
3D pomegranate-like TiN@graphene composites with electrochemical reaction chambers as sulfur hosts for ultralong-life lithium–sulfur batteries†
Rongjie
Luo
ab,
Qiuhong
Yu
ab,
Yang
Lu
 ab,
Mengjie
Zhang
ab,
Tao
Peng
ab,
Hailong
Yan
ab,
Xianming
Liu
c,
Jang-Kyo
Kim
d and
Yongsong
Luo
*ab
ab,
Mengjie
Zhang
ab,
Tao
Peng
ab,
Hailong
Yan
ab,
Xianming
Liu
c,
Jang-Kyo
Kim
d and
Yongsong
Luo
*ab
aSchool of Physics and Electronic Engineering, Xinyang Normal University, Xinyang 464000, P. R. China. E-mail: ysluo@xynu.edu.cn; Fax: +86 376 6390801; Tel: +86 376 6390801
bKey Laboratory of Microelectronic and Energy of Henan Province, Xinyang Normal University, Xinyang 464000, P. R. China
cCollege of Chemistry and Chemical Engineering, Luoyang Normal University, Luoyang 471934, P. R. China
dDepartment of Mechanical and Aerospace Engineering, Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, P. R. China
First published on 6th December 2018
Abstract
The low loading and poor cycling performance of sulfur cathodes are among the critical barriers restricting the practical application of lithium–sulfur (Li–S) batteries. The rational design of composites consisting of transition metals and conductive nanocarbon is considered an effective strategy to construct cathode materials for Li–S batteries with excellent cycling stability and rate capability. Herein, we propose a spray drying method to fabricate 3D pomegranate-like titanium nitride (TiN)@graphene composites as hosts for sulfur cathodes. The hollow spheres are coated with graphene layers to form a shell, serving as a highly efficient electrochemical reaction chamber and a reservoir for polysulfides. The TiN@graphene/S electrode exhibits an excellent capacity of 810 mA h g−1 after 200 cycles at 0.5C. The cathodes with high areal sulfur loadings of 2.8 and 3.6 mg cm−2 maintained remarkable capacities of 568 and 515 mA h g−1, respectively, after 500 cycles. The TiN hollow spheres not only accommodate the large volume expansion of sulfur but also improve the conversion of polysulfides during the discharge/charge process. The excellent electrical conductivity of the few-layered graphene shell facilitates electron transport and maintains structural stability. This work offers a strategy to combine inorganic compounds and nanocarbon as sulfur hosts to improve the electrochemical properties of Li–S batteries.
Conceptual insightsInorganic compounds including transition-metal oxides, sulfides, and carbides have received special attention as some of the most promising host materials for lithium–sulfur batteries. Titanium nitride has high polarity and excellent conductivity, which can offer abundant adsorption sites to chemically adsorb polysulfides and block the diffusion of LiPSs more efficiently. Herein, we employ a simple spray drying method for the successful preparation of 3D pomegranate-like TiN@graphene composites, combining a 3D graphene framework with highly conductive, polar TiN nanospheres. The hollow TiN nanospheres on the graphene framework can be used as electrochemical reaction chambers to trap soluble polysulfides and to improve the kinetics of polysulfide redox reactions. This work offers a facile method for fabricating sulfur cathodes, enhancing the commercial viability of LSBs. |
Lithium–sulfur (Li–S) batteries have attracted increasing attention during the last decade due to their high theoretical capacity (1672 mA h g−1) and high theoretical energy density (2600 W h kg−1).1,2 Compared to transition metal oxide cathodes, pristine sulfur is considered to be the most promising candidate for fabrication of cost-effective electrode materials for safe, high energy batteries, thanks to the low cost, environmental benignity, and natural abundance of sulfur. In fact, the high theoretical capacity of sulfur has rarely been achieved due to its poor electronic conductivity of 5 × 10−28 S m−1, its large volume expansion of 79%, and the solubility of polysulfide species during cycles.3–5 These problems cause low active material utilization, poor cycling stability, and low coulombic efficiency (CE), which hamper the widespread application of sulfur in energy storage systems.6,7
To solve these issues, extensive efforts have been dedicated towards the construction of novel sulfur host materials so as to improve the electrochemical properties of Li–S systems. For example, carbon-based materials have been extensively studied due to their well-designed porous structures and high electrical conductivities. Infusing sulfur into carbon-based materials such as graphene, carbon nanotubes (CNTs) and porous carbon improved the conductivity of sulfur electrodes and mitigated the migration of polysulfides to the anode, the so-called polysulfide shuttling.8–10 Unfortunately, the shuttling effect has not been fully alleviated even with the carbon materials because of the low affinity between the carbon and polar, high-order lithium polysulfides (LiPSs).10–12
Much effort has been focused on overcoming the shuttling effect by suppressing the diffusion of LiPSs, especially using polar materials to enhance the chemical affinity through a polar–polar interaction. Polar inorganic compounds, including transition-metal oxides,13,14 sulfides,15,16 carbides,17 and nitrides,18–22 have been used as sulfur hosts or additive materials, which offered abundant active sites for adsorption of polysulfides and improved the electrochemical performance of the batteries. In addition, the fast transformation of LiPSs is also a key to suppressing LiPS shuttling and mitigating the loss of active materials. Various transition-metal oxides have been successfully used to increase the redox reaction rates of Li–S batteries due to their high catalytic activity.11 However, these transition-metal oxides possess poor electrical conductivities, small surface areas, and small porosities. These characteristics lead to inefficient confinement of LiPSs in the cathode, and result in sluggish redox reaction kinetics, thereby decreasing sulfur utilization and rate capability.15 Hence, a reasonable combination of inorganic compounds and carbon materials, such as CNTs and graphene, can effectively enhance the electron transfer and thus improve the catalytic conversion of LiPSs.
