
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of sterically hindered 4,5-diarylphenanthrenes via acid-catalyzed bisannulation of benzenediacetaldehydes with alkynes†
Yuanming
Li
 a,
Akiko
Yagi
ab and
Kenichiro
Itami
*abc
a,
Akiko
Yagi
ab and
Kenichiro
Itami
*abc
aInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Chikusa, Nagoya 464-8602, Japan. E-mail: itami@chem.nagoya-u.ac.jp
bGraduate School of Science, Nagoya University, Chikusa, Nagoya 464-8602, Japan
cJST-ERATO, Itami Molecular Nanocarbon Project, Nagoya University, Chikusa, Nagoya 464-8602, Japan
First published on 17th April 2019
Abstract
The synthesis of sterically hindered phenanthrenes via acid-catalyzed bisannulation reaction is described. Treatment of 1,4-benzenediacetaldehyde with terminal aryl alkynes in the presence of B(C6F5)3 provides 4,5-diarylphenanthrenes in good yields with excellent regioselectivity. The use of internal alkyne substrates enabled the synthesis of sterically hindered 3,4,5,6-tetrasubstituted phenanthrenes displaying augmented backbone helicity. Furthermore, 1,5-disubstituted, 1,8-disubstituted, 1,2,5,6-tetrasubstituted, and 1,2,7,8-tetrasubstituted phenanthrenes can be obtained through the reaction of alkynes with 1,3-benzenediacetaldehyde or 1,2-benzenediacetaldehyde disilyl acetal.
Introduction
Angularly fused polycyclic aromatics (AF-PAHs; e.g., phenacenes and helicenes) are unique structural motifs that are utilized in a wide array of materials ranging from organic electronics1 to medicinal chemistry.2 Phenanthrene, the simplest AF-PAH, is the distinct fundamental subunit of higher AF-PAHs and the development of synthetic methods for the preparation of functionalized phenanthrenes has received extensive attention over the years (Fig. 1a). The Mallory cyclization of stilbene derivatives (i)3 and the annulation of alkynylated biaryls (ii)4 are useful methods for the synthesis of phenanthrene derivatives. Additionally, transition metal catalyzed annulations of 2-substituted biphenyl with alkynes have also been reported (iii).5 Other methods, such as ring-closing olefin metathesis,6 carbonyl-olefin metathesis,7 McMurry cyclization,8 and carbene dimerization,9 have also been used (iv). However, synthetic methods for the construction of sterically hindered 4,5-disubstituted phenanthrenes are particularly limited.10 For example, known preparations of 4,5-diphenylphenanthrene require either a four- or five-step synthesis and provide the product in low overall yields (Fig. 1b).11 Herein, we report the acid-catalyzed bisannulation reaction of 1,4-benzenediacetaldehyde with aryl alkynes that can access sterically hindered, twisted 4,5-disubstituted and 3,4,5,6-tetrasubstituted phenanthrenes. Furthermore, reactions of isomeric 1,3-benzenediacetaldehyde and 1,2-benzenediacetaldehyde disilyl acetal with aryl alkynes gave 1,5-disubstituted, 1,8-disubstituted, 1,2,5,6-tetrasubstituted and 1,2,7,8-tetrasubstituted phenanthrenes, respectively.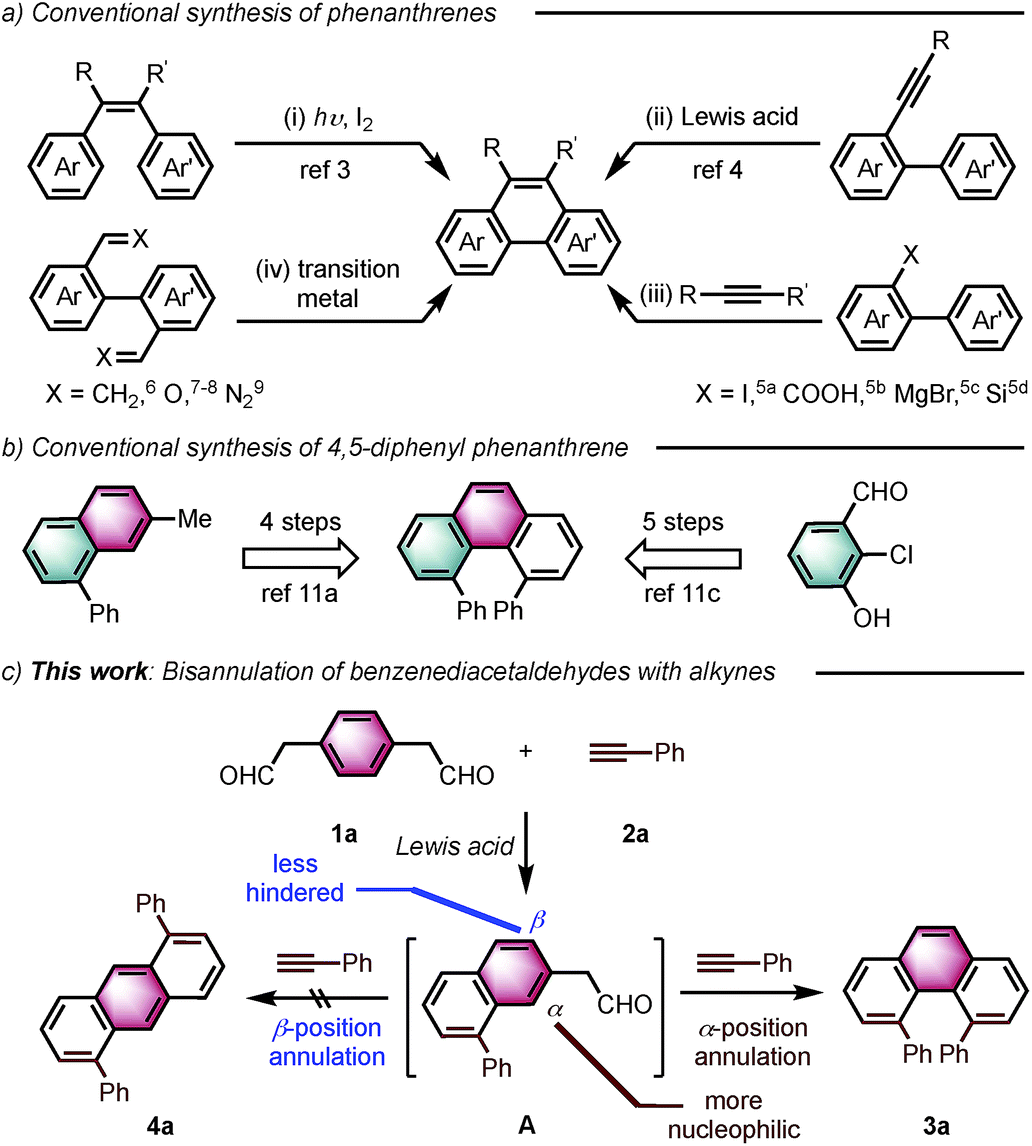 | ||
| Fig. 1 Synthetic strategies leading to phenanthrenes. | ||
Inspired by the synthesis of naphthalene derivatives through the annulation of phenylacetaldehydes with alkynes,12 we envisioned that the acid-catalyzed reaction of 1,4-benzenediacetaldehyde (1a)13 with phenylacetylene (2a) could provide the intermediate naphthalene A (Fig. 1c).
