
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the interactions of decavanadate with thaumatin, lysozyme, proteinase K and human serum proteins by its coordination to a pentaaquacobalt(II) complex cation†
Lukáš
Krivosudský‡
 a,
Alexander
Roller
b and
Annette
Rompel
*a
a,
Alexander
Roller
b and
Annette
Rompel
*a
aUniversität Wien, Fakultät für Chemie, Institut für Biophysikalische Chemie, Althanstraße 14, Wien 1090, Austria. E-mail: annette.rompel@univie.ac.at; Web: http://www.bpc.univie.ac.at
bUniversität Wien, Fakultät für Chemie, Zentrum für Röntgenstrukturanalyse, Währinger Straße 42, 1090 Wien, Austria
First published on 28th October 2019
Abstract
The decavanadate anion, HxV10O28(6−x)− (V10), is one of the most studied vanadium polyoxometalate species. In recent decades several works have pointed to its biological relevance coming mainly from its ability to bind to proteins (such as actin, myosin or ion pumps). On the other hand, non-functional binding was observed in several protein crystal structures, where V10 was incorporated “accidentally” resulting from the presence of Na3VO4 as a phosphatase inhibitor. In this work we broaden the potential biological applications of V10 by presenting the synthesis and characterization of two decavanadate species where the anion acts as a ligand: (2-hepH)(NH4)[{Cu(H2O)2(2-hep)}2V10O28]·4H2O (V10Cu) and (2-hepH)2[{Co(H2O)5}2V10O28]·4H2O (V10Co) (2-hep = 2-hydroxyethylpyridine). Unlike free decavanadate, the complex anions stay intact in model buffer solutions (0.1 M 2-(N-morpholino)ethanesulfonic acid, 0.5 M NaCl, pH = 5.8 and 8.0). It has been shown that V10Co is stable also in the presence of proteins and for the first time it was possible to study the interaction of decavanadate with proteins without the interference of lower vanadate oligomers. This allowed comparison of interactions of V10 and V10Co with the model proteins thaumatin, lysozyme, proteinase K, human serum albumin and transferrin under conditions close to biological ones (0.1 M 2-(N-morpholino)ethanesulfonic acid, 0.5 M NaCl, pH = 5.8). The linewidths of the signals at half-height in 51V NMR spectra reflect the strength of interaction of a vanadium species with a protein, and thus it was shown that V10 and V10Co both bind strongly to thaumatin, V10 binds to lysozyme and V10Co binds to proteinase K. V10 interacts with both human serum albumin and transferrin, but surprisingly V10Co exhibits high affinity to transferrin but does not interact with albumin.
1 Introduction
Polyoxometalates (POMs) are an important group of metal oxide clusters1 exhibiting diverse archetypal structures of, particularly, vanadates, molybdates and tungstates.2–4 The tremendous variability of POMs has given rise to their application in distinct areas of materials science,5,6 catalysis,7–10 electrochemistry and redox processes,11,12 photochemistry13–15 and magnetism.16–18 In recent years, the roles of POMs in biological systems have been intensively investigated. Such studies may be divided into two groups: functional binding and interaction of POMs in biological systems widening the borders of medicinal chemistry;19–24 and non-functional interaction with biomolecules, such as proteins, enhancing the current possibilities in macromolecular crystallography by promoting the crystallization or being useful in obtaining the initial phases while solving the structures of proteins.25–28Decavanadate, HxV10O28(6−x)− (V10), is the predominant species formed in vanadate solutions at vanadium(V) concentrations above 1 mM in the pH range of ≈2–6.29,30 The structure of V10 consists of ten face-sharing octahedra (Scheme 1). The symmetrically non-equivalent vanadium atoms VA, VB and VC give rise to three different signals in 51V NMR spectra depending on the conditions: while the low-field signal of VA atoms stays in a narrow region around −425 ± 3 ppm, the other two peaks representing VB and VC atoms are more sensitive to changes in acidity and exhibit signals at approximately −505 ± 10 ppm and −525 ± 10 ppm, respectively. The oxygen atoms OB and OC are potential sites for protonation, and the atoms OC, OF, OG and OD are the most potential sites for ligation to transition metal ions.31,32 As such, changes in 51V NMR parameters reflect the versatility of structural modifications of V10. While protonation and coordination of V10 manifest mostly in peaks’ movement, the interaction and binding to proteins result in significant peak broadening defined by linewidths at half-height of the signals, W1/2. This is caused by the ligand bulkiness and decreased symmetry of the V10 species upon interaction.32
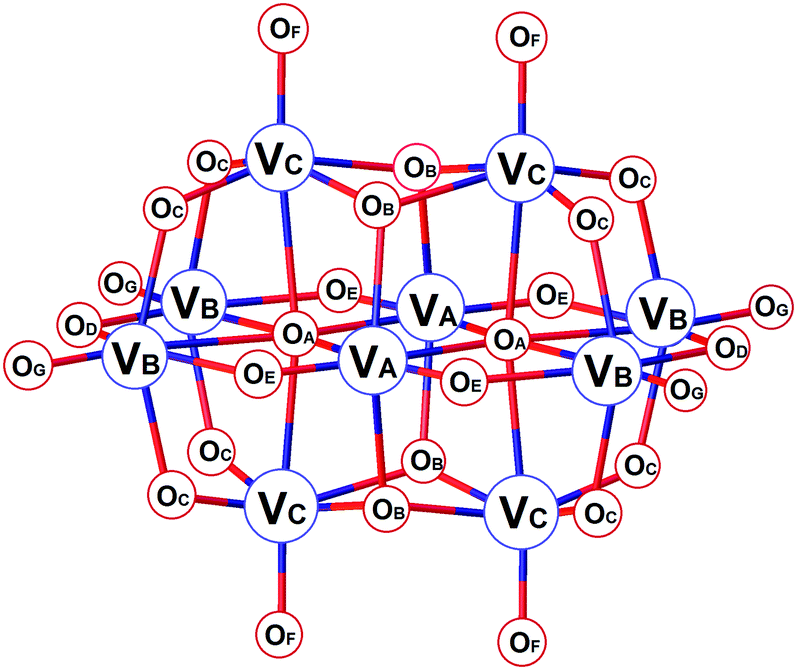 | ||
| Scheme 1 The structure of [V10O28]6− showing different types of vanadium and oxygen atoms. | ||
Vanadium is naturally omnipresent in biological matrices33,34 in a few enzymes such as vanadium dependent haloperoxidases and nitrogenases35 or acts as a crucial component in the energetic metabolism of Ascidians.36 Some artificial vanadium compounds, on the other hand, exhibit insulin mimetic properties, antitumor activity, antibacterial activity or anti-HIV activity.35–39
Specifically decavanadate itself has also been studied with respect to many biological aspects,40–43 and it was shown that V10 binds to several proteins such as actin,44 myosin,45 ion pump Ca2+–ATPase,46 bovine serum albumin and gelatine,47 and microtubule-associated proteins.48 Protein crystallography revealed the presence of V10 in the crystal structures of acid phosphatase A (F. tularensis),49 human activated receptor tyrosine kinase,50 NTPDase1 (L. pneumophila),51 NTPDase1 (R. norvegicus),52 and human TRPM4 channel.53 In all cases, V10 was formed from the initially employed Na3VO4 (used as a phosphatase inhibitor) and its role may be explained as stabilization and rigidification of the protein structure.25
In this work we compare the interaction of both free and ligated decavanadate with commercially available model proteins thaumatin, lysozyme, proteinase K, as well as human serum albumin and transferrin. We utilize 51V NMR spectroscopy as a powerful tool to investigate the stability of two decavanadates coordinated to metal centres Cu(II) and Co(II) under conditions usually used for protein crystallization.
2 Experimental
2.1 Materials and methods
All chemicals were of analytical grade and used as received without further purification. All proteins were supplied as lyophilized powders (supplier, reference code): thaumatin from Thaumatococcus danielii (Sigma, T7638; a mixture of thaumatin I and thaumatin II with traces of other sweet proteins), proteinase K from Tritirachium album (Sigma, P6556), lysozyme from chicken eggwhite (Carl Roth GmbH & Co. KG, 8259.3), albumin from human serum (Sigma, A1653) and apo-transferrin from human serum (Sigma, T1147). The determination of C/H/N was carried out by using an ‘EA 1108 CHNS-O’ elemental analyzer by Carlo Erba Instruments at the Mikroanalytisches Laboratorium, University of Vienna. Metal elements’ analyses were performed in aqueous solutions containing 2% HNO3 using inductively coupled plasma mass spectrometry (PerkinElmer Elan 6000 ICP MS) for Mo and V, and atomic absorption spectroscopy (PerkinElmer 1100 Flame AAS) for Cu and Co. Standards were prepared from single-element standard solutions of concentration 1000 mg L−1 (Merck, Ultra Scientific and Analytika Prague). FT-IR spectroscopy was performed on a Bruker Vertex 70 IR Spectrometer equipped with a single reflection diamond-ATR (attenuated total reflectance) unit in the range of 4000–100 cm−1.2.2 51V NMR spectroscopy
51V nuclear magnetic resonance spectroscopy measurements of aqueous solutions were performed on a Bruker Avance II 500 MHz instrument operating at 131.60 MHz for 51V nucleus (2000 scans, accumulation time 0.05 s, relaxation delay 0.01 s). Chemical shift values are given with reference to VOCl3 (δ = 0 ppm) as a standard. The solutions were in general prepared by dissolving 0.01 mmol of the given decavanadate in 700 μL of the buffer solution (0.11 M MES, 0.55 M NaCl, pH = 5.8; MES = 2-(N-morpholino)ethanesulfonic acid) and addition of 100 μL of D2O used for locking. Next, the solution was either made up to 1000s μL with the buffer (Section 3.3.1) or a solution of a protein in the same buffer was slowly added. The protein solutions were prepared by dissolution of the given amount of solid protein in 200 μL of the buffer solution. The solution of proteinase K was prepared as 3.5 M solution in 0.1 M TRIS (tris(hydroxymethyl)aminomethane) buffer (pH = 7.0), and subsequently 1 μL of this solution was diluted in 200 μL of the MES buffer solution. The spectra were collected at RT one hour after preparation of the solutions.2.3 X-ray diffraction on single crystals
The X-ray diffraction data were collected on Bruker X8 APEXII (V10Cu, Mo Kα) and Bruker D8 Venture (V10Co, Cu Kα) instruments equipped with multilayer monochromators, Incoatec Microfocus sealed tubes, and Kryoflex and Oxford cooling devices. The structures were solved by direct methods and refined by full-matrix least-squares. Nonhydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were inserted at calculated positions and refined with riding coordinates except for solvent molecules and H atoms in NH4+ in V10Cu. This was not done because of disorder and partial occupancies of O and N atoms in question. The following software was used for the structure solving procedure: Bruker SAINT software package54 (frame integration, cell refinement), SADABS55 (absorption correction), SHELXS-201356 (structure solution), SHELXL-201357 (structure refinement), OLEX258 (user interface and publication material) and Diamond59 (graphics). Experimental data and CCDC-codes can be found in Table 2.2.4 Syntheses
(NH4)6[V10O28]·6H2O (V10): the compound was prepared as per the previously described procedure60 and the composition was confirmed by FT-IR spectroscopy and elemental analysis. Analytical data for V10O34N6H36 in % (calc.): V 46.3 (43.4). IR: [V10O28]6−: 958vs, 816m, 727s, 588m, 516s, 452s, 392vs; NH4+: 1405s, 3151sh, 3009sh, 2008sh.(2-hepH)(NH4)[{Cu(H2O)2(2-hep)}2V10O28]·4H2O (V10Cu): NH4VO3 (0.468 g, 4 mmol) was dissolved in 30 mL of distilled water by heating. After cooling to RT, the pH of the solution was adjusted to 4.6 with 5 M HCl. Then, 2-hep (2-hep = 2-hydroxyethylpyridine) was added (0.45 mL, 4 mmol) and the pH was adjusted to 4.6 with 5 M HCl again. Finally, Cu(NO3)2·6H2O was added (0.484 g, 2 mmol). After dissolution, the pH dropped to 4.0. The clear orange-green solution was left to crystallize at 18 °C. After three days, green crystals of V10Cu were isolated. Yield 250 mg (39% based on V). Analytical data for V10Cu2O39C21H48N4 in % (calc.): V 30.7 (31.5), Cu 7.76 (7.86), C 15.36 (15.60), H 3.02 (3.00), N 3.41 (3.46). IR: [V10O28]6−: 983s, 959vs, 937s, 850s, 793s, 721s, 512s, 429s, 386s; 2-hep: 1609m, 1488m, 1313m, 1248w, 1109w, 1073w, 1051w, 1025w; NH4+: 1439s, 3210sh.
(2-hepH)2[{Co(H2O)5}2V10O28]·4H2O (V10Co): a procedure similar to that in the previous synthesis was employed, except that upon addition of Co(NO3)2·6H2O (0.596 g, 2 mmol) it was necessary to adjust the pH of the solution to 4.0 with 5 M HCl. The clear red solution was left to crystallize at RT. After three days, red crystals of V10Co were isolated. Yield 400 mg (63% based on V). Analytical data for V10Co2O44C14H48N2 in % (calc.): V 31.7 (32.3), Co 7.17 (7.48), C 10.55 (10.67), H 3.11 (3.07), N 1.82 (1.78). IR: [V10O28]6−: 989s, 967vs, 921s, 811s, 724s, 596s, 554s, 505s, 446m, 398s; 2-hep: 1608m, 1772m, 1248w, 1024w. Crystals suitable for X-ray diffraction were grown by prolonged crystallization at 18 °C in a flask closed with perforated Parafilm.
3 Results and discussion
3.1 Syntheses
To the best of our knowledge, the work of Schwendt et al.61 presenting the synthesis and characterization of (2-hepH)2[{Cu(H2O)2(2-hep)}2V10O28]·6H2O is the only one reporting a coordination compound of decavanadate that stays intact in aqueous solution. Inspired by this work, the synthetic protocol was modified in order to prepare new stable complexes of decavanadate. A reversed addition of the individual reaction components and stepwise acidification of the solution prevented formation of heavy precipitates that were described in the original procedure61 and such an approach resulted in successful isolation of V10Cu containing the [{Cu(H2O)2(2-hep)}2V10O28]2− complex anion and a new compound V10Co composed of the purely inorganic complex anion [{Co(H2O)5}2V10O28]2−. In this case, 2-hep did not coordinate to the cobalt(II) center and acted only as a cation. Syntheses employing NiII, ZnII, MnII, LaIII and CeIII nitrates were not successful. Despite the systematic study it was possible to isolate only two complexes of decavanadate, and even they differed in the coordination mode of decavanadate and in the structure of the coordinated transition metal. Table 1 summarizes 22 compounds where decavanadate coordinates to transition metal cations. There are ten CuII, four ZnII, four MnII and two AgI complexes. Compound V10Co is the first complex of decavanadate involving CoII. There are about 100 salts of decavanadate and some of them contain a transition metal complex in the cationic part, including the [Co(H2O)6]2+ complex cation.62a–c Therefore, in general it is still not clear what conditions are required for decavanadate to act as a ligand.| Compound | Type of coordinated O atoma | Ref. |
|---|---|---|
| a As in Scheme 1. Abbreviations: 2,2′-bipy – 2,2′-bipyridine, btx – 1,4-bis(triazol-1-ylmethyl)benzene, ppz – piperazine, pz – pyrazole, im – imidazole, mim – 1-methylimidazole, trz - 1,2,4-triazole, en – ethylenediamine, amp – 2-(aminomethyl)pyridine, 2-hep = 2-hydroxyethylpyridine, L – 5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane. | ||
| [Cu(2,2′-bipy)2]2[H2V10O28]·(2,2′-bipy)·H2O | OC | 63 |
| {[(CuL)0.5(H2L)1.5][H2V10O28]·6H2O}n | OF | 64 |
| (NH4)2[Cu2(NH3CH2CH2COO)4(V10O28)]·10H2O | OG | 65 |
| (Hpz)2[{Cu(pz)4}2V10O28]·2H2O | OG, OF | 66 |
| {Cu(pz)}4[{Cu(pz)3}2V10O28] | OG, OC | 66 |
| (2-hepH)2[{Cu(H2O)2(O,N-2-hep)}2(V10O28)]·6H2O | OC | 61 |
| (2-hepH)(NH4)[{Cu(H2O)2(2-hep)}2V10O28]·4H2O | OC | This work |
| [Cu(2-amp)2(H2O)]2H2V10O28·4H2O | OC | 61 |
| (H3O)2[{Cu(en)2(H2O)}2V10O28]·3H2O | OC, OD | 67 |
| [{Cu(en)2}3(V10O28)]·6H2O | OC, OD | 67 |
| [Zn2(H2O)14(V10O28)]·H2ppz | OD | 68 |
| [Zn(H2O)6][Zn2V10O28(H2O)10]·6H2O | OF | 69 |
| [Zn(en)2]3[V10O28]·5H2O | OC | 70 |
| [Zn(im)2(DMF)2]2[H2V10O28]·im·DMF | OC | 71 |
| {[Zn3(trz)3(H2O)4(DMF)]2[V10O28]·4H2O}n | OC | 71 |
| (2-hepH)2[{Mn(H2O)5}2V10O28]·4H2O | OF | 72 |
| {[Mn(mim)4]2[H2V10O28]} | OC, OF | 73 |
| (NMe4)2[{Mn(H2O)5}2V10O28]·5H2O | OF | 76 |
| [NH3C(CH2OH)3]2[{Mn(H2O)5}2V10O28]·2H2O | OF | 76 |
| [Ag(btx)}4H2V10O28]·2H2O | OC | 74 |
| [{Na3(H2O)8(μ2-H2O)6Ag2}HV10O28]·6H2O | OC, OG | 75 |
| (2-hepH)2[{Co(H2O)5}2V10O28]·4H2O | OF | This work |
3.2 Crystal structures
The structure solution and refinement details for compounds V10Cu and V10Co are summarized in Table 2. The crystal structure refinement of V10Cu revealed the presence of the complex anion [{Cu(H2O)2(2-hep)}2V10O28]2− that has been already reported (Fig. 1).61 The decavanadate anion [V10O28]6− is a bridging ligand between two {Cu(H2O)2(2-hep)}2+ fragments and binds to the Cu1 atom through oxygen atom O12 (OC in Scheme 1) at 1.9612(8) Å. The organic ligand is chelating the Cu1 atom by the N atom of the pyridine ring [N1–Cu1 1.9802(11) Å] and the O atom of the hydroxyl group [O13–Cu1 1.9857(9) Å]. The tetragonal pseudoplane around the atom Cu1 is completed by a water molecule [O15–Cu1 1.9824(10) Å]; the water molecule in the axial position completes the pyramidal geometry [O14–Cu1 2.2860(10) Å].| V10Cu | V10Co | |
|---|---|---|
| CCDC No. | 1909251 | 1909250 |
| Empirical formula | V10Cu2O39C21H48N4 | V10Co2O44C14H48N2 |
| M r | 1605.02 | 1575.80 |
| Crystal system, space group | Monoclinic, C2/c | Triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| Temperature (K) | 100 | 100 |
| a (Å) | 27.0345(15) | 9.4219(5) |
| b (Å) | 9.9170(5) | 11.2451(6) |
| c (Å) | 20.5851(12) | 11.9820(7) |
| α (°) | 78.5909(17) | |
| β (°) | 119.341(5) | 75.1180(17) |
| γ (°) | 66.3817(17) | |
| V (Å3) | 4810.9(5) | 1117.66 (11) |
| Z | 4 | 1 |
| Radiation type | Mo Kα | Cu Kα |
| μ (mm−1) | 2.82 | 23.51 |
| Absorption correction | Multi-scan | Multi-scan |
| T min, Tmax | 0.505, 0.747 | 0.534, 0.754 |
| F(000) | 3160 | 782 |
| Theta range | 2.2–35.7° | 5.7–72.6° |
| Crystal size in mm | 0.45 × 0.25 × 0.22 | 0.11 × 0.11 × 0.04 |
| Index ranges | h = −44 → 44 | h = −11 → 11 |
| k = −16 → 16 | k = −13 → 13 | |
| l = −33 → 33 | l = −12 → 14 | |
| No. of measured, independent and observed [I > 2s(I)] reflections | 343![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 398, 11216, 9986 398, 11216, 9986 |
16530, 4079, 3882 |
| R int | 0.062 | 0.127 |
| No. of reflections, parameters, restraints | 11216, 421, 9 | 4079, 337, 20 |
| Goodness-of-fit on F2 | 1.10 | 1.16 |
| R[F2 > 2s(F2)] | 0.024 | 0.098 |
| wR(F2) | 0.071 | 0.222 |
| Largest diff. peak and hole | 0.79 e Å−3, −0.65 e Å−3 | 1.79 e Å−3, −1 e Å−3 |
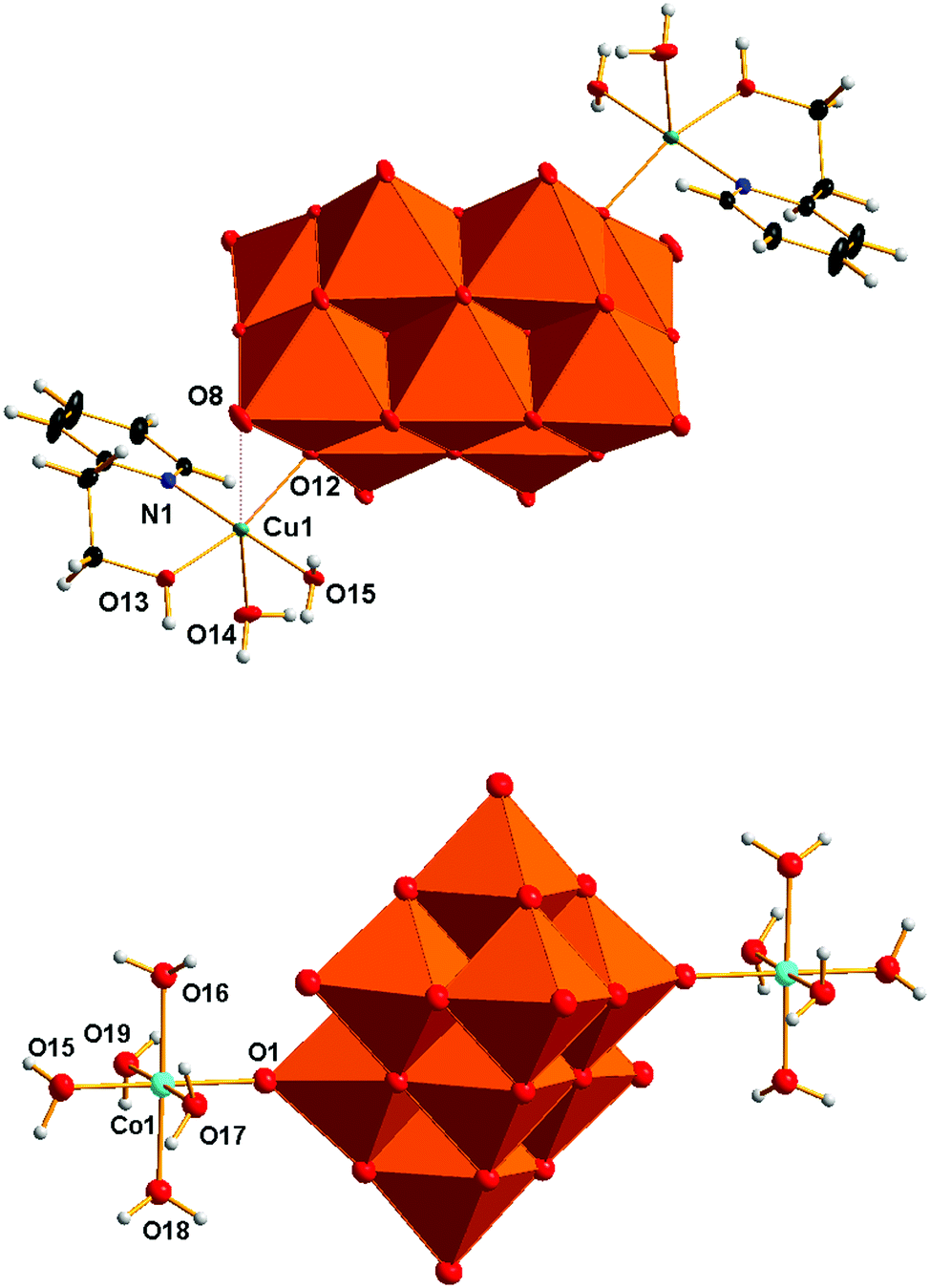 | ||
| Fig. 1 Molecular structures of the anions [{Cu(H2O)2(2-hep)}2V10O28]2− (V10Cu) and [{Co(H2O)5}2V10O28]2− (V10Co) as revealed by X-ray crystallography showing atom labelling. Colour code: orange octahedra {VO6}, green Cu, light blue Co, blue N, red O, black C, gray H. Ellipsoids are displayed at a 30% probability level. | ||
The closest contact in the trans position towards this water molecule is oxygen atom O8 of [V10O28]6− (OG in Scheme 1). The short distance O8–Cu1 2.889 Å indicates some weak attraction. The crystal structure of V10Cu differs from the previously described one61 in the presence of the NH4+ cation that was confirmed by elemental analysis and IR spectroscopy.
The asymmetric unit of V10Co contains one half of the centrosymmetric anion [{Co(H2O)5}2V10O28]2− (Fig. 1), one molecule of (2-hepH)+ balancing its charge and two water molecules of crystallization. The decavanadate anion [V10O28]6− is acting as a bridging ligand for two {Co(H2O)5}2+ fragments. The slightly irregular octahedral coordination sphere of the Co1 atom is completed by one oxygen atom O1 coming from the terminal V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of the decavanadate (atom OF in Scheme 1). Such a coordination fashion is not unknown and was already reported for manganese(II)72,76 and zinc(II) derivatives.68,69 The bond length O1–Co1 2.089(3) Å indicates a relatively strong coordination bond of decavanadate to the metal centre. The bond lengths of the Co1 atom and oxygen atoms of the coordinated water molecules are in the range of 2.105 – 2.115 Å for the equatorial ligands and 2.057(4) Å for the Co1–O15 bond of the water molecule in the trans position towards the decavanadate ligand.
O group of the decavanadate (atom OF in Scheme 1). Such a coordination fashion is not unknown and was already reported for manganese(II)72,76 and zinc(II) derivatives.68,69 The bond length O1–Co1 2.089(3) Å indicates a relatively strong coordination bond of decavanadate to the metal centre. The bond lengths of the Co1 atom and oxygen atoms of the coordinated water molecules are in the range of 2.105 – 2.115 Å for the equatorial ligands and 2.057(4) Å for the Co1–O15 bond of the water molecule in the trans position towards the decavanadate ligand.
The supramolecular structure of V10Co is stabilized by a rich network constructed from hydrogen bonds in which all the individual components are involved. Due to the presence of {Co(H2O)5}2+ fragments the most prominent hydrogen bonds are formed between the adjacent [{Co(H2O)5}2V10O28]2− anions. Three oxygen atoms of the decavanadate O5, O6 and O11 are connected with hydrogen bonds to three water molecules of {Co(H2O)5}2+ at contact distances O5⋯O17 2.636 Å, O6⋯O17 2.636 Å and O11⋯O15 2.744 Å.
3.3 Solution studies
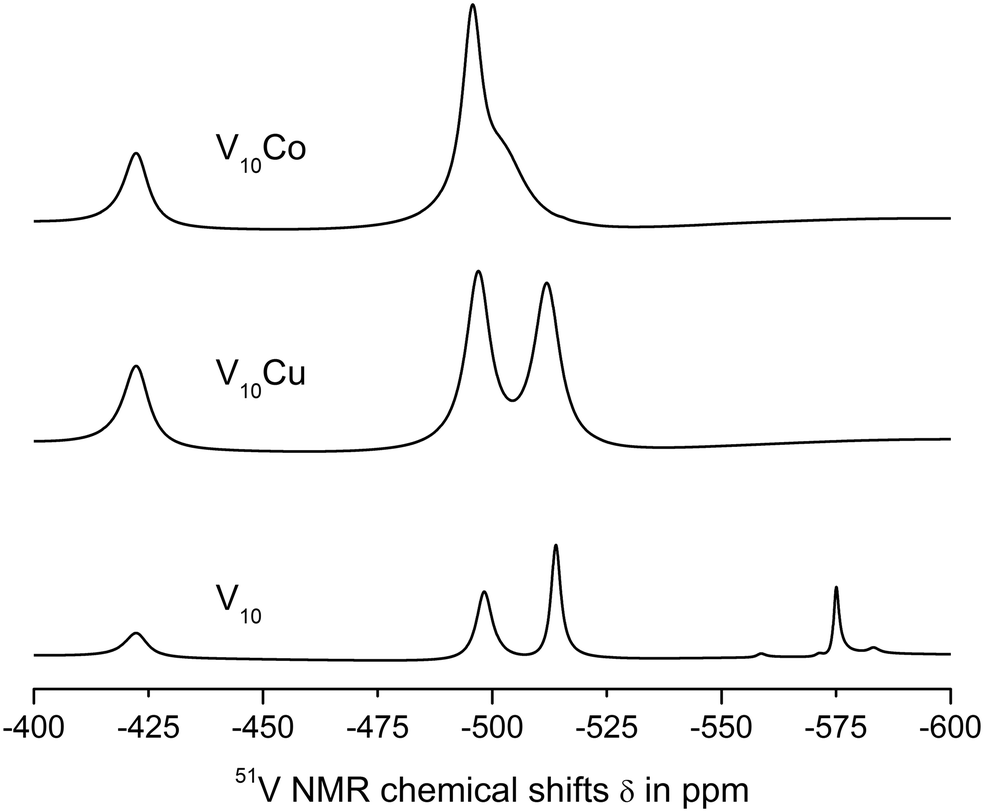 | ||
| Fig. 2 51V NMR spectra of 1 mM aqueous solutions of V10, V10Cu and V10Co in 0.1 M MES buffer, 0.5 M NaCl at pH = 5.8. | ||
The spectra of V10Cu and V10Co exhibit only peaks that can be assigned to vanadium atoms arising from decavanadate: −422.3, −496.9 and −511.9 ppm for V10Cu and −422.3 and −495.9 ppm (broad peak) for V10Co. This is the first indication that the complex anions stay intact and do not dissociate off the Cu(II) and Co(II) centres. As a matter of fact, the presence of free decavanadate would evoke vanadate self-condensation reactions and the overall picture of the present species should be similar to that of V10. Next, similar to Schwendt et al.,61 we observed significant movement of the high-field signal of the VC atom in V10Cu to −511.9 ppm compared to −513.8 ppm in V10, and the signal of the VB atom is shifted by 1.3 ppm. For V10Co, the shift of the peak is much more obvious and instead of two independent signals for VB and VC atoms we observe only a broad peak with the maximum at −495.9 ppm having a high-field shoulder. Based on peak integration, the integral intensities of the two present signals are in the ratio 2:8. In addition, significant peak broadening of the individual signals was observed (Table 3). This can be explained by at least two factors. Firstly, vanadates usually provide narrower lines in comparison to common vanadium(V) complexes because of higher symmetry (i.e. D2h for ideal [V10O28]6−). Thus, peak broadening originates in the decrease of symmetry of the coordinated decavanadates. It is also important to note that for V10Co the low field signal is broadened by only 10% in comparison to free decavanadate, while the second signal is broader by more than 220% compared to the sum of individual signals of VB and VC in V10. This difference is naturally caused by the fact that the VC atoms are the closest ones to the Co(II) atom and the inner VA atoms are less affected by the coordination. The compound V10Cu formed a cloudy precipitate in the buffer solution which might have also influenced the peaks’ width, but despite this, similar peak broadening as for V10Co was observed – 14% and 200%, respectively. This leads to the conclusion that the peak coalescence is a consequence of its coordination to the Cu(II) center and not the presence of a precipitate. However, we excluded V10Cu from further examination to prevent misinterpretation of protein interaction experiments.
| HxV10O28(6−x)− | H2VO4− | H2V2O72− | V4O124− | V5O155− | |||
|---|---|---|---|---|---|---|---|
| VA | VB | VC | |||||
| a The linewidth was calculated from the half-height for the low-field and high-field components of the merged broad signal. | |||||||
| V10 | −422.5 | −498.2 | −513.8 | −558.5 | −571.2 | −575.0 | −583.0 |
| 636.22 | 483.86 | 309.16 | 176.77 | ||||
| V10Cu | −422.3 | −496.9 | −511.9 | — | — | — | — |
| 727.51 | 775.17 | 806.75 | |||||
| V10Co | −422.3 | −495.9 | — | — | — | — | |
| 702.38 | 1794.05a | ||||||
On the other hand, peak broadening may also originate in the presence of paramagnetic centres Cu(II) (d9) and Co(II) (d7). The experience has shown, however, that if an extraneous paramagnetic species interferes in a 51V NMR experiment, this manifests itself also in a lower signal-to-noise ratio and uneven lines (this was not the case).
At pH = 8.0 (0.1 M MES, 0.5 NaCl) profound decomposition of V10 into lower vanadates was observed, V10Cu dissociated off the Cu(II) complex cation, while V10Co was still the only species present (see Fig. S1 and Table S1, ESI†).
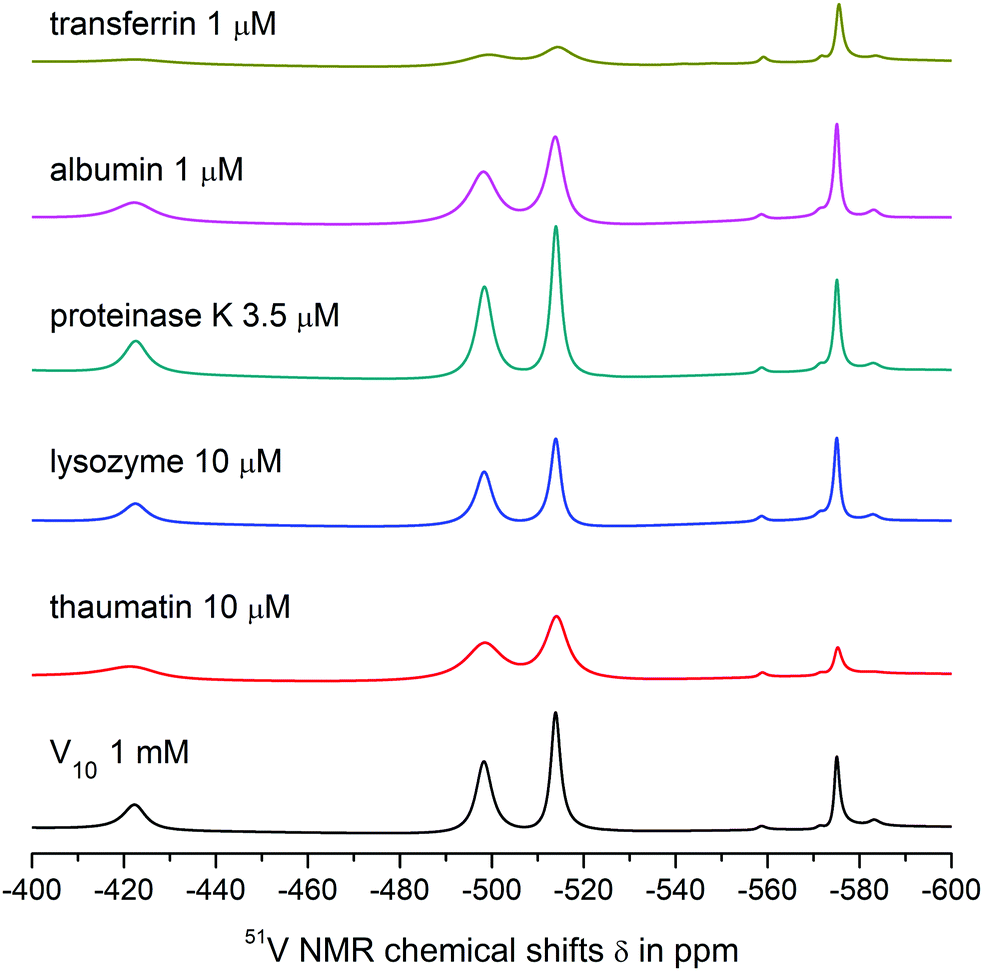 | ||
| Fig. 3 51V NMR spectra showing interactions of 1 mM aqueous solutions of V10 with model proteins under given concentrations in 0.1 M MES buffer, 0.5 M NaCl at pH = 5.8. The black line represents the spectrum of the referent V10Co and the coloured lines show the spectra of solutions of V10Co in the presence of different proteins at given concentrations. | ||
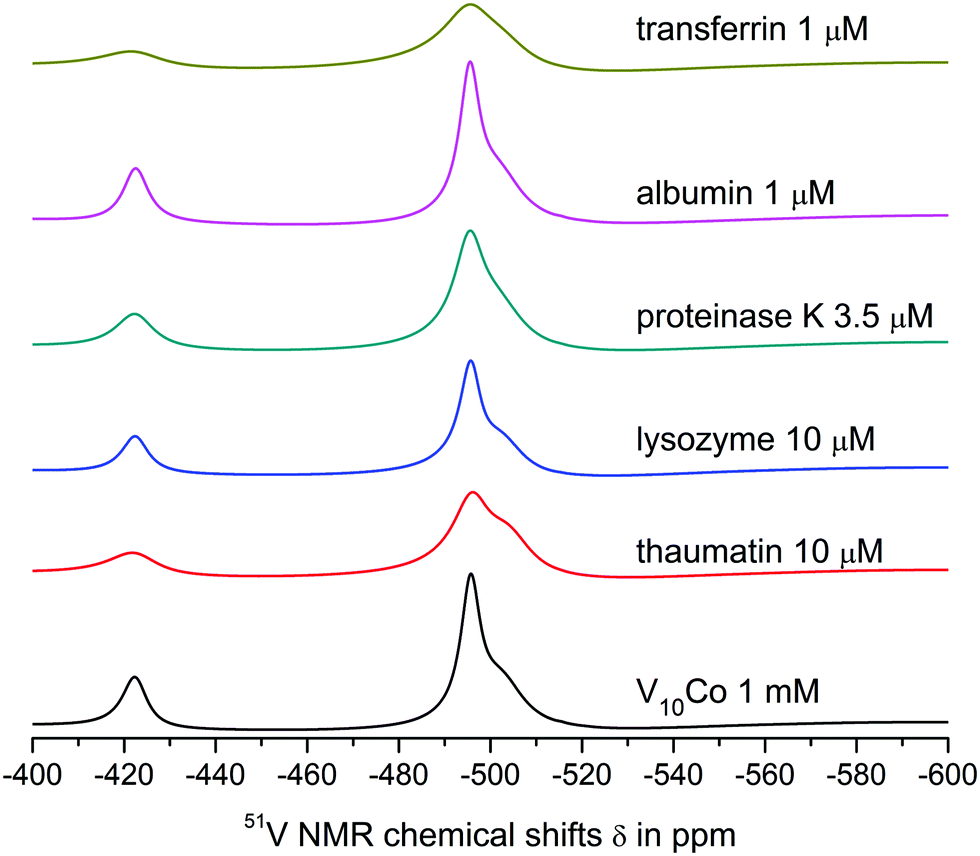 | ||
| Fig. 4 51V NMR spectra showing interactions of 1 mM aqueous solutions of V10Co with model proteins under given concentrations in 0.1 M MES buffer, 0.5 M NaCl at pH = 5.8. The black line represents the spectrum of the referent V10Co and the coloured lines show the spectra of solutions of V10Co in the presence of different proteins at given concentrations. | ||
| Protein | Decavanadate | HxV10O28(6−x)− | V4O124− | |||
|---|---|---|---|---|---|---|
| VA | VB | VC | ||||
| a The linewidth was calculated from the half-height for the low-field and high-field components of the merged broad signal. | ||||||
| None | V10 | −422.5 | −498.2 | −513.8 | −575.0 | |
| 636.22 | 483.86 | 309.16 | 176.77 | |||
| V10Co | −422.3 | −495.9 | — | |||
| 702.38 | 1794.05a | |||||
| Thaumatin 10 μM | V10 | −421.5 | −498.5 | −514.1 | −575.2 | |
| 1144.08 | 1198.98 | 746.33 | 274.76 | |||
| V10Co | −421.8 | −495.9 | — | |||
| 1265.82 | 2254.31a | |||||
| Lysozyme 10 μM | V10 | −422.5 | −498.3 | −4513.9 | −575.0 | |
| 580.67 | 530.95 | 342.1 | 199.5 | |||
| V10Co | −422.4 | −495.7 | — | |||
| 732.6 | 1810.91a | |||||
| Proteinase K 3.5 μM | V10 | −422.6 | −498.4 | −513.9 | −575.1 | |
| 653.95 | 510.83 | 326.98 | 194.65 | |||
| V10Co | −422.3 | −495.6 | — | |||
| 977.23 | 2041.13a | |||||
| Albumin 1 μM | V10 | −422.3 | −498.2 | −513.8 | −575.0 | |
| 943.72 | 851.65 | 532.05 | 192.97 | |||
| V10Co | −422.5 | −495.6 | — | |||
| 709.7 | 1629.61a | |||||
| Transferrin 1 μM | V10 | −422.6 | −499.5 | −513.8 | −575.5 | |
| 967.87 | — | 983.55 | 230.59 | |||
| V10Co | −421.6 | −495.6 | — | |||
| 1268.63 | 2404.42a | |||||
Both V10 and V10Co bind strongly to thaumatin resulting in significant peak broadening. In the case of lysozyme, very weak binding to V10 may be deduced from slightly broadened higher-field peaks, but no obvious interaction was observed for V10Co. Finally, proteinase K seemed not to interact with free decavanadate, but binds strongly to V10Co. In this case we used about 3 times lower protein concentration; therefore, after extrapolation of the data it can be assumed that the binding of V10Co to proteinase K is comparatively stronger than to thaumatin.
O groups of decavanadate occupy the binding sites for interaction of decavanadate with this protein and block the interaction. In fact, this was the only case where the interaction was observed only for the free decavanadate but not for the coordinated one. The interaction of V4O124− with human serum albumin is much weaker than that of decavanadate based on only about 10% increase in the peak width.
The interaction of V10 with transferrin is even stronger, and a significant binding to V4O124− was also observed. The shift of the peak corresponding to the VB atom of the decavanadate by 1 ppm made it impossible to determine its half-height width. As expected, the presence of Co(II) causes extremely strong binding of V10Co to transferrin, resulting in peak broadening comparable to that observed in the case of thaumatin, but at 100× lower protein concentration.
The stability of V10Co, V10Cu and potentially other coordinated decavanadates in the examined medium at 10 mM total vanadium concentration, in line with the potential of functionalized decavanadates to interact with various proteins, open new possibilities for the investigation of decavanadate's effects in biological systems – for the first time without the side effects of the always present lower oligovanadates (decavanadate may be the only species present in multicomponent solvents).77 On the other hand, at physiological concentrations (cV = 1 μM and less), vanadate exists only as a monomeric species (VO2+, H2VO4−, HVO4− depending on the pH).32 We therefore checked the stability of V10Co at 1 μM concentration (10 μM total vanadium) by 51V NMR (Fig. S2, ESI†). The chemical shifts corresponding to the decavanadate species (−421.2, −497.9 and −513.1) represent an undisturbed anion indicating that the Co(II) centers are no longer involved in coordination.
4 Conclusions
In this work, we showed that the complex anions [{Cu(H2O)2(2-hep)}2V10O28]2− and [{Co(H2O)5}2V10O28]2− involving coordinated decavanadate are stable in aqueous solution and do not decompose in the buffer solution compatible with proteins (0.1 M MES, 0.5 NaCl) at pH = 5.8 making them promising candidates for biological studies. For the first time, an interaction between modified decavanadate and biomolecules without the participation of lower oligovanadates was performed. The pilot interaction studies with several proteins used in model protein crystallization research showed that V10 and V10Co bind to thaumatin, V10 binds also to lysozyme and V10Co binds to proteinase K. As expected, V10 interacts with human serum albumin and transferrin, but surprisingly V10Co exhibits high affinity to transferrin but does not interact with albumin. The isolation and structural characterization of the crystalline products is the ultimate goal necessary for more precise understanding of the interaction between POMs and proteins. It is expected that, in addition to electrostatic interaction of V10 with proteins, the presence of heterometals may induce a complementary interaction – even a covalent bond – when the coordinated transition metal contains labile ligands such as water molecules (as in V10Co and V10Cu) or a vacant accessible coordination position (as in V10Cu). Furthermore, both V10Cu and V10Co contain biogenic transition metals that are known to interact with biomolecules to a great extent. In conclusion, the high potential of ligated decavanadate in medicinal chemistry and protein crystallography necessitates the challenging development of synthetic methods leading reliably to stable complexes of decavanadate (i.e. decavanadato complexes).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Austrian Science Fund (FWF): M2200 (LK) and P27534 (AR) and the University of Vienna. The authors are grateful to Dr Marek Bujdoš (Comenius University in Bratislava, Faculty of Natural Sciences, Institute of Laboratory Research on Geomaterials, Mlynská dolina, Ilkovičova 6, Bratislava, 84215, Slovakia) for elemental analyses and Ao. Univ.-Prof. Dr Markus Galanski (Universität Wien, Fakultät für Chemie, Institut für Anorganische Chemie, Währinger Straße 42, 1090 Wien, Austria) for great support with 51V NMR measurements.References
- M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer-Verlag Berlin Heidelberg, 1983 Search PubMed.
- M. T. Pope, M. Sadakane and U. Kortz, Eur. J. Inorg. Chem., 2019, 340–342, DOI:10.1002/ejic.201801543.
- J. F. Keggin, Nature, 1933, 131, 908–909, DOI:10.1038/131908b0.
- M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34–48, DOI:10.1002/anie.199100341.
- E. Coronado and C. J. Gómez-García, Chem. Rev., 1998, 98, 273–296, DOI:10.1021/cr970471c.
- Y.-F. Song and R. Tsunashima, Chem. Soc. Rev., 2012, 41, 7384–7402, 10.1039/c2cs35143a.
- C. L. Hill and C. M. Prosser-McCartha, Coord. Chem. Rev., 1995, 143, 407–455, DOI:10.1016/0010-8545(95)01141-B.
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962, DOI:10.1021/cr500390v.
- H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572–7589, 10.1039/c2cs35292c.
- N. Mizuno, K. Yamaguchi and K. Kamata, Coord. Chem. Rev., 2005, 249, 1944–1956, DOI:10.1016/j.ccr.2004.11.019.
- M. Sadakane and E. Steckhan, Chem. Rev., 1998, 98, 219–238, DOI:10.1021/cr960403a.
- N. I. Gumerova and A. Rompel, Nat. Rev. Chem., 2018, 2, 0112, DOI:10.1038/s41570-018-0112.
- C. Streb, K. Kastner and J. Tucher, Phys. Sci. Rev., 2019, 4 DOI:10.1515/psr-2017-0177.
- C. Streb, Dalton Trans., 2012, 41, 1651–1659, 10.1039/C1DT11220A.
- J. Tucher, Y. Wu, L. C. Nye, I. Ivanovic-Burmazovic, M. M. Khusniyarov and C. Streb, Dalton Trans., 2012, 41, 9938–9943, 10.1039/C2DT30304C.
- M. A. Al-Damen, J. M. Clemente-Juan, E. Coronado, C. Martí-Gastaldo and A. Gaita-Ariño, J. Am. Chem. Soc., 2008, 130, 8874–8875, DOI:10.1021/ja801659m.
- J. M. Clemente-Juan, E. Coronado and A. Gaita-Ariño, Chem. Soc. Rev., 2012, 41, 7464–7478, 10.1039/c2cs35205b.
- J. M. Clemente-Juan and E. Coronado, Coord. Chem. Rev., 1999, 193–195, 361–394, DOI:10.1016/S0010-8545(99)00170-8.
- J. T. Rhule, C. L. Hill, D. A. Judd and R. F. Schinazi, Chem. Rev., 1998, 98, 327–358, DOI:10.1021/cr960396q.
- L. S. Van Rompuy and T. N. Parac-Vogt, Curr. Opin. Biotechnol., 2019, 58, 92–99, DOI:10.1016/j.copbio.2018.11.013.
- D. A. Judd, J. H. Nettles, N. Nevins, J. P. Snyder, C. D. Liotta, J. Tang, J. Ermolieff, R. F. Schinazi and C. L. Hill, J. Am. Chem. Soc., 2001, 123, 886–897, DOI:10.1021/ja001809e.
- A. Bijelic, M. Aureliano and A. Rompel, Chem. Commun., 2018, 54, 1153–1169, 10.1039/c7cc07549a.
- H.-J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637–643, DOI:10.1002/cbic.200301023.
- A. Bijelic, M. Aureliano and A. Rompel, Angew. Chem., Int. Ed., 2019, 58, 2980–2999, DOI:10.1002/anie.201803868 and 10.1002/ange.201803868.
- A. Bijelic and A. Rompel, Coord. Chem. Rev., 2015, 299, 22–38, DOI:10.1016/j.ccr.2015.03.018.
- A. Bijelic and A. Rompel, Acc. Chem. Res., 2017, 50, 1441–1448, DOI:10.1021/acs.accounts.7b00109.
- C. Molitor, A. Bijelic and A. Rompel, IUCrJ, 2017, 4, 734–740, DOI:10.1107/S2052252517012349.
- A. Bijelic and A. Rompel, ChemTexts, 2018, 4, 10, DOI:10.1007/s40828-018-0064-1.
- L. Pettersson, B. Hedman, A. M. Nennen and I. Andersson, Acta Chem. Scand., Ser. A, 1985, 39, 499–506, DOI:10.3891/acta.chem.scand.39a-0499.
- H. Schmidt, I. Andersson, D. Rehder and L. Pettersson, Chem. – Eur. J., 2001, 7, 251–257, DOI:10.1002/1521-3765(20010105)7:1<251::AID-CHEM251>3.0.CO;2-9.
- A. S. Tracey, G. R. Willsky and E. S. Takeuchi, Vanadium. Chemistry, Biochemistry and Practical Applications, CRC Press, 2007 Search PubMed.
- D. Rehder, Bioinorganic Vanadium Chemistry, John Wiley & Sons, Chichester, 2008 Search PubMed.
- D. Rehder, Metallomics, 2015, 7, 730–742, 10.1039/c4mt00304g.
- D. Rehder, Met. Ions Life Sci., 2013, 13, 139–169, DOI:10.1007/978-94-007-7500-8-5.
- D. C. Crans, J. J. Smee, E. Gaidamauskas and L. Yang, Chem. Rev., 2004, 104, 849–902, DOI:10.1021/cr020607t.
- T. Ueki, N. Yamaguchi, Y. Romaidi, Y. Isago and H. Tanahashi, Coord. Chem. Rev., 2015, 301–302, 300–308, DOI:10.1016/j.ccr.2014.09.007.
- J. C. Pessoa, S. Etcheverry and D. Gambino, Coord. Chem. Rev., 2015, 301–302, 24–48, DOI:10.1016/j.ccr.2014.12.002.
- D. Rehder, Future Med. Chem., 2012, 4, 1823–1827, DOI:10.4155/fmc.12.103.
- S. Y. Wong, R. W.-Y. Sun, N. P. Chung, C. L. Lin and C. M. Che, Chem. Commun., 2005, 3544–3546, 10.1039/B503535J.
- S. Treviño, A. Díaz, E. Sánchez-Lara, B. L. Sanchez-Gaytan, J. M. Perez-Aguilar and E. González-Vergara, Biol. Trace Elem. Res., 2019, 56, 10893–10903, DOI:10.1007/s12011-018-1540-6.
- M. Aureliano and D. C. Crans, J. Inorg. Biochem., 2009, 103, 536–546, DOI:10.1016/j.jinorgbio.2008.11.010.
- M. Aureliano, Oxid. Med. Cell. Longevity, 2016, 6103457, DOI:10.1155/2016/6103457.
- M. Aureliano and R. M. C. Gândara, J. Inorg. Biochem., 2005, 99, 979–985, DOI:10.1016/j.jinorgbio.2005.02.024.
- M. P. M. Marques, D. Gianolio, S. Ramos, L. A. E. B. D. Carvalho and M. Aureliano, Inorg. Chem., 2017, 56, 10893–10903, DOI:10.1021/acs.inorgchem.7b01018.
- T. Tiago, P. Martel, C. Gutiérrez-Merino and M. Aureliano, Biochim. Biophys. Acta, Gen. Subj., 2007, 1774, 474–480, DOI:10.1016/j.bbapap.2007.02.004.
- M. Aureliano, G. Fraqueza and C. A. Ohlin, Dalton Trans., 2013, 42, 11770–11777, 10.1039/C3DT50462J.
- S. M. Ashraf Rajesh and S. Kaleem, Anal. Biochem., 1995, 230, 68–74, DOI:10.1006/abio.1995.1439.
- S. Lobert, N. Isern, B. S. Hennington and J. J. Correia, Biochemistry, 1994, 33, 6244–6252, DOI:10.1021/bi00186a026.
- R. L. Felts, T. J. Reilly and J. J. Tanner, J. Biol. Chem., 2006, 281, 30289–30298, DOI:10.1074/jbc.M606391200.
- J. H. Bae, E. D. Lew, S. Yuzawa, F. Tomé, I. Lax and J. Schlessinger, Cell, 2009, 138, 514–524, DOI:10.1016/j.cell.2009.05.028.
- M. Zebisch, M. Krauss, P. Schäfer, P. Lauble and N. Sträter, Structure, 2013, 21, 1460–1475, DOI:10.1016/j.str.2013.05.016.
- M. Zebisch, M. Krauss, P. Schäfer and N. Sträter, J. Mol. Biol., 2012, 415, 288–306, DOI:10.1016/j.jmb.2011.10.050.
- P. A. Winkler, Y. Huang, W. Sun, J. Du and W. Lü, Nature, 2017, 552, 200–204, DOI:10.1038/nature24674.
- Bruker SAINT, V8.32B Copyright 2005–2015, Bruker AXS, 2013 Search PubMed.
- G. M. Sheldrick, SADABS, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122, DOI:10.1107/S0108767307043930.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8, DOI:10.1107/S2053229614024218.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341, DOI:10.1107/S0021889808042726.
- Diamond – Crystal and Molecular Structure Visualization. Crystal Impact – Dr H. Putz & Dr K. Brandenburg GbR, Kreuzherrenstr. 102, 53227 Bonn, Germany. http://www.crystalimpact.com/diamond.
- G. G. Long, R. L. Stanfield and F. C. Hentz Jr., J. Chem. Educ., 1979, 56, 195–196, DOI:10.1021/ed056p195.
- L. Bartošová, Z. Padělková, E. Rakovský and P. Schwendt, Polyhedron, 2012, 31, 565–569, DOI:10.1016/j.poly.2011.10.042.
- (a) C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179, DOI:10.1107/S2052520616003954; (b) D. C. Crans, B. J. Peters, X. Wu and C. C. McLauchlan, Coord. Chem. Rev., 2017, 344, 115–130, DOI:10.1016/j.ccr.2017.03.016; (c) S. R. Amanchi and S. K. Das, Front. Chem., 2018, 6, 469, DOI:10.3389/fchem.2018.00469.
- T. H. Li, J. Lü, S. Gao, F. Li, F. Li and R. Cao, Chem. Lett., 2007, 36, 356–357, DOI:10.1246/cl.2007.356.
- G. C. Ou, L. Jiang, X. L. Feng and T. B. Lu, Dalton Trans., 2009, 71–76, 10.1039/B810802A.
- L. Klištincová, E. Rakovský and P. Schwendt, Inorg. Chem. Commun., 2008, 11, 1141–1142, DOI:10.1016/j.inoche.2008.06.020.
- J. Thomas, M. Agarwal, A. Ramanan, N. Chernova and M. S. Whittingham, CrystEngComm, 2009, 11, 625–631, 10.1039/B815840A.
- M. V. Pavliuk, V. G. Makhankova, O. V. Khavryuchenko, V. N. KokozayaIrin, I. V. Omelchenko, O. V. Shishkin and J. Jezierska, Polyhedron, 2014, 81, 597–606, DOI:10.1016/j.poly.2014.06.044.
- L. Wang, X. Sun, M. L. Liu, Y. Q. Gao, W. Gu and X. Liu, J. Cluster Sci., 2008, 19, 531–542, DOI:10.1007/s10876-008-0196-3.
- M. Graia, R. Ksiksi and A. Driss, J. Chem. Crystallogr., 2008, 38, 855–859, DOI:10.1007/s10870-008-9410-2.
- H. Pang, X. Meng, H. Ma, B. Liu and S. Li, Z. Naturforsch., 2012, 67b, 855–859, DOI:10.5560/ZNB.2012-0120.
- W. Xu, F. Jiang, Y. Zhou, K. Xiong, L. Chen, M. Yang, R. Feng and M. Hong, Dalton Trans., 2012, 41, 7737–7745, 10.1039/c2dt30532a.
- L. Klištincová, E. Rakovský and P. Schwendt, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, m97–m99, DOI:10.1107/S0108270109001917.
- J.-K. Li, C.-P. Wei, Y.-Y. Wang, M. Zhang, X.-R. Lv and C.-W. Hu, Inorg. Chem. Commun., 2018, 87, 5–7, DOI:10.1016/j.inoche.2017.11.009.
- Y. Qi, E. Wang, J. Li and Y. Li, J. Solid State Chem., 2009, 182, 2640–2645, DOI:10.1016/j.jssc.2009.07.022.
- S. Y. Samar and K. Das, J. Mol. Struct., 2017, 1146, 23–31, DOI:10.1016/j.molstruc.2017.05.097.
- P. Maurício, A. L. Franco, J. Rüdiger, J. F. Soares, G. G. Nunes and D. L. Hughes, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2015, 71, 146–150, DOI:10.1107/S2056989014028230.
- C. Slebodnick and V. L. Pecoraro, Inorg. Chim. Acta, 1998, 283, 37–43, DOI:10.1016/S0020-1693(98)00088-7.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1909250 and 1909251. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9nj02495f |
| ‡ Comenius University in Bratislava, Faculty of Natural Sciences, Department of Inorganic Chemistry, Ilkovičova 6, 842 15 Bratislava, Slovakia. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |