
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvances in porous and nanoscale catalysts for viable biomass conversion
Putla
Sudarsanam
 *a,
Elise
Peeters
a,
Ekaterina V.
Makshina
a,
Vasile I.
Parvulescu
b and
Bert F.
Sels
*a
*a,
Elise
Peeters
a,
Ekaterina V.
Makshina
a,
Vasile I.
Parvulescu
b and
Bert F.
Sels
*a
aCentre for Surface Chemistry and Catalysis, Faculty of Bioscience Engineering, KU Leuven, Celestijnenlaan 200f, 3001, Heverlee, Belgium. E-mail: sudarsanam.putla@kuleuven.be; putla2007@gmail.com; bert.sels@kuleuven.be; Fax: +32 16 321 998; Tel: +32 16 321 610
bUniversity of Bucharest, Department of Organic Chemistry, Biochemistry and Catalysis, B-dul Regina Elisabeta 4-12, Bucharest 030016, Romania
First published on 20th February 2019
Abstract
Heterogeneous catalysis is a promising technology for the valorization of renewable biomass to sustainable advanced fuels and fine chemicals. Porosity and nanostructure are the most versatile features of heterogeneous solid catalysts, which can greatly determine the accessibility of specific active sites, reaction mechanisms, and the selectivity of desirable products. Hence, the precise tuning of porosity and nanostructure has been a potential strategy towards developing novel solid catalysts with indispensable characteristics for efficient biomass valorization. Herein, we present a timely and comprehensive review of the recent advances in catalytic biomass conversions over microporous zeolites, mesoporous silicas, and nanostructured metals/metal oxides. This review covers the catalytic processing of both edible (lipids and starch) and non-edible (lignocellulose) biomass as well as their derived compounds, along with a systematic evaluation of catalyst reusability/kinetic/mechanistic aspects in the relevant processes. The key parameters essential for tailoring particle size, morphology, porosity, acid–base, and redox properties of solid catalysts are emphasized, while discussing the ensuing catalytic effects towards the selective conversion of biomass into desirable chemicals. Special attention has been drawn to understand the role of water in liquid phase biomass conversions as well as the hydrothermal stability and the deactivation of nanoporous catalysts. We believe this comprehensive review will provide new insights towards developing state-of-the-art solid catalysts with well-defined porosity and nanoscale properties for viable biomass conversion.
 Putla Sudarsanam | Putla Sudarsanam (1986), currently Marie Curie Postdoctoral Fellow at KU Leuven (Belgium), obtained his PhD degree in Chemistry (2015) from CSIR-Indian Institute of Chemical Technology, India (Supervisor: Dr B. Mahipal Reddy). Before joining at KU Leuven, he did postdoctoral work with Prof. Suresh Bhargava (RMIT University, Melbourne, 2014–2016) and with Prof. Angelika Brückner (Leibniz Institute for Catalysis, Germany, 2017). His research focuses on developing heterogeneous solid catalysts for biomass valorization, CO2 upgrading, green chemistry, and environmental protection. He received several awards/fellowships, including Marie Skłodowska-Curie Fellowship-2016, Leibniz-DAAD Postdoc Fellowship-2016, Best PhD Thesis Award-2015 (The Catalysis Society of India), EuropaCat PhD Student Award-2013 (XIth European Congress on Catalysis, France) and Endeavour Research Fellowship-2013 (Australia). He authored about 50 peer-reviewed journal articles with an h-index of 25 and more than 1550 citations. |
 Elise Peeters | Elise Peeters obtained her MSc degree in Bioscience Engineering (Catalytic Technology) at KU Leuven (Belgium) in 2017. She completed her master's thesis at the Centre for Surface Chemistry and Catalysis under the supervision of Prof. Bert Sels, where she studied the hydrodeoxygenation of (ligno)cellulose into liquid short alkanes. She is currently pursuing a PhD in the same group as an FWO-funded fellow. Her current research topic focuses on the efficient conversion of biomass-derived molecules by Sn-BEA catalysis. |
 Ekaterina V. Makshina | Dr Ekaterina V. Makshina obtained her PhD degree in 2007 in the frame of a cooperation agreement (Co-tutelle) of Moscow State University (Russia) and Littoral University (France) working on the topic of methanol and VOCs oxidation over supported oxide catalysts. In 2010, she joined the group of Prof. Sels at the Centre for Surface Chemistry and Catalysis (KU Leuven). Her research expertise covers the development of the solid catalysts and new processes targeting biomass transformation into commodity chemicals. |
 Vasile I. Parvulescu | Vasile I. Parvulescu received his PhD from the Polytechnic University of Bucharest in 1986 with a thesis investigating the selectivity of bi- and multi-metal catalysts in hydrogenation of aromatic hydrocarbons. After several years as high-signor researcher at the Institute of Inorganic and Rare Metals, in 1992 he joined the University of Bucharest, where he becomes full professor in 1999. He is currently director of the Department of Organic Chemistry, Biochemistry and Catalysis. His scientific interest concerns the study of heterogeneous catalysts for green and fine chemistry and environmental protection. He authored more than 380 papers, 27 patents, and 7 books. He was awarded by the Romanian Academy (“Nicolae Teclu” Award, 1990), Romanian President (Knight for Merits, 2008) and The National Grand Lodge of Romania (“Grigore Moisil“ award for exact sciences, 2015). |
 Bert F. Sels | Bert F. Sels (1972), currently full professor at KU Leuven, obtained his PhD degree in 2000 in the field of heterogeneous oxidation catalysis. He was awarded the DSM Chemistry Award in 2000, the Incentive Award by the Belgian Chemical Society in 2005, and the Green Chemistry Award in 2015. He is currently director of the Centre for Surface Chemistry and Catalysis, designing heterogeneous solid catalysts for future challenges in industrial organic and environmental catalysis. His expertise includes heterogeneous catalysis in bio-refineries, design of hierarchical zeolites and carbons and the spectroscopic and kinetic study of active sites for small-molecule activation. He authored about 310 peer-reviewed journal articles with an h index of 75 and more than 18100 citations, and filed 25 patents. |
1. Introduction
Escalating energy demand, depletion of fossil fuels, and abnormal climate change are the major problems facing humankind in the 21st century.1–5 Non-renewable fossil fuels (coal, crude oil, and natural gas) currently provide more than 90% of our energy needs. Nevertheless, the surplus consumption of fossil fuels is the primary reason for increasing levels of atmospheric CO2 and hence, global warming, a major threat to all living organisms. The utilization of renewable energy sources (wind, solar, geothermal, hydropower, and biomass) as alternatives to fossil fuels is one of the promising solutions for a more sustainable society. The valorization of biomass (an abundant non-fossil carbon source) has particularly attracted a lot of attention because of its ability to provide a wide range of valuable functionalized chemicals and high-energy density fuels (Fig. 1). Biomass is the fourth largest source of energy, after crude oil, coal, and natural gas and has been used to generate heat and power over the last few decades.6 Nature itself produces huge amounts of biomass via photosynthesis using water, sunlight, and atmospheric CO2 (Fig. 2).6 Hence, the efficient valorization of biomass could not only provide useful fuels and chemicals, but also assist to reduce the net emissions of CO2 by an interplay chemistry of photosynthesis and biorefinery processes. Consequently, biofuel share is predicted to reach about 9% in the transport sector by 2030.4 The utilization of naturally abundant biomass as a feedstock for chemicals production can also boost the principles of green chemistry within the context of sustainability.7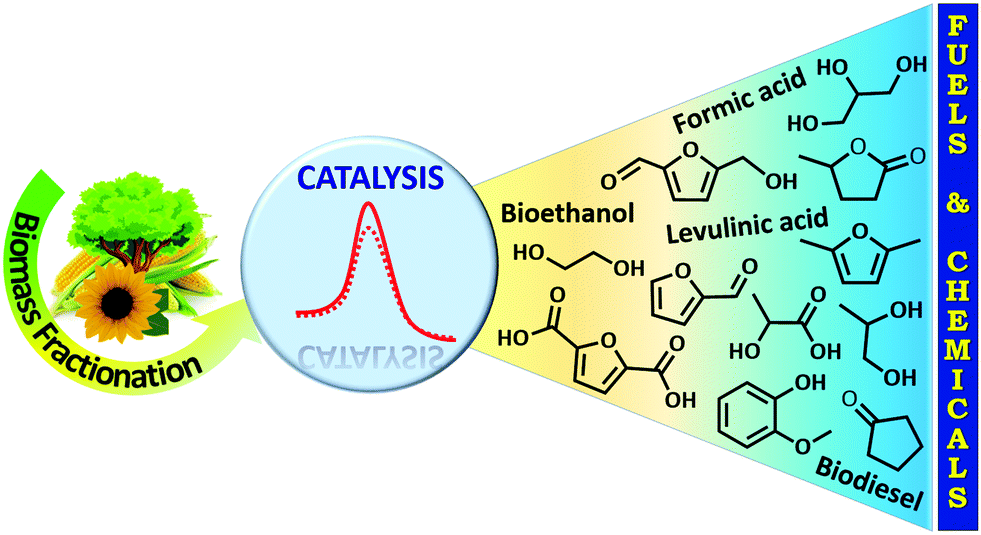 | ||
| Fig. 1 Catalytic biomass upgrading into high-value fuels and fine chemicals. | ||
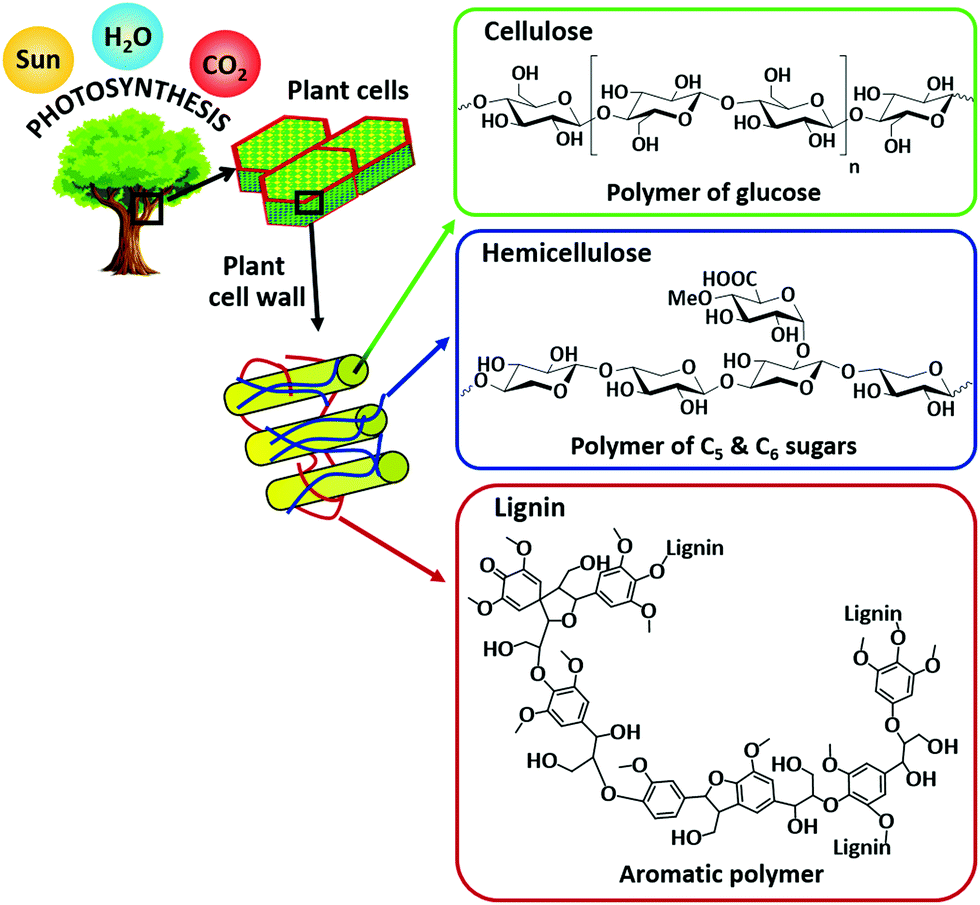 | ||
| Fig. 2 Structure of lignocellulose biomass with the major components: cellulose, hemicellulose, and lignin. | ||
Biomass can be largely derived from lignocellulose, lipids, and starch crops.8,9 A number of feasible processes and technologies, mostly catalytic based routes, have been developed for biomass valorization. These include biochemical (enzymatic hydrolysis and fermentation), thermochemical (combustion, pyrolysis, and gasification), hydroprocessing, oxidation, (trans)esterification, isomerization, dehydration, hydrolysis, and many others. For instance, bioethanol is produced via fermentation of edible biomass feedstocks (sugar cane or corn).10,11 Vegetable oils, obtained from food sources (soybeans, palm or sunflower), are the raw materials for biodiesel production via catalytic transesterification reaction.12,13 By using thermochemical methods, all plant-based residues can be converted into bio-oil or syngas.9,14 Catalytic hydroprocessing methods are essential for upgrading biomass-derived platform molecules to fuel grade chemicals. On the other hand, biopolymer building blocks can be produced via catalytic oxidation of biomass-derived compounds. Recently, the reductive catalytic fractionation process has attracted much attention as this strategy converts non-edible lignocellulosic biomass into two valuable fractions: (i) lignin-based phenolic compounds and (ii) (hemi)cellulose pulp.15–17Fig. 2 shows the structure of lignocellulose with the major components: cellulose, hemicellulose, and lignin. In many of these biomass conversions, selectively directing a reaction pathway towards desirable chemicals remains a great challenge due to uncontrolled interactions between the catalyst active sites and the biomass molecules.
The application of heterogeneous solid catalysts, alternative to homogeneous liquid catalysts, in biomass valorization is of great importance because of their versatile physicochemical properties, high hydrothermal stability, and efficient catalyst recovery/reusability.18,19 Indeed, heterogeneous catalysis field contributes to about 90% of chemical production processes and to more than 20% of all industrial products.3 The most notable features of a solid catalyst that can play a beneficial role in catalytic reactions are particle size, shape, and porosity. For example, the reduction of a catalyst particle size to nanoscale (1–100 nm size in at least one dimension) can lead to the generation of novel catalytic materials with enriched surface active sites, modified interaction specificities, and improved redox properties that could be substantially different from their bulk counterparts.20–23 The specific surface area of nanosized catalysts can also be largely improved due to higher surface to volume ratio (Fig. 3), enabling facile accessibility of surface active sites to biomass molecules.21 The use of metal nanoparticles (Ag NPs and Pt NPs) in catalysis has already been attempted in the 19th century for photography as well as for H2O2 decomposition, respectively.24 In 1940s, the Nord group reported the catalytic application of nanomaterials for nitrobenzene reduction.25,26 In 1970s and 1980s, Parravano et al.27 and Haruta et al.28 developed various supported nano-Au catalysts for hydrogen transfer and for low temperature CO oxidation, respectively. On the other hand, fine-tuning the particle morphology of a catalyst, especially of metals or metal oxides, will optimize the electronic and geometrical properties at the catalyst surface. Shape-controlled nanoscale metal oxides can preferentially expose large amounts of surface-active crystal facets. As a result, the interactions between the active sites and the reactive species could be enhanced, which is a key driving force for obtaining improved reaction rates in biomass valorization.
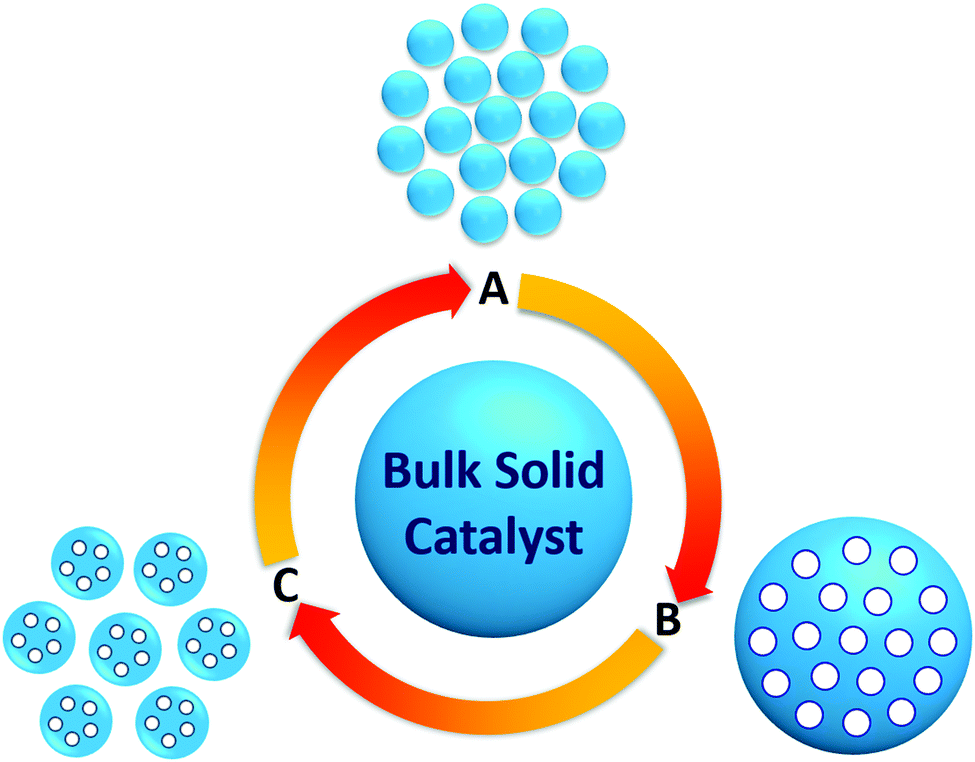 | ||
| Fig. 3 Exemplary routes to improve surface area and diffusion properties of bulk solid catalysts: (A) reduction of particle size with shape-control, (B) introduction of pores, and (C) combination of both particle size reduction and introduction of pores. | ||
In addition to particle size and morphology, controlling the porosity of solid catalysts is also indispensable for improving catalyst activity and product selectivity (Fig. 3). The pore dimensions of solid catalysts can directly influence the diffusion properties of reactive species during biomass conversion reactions.21,29 Moreover, the use of well-defined porous materials as catalyst supports can provide specific adsorption sites for stabilizing catalytic active phases.22 Microporous zeolites and mesoporous silicas are widely studied porous materials in catalytic biomass conversions.30–34 A major challenge in using these single-mode porous materials as catalysts for biomass-related reactions is the mass transport limitations because biomass molecules are complex and highly viscous. For instance, active sites (e.g., acid sites) are predominantly located inside the micropores of the zeolites, making the diffusion of larger biomass molecules through the confined micropores more difficult. This will lead to low conversion rates, uncontrolled product selectivity, and rapid catalyst deactivation. In this respect, hierarchically porous materials containing multiple-scale pores (micro, meso, or macro) have attracted great attention because of their favourable catalytic properties compared to single-mode porous analogues.35,36 These unique porous materials not only combine the advantages of active site accessibility and efficient mass transport effects, but also endow ample space for the incorporation of catalytically active phases, including metal nanoparticles (Au, Pd, Ru, Rh, Cu, Ni, Fe, etc.) or acid–base species (–SO3H, –NH2, etc.).