We initially anticipated that the second annulation with another equivalent of alkyne could take place at the less hindered β-position of A to give 1,5-diphenylanthracene (4a). Surprisingly, however, we observed instead an unprecedented α-position annulation to give isomeric 4,5-diphenylphenanthrene (3a), which is an even more interesting, otherwise difficult-to-access AF-PAH.
Results and discussion
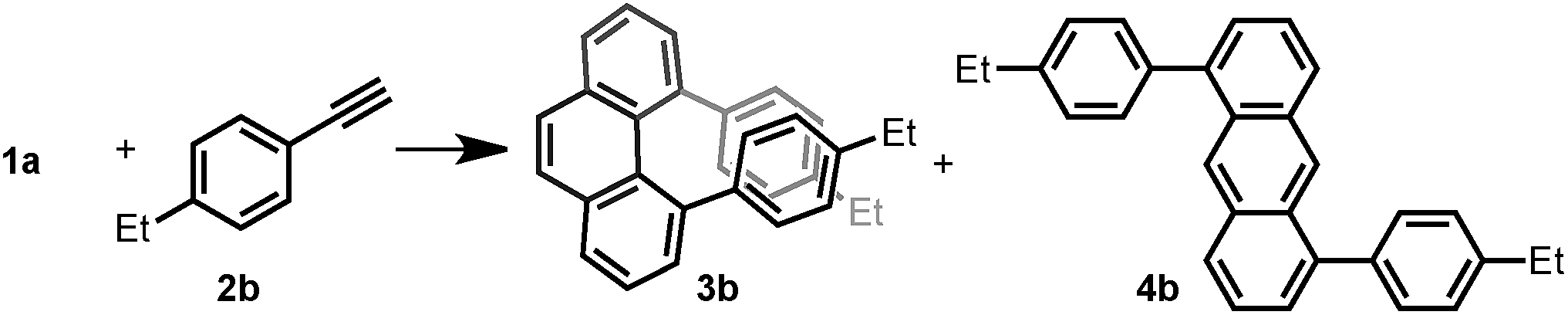
Thus, we investigated the effect of acids and other reaction parameters in the reaction of 1a and 1-ethyl-4-ethynylbenzene (2b) as a model system (Table 1). Though the use of Brønsted acid HNTf2,12g Lewis acids CuCl2/AgSbF6 (ref. 12c) or TiCl4 (ref. 12b) failed to give more than trace amounts of target products (entries 1–3), FeCl3 (ref. 12d) and GaCl3 (ref. 12a) were able to achieve the phenanthrene transformation in 16% and 35% yield, respectively (entries 4 and 5). A catalytic amount of BF3·Et2O12f led to the formation of 3b in 41% yield at 80 °C (entry 6). Subsequently, B(C6F5)3 was found to give a higher yield (45%) under milder conditions (entry 7). The best result was achieved upon reacting 1a (1.0 equiv.) with 2b (2.5 equiv.) in the presence of 20 mol% B(C6F5)3 and MS4Å in dichloromethane (DCM) at room temperature for 15 h, giving 3b in 65% isolated yield with excellent regioselectivity (entry 8). Notably, the anthracene product 4b was not detected under any of the conditions tested. However, the insoluble material formed during the reaction was isolated, and we presume that the self-polymerization of 1a is the major side reaction (see ESI†). The use of MS4Å was necessary to suppress the hydration of the alkyne. A solvent screening revealed that the initially tested DCM was the optimal medium for this reaction (entries 9–12).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Additive (equiv.) | Temp (°C) | Time (h) | Solvent | 3b (%) | 4b (%) |
| a Reaction conditions: 1a (0.10 mmol) and 2b (0.25 mmol) in solvent (2.5 mL). b CuCl2 (0.08)/AgSbF6 (0.16). c 100 mg MS4Å (1/16) were added. d Yield was determined by 1H NMR spectroscopy. n.d. = not detected. rt = room temperature. | ||||||
| 1 | HNTf2 (0.15) | rt | 15 | DCE | n.d. | n.d. |
| 2 | CuCl2/AgSbF6b | 55 | 22 | DCM | n.d. | n.d. |
| 3 | TiCl4 (2.0) | rt | 3 | DCM | Trace | n.d. |
| 4 | FeCl3 (0.10) | 80 | 15 | DCE | 16 | n.d. |
| 5 | GaCl3 (0.20) | 40 | 6 | DCM | 35 | n.d. |
| 6 | BF3·Et2O (0.05) | 80 | 15 | DCE | 41 | n.d. |
| 7 | B(C6F5)3 (0.20) | rt | 15 | DCM | 45 | n.d. |
| 8c | B(C6F5)3 (0.20) | rt | 15 | DCM | 67 | n.d. |
| 9c | B(C6F5)3 (0.20) | rt | 15 | DCE | 59 | n.d. |
| 10c | B(C6F5)3 (0.20) | rt | 15 | toluene | 48 | n.d. |
| 11c | B(C6F5)3 (0.20) | rt | 15 | CHCl3 | 7 | n.d. |
| 12c | B(C6F5)3 (0.20) | rt | 15 | THF | Trace | n.d. |
Based on the reported reaction mechanism of phenylacetaldehydes with alkynes,12 we suggest the following mechanism (Scheme 1, for details, see ESI†). Initially, B(C6F5)3 coordinates with the carbonyl oxygen,14 which would trigger the nucleophilic attack of the alkyne partner to give the vinyl carbocation A′. Subsequently, intermediate A′ undergoes an intramolecular electrophilic aromatic substitution, followed by aromatization to provide the naphthalene A. A DFT calculation revealed that the Mulliken atomic charge at α-position is actually more negative than β-position in the naphthalene A (−0.275 compared to −0.176, see ESI† for details). The electronic bias of the C–C bond formation is overcoming the steric hindrance between the two phenyl rings in C, providing α-position annulation product 3a exclusively.
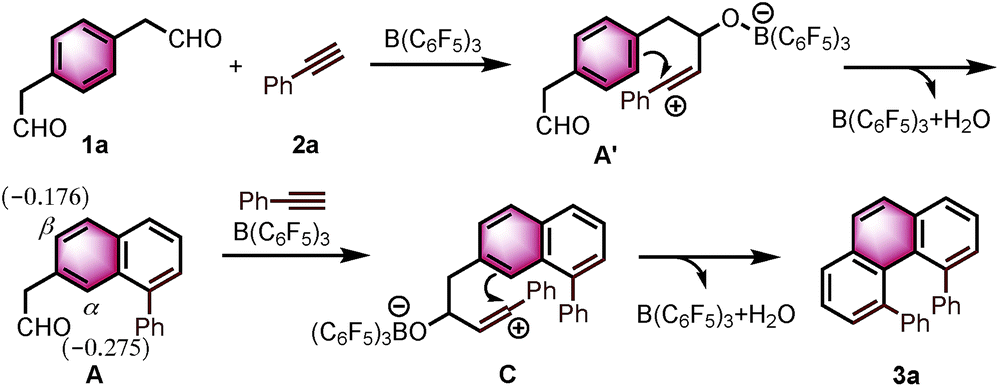 | ||
| Scheme 1 A possible reaction mechanism. | ||
With the optimized conditions in hand, we investigated the substrate scope with respect to the alkyne coupling partner (Scheme 2). Installing electron-donating groups on the para-position of the phenylacetylene aromatic ring did not significantly alter the reaction outcome (3a–d). Interestingly, the reaction of p-ethynyltoluene (containing both terminal and disubstituted alkyne moieties) with 1a selectively gave 3e in 56% yield. With the dimethylamino substituent, the desired product 3f was not formed. This may be due to the formation of N/B frustrated Lewis pairs.15 However, acetamide substituent was tolerated and target product 3g was obtained in 27% yield. Under the optimized reaction conditions, the reaction of 1a with alkyne bearing electron-withdrawing groups at the para-position gave the target products 3h–j in low yield. Further optimization revealed that the yields of 3h–j increased to 44%, 37%, and 34%, respectively, in the presence of 0.25 mL hexafluoroisopropanol (HFIP). This is likely because of the effect of HFIP on the stabilization of vinyl cations (see the proposed mechanism).16 An aryl alkyne bearing a strong electron-withdrawing trifluoromethyl group in the para-position did not afford the desired product 3k. The para-boronic acid pinacol (Bpin) ester-substituted aryl alkyne provided the product 3l in 21% yield. The meta-substituents of the aryl acetylene moiety were also tolerated to give the corresponding products 3m–p. To our delight, the ortho-methyl-substituted aryl alkyne afforded the product 3q in good yield as well. Due to the higher rotational barrier,17 product 3q exists in solution as a mixture of multiple rotamers as confirmed by variable temperature 1H NMR and 1D NOE experiments.18 Even the more hindered substrate (2,4,6-trimethylphenyl)acetylene underwent bisannulation to give the target product 3r in 7% yield. Moreover, the reactions of 1a with heteroaromatic alkynes such as 2-ethynylthiophene and 3-ethynylthiophene gave the products 3s and 3t in 47% and 61%, respectively. Unfortunately, aliphatic alkynes (1-octyne) and methyl acetylenecarboxylate were not tolerated under the reaction conditions and the corresponding products 3u and 3v were not detected, which emphasized the effect of the aromatic moiety of the phenylacetylenes on the stabilization of vinyl cations (see the proposed mechanism). However, the reaction of 1a with ethoxyacetylene provided a trace amount of 4,5-diethoxyphenanthrene (3w), and the mono-annulation product 2-(8-ethoxynaphthalen-2-yl)acetaldehyde was obtained in 8% yield. To show practicality, we ran a 2.0 mmol large-scale experiment with 2b to give 3b in 55% isolated yield (425 mg prepared). To our delight, the 2.0 mmol large-scale reaction of 2l with 1a gave the target product 3l at comparable yield (25%) using 2.0 equivalent BF3·Et2O instead of 20 mol% B(C6F5)3 (for details, see ESI†).
