science Foundation (315230_159783). The authors thank PD Dr med. Matthias Steinwachs for providing the healthy adult cartilage samples. We acknowledge Dr Emma Cavalli (ETH, Zurich) for isolating the hACs and expert surgical assistance. We also thank Philipp Fisch (ETH, Zurich) for sharing his knowledge on nanoindentation tests. The authors acknowledge the facilities and scientific and technical assistance of Scientific Centre for Optical and Electron Microscopy (ScopeM), ETH Zürich.References
- M. Krill, N. Early, J. S. Everhart and D. C. Flanigan, JBJS Rev., 2018, 6, e5 CrossRef PubMed.
- Z. Ximu, Z. Wei and Y. Maobin, Curr. Stem Cell Res. Ther., 2018, 13, 497–516 CrossRef.
- J. Yang, Y. S. Zhang, K. Yue and A. Khademhosseini, Acta Biomater., 2017, 57, 1–25 CrossRef CAS PubMed.
- M. Liu, X. Zeng, C. Ma, H. Yi, Z. Ali, X. Mou, S. Li, Y. Deng and N. He, Bone Res., 2017, 5, 17014 CrossRef CAS PubMed.
- K. M. C. Tsang, N. Annabi, F. Ercole, K. Zhou, D. J. Karst, F. Li, J. M. Haynes, R. A. Evans, H. Thissen, A. Khademhosseini and J. S. Forsythe, Adv. Funct. Mater., 2015, 25, 977–986 CrossRef CAS PubMed.
- N. Annabi, J. W. Nichol, X. Zhong, C. Ji, S. Koshy, A. Khademhosseini and F. Dehghani, Tissue Eng., Part B, 2010, 16, 371–383 CrossRef CAS PubMed.
- L. Abune, N. Zhao, J. Lai, B. Peterson, S. Szczesny and Y. Wang, ACS Biomater. Sci. Eng., 2019, 5, 2382–2390 CrossRef CAS PubMed.
- K. J. De France, F. Xu and T. Hoare, Adv. Healthcare Mater., 2018, 7, 1700927 CrossRef PubMed.
- I. K. Ko, S. J. Lee, A. Atala and J. J. Yoo, Exp. Mol. Med., 2013, 45, e57 CrossRef PubMed.
- D. R. Griffin, W. M. Weaver, P. O. Scumpia, D. Di Carlo and T. Segura, Nat. Mater., 2015, 14, 737 CrossRef CAS PubMed.
- Y. Du, E. Lo, S. Ali and A. Khademhosseini, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9522–9527 CrossRef CAS PubMed.
- E. Jain, K. M. Scott, S. P. Zustiak and S. A. Sell, Macromol. Mater. Eng., 2015, 300, 823–835 CrossRef CAS.
- X. Zhao, S. Liu, L. Yildirimer, H. Zhao, R. Ding, H. Wang, W. Cui and D. Weitz, Adv. Funct. Mater., 2016, 26, 2809–2819 CrossRef CAS.
- A. S. Caldwell, G. T. Campbell, K. M. T. Shekiro and K. S. Anseth, Adv. Healthcare Mater., 2017, 6, 1700254 CrossRef PubMed.
- A. Khademhosseini and R. Langer, Biomaterials, 2007, 28, 5087–5092 CrossRef CAS.
- R. M. Staruch, G. E. Glass, R. Rickard, S. P. Hettiaratchy and P. E. Butler, Tissue Eng., Part B, 2017, 23, 183–198 CrossRef CAS PubMed.
- D. Sivakumaran, D. Maitland and T. Hoare, Biomacromolecules, 2011, 12, 4112–4120 CrossRef CAS PubMed.
- K. Wang, S. Lin, K. Nune and R. Misra, J. Biomater. Sci., Polym. Ed., 2016, 27, 441–453 CrossRef CAS PubMed.
- C. Yu, J. Liu, G. Lu, Y. Xie, Y. Sun, Q. Wang, J. Liang, Y. Fan and X. Zhang, J. Mater. Chem. B, 2018, 6, 5164–5173 RSC.
- L. D. Solorio, E. L. Vieregge, C. D. Dhami and E. Alsberg, Tissue Eng., Part B, 2012, 19, 209–220 CrossRef.
- C. Luo, J. Zhao, M. Tu, R. Zeng and J. Rong, Mater. Sci. Eng., C, 2014, 36, 301–308 CrossRef CAS PubMed.
- F. Li, V. X. Truong, H. Thissen, J. E. Frith and J. S. Forsythe, ACS Appl. Mater. Interfaces, 2017, 9, 8589–8601 CrossRef CAS PubMed.
- F. Li, V. X. Truong, P. Fisch, C. Levinson, V. Glattauer, M. Zenobi-Wong, H. Thissen, J. S. Forsythe and J. E. Frith, Acta Biomater., 2018, 77, 48–62 CrossRef CAS PubMed.
- L. Riley, L. Schirmer and T. Segura, Curr. Opin. Biotechnol., 2019, 60, 1–8 CrossRef CAS PubMed.
- J. E. Mealy, J. J. Chung, H.-H. Jeong, D. Issadore, D. Lee, P. Atluri and J. A. Burdick, Adv. Mater., 2018, 30, 1705912 CrossRef PubMed.
- E. Sideris, D. R. Griffin, Y. Ding, S. Li, W. M. Weaver, D. Di Carlo, T. Hsiai and T. Segura, ACS Biomater. Sci. Eng., 2016, 2, 2034–2041 CrossRef CAS.
- A. Sheikhi, J. de Rutte, R. Haghniaz, O. Akouissi, A. Sohrabi, D. Di Carlo and A. Khademhosseini, Biomaterials, 2019, 192, 560–568 CrossRef CAS PubMed.
- S. Xin, O. M. Wyman and D. L. Alge, Adv. Healthcare Mater., 2018, 7, 1800160 CrossRef PubMed.
- B. J. Huang, J. C. Hu and K. A. Athanasiou, Biomaterials, 2016, 98, 1–22 CrossRef CAS PubMed.
- A. Barbero, S. Grogan, D. Schäfer, M. Heberer, P. Mainil-Varlet and I. Martin, Osteoarthr. Cartil., 2004, 12, 476–484 CrossRef PubMed.
- M. Schnabel, S. Marlovits, G. Eckhoff, I. Fichtel, L. Gotzen, V. Vécsei and J. Schlegel, Osteoarthr. Cartil., 2002, 10, 62–70 CrossRef CAS PubMed.
- D. Studer, E. Cavalli, F. A. Formica, G. A. Kuhn, G. Salzmann, M. Mumme, M. R. Steinwachs, L. A. Laurent-Applegate, K. Maniura-Weber and M. Zenobi-Wong, J. Tissue Eng. Regener. Med., 2017, 11, 3014–3026 CrossRef CAS.
- E. Cavalli, C. Levinson, M. Hertl, N. Broguiere, O. Brück, S. Mustjoki, A. Gerstenberg, D. Weber, G. Salzmann, M. Steinwachs, G. Barreto and M. Zenobi-Wong, Sci. Rep., 2019, 9, 4275 CrossRef PubMed.
- V. Claire, B. Carine, M. Christophe, G. Jan, B. Jean-Marc, J. Christian, W. Pierre, G. Jerome and N. Daniele, Curr. Stem Cell Res. Ther., 2009, 4, 318–329 CrossRef.
- D. Studer, C. Millan, E. Ozturk, K. Maniura-Weber and M. Zenobi-Wong, Eur. Cells Mater., 2012, 24, 118–135 CrossRef CAS PubMed.
- Y. Liu, G. Zhou and Y. Cao, Engineering, 2017, 3, 28–35 CrossRef CAS.
- S. Darwiche, C. Scaletta, W. Raffoul, D. P. Pioletti and L. A. Applegate, Cell Med., 2012, 4, 23–32 CrossRef.
- N. Broguiere, E. Cavalli, G. M. Salzmann, L. A. Applegate and M. Zenobi-Wong, ACS Biomater. Sci. Eng., 2016, 2, 2176–2184 CrossRef CAS.
- J. Garcia, C. Mennan, H. S. McCarthy, S. Roberts, J. B. Richardson and K. T. Wright, Stem Cells Int., 2016, 2016, 11 Search PubMed.
- S. Ma, M. Natoli, X. Liu, M. P. Neubauer, F. M. Watt, A. Fery and W. T. S. Huck, J. Mater. Chem. B, 2013, 1, 5128–5136 RSC.
- Y. Hou, W. Xie, K. Achazi, J. L. Cuellar-Camacho, M. F. Melzig, W. Chen and R. Haag, Acta Biomater., 2018, 77, 28–37 CrossRef CAS.
- A. C. Newby, Cardiovasc. Res., 2006, 69, 614–624 CrossRef CAS PubMed.
- A. J. Sophia Fox, A. Bedi and S. A. Rodeo, Sports Health, 2009, 1, 461–468 CrossRef PubMed.
- W. Dessau, H. von der Mark, K. von der Mark and S. Fischer, J. Embryol. Exp. Morphol., 1980, 57, 51–60 CAS.
- I. E. Erickson, S. R. Kestle, K. H. Zellars, M. J. Farrell, M. Kim, J. A. Burdick and R. L. Mauck, Acta Biomater., 2012, 8, 3027–3034 CrossRef CAS PubMed.
- P. A. Levett, D. W. Hutmacher, J. Malda and T. J. Klein, PLoS One, 2014, 9, e113216 CrossRef PubMed.
- J. B. Richardson and A. M. Bhosale, Br. Med. Bull., 2008, 87, 77–95 CrossRef.
- J. D. Hayes, R. L. Brower and K. J. John, Clin. Podiatr. Med. Surg., 2001, 18, 35–53 Search PubMed.
- T. Kirsch and K. von der Mark, Bone Miner., 1992, 18, 107–117 CrossRef CAS.
- Y. S. Bland and D. E. Ashhurst, Anat. Embryol., 1996, 194, 607–619 CrossRef CAS.
- C. I. Lorda-Diez, J. A. Montero, C. Martinez-Cue, J. A. Garcia-Porrero and J. M. Hurle, J. Biol. Chem., 2009, 284, 29988–29996 CrossRef CAS.
- T. Rice, J. E. Larsen, H. Li, R. K. Nuttall, P. H. Larsen, S. Casha, J. Hurlbert, D. Edwards and V. W. Yong, Neuroimmunol. Neuroinflammation, 2017, 4, 243–253 CrossRef CAS.
- D. Studer, S. Lischer, W. Jochum, M. Ehrbar, M. Zenobi-Wong and K. Maniura-Weber, Tissue Eng., Part C, 2012, 18, 761–771 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm01524h |
| ‡ These authors contributed equally. |
| § Present address: Murdoch Children's Research Institute, The Royal Children's Hospital, 50 Flemington Road, Parkville, VIC 3052, Australia. |
| This journal is © The Royal Society of Chemistry 2020 |