Various biomaterials with unique natural, hierarchical structures deserve to be studied and imitated.23–25 Among the biological models, the pomegranate structure offers a good reference for study due to its multi-seed nature (Fig. 1a). We mimic the pomegranate structure in this study to propose a similar microsphere structure, as shown in Fig. 1b.
 | ||
| Fig. 1 (a) Optical photograph of a pomegranate; (b) schematic of a pomegranate-like structure; (c) schematic of the synthesis process of TiN@graphene and TiN@graphene/S. SEM images of (d) sPS@TiO2, (e) sPS@TiO2@GO, and (f) TiN@graphene; and (g) TEM image of TiN@graphene/S. | ||
Here, we report that a 3D pomegranate microsphere consisting of a number of hollow titanium nitride (TiN) spheres and a graphene shell, which serve as electrochemical reaction chambers for sulfur and a conductive layer, respectively. The TiN@graphene (PTG) composite was prepared by a simple spray drying technique combined with a templated synthesis method. The sulfur-loaded TiN@graphene (PTG/S) electrodes exhibited excellent cycling stability with an exceptionally low capacity fading rate of 0.041% per cycle at 1C. The PTG composite has several desirable functional and structural features: (1) TiN spheres with a high electrical conductivity and chemical stability provide fast electron transport and rapid lithium diffusion, which enable high active material utilization; (2) the densely-packed hollow TiN spheres contribute to stable interparticle contact during cycles; (3) the hollow TiN spheres with large void spaces enable high sulfur loading and buffer volume variations; (4) TiN could serve as an electrocatalyst, thus greatly improving the kinetics of polysulfide redox reactions, especially when transforming soluble LiPSs into insoluble Li2S2 and Li2S; (5) the strong physicochemical adsorption between the highly polar TiN and polysulfides can enhance the utilization of active materials; and (6) graphene can serve as an efficient pathway for fast electron transport and as a protective layer to maintain the structural stability. Thanks to these advantages, the PTG/S cathodes exhibit high specific capacities and excellent long-term cycling stability.
Material synthesis
Synthesis of sulfonated polystyrene (sPS) spheres
Monodisperse polystyrene spheres were synthesized by free radical emulsion polymerization followed by a precipitation method.26 In a typical procedure, 8 mL of styrene was first added to a 250 mL flask containing 82 mL of deionized water. Then, the reaction flask was heated to 80 °C in an Ar atmosphere. After stirring for 30 min, 10 mL of deionized (DI) water with 0.2 g of potassium persulfate (KPS) dissolved was added. After mixing for 10 h, the product was centrifuged with DI water several times and dried in an oven overnight. The dried polystyrene powder (3.0 g) was immersed in 30 mL of concentrated sulfuric acid (98%), and the mixture was stirred for 24 h at room temperature. The sulfonated polystyrene (sPS) nanospheres were washed with ethanol and freeze dried.Synthesis of sPS@TiO2
1 g of sPS was ultrasonically dispersed in 50 mL ethanol for 1 h to form a homogeneous solution, and then 1.25 mL tetrabutyl titanate was added and the mixture was magnetically stirred for 30 min. 1.25 mL of water was added dropwise for 20 min. After 8 h of storage at room temperature, the product was centrifuged at 8000 rpm for 10 min, and washed with ethanol three times. The synthesized sPS@TiO2 spheres were dispersed in 50 mL of water.Synthesis of TiN@graphene and TiN@graphene/S composites
Graphene oxide (GO) was prepared from graphite nanoflakes through a modified Hummers method. 50 mL sPS@TiO2 was added to 50 mL of GO dispersion (1.5 mg mL−1) under vigorous stirring. The resulting solution was subjected to spray drying in air to obtain light yellow powders, where the temperatures at the inlet and outlet of the spray dryer were 140 °C and 80 °C, respectively. The obtained powders were transferred to a tubular furnace and heated at 300 °C for 2 h in an Ar atmosphere followed by annealing at 800 °C for 2 h at a heating rate of 2 °C min−1 in an NH3 atmosphere to collect black TiN@graphene composites. The TiN@graphene/S composites were prepared by mixing sulfur and TiN@graphene powders, which were sealed in a glass tube under vacuum and kept at 155 °C for 12 h.Characterization
The crystalline structure and phase of the composites were identified by X-ray diffraction (XRD, D8 Advance (Bruker) automated X-ray diffractometer system) using Cu-Kα (λ = 1.5418 Å) radiation at 40 kV and 40 mA, with 2θ between 10° and 80° at room temperature. The morphologies were examined using a field emission scanning electron microscope (FESEM, Hitachi S-4800) with an energy dispersive X-ray (EDX) detector and a transmission electron microscope (TEM, JEOL JEM-2010). X-ray photoelectron spectroscopy (XPS) was used to study the surface chemistry of materials on an XPS system (Perkin-Elmer model PHI 5600) at 0.3–0.5 eV from a monochromated aluminum anode X-ray source. The optical images were taken using a Sony camera. The UV-vis absorption test was performed on a UV-1901 spectrophotometer (Beijing Puxi). The surface area analysis was carried out using a Brunauer–Emmett–Teller surface area analyzer (BET, ASAP2460).Electrochemical measurements
The TiN@graphene/S composite was mixed with acetylene black and polyvinylidene difluoride (PVDF) binder at a weight ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10, with N-methyl-2-pyrrolidone (NMP) as a solvent. The slurry was pressed onto an aluminum foil and dried at 60 °C overnight to prepare the cathode, which was cut into pellets with a diameter of 1.0 cm and dried for 12 h. The electrochemical properties of Li–S batteries were measured using CR2032 coin cells, which were prepared using a lithium metal sheet as the counter- and the reference electrode and a Celgard 2400 film as the separator. The electrolyte was prepared using 1.0 M LiTFSI in a mixture of DOL/DME (1:1 by volume) with 1.0% LiNO3. The cyclic voltammetry (CV) test was carried out at cut-off potentials between 1.7 and 2.8 V at a scanning rate of 0.1 mV s−1 on a CHI650B electrochemical workstation. Galvanostatic charge/discharge (GCD) experiments were conducted using a Neware battery test system in the range of 1.7–2.8 V versus Li+/Li. The capacities were calculated based on the mass of sulfur. The sulfur content of TiN@graphene/S composites was found to be 79.3 wt% according to the TGA, corresponding to 63.4 wt% of the electrode mix with a typical sulphur mass loading of 0.8–3.6 mg cm−2. The electrochemical impedance spectroscopy (EIS) was performed at frequencies ranging from 100 kHz to 10 mHz on a CHI650B electrochemical workstation at 25 °C.
:10:10, with N-methyl-2-pyrrolidone (NMP) as a solvent. The slurry was pressed onto an aluminum foil and dried at 60 °C overnight to prepare the cathode, which was cut into pellets with a diameter of 1.0 cm and dried for 12 h. The electrochemical properties of Li–S batteries were measured using CR2032 coin cells, which were prepared using a lithium metal sheet as the counter- and the reference electrode and a Celgard 2400 film as the separator. The electrolyte was prepared using 1.0 M LiTFSI in a mixture of DOL/DME (1:1 by volume) with 1.0% LiNO3. The cyclic voltammetry (CV) test was carried out at cut-off potentials between 1.7 and 2.8 V at a scanning rate of 0.1 mV s−1 on a CHI650B electrochemical workstation. Galvanostatic charge/discharge (GCD) experiments were conducted using a Neware battery test system in the range of 1.7–2.8 V versus Li+/Li. The capacities were calculated based on the mass of sulfur. The sulfur content of TiN@graphene/S composites was found to be 79.3 wt% according to the TGA, corresponding to 63.4 wt% of the electrode mix with a typical sulphur mass loading of 0.8–3.6 mg cm−2. The electrochemical impedance spectroscopy (EIS) was performed at frequencies ranging from 100 kHz to 10 mHz on a CHI650B electrochemical workstation at 25 °C.
Adsorption properties of lithium polysulfides
Li2S6 solution was used as the stock solution for adsorption measurements and was prepared with Li2S and sulfur at a molar ratio of 1:5 using 1:1 (v/v) DOL/DME as a solvent. The concentration of the Li2S6 solution prepared was 5 mmol L−1. 20 mg PG, TiN and PTG were each added to 5 mL of the lithium polysulfide stock solution and the mixtures were vigorously stirred to facilitate adsorption before monitoring color changes.
Results and discussion
The synthesis process of pomegranate-like TiN@graphene (PTG) and TiN@graphene/S (PTG/S) composites is schematically shown in Fig. 1c. We utilized sulfonated polystyrene (sPS) nanospheres with an average size of ∼400 nm as the initial templates (Fig. S1, ESI†). Firstly, a TiO2 layer was coated on the surfaces of the sulfonated polystyrene (sPS) spheres by a precipitation method26 (Fig. 1d). Secondly, GO layers were applied onto the obtained sPS@TiO2 by spray drying to form a close-packed cluster. Thirdly, the product was heat-treated at 300 °C for 2 h to remove sPS. Fourthly, the TiN@graphene spheres were annealed in an NH3 atmosphere at 800 °C to transform TiO2 into TiN and reduce GO to N-doped reduced graphene oxide (rGO). For comparison, samples without TiN spheres were also prepared, denoted as graphene sphere clusters (PG). In the final step, sulfur was encapsulated into the TiN@graphene spheres via a melt-diffusion process to obtain the TiN@graphene/S composite (PTG/S).The morphologies of the samples were characterized using scanning electron microscopy (SEM), and typical images are shown in Fig. 1e and f. The SEM images of the initial precursor before carbonization exhibit a 3D pomegranate-like architecture (Fig. 1e). After carbonization in NH3, PG and PTG inherited the unique spherical 3D structure with a diameter ranging from 1 to 5 μm, as shown in Fig. 1f and Fig. S2c, d (ESI†). The PG and PTG microspheres consist of primary spheres of uniform sizes ∼400 nm (Fig. S2b and d, ESI†). Collapsed spheres are seen on the surfaces of PG, while the TiN spheres of PTG are encapsulated by rGO shells. Their inner structures were examined using TEM. The TEM images shown in Fig. 2a–c confirm that PG possess uniform spherical void spaces with a diameter of approximately 400 nm, arising from the decomposition of sPS. Fig. 2d and e show that PTG consists of graphene and hollow TiN spheres of the same diameter. The TiN shell ∼15 nm in thickness was covered with graphene sheets (Fig. 2e and f), indicating that the TiN spheres were successfully encapsulated into the irregular sheet-like graphene structure. The TiN crystal structure was clearly identified in the high resolution TEM images (Fig. S3a, ESI†), where the TiN crystallite exhibits well-resolved lattice fringes with an interlayer distance of 0.212 nm, which correspond to the (200) crystal plane of cubic TiN.
 | ||
| Fig. 2 (a–c) TEM images of PG; (d and e) TEM images and (f) HRTEM image of PTG composites. (g) TEM image and (h–k) corresponding elemental mappings of PTG/S composites. | ||
Sulfur was infiltrated into the cluster matrix via a melt-diffusion process. The presence of a large number of particles inside the titanium nitride sphere means that sulfur is well encapsulated in PTG (Fig. 1g and Fig. S3b, ESI†). The elemental mapping by energy-dispersive X-ray spectroscopy (EDX) confirms the presence of S, Ti, C and N elements in the PTG/S superstructures (Fig. 2h–k).
Fig. 3a shows the XRD patterns of the PG and PTG composites. A peak was observed at 2θ ∼ 22.7° and 28.5° in PG and PTG, respectively, which is assigned to the (002) reflections in the graphene nanosheets. The XRD pattern of PTG shows five characteristic peaks at 2θ = 36.8°, 42.6°, 61.9°, 74.1°, and 78.0°, corresponding to the (111), (200), (220), (311), and (222) lattice planes, respectively. These peaks match well with the JCPDS card (no: 06-0642) file, signifying the formation of cubic phase TiN. The size of TiN crystals calculated by the Scherrer formula is 7.5 nm, which is consistent with the grain size of TiN seen in the TEM images (Fig. 2f and Fig. S3a, ESI†). The sulfur (JCPDS card no: 77-0145) in the PTG/S composite exhibits a crystalline state with typical sharp peaks, as shown in Fig. S4 (ESI†).