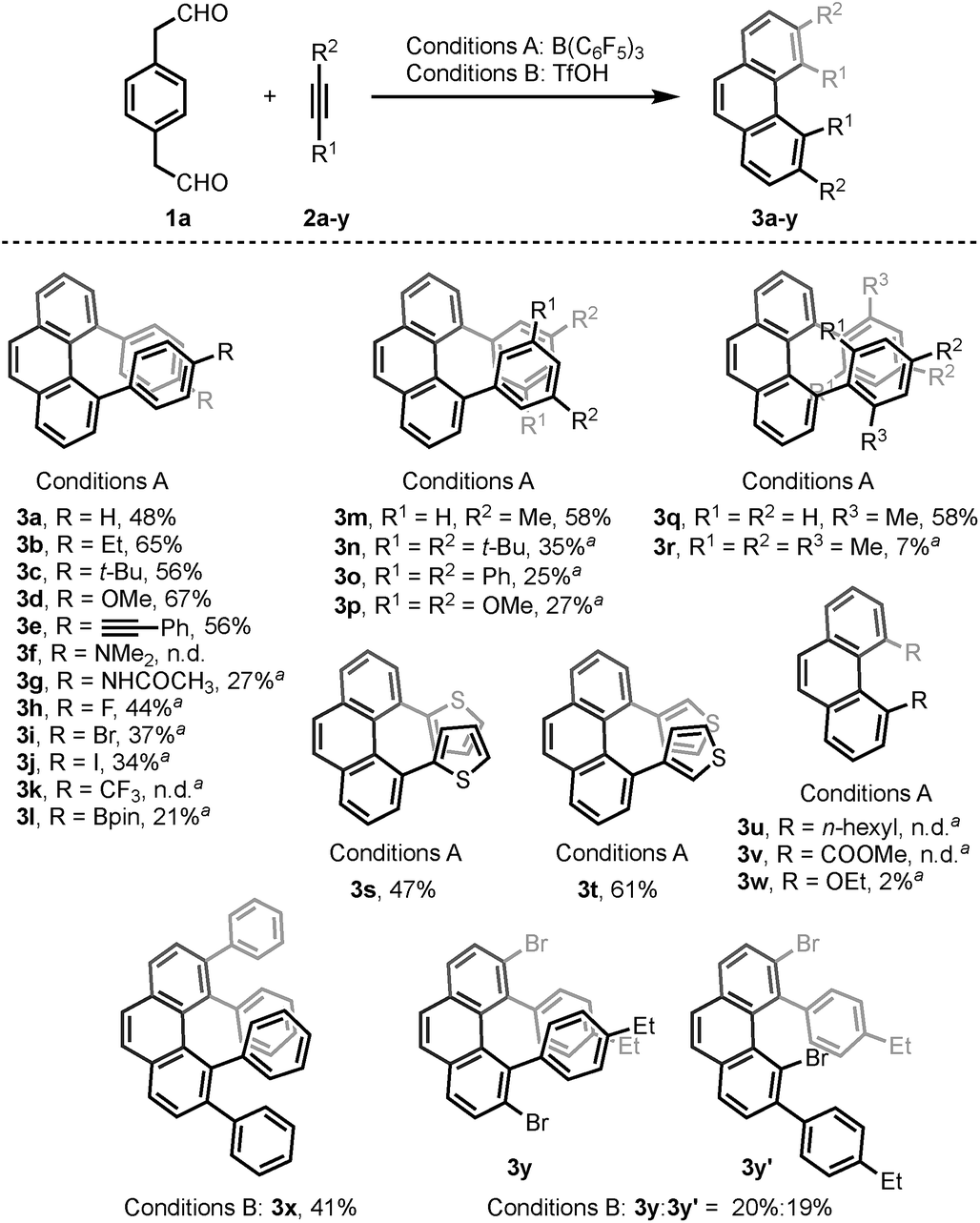 | ||
Scheme 2 The reaction of 1a with alkynes. Reaction conditions: 1a (0.10 mmol), 2 (0.25 mmol). Conditions A: 20 mol% B(C6F5)3/MS4Å, DCM, rt, 15 h. Conditions B: 150 mol% TfOH, CF3Ph/HFIP = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, −30 °C, 15 h. Yields given correspond to isolated yields. Note: aSolvent:DCM/HFIP = 10:1. :1, −30 °C, 15 h. Yields given correspond to isolated yields. Note: aSolvent:DCM/HFIP = 10:1. | ||
The scope of the bisannulation reaction with internal alkyne substrates such as diphenylacetylene is also shown in Scheme 2. Further optimization revealed that this transformation is better conducted in the presence of 150 mol% triflic acid (TfOH) using CF3Ph and HFIP as solvent at −30 °C for 15 h. Sterically hindered 3,4,5,6-tetraphenylphenanthrene (3x) was thus readily synthesized in 41% yield. This protocol could be extended to the use of bromide-substituted phenylacetylene to give a 1.1:1.0 regioisomeric mixture of 3y and 3y′ (for details, see ESI†).
The structures of 3c and 3x were unambiguously determined by X-ray crystallographic analysis (Fig. 2). The torsion angle of the phenanthrene moiety of 3c was 27.5(2)° (measured through the C4–C4a–C4b–C5 torsion). The two para-tert-butyl substituted phenyl rings (ring D and E) are oriented roughly parallel to each other (see Fig. 2a, side view, within 13.2°). On the other hand, the increased steric demands caused by the 3,4,5,6-tetraphenyl substitution in 3x led to greater twisting of the phenanthrene backbone with a C4–C4a–C4b–C5 torsion angle of 35.21(16)°. In addition, the two phenyl rings (ring D and E) are oriented roughly parallel to each other as well (see Fig. 2b, side view, within 11.1°). These nonplanar features of overcrowded phenanthrenes should be beneficial in some of device-oriented applications, such as organic light emitting diodes, where the strong π–π intermolecular interactions cause detrimental effects.
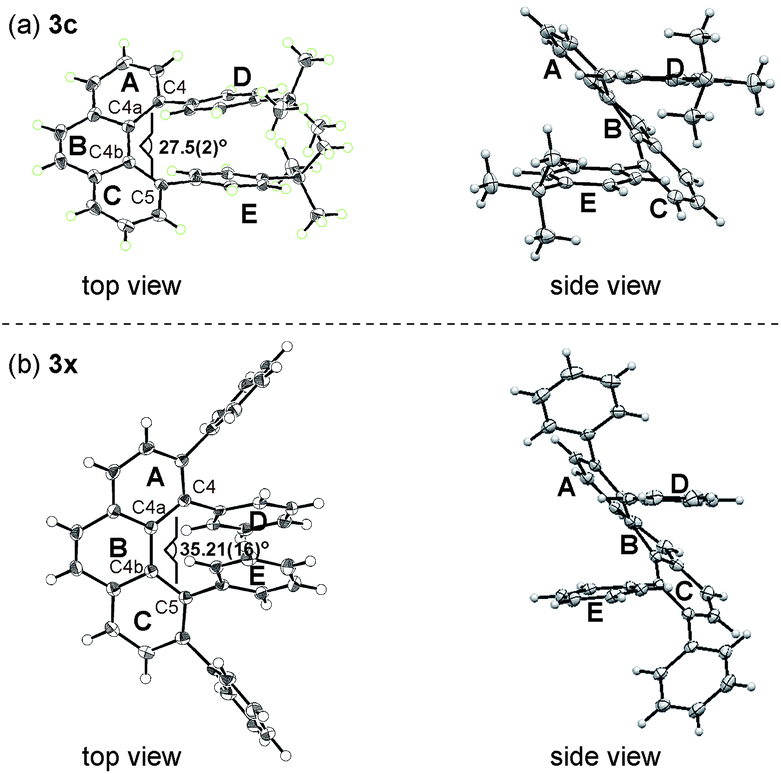 | ||
| Fig. 2 X-ray structure of (a) 3c and (b) 3x at 50% thermal probability. | ||
Furthermore, the chiral resolution of 3b, 3n, and 3x were achieved by HPLC at 25 °C. The racemization of 3b and 3x was observed at elevated temperature (see ESI†). The configurational stability of 4,5-diarylphenanthrene with ortho- or meta-substituents in the aryl moiety (such as 3m–r) are comparatively higher than 3a (Scheme 3).17 A kinetic study of the thermal racemization of 3n was performed, corresponding to a racemization barrier of 126 kJ mol−1 at 100 °C. Using the solution of highly enantioenriched 3n in 1,2-dichloroethane, only a small drop in e.r. was observed after heating at 85 °C for 3 h (from 96.8:3.2 to 94.0:6.0). These results (for details, see ESI†) demonstrate that the configurational stability of these helically chiral compounds can in fact be tuned by judicious substituent choice – an important advantage with respect towards their potential use as chiral materials. The preliminary investigation of the direct asymmetric bisannulation of 1,4-benzenediacetaldehyde with alkynes were conducted as well. However, the use of BINOL-phosphoric acid and DL-10-camphorsulfonic acid failed to give the target products (for details, see ESI†).
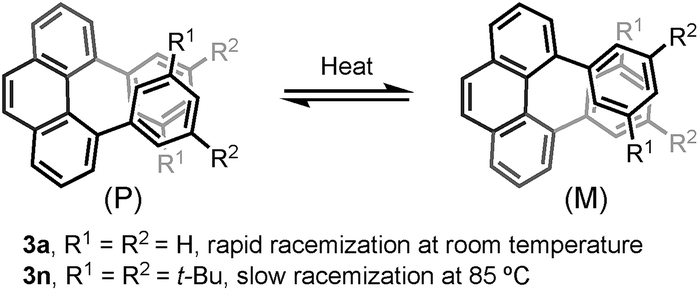 | ||
| Scheme 3 The racemization of 4,5-diarylphenanthrenes. | ||
Interestingly, we observed a different outcome when using other benzenediacetaldehyde positional isomers (e.g., 1,3-benzenediacetaldehyde (1b)) as substrates in the bisannulation reaction (Scheme 4). Under the optimized reaction conditions, the reaction of 1b with terminal alkynes provided unsymmetrical 1,5-disubstituted phenanthrenes 5a and 5b with excellent regioselectivity.19 The reaction with diphenylacetylene still occurred with excellent regioselectivity to give 1,2,5,6-tetraphenylphenanthrene (5c) in 23% yield. The corresponding 2,6-dibromo-1,5-bis(4-ethylphenyl)phenanthrene (5d) was obtained in 46% yield with excellent regioselectivity. Noteworthy, 5d could serve as a valuable building block in cross-coupling reactions to prepare multisubstituted phenanthrenes. The observed outcome is consistent with the reaction going through an intermediate 1,2,6-trisubstituted naphthalene (Scheme 4, Intermediate B), where the α-position (5 position) again presents a more favorable pathway for the second annulation than the β-position (7 position).