1.1 Scope of the review
Over the last ten years, tremendous research endeavors have been undertaken towards developing a variety of novel heterogeneous solid catalysts for the efficient conversion of biomass to sustainable advanced fuels and fine chemicals. These efforts mainly devoted towards fine-tuning of particle size, shape, surface structure, and porosity of solid catalysts with the aim of enriching the surface active sites both electronically and geometrically. This will control the interactions of catalytically active sites with the biomass molecules, directing a preferential reaction pathway to obtain desirable products. These developments have been made largely in hydroprocessing, oxidation, hydrolysis, (trans)esterification, isomerization, and dehydration of various biomass feedstocks and their derived compounds. Consequently, several review articles have been published on the topic of catalytic biomass valorization. For instance, the Pierre Gallezot group provided a review article on biomass conversions over metal-based catalysts.37 Wang et al.34 summarized the catalytic applications of nanoporous materials for biomass valorization. In 2016, the Sels group published a critical review on potential and challenges of zeolites for catalytic biomass upgrading.8 A few reviews have been also published on the topic of designing mesoporous silica materials for various applications.33,38 However, the catalytic applications of mesoporous silica materials including for biomass valorization has not yet been summarized in detail. As well, many interesting results have been recently published on catalytic biomass conversion over microporous zeolites and nanoscale metals/metal oxides. Hence, a timely comprehensive review, highlighting the importance of both porosity and nanoscale properties of solid catalysts for biomass valorization, is essential for the benefit of both academia and industry researchers. Based on the above background, this review aims to provide a detailed summary of the recent advances in the catalytic applications of microporous zeolites, mesoporous silica materials, and nanoscale metals/metal oxides for biomass valorization (Fig. 4). In each section, several feasible approaches developed for fine-tuning the particle size, morphology, and the porosity of above-mentioned catalysts are emphasized, while discussing the role of ensuing catalytic effects in biomass conversions. The catalytic processing of both edible (lipids and starch) and non-edible (lignocellulose) biomass substrates and their derived compounds are summarized in detail. We believe that this comprehensive review will act as a useful source to guide researchers towards developing state-of-the-art solid catalysts with well-defined nanoscale and porosity properties for a viable bio-refinery industry. | ||
| Fig. 4 Schematic overview of this review. | ||
2. Zeolite-based catalysts for biomass valorization
Zeolites are the most extensively used solid catalysts in chemical industry, for instance in catalytic cracking of crude oil, Friedel–Crafts reactions, acid-catalyzed chemical transformations, and recently for biomass conversion.30,39,40 The application of zeolites in oil refinery resulted in about 30% increase in gasoline yield, indicating their importance in petrochemical industry.39 The unique ability of zeolites to adsorb water and release it upon heating has paved the way for the development of synthetic zeolites A, B, and X (Linde division of Union Carbide, 1940).41 Subsequently, the scientific community has recognized the exceptional role of zeolites in shape-selective catalysis. Zeolites are crystalline microporous aluminosilicates, with pore sizes normally in the range of 0.3–2 nm. They typically contain three-dimensional networks of MO4 tetrahedral units (M = Al, Si), connected via corner-sharing oxygen atoms to form highly ordered interconnected pores (Fig. 5). The porous structure of zeolites can accommodate several cations, such as H+, Na+, K+, and Mg2+, along with abundant water molecules. Hence, zeolites can be represented by an empirical formula, namely M2/nO·Al2O3·xSiO2·yH2O, where M = heterocation, n = cation valence, x = 2–200, and y = number of water molecules. The substituted cations are predominantly located in relatively large cavities formed by negatively charged [Si1−xAlxO2]x− framework, where x ≤ 0.5 (Lowenstein's rule).42 The International Zeolite Association has recognized over 200 zeolite frameworks so far, but only about 10 types have found practical applications.40 The value of global zeolite market was about 1.30 billion USD in 2013 and is estimated to reach 1.76 billion USD in 2020, signifying the continuous growth of zeolites industry.39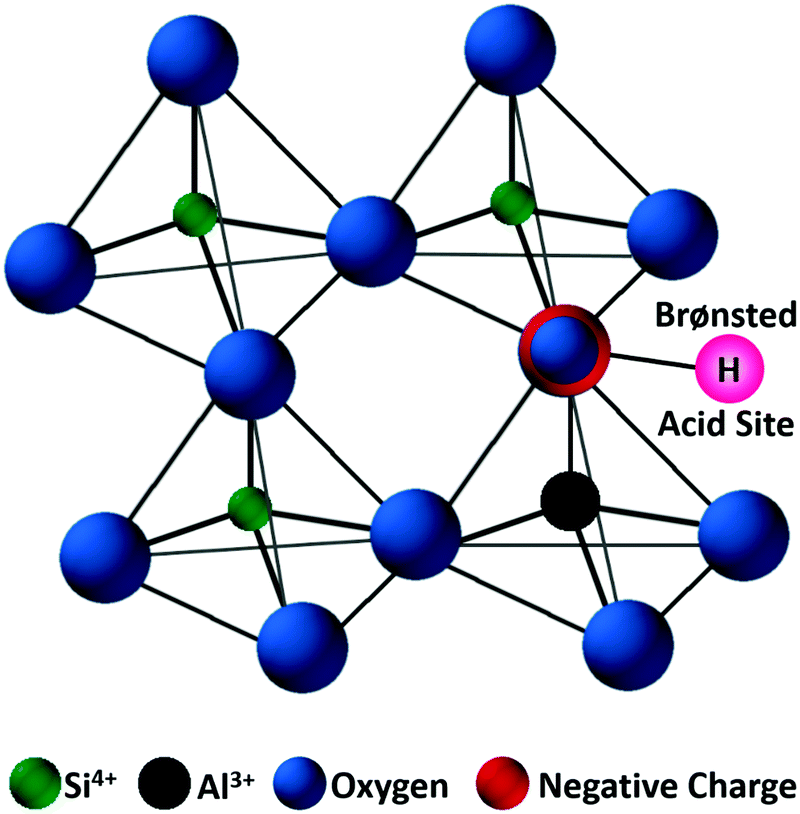 | ||
| Fig. 5 Representative zeolite framework of a tetrahedral [SiO4]4−, with generation of a negative charge due to the incorporation of Al3+ and substitution of a proton to maintain the charge balance. Adapted with permission from ref. 45. Copyright (2002) Elsevier. | ||
2.1 Catalytic role of zeolite properties in biomass valorization
Zeolite-based catalysts are of great interest for biomass valorization because of their unique porous structure, shape selectivity, and strong acidity.8,41,43 Especially, the tunable Brønsted/Lewis acidity is a vital feature of zeolites that can significantly determine their catalytic activity and selectivity in biomass upgrading.40,44 The substitution of Si4+ by Al3+ in the tetrahedral framework of the zeolite is the driving force for the formation of Brønsted acid sites.30,45 This is because incorporation of Al3+ into the zeolite framework generates a negative charge (Fig. 5), that needs to be compensated by cations to maintain the charge balance.45 If the substituted cation is a proton (H+), the resulting zeolite contains strong Brønsted acidity (Fig. 5). It is therefore obvious that with the increase of Al/Si ratio (or decrease of Si/Al ratio) the number of Brønsted acid sites in the zeolite framework increases if all the cation sites are occupied by protons. Nevertheless, when the amount of Al reaches a certain limit, weak Brønsted acid sites can be formed due to the accumulation of excess protons in the zeolite pores. On the other hand, the Lewis acidity of zeolites is dependent on the extra-framework Al and/or the substituted heterocations.44,46,47 The extra-framework Al can present as isolated Al2O3 particles in the zeolite structure, which are usually formed during the calcination of zeolites at higher temperatures. Sometimes, these isolated Al2O3 particles can also be formed during the synthesis process due to unsuccessful incorporation of excess Al into the zeolite framework. Besides, metals (e.g., Ti, Zr, Hf, Nb, or Ta) incorporated into the zeolite frameworks can act as Lewis acidic sites. The improved hydrophobic nature is an added benefit of these metallozeolites, which is an important criterion for their catalytical applications in hydrothermal biomass conversions.Shape-selectivity of zeolites, originated by precisely defined pore systems, plays a crucial role in promoting a specific reaction pathway for obtaining desirable chemicals.40,41,48,49 The different pore window size of zeolites can cause selective active site accessibility and mass transfer effects as shown in Fig. 6.50 For instance, only reactant molecules with suitable sizes and configurations relative to the pore window size of zeolites can reach internal active sites (reactant shape selectivity, Fig. 6A).49,50 Similarly, zeolite pores can impose steric hindrance to the reaction and determine the formation of transition state molecules (transition state shape selectivity, Fig. 6B). Simultaneously, products with smaller size than the pore sizes of the zeolite are selectively formed (product shape selectivity, Fig. 6C).
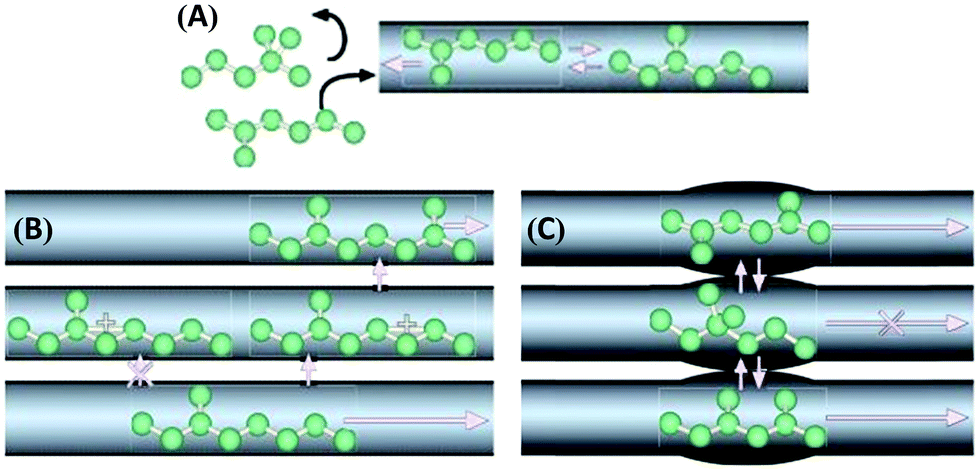 | ||
| Fig. 6 Different types of shape selectivity in zeolites: (A) reactant shape selectivity, (B) transition state shape selectivity, and (C) product shape selectivity. Reproduced with permission from ref. 50. Copyright (2008) Springer Nature. | ||
Catalytically active (acid) sites are predominantly present inside the micropores of zeolites. In the case of upgrading larger biomass molecules (polysaccharides, disaccharides, lignin, triglycerides, etc.), active site accessibility is a key concern because of severe mass transfer constraints imposed by micropores, which may lead to low conversion rates. For instance, zeolites having a free pore aperture of 0.74 nm do not allow the diffusion of larger sucrose molecules (diameter is 0.88 nm) to reach the internal acid sites.51 To circumvent this problem, the development of hierarchical porous zeolites containing a multimodal pore size distribution has attracted a lot of attention, as this strategy combines the advantages of micropores (provide active sites and determine shape-selectivity) as well as mesopores (facilitate rapid mass transfer properties).31,52 New active sites can also be formed in hierarchical zeolites due to structural modifications and perturbations that occur during their synthesis. For example, the alkaline treatment of zeolites led to the formation of a new type of surface basic sites, along with innate acid–base sites, which may exhibit favourable catalytic effects in biomass valorization (Fig. 7).31,52
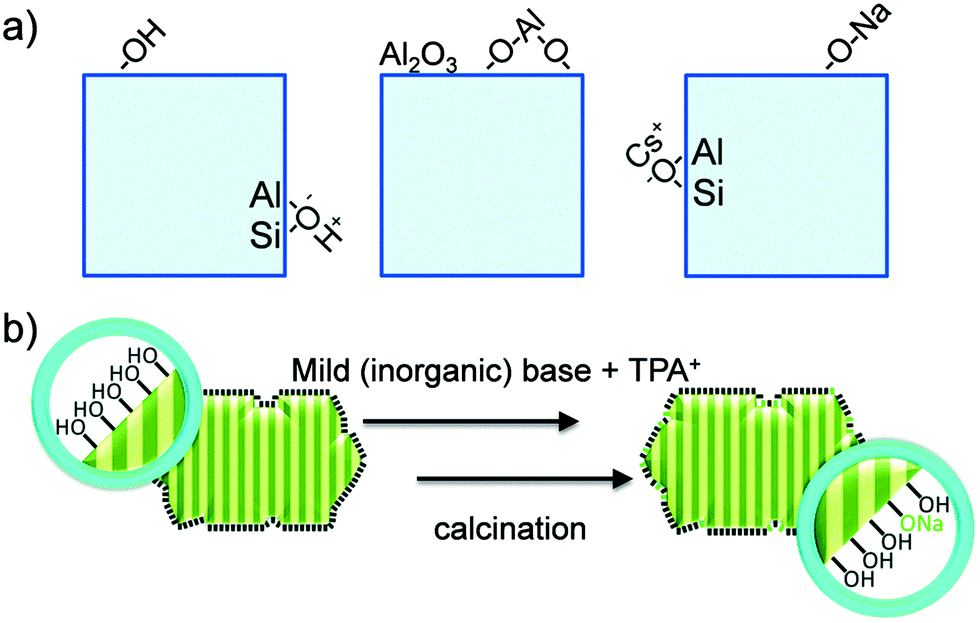 | ||
| Fig. 7 Formation of various active sites in hierarchical zeolites. (a) Typical Brønsted acid sites and surface hydroxyl groups (left), Lewis acid sites derived from steaming and alkaline-induced surface realumination (middle), and basic sites generated by the framework or by the formation of surface-bound alkali cations (right). (b) Generation of novel surface basic sites by tailored alkaline treatment. Reproduced with permission from ref. 31 and 52. Copyright (2016) Royal Society of Chemistry and Copyright (2014) John Wiley and Sons, respectively. | ||
Owing to their high specific surface area, zeolites are efficient support materials for the stabilization/dispersion of active phase metals/metal oxides (Pd, Pt, Au, Ru, Ag, Ni and V2O5).30,53 Advantages of acid properties of zeolites coupled with the redox properties of metals can lead to the generation of novel, versatile catalysts with improved activities and selectivities. The incorporated heterocations (e.g., Cu) in zeolite structures can also play a vital role in modifying the chemical and surface properties of zeolite supported metal oxide (V2O5) catalysts, resulting in optimized host–guest interaction that will direct the formation of desirable bio-based chemicals (e.g., 2,5-diformylfuran).53 Moreover, the presence of secondary voids in hierarchical zeolites can offer an ideal space for the deposition of adequate amounts of catalytically active metals or metal oxides.