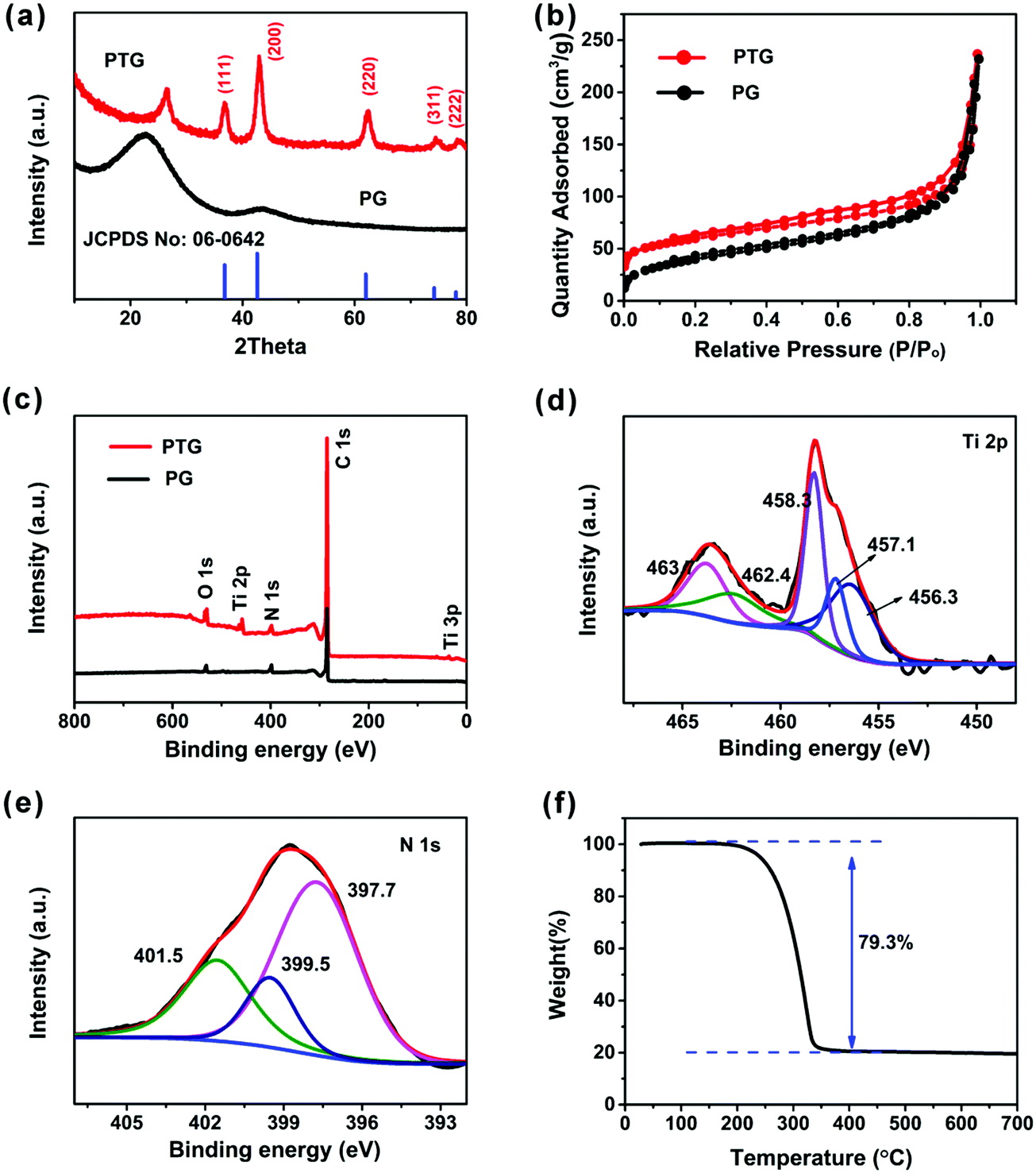 | ||
| Fig. 3 (a) XRD patterns of PG and PTG composites; (b) N2 adsorption–desorption isotherms of PG and PTG composites; (c) XPS survey spectra of PG and PTG; deconvoluted XPS spectra of (d) Ti 2p and (e) N 1s in PTG composites; and (f) TGA curve of PTG/S obtained in a nitrogen atmosphere. | ||
As illustrated in Fig. 3b, the N2 adsorption–desorption analysis was performed to further investigate the porous structure of the PTG composites. The Brunauer–Emmett–Teller (BET) surface area of PTG was measured to be 204 m2 g−1. The BJH pore size distribution of PTG is shown in Fig. S5a (ESI†), which indicates that the mesopore size is mainly concentrated at the 3–4 nm length scale. The large specific surface area and abundant mesopores can efficiently inhibit polysulfide dissolution and improve the electrochemical performance of Li–S batteries.
X-ray photoelectron spectroscopy (XPS) was used to evaluate the surface chemistries of the PG and PTG composites, as shown in Fig. 3c–e, and the corresponding elemental compositions are given in Table S1 (ESI†). Major differences between the spectra of PG and PTG are the presence of Ti 2P and Ti 3p in the PTG composite (Fig. 3c). Table S1 (ESI†) indicates significant oxygen contents in both PG and PTG. The oxygen in PG arose mainly from the functional groups, such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and O–CO, in the partly reduced GO, whereas the oxygen species in PTG were present in the form of TiO2 or TiOxNy in addition to the oxygenated functional groups on GO.27 This observation is partly confirmed by the deconvoluted Ti 2p spectrum (Fig. 3d). The strong Ti 2p signal at binding energies ranging 454–467 eV can be divided into five peaks, namely Ti–N (2p3/2 at 456.3 eV and 2p1/2 at 462.4 eV), Ti–N–O (2p3/2 at 457.1 eV and 2p1/2 at 463.1 eV), and Ti–O (2p3/2 at 458.3 eV).28,29 The deconvoluted N 1s spectrum (Fig. 3e) is divided into three peaks, which are pyridinic-N (at 397.7 eV), pyrrolic-N (at 399.5 eV) and graphitic-N (at 401.5 eV). The majority of nitrogen in PTG belongs to the pyridinic-N, which could enhance the chemical affinity to sulfur, resulting in improved cycling stability.30 The sulfur content in the PTG host was determined to be 79.3 wt% by thermogravimetric analysis (TGA) (Fig. 3f).
O and O–CO, in the partly reduced GO, whereas the oxygen species in PTG were present in the form of TiO2 or TiOxNy in addition to the oxygenated functional groups on GO.27 This observation is partly confirmed by the deconvoluted Ti 2p spectrum (Fig. 3d). The strong Ti 2p signal at binding energies ranging 454–467 eV can be divided into five peaks, namely Ti–N (2p3/2 at 456.3 eV and 2p1/2 at 462.4 eV), Ti–N–O (2p3/2 at 457.1 eV and 2p1/2 at 463.1 eV), and Ti–O (2p3/2 at 458.3 eV).28,29 The deconvoluted N 1s spectrum (Fig. 3e) is divided into three peaks, which are pyridinic-N (at 397.7 eV), pyrrolic-N (at 399.5 eV) and graphitic-N (at 401.5 eV). The majority of nitrogen in PTG belongs to the pyridinic-N, which could enhance the chemical affinity to sulfur, resulting in improved cycling stability.30 The sulfur content in the PTG host was determined to be 79.3 wt% by thermogravimetric analysis (TGA) (Fig. 3f).
Electrochemical performance
To evaluate the electrochemical performance of the PG and PTG/S composites, Li coin cells (2032-type) were fabricated with lithium foil as the counter electrode. The cells were cycled within a potential range of 1.7 and 2.8 V (vs. Li/Li+) on a Neware battery test system, and the specific capacity was calculated based on the sulfur content in the composites. Cyclic voltammetry (CV) measurements were conducted on a CHI650B electrochemical workstation. As shown in Fig. 4a, the CV curves exhibit typical electrochemical behavior of a sulfur cathode. Two cathodic peaks at 2.32 and 2.04 V are observed at a scan rate of 0.1 mV s−1, which agree well with previous reports regarding the two main stage reduction reactions.20,31 The peak located at 2.32 V should be related to the conversion of sulfur to long-chain soluble lithium polysulfides (Li2Sn, 4 ≤ n ≤ 8), and the peak at 2.04 V corresponds to the reduction of the polysulfides to insoluble short-chain Li2S and Li2S2. A broad anodic peak located between 2.32 and 2.40 V could be ascribed to the oxidation of Li2S2 and Li2S, forming long-chain lithium polysulfides or S8. It is worth noting that the PTG/S cathode delivers a higher current density than the PG/S cathode, especially at high sweep rates. This finding suggests that TiN plays an important role in reducing polarization and accelerating the redox kinetics in polysulfides. | ||
| Fig. 4 (a) CV curves of PG/S and PTG/S cathodes in the range of 1.7–2.8 V at a sweep rate of 0.1 mV s−1; and (b and c) CV curves and (d) the corresponding area II/I ratios of PTG/S and PG/S cathodes at different sweep rates. | ||
The CV tests were performed at different sweep rates to study the electrocatalytic behavior of the sulfur cathodes, as shown in Fig. 4b and c. We quantified the ratios of the areas under the CV curves, and the area II/I ratios are shown in Fig. 4d for the two cathodes. The area ratios of the PTG/S cathode were consistently higher than those of the PG/S counterpart as a reflection of the catalytic properties of TiN during the reduction of polysulfides and a slow redox reaction in the PG/S cathode. This result further confirms that the presence of TiN could accelerate charge transfer and promote conversion of LiPSs into insoluble Li2S. When the sweep rate was increased from 0.1 to 1.5 mV s−1, the area ratio gradually decreased for both the PTG and PG/S cathodes, indicating lower utilization of sulfur at a higher sweep rate. The lithium ion diffusion characteristics of the cathodes were evaluated according to the CV curves obtained at different sweep rates, and both the calculation procedure and the results are presented in Fig. S6 (ESI†). The slopes of the curves are directly correlated with the lithium ion diffusion of the electrodes. Although the slope of peak (I) is marginally lower, those of peaks (II) and (III) of the PTG/S cathode are much higher than those of the PG/S counterpart. This finding indicates that the presence of TiN could effectively ameliorate the lithiation kinetics, especially for the conversion of long-chain soluble lithium polysulfides to Li2S/Li2S2.
The GDC profiles for both cathodes at different current rates are shown in Fig. 5a and b. The discharge profiles contain two plateaus, which correspond to the oxidation of S8 to form Li2S4 and from Li2S4 to Li2S, respectively. As expected, the PTG/S cathode exhibits higher discharge capacities and better rate capabilities due to its higher conductivity and more efficient utilization of sulfur than the PG/S counterpart. The reduced voltage hysteresis in PTG/S led to an improved energy efficiency compared to the PG/S cathode, as shown in Fig. S7 (ESI†). It is found that the voltage gap of 0.21 V between the charge and discharge plateaus of the PTG/S electrode was lower than that of the PG/S electrode (0.26 V). The lower voltage gap and higher energy efficiency (92%) of the PTG/S cathode are attributed to enhanced redox kinetics due to the addition of TiN.32
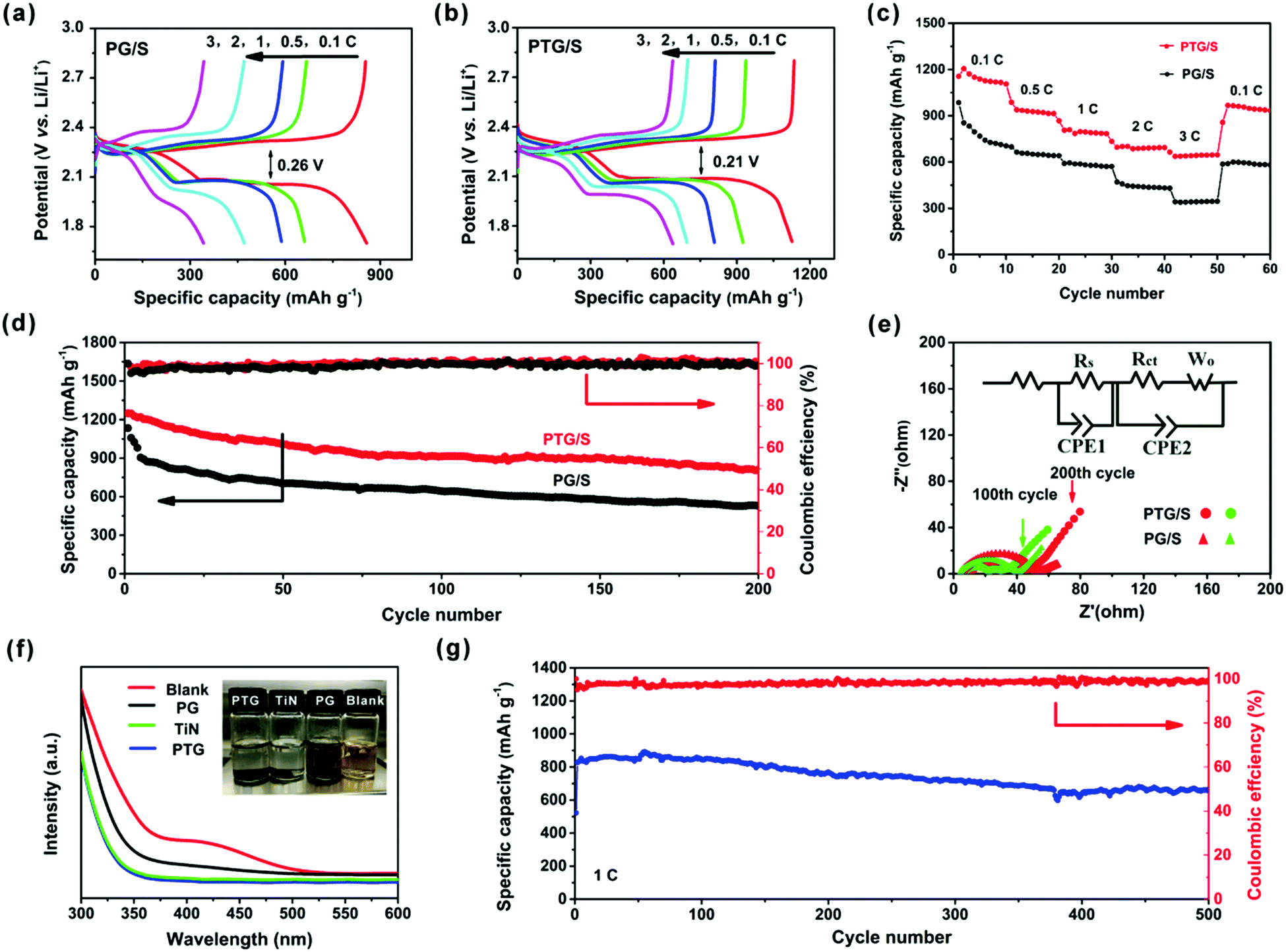 | ||
| Fig. 5 (a and b) Galvanostatic discharge/charge profiles of PG/S and PTG/S cathodes at different current rates of 0.1, 0.5, 1.0, 2.0 and 3.0C; (c) rate performance and (d) cycling performance and coulombic efficiencies of PTG/S and PG/S cathodes at 0.5C; (e) Nyquist plots of PTG/S and PG/S cathodes after 100 and 200 cycles; (f) UV-vis spectra (300–600 nm) and the corresponding photographs of the Li2S6 solution after 10 h upon contact with PTG, TiN, blank and PG; and (g) long-term cycling stability and coulombic efficiencies of PTG/S cathodes at 1C with an areal S loading of 1.2 mg cm−2. | ||
Fig. 5c shows the rate capabilities of the PTG/S electrode at various current densities. The discharge capacities of the PTG/S electrode are 1205, 1106, 806, 695, and 664 mA h g−1 at 0.1, 0.5, 1, 2, and 3C, respectively. When the current density was reverted to 0.1C, a high discharge capacity of 967 mA h g−1 was achieved, indicating the robustness and superior stability of the cathode. In contrast, the PG/S electrode exhibited lower discharge capacities at various C-rates ranging from 0.1 to 3C.
The cycling stability profiles of the two cathodes at 0.5C are shown in Fig. 5d. The initial discharge capacity of the PTG/S cathode at 0.5C is 1249 mA h g−1 with 75% sulfur utilization. The discharge capacity is very stable and is maintained at 810 mA h g−1 after 200 cycles. In contrast, the PG/S composite cathode shows a much lower specific capacity with a remaining capacity of 527 mA h g−1 after 200 cycles, which is equivalent to a capacity retention of about 47% of the initial value. The tested cells were disassembled after 200 cycles and the separators in the cells were examined, as shown in Fig. S8 (ESI†). Both sides of the separators of the PG/S electrode exhibit a distinct yellow color, while the reverse side of the separators in the PTG/S electrode becomes light yellow, indicating mitigated polysulfide shuttling in the latter cathode.
Fig. 5e and Fig. S9 (ESI†) show the electrochemical impedance spectra (EIS) of the PTG/S and PG/S cathodes. Impedance parameters were determined from the Nyquist plots fitted with an equivalent circuit, and the results are shown in Table S2 (ESI†). Before the cycling test, the difference in charge transfer resistance (Rct) between the fresh PTG/S and PG/S cathodes is insignificant (Fig. S9, ESI†). This finding signifies the beneficial role of the graphene skeleton for charge transfer, and also indicates that the introduction of TiN did not have a negative effect on charge transfer. The semicircles in the mid-frequency region reflect the resistances of SEI films formed on the electrode surface (Rs).33–39 The Rs value of the PTG/S cell decreased significantly after cycles due to the sufficient penetration of the electrolyte as well as dissolution and subsequent re-distribution of the active materials.37 In addition, the PTG/S cell exhibits relatively smaller Rct and Ro values than the PG/S electrode, indicating faster electrochemical kinetics of the former.
The polysulfide adsorption capabilities of PTG, TiN and PG were examined by monitoring the color changes of the Li2S6 solution and the UV-vis spectra were obtained after tests, as shown in Fig. 5f. All Li2S6 solutions containing PTG and TiN became nearly colorless after 10 h of contact, while the solution with PG presented a negligible change in color, indicating that some Li2S6 still remains in the solution. Further, the overall intensities of the UV-vis spectra were significantly reduced and the absorption peaks at 410 nm completely disappeared for the solutions with PTG and TiN, compared to the blank solution or the solution containing PG. These results show that the adsorption of the Li2S6 species by PTG and TiN is much stronger than PG.
The long-term cycling performance of the PTG/S electrode at 1C is shown in Fig. 5g. The discharge capacity is maintained as high as 663 mA h g−1 after 500 continuous cycles. It is worth stressing that the corresponding capacity retention was ∼78%, equivalent to a remarkable capacity fading rate of 0.041% per cycle (calculated after the second cycle). The corresponding CE of the PTG/S cathode is ∼99% after 500 cycles. The electrochemical performances of the PTG/S cathodes with different sulfur loadings were also investigated at 1C (Fig. 6a). The PTG/S cathodes with high areal sulfur loadings of 2.8 and 3.6 mg cm−2 deliver excellent capacities of 568 and 515 mA h g−1 after 500 cycles, respectively. Fig. 6b and 6c show the discharge/charge mechanisms of the PTG/S and PG/S electrodes during cycles, respectively. The encapsulation of TiN spheres by the conductive graphene layers effectively limits the shuttling of LiPSs through physical confinement and chemisorption (Fig. 6b). In contrast, the PG/S cathode without TiN spheres suffers from LiPS shuttling and loss of the active materials due to poor interactions between the carbon and polysulfides (Fig. 6c), resulting in rapid capacity decay. It follows then that the excellent electrochemical performance of the PTG/S electrode can be attributed to the synergistic effect arising from the 3D pomegranate-like TiN spheres and the graphene layers on them.
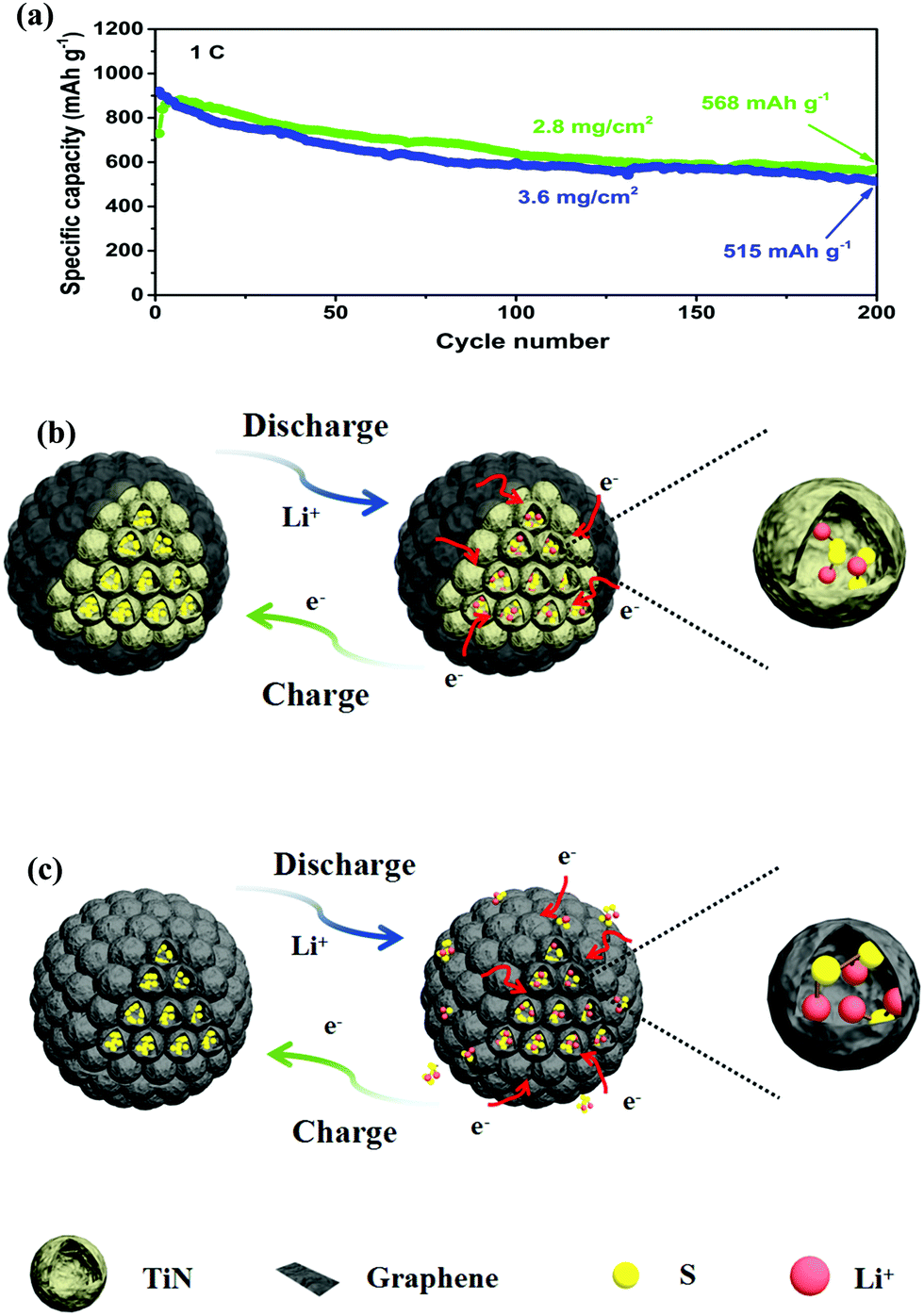 | ||
| Fig. 6 (a) Long-term cycling stability of PTG/S cathodes at 1C with areal S loadings of 2.8 and 3.6 mg cm−2; and (b and c) schematics of reversible discharge–charge mechanisms of PTG/S and PG/S cathodes. | ||
Conclusions
In summary, a pomegranate-like 3D TiN@graphene (PTG) composite was prepared by a spray drying method. Experimental results show that the composite is an efficient sulfur host for use in Li–S batteries with long lifetimes. Thanks to the synergistic effect of the 3D graphene skeleton and the polarity of the TiN spheres in the composite, the rationally designed PTG/S electrode delivered an excellent initial discharge capacity of 1249 mA h g−1, which remained at 810 mA h g−1 after 200 cycles at 0.5C. In addition, its discharge capacity was 663 mA h g−1 after 500 continuous cycles at 1C, which corresponds to a capacity retention of ∼78%, equivalent to a remarkable capacity loss rate of 0.041% per cycle. This work demonstrates that the highly conductive TiN spheres and the 3D graphene structure offer physical constraints, facilitate strong LiPS chemisorption, and promote conversion of LiPSs into insoluble Li2S, leading to an exceptional electrochemical performance of the PTG/S cathode.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 61874093, 51502257, and 61574122), the Program for Science & Technology Innovation Talents in Universities of Henan Province (No. 15HASTIT018), and the Natural Science Foundation of Henan Province (182300410283).References
- W. G. Chong, J. Q. Huang, Z. L. Xu, X. Y. Qin, X. Wang and J. K. Kim, Adv. Funct. Mater., 2017, 27, 1604815 CrossRef.
- X. Liang, C. Hart, Q. Pang, A. Garsuch, T. Weiss and L. F. Nazar, Nat. Commun., 2015, 6, 5682 CrossRef.
- Q. H. Yu, Y. Lu, T. Peng, X. Y. Hou, R. J. Luo, Y. G. Wang, H. L. Yan, X. M. Liu, J. K. Kim and Y. S. Luo, J. Phys. D: Appl. Phys., 2017, 50, 115002 CrossRef.
- B. Zhang, X. Qin, G. R. Li and X. P. Gao, Energy Environ. Sci., 2010, 3, 1531–1537 RSC.
- A. Manthiram, S. Chung and C. Zu, Adv. Mater., 2015, 27, 1980–2006 CrossRef CAS PubMed.
- Z. L. Xu, J. Q. Huang, W. G. Chong, X. Qin, X. Wang, L. M. Zhou and J. K. Kim, Adv. Energy Mater., 2017, 7, 1602078 CrossRef.
- M. Wild, L. O’Neill, T. Zhang, R. Purkayastha, G. Minton, M. Marinescub and G. J. Offer, Energy Environ. Sci., 2015, 8, 3477–3494 RSC.
- R. J. Luo, Y. Lu, X. Y. Hou, Q. H. Yu, N. T. Wu, T. Peng, H. L. Yan, X. M. Liu, J. K. Kim and Y. S. Luo, ChemistrySelect, 2018, 3, 3952–3957 CrossRef CAS.
- R. P. Fang, S. Y. Zhao, S. F. Pei, X. T. Qian, P. X. Hou, H. M. Cheng, C. Liu and F. Li, ACS Nano, 2016, 10, 8676–8682 CrossRef CAS PubMed.
- C. Tang, Q. Zhang, M. Q. Zhao, J. Q. Huang, X. B. Cheng, G. L. Tian, H. J. Peng and F. Wei, Adv. Mater., 2014, 26, 6100–6105 CrossRef CAS PubMed.
