
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism unravelling for ultrafast and selective 99TcO4− uptake by a radiation-resistant cationic covalent organic framework: a combined radiological experiment and molecular dynamics simulation study†
Linwei
He‡
a,
Shengtang
Liu‡
a,
Long
Chen‡
a,
Xing
Dai
a,
Jie
Li
a,
Mingxing
Zhang
ab,
Fuyin
Ma
a,
Chao
Zhang
c,
Zaixing
Yang
 *a,
Ruhong
Zhou
a,
Zhifang
Chai
a and
Shuao
Wang
*a
*a,
Ruhong
Zhou
a,
Zhifang
Chai
a and
Shuao
Wang
*a
aState Key Laboratory of Radiation Medicine and Protection, School of Radiation Medicine and Protection, Collaborative Innovation Center of Radiological Medicine of Jiangsu Higher Education Institutions, Soochow University, Suzhou 215123, China. E-mail: shuawang@suda.edu.cn; zxyang@suda.edu.cn
bShanghai Institute of Applied Physics, Chinese Academy of Sciences, No. 2019 Jialuo Rd., Jiading Dist., Shanghai, 201800, China
cSchool of Materials Science and Engineering, Anhui University of Science and Technology, Huainan 232001, China
First published on 19th February 2019
Abstract
99Tc is one of the most problematic fission products in the nuclear fuel cycle owing to its large inventory in used nuclear fuel, long half-life, potential radiation hazard, high environmental mobility of its major species 99TcO4−, and its redox-active nature. Ideally, 99TcO4− should be removed at the first stage, when the used fuel rods are dissolved in highly concentrated nitric acid solution, which can substantially reduce its interference with the solvent extraction process through catalytic redox reactions with the key actinides and diminish the chance of discharge into the environment as the volatile species during the waste vitrification process. However, this task cannot be achieved by any of the reported anion-scavenging materials including traditional polymeric anion-exchange resins, inorganic cationic framework materials, and recently developed cationic metal–organic framework materials, because they either are not stable under the extreme conditions of the combined high acidity and strong radiation field or do not possess the required uptake selectivity towards 99TcO4− in the presence of a huge excess of competing anions such as NO3− and SO42−. Herein, we present the first study of 99TcO4− removal under extreme conditions by a two-dimensional conjugated cationic covalent organic framework material, SCU-COF-1. This material exhibits ultrahigh acid stability, great resistance towards both large-dose β and γ irradiation and unprecedented 99TcO4− uptake capabilities including extremely fast sorption kinetics (sorption equilibrium can be reached within 1 min), ultrahigh uptake capacity (702.4 mg g−1 for the surrogate ReO4− at a slightly elevated temperature), and good anion-exchange selectivity towards 99TcO4−. These excellent features endow SCU-COF-1 with the practical capabilities of separating 99TcO4− from both simulant highly acidic fuel reprocessing solutions (3 M nitric acid) and low-activity waste streams at the US legacy nuclear site. The anion-exchange mechanism and the 99TcO4− uptake selectivity are further demonstrated and clearly visualized by the molecular dynamics simulation investigations.
Introduction
Technetium is the lightest element in the periodic table that is comprised of only unstable isotopes such as 97Tc, 98Tc, 99Tc, 99mTc, and 101Tc.1 Among these, 99mTc has been extensively utilized in medical imaging and diagnosis,2 while 99Tc represents a significant issue in the nuclear fuel cycle and the environmental system at the legacy nuclear sites.3 It is estimated that 1990 kg of 99Tc had been generated from 1943 to 1987 since the nuclear weapon production at Hanford, Washington State, a legacy nuclear site in the United States.4 With a high fission yield of 6.06%, about 21 kg of 99Tc is produced in a 1 GWe reactor each year.5 According to the year-end total net electrical capacity determined by International Atomic Energy Agency (IAEA),6 the estimated accumulation of 99Tc from 1998 to 2017 is 154![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 539 kg, and this number would continue to rapidly increase due to active nuclear power production and the fast development of nuclear power plants in several countries, especially China. 99Tc is an extremely long-lived (t1/2 = 2.13 × 105a) β-emitting radionuclide (E = 294 keV) that is both chemical toxic and a radiation hazard.4,7 The inhalation of vapor or dust contaminated with 99Tc can pose a significant cancer risk,5 while 99Tc can accumulate in the mammary tissue and thyroid after digestion.8
539 kg, and this number would continue to rapidly increase due to active nuclear power production and the fast development of nuclear power plants in several countries, especially China. 99Tc is an extremely long-lived (t1/2 = 2.13 × 105a) β-emitting radionuclide (E = 294 keV) that is both chemical toxic and a radiation hazard.4,7 The inhalation of vapor or dust contaminated with 99Tc can pose a significant cancer risk,5 while 99Tc can accumulate in the mammary tissue and thyroid after digestion.8
99Tc is predominately present in the nuclear fuel cycle and in aqueous environments as the pertechnetate anion (99TcO4−). Unlike its congener MnO4−, 99TcO4− is not a strong oxidant, giving rise to its high stability under a variety of environmental conditions. The high symmetry (Td) and low charge density of 99TcO4− lead to its non-complexing nature and extremely high solubility (11.3 M at 20 °C).9 Therefore, 99TcO4− can facilely migrate in the natural water system and its transportation velocity is not affected by the majority of natural materials. Currently, vitrification followed by geological disposal has been established to be the only acceptable strategy for treating the high levels of nuclear waste in several countries.10 However, this strategy is not applicable to 99Tc because it would easily escape from the vitrification glass as the volatile 99Tc2O7.11 This issue along with improper waste management further results in the identified contamination of 99Tc in the groundwater at a variety of legacy nuclear sites, with 99Tc being the major radioactivity contributor in the vadose zone.12 Another significant issue for 99Tc is its catalytic redox capability with uranium, neptunium, and plutonium during used fuel reprocessing (e.g. the plutonium–uranium redox extraction (PUREX) process), hindering the precise valence state control of these key actinides, even when 99Tc is present in very low concentrations.13 Therefore, it would be highly desirable for 99TcO4− to be first separated when used nuclear fuel rods are dissolved in concentrated nitric acid prior to the PUREX process. This would be beneficial not only for the effective extraction of actinides but also in the elimination of 99Tc discharge into the environment during the following waste disposal processes.
Up to now, there have only been a handful of materials, including inorganic cationic framework materials,14 polymeric networks and anion-exchange resins,15 as well as cationic metal–organic frameworks (MOFs),16 reported to exhibit TcO4− separation capability from nuclear waste solutions. Among these, crystalline inorganic cationic framework materials, such as layered double hydroxides (LDHs),14h,i Y2(OH)5Cl,14f and NDTB-114a suffer from the clear demerits of poor selectivity, low capacity and slow uptake kinetics. Especially, poor selectivity makes these materials inapplicable because, either in nuclear waste solutions or natural water systems, coexisting anions such as NO3−, SO42−, CO32−, and PO43− that can induce anion-exchange competition with TcO4− are all present in 100- to 10000-fold of excess. Commercially available polymeric anion-exchange resins show a notable advantage in 99TcO4− uptake selectivity, but their poor radiation resistance impedes their practical applications in the radiological field especially in used fuel repossessing, where strong radiation fields (high fluxes of α, γ, β, and neutron irradiations) are present at extremely high dose rates. It was reported that the 99TcO4− uptake capacity would gradually decrease as the receiving radiation doses increase.15f,17 This critical issue is further amplified given that the 99TcO4− uptake kinetics by these materials are rather slow, leading to a significantly higher dosage received during the anion-exchange process. The recently developed systems based on cationic MOFs do offer solutions to the aforementioned drawbacks and find extensive application potentials in the remediation of 99TcO4− from contaminated water systems under relatively neutral pHs.16 However, none of these cationic MOFs can survive the highly acidic conditions (e.g. 3 M nitric acid) that are required for used fuel reprocessing.
Covalent organic frameworks (COFs) are a class of emerging crystalline polymers with periodic structures and well-defined nanopores.18 Importantly, all these structural features are predictable and highly tunable and can be tailored for specific applications, similar to the MOF system. Different from the common COFs with neutral skeletons, ionic COFs (iCOFs) are a subgroup of COFs with charged linkages in the framework or attachments in the side chain, which would promote particular properties such as selective ion-exchange, compared with neutral COFs.19 We initially proposed that the system of iCOFs may overcome the challenge for TcO4− separation under the combined extreme conditions of high acidity, strong radiation field, and high ionic strength for several reasons. First, compared with the majority of MOF materials, COFs exhibit clear elevations in the stabilities under different types of extreme conditions such as high acidity, alkalinity, and salinity, owing to the increased robustness of the covalent bond-based linkages for building the structure.18d,e,g Second, many COFs are highly conjugated and can substantially stabilize the radical-based intermediates generated by radiation, a merit not possessed by traditional polymeric anion-exchange resins. This may lead to excellent radiation resistance for these COF materials, one of the most important prerequisites for nuclear applications. Third, the porous nature and ordered channels may aid in the rapid transportation of 99TcO4− in the COF structure, which is advantageous for accelerating the anion-exchange process, in comparison to amorphous polymeric materials. Finally, the local hydrophobic environment in the COFs is expected to enhance the uptake selectivity for relatively hydrophobic 99TcO4−, compared to coexisting anions with higher charge densities, a design rule that has already been applied in selective polymeric anion-exchange resins.15a Recently, two 3D cationic COF materials 3D-ICOF-1 and 3D-ICOF-2 were tested for the removal of MnO4− under neutral conditions by Qiu et al.,19f which initially suggests the promise of 99TcO4− remediation using these types of materials, although the capability may be overestimated owing to the partial chemical reduction of MnO4− during the sorption experiment. Despite that, to the best of our knowledge, there has been no study on the separation and remediation of 99TcO4− by COFs through the direct radiological test.
iCOFs are in general very challenging to build because the charge repulsion between the adjacent building blocks impedes their ordered connection, often leading to poor crystallinity or an amorphous structure.19g Herein, we provide a new strategy to constitute cationic COFs with good crystallinity based on positively charged linkers and neutral nodes via irreversible tautomerism. The synthesized two-dimensional viologen-based iCOF (denominated as SCU-COF-1) displays an unusual ABC-staggered stacking model and unprecedented 99TcO4− uptake capability including large uptake capability, fast sorption kinetics, high anion-exchange selectivity towards 99TcO4−, great acid stability, and decent radiation resistance. These excellent features endow SCU-COF-1 with the promising ability to remove 99TcO4− from 3 M nitric acid solution, a key step for potentially establishing a new process aiming at solving the “technetium issue” in the nuclear fuel cycle.
Results and discussion
SCU-COF-1 is synthesized from an aminated viologen and commercially available 2,4,6-triformylphloroglucinol (Tp). After acid-catalyzed solvothermal synthesis and tautomerization from the enol form to the keto form,20 the dark brown crystalline product is obtained (Fig. 1a). The scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of SCU-COF-1 show the layered structure of the material (Fig. S1 and S2†).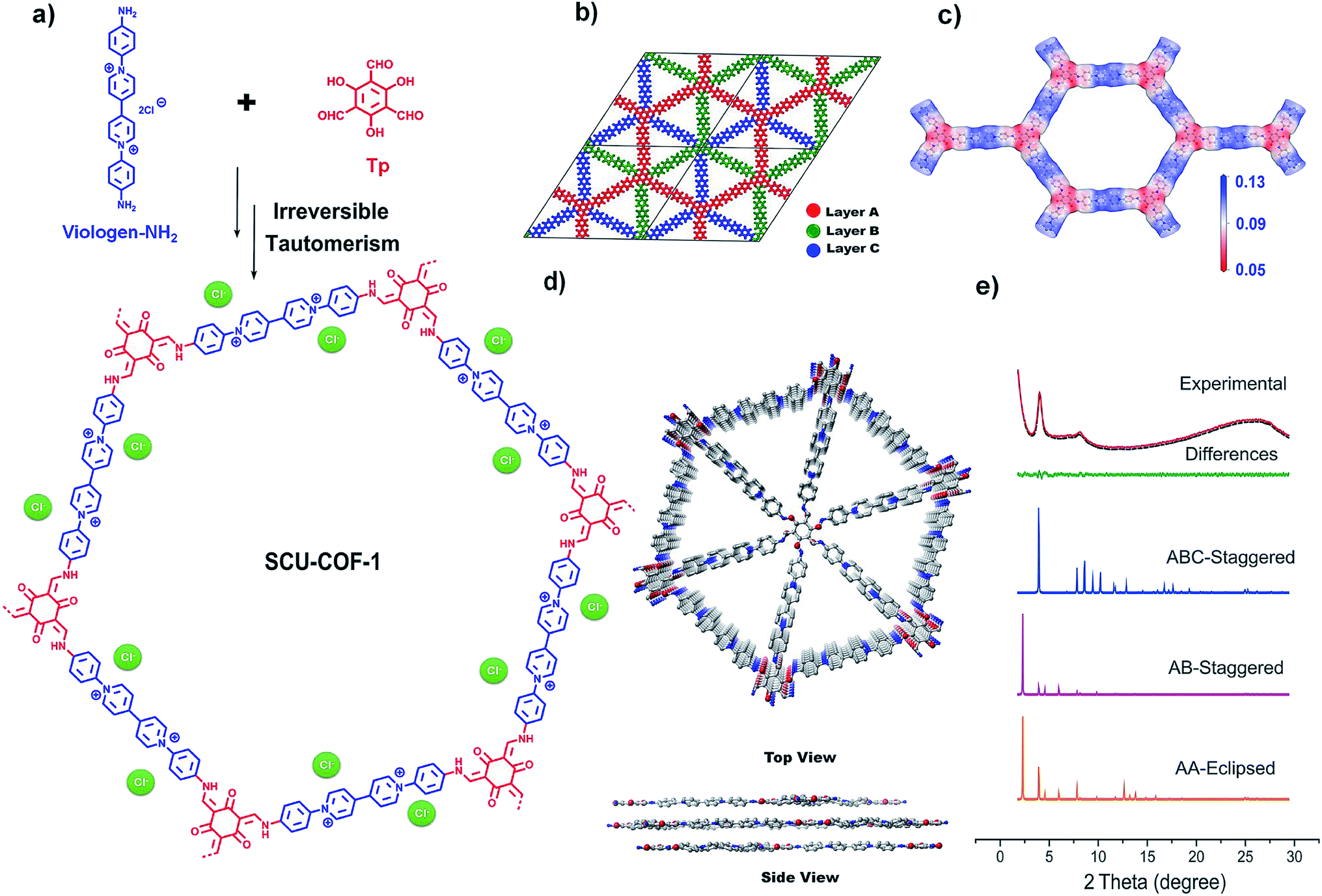 | ||
| Fig. 1 (a) Synthesis of SCU-COF-1. (b) The expanded R3 symmetric unit cell matching with the ABC-staggered stacking model (red for layer A, green for layer B, and blue for layer C). (c) Electrostatic potential distribution mapping of SCU-COF-1. (d) Top view and side view of SCU-COF-1 (the chloride ions and hydrogen atoms are omitted for clarity). (e) Experimental PXRD pattern of SCU-COF-1 (red line), Pawley refined pattern (black dots), difference curve between experimental and refined pattern (green line), AA-eclipsed pattern (orange line), AB-staggered pattern (purple line), and ABC-staggered pattern (blue line). | ||
The powder X-ray diffraction (PXRD) pattern of SCU-COF-1 (Fig. 1e) identifies an ordered structure, where a strong low angle peak at 2θ = 4.02° is observed. Two other prominent peaks at 8.23° and 25.59° were also identified. The peak at 4.02° corresponds to the (100) plane of the structure. The broad feature at 8.23° is recognized as a superposition of peaks at 7.85° and 8.60°, which are attributed to the (110) and (200) planes, respectively. Additionally, the presence of another broad peak at 25.59°, which should be assigned to the (001) plane, suggests highly ordered π–π stacking existing between the adjacent layers and the good planarity of the 2D layers.21
To obtain the simulated crystal structure of SCU-COF-1, theoretical simulations were conducted using Materials Studio. Three stacking models were generated and optimized: the eclipsed stacking (AA-stacking) model, the AB-staggered stacking model, and the ABC-staggered stacking model (see ESI† for details). The simulated PXRD patterns corresponding to the three different models are shown in Fig. 1e for comparison. The experimental pattern matches well with the simulated pattern derived from the unique ABC-stacking model. The features of stacking are shown in the top and side views of SCU-COF-1 (Fig. 1d). In order to distinguish the three distinct layers more clearly, we marked out layer A, layer B, and layer C with different colours in a periodic unit cell, respectively (Fig. 1b). After DFT optimization and Pawley refinement with good agreement factors (Rp = 1.55%, Rwp = 1.98%), we verified the structure in the R3 space group with the unit cell parameters of a = b = 43.47 Å, c = 10.77 Å, α = β = 90°, γ = 120°. In contrast, the simulated patterns of the AA-stacking model and the AB-staggered stacking model in the P6 space group clearly deviate from the experimental pattern.
Overall, the spatial structure of SCU-COF-1 can be described as parts of two reacted ligands being connected sequentially into a flat cationic 2D layer with a large hexagonal channel (∼4.34 nm). These cationic layers are further stacked via π–π interaction in the ABC-staggered manner, which in fact minimizes the charge repulsion between the adjacent layers. A series of shrunken triangular channels (∼1.44 nm) along the c-axis can be still observed after the stacking. We further simulated the electrostatic potential (ESP) distribution of the cationic layer (Fig. 1c). The results show that the regions close to the N atoms in the pyridine rings provide the most positive ESP (blue areas in the hexatomic ring), which are appropriate for accommodating the charge-balancing Cl− anion, whose existence can be identified through EDS analysis (Fig. S3†). In addition, the positive charges are delocalized on the pyridine ring, which is expected to play a leading role in the anion-exchange process.
The porosity of SCU-COF-1 is probed by the nitrogen gas adsorption isotherm measured at 77 K (Fig. 2a, red dots and lines). The calculated Brunauer–Emmett–Teller (BET) surface area of 22.98 m2 g−1 indicates moderate porosity, which partially originates from the staggered stacking of layers that may hinder the efficient transportation of nitrogen in the structure. A similar phenomenon was observed in other 2D COF materials with the staggered stacking.19h,j,21 On the other hand, the presence of charge-balancing Cl− in the pore may further block the entrance of guest gas molecules. However, this relatively small surface area is not an issue for rapid anion-exchange as discussed below. In addition, the nonlocal density functional theory (NLDFT) analysis gives an average pore width of 1.37 nm, which is in good agreement with the simulated crystal structure of SCU-COF-1 (Fig. 2b).
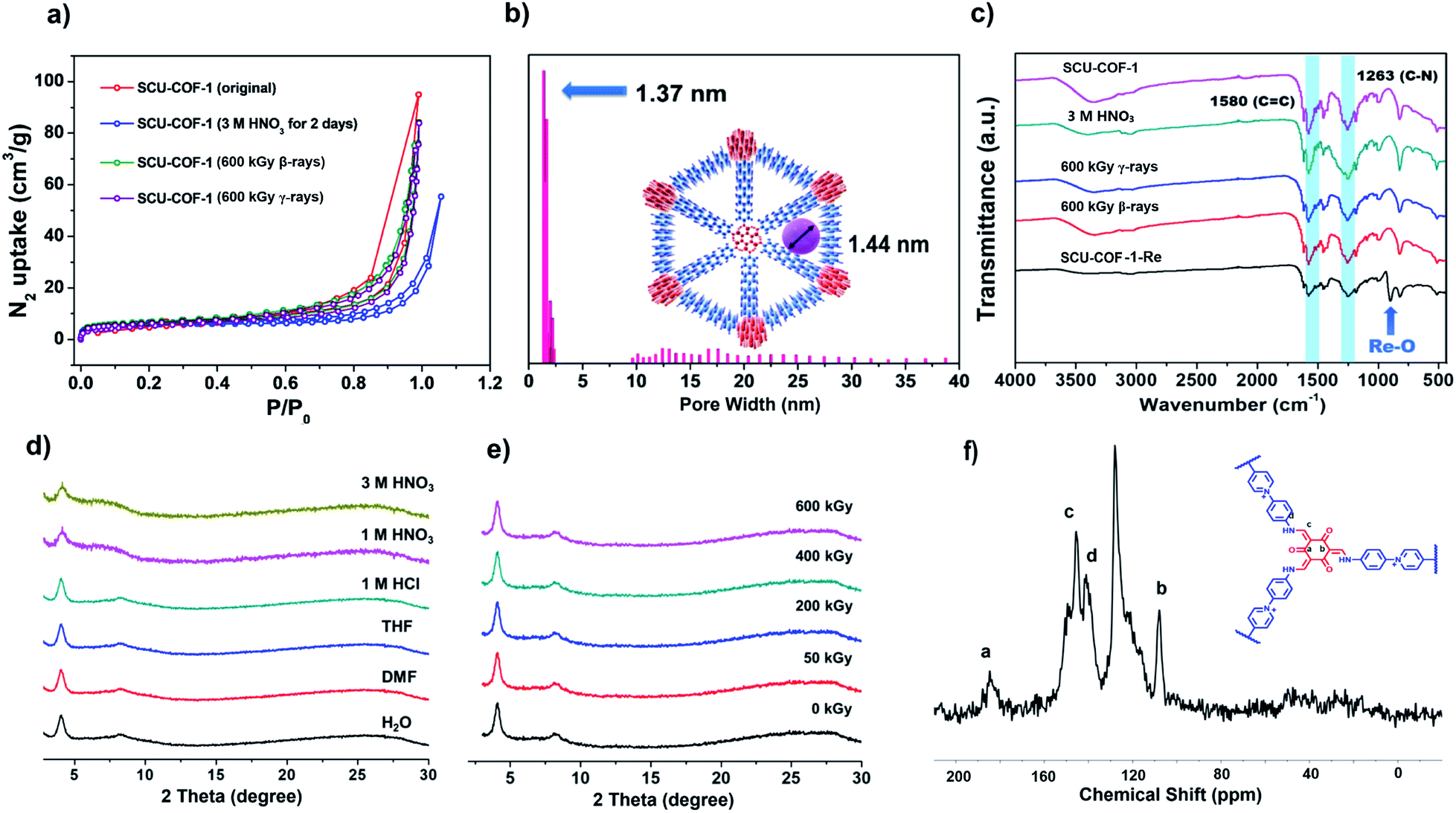 | ||
| Fig. 2 (a) N2 sorption isotherms of SCU-COF-1 after different treatments with 3 M HNO3, 600 kGy β-rays, and 600 kGy γ-rays at 77 K. (b) Pore-size distribution of SCU-COF-1 calculated by NLDFT modelling based on N2 adsorption data. (c) IR spectra of SCU-COF-1 and SCU-COF-1 after treatment with 3 M HNO3, 600 kGy γ-irradiation, 600 kGy β-irradiation, and stock solution containing ReO4−. (d) PXRD patterns of SCU-COF-1 after treatments in various solvents and acids for 48 h. (e) PXRD patterns of SCU-COF-1 after being irradiated with various doses of β-irradiation (50 kGy, 200 kGy, 400 kGy, 600 kGy). (f) 13C solid NMR spectrum of SCU-COF-1. | ||
To elevate the thermal stability of SCU-COF-1, thermogravimetric analysis (TGA) was measured from 25 °C to 900 °C in N2 atmosphere. The weight loss before 100 °C could be assigned to the loss of lattice water, and SCU-COF-1 was thermally stable up to 225 °C (Fig. S4†). The chemical stabilities of SCU-COF-1 were investigated by dispersing fresh samples in various solvents or acids including H2O, DMF, THF, aqueous HCl (1 M), aqueous HNO3 (1 M and 3 M), and aqueous NaOH (1 M) solutions for 48 h. In various solvents (H2O, DMF, THF) or 1 M HCl, the PXRD patterns were almost identical to that of the original sample, indicating that SCU-COF-1 can retain its crystalline structure in these solvents. Even in 1 M HNO3 and 3 M HNO3, the crystallinity of SCU-COF-1 slightly decreased but maintained the ordered structure (Fig. 2d). The unchanged characteristic vibrational peaks in the IR spectra (Fig. 2c) and a BET surface area of 18.25 m2 g−1 (Fig. 2a, blue dots and lines) for samples treated with 3 M HNO3 further underscored the acid