2.2 Pristine and substituted zeolites
Zeolite-Y, developed by Union Carbide (1959), was the first zeolite applied (as isomerization catalyst) in chemical industry and later, zeolite-X (Mobil Oil, 1962) was used as hydrocarbon cracking catalyst.54 Owing to attractive acid and porous properties, the use of both pristine and modified zeolites has gained paramount attention for catalytic biomass conversions.55The catalytic pyrolysis of cellulose, cellobiose, lignin, and switch grass was strongly dependent on the loading of Fe (1.4, 2.8 and 4.2 wt%) in Fe-modified HZSM-5 zeolites synthesized by partial ion exchange of NH4ZSM-5 with Fe(II).65 Especially, the 1.4 wt% Fe loaded HZSM-5 was demonstrated to be efficient towards aromatics production (mainly benzene and naphthalene). Like Fe, addition of Zn, Ga, and Ni to HZSM-5 has a profound effect in the CFP of eucalyptus urophylla.66 Among those, the Ga-modified zeolite gave the highest amount of aromatics (especially xylenes), whereas low yields were achieved over Ni-modified HZSM-5 catalyst. Hydrocarbon yield and heating value of dry oil were largely improved in the CFP of ground corncob catalyzed by ZSM-5 zeolite (Si/Al = 25) coated ceramic honeycomb (ZCCH).67 In addition, the ZCCH catalyst can prevent the formation of undesirable products including light oxygenates and furans. In USY zeolite catalyzed CFP of de-oiled Millettia (Pongamia) pinnata seed waste, selectivity of monocyclic aromatic hydrocarbons was found to increase from 4.7–17.4% to 52.5–56.5% when the ratio of biomass![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :catalyst increased from 1:1 to 1:5 at 500 °C, respectively.68 Moreover, zeolite catalyst played a crucial role in selective cleavage of carbon chain towards production of only shorter hydrocarbons (C5–12). The Si/Al ratio was found to be a key factor to tune the catalytic performance of MCM-22 zeolite for the CFP of acid-washed wheat straw.44 The MCM-22 zeolite with the lowest Al content (Si/Al = 40) provided higher bio-oil deoxygenation activity at 450 °C pyrolysis temperature. Interestingly, an increase in the pyrolysis temperature led to the deposition of a lower amount of coke on the zeolite. On the other hand, reducing the Si/Al ratio (which creates abundant acid sites) had a negative effect on product selectivity since acid sites favored the undesirable side reactions, leading to formation of gaseous hydrocarbons and higher coke deposits. A steel slag-derived zeolite (FAU-SL) catalyst afforded maximum yield (47 wt%) of bio-oil from the pyrolysis of oil palm mesocarp fiber, which was carried out in a fixed-bed reactor.69 High mesoporosity and strong surface acidity of FAU-SL were the primary factors to obtain better activity results in this study. Zeolite structure and its acidity were found to show a great effect in the production of phenols from CFP of lignin.70 Notably, large-pore-size zeolites (e.g., FAU) with Si/Al = 15 are highly selective towards phenols formation.
:catalyst increased from 1:1 to 1:5 at 500 °C, respectively.68 Moreover, zeolite catalyst played a crucial role in selective cleavage of carbon chain towards production of only shorter hydrocarbons (C5–12). The Si/Al ratio was found to be a key factor to tune the catalytic performance of MCM-22 zeolite for the CFP of acid-washed wheat straw.44 The MCM-22 zeolite with the lowest Al content (Si/Al = 40) provided higher bio-oil deoxygenation activity at 450 °C pyrolysis temperature. Interestingly, an increase in the pyrolysis temperature led to the deposition of a lower amount of coke on the zeolite. On the other hand, reducing the Si/Al ratio (which creates abundant acid sites) had a negative effect on product selectivity since acid sites favored the undesirable side reactions, leading to formation of gaseous hydrocarbons and higher coke deposits. A steel slag-derived zeolite (FAU-SL) catalyst afforded maximum yield (47 wt%) of bio-oil from the pyrolysis of oil palm mesocarp fiber, which was carried out in a fixed-bed reactor.69 High mesoporosity and strong surface acidity of FAU-SL were the primary factors to obtain better activity results in this study. Zeolite structure and its acidity were found to show a great effect in the production of phenols from CFP of lignin.70 Notably, large-pore-size zeolites (e.g., FAU) with Si/Al = 15 are highly selective towards phenols formation.
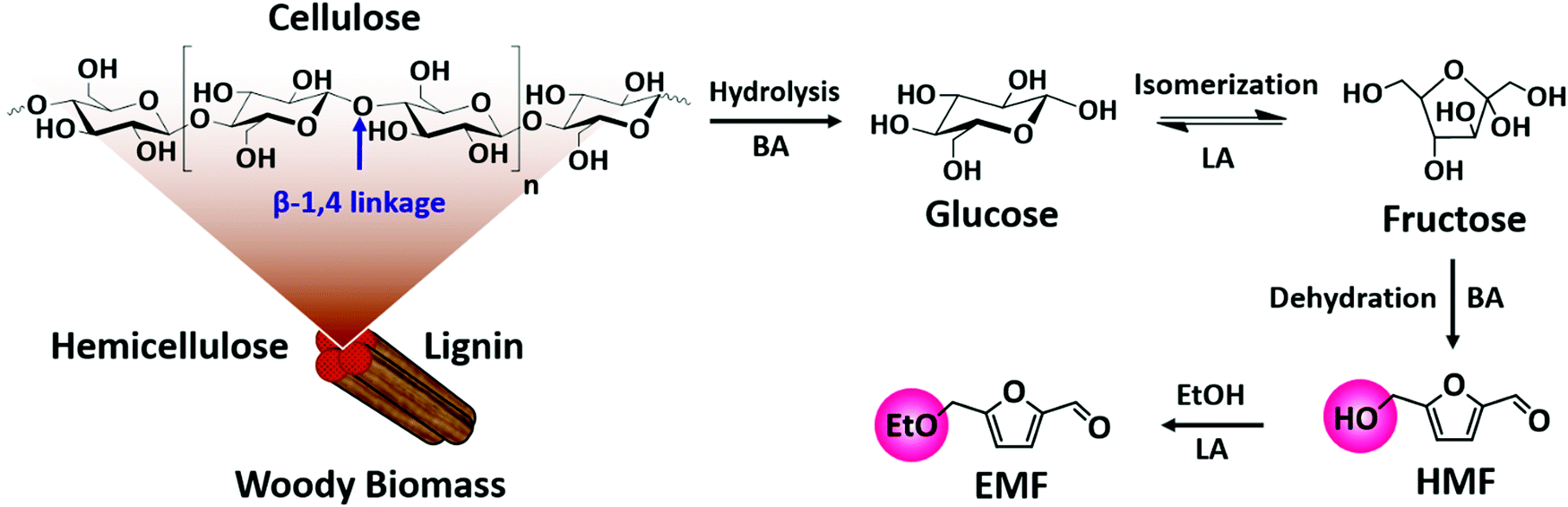 | ||
| Fig. 8 Upgrading cellulose into various high-value products: glucose, fructose, 5-hydroxymethylfurfural (HMF) and 5-ethoxymethylfurfural (EMF): BA – Brønsted acid and LA – Lewis acid. | ||
5-Hydroxymethylfurfural (HMF) is a vital platform molecule for future biorefineries. The conversion of glucose to HMF (Fig. 8) involves glucose isomerization to fructose (Lewis acid catalyzed step) and its subsequent dehydration to HMF (Brønsted acid catalyzed step). It is obvious that glucose-to-HMF transformation requires both Lewis and Brønsted acid sites. An overview of the most significant catalytic results achieved in liquid phase carbohydrates-to-HMF transformation over porous catalysts is presented in Table 1. The conversion of glucose to HMF in liquid phase was investigated using various ZSM-5 based zeolites (H-, Fe- and Cu-ZSM-5).73 About 80% glucose conversion with 42% HMF yield was obtained over H-ZSM-5, which was attributed to optimum molar ratio of Lewis/Brønsted acidity (entry 1, Table 1). The importance of Lewis–Brønsted acidity for glucose-to-HMF transformation was also observed over Al-rich β zeolites (entry 2, Table 1),74 [Sn, Al]-β zeolites (entry 3, Table 1)75 and H-β-25 zeolite.76 For fructose dehydration, narrow pore zeolites exhibited superior HMF selectivity (about 90%) at 85–94% fructose conversions (entry 4, Table 1), whereas larger pore zeolites promoted undesirable HMF oligomerization towards humins production.77 Owing to strong acid properties, HY zeolite was found to be active for fructose-to-HMF transformation.78 The subsequent hydrogenation of HMF to 2,5-dihydroxymethylfuran or 2,5-dimethylfuran was performed using a Cu/ZnO/Al2O3 catalyst. One-pot synthesis of 5-ethoxymethylfurfural (EMF) from glucose involves multiple steps as shown in Fig. 8. EMF is a potential next-generation biofuel with a high energy density (8.7 kilowatt-hours per litre, kW h L−1), greater than that of bioethanol (6.1 kW h L−1) and comparable to that of gasoline (8.8 kW h L−1) and diesel (9.7 kW h L−1).79,80 EMF can be transformed to 5-ethoxymethylfurfuryl alcohol (a promising diesel additive) and to esters of 2,5-furandicarboxylic acid (an ideal alternative to terephthalic acid for the polymer industry). The dealuminated β zeolite gave reasonably good yields of EMF (37%) from glucose.81 Interestingly, the combination of H-form zeolite and Amberlyst-15 led to efficient transformation of glucose to EMF (46%). The production of EMF from various mono-, di- and poly-saccharides was also examined in this work. The acidity, adsorption ability, and the internal pore dimensions of zeolites were the crucial factors for the etherification of HMF with isobutene to produce tert-butoxymethylfurfural.82 HY zeolite with a SiO2/Al2O3 mole ratio of 12 is highly selective towards bio-ether product (94%) at 59% HMF conversion. A simple calcination step in air was performed to regenerate the catalyst activity for the next cycle.
| Entry | Catalyst | Biomass substrate | Reaction conditions | Catalytic activity | Key factors for the catalyst activity | Ref. | |
|---|---|---|---|---|---|---|---|
| Conv. (%) | Yield (%) | ||||||
| a MIBK – methyl isobutyl ketone. b Calcination at 500 °C. c DMSO – dimethyl sulfoxide. d THF – tetrahydrofuran. e 15 wt% Nafion loading. | |||||||
| 1 | H-ZSM-5 zeolite | Glucose | 3:1 wt% of glucose:catalyst, H2O/MIBKa, 195 °C, 30 min |
80 | 42 | Optimum amounts of Lewis–Brønsted acid sites | 73 |
| 2 | Al-rich beta zeoliteb | Glucose | 0.1 g catalyst, 0.67 mmol glucose, H2O, DMSOc, THFd, 180 °C, 3 h | 86 | 56 | Optimum amounts of Lewis–Brønsted acid sites | 74 |
| 3 | [Sn,Al]-beta zeolite | Glucose | 0.05 g catalyst, 10 wt% glucose solution, DMSO, 160 °C, 4 h | 60 | 37 | Suitable Sn/Al molar ratio and optimum amounts of Lewis–Brønsted acid sites | 75 |
| 4 | ZSM-5 zeolite | Fructose | 0.033 g catalyst, 1 g fructose, 190 °C, 80 min, H2O-MIBK mixture | 85 | 74.8 | Narrow pore dimensions | 77 |
| 5 | Mesoporous AlSiO-20 | Glucose | 0.2 g catalyst, 0.2 g glucose, THF/H2O–NaCl, 160 °C, 90 min | 91.7 | 63 | High BET surface area and suitable ratio of Brønsted/Lewis acidity | 178 |
| 6 | 10Al-MCM-41 silica | Glucose | 0.05 g catalyst, 0.15 g glucose, 195 °C, 30 min, NaClaq/MIBK | 96 | 63 | Mesoporous structure and optimum amounts of acid sites | 185 |
| 7 | Functionalized hierarchical porous SiO2 | Cellulose | 0.04 g catalyst, 120 °C, 30 min, DMSO/H2O | — | 44.5 | Hierarchical porous structure, high BET surface area, and abundant acid–base sites | 193 |
| 8 | Nafion/mesocellular SiO2e | Fructose | Catalyst (acid amount: 0.06 mmol H+), fructose in DMSO (3 wt%, 12.5 g), 90 °C, 2 h | 94 | 89.3 | Strong acid strength and open ultra-large pore system | 211 |
| 9 | Sulfated MCM-41 silica | Fructose | 0.033 g catalyst, 1 g fructose, 190 °C, 80 min, H2O-MIBK mixture | 94 | 84.6 | Mesoporous structure and weak acidity | 77 |
| 10 | Sulfated KIT-5 silica | Fructose | 0.04 g catalyst, 1 mmol fructose, DMSO, 125 °C, 45 min | — | 94 | Brønsted acid sites | 233 |
Acid-catalyzed dehydration of C5 sugars, such as xylose and arabinose gives furfural, a very important chemical for the production of fuel additives and polyester monomers.83–85 Cui et al.83 indicated that the co-operative interaction of Hβ zeolite with γ-butyrolactone–water solvent system enables both selective C–C bond cleavage of acyclic hexoses into pentoses and the subsequent dehydration into furfural. The obtained furfural yields from fructose, glucose, and cellulose were 63.5, 56.5, and 38.5%, respectively. Both Lewis and Brønsted acid sites in Sn–Al-β zeolite are essential for the catalytic transfer hydrogenation and hydrolysis of furfural to γ-valerolactone.86 The degree of dealumination and Sn-incorporation significantly influenced the acid properties of zeolite and consequently its catalytic efficiency. Magnetically recoverable β-zeolites exchanged with transition metals (Pd, Fe and Ir) were demonstrated to be catalytically active for upgrading furfural and furfuryl alcohol into bio-based chemicals.87 Particularly, Pd-exchanged zeolite showed the best catalytic performance towards isopropyl levulinate production from both furfural and furfuryl alcohol. H-ZSM-5 zeolite is a versatile catalyst for the transformation of furfural to indoles via thermo-catalytic route, followed by ammonization.88 A possible reaction pathway was proposed in which furfural initially reacts with NH3 to give furfural-imine, subsequent cracking to furan, followed by its conversion to pyrrole and finally to indole. H-ZSM-5 zeolite can also efficiently catalyze furfuryl alcohol hydrolysis to obtain levulinic acid (LA).89 The remarkable hydrophobic nature of H-ZSM-5 alters the internal solvent microenvironment within the zeolite framework to achieve more than 70% yields of LA. Mahmoud et al.90 studied Diels–Alder reaction of furan and methyl acrylate (or acrylic acid) using Lewis acidic zeolites (Hf-, Zr-, and Sn-β). All zeolites selectively catalyze the thermodynamically controlled reaction of furans with acrylates with higher turnover frequencies compared to other homogeneous and heterogeneous Lewis acid catalysts. The resulting product i.e. oxanorbornene was dehydrated to produce methyl benzoate in the presence of methanesulfonic acid and acetic anhydride. The integrated reduction-acid conversion of furfural to valuable bio-products was carried out over a β zeolite possessing Zr and Al sites.91 The existence of Zr-sites was crucial for initiating the reduction of furfural to furfuryl alcohol as well as for the reduction of alkyl levulinate esters to γ-valerolactone. The co-presence of Al-sites promoted acid-catalyzed steps. DFT calculations suggested three types of reaction pathways for H-ZSM-5 catalyzed furfural decarbonylation to form furan product.92 Among them, the protonation of α-carbon to the arenium-ion like intermediate was found to be the preferred pathway for furfural decarbonylation as this pathway requires low activation energy for the rate-determining step. Owing to well-balanced strong Lewis acid sites and an optimal pore size, H-β-zeolite was able to catalyze the transformation of γ-valerolactone to ethyl-4-ethoxy pentanoate, a potential bio-ether fuel.93 About 86% product yield with 97% selectivity was obtained at optimized reaction conditions. The vapor phase decarbonylation of lactic acid to acetaldehyde was investigated using HMCM-22, HZSM-5 and NaZSM-5 zeolites.94 Interestingly, the NaZSM-5 zeolite, which contains intermediate strength of Lewis acid sites (Brønsted/Lewis acid sites ratio is 0.4) is highly selective towards acetaldehyde production (yield of acetaldehyde + CO = 96%) by suppressing the formation of lactic acid oligomers and coke (Fig. 9). In contrast, the strong Brønsted acidic zeolites (HMCM-22 and HZSM-5) gave only 39.7% and 57% yields of decarbonylation products (acetaldehyde + CO), respectively, because of the formation of considerable amounts of lactic acid oligomers.