- D. H. Liu, C. Zhang, G. M. Zhou, W. Lv, G. W. Ling, L. J. Zhi and Q. H. Yang, Adv. Sci., 2017, 1700270 Search PubMed.
- H. J. Peng and Q. Zhang, Angew. Chem., Int. Ed., 2015, 54, 11018–11020 CrossRef CAS.
- M. W. Xiang, H. Wu, H. Liu, J. Huang, Y. f. Zheng, L. Yang, P. Jing, Y. Zhang, S. X. Dou and H. K. Liu, Adv. Funct. Mater., 2017, 27, 1702573 CrossRef.
- J. Y. Li, B. Ding, G. Y. Xu, L. R. Hou, X. G. Zhang and C. Z. Yuan, Nanoscale, 2013, 5, 5743 RSC.
- T. Chen, L. B. Ma, B. R. Cheng, R. P. Chen, Y. Hu, G. Y. Zhu, Y. R. Wang, J. Liang, Z. X. Tie, J. Liu and Z. Jin, Nano Energy, 2017, 38, 239–248 CrossRef CAS.
- T. Chen, Z. W. Zhang, B. R. Cheng, R. P. Chen, Y. Hu, L. B. Ma, G. Y. Zhu, J. Liu and Z. Jin, J. Am. Chem. Soc., 2017, 139, 12710–12715 CrossRef CAS.
- F. Zhou, Z. Li, X. Luo, T. Wu, B. Jiang, L. L. Lu, H. B. Yao, M. Antonietti and S. H. Yu, Nano Lett., 2018, 18, 1035–1043 CrossRef CAS PubMed.
- J. Zhang, C. Y. You, W. H. Zhang, J. Wang, S. Guo, R. Yang and Y. H. Xu, Electrochim. Acta, 2017, 250, 159–166 CrossRef CAS.
- Z. X. Hao, L. X. Yuan, C. J. Chen, J. W. Xiang, Y. Y. Li, Z. M. Huang, P. Hu and Y. H. Huang, J. Mater. Chem. A, 2016, 4, 17711–17717 RSC.
- Z. M. Cui, C. X. Zu, W. D. Zhou, A. Manthiram and J. B. Goodenough, Adv. Mater., 2016, 28, 6926–6931 CrossRef CAS PubMed.
- X. Li, K. Ding, B. Gao, Q. W. Li, Y. Y. Li, J. J. Fu, X. M. Zhang, P. K. Chu and K. F. Huo, Nano Energy, 2017, 40, 655–662 CrossRef CAS.
- B. Qi, X. S. Zhao, S. G. Wang, K. Chen, Y. J. Wei, G. Chen, Y. Gao, D. Zhang, Z. H. Sun and F. Li, J. Mater. Chem. A, 2018, 6, 14359–14366 RSC.
- H. B. Yao, H. Y. Fang, X. H. Wang and S. H. Yu, Chem. Soc. Rev., 2011, 40(7), 3764–3785 RSC.
- S. Sotiropoulou, Y. Sierra-Sastre, S. S. Mark and C. A. Batt, Chem. Mater., 2008, 20(3), 821–834 CrossRef CAS.
- H. B. Yao, G. Y. Zheng, W. Y. Li, M. T. McDowell, Z. W. Seh, N. Liu, Z. D. Lu and Y. Cui, Nano Lett., 2013, 13(7), 3385–3390 CrossRef CAS PubMed.
- S. H. Ahn, D. J. Kim, W. S. Chi and J. H. Kim, Adv. Funct. Mater., 2014, 24, 5037–5044 CrossRef CAS.
- L. Wang, J. G. Sun, R. R. Song, S. B. Yang and H. H. Song, Adv. Energy Mater., 2016, 6, 1502067 CrossRef.
- P. Sun, R. Lin, Z. L. Wang, M. J. Qiu, Z. S. Chai, B. D. Zhang, H. Meng, S. Z. Tan, C. X. Zhao and W. J. Mai, Nano Energy, 2017, 31, 432–440 CrossRef CAS.
- X. H. Lu, G. M. Wang, T. Zhai, M. H. Yu, S. L. Xie, Y. C. Ling, C. L. Liang, Y. X. Tong and Y. Li, Nano Lett., 2012, 12(10), 5376–5381 CrossRef CAS PubMed.
- Y. F. Dong, B. L. Wang, K. N. Zhao, Y. H. Yu, X. D. Wang, L. Q. Mai and S. Jin, Nano Lett., 2017, 17, 5740–5746 CrossRef CAS PubMed.
- Y. Q. Tao, Y. J. Wei, Y. Liu, J. T. Wang, W. M. Qiao, L. C. Ling and D. H. Long, Energy Environ. Sci., 2016, 9(10), 3230–3239 RSC.
- Z. Yuan, H. J. Peng, T. Z. Hou, J. Q. Huang, C. M. Chen, D. W. Wang, X. B. Cheng, F. Wei and Q. Zhang, Nano Lett., 2016, 16, 519–527 CrossRef CAS.
- M. Xiao, M. Huang, S. Zeng, D. Han, S. Wang, L. Sun and Y. Meng, RSC Adv., 2013, 3, 4914–4916 RSC.
- L. Xiao, Y. Cao, J. Xiao, B. Schwenzer, M. H. Engelhard, L. V. Saraf, Z. Nie, G. J. Exarhos and J. Liu, Adv. Mater., 2012, 24, 1176–1181 CrossRef CAS PubMed.
- L. Miao, W. Wang, K. Yuan, Y. Yang and A. Wang, Chem. Commun., 2014, 50, 13231–13234 RSC.
- S. Evers and L. F. Nazar, Acc. Chem. Res., 2012, 46, 1135–1143 CrossRef PubMed.
- X. X. Gu, C. J. Tong, C. Lai, J. X. Qiu, X. X. Huang, W. L. Yang, B. Wen, L. M. Liu, Y. L. Hou and S. Q. Zhang, J. Mater. Chem. A, 2015, 3, 16670–16678 RSC.
- A. Abouimrane, D. Dambournet, K. W. Chapman, P. J. Chupas, W. Weng and K. Amine, J. Am. Chem. Soc., 2012, 134, 4505–4508 CrossRef CAS PubMed.
- J. Yang, J. Xie, X. Zhou, Y. Zou, J. Tang, S. Wang, F. Chen and L. Wang, J. Phys. Chem. C, 2014, 118, 1800–1807 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nh00343b |
| This journal is © The Royal Society of Chemistry 2019 |