stabilities of SCU-COF-1. Note that the slightly reduced surface area may partially originate from the larger ionic radii of NO3− exchanged into the structure compared with the original Cl−, which may further block the transportation pathway of gas molecules. SCU-COF-1 is unstable in highly basic solutions (1 M NaOH). This can be explained by the presence of the cationic pyridine hexatomic rings, which are easily attacked by the hydroxyl as the nucleophile in solution.22 More impressively, the highly conjugated structure of SCU-COF-1 protects the functional cationic pyridinium rings from high doses (200 kGy, 400 kGy, and 600 kGy) of β-irradiation provided by an electron accelerator (1.5 MeV, dose rate of 2250 kGy h−1) or γ-irradiation from a 60Co source (92.42 PBq, dose rate of 3.125 kGy h−1). The PXRD patterns of the irradiated samples confirm that SCU-COF-1 fully retains its crystallinity after irradiation (Fig. 2e and S5†). In addition, the nearly unchanged IR spectra (Fig. 2c) and BET surface areas after being exposed to β- (20.01 m2 g−1) or γ- (19.00 m2 g−1) irradiations (Fig. 2a, green and purple dots) of SCU-COF-1 synergistically demonstrate its excellent radiation resistance. Moreover, the radiation resistance can be further confirmed by the anion-exchange capacity experiment using the irradiated sample (see discussion below). In the Fourier-transform infrared (FT-IR) spectrum, the strong peaks at 1580 and 1263 cm−1 originate from the stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–N, respectively, confirming the existence of the cis-keto form (Fig. 2c). Furthermore, the 13C nuclear magnetic resonance (NMR) spectrum exhibits four characteristic peaks. The signal at ∼185 ppm, which is assigned as the C atoms of ketone, demonstrates the formation of the cis-keto structure of SCU-COF-1 (Fig. 2f).20b
C and C–N, respectively, confirming the existence of the cis-keto form (Fig. 2c). Furthermore, the 13C nuclear magnetic resonance (NMR) spectrum exhibits four characteristic peaks. The signal at ∼185 ppm, which is assigned as the C atoms of ketone, demonstrates the formation of the cis-keto structure of SCU-COF-1 (Fig. 2f).20b
To investigate the sorption kinetics of SCU-COF-1 towards 99TcO4− or ReO4− as the non-radioactive surrogate of 99TcO4− owing to their almost identical charge densities and therefore similar anion-exchange properties,4 20 mg of solid materials was added into 20 mL of aqueous solution containing 28 ppm ReO4− or 14 ppm 99TcO4− at 300 K. As shown in Fig. 3a, the concentrations of 99TcO4− as a function of contact time were measured by its characteristic peak at 290 nm in the UV-vis spectra. In addition, the kinetics curve can be also confirmed by measuring the time-dependent radioactivity of the solution through a liquid scintillation counting (LSC) technique (Fig. 3b). The results show that SCU-COF-1 can almost quantitatively remove 99TcO4− from solution within an extremely short of time of ∼30 s, which is, in fact, the shortest time required for experimental operation. Therefore, it is difficult to establish a sorption kinetics model, as the first data point has already reached the sorption equilibrium. The results of ReO4− removal kinetics experiment are identical with those of 99TcO4− (Fig. 3c). This ultrafast anion-exchange kinetics sets SCU-COF-1 as the record-holding material along with the recently reported SCU-CPN-1 and is significantly faster than those of other types of anion-exchange materials (Table S1†). For inorganic cationic framework materials and commercial polymeric anion-exchange resins, the anion-exchange equilibrium would require at least hours (up to days) to establish. For example, the first purely inorganic 3D cationic framework NDTB-1 can remove approximately 72% of ReO4−/99TcO4− in more than 36 h.14b The anion-exchange resins Purolite A532E and A530E resins15a require at least 2.5 h to reach equilibrium. Only 32% and 38% of ReO4−, respectively, can be removed from the solution in a short time (5 min).15c Recently developed cationic MOFs show progress in this regards, but still require at least 10 min to quantitatively remove 99TcO4− from solution.16b We propose that this excellent feature is a result of ordered channels in the structure of SCU-COF-1 that allows for the efficient transportation of guest anions and the hydrophobic pore that leads to the fast exchange-out of relatively hydrophilic Cl− back into the aqueous solution (more profound discussions are provided in the section of the sorption mechanism). The advantage in anion-exchange kinetics is the benefit of the rapid remediation of anionic radionuclide contamination during an emergency event as well as reducing the received radiation dosage when dealing with highly radioactive systems such as used fuel reprocessing to minimize the radiation-induced degradation of materials. Moreover, the distribution coefficient (Kd) of SCU-COF-1 calculated from the ReO4− sorption kinetics experiment is 3.89 × 105 mL g−1, which is higher than most reported materials (Table S2†), exhibiting a promising removal depth for the remediation task. FT-IR (Fig. 2c) and EDS mapping measurements (Fig. S3†) also confirm the successful substitution of Cl− by ReO4− in SCU-COF-1. The sorption isotherm experiment was carried out at 300 K to gain insight into the anion-exchange capacity of SCU-COF-1 for trapping ReO4−/99TcO4−. From the curve, the saturated sorption capacity calculated based on the Langmuir model is 367.6 mg ReO4− per g sorbents (Fig. 3d and Table S3†). This value is notably lower than the theoretic sorption capacity (872.7 mg g−1) calculated by assuming Cl− anions are completely substituted, suggesting the presence of a kinetic barrier for the complete anion-exchange, which can be confirmed by the theory study discussed below. We therefore checked the sorption capacity at an elevated temperature of 373 K. Upon heating and refluxing for 24 h, the maximum ReO4− uptake capacity reached 702.4 mg g−1, as the actual experimental value, being quite close to the theoretic value. This sets SCU-COF-1 as a significant anion-exchange material with one of the highest reported exchange capacities. In comparison with inorganic materials such as Yb3O(OH)6Cl (48.6 mg g−1),14d,16c NDTB-1 (49.4 mg g−1)14a,16c and LDH (130.2 mg g−1),14h,15a polymeric anion-exchange resins such as Purolite A532E (446 mg g−1),16a,c and cationic MOFs including UiO-66-NH3+ (159 mg g−1),16e SCU-100 (541 mg g−1),16c and SCU-101 (247 mg g−1),16b SCU-COF-1 exhibits a notably larger sorption capacity towards ReO4−.