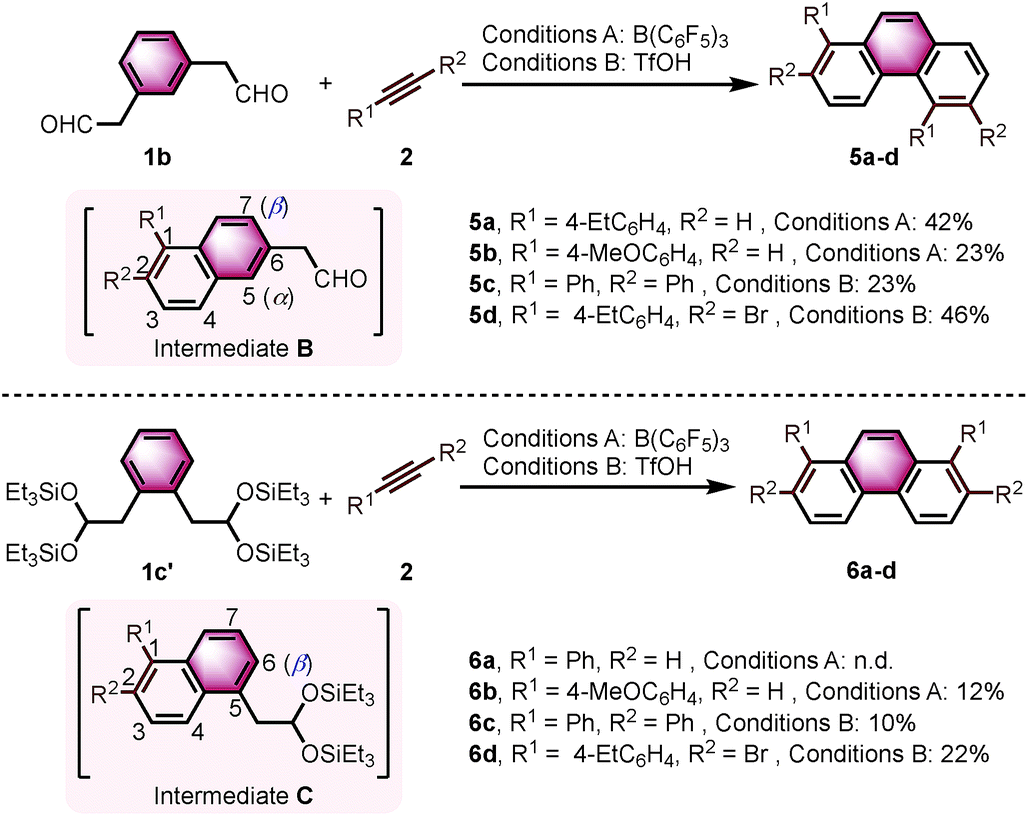 | ||
| Scheme 4 The reaction of 1b and 1c′ with alkynes. Reaction conditions: 1b or 1c′ (0.10 mmol), 2 (0.25 mmol). Conditions A: 20 mol% B(C6F5)3/MS4Å, DCM/HFIP = 10:1, rt, 15 h. Conditions B: 150 mol% TfOH, CF3Ph/HFIP = 10:1, −30 °C, 15 h. Yields given correspond to isolated yields. n.d. = not detected. | ||
We further continued our studies by investigating the scope with 1,2-benzenediacetaldehyde (1c). However, 1c is unstable and cannot be isolated. The disilyl acetal 1c′, which is the precursor of 1c, was used as its synthetic equivalent (Scheme 4). The reactions of 1c′ with 2a did not provide 1,8-diphenylphenanthrene (6a). With more reactive alkyne (4-ethynylanisole), the target product 6b was isolated in 12% yield. Diphenylacetylene was also prone to react to give the target product 1,2,7,8-tetraphenylphenanthrene (6c) in 10% yield. The reaction with bromide-substituted phenylacetylene gave 6d in 22% yield with excellent regioselectivity. The second annulation of the intermediate 1,2,5-trisubstituted naphthalene (Scheme 4, Intermediate C) with alkynes only underwent through the β-position (6 position).12d Some α-aryl-substituted diacetaldehyde and diketones were also tested, but no corresponding products were formed under the current conditions (see ESI†).
The multisubstituted phenanthrenes derived from bisannulation reactions can serve as useful building blocks for the synthesis of organic materials and ligands. To demonstrate the utility of this method, the new helical, chiral diphosphine ligand203z was prepared via the coupling reaction of 3j with HPPh2 (Scheme 5). When treating 3z with [PdCl2(CH3CN)2], interesting binuclear palladium complex 7a was generated in 90% yield.21 The X-ray structural analysis showed that 7a consists of two molecules of 3z with same chiral sense. There is no bonding between two Pd atoms and the complex features a trans-spanned chelating coordination with slightly distorted Cl–Pd–Cl angles (171.38(4)° and 170.76(4)°). The P1–P2 distance (6.9277(12) Å) is much larger than the P1–P4 distance (4.6528(12) Å). Such a large P–P distance and the helical structure of 3z might provide the opportunity to interact with metal ions22 in unique coordination patterns to give a good catalytic activity.23
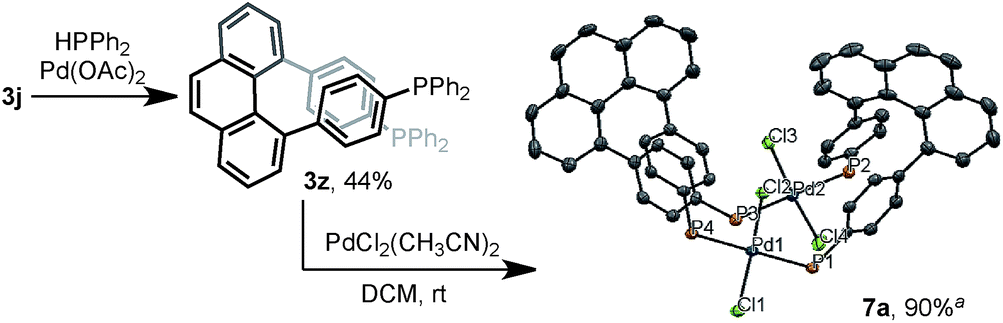 | ||
| Scheme 5 The synthesis of a unique chiral diphosphine ligand 3z and binuclear Pd complex 7a. Note: aX-ray structure of binuclear Pd complex 7a at 50% thermal probability. Hydrogen atoms, phenyl groups, and solvent molecules are omitted for clarity. | ||
Conclusions
In summary, we have developed a concise acid-catalyzed cascade annulation to prepare sterically hindered 4,5-disubstituted phenanthrenes with high regioselectivity from readily available 1,4-benzenediacetaldehyde and terminal aryl alkynes. However, the terminal aryl alkynes with strong electron-withdrawing substituents are not reactive. With internal aryl alkynes, 3,4,5,6-tetrasubstituted phenanthrenes are provided in moderate yield. Furthermore, this method could be extended to reactions of alkynes with isomeric 1,3-benzenediacetaldehyde and 1,2-benzenediacetaldehyde disilyl acetal to provide regioselective multisubstituted phenanthrenes. In addition, a new helical chiral diphosphine ligand was prepared. The reaction presented here is not only interesting in terms of the regioselective annulation, but it is also valuable for the preparation of twisted phenanthrenes and multisubstituted phenanthrenes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ERATO program from JST (JPMJER1302 to K. I.). We thank Dr Yasutomo Segawa, Dr Kenta Kato and Mr Wataru Matsuoka for X-ray analysis, Dr Kenta Kato for DFT calculation, Dr David R. Levine and Dr Chaolumen for constructive criticism of the manuscript, Dr Shin Suzuki and Dr Guillaume Povie for providing some starting materials. JSPS is greatly acknowledged for the postdoctoral fellowship to Y. L. ITbM is supported by the World Premier International Research Center Initiative (WPI), Japan.Notes and references
- (a) J. Li, G. Hu, N. Wang, T. Hu, Q. Wen, P. Lu and Y. Wang, J. Org. Chem., 2013, 78, 3001–3008 CrossRef CAS PubMed; (b) Z. He, X. Xu, X. Zheng, T. Ming and Q. Miao, Chem. Sci., 2013, 4, 4525–4531 RSC; (c) S. Wang, X. Yan, Z. Cheng, H. Zhang, Y. Liu and Y. Wang, Angew. Chem., Int. Ed., 2015, 54, 13068–13072 CrossRef CAS PubMed.
- (a) W. T. Colwell, V. Brown, P. Christie, J. Lange, C. Reece, K. Yamamoto and D. W. Henry, J. Med. Chem., 1972, 15, 771–775 CrossRef CAS PubMed; (b) M. G. Banwell, A. Bezos, C. Burns, I. Kruszelnicki, C. R. Parish, S. Su and M. O. Sydnes, Bioorg. Med. Chem. Lett., 2006, 16, 181–185 CrossRef CAS PubMed; (c) X.-M. Zhou, C.-J. Zheng, L.-S. Gan, G.-Y. Chen, X.-P. Zhang, X.-P. Song, G.-N. Li and C.-G. Sun, J. Nat. Prod., 2016, 79, 1791–1797 CrossRef CAS PubMed.
- (a) F. B. Mallory and C. W. Mallory, Org. React., 1984, 30, 1 CAS; (b) Y.-g. Shi, S. K. Mellerup, K. Yuan, G.-F. Hu, F. Sauriol, T. Peng, N. Wang, P. Chen and S. Wang, Chem. Sci., 2018, 9, 3844–3855 RSC.
- (a) T. Yao, M. A. Campo and R. C. Larock, Org. Lett., 2004, 6, 2677–2680 CrossRef CAS PubMed; (b) V. Mamane, P. Hannen and A. Fürstner, Chem.–Eur. J., 2004, 10, 4556–4575 CrossRef CAS PubMed; (c) C. A. Witham, W. Huang, C.-K. Tsung, J. N. Kuhn, G. A. Somorjai and F. D. Toste, Nat. Chem., 2009, 2, 36–41 CrossRef PubMed; (d) T.-A. Chen, T.-J. Lee, M.-Y. Lin, S. M. A. Sohel, E. W.-G. Diau, S.-F. Lush and R.-S. Liu, Chem.–Eur. J., 2010, 16, 1826–1833 CrossRef CAS PubMed; (e) K. Komeyama, R. Igawa and K. Takaki, Chem. Commun., 2010, 46, 1748–1750 RSC.
- (a) R. C. Larock, M. J. Doty, Q. Tian and J. M. Zenner, J. Org. Chem., 1997, 62, 7536–7537 CrossRef CAS; (b) C. Wang, S. Rakshit and F. Glorius, J. Am. Chem. Soc., 2010, 132, 14006–14008 CrossRef CAS PubMed; (c) A. Matsumoto, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 6557–6559 CrossRef CAS PubMed; (d) K. Ozaki, K. Murai, W. Matsuoka, K. Kawasumi, H. Ito and K. Itami, Angew. Chem., Int. Ed., 2017, 56, 1361–1364 CrossRef CAS PubMed.
- A. Iuliano, P. Piccioli and D. Fabbri, Org. Lett., 2004, 6, 3711–3714 CrossRef CAS PubMed.
- C. C. McAtee, P. S. Riehl and C. S. Schindler, J. Am. Chem. Soc., 2017, 139, 2960–2963 CrossRef CAS PubMed.
- J. E. McMurry, T. Lectka and J. G. Rico, J. Org. Chem., 1989, 54, 3748–3749 CrossRef CAS.
- Y. Xia, Z. Liu, Q. Xiao, P. Qu, R. Ge, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 5714–5717 CrossRef CAS PubMed.
- (a) R. A. Pascal, Chem. Rev., 2006, 106, 4809–4819 CrossRef CAS PubMed; (b) K. K. Wang, Twisted Arenes, in Polyarenes I, ed. J. S. Siegel and Y.-T. Wu, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014, pp. 31–61 Search PubMed; (c) T. Fujikawa, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2016, 138, 3587–3595 CrossRef CAS PubMed; (d) W. Yang, G. Longhi, S. Abbate, A. Lucotti, M. Tommasini, C. Villani, V. J. Catalano, A. O. Lykhin, S. A. Varganov and W. A. Chalifoux, J. Am. Chem. Soc., 2017, 139, 13102–13109 CrossRef CAS PubMed.
- (a) A. H. A. Tinnemans and W. H. Laarhoven, J. Am. Chem. Soc., 1974, 96, 4617–4622 CrossRef CAS; (b) H. Li, J. L. Petersen and K. K. Wang, J. Org. Chem., 2001, 66, 7804–7810 CrossRef CAS PubMed; (c) J. Carreras, G. Gopakumar, L. Gu, A. Gimeno, P. Linowski, J. Petuškova, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2013, 135, 18815–18823 CrossRef CAS PubMed.
- (a) G. S. Viswanathan, M. Wang and C.-J. Li, Angew. Chem., Int. Ed., 2002, 41, 2138–2141 CrossRef CAS; (b) G. W. Kabalka, Y. Ju and Z. Wu, J. Org. Chem., 2003, 68, 7915–7917 CrossRef CAS PubMed; (c) R. Balamurugan and V. Gudla, Org. Lett., 2009, 11, 3116–3119 CrossRef CAS PubMed; (d) X. Bu, L. Hong, R. Liu, J. Hong, Z. Zhang and X. Zhou, Tetrahedron, 2012, 68, 7960–7965 CrossRef CAS; (e) R. Umeda, S. Nishi, A. Kojima, K. Kaiba and Y. Nishiyama, Tetrahedron Lett., 2013, 54, 179–182 CrossRef CAS; (f) S. Xiang, H. Hu, J. Ma, Y. Li, B. Wang, C. Feng, K. Zhao, P. Hu and X. Chen, Sci. China: Chem., 2013, 56, 945–951 CrossRef CAS; (g) S. Ponra, M. R. Vitale, V. Michelet and V. Ratovelomanana-Vidal, J. Org. Chem., 2015, 80, 3250–3257 CrossRef CAS PubMed.
- D. Bézier, S. Park and M. Brookhart, Org. Lett., 2013, 15, 496–499 CrossRef PubMed
 .
. - (a) G. Erker, Dalton Trans., 2005, 1883–1890 RSC; (b) R. L. Melen, Chem. Commun., 2014, 50, 1161–1174 RSC; (c) M. M. Hansmann, A. López-Andarias, E. Rettenmeier, C. Egler-Lucas, F. Rominger, A. S. K. Hashmi and C. Romero-Nieto, Angew. Chem., Int. Ed., 2016, 55, 1196–1199 CrossRef CAS PubMed; (d) Y. Soltani, L. C. Wilkins and R. L. Melen, Angew. Chem., Int. Ed., 2017, 56, 11995–11999 CrossRef CAS PubMed.
- T. Voss, T. Mahdi, E. Otten, R. Fröhlich, G. Kehr, D. W. Stephan and G. Erker, Organometallics, 2012, 31, 2367–2378 CrossRef CAS.
- I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem., 2017, 1, 0088 CrossRef CAS.
- A. H. A. Tinnemans and W. H. Laarhoven, Tetrahedron, 1979, 35, 1537–1541 CrossRef CAS.
- D. X. Hu, P. Grice and S. V. Ley, J. Org. Chem., 2012, 77, 5198–5202 CrossRef CAS PubMed.
- Based on the crude 1H NMR of the reaction mixture, the multisubstituted anthracene was not observed..
- M. T. Reetz, E. W. Beuttenmüller and R. Goddard, Tetrahedron Lett., 1997, 38, 3211–3214 CrossRef CAS.
- (a) L. Chahen, B. Therrien and G. Süss-Fink, J. Organomet. Chem., 2006, 691, 4257–4264 CrossRef CAS; (b) Z. Freixa and P. W. N. M. van Leeuwen, Coord. Chem. Rev., 2008, 252, 1755–1786 CrossRef CAS.
- (a) C. A. Bessel, P. Aggarwal, A. C. Marschilok and K. J. Takeuchi, Chem. Rev., 2001, 101, 1031–1066 CrossRef CAS PubMed; (b) R. C. Smith and J. D. Protasiewicz, Organometallics, 2004, 23, 4215–4222 CrossRef CAS.
- (a) X. Wang, P. Guo, Z. Han, X. Wang, Z. Wang and K. Ding, J. Am. Chem. Soc., 2014, 136, 405–411 CrossRef CAS PubMed; (b) M. T. Reetz and S. Sostmann, J. Organomet. Chem., 2000, 603, 105–109 CrossRef CAS.
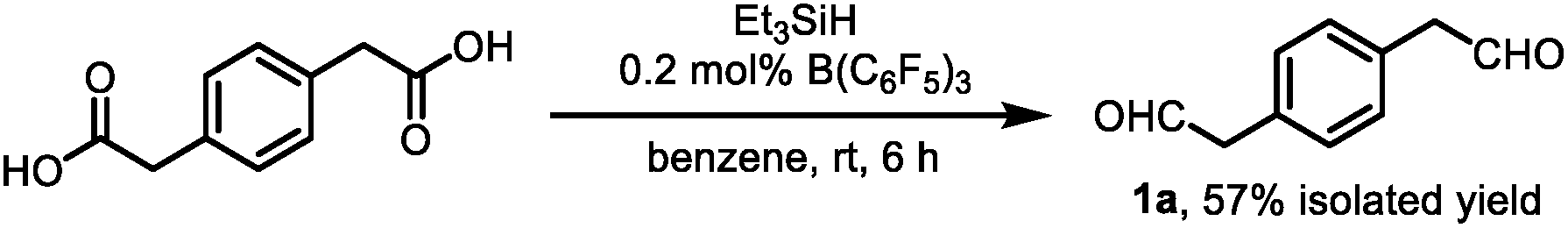
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses, NMR, UV-vis-near IR absorption, CV and crystallographic table. CCDC 1865671 (3c), 1865672 (3x), 1865673 (7a). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00334g |
| This journal is © The Royal Society of Chemistry 2019 |