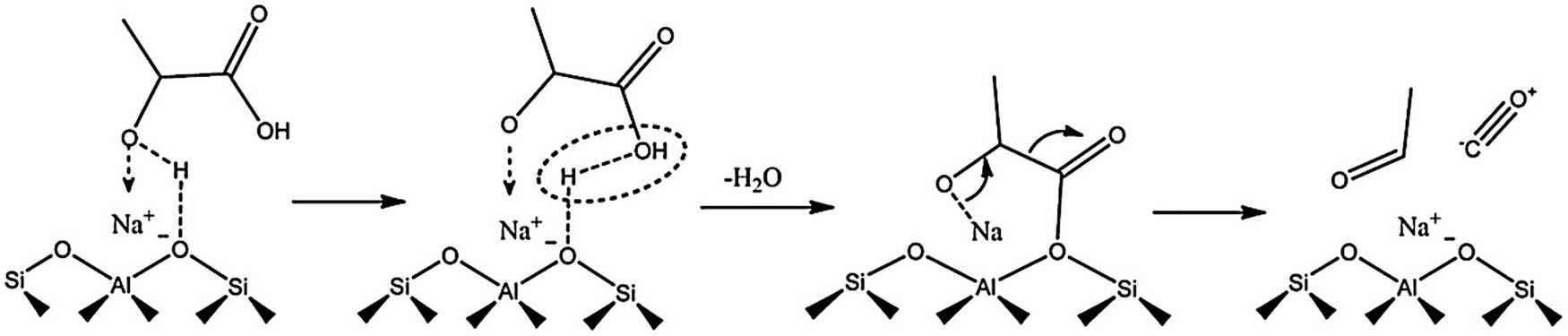 | ||
| Fig. 9 Probable reaction mechanism for the decarbonylation of lactic acid to acetaldehyde over Na-ZSM5 zeolite. Reproduced with permission from ref. 94. Copyright (2018) Elsevier. | ||
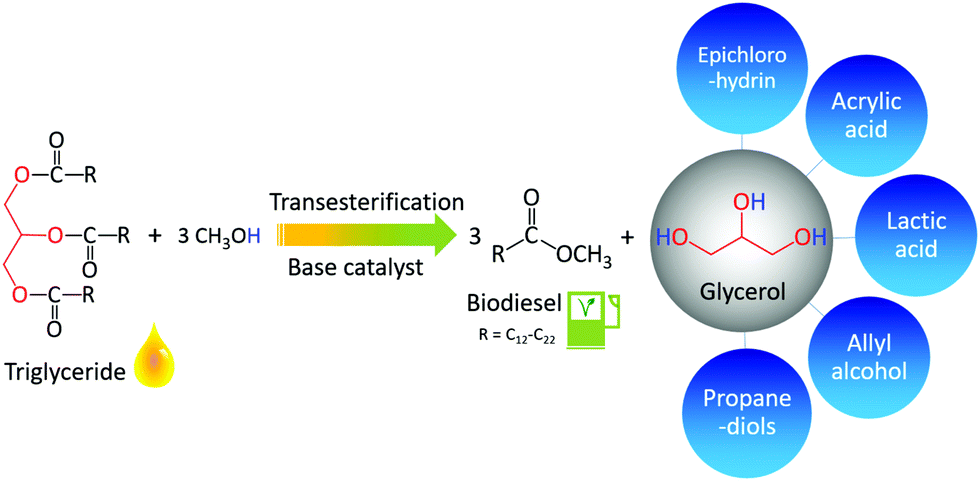 | ||
| Fig. 10 Catalytic transformation of triglycerides (oils and fats) to produce biodiesel with glycerol as a byproduct, which can be upgraded to a variety of drop-in chemicals. | ||
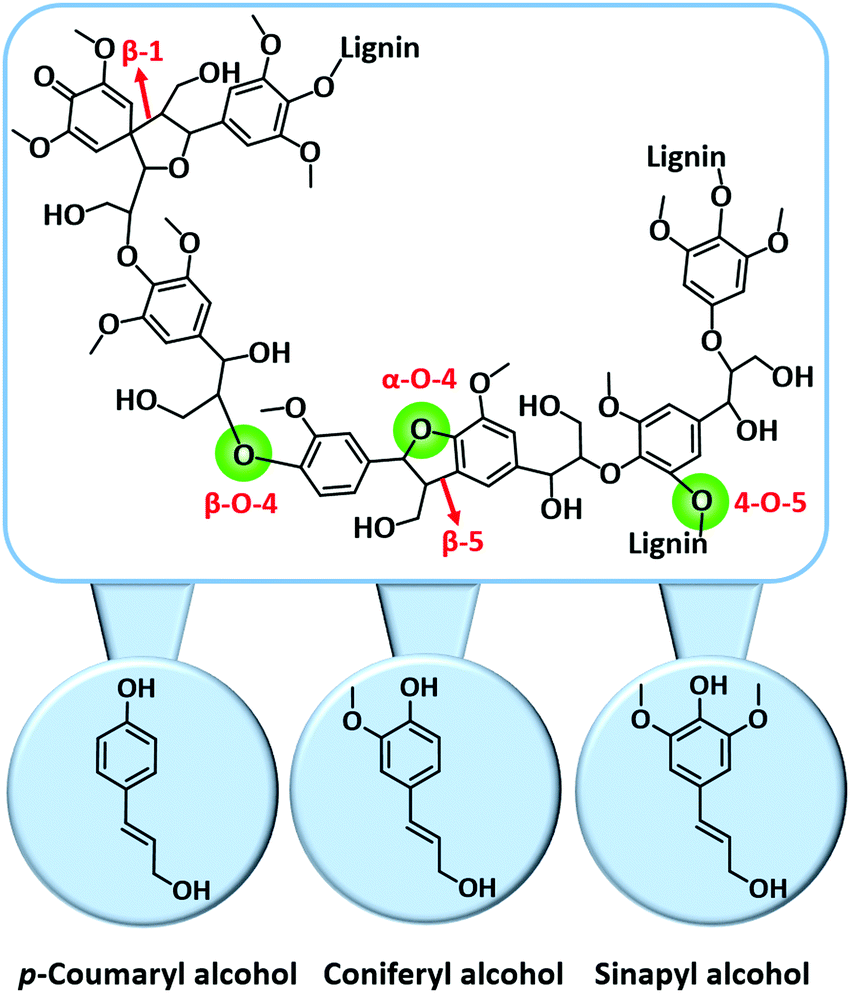 | ||
| Fig. 11 Key C–O and C–C linkages in lignin and its key building blocks. Adapted with permission from ref. 18. Copyright (2018) Royal Society of Chemistry. | ||
Incorporation of boron had a considerable effect on the acidity of zeolites: the amount of weak acid sites increases proportionally with the amount of incorporated boron.115 Borosilicalite-1 zeolites synthesized in the presence of alkali hydroxide were catalytically more active in steam catalytic cracking of dodecane due to the formation of an active trigonal coordination system (boron site). The structure–activity properties of four different zeolites, namely MCM-22, MCM-49, MCM-56 and Ce/MCM-22, before and after water treatment, were investigated in the liquid phase tetrahydropyranylation of n-propanol or tert-butanol with 3,4-dihydro-2H-pyran.55 Water treatment of zeolites at 160 °C for 48 h caused a reduction in both acid site concentration and micropore volume. Except for Ce/MCM-22, all zeolites showed higher catalytic performance in the tetrahydropyranylation of alcohols. Low activity of Ce/MCM-22 was due to the pore blockage by Ce species. The HZSM-5 framework remains unaffected after alkali treatment (NaOH), with a favorable improvement in the pore size and acidity at low NaOH concentrations (<0.5 M).116 On the other hand, the zeolite structure can be degraded with a significant reduction of micropore size and acidity concentration at higher NaOH concentrations. Results obtained from gas phase oligomerization of biomass-derived ethylene revealed that the proper NaOH treatment not only favors ethylene conversion and product yield, but also improves catalyst stability. The selective aldol condensation of acetaldehyde to C4 products was examined over NaX and NaY zeolites in liquid phase.117 Effects of several parameters, such as number and location of exchangeable cations, nature of cations, and post-synthesis modifications were studied towards product selectivity. NaY catalyst displayed higher C4/(C6 + C8) product ratio than NaX, attributed to the presence of optimized basic sites in NaY catalyst.
The acidic sites strength in HZSM-5 based zeolites exhibited a significant effect in the gas phase conversion of bio-ethanol to propylene, which is an important starting chemical for the synthesis of propylene oxide and polypropylene.118,119 For instance, medium strength acid sites favor the formation of propylene, while weak acid sites are selective towards ethylene formation.118 On the other hand, strong acid sites promote the formation of other paraffins, C5+ aliphatics, and aromatics. The highest yield of propylene obtained over HZSM-5 with Si/Al2 ratio of 80 was 32%. However, this zeolite suffered from severe catalytic deactivation after 4 h time-on-stream study. Doping of HZSM-5 with zirconium was suggested to improve its stability for propylene production. In another work, a hydrothermally synthesized HZSM-5/SAPO-34 composite was found to show high propylene yield and improved catalytic stability than the parent HZSM-5 and mechanically mixed HZSM-5/SAPO-34 in gas phase ethanol-to-propylene transformation.120 Interestingly, a high yield of propylene (∼50%) was achieved over a β zeolite-In2O3 composite, while pure In2O3 provided only 32% propylene yield.121 The improved catalytic performance of β zeolite-In2O3 composite was attributed to the promoting role of β zeolite in the conversion of acetone intermediate to propylene via an additional reaction pathway. The conversion of bio-ethanol to butadiene is an industrially important process, aiming at bio-based production of butadiene rubber. Butadiene synthesis from ethanol is a complex cascade reaction that typically requires well-balanced amounts of catalytic active sites.122,123 Promising results in terms of butadiene production were achieved over Zr-containing β zeolites doped with 1 wt% Ag, particularly over the catalyst prepared using a post-synthesis method.124 The presence of Zr(OSi)3OH open sites (Fig. 12) with high Lewis acidity is key for improved conversion of ethanol to butadiene in continuous-flow conditions. As a result, Ag/Zr-β zeolite (Si/Al ratio of 75 in parent zeolite and 3.5 wt% Zr) afforded higher production rate of butadiene (3 μmol g−1 s−1).
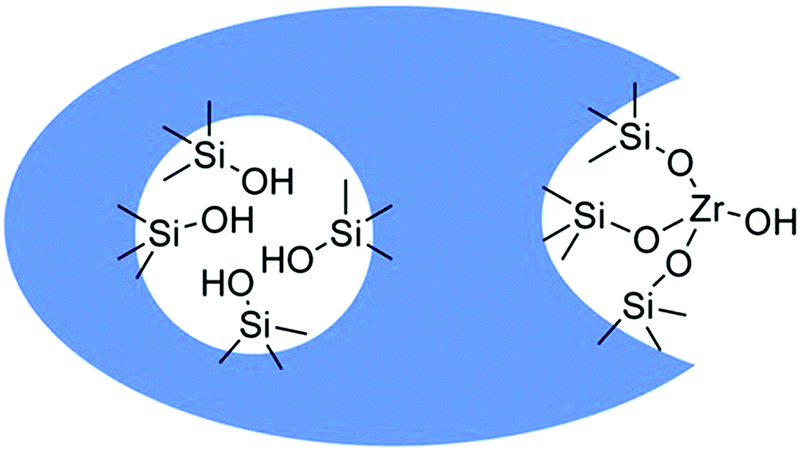 | ||
| Fig. 12 Schematic representation of Zr grafting by treatment of dealuminated zeolites with ZrOCl2 in DMSO. Reproduced with permission from ref. 124. Copyright (2016) John Wiley and Sons. | ||
2.3 Zeolite-supported catalysts
The presence of unique pore dimensions and structures allow the zeolites to accommodate a wide range of catalytically active metals and metal oxides. This adds multifunctional properties to the final catalytic materials with synergistic interactions, which may have important applications in biomass conversion.Modification of HZSM-5 using a mesoporous silica (MCM-41) was attempted to improve its porous structure and acidity and consequently the catalytic efficiency of Ni/HZSM-5 for the transformation of sorbitol into aromatic compounds in gas phase.133 The acid sites of zeolite and its shape selectivity promoted the dehydration and aromatization of sorbitol, whereas Ni metal centres catalyzed the hydrogenation and cracking steps. The catalyst acidity determines the carbon distribution of products, resulting in 66.3% carbon yield in aromatic compounds at full conversion of sorbital. For the hydrogenation of ethyl levulinate, the 1% Pt/ZSM-35 catalyst provided 100% ethyl levulinate conversion and 99% γ-valerolactone selectivity, outperforming Pt catalysts supported on SAPO-34, ZSM-35, ZSM-5, HY, USY, and MOR zeolites.134 Abundant metallic Pt NPs and optimum surface acidity of ZSM-35 were the primary reasons for achieving high yields of γ-valerolactone. The synergistic effect of metal sites and zeolite acid sites was also observed in Zn/ZSM-5 catalysts for the conversion of γ-valerolactone to aromatic compounds.135 A Cu–MOR zeolite, synthesized by a one-pot template-free route, was used as an efficient support for stabilizing vanadium oxide species.53 The investigated aerobic oxidation of HMF over the 10V2O5@Cu–MOR(125) catalyst demonstrated a high yield of 2,5-diformylfuran (91.5%) and excellent catalyst reusability performance (entry 1, Table 5). The synergy between the guest V sites and the framework Cu species contributed to the exceptional performance of 10V2O5@Cu–MOR(125) catalyst in the liquid phase oxidation of HMF. Owing to high dispersion of Ru NPs on NaY zeolite, the 2 wt% Ru/NaY catalyst displayed about 78% yield of 2,5-dimethylfuran at nearly 100% HMF conversion during liquid phase HMF hydrogenation (entry 1, Table 4).136 Furthermore, no considerable loss in HMF conversion and 2,5-dimethylfuran yield was found even after fifth recycle of the catalyst. Pd NPs encapsulated inside of silicalite-1 zeolite (Pd@S-1-OH) exhibited high selectivity to furan (>99.9%) at complete conversion of furfural, outperforming the conventional Pd/zeolite catalysts (entry 1, Table 3).137 This superior catalytic efficiency was due to the controlled diffusion of molecules within the hydrophilic micropores of zeolite, enabling efficient adsorption of furfural and by-products as well as facile diffusion of furan product.
| Entry | Catalyst | Reactant | Reaction conditions | Product | Catalytic activity | Key factors for the catalyst activity | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Conv. (%) | Yield (%) | |||||||
|
a BTX – benzene, toluene, xylene.
b Ce/Si = 0.03.
c WHSV – weight hour space velocity.
d TOF – turnover frequency.
e Ni:Co = 1 (20 wt% total loading).
f Ru = 5 wt% and Zr:La = 2.
|
||||||||
| 1 | 1 wt% Pt/H-MFI-90 | Guaiacol | Liquid phase, 0.05 g catalyst, 2 mmol substrate, n-hexadecane, 180 °C, 5 h, 50 bar H2 | Cyclohexane | 100 | 93 | Pt reducibility and acidic strength of zeolite | 138 |
| 2 | Fe–Ni/H-beta | Anisole | Vapor phase, 0.1 g/min feeding rate, 18 g catalyst, 0.32 L min−1 flow rate of H2, 350 °C | BTXa | 100 | 24.9 | Pore size, BET surface area, and acid sites | 140 |
| 3 | 5 wt% Ni/Ce-SBA-15b | Anisole | Vapor phase, H2/feedstock ratio (300 Nm3 m−3), 290 °C, 7 bar H2, WHSVc (163.2 h−1), 6 h | Benzene | 7.9 × 103 (TOF, s−1)d | — | High metal dispersion and specific Ni sites in contact with Ce surface species | 174 |
| 4 | 5 wt% Ni/Al-SBA-15 | Anisole | Liquid phase, 50 mL of a solution containing 3 wt% anisole, 220 °C, 50 bar H2, 2 h | Cyclohexane | 99.8 | 95 | Acidity of support and synergy of metallic and acid sites | 226 |
| 5 | CuNi/Ti-MCM-41 | Guaiacol | Liquid phase, 0.1 g catalyst, 3 wt% guaiacol of heptane, 100 bar H2, 260 °C, 6 h | Cyclohexane | 90 | 45 | Abundant acid/redox sites and high metal dispersion | 228 |
| 6 | Co/Al-MCM-41 | Guaiacol | Vapor phase, 1 atm, 1.67 h contact time, 30 min, 300 °C H2/guaiacol molar ratio = 25 | Phenol | 96 | 39 | Optimum amounts of acid and metal sites | 229 |
| 7 | MoO3 | phenol | Liquid phase, 1 g catalyst, octane, 340 °C, 5 bar H2 + 30 bar N2 | Benzene | 98 | 97 | Oxygen vacancies in MoO3 | 392 |
| 8 | 10.5 wt% MoOx/Al2O3 | m-Cresol | Liquid phase, 0.12 g catalyst, 340 °C, 40 bar H2 | Toluene | 20 ± 1 (TOF, h−1) | — | Reducibility of Mo species | 394 |
| 9 | 10 wt% Co/TiO2 | Eugenol | Liquid phase, 1 mmol eugenol, 0.065 g catalyst, n-dodecane, 10 bar H2, 200 °C, 2 h | Propylcyclohexanol | 100 | 99.9 | High dispersion of Co and strong metal–support interaction | 395 |
| 10 | 0.4 wt% Pt/20Nb–Al2O3 | Diphenyl ether | Vapor phase, W/F = 15.2 min, 0.05 g catalyst, 30 bar H2, H2/oil = 300, 240 °C | Cyclohexane | 100 | 99.2 | Abundant water-tolerant Lewis acid sites | 397 |
| 11 | NiCoe/γ-Al2O3 | Guaiacol | Liquid phase, 5 g guaiacol, 45 g water, 2 wt% of catalyst, 8 h, 200 °C, 50 bar initial H2 | Cyclohexanol | 96 | 65.2 | High acidity and strong metal–support interaction | 398 |
| 12 | Ti2.0Pd/SiO2 | Guaiacol | Vapor phase, 0.20 g catalyst, 3 wt% guaiacol in n-dodecane, 300 °C, 20 bar H2 | Cyclohexane | 100 | 80 | Smaller Pd particles and high deoxygenation activity of TiO2 | 400 |
| 13 | Ruf/ZrO2–La(OH)3 | Guaiacol | Liquid phase, 5 mmol guaiacol, 0.10 g catalyst, H2O, 40 bar H2, 4 h | Cyclohexanol | 100 | 91.6 | High metal dispersion and basic strength of the support | 403 |
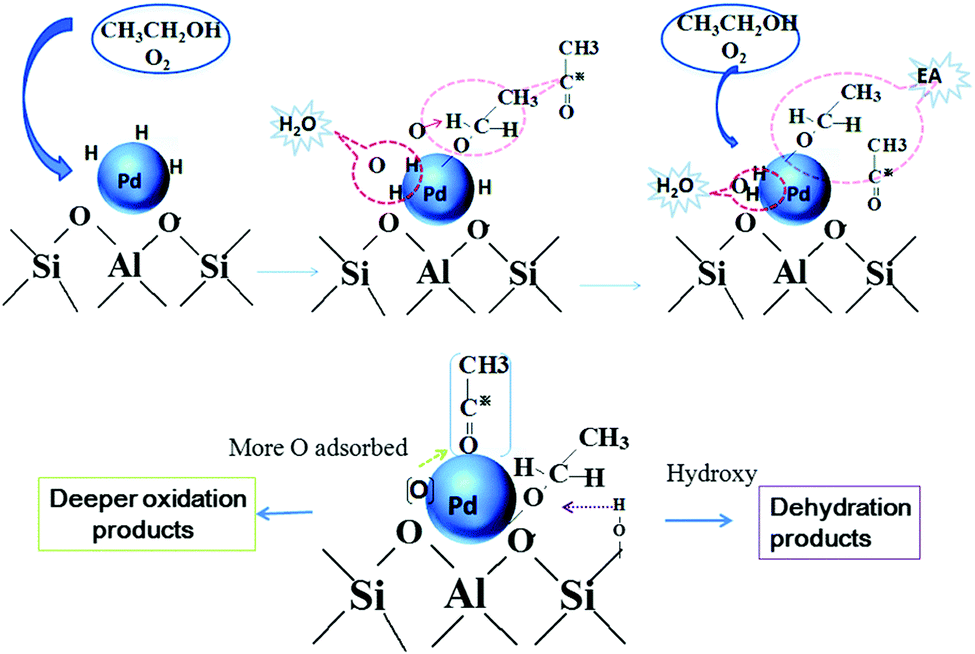 | ||
| Fig. 13 Mechanism of ethyl acetate formation from ethanol through reverse hydrogen spillover process and the mechanism of dehydration and deeper oxidation over the Pd catalyst. Reproduced with permission from ref. 146. Copyright (2016) Royal Society of Chemistry. | ||
2.4 Hierarchical porous zeolites
Hierarchical porous zeolites, having at least two levels of porosity, are of increasing research interest for catalytic biomass valorization because of their favorable mass transport properties compared to that of conventional microporous zeolites.30,52 As shown in Fig. 14 (right side, hierarchical zeolite), the introduction of secondary pores into the zeolite structures can provide three key advantages: (1) enhanced active site accessibility due to controllable diffusion of molecules (orange and green spheres).8 (2) Improved product selectivity (green spheres) driven by the presence of larger external surface. (3) A higher number of active sites (i.e. pore mouths) available for the reaction of bulky molecules (red spheres). Various facile strategies have been developed for the synthesis of hierarchical porous zeolites, including removal of framework atoms, surfactant-assisted procedure, hard-templating, zeolitization of preformed solids, and organosilane-based methods.8,30,52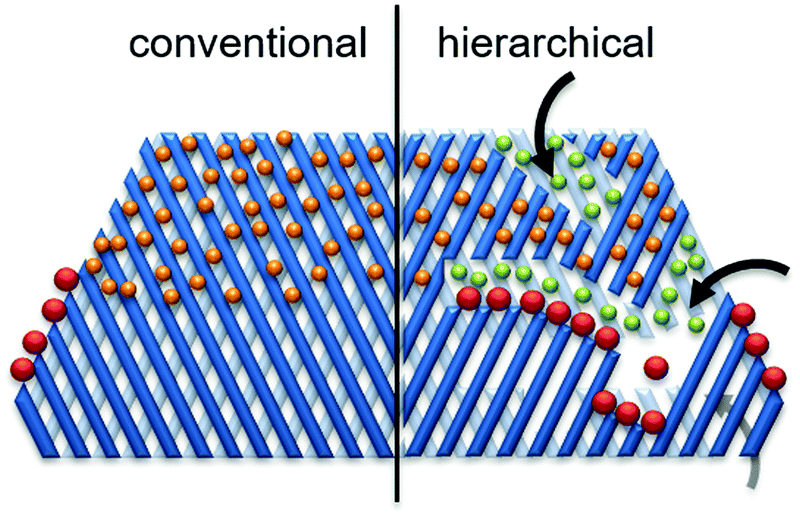 | ||
| Fig. 14 Schematic representation of advantages of hierarchical zeolites towards active sites accessibility and transport/diffusion of reactants/products. Reproduced with permission from ref. 8. Copyright (2016) Royal Society of Chemistry. | ||
 | ||
| Fig. 15 TEM images for (A) Com-ZSM-5, (B) Micro-ZSM-5, (C) Nano-ZSM-5-10%, and (D) Hier-ZSM-5–10%. Reproduced with permission from ref. 102. Copyright (2017) John Wiley and Sons. | ||
3. Mesoporous SiO2 based catalysts for biomass valorization
Over the last two decades, mesoporous materials have been tremendously used in heterogeneous catalysis because of their exceptional porosity and surface area properties.33,163,164 Mesoporous materials have also found plentiful applications in adsorption, sensing, drug delivery, nanodevices, and lithium-ion batteries. According to IUPAC, mesoporous materials (the Greek prefix ‘meso’ means ‘in between’) are defined as those having pore diameters (i.e. 2–50 nm) in between micropores (<2 nm) and macropores (>50 nm).165 Two types of synthesis methods, namely soft-templating and hard-templating, are often used for the production of mesoporous materials with monodispersed pores in a long-range ordered structure (Fig. 16).166 The strategic motivation to develop mesoporous materials was to overcome the mass transport limitations associated with the zeolites whose pore dimensions are usually below 2 nm. Porous materials with similar textural and acid characteristics to zeolites, but with larger pores could mitigate the barrier for the diffusion of bulky molecules, leading to improved reaction rates.165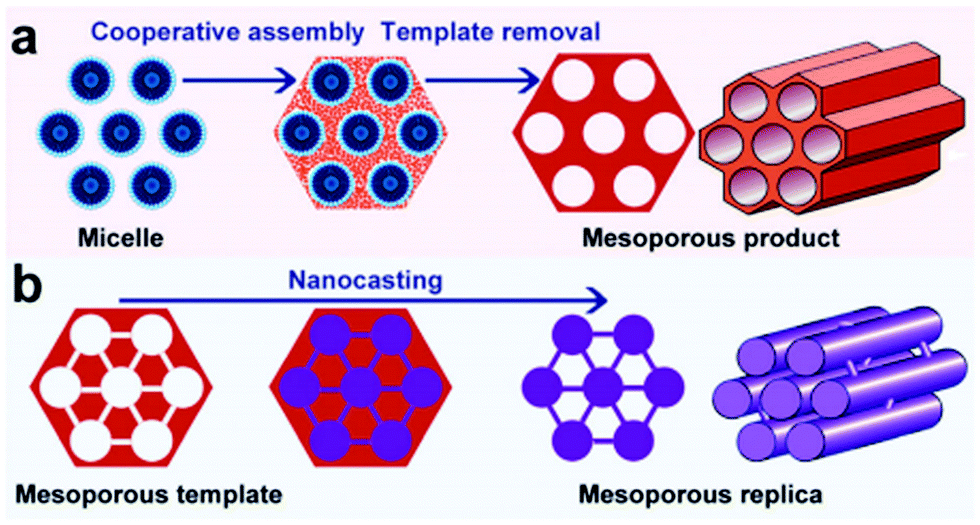 | ||
| Fig. 16 (a) soft-templating and (b) hard-templating synthesis routes for the design of ordered mesoporous materials. Reproduced with permission from ref. 166. Copyright (2011) Royal Society of Chemistry. | ||
The pioneering work of Mobil's scientists in the 1990s led to the discovery of mesoporous silica materials, popularly known as the M41S family.32,33,164,167 This breakthrough finding has inspired the scientific community towards developing a variety of novel mesoporous materials, including non-silica metal oxides (Al2O3, ZrO2, Co3O4, NiO, Fe2O3, and Mn3O4), mesoporous carbons, mesoporous organic polymers, and mesoporous inorganic non-oxide materials (metal nitrides, metal chalcogenides, carbides, and fluorides). The use of supramolecular aggregates as structure-directing agents in a sol–gel synthesis medium plays a crucial role in the formation of ordered mesoporous silica structures, with controlled pore size distribution in hexagonal, cubic or lamellar structures (Fig. 17).32 By modifying synthesis methodologies and self-assembly mechanisms, a wide variety of mesoporous silica materials have been developed, including SBA (Santa Barbara Amorphous, University of California, Santa Barbara),168 KIT (Korean Institute of Technology, currently known as Korea Advanced Institute of Science and Technology),169 MSU (Michigan State University, USA)170 and COK (Centre for Surface Chemistry and Catalysis, KU Leuven, Belgium)171 families.
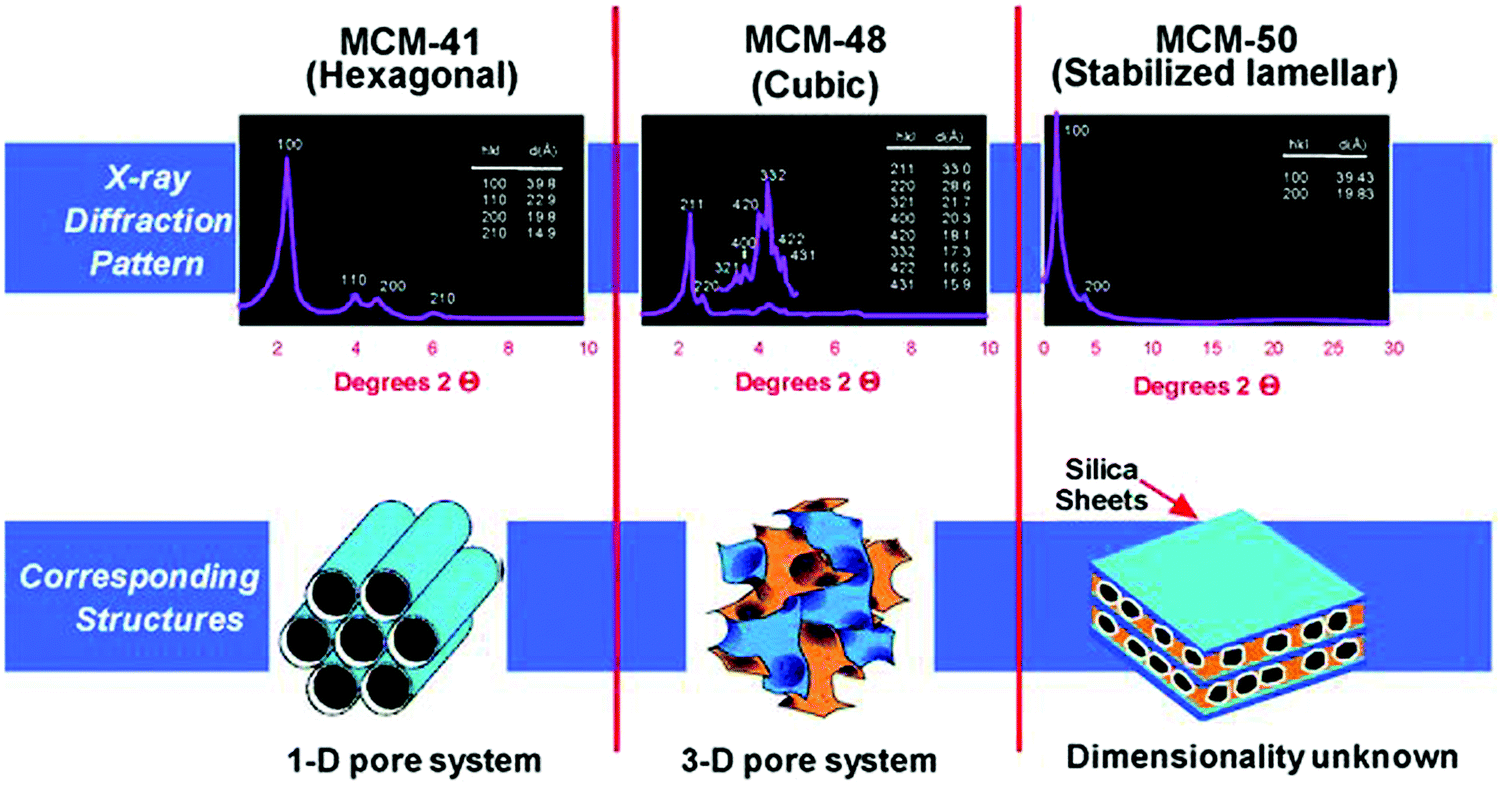 | ||
| Fig. 17 Powder XRD patterns and structures of mesoporous SiO2 materials (MCM-41, MCM-48, and MCM-50). MCM – Mobil Composition of Matter or Mobil Crystalline Material. Reproduced with permission from ref. 32. Copyright (2013) Royal Society of Chemistry. | ||
3.1 Catalytic role of mesoporous SiO2 properties in biomass valorization
Mesoporous silica materials exhibit a broad spectrum of fascinating features, such as unique textural properties, negligible mass transport limitations, and diversified functionalization.29 Notably, they show tailorable pore diameters (2–50 nm), large pore volumes, and high specific surface areas (400–1400 m2 g−1). Owing to these exceptional characteristics, mesoporous silica can efficiently interact with atoms, molecules, or ions, not only at the external surface, but also through the internal pores.33,164 The high specific surface area of mesoporous silica enables homogeneous dispersion of catalytically active metal species (Ru, Ni, Cu, Co, Mn, Pt, Pd, Fe, etc.), which could be readily accessible throughout the catalyst matrix. The ordered mesoporous silica materials with relatively large pores are particularly important for catalytic biomass upgrading as they can allow the bulky biomass molecules to access the active sites inside the pores (Fig. 18).172 The significance of hierarchical SiO2 materials, having interconnected mesopore–macropore networks and sometimes micropores, is also recognized in biomass valorization. The key advantage of hierarchical porous materials in catalysis is that micro/mesopores can offer active sites for reactants, whereas macropores can provide feasible routes for reactants/intermediates accessing the active sites, as well as for the facile desorption of products, resulting in high turnover frequency rates.36 These multimodal porous structures can tremendously facilitate mass-transport in viscous liquid phase biomass conversions, such as biodiesel synthesis.173 Morphology of mesoporous silica materials is also a pivotal parameter, together with pore structure and surface area, for fine-tuning their activities and selectivities in biomass upgrading. Precise shape control of catalyst particles allows preferential exposure of active species on the catalyst surface, which is a vital criterion for heterogeneous catalysis. In addition, the surface-exposed active species can provide an advantageous basis for the systematic investigation of structure–activity relationships, which could provide new insights for the design of high-performing shape-controlled mesoporous catalysts for efficient biomass conversion. | ||
| Fig. 18 (a) Disordered and (b) ordered mesoporous structures. Reproduced with permission from ref. 172. Copyright (2013) American Chemical Society. | ||
In order to improve the catalytic activity of mesoporous silica, incorporation of metals into the silica framework has been frequently attempted.174–177 Aluminium was the first metal incorporated into the framework of mesoporous silica (e.g., MCM-41) to develop a potential alternative to zeolites for petrochemical industry.177 Since then, various metals including Zr, Co, and Sn have been incorporated in order to obtain high-performing mesoporous silica catalysts. For example, by carefully varying the composition of Al/Si ratio, new mesoporous silica–alumina catalysts were developed with high BET surface area and abundant acid properties. The resultant composites showed excellent catalytic activities in the production of HMF from carbohydrates.178 Incorporation of acid or base functional species in the framework of mesoporous silica is also a promising strategy to multiply its applications in catalysis. The large pore volume of mesoporous silica provides ample room to accommodate these functional species.179 For instance, acid functionalized (–SO3H, –SO3H/C2H5, –COOH, and –CN) SBA-15 catalysts were found to show high performance for the esterification of fatty acids as well as for the transesterification of triglycerides to produce biodiesel fuel. Therefore, by controlling the surface and the internal porous structures, a variety of advanced mesoporous silica materials have been developed and their catalytic applications in biomass conversions are reviewed in the following paragraphs.
3.2 Pristine and doped mesoporous SiO2 catalysts
Mesoporous silica-based materials can exhibit Lewis acidity originating from Si as well as from incorporated metals. Hence, the Lewis acidity and ordered mesoporous structures are the key factors for the catalytic efficiency of mesoporous silicas, mainly for acid-catalyzed biomass conversions.000 h−1. Furthermore, MCM-41 catalyst was active for more than 50 h time-on-stream study, with a remarkable resistance to water, allowing the use of crude ethanol without further purification.
3.3 Shape-controlled and hierarchical porous SiO2-based catalysts
Mesoporous silica-based materials with well-defined shapes have attracted paramount attention for catalytic biomass upgrading, as reaction rates and product selectivity can be controlled by shape-dependent properties.178,192 As well, hierarchical porous silica materials have been featured in forefront research towards catalytic biomass conversions because of their synergistic meso–macro porous structures.36,179 Several studies also reported the cooperative effect of shape-control and pore hierarchy in enhancing the catalytic performance of porous silica materials. In this section, we focus on the catalytic applications of shape-controlled and hierarchical porous SiO2-based materials for various biomass conversions.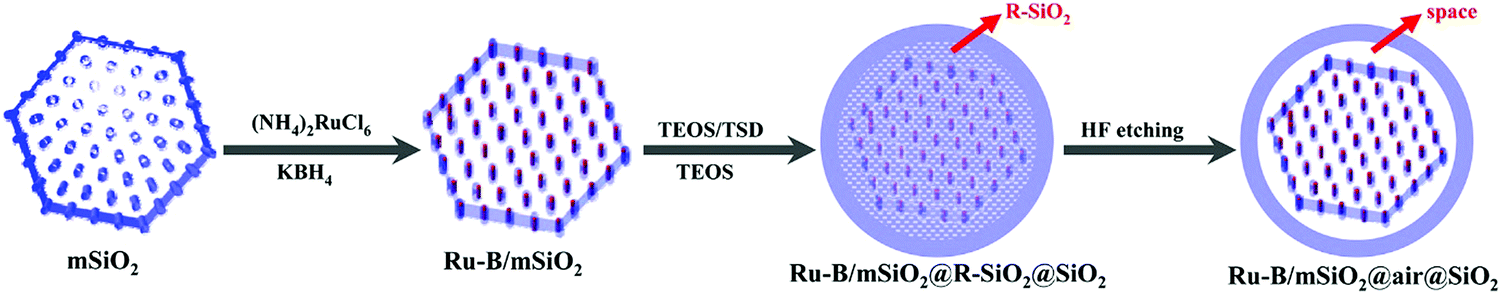 | ||
| Fig. 19 Illustration of the synthesis process of yolk–shell structured Ru–B/mSiO2@air@SiO2 catalyst. Reproduced with permission from ref. 192. Copyright (2014) American Chemical Society. | ||
A propylsulfonic acid-functionalized SBA-15, containing fiber-like domains with an average particle size of 430 nm aggregated into macrostructures (Fig. 20), was investigated for the condensation of furfuryl alcohol with 1-butanol to give butyl levulinate.196 The successful incorporation of sulfonic acid groups into the SBA-15 structure was confirmed by FT-IR analysis. This catalyst showed about 96% yield (and 100% selectivity) of butyl levulinate and no leaching of the active species was noticed. A strong relationship of surface-decorated active species with the catalytic activity of Co–Mn oxide/nanospherical porous silica was found in liquid phase, base-free oxidation of HMF to 2,5-furandicarboxylic acid.197 The optimized catalyst (Mn:Co = 1:2) displayed ∼100% HMF conversion with a 72.4% yield of 2,5-furandicarboxylic acid as well as a durable catalytic activity for several recycles (entry 2, Table 5). Uniformly dispersed and thermally stable Ru nanoclusters were found within mesoporous zirconium silica nanospheres, which are the primary factors for achieving improved catalytic results in the aqueous-phase hydrogenation of furan derivatives.199 The reaction temperature also played a determinant role in controlling product selectivity over the Ru cluster catalyst. A plausible mechanism was proposed for the channel-length effect of mesoporous silica on the size of Ru clusters (Fig. 21).
 | ||
| Fig. 20 SEM images of SBA-15-SO3H catalyst. Reproduced with permission from ref. 196. Copyright (2014) John Wiley and Sons. | ||
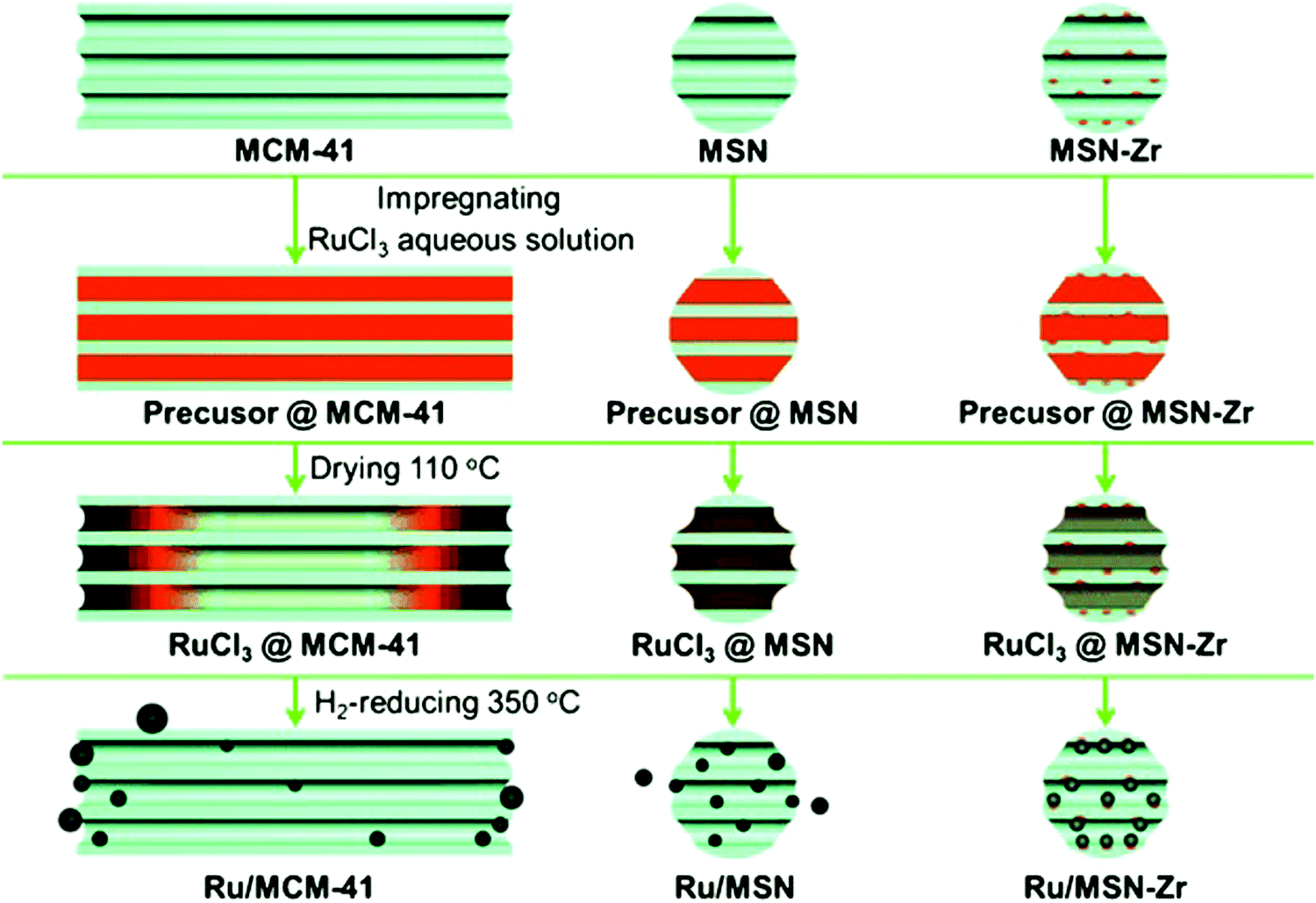 | ||
| Fig. 21 Mechanism for the channel-length effect of mesoporous silica on the synthesis of supported Ru catalysts. Reproduced with permission from ref. 199. Copyright (2013) John Wiley and Sons. | ||
3.4 Mesoporous SiO2 supported catalysts
The dispersion of metal NPs into the framework of porous materials is a widely used strategy for the development of highly stable, efficient supported metal catalysts. As a result, aggregation and leaching of active phase particles can be prevented, while maintaining active site accessibility to reactants/intermediates.177 Mesoporous silica is regarded as an ideal supporting carrier for stabilizing metal particles because of its unique characteristics, such as large surface area and suitable pore dimensions.177,197Hydrogen is one of the highly appealing energy sources for our modern world. However, it is mainly produced from traditional carbon-sources via steam reforming of natural gas or coal gasification. As an alternative process, the direct conversion of cellulose to hydrogen rich gas was performed using Ni catalysts supported on different mesoporous silica materials, such as SBA-15, SBA-16, KIT-6, and MCM-41.205 Among them, Ni/SBA-15 and Ni/KIT-6 catalysts afforded the highest hydrogen yield, outperforming the Ni catalyst supported on commercial silica. The presence of high BET surface area, pore volume, and larger pore diameter facilitates the easy diffusion of complex reactant/intermediate molecules towards the active sites, hence increasing hydrogen formation. For selective hydrogenation of glucose to sorbitol, comparable activity results were achieved over Ru/MCM-48 catalyst with respect to Ru/TiO2 and Ru/C catalysts.180 In recent work, bimetallic Ru–Ni particles (total metal loading is 3 wt%) supported on MCM-48 were demonstrated to be efficient catalysts for the hydrogenation of glucose to sorbitol.206 Importantly, catalysts with Ru:Ni ratios higher than 0.45 displayed the best performance compared to monometallic Ni/MCM-48 catalyst. The Ru–Ni/MCM-48 catalyst was reused for three cycles without much loss in catalytic activity.
García-Sancho et al.207 demonstrated a considerable effect of Nb2O5 on the catalytic performance of Nb2O5/MCM-41 for the dehydration of xylose to furfural. About 74.5% conversion with a 36.5% furfural yield was achieved over a 16 wt% loaded Nb2O5 catalyst. Moreover, the optimized catalyst was reused for three runs with no significant loss of catalytic activity. Catalytic hydrogenation of furfural can provide several valuable chemicals and solvents, including furan, furfuryl alcohol, and tetrahydrofuran. The most significant results obtained for furfural hydrogenation catalyzed by porous and nanoscale materials are presented in Table 3. For instance, bimetallic Co–Cu particles supported on SBA-15 with a fixed Cu loading (10 wt%) and various Co loadings (2.5, 5, and 10 wt%) were developed for the liquid phase hydrogenation of furfural to furfuryl alcohol.208 The effect of several reaction parameters, such as catalyst dosage, pressure, temperature, and furfural concentration was evaluated for this reaction. Compared to the catalysts supported on conventional silica, the Co–Cu/SBA-15 catalyst (Cu 10 wt% and Co 10 wt%) was found to show a high catalytic performance with 99% furfural conversion and 80% furfuryl alcohol selectivity, which was attributed to high dispersion of active Cu sites (entry 2, Table 3). The beneficial effect of both metal–metal and metal–support interactions was noticed for the liquid phase hydrogenation of furfural to tetrahydrofurfuryl alcohol.209 The reaction was carried out over Ni–Co catalysts supported on short channeled SBA-15, MCM-41, MCM-4, MCF-2 and conventional SBA-15. Interestingly, Ni–Co catalyst (Ni/Co molar ratio = 0.67) supported on short channeled SBA-15 showed a higher yield of tetrahydrofurfuryl alcohol (92%) at full conversion of furfural, due to the unique structure of the short channels as well as the appropriate and uniform pore size distribution (entry 3, Table 3). The formation of NiCo2O4 species favored the homogenous distribution of active sites (Ni and CoO) in bimetallic catalysts, which also contributed greatly to obtain improved product yields in the hydrogenation of furfural. A series of Cu/mesoporous SiO2 catalysts (Cu loadings from 2.5 to 20 wt%) were evaluated for the gas phase hydrogenation of furfural.210 The catalyst with 10 wt% Cu loading provided an appropriate balance between the tendency to form carbonaceous deposits and the resistance to particle sintering, giving good yields of 2-methylfuran (95%) after 5 h (entry 4, Table 3).
| Entry | Catalyst | Reaction conditions | Product | Catalytic activity | Key factors for the catalyst performance | Ref. | |
|---|---|---|---|---|---|---|---|
| Conv. (%) | Yield (%) | ||||||
| a (Cu – 10 wt% and Co – 10 wt%). b FOL – furfuryl alcohol. c (Ni/Co molar ratio = 0.67). d THFA – tetrahydrofurfuryl alcohol. e MF – 2-methylfuran. f (Cu – 10 wt% and Fe – 10 wt%). g LHSV – liquid hourly space velocity. h GHSV – gas hourly space velocity. | |||||||
| 1 | Pd NPs@silicalite-1 zeolite | Gas phase, 0.1 g catalyst, 5 wt% furfural (mass ratio of furfural/n-butyl alcohol at 1/19), feed rate at 0.5 mL h−1, 10% H2/Ar at 10 mL min−1, 250 °C, atmospheric pressure | Furan | 100 | 99.9 | Controlled diffusion of molecules within the hydrophilic zeolite micropores | 137 |
| 2 | Co–Cu/SBA-15a | Liquid phase, 1 g catalyst, 170 °C, 20 bar H2, furfural-to-isopropanol molar ratio of 1:7, 4 h |
FOLb | 99 | 80 | High dispersion of active Cu sites | 208 |
| 3 | Ni–Co/SBA-15c | Liquid phase, 0.8 g catalyst, 5.82 g furfural, ethanol, 90 °C, 50 bar initial H2, 2 h | THFAd | 100 | 92 | Short pore channels, appropriate and uniform pore size distribution of SBA-15, uniform and high dispersion of Ni0 and CoO | 209 |
| 4 | 10 wt% Cu/mesoporous SiO2 | Gas phase, 5 h, 210°C, H2/furfural molar ratio of 11.5, weight hourly space velocity of 1.5 h−1 |
MFe | — | 95 | Higher amounts of accessible active Cu sites as well as appropriate balance between the tendency to form carbonaceous deposits and resistance to particle sintering | 210 |
| 5 | Pd–Pt/TiO2 | Liquid phase, 0.3 M furfural, substrate/metal molar ratio = 500, 3 bar H2, 30 °C, 240 min | THFA | 100 | 95 | Synergistic Pd–Pt interaction | 336 |
| 6 | CuNi/MgAlO (Cu/Ni = 1:1) |
Liquid phase, 5 mmol furfural, methanol, 0.01 g catalyst, 100 °C, 4 h, 4 MPa H2 | FOL | 100 | 99 | High metal dispersion, homogeneous composition of CuNi alloys, and catalyst basicity | 337 |
| 7 | CuZnAl mixed oxide (Cu/Zn = 0.5) | Liquid phase, 0.2 g catalyst, 5 mmol furfural, 150 °C, 4 MPa H2, 6 h, H2O | Cyclopentanone | 97.9 | 62 | Smaller sized Cu particle with homogeneous dispersion and high specific surface area | 339 |
| 8 | CuFef/γ-Al2O3 | Gas phase, 1 mL catalyst, 175 °C, LHSVg 1 h−1 with respect to furfural and GHSVh 1200 h−1 with respect to H2, 1 atm. pressure | FOL | 93 | 91 | Synergistic bimetallic particles and abundant oxygen vacancies in iron oxide | 340 |
The combination of mesocellular silica foam (MCF) with Nafion-resin nanoparticles was found to show good catalytic performance in the liquid phase dehydration of fructose to HMF (entry 8, Table 1).211 The effects of Nafion loading, reaction temperature, time, and solvent were investigated for this reaction. A 89.3% yield of HMF was obtained over a Nafion(15)/MCF catalyst, which was attributed to strong acid strength and open large pore system. A possible reaction mechanism was proposed for fructose dehydration to HMF over Nafion-modified MCF catalysts as shown in Fig. 22. A three-phase catalytic system, including a solid acid catalyst (nanostructured vanadium phosphate and mesostructured cellular silica foam) was developed for the conversion of fructose to HMF.212 Pyridine-adsorption FT-IR studies revealed the presence of both Brønsted and Lewis acidic sites in the developed catalyst. A high selectivity to HMF (91%) at 89% fructose conversion was obtained over this catalytic system because of its ability to inhibit undesired side reactions of HMF in the presence of H2O. A direct correlation of the γ-valerolactone production rate with the dispersion state of Zr species was observed for the catalytic transfer hydrogenation of levulinic acid and its esters over ZrO2/SBA-15 catalysts.213 A plausible reaction mechanism was proposed, in which basic Zr–OH sites promote the catalytic transfer hydrogenation reaction involving a six-membered ring transition state.
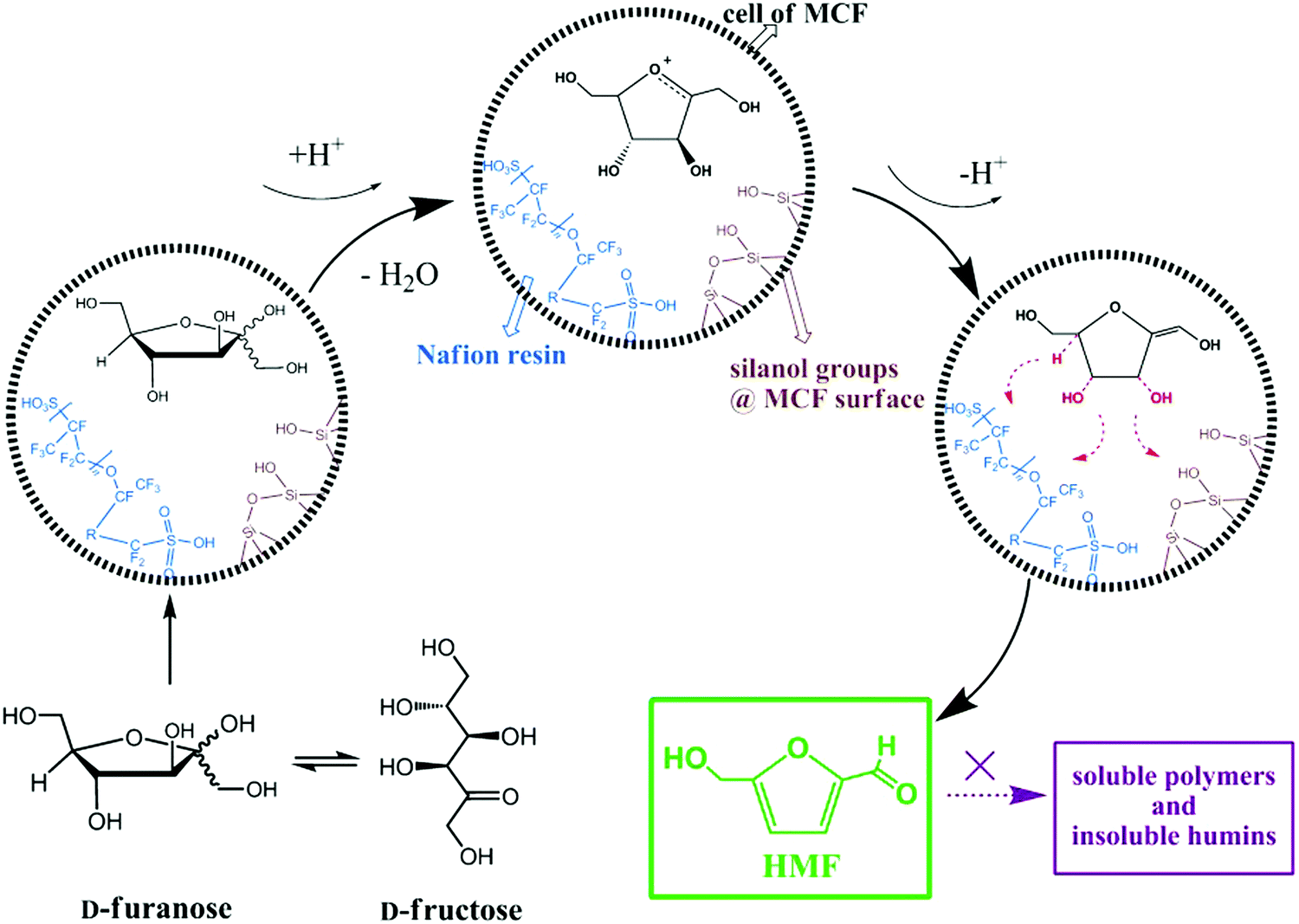 | ||
| Fig. 22 Mechanism for the dehydration of fructose to HMF over Nafion-modified MCF catalysts. Reproduced with permission from ref. 211. Copyright (2013) John Wiley and Sons. | ||
:H-site ratio, the α-MoC1−x/SBA-15 catalyst showed excellent decarbonylation selectivity in the HDO of acetic acid. Compared to Ru particles supported on conventional SiO2, Ru/SBA-15 catalyst (0.2–2 wt% Ru) was found to display a good activity for the aqueous phase hydrogenation of three bio-based molecules: acetol, 2-butanone, and methyl 2-acetamidoacrylate.223 High dispersion of Ru NPs, unique structure of SBA-15, along with improved reducibility of the oxidized surface layer formed at the surface of Ru metal nanocrystals are vital for achieving high product yields from all three bio-based molecules.
Various aromatic chemicals, including benzene, toluene, xylenes, phenol, cresols, and alkyl phenols were obtained from the hydroprocessing of lignin pyrolysis vapors carried out over a mesostructured Fe/SiO2 catalyst.224 Owing to larger mesoporous dimensions, Fe/SiO2 catalyst inhibited the accumulation of carbon particles, whereas coke deposits completely blocked the micropores in Fe/activated carbon catalyst. The activity of Fe/SiO2 catalyst could be regenerated by a controlled oxidation process, which can remove carbon deposits and reduce iron in sequential fixed or fluidized beds. The Co/SBA-15 catalyst is selectively promoted the hydrogenolysis of methoxy functional group of anisole.225 In contrast, hydrogenation of aromatic ring of anisole to methoxycyclohexane was found in the presence of Ni/SBA-15 catalyst because of high hydrogenation ability of nickel species. Yang et al.174 revealed the effect of Ce/Si molar ratio (from 0 to 0.08) on the efficiency of Ni/Ce-SBA-15 catalyst for the vapor phase HDO of anisole. Incorporation of Ce enhances the Ni dispersion and the specific active sites in 5% Ni/Ce-SBA-15 (Ce/Si = 0.03), resulting in improved yields of benzene (entry 3, Table 2). Both the metal dispersion (Co and Ni) and the metal–support interaction were strongly dependent on the support nature (SBA-15 and Al-SBA-15).226 Compared with Co-based catalysts, 5 wt% Ni catalyst supported on Al-SBA-15 provided higher anisole conversion (99.8%) with 95% selectivity to cyclohexane (entry 4, Table 2). The strong interaction of Co with the support hinders the total Co reduction prior to the reaction, and hence a lower activity in the HDO of anisole.
Surprisingly, monometallic Co/Al-MCM-41 catalyst was found to show high activity in terms of deoxygenation products yield for the vapor phase HDO of guaiacol, compared to bimetallic Co–Ni/Al-MCM-41 catalyst (10 wt% of total metal loading).227 This was attributed to nickel's ability to promote multiple hydrogenolysis of guaiacol C–C bonds, leading to carbon loss via methanation, especially at high temperatures. On the other hand, Ambursa et al.228 demonstrated the favorable effect of both bimetallic interactions and incorporated metal for the liquid phase HDO of guaiacol over Cu–Ni/Ti-MCM-41 catalysts (entry 5, Table 2). The incorporation of Ti into the catalyst matrix led to abundant acid-redox properties and enhanced metal dispersion. As a result, 10 mol% CuNi/TiMCM-41 (Cu:Ni = 3:1) catalyst presented 90% guaiacol conversion and 50% cyclohexane selectivity, outperforming the unpromoted CuNi/MCM-41 catalyst. Hamid et al.175 also investigated the effect of Ti amount (Ti/Si = 10, 20 and 30%) on the structure–activity properties of bimetallic Cu–Ni/Ti-MCM-41 catalysts for the liquid phase HDO of guaiacol. Although Ti loading indicated an adverse effect on the BET surface area, large amounts of acid sites and better redox properties were found for a 20% Ti loaded Cu–Ni/Ti-MCM-41 catalyst, which gave 74.2% guaiacol conversion with 48.8% cyclohexane selectivity. The authors propose that acid sites initiate the removal of oxygen via dehydration, while redox sites promote hydrogenation of guaiacol to cyclohexane. A continuous-flow strategy was developed for the vapor phase HDO of guaiacol over M/Al-MCM-41 (M = Co and Ni) catalysts (entry 6, Table 2).229 The acidic Al-MCM-41 support stimulates the methyl transfer reaction to obtain methylated products. The Co/Al-MCM-41 catalyst is particularly selective towards C–O hydrogenolysis of guaiacol to obtain phenol. Compared with a conventional Ni/γ-Al2O3 catalyst, the 10 wt% Ni catalyst supported on mesoporous SiO2 (broad pore size distribution) exhibited high hydrocarbon yields and excellent catalytic stability for the liquid phase HDO of lignin-derived compounds.230 The Ni/SiO2 catalyst preferentially promotes the hydrogenation of aromatic rings to yield fuel grade hydrocarbons.
3.5 Sulfated mesoporous silica catalysts
Sulfated mesoporous silica catalysts not only combine the unique characteristics of individual candidates, but also endow new catalytically active sites, depending on the configurations of sulfate species on porous channels of silica. Particularly, these materials can show both Lewis and Brønsted acid properties due to the existence of metal centers and the immobilized –SO3H species, respectively, which could play a vital role in acid-catalyzed biomass conversions. Impregnation, grafting or immobilization have been largely used for the synthesis of these type of materials.179Many efforts have been undertaken to develop efficient sulfated mesoporous silica catalysts for the production of various bio-based chemicals, including 5-hydroxymethylfurfural (HMF). For instance, Cheng et al.77 revealed the beneficial role of a sulfated mesoporous silica, synthesized by impregnation of H2SO4 on MCM-41, followed by calcination at 550 °C, for the dehydration of fructose to HMF. Owing to well-balanced pore size and acidity, the sulfated mesoporous silica afforded about 90% HMF selectivity at 94% fructose conversion (entry 9, Table 1). The combination of a –SO3H functionalized mesoporous silica and an immobilized enzyme with –NH2 functionalized mesoporous silica is greatly useful for the direct conversion of glucose to HMF.231 About 61% and 30% yields of fructose and HMF, respectively, were obtained in a monophasic solvent system composed of tetrahydrofuran and H2O (4:1 v/v). Catalyst preparation method exhibited a great effect on the performance of sulfated mesoporous silica for fructose-to-HMF transformation.232 Markedly, the sulfated MCM-41 catalyst prepared by H2SO4 impregnation, followed by calcination at 550 °C provided 98% and 77% fructose conversions with 94% and 88% HMF selectivities when using 10% and 30% concentrations of fructose, respectively. Owing to abundant Brønsted acid sites, KIT-5-SO3H catalyst gave higher yields of HMF (94%) from fructose dehydration in liquid phase, while the Al incorporated KIT-5 catalyst afforded a 88.3% HMF yield (entry 10, Table 1).233
Several mesoporous silica materials coated by functionalized carbon with –SO3H or –COOH species were investigated for the production of useful bio-based chemicals.234 The resulting catalysts with abundant acid site content and strength displayed good yields of furanic ethers and levulinate esters from HMF, fructose, and furfuryl alcohol with ethanol. Owing to favorable textural properties and enhanced active site accessibility, these mesoporous catalysts showed higher reaction rates than Amberlyst™-15 (on the same catalyst mass basis), carbon based catalyst, aluminosilicate, non-porous silica and H-β zeolite. Manayil et al.235 developed octyl co-grafted PrSO3H/SBA-15 solid acids for the esterification of acetic acid with methanol. Results revealed that turnover frequencies for the targeted reaction improved with the increase of both surface acid site density and hydrophobicity of the catalysts. The nature of the acidic group (–SO3H, –SO3H/C2H5, –COOH, and –CN) has a huge effect on the activity of mesoporous silica for the transesterification of triglycerides to produce biodiesel.236 Among them, SBA-15/SO3H catalyst was found to be highly efficient for biodiesel production, which was attributed to high concentration of acid sites.
4. Nanoscale metal/metal oxide based catalysts for biomass valorization
Metals, metal oxides, or combinations of both are the most widely used heterogeneous catalysts in several industrially important applications, including auto-exhaust purification,237,238 petrochemicals production, as well as in biomass valorization.23,37,239 The ability of metals to exhibit multiple stable oxidation states is the primary factor for their widespread catalytic applications, especially in oxidation and hydrogenation processes. The coupling of a metal with another metal or metal oxide could lead to improved redox properties due to the synergistic metal–metal interactions, stimulated by the interplay of electron transfer at the interface structures. The addition of Cu, Ni or Co to Mo/Al2O3 catalyst, for instance, led to an enhanced reducibility, which played a crucial role for the formation of allyl alcohol with respect to hydroxyacetone in glycerol dehydration reaction.240The performance of a solid catalyst is greatly dependent on its outermost surface composition and geometry.241 Hence, fine-tuning the energy distribution and electronic structure of the catalyst surface could control the activation energy barrier and determine the catalyst activity/selectivity.242 Several crucial parameters have been suggested for precise modification of the catalyst surface, both electronically and geometrically (Fig. 23).23 Particularly, when the size of a catalyst particle is reduced to nanoscale range (1–100 nm in at least one dimension), the textual and redox properties at the catalyst surface can be greatly augmented, which can provide unusual catalytic activities. A classic example to illustrate this size-dependent effect is gold, which is considered as catalytically inactive in the bulk state, due to its insignificant chemisorption and redox properties. In contrast, uniformly dispersed gold nanoparticles (2–5 nm) on a metal oxide exhibit outstanding catalytic activity for several oxidation reactions.243–249 This pronounced size-dependent catalytic performance was evaluated based on the enhanced surface electronic and geometric properties of the smaller sized gold nanoparticles.250 In addition, shape-controlled nanoscale metals/metal oxides can provide exceptional catalytic properties because of the co-operative effect of size- and shape-dependent properties.250,251 In this section, the most important properties of metals, metal oxides, and metal oxide supported metals are highlighted, while discussing their ensuing catalytic effects in biomass valorization. For a deeper understanding, the catalytic performance of nanoscale metals/metal oxides achieved in biomass conversions is compared with that of the corresponding bulk analogues.
 | ||
| Fig. 23 Illustration of key parameters for the synthesis of nanoscale metal catalysts. Reproduced with permission from ref. 23. Copyright (2016) Royal Society of Chemistry. | ||
4.1 Nanostructured metals
Mono- and bi-metallic nanoparticles have gained unprecedented importance in catalysis because of their energetic structural and redox properties that could be substantially differ from their bulk counterparts.14,23,239,252,253 An active site in a metal catalyst could be a single atom or a collection of atoms that directly participate in a catalytic reaction through chemical bond cleavage and formation, during which the neighboring atoms could influence the energetics of these processes.254 The variation of particle size and surface structure of a metal catalyst can markedly affect the coordination number of surface metal atoms.255 Consequently, nanoscale metal particles contain a high percentage of coordinatively unsaturated atoms located preferentially at the surface, edges, or corners. Low coordinated atoms are highly energetic and can readily participate in catalytic redox processes because of their strong tendency to coordinate with reactant molecules in their vicinity.255Catalytic oxidation is one of the widely used methods for biomass valorization.256,257 The catalytic activity of metals in oxidation reactions is mainly determined by their affinity towards adsorbing molecular oxygen or other oxidants (e.g., H2O2). The estimated adsorption energy profiles revealed that oxygen could make strong bonds with metals present on the left side of transition metal series. Moreover, oxygen shows a strong affinity towards 3d metals compared to 4d and 5d metals.258 Accordingly, Ru binds the oxygen much stronger than Pd and Ag, while Au exhibits weak binding energy per oxygen atom, which is even less than the binding energy of O2 molecule.259 However, by reducing the size of the metal to nanoscale, the oxygen adsorption efficiencies can be improved, as observed perceptibly in the case of Au NPs.28,260,261 Hence, the selection of a metal catalyst for oxidation reactions not only depends on its position in the Periodic table, but also on its size-dependent properties.
The coordination of a metal with another metal can lead to new, selective catalytically active sites due to synergistic metal–metal interactions.262 In addition, the presence of an another metal can improve catalyst stability against adverse conditions, including low or high pH and aqueous or oxygen environments. Such synergistic interactions could be more pronounced in nanoalloy catalysts, as the probability of metal–metal interactions increases with enhanced electron sharing and larger surface areas.262,263 Based on the mixing pattern and geometric structure, nanoalloys can be classified as core–shell (A), subcluster segregated (B), mixed (C), and three shell (D) nanoalloys (Fig. 24).263 Interestingly, the particle size also influences the ability to form nanoalloys. Fe and Ag, for example, are immiscible in the bulk state, but they readily form alloys in a finite nanocluster state.263
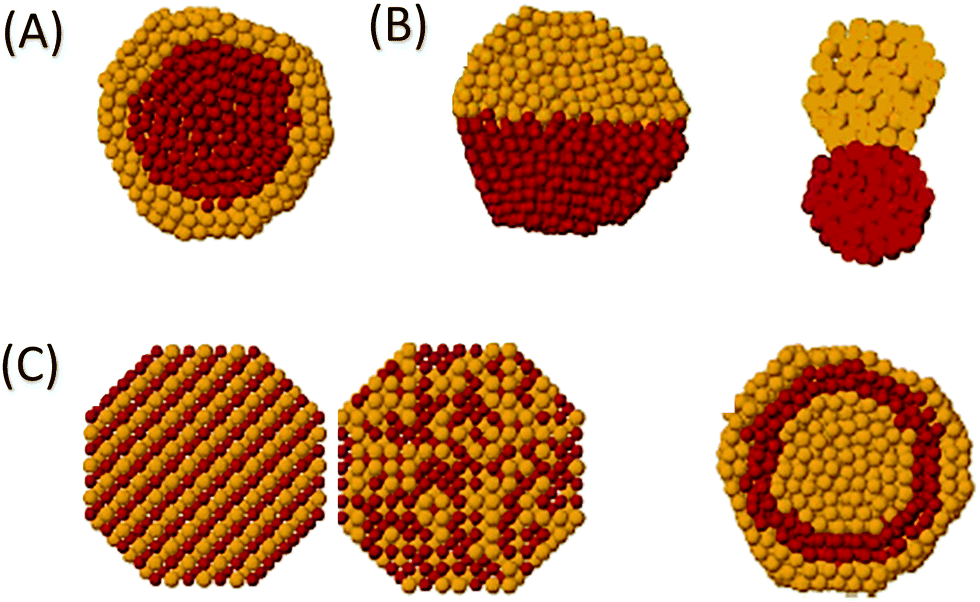 | ||
| Fig. 24 Schematic representation of some possible mixing patterns of nanoalloys: (A) core–shell, (B) subcluster segregated, (C) mixed, and (D) three shell. The pictures show cross sections of the clusters. Reproduced with permission from ref. 263. Copyright (2008) American Chemical Society. | ||
4.2 Nanostructured metal oxides
Metal oxides, that can play the role of both active phase as well as catalyst support, are essential components for the development of several industrial heterogeneous catalysts.264–266 Particularly, transition and inner transition metal oxides occupy a paramount place in heterogeneous catalysis as they exibit vastly diverse configurations and fascinating properties. The catalytic activity of metal oxides is dependent mainly on three parameters: (1) acid–base strength, (2) oxygen vacancy defects, and (3) hydrothermal stability.264,267,268 The acid- or base-strength of metal oxides is largely dependent on charge-to-radius ratio of cations and the metal oxide phase. For example, Ce4+ exhibits a higher acidic-strength than Ce3+ because of the higher charge-to-radius ratio (Ce4+), and hence pure ceria (CeO2) is acidic in nature.268–270 Metal oxide phase can also influence the acid–base strength and the catalytic performance as evidenced for TiO2 and ZrO2 catalysts.271 Anatase TiO2 containing adeque amounts of acid–base sites was able to catalyze glucose-to-HMF reaction, while rutile TiO2 has no significant effect at similar reaction conditions. In the case of ZrO2, monoclinic/tetragonal mixed phase exhibited good catalytic activity in isomerization of glucose to fructose, due to the presence of abundant basic sites in mixed phase oxide.The presence of oxygen vacancies in metal oxides can influence their catalytic activity/selectivity in biomass upgrading. Oxygen vacancy defects can be generated during synthesis process, catalytic reaction, or by post-synthesis hydrogen treatment.272–274 Oxygen vacancies and the corresponding reduced metal centers could play a beneficial role in lowering the activation energy barriers and stabilizing reaction intermediates, leading to improved biomass conversion rates. Xiao et al.273 demonstrated that oxygen vacancies present in reducible metal oxides could act as the active sites for biomass deoxygenation reactions. They found that oxidized zinc (without oxygen vacancies) was almost catalytically inactive (Fig. 25a), while the presence of oxygen vacancies led to an enhanced deoxygenation of bio-derived acetic acid to acetaldehyde (Fig. 25b). The significance of oxygen vacancies in biomass valorization is also noticed over other metal oxides, such as TiO2 and ZrO2,274 MoO3,275 CeO2,276 and CeO2–ZrO2.277 An added benefit of oxygen vacancies is their mild acidic strength that can direct a specific reaction pathway towards the formation of desirable bio-based chemicals. For example, compared to the hydrogenation route the direct deoxygenation of lignin-derived compounds is greatly favored over oxygen vacancy sites (acid sites) of MoO3.278
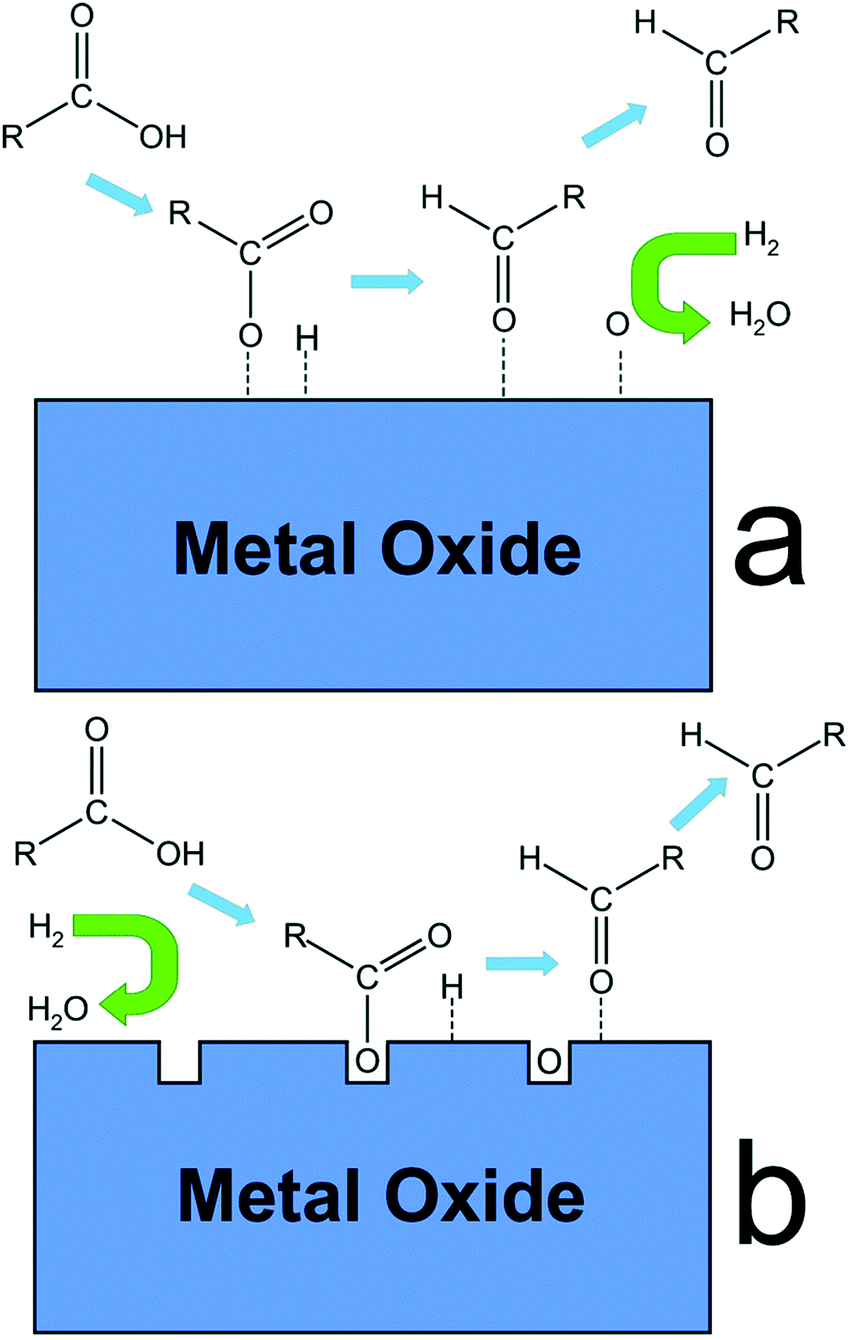 | ||
| Fig. 25 Mechanisms for the deoxygenation of bio-derived acetic acid over zinc oxide. (a) Surface-driven mechanism: acetic acid molecules adsorb on the catalyst surface to react with hydrogen. (b) Vacancy-driven mechanism: catalyst facilitates the dissociative adsorption of the acetic acid molecules through the vacant site on the catalyst surface. Reproduced with permission from ref. 273. Copyright (2018) Royal Society of Chemistry. | ||
Mixed oxides composed of two or more different metals can exhibit unusual catalytic efficiencies compared to individual metal oxides. This is because of their attractive acid or base, redox, and surface area properties, enriched by cooperative metal–metal interactions.264,268,278 As stated previously, pristine ceria contains a high number of acid sites. However, incorporation of lower valent cations (e.g., La3+ or Mg2+) into the ceria lattice can generate considerable amounts of basic sites due to the variation in the charge-to-ionic radii of Ce4+ (0.097 nm) and La3+ (1.10 nm) or Mg2+ (0.072 nm). Concurrently, it must be noted that the strong basicity of the doped species may show a negative effect on catalyst's performance, since the basic centers often tend to donate their electrons to the adjacent electron acceptors (acidic centers). As a result, the oxidizing ability of the active components will decrease, leading to low catalytic activity in biomass oxidation reactions. Mixed oxides can also show enhanced oxygen mobility and textural properties (particle size and surface area) due to the synergistic interactions of heteroatoms, as noticed in the case of CeO2–ZrO2 mixed oxide.245,277,279–281 Consequently, CeO2–ZrO2 mixed oxides were found to be catalytically active in various biomass upgrading reactions, such as ketonization of carboxylic acids and esters,279 hydrodeoxygenation of guaiacol,277 and deoxygenation of oleic acid.282
Hydrothermal stability of metal oxides is another attractive property that boosts their catalytic applications in biorefinery research since most biomass conversions are often carried out at harsh hydrothermal conditions and sometimes in sub- or super-critical liquid environments.283,284 Xiong et al.284 evaluated the stability of various solid materials including metal oxides under superheated steam conditions (Fig. 26). It was demonstrated that TiO2, ZrO2 and CeO2–ZrO2 exhibit higher hydrothermal stabilities for biomass gasification in supercritical water, while silica and activated carbon were unsuitable for this reaction.285 The improved hydrothermal stability of mixed oxides (silica–alumina) in comparison to pristine metal oxides was also noticed in cellobiose hydrolysis at 200 °C.286
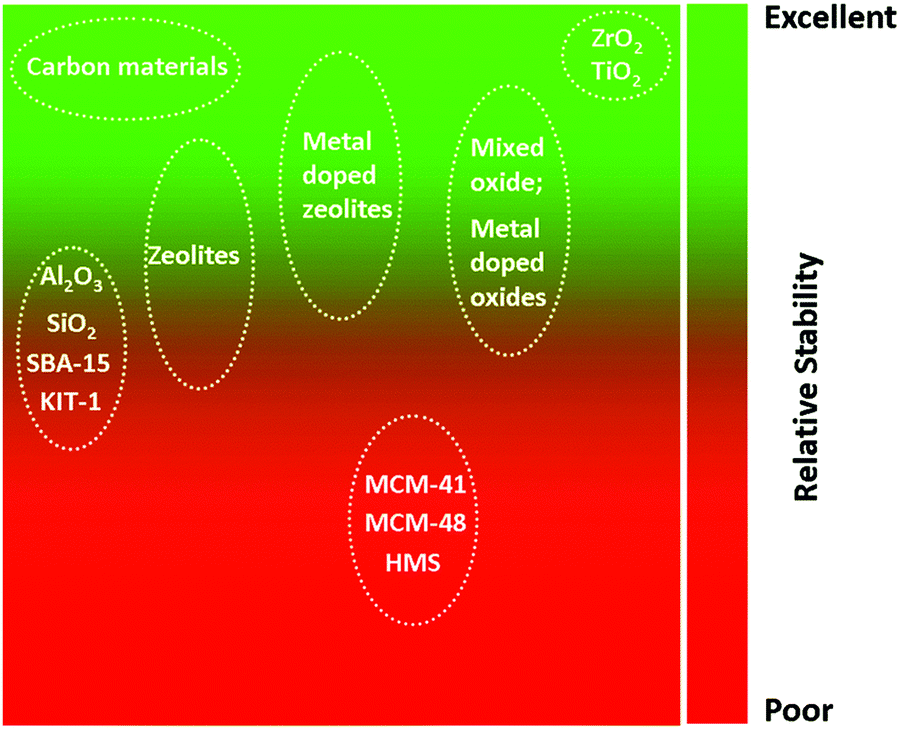 | ||
| Fig. 26 Hydrothermal stability of different heterogeneous solid catalysts in superheated steam conditions (T > 400 °C). Reproduced with permission from ref. 284. Copyright (2014) Royal Society of Chemistry. | ||
4.3 Nanostructured metal/metal oxide catalysts
Despite of their interesting structural and redox properties, it remains a great challenge to control the aggregation of metal nanoparticles at high temperature conditions during catalyst synthesis or catalytic reaction, resulting in lower catalytic efficiencies. A plausible solution to circumvent this problem is to deposit metal nanoparticles on a high surface area metal oxide.287–290 The aim of using this route is to obtain uniformly dispersed metal nanoparticles and to improve their stability via strong metal–support interactions.287 Tauster et al.291 introduced this metal–support interaction concept and emphasized the importance of the support role in enhancing the properties of metal nanoparticles.In addition to specific surface area, the morphology of metal oxides can play a significant role in tuning the structure–activity performance of supported metal catalysts. For example, ceria can form different nanosized shapes with preferentially exposed crystal facets, such as nanocubes ({100}), nanorods ({110} & {100}), and nanopolyhedra ({111} & {100}).292–297 These shape-dependent crystal facets can selectively influence the surface active sites, such as oxygen vacancies and redox couple (Ce4+/Ce3+). Hence, unusual catalytic activities can be achieved when a transition metal is deposited on these shape-controlled metal oxides. The importance of these shape-controlled ceria supports is evident in various biomass upgrading reactions, such as production of acrylic acid from glycerol,292 oxidative transformation of 1,3-propanediol into methyl esters293 and hydroprocessing of rice straw biomass.276
Heterogeneity of particle size distribution is a key concern of conventionally synthesized supported metal catalysts, which not only adversely affects the atom efficiency, but also enables unwanted side reactions.245,246,248,298,299 Uniquely identifying and controlling the location of active metal centers is also quite difficult in conventional supported metal catalysts, however it is essential to promote a specific reaction pathway towards desirable chemicals. In this context, the development of heterogeneous single-atom catalysts, also denoted as SACs, consisting of isolated metal atoms singly dispersed on supports, has recently attracted much attention for several catalytic applications including biomass upgrading. These novel catalytic systems can maximize the efficiency of metal atoms and offer great potential for achieving exceptional catalytic activity/selectivity compared with that of conventional supported metal catalysts.299
4.4 Valorization of lignocellulose biomass
Reductive catalytic fractionation (RCF) is a promising process for the full exploitation of lignocellulosic biomass to lignin oil (phenolic monomers, dimers, or oligomers) as well as a solid hemi(cellulose) pulp for paper, chemicals or fuels production. The Sels group has made substantial advances in RCF process to improve lignin oil yield and selectivity of the phenolic monomers.300 For instance, they developed a potential RCF process for the valorization of birch wood in supercritical methanol using Ni–Al2O3 catalyst pellets.301 The catalyst was placed in a reactor basket to allow facile catalyst recovery/reusability and hence, catalyst-free pulp production. About 90% delignification of birch wood was achieved and the obtained lignin oil contained over 40% phenolic monomers with a high selectivity to 4-n-propanolguaiacol and 4-n-propanolsyringol products. The catalytic efficiency of spent Ni–Al2O3 pellets could be regenerated by applying a simple thermal H2-treatment. The delignified carbohydrate pulp was used to produce bioethanol (73% yield) through simultaneous enzymatic saccharification and yeast fermentation. RANEY® nickel or spongy nickel, a fine-grained nickel solid derived from a Ni-Al alloy, showed good activity in RCF process of lignocellulose using 2-PrOH as a hydrogen-donor.302 The key advantage of this process is that RANEY® Ni can be efficiently removed from the reaction mixture using a magnet. The catalyst-free carbohydrates pulp can thus be subsequently used for the production of glucose and xylose via enzymatic hydrolysis. In another work, the combination of RANEY® Ni with NaOH is used for the transformation of various biomass sources (fir, pine, bamboo, and corncob) into bio-oil in a 2-propanol/H2O system.303 The conversion of biomass could reach up to 97–99% at optimized reaction conditions. In the case of fir feedstock, about 64% yield of bio-oil and a heating value of 30.1 MJ/kg were achieved. Results revealed that NaOH is crucial to achieve higher yields of bio-oil and RANEY® Ni can catalyze the subsequent removal of oxygen species in bio-oil via hydrodeoxygenation reaction.Tathod et al.304 observed the beneficial effect of Sn in Pt/γ-Al2O3 catalyst towards sugar alcohols production from agricultural waste (bagasse, rice husk, and wheat straw). Compared to carbon support, high yields of sugar alcohols were obtained over γ-Al2O3 supported PtSn catalysts. The PtSn/γ-Al2O3 catalyst was also able to convert mono- (glucose, fructose, xylose, arabinose, and galactose) and poly-saccharides (xylan, arabinogalactan, and inulin) into sugar alcohols. The presence of electron deficient Sn species, which helped in polarizing carbonyl bonds of sugars, was the key factor for improved performance of supported PtSn catalysts. He et al.305 proposed a potential strategy for the direct conversion of raw corn stover to lactic acid using ethanol/H2O and oxalic acid in the presence of MgO catalyst. Results revealed that ethanol/H2O solvent system could efficiently fractionate corn stover into 88wt% of hemicellulose and 89.2wt% of lignin, while retaining a stable cellulose pulp. Oxalic acid played a favourable role in the cleavage of intermolecular linkages between hemicellulose and lignin. On the other hand, MgO catalyzed the conversion of dissolved hemicellulose derivatives to lactic acid with 79.6wt% yield and 90% selectivity. In recent work, the hydro-liquefaction of rice stalk to bio-crude oil was carried out by means of hydrogen generated in situ during the oxidation of aluminium (gives Al2O3) under supercritical ethanol conditions.306 The generated Al2O3 can also act as a catalyst for the transformation of ethanol to provide additional H2 for the hydro-liquefaction of rice stalk. About 75% energy recovery and 33 wt% yield of bio-oil were achieved at optimized conditions. Ni nanoparticles dispersed on