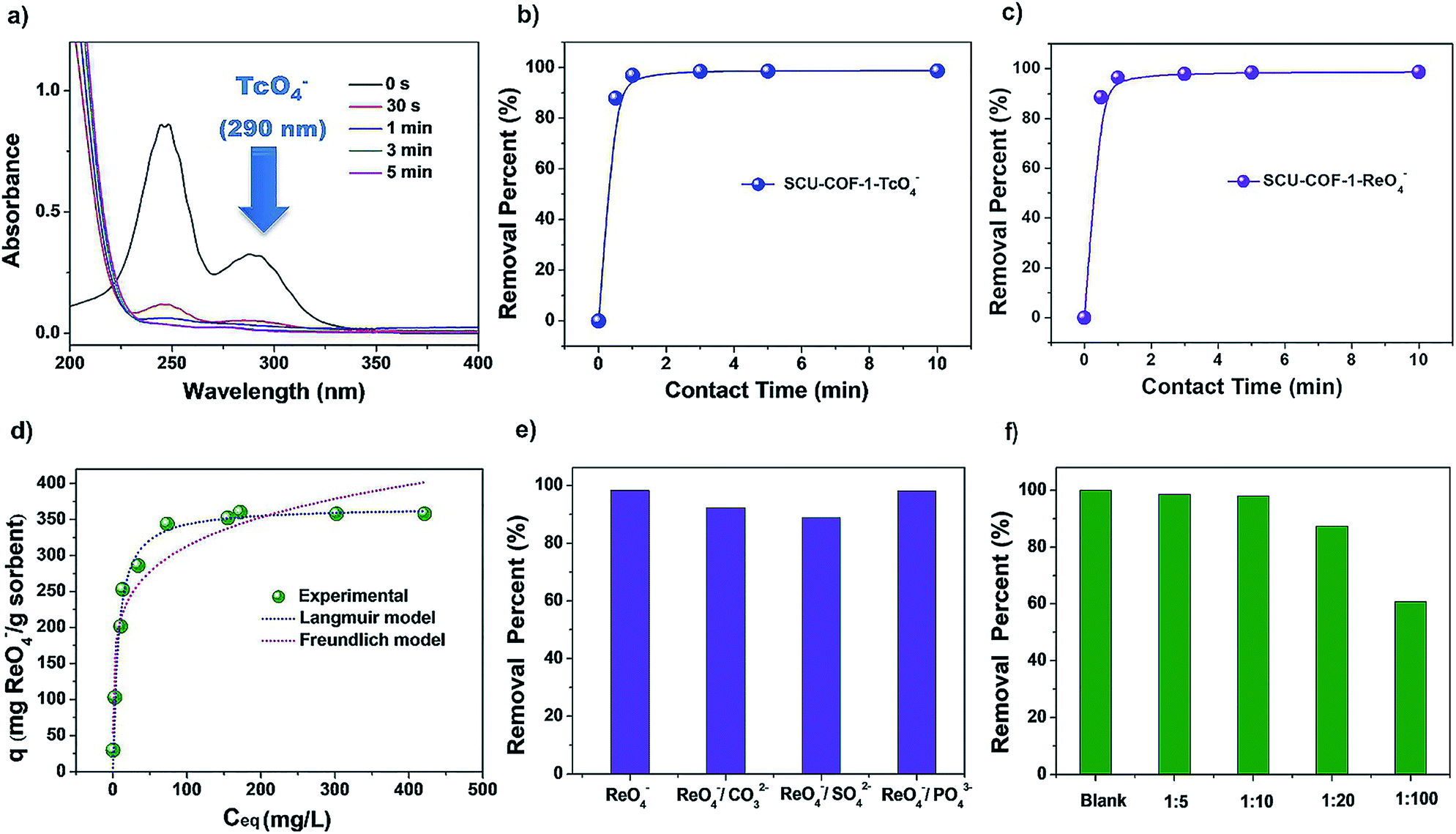 | ||
| Fig. 3 (a) UV-vis absorption spectra of 99TcO4− with various contact times at 300 K (initial concentration of TcO4− = 14 ppm). (b) Sorption kinetics curve of 99TcO4− by SCU-COF-1 at 300 K (initial concentration of 99TcO4− = 14 ppm). (c) Sorption kinetics curve of ReO4− by SCU-COF-1 at 300 K (initial concentration of ReO4− = 28 ppm). (d) Sorption isotherm of SCU-COF-1 for ReO4− uptake at 300 K. (e) Effect of competing oxo-anions on the removal rate of ReO4− (molar ratio = 1:1, initial concentration of ReO4− = 28 ppm). (f) Effect of excessive NO3−on the removal rate of ReO4− (molar ratio = 1:5, 1:10, 1:20, 1:100, initial concentration of ReO4− = 28 ppm). | ||
Since anions such as NO3− and SO42− often coexist in huge excess with 99TcO4− both in highly radioactive waste solutions (e.g. nitric acid solution) and under environmentally relevant conditions (such as in groundwater), the anion-exchange selectivity of sorbents is critically required for practical applications. Because those anions with higher charge density often exhibit stronger electrostatic interactions with the cationic host, they are often captured with priority by typical anion-exchange materials, which is known as Hofmeister bias selectivity, unless other types of driving forces are present.23 SO42−, CO32− and PO43− (one equivalent) were therefore chosen to evaluate the selectivity for ReO4− removal (Fig. 3e). In competing with different anions, SCU-COF-1 exhibits excellent uptake selectivity, and the removal percentages remain in the range of 85% to 99%. Even in the presence of a high concentration of NO3− (NO3−:ReO4− = 100:1, molar ratio), the removal percentage of ReO4− still reached 60% (Fig. 3f). The high selectivity for ReO4− is likely owing to the hydrophobic nature of the COF skeleton, which provides a remarkable affinity for relatively hydrophobic anions with lower charge densities.15c Some commercial resins, for example SuperLig-639,24 with a high uptake capacity towards 99TcO4−/ReO4−, have obvious drawbacks including low selectivity in the presence of a large excess of NO3− anions.3 More impressively, experiments for removing radioactive 99TcO4− were conducted in a simulated Hanford low-activity waste (LAW) melter recycle stream (Table S4†), which contains far higher amounts of NO3−, NO2− and Cl− (at least 300-fold in excess for each anion) than 99TcO4−. The results show that SCU-COF-1 can remove 20.9% and 62.8% of 99TcO4− at a solid/liquid ratio of 1 and 5, respectively (Table S5†).
To probe the uptake capability of SCU-COF-1 in acidic and alkaline conditions, solutions containing 400 ppm ReO4− in various pH values were used in the experiments. The uptake capacities of SCU-COF-1 were almost unchanged through a wide pH range from 4 to 10 (Fig. 4a). More significantly, adsorption experiments in extreme conditions were conducted to estimate its potential application in highly acidic conditions that is relevant to used fuel reprocessing. We further prepared a 1 M HNO3 solution containing ReO4− (c0 = 28 ppm or c0 = 338 ppm) and a 3 M HNO3 solution containing ReO4− (c0 = 253 ppm, NO3−:ReO4− = 2922, molar ratio). The latter system simulates the real situation of used fuel reprocessing. Impressively SCU-COF-1 exhibits superior extraction performance, as approximately 40% of ReO4− are removed at a solid–liquid ratio of 1:20 in the solution containing 338 ppm ReO4− (Fig. 4b). Under the conditions of 3 M HNO3, SCU-COF-1 can still remove 52.3% of ReO4− at a solid–liquid ratio of 1:40 in the solution (Fig. 4c). These significant results indicate the SCU-COF-1 are indeed feasible for 99Tc separation at the first stage of used fuel reprocessing prior to the PUREX process.
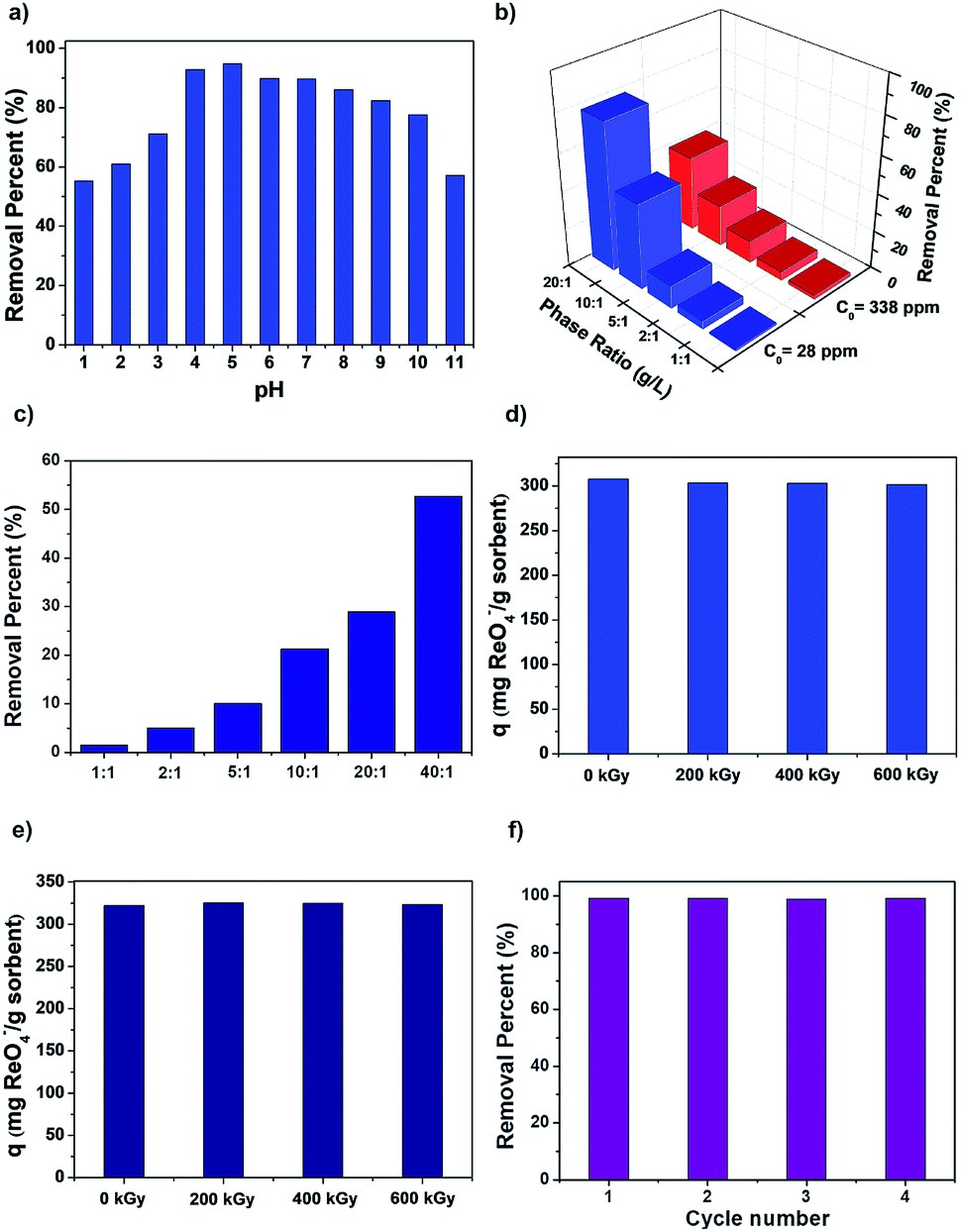 | ||
| Fig. 4 (a) pH effect on the sorption capacity of ReO4− by SCU-COF-1 (initial concentration of ReO4− = 400 ppm). (b) Removal experiment of ReO4− by SCU-COF-1 with various solid–liquid ratios in 1 M HNO3 solution (initial concentration of ReO4− = 28 ppm or 338 ppm). (c) Removal experiment of ReO4− by SCU-COF-1 with various solid–liquid ratios in 3 M HNO3 solution (initial concentration of ReO4− = 253 ppm). (d) Sorption properties of SCU-COF-1 after being irradiated with different doses of β-irradiation (initial concentration of ReO4− = 2000 ppm). (e) Sorption properties of SCU-COF-1 after being irradiated with different doses of γ-irradiation (initial concentration of ReO4− = 2000 ppm). (f) Reversibility of SCU-COF-1 for removing ReO4− at pH 7 (initial concentration of ReO4− = 28 ppm). | ||
After treatment in different doses of β- or γ-irradiation, the uptake for ReO4− by SCU-COF-1 is uninfluenced. The removal capacity at room temperature for ReO4− was completely unchanged after irradiation (Fig. 4d and e). In comparison, two commercial resins Purolite A532E and A530E have reductions of 9% and 18% of sorption capacities after exposure to 600 kGy β-irradiation (or γ-irradiation), respectively.15c Additionally, SCU-COF-1 is also a reversible anion-exchange material for multiple sorption/desorption trials. More than 95% of ReO4− could be desorbed back to solution after treatment with 3 M NaCl solution at 300 K (Table S6†). Even after 4 cycles of sorption/desorption, the sorption performance of SCU-COF-1 was not affected (Fig. 4f). More impressively, the COF can keep almost the same regeneration property after removing ReO4− in 3 M HNO3 (Fig. S6†), which can significantly reduce the cost for practical applications.
We further explored the adsorption dynamics of 99TcO4− anions into the structure of SCU-COF-1 and the underlying uptake mechanism using all-atom molecular dynamics (MD) simulations (Fig. 5a). Impressively, after placing the solid structure of SCU-COF-1 into the aqueous 99TcO4− solution, a large number of 99TcO4− anions initially diffused in aqueous solution were rapidly sorbed into the interior of SCU-COF-1. A video revealing the anion-exchange process is provided as a ESI file.† During this process, many Cl− anions originally dwelling in the pores of SCU-COF-1 were desorbed from the solid structure (Fig. 5b, also see the Movie S1 in ESI† for more intuitive illustration). To quantitatively describe this dynamics process, the adsorption rate of 99TcO4− anions from the external solution environment (Fig. 5c red curve) and the residence rate of original Cl− anions in SCU-COF-1 (Fig. 5c green curve) along with the simulation time were calculated. As shown in Fig. 5c, a very clear crossover occurs at ∼26 ns. After this time point, the amount of 99TcO4− anions entering SCU-COF-1 exceeds the amount of Cl− anions that still remain in SCU-COF-1. After ∼40 ns, a dynamic equilibrium is reached, i.e., the 99TcO4− adsorption rate is nearly stabilized at a constant value of (60.6 ± 2.3)%. Meanwhile, the reserving rate of Cl− anions is nearly unchanged at (34.1 ± 2.0)%. In addition, another control simulation in pure water demonstrates that in the absence of 99TcO4−, nearly (87.2 ± 1.4)% of Cl− anions could be retained in SCU-COF-1 throughout the full simulation period (Fig. 5c blue curve). This suggests that the sorption of 99TcO4− anions initiates and further complicates the following anion-exchange process.
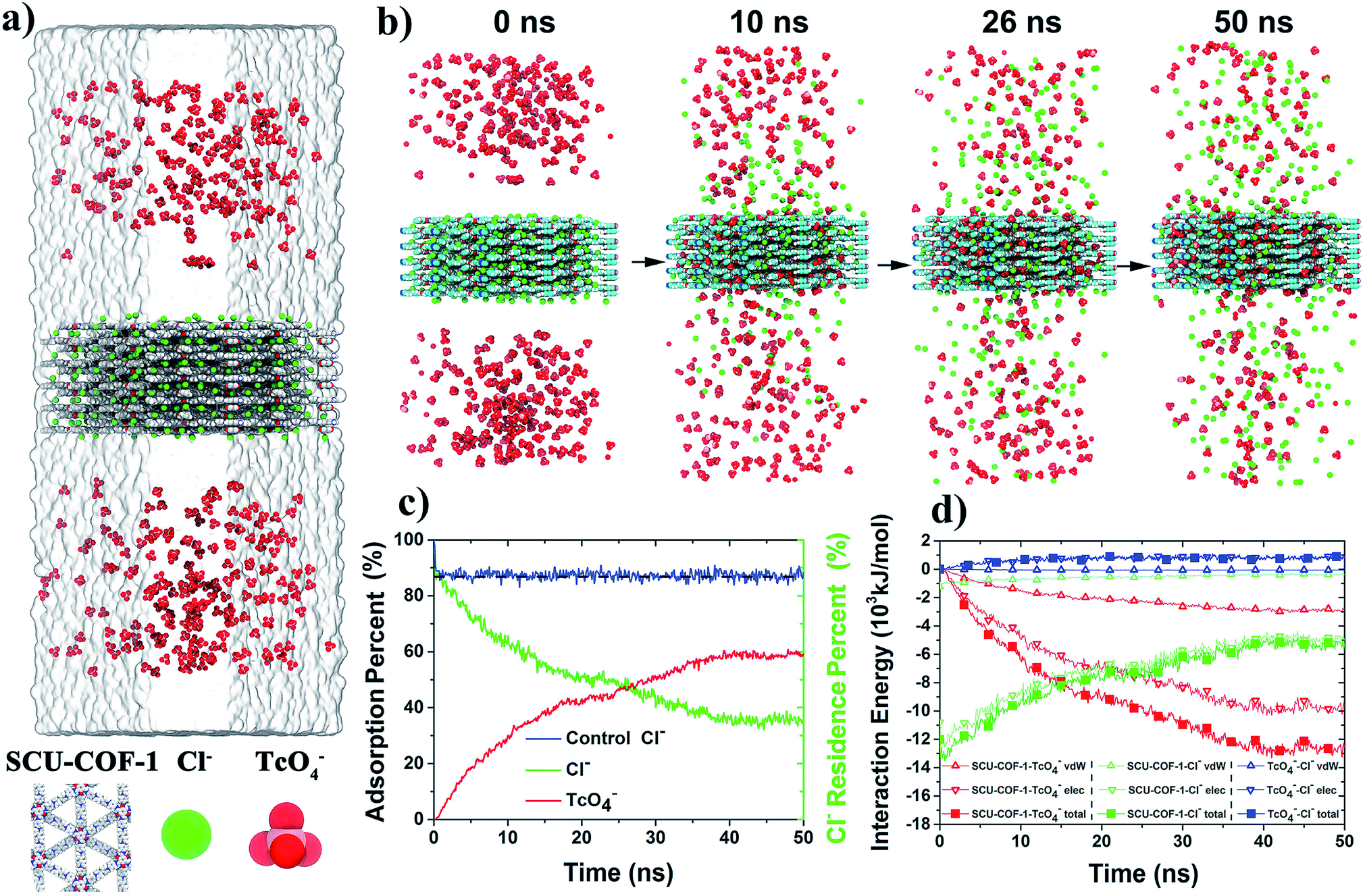 | ||
| Fig. 5 MD simulations on the adsorption of 99TcO4− anions into SCU-COF-1. (a) The initial configuration for MD simulations. Here, all composing atoms, and Cl− anions are represented with van der Waals (vdW) balls, water with a transparent surface. NH4+ ions are not shown for clarity. (b) A time-series of snapshots to show the adsorbing dynamics of 99TcO4− anions into SCU-COF-1. The blue framework is the simulation box edge. (c) Time evolution of the adsorption rate of 99TcO4− anions (red) and the residence rate of Cl− anions in SCU-COF-1 (green); as well as the residence rate of Cl− anions in SCU-COF-1 with pure water as the control (blue curve). (d) The nonbonded interaction energies (including electrostatic, vdW, and their sum) of (SCU-) COF-99TcO4−, COF-Cl−, and 99TcO4−-Cl− as a function of simulation time. | ||
To uncover the driving forces for the anion-exchange process, the time evolution of the nonbonded interaction energies of COF-99TcO4−, COF-Cl−, and 99TcO4−-Cl− are calculated and analyzed, and they were further deconvoluted into the contributions from the electrostatic and van der Waals (vdW) interactions as well (Fig. 5d). During the main 99TcO4− uptake process (<40 ns; after 40 ns, simulations are converged), the nonbonded interaction energy between 99TcO4− anions and SCU-COF-1 sharply decreases by ∼13000 kJ mol−1 (decreases from 0 to −13000 kJ mol−1; Fig. 5d solid red square) (i.e., more favorable). The contribution from the electrostatic part is ∼10000 kJ mol−1 (Fig. 5d hollow red inverted triangle), which is about 3.3-fold of that from the vdW part (∼3000 kJ mol−1) (Fig. 5d hollow red triangle). During the same period, many Cl− anions were desorbed from SCU-COF-1. The nonbonded interaction energy between Cl− anions and SCU-COF-1 increases by ∼8000 kJ mol−1 (increases from −13000 to −5000 kJ mol−1; Fig. 5d solid green square) (less favorable), of which the electrostatic part contributes ∼7400 kJ mol−1 (Fig. 5d hollow green inverted triangle) and the vdW part contributes ∼600 kJ mol−1 (Fig. 5d hollow green triangle). Meanwhile, the nonbonded interaction energy between the Cl− anions and 99TcO4− anions increased by ∼850 kJ mol−1 (increases from 0 to 850 kJ mol−1; Fig. 5d solid blue square) (less favorable), with the electrostatic part increasing by 900 kJ mol−1 (repulsive) (Fig. 5d hollow blue inverted triangle), whereas the vdW part decreases by ∼50 kJ mol−1 (attractive) (Fig. 5d hollow blue triangle). These results suggest that the strong direct nonbonded interaction (particularly the electrostatic interactions) between 99TcO4− anions and SCU-COF-1 intensively drives this vigorous 99TcO4− uptake process. During this process, the 99TcO4− anions gained considerable energy, which can overcompensate for the energetic penalty induced by the desorption of Cl− anions. Moreover, the direct electrostatic repulsive interaction between 99TcO4− anions and Cl− anions further promotes the desorption of Cl− anions. To gain a more microscopic insight into the substituting procedure of Cl− anions by 99TcO4− anions, we further compared the binding free energy of a single 99TcO4− anion to the binding site that is most energetic favorable for the Cl− anion. This binding site is determined by the analysis of the Cl− anions residing probability distribution in the control simulation (for a better sampling). It was demonstrated that Cl− ions prefer to be located near the pyridine ring on the inner wall of SCU-COF-1 (Fig. 6a), where the most positive charges are distributed (Fig. 1f). The potential of mean force (PMF) data demonstrates that the binding free energies are −9.9 kJ mol−1 and −13.7 kJ mol−1 for the Cl− anion and 99TcO4− anion, respectively, indicating that the 99TcO4− anion can successfully outcompete the Cl− anion in binding behavior (Fig. 6b). In other words, when one Cl− anion (at it most energetic favorable binding site) is exchanged with one 99TcO4− anion, it can gain 3.8 kJ mol−1 of energy total, which can be attributed to the preferential binding of 99TcO4− anions to SCU-COF-1 over Cl− anions. Therefore, an entire picture for the anion-exchange process can be revealed: on one hand, the much stronger binding free energy between the 99TcO4− anion and SCU-COF-1 leads to successful substitution of Cl− by taking over the anion binding site in SCU-COF-1 as time progresses; on the other hand, the occupation of the 99TcO4− anion significantly weakens the direct interactions between the SCU-COF-1 and Cl− anion and gradually excludes Cl− anions out of the SCU-COF-1 due to the direct electrostatic repulsive interactions between the 99TcO4− anion and Cl− anion.
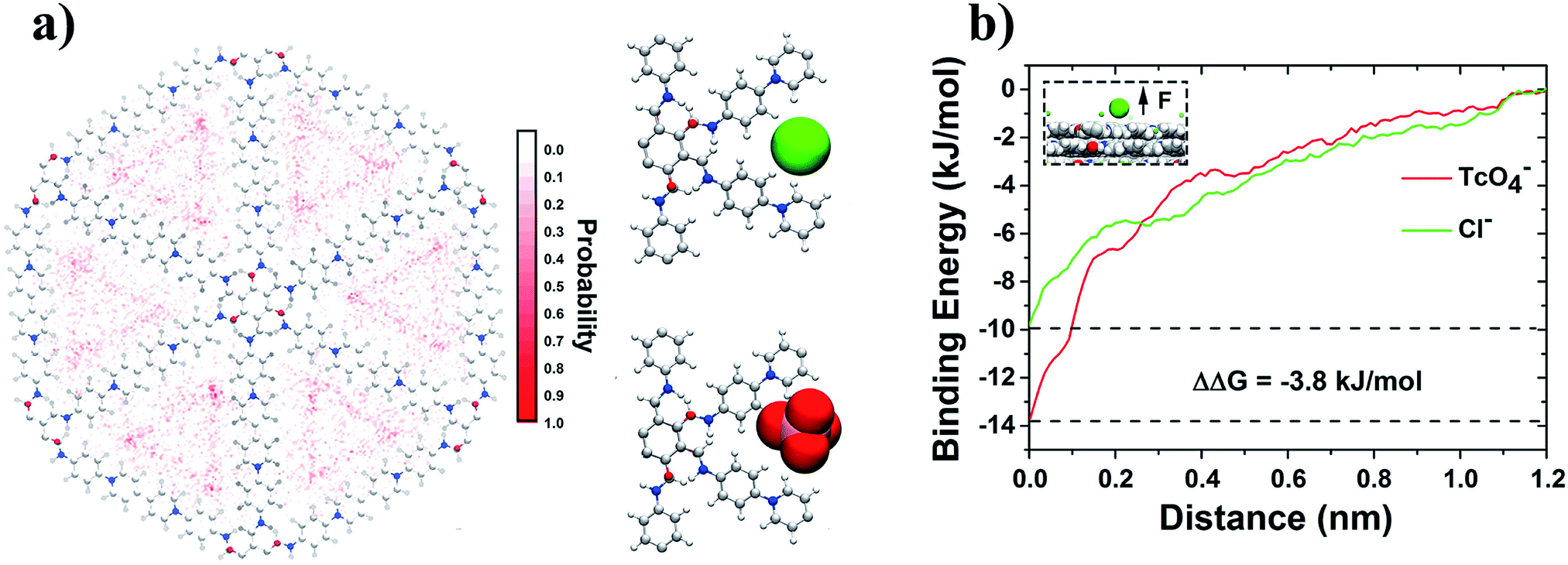 | ||
| Fig. 6 Binding energies of Cl− and 99TcO4− with SCU-COF-1. (a) The Cl− residence probability distribution within SCU-COF-1 in the control simulations (left) and the most preferential binding configuration of Cl− to SCU-COF-1 (right top). The 99TcO4− binding configuration yielded by substituting Cl− with 99TcO4− (right bottom). (b) Binding energies of Cl− (green) and 99TcO4− (red) with SCU-COF-1 by computing their potential of mean force (PMF) profiles moving from their binding sites to the bulk water. The inset figure displays the reaction coordinate in the PMF calculation. | ||
Conclusion
In summary, we herein present the first study on 99TcO4− remediation and separation using a cationic COF material (SCU-COF-1), showing promises in material stabilities under the extreme conditions of concentrated acidic solutions and strong radiation fields and uptake capabilities including sorption kinetics, capacity, selectivity, and reversibility. Another attractive advantage for the cationic COF material is the meeting of the CHON-only rule,25 which gives rise to potential combustibility that may be required for certain applications during nuclear waste management. The combination of these advantages cannot be achieved by the reported anion-exchange materials such as inorganic cationic framework materials, polymeric anion-exchange resins, and cationic MOF materials. Given that these merits are now integrated into a single material, the possibility for solving the “technetium issue” in the nuclear fuel cycle becomes visible. Furthermore, the overcoming of other challenges of radionuclide partitioning during nuclear waste disposal such as trivalent lanthanide/actinide separation and caesium strontium uptake using highly robust and designable COF materials can be expected in the future.Methods
Security declaration
Caution! 99Tc is a β-emitter with a long half-life (t1/2 = 2.13 × 105 years) and presents a significant health risk once inhaled or digested. All experiments involving 99Tc must be carried out in a licensed laboratory that is dedicated to radiological studies.Reagents and characterizations
4,4-Bipyridine and 1-chloro-2,4-dinitrobenzene were provided by Aladdin Industrial Co., Ltd. 1,4-Diaminobenzene and 2,4,6-triformylphloroglucinol (Tp) were purchased from Adamas Reagent Co., Ltd and Beijing HWRK Chem Co., Ltd, respectively. All solvents were purchased from Chinasun Specialty Products Co., LTD. [4,4′-Bipyridine]-1,1′-diium dichloride (BDB) and viologen-NH2 was synthesized according to the reference.26 NaReO4, HCl, NaOH, and HNO3 were analytically pure and used. 99TcO4− stock solutions were prepared by dissolving a desired amount of NH499TcO4 (99%) in deionized water. The 1H and 13C nuclear magnetic resonance (NMR) spectra were acquired on Avance III HD-400 and Abance III WB-400 instruments, respectively. Fourier-transform infrared spectroscopy (FT-IR) spectra in the range of 4000–400 cm−1 region were carried out on a Thermo Nicolet iS50 spectrometer. Thermogravimetric analyses (TGA) were performed using a NETZSCH STA 449F3 instrument under nitrogen flow from 25 °C to 900 °C. Nitrogen adsorption was performed under the liquid nitrogen bath at 77 K. Powder X-ray diffraction (PXRD) of the COFs were carried out on a Bruker D8 Advance diffractometer with a radiation source (Cu Kα, λ = 1.54056 Å), and the patterns were collected from 2° to 30°. SEM images were obtained by a FEI Quanta 200FEG scanning electron microscope (SEM) with the energy of the electron beam being 20 keV. The morphology was observed by using a FEI Tecnai G2 field emission transmission electron microscope (TEM) under a voltage of 120 kV. Nitrogen adsorption experiments were carried out at 77 K in a liquid nitrogen bath with the pressure ranging from 0 to 760 Torr by a Quantachrome Autosorb Gas Sorption analyzer IQ2. The concentrations of ReO4− stock solutions were measured by inductively coupled plasma-atomic emission spectrometry (ICP-AES, Thermo Fisher Scientific iCAP 7000). A UV-vis spectrometer (Varian Cary 6000i) was used to measure the concentration of 99TcO4− by identifying the adsorption peak at 290 nm. Multiple scans were performed by the Cary instrument as the default setting, and the averaged spectra were provided automatically. Additionally, the radioactivity of the 99TcO4− solution was measured by a liquid scintillation counting (LSC) system (Perkin Elmer Quantulus 1220). The source of β-irradiation (1.5 MeV) was provided by an electron accelerator. The γ-irradiation was provided by a 60Co source (92.42 PBq).Synthesis of BDB
[4,4′-Bipyridine]-1,1ʹ-diium dichloride (BDB) was synthesized using the Zincke reaction.21 4,4-Bipyridine (1.0 g, 6.4 mmol), 1-chloro-2,4-dinitrobenzene (4.5 g, 19.2 mmol), and 80 mL of absolute ethanol were added to a flask under reflux conditions for 72 h. Upon full conversion, the solution was removed by suction filtration and washed with 150 mL of heated ethanol. The yellow solid was collected after drying in a vacuum oven at 50 °C for 4 h (45% yield, Scheme S1†).Synthesis of viologen-NH2
BDB (500 mg, 0.89 mmol) and 1,4-diaminobenzene (289 mg, 2.67 mmol) were dissolved in 250 mL of absolute ethanol in a flask. The mixture was next refluxed for 72 h. The reaction process was followed by thin-layer chromatography (TLC). The solution was then concentrated to 100 mL using a rotary evaporator. The mixture was poured into 750 mL of THF, and after 2.5 h the dark brown solid was collected by suction filtration and washed with THF (10 mL × 5). The precipitate was dried in an oven under vacuum at 45 °C for 5 h (70% yield, Scheme S1†). 1H NMR (D2O, 400 MHz): δ (ppm) = 9.26 (d, 4H, 4.0 Hz), 8.68 (d, 4H, 2.0 Hz), 7.60 (d, 4H, 8.0 Hz), 6.05 (d, 4H, 4.0 Hz) (Scheme S1 and Fig. S8†).Synthesis of SCU-COF-1
To a 10 mL Pyrex tube containing viologen-NH2 (18.51 mg, 0.045 mmol) and Tp (6.30 mg, 0.03 mmol), a mixture of 1.9 mL of o-DCB, 0.1 mL of BuOH, and 0.2 mL of acetic-acid catalyst (19:1:2, by vol.) were added. Then, the tube was degassed by three-pump-thaw for three cycles. The sealed tube was heated at 120 °C for 6 d (Fig. 1a). The precipitate was collected after filtration and washed with THF (5 mL × 3) and absolute methanol (5 mL × 5) to ensure the removal of the residual oligomers and unreacted ligands. The dark brown SCU-COF-1 was gained after drying in vacuum at 40 °C for 5 h.
Batch experiments
The sorption experiments for ReO4− and 99TcO4− were first performed at 300 K according to the batch sorption method with a solid/liquid ratio of 1 g L−1. Typically, 5 mg of solid material (SCU-COF-1) was added to a stock solution of ReO4−/99TcO4− and shaken on an automatic shaker. After a specific contact time, the mixture was separated with a 0.22 μm nylon membrane filter. The concentrations of ReO4− were measured by ICP-AES. The concentrations and activity of 99TcO4− were determined by UV-vis spectrometry and the LSC technique.Sorption kinetics study of SCU-COF-1
20 mg of SCU-COF-1 was added into 20 mL of stock solution containing 28 ppm ReO4− or 14 ppm 99TcO4−, respectively. The mixture was then shaken for a specific contact time (30 s, 1 min, 3 min, 5 min, 10 min) on an automatic shaker. The desired time refers to the solid–liquid contact time and the time for acquiring the spectroscopic data is not included. After separating them by a 0.22 μm nylon membrane filter, the concentrations of ReO4− at different contact times were measured by ICP-AES. The concentrations of 99TcO4− were determined by UV-vis spectra and LSC.Sorption isotherm experiments
The sorption isotherm studies for ReO4− were performed using a series of stock solutions containing various ReO4− concentrations (28 to 1000 ppm) with the same ratio of solid–liquid. The resulting mixture was shaken for 14 h to reach the adsorption equilibrium (24 h for samples in heating sorption experiments) and then separated by a 0.22 μm nylon membrane filter. To overcome the kinetic barrier for the complete exchange process, an additional sorption experiment at an elevated temperature was conducted by adding 5 mg of SCU-COF-1 to a 5 mL solution containing 2000 ppm ReO4− at 373 K under reflux conditions. The concentrations of ReO4− after sorption were measured by ICP-AES.pH effect study
ReO4− uptake at various pH values were evaluated in different stock solutions containing 400 ppm ReO4−. The range of pH values is from 1 to 11. After being shaken for 14 h, the concentrations then were determined by ICP-AES.Anion-exchange selectivity experiments
The effect of NO3− was studied by adding NaNO3 with various concentrations (0.15 mM, 1.5 mM, 3 mM, and 15 mM) into a 0.15 mM ReO4− solution, respectively. The competing effects of other different anions (CO32−, SO42−, and PO43−) were performed by adding concentrations of Na2SO4, Na2CO3, or NaH2PO4 (0.15 mM) into a 0.15 mM ReO4− solution, respectively. The powder of SCU-COF-1 was added in each solution mentioned above, respectively. Experiments in simulated nuclear waste (Hanford Low Activity Waste Melter Recycle Stream) were conducted in a similar method with determined conditions according to reported protocol.14a 5 mg of COF materials were added to a 5 mL prepared solution (solid/liquid = 1), and 10 mg of materials were added to a 2 mL prepared solution (solid/liquid = 5). After being shaken for 14 h to ensure reaching equilibrium, the mixture was separated by a 0.22 μm nylon membrane filter for ICP analysis.Batch experiments in 1 M and 3 M HNO3
2 mg, 4 mg, 10 mg, 20 mg, or 40 mg of solid materials were soaked into 2 mL of 1 M HNO3 solution containing 28 ppm ReO4− and were respectively shaken for 14 h. Additionally, 2 mL of 1 M HNO3 solution containing 338 ppm ReO4− was used following the same procedure. The concentrations were then measured after sorption. A 3 M HNO3 solution containing 253 ppm ReO4− was prepared according to the same method. The suspension was shaken for 14 h to reach the adsorption equilibrium and was then separated by a 0.22 μm nylon membrane filter. The concentrations were determined by ICP-AES.Radiation-resistance experiments
SCU-COF-1 was exposed to the β- or γ-irradiations for four doses (50 kGy, 200 kGy, 400 kGy, 600 kGy). Then, the PXRD patterns were obtained, and the adsorption experiments were performed by using a solution containing 350 ppm ReO4−. After being shaken for 14 h, the suspension was separated by a 0.22 μm nylon membrane filter for ICP analysis.Regeneration study
130 mg of SCU-COF-1 material sorbed with 130 mL of 28 ppm ReO4− (at pH = 7) was immersed into a 1 M NaCl solution, followed by washing with water. Multiple sorption/desorption trials of up to 4 runs were performed either in a pH 7 water solution or in a 3 M nitric acid solution.Computational methods
The SCU-COF-1 model membrane was parallelly stacked by 15 unit cells (sheets) that were obtained from our first-principle calculations based on the density functional theory (DFT) (Fig. S11†), with an initial inter-sheet distance of 0.35 nm. Each sheet consists of 336 carbon atoms, 48 nitrogen atoms, 24 oxygen atoms, 240 hydrogen atoms, and 24 anions Cl−, which gives a total of 672 atoms. The partial charges for COF atoms were obtained from DFT calculations on the unit cell of COF. The Lennard-Jones (L-J) parameters for the COF atoms were obtained from all-atom optimized potentials for liquid simulations (OPLS-AA) force field (FF).27 The Lorentz–Berthelot combining rules were used for calculating cross L-J interaction parameters. Many theoretical simulation results have demonstrated good agreement with those in experimental studies.28 The reasonable accuracy of OPLS-AA FF was also verified in characterizing microscopic insights for the adsorption or diffusion of a wide range of contaminants in various porous materials.28a,b,29 The FF parameters of the 99TcO4− anions were taken from a previous simulation study by Carbone and co-workers,28d in which the hydration free energies, hydrated radii, and diffusion coefficients of 99TcO4− were studied and showed good agreement with experimental results. The COF membrane was placed into the aqueous 99TcO4− solution to study the adsorption behaviors of 99TcO4−. The simulation box has a size of (8.71 nm × 7.54 nm × 17.61 nm), which contained a COF membrane, 360 TcO4− anions, 360 NH4+ (for neutralizing the simulation system), and 65646 water molecules, thus giving a total of 210618 atoms. As a control, the COF was also solvated into a pure water box with a size of (8.71 nm × 7.54 nm × 17.61 nm) containing 70078 water molecules, which gave a total of 220314 atoms, and served as the control system to check the residence of Cl−.
All MD simulations were performed with the GROMACS software package30 (version 4.6.7). VMD31 software (version 1.9.3) was applied for trajectory visualization and analysis. The SPC/E water model32 was used for water. In the production run, a time step of 2.0 fs was used, and the data were collected every 10 ps. For each system, three independent trajectories of 50 ns were generated, which resulted in a total aggregate simulation time of 300 ns. All production runs were under isothermal-isochoric (NVT) ensemble; the temperature was maintained at 300 K using a υ-rescale thermostat.33 The particle-mesh Ewald (PME) method34 was used for long-range electrostatic interactions. A cutoff distance of 1.2 nm was used for the calculation of van der Waals (vdW) interactions. All solute bonds between heavy atoms and hydrogens were constrained with the LINCS algorithm.35 Periodic boundary conditions were applied in all directions, and the COF were frozen throughout the simulation process. We used umbrella-sampling simulations to investigate the potential of mean force (PMF) of a single 99TcO4− and Cl−. The reaction path for the PMF calculation was defined by pulling center of mass of the anion (99TcO4− or Cl−) from the most favourable binding site of Cl− along the upper surface of COF membrane to the bulk water (from ∼0.0 nm to ∼1.2 nm of the COF upper surface) with a harmonic force of 2000 kJ mol−1 nm−2. The reaction coordinate was divided into 12 windows, with each window simulated for 3 ns and thus accumulating a total simulation of 36 ns. The weighted histogram analysis method36 (WHAM) was applied to calculate the free energy. All PMF profiles were normalized to zero at a distance of 1.2 nm.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (21825601, 21790374, 21806117, and 11404233) and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).Notes and references
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press/Taylor and Francis Group, Boca Raton, FL, 87th edn, 2006 Search PubMed.
- (a) S. S. Jurisson and J. D. Lydon, Chem. Rev., 1999, 99, 2205–2218 CrossRef CAS PubMed; (b) J. R. Dilworth and S. J. Parrott, Chem. Soc. Rev., 1998, 27, 43–55 RSC; (c) M. D. Bartholomä, A. S. Louie, J. F. Valliant and J. Zubieta, Chem. Rev., 2010, 110, 2903–2920 CrossRef PubMed.
- D. Banerjee, D. Kim, M. J. Schweiger, A. A. Kruger and P. K. Thallapally, Chem. Soc. Rev., 2016, 45, 2724–2739 RSC.
- J. G. Darab and P. A. Smith, Chem. Mater., 1996, 8, 1004–1021 CrossRef CAS.
- J. P. Icenhower, N. P. Qafoku, J. M. Zachara and W. J. Martin, Am. J. Sci., 2010, 310, 721–752 CrossRef CAS.
- International Atomic Energy Agency, Nuclear Power Reactors in the World, Reference Data Series No. 2, IAEA, Vienna, 2018 Search PubMed.
- K. H. Lieser, Radiochim. Acta, 1993, 63, 5–8 CAS.
- R. Carmody and J. H. Highman, Br. J. Radiol., 1975, 48, 63–64 CrossRef CAS PubMed.
- (a) R. Alberto, G. Bergamaschi, H. Braband, T. Fox and V. Amendola, Angew. Chem., Int. Ed., 2012, 51, 9772–9776 CrossRef CAS PubMed; (b) K. Ito and A. Yachidate, Carbon, 1992, 30, 767–771 CrossRef CAS; (c) B. Gu and K. E. Dowlen, An Investigation of Groundwater Organics, Soil Minerals, and Activated Carbon on the Complexation, Adsorption, and Separation of Technetium-99, Publication No. 4502, Environmental Sciences Division, ORNL/TM-13154, Oak Ridge, Tennessee, 1996 Search PubMed; (d) S. V. Mattigod, R. J. Serne and G. E. Fryxell, Selection and Testing of “Getters” for Adsorption of Iodine-129 and Technetium-99: A Review, PNNL-14208, 2003 CrossRef; (e) B. Gu, K. E. Dowlen, L. Liang and J. L. Clausen, Sep. Technol., 1996, 6, 123–132 CrossRef CAS.
- S. Gin, A. Abdelouas, L. J. Criscenti, W. L. Ebert, K. Ferrand, T. Geisler, M. T. Harrison, Y. Inagaki, S. Mitsui, K. T. Mueller, J. C. Marra, C. G. Pantano, E. M. Pierce, J. V. Ryan, J. M. Schofield, C. I. Steefel and J. D. Vienna, Mater. Today, 2013, 16, 243–248 CrossRef CAS.
- B. C. Childs, F. Poineau, K. R. Czerwinski and A. P. Sattelberger, J. Radioanal. Nucl. Chem., 2015, 306, 417–421 CrossRef CAS.
- (a) J. Eagling, P. J. Worsfold, W. H. Blake and M. J. Keith-Roach, Environ. Sci. Technol., 2012, 46, 11798–11803 CrossRef CAS PubMed; (b) C. I. Pearce, J. P. Icenhower, R. M. Asmussen, P. G. Tratnyek, K. M. Rosso, W. W. Lukens and N. P. Qafoku, ACS Earth Space Chem., 2018, 2, 532–547 CrossRef CAS; (c) C. L. Corkhill, J. W. Bridge, X. C. Chen, P. Hillel, S. F. Thornton, M. E. Romero-Gonzalez, S. A. Banwart and N. C. Hyatt, Environ. Sci. Technol., 2013, 47, 13857–13864 CrossRef CAS PubMed.
- (a) X. Zhou, G.-a. Ye, H. Zhang, L. Li, F. Luo and Z. Meng, Radiochim. Acta, 2014, 102, 111–116 CAS; (b) V. I. Marchenko, K. N. Dvoeglazov and V. I. Volk, Radiochemistry, 2009, 51, 329–344 CrossRef CAS.
- (a) S. Wang, P. Yu, B. A. Purse, M. J. Orta, J. Diwu, W. H. Casey, B. L. Phillips, E. V. Alekseev, W. Depmeier, D. T. Hobbs and T. E. Albrecht-Schmitt, Adv. Funct. Mater., 2012, 22, 2241–2250 CrossRef CAS; (b) S. Wang, E. V. Alekseev, J. Diwu, W. H. Casey, B. L. Phillips, W. Depmeier and T. E. Albrecht-Schmitt, Angew. Chem., Int. Ed., 2010, 49, 1057–1060 CrossRef CAS PubMed; (c) P. Yu, S. Wang, E. V. Alekseev, W. Depmeier, D. T. Hobbs, T. E. Albrecht-Schmitt, B. L. Phillips and W. H. Casey, Angew. Chem., Int. Ed., 2010, 49, 5975–5977 CrossRef CAS PubMed; (d) H. V. Goulding, S. E. Hulse, W. Clegg, R. W. Harrington, H. Y. Playford, R. I. Walton and A. M. Fogg, J. Am. Chem. Soc., 2010, 132, 13618–13620 CrossRef CAS PubMed; (e) S. R. J. Oliver, Chem. Soc. Rev., 2009, 38, 1868–1881 RSC; (f) L. J. McIntyre, L. K. Jackson and A. M. Fogg, Chem. Mater., 2008, 20, 335–340 CrossRef CAS; (g) K. H. Goh, T. T. Lim and Z. Dong, Water Res., 2008, 42, 1343–1368 CrossRef CAS PubMed; (h) Y. Wang and H. Gao, J. Colloid Interface Sci., 2006, 301, 19–26 CrossRef CAS PubMed; (i) H. W. Olfs, L. O. Torres-Dorante, R. Eckelt and H. Kosslick, Appl. Clay Sci., 2009, 43, 459–464 CrossRef CAS.
- (a) J. Li, L. Zhu, C. Xiao, L. Chen, Z. Chai and S. Wang, Radiochim. Acta, 2018, 106, 581–591 CAS; (b) P. Samanta, P. Chandra, S. Dutta, A. V. Desai and S. K. Ghosh, Chem. Sci., 2018, 9, 7874–7881 RSC; (c) J. Li, X. Dai, L. Zhu, C. Xu, D. Zhang, M. A. Silver, P. Li, L. Chen, Y. Li, D. Zuo, H. Zhang, C. Xiao, J. Chen, J. Diwu, O. K. Farha, T. E. Albrecht-Schmitt, Z. Chai and S. Wang, Nat. Commun., 2018, 9, 3007 CrossRef PubMed; (d) J. Zu, M. Ye, P. Wang, F. Tang and L. He, RSC Adv., 2016, 6, 18868–18873 RSC; (e) D. Banerjee, S. K. Elsaidi, B. Aguila, B. Li, D. Kim, M. J. Schweiger, A. A. Kruger, C. J. Doonan, S. Ma and P. K. Thallapally, Chem.–Eur. J., 2016, 22, 17581–17584 CrossRef CAS PubMed; (f) B. Gu, G. M. Brown, P. V. Bonnesen, L. Liang, B. A. Moyer, R. Ober and S. D. Alexandratos, Environ. Sci. Technol., 2000, 34, 1075–1080 CrossRef CAS; (g) P. V. Bonnesen, G. M. Brown, S. D. Alexandratos, L. B. Bavoux, D. J. Presley, V. Patel, R. Ober and B. A. Moyer, Environ. Sci. Technol., 2000, 34, 3761–3766 CrossRef CAS.
- (a) L. Zhu, C. Xiao, X. Dai, J. Li, D. Gui, D. Sheng, L. Chen, R. Zhou, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Environ. Sci. Technol. Lett., 2017, 4, 316–322 CrossRef CAS; (b) L. Zhu, D. Sheng, C. Xu, X. Dai, M. A. Silver, J. Li, P. Li, Y. Wang, Y. Wang, L. Chen, C. Xiao, J. Chen, R. Zhou, C. Zhang, O. K. Farha, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, J. Am. Chem. Soc., 2017, 139, 14873–14876 CrossRef CAS PubMed; (c) D. Sheng, L. Zhu, C. Xu, C. Xiao, Y. Wang, Y. Wang, L. Chen, J. Diwu, J. Chen, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Environ. Sci. Technol., 2017, 51, 3471–3479 CrossRef CAS PubMed; (d) A. V. Desai, B. Manna, A. Karmakar, A. Sahu and S. K. Ghosh, Angew. Chem., 2016, 128, 7942–7946 CrossRef; (e) D. Banerjee, W. Xu, Z. Nie, L. E. Johnson, C. Coghlan, M. L. Sushko, D. Kim, M. J. Schweiger, A. A. Kruger, C. J. Doonan and P. K. Thallapally, Inorg. Chem., 2016, 55, 8241–8243 CrossRef CAS PubMed; (f) H. Fei, M. R. Bresler and S. R. J. Oliver, J. Am. Chem. Soc., 2011, 133, 11110–11113 CrossRef CAS PubMed; (g) R. J. Drout, K. Otake, A. J. Howarth, T. Islamoglu, L. Zhu, C. Xiao, S. Wang and O. K. Farha, Chem. Mater., 2018, 30, 1277–1284 CrossRef CAS.
- (a) K. K. S. Pillay, J. Radioanal. Nucl. Chem., 1986, 102, 247–268 CrossRef CAS; (b) K. M. Long, G. S. Goff, S. D. Ware, G. D. Jarvinen and W. H. Runde, Ind. Eng. Chem. Res., 2012, 51, 10445–10450 CrossRef CAS.
- (a) A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed; (b) T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 CrossRef CAS PubMed; (c) P. J. Waller, F. Gandara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS PubMed; (d) J. Jiang, Y. Zhao and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 3255–3265 CrossRef CAS PubMed; (e) Y. Song, Q. Sun, B. Aguila and S. Ma, Adv. Sci., 2019, 6, 1801410 CrossRef PubMed; (f) M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef; (g) S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS PubMed; (h) S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed; (i) J. L. Segura, M. J. Mancheno and F. Zamora, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC; (j) N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS; (k) X. H. Liu, C. Z. Guan, D. Wang and L. J. Wan, Adv. Mater., 2014, 26, 6912–6920 CrossRef CAS PubMed; (l) X. Zou, H. Ren and G. Zhu, Chem. Commun., 2013, 49, 3925–3936 RSC; (m) S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- (a) O. Yahiaoui, A. N. Fitch, F. Hoffmann, M. Froba, A. Thomas and J. Roeser, J. Am. Chem. Soc., 2018, 140, 5330–5333 CrossRef CAS PubMed; (b) A. Mal, R. K. Mishra, V. K. Praveen, M. A. Khayum, R. Banerjee and A. Ajayaghosh, Angew. Chem., Int. Ed., 2018, 57, 8443–8447 CrossRef CAS PubMed; (c) Q. Lu, Y. Ma, H. Li, X. Guan, Y. Yusran, M. Xue, Q. Fang, Y. Yan, S. Qiu and V. Valtchev, Angew. Chem., Int. Ed., 2018, 57, 6042–6048 CrossRef CAS PubMed; (d) Y. Zhang, J. Duan, D. Ma, P. Li, S. Li, H. Li, J. Zhou, X. Ma, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2017, 56, 16313–16317 CrossRef CAS PubMed; (e) J. Roeser, D. Prill, M. J. Bojdys, P. Fayon, A. Trewin, A. N. Fitch, M. U. Schmidt and A. Thomas, Nat. Chem., 2017, 9, 977–982 CrossRef CAS PubMed; (f) Z. Li, H. Li, X. Guan, J. Tang, Y. Yusran, Z. Li, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, J. Am. Chem. Soc., 2017, 139, 17771–17774 CrossRef CAS PubMed; (g) N. Huang, P. Wang, M. A. Addicoat, T. Heine and D. Jiang, Angew. Chem., Int. Ed., 2017, 56, 4982–4986 CrossRef CAS PubMed; (h) S.-B. Yu, H. Lyu, J. Tian, H. Wang, D.-W. Zhang, Y. Liu and Z.-T. Li, Polym. Chem., 2016, 7, 3392–3397 RSC; (i) Q. Sun, B. Aguila, J. Perman, N. Nguyen and S. Ma, J. Am. Chem. Soc., 2016, 138, 15790–15796 CrossRef CAS PubMed; (j) S. Mitra, S. Kandambeth, B. P. Biswal, M. A. Khayum, C. K. Choudhury, M. Mehta, G. Kaur, S. Banerjee, A. Prabhune, S. Verma, S. Roy, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2016, 138, 2823–2828 CrossRef CAS PubMed; (k) H. Ma, B. Liu, B. Li, L. Zhang, Y. G. Li, H. Q. Tan, H. Y. Zang and G. Zhu, J. Am. Chem. Soc., 2016, 138, 5897–5903 CrossRef CAS PubMed; (l) Y. Du, H. Yang, J. M. Whiteley, S. Wan, Y. Jin, S. H. Lee and W. Zhang, Angew. Chem., Int. Ed., 2016, 55, 1737–1741 CrossRef CAS PubMed; (m) Y. Yuan, F. Sun, F. Zhang, H. Ren, M. Guo, K. Cai, X. Jing, X. Gao and G. Zhu, Adv. Mater., 2013, 25, 6619–6624 CrossRef CAS PubMed.
- (a) G.-H. Ning, Z. Chen, Q. Gao, W. Tang, Z. Chen, C. Liu, B. Tian, X. Li and K. P. Loh, J. Am. Chem. Soc., 2017, 139, 8897–8904 CrossRef CAS PubMed; (b) X. Han, J. Zhang, J. Huang, X. Wu, D. Yuan, Y. Liu and Y. Cui, Nat. Commun., 2018, 9, 1294 CrossRef PubMed; (c) S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524–19527 CrossRef CAS PubMed; (d) S. Karak, S. Kumar, P. Pachfule and R. Banerjee, J. Am. Chem. Soc., 2018, 140, 5138–5145 CrossRef CAS PubMed.
- G. Das, T. Skorjanc, S. K. Sharma, F. Gandara, M. Lusi, D. S. S. Rao, S. Vimala, S. K. Prasad, J. Raya, D. S. Han, R. Jagannathan, J. C. Olsen and A. Trabolsi, J. Am. Chem. Soc., 2017, 139, 9558–9565 CrossRef CAS PubMed.
- (a) F. Gu, H. Dong, Y. Li, Z. Si and F. Yan, Macromolecules, 2013, 47, 208–216 CrossRef; (b) M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8, 513–523 CrossRef CAS PubMed.
- R. Custelcean and B. A. Moyer, Eur. J. Inorg. Chem., 2007, 1321–1340 CrossRef CAS.
- W. R. Wilmarth, G. J. Lumetta, M. E. Johnson, M. R. Poirier, M. C. Thompson, P. C. Suggs and N. P. Machara, Solvent Extr. Ion Exch., 2011, 29, 1–48 CrossRef CAS.
- K. L. Nash and G. J. Lumetta, Advanced separation techniques for nuclear fuel reprocessing and radioactive waste treatment, Woodhead Publishing Limited, Cambridge, UK, 2011 Search PubMed.
- F. Biedermann and O. A. Scherman, J. Phys. Chem. B, 2012, 116, 2842–2849 CrossRef CAS PubMed.
- (a) W. L. Jorgensen and J. Tiradorives, J. Am. Chem. Soc., 1988, 110, 1657–1666 CrossRef CAS PubMed; (b) W. L. Jorgensen, D. S. Maxwell and J. TiradoRives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- (a) T. Zheng, Z. X. Yang, D. X. Gui, Z. Y. Liu, X. X. Wang, X. Dai, S. T. Liu, L. J. Zhang, Y. Gao, L. H. Chen, D. P. Sheng, Y. L. Wang, J. Diwu, J. Q. Wang, R. H. Zhou, Z. F. Chai, T. E. Albrecht-Schmitt and S. Wang, Nat. Commun., 2017, 8, 1354 CrossRef PubMed; (b) Y. Li, Z. Yang, Y. Wang, Z. Bai, T. Zheng, X. Dai, S. Liu, D. Gui, W. Liu, M. Chen, L. Chen, J. Diwu, L. Zhu, R. Zhou, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Nat. Commun., 2017, 8, 1354 CrossRef PubMed; (c) L. N. Li, W. Ma, S. S. Shen, H. X. Huang, Y. Bai and H. W. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 31032–31041 CrossRef CAS PubMed; (d) C. D. Williams and P. Carbone, J. Chem. Phys., 2015, 143, 174502 CrossRef PubMed.
- (a) Q. Y. Yang and C. L. Zhong, J. Phys. Chem. B, 2006, 110, 17776–17783 CrossRef CAS PubMed; (b) Q. Y. Yang, C. Y. Xue, C. L. Zhong and J. F. Chen, AIChE J., 2007, 53, 2832–2840 CrossRef CAS.
- (a) B. Hess, C. Kutzner, D. Van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed; (b) D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14, 33–38 CrossRef CAS.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- (a) S. Kumar, D. Bouzida, R. H. Swendsen, P. A. Kollman and J. M. Rosenberg, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS; (b) J. S. Hub, B. L. de Groot and D. Van der Spoel, J. Chem. Theory Comput., 2010, 6, 3713–3720 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00172g |
| ‡ These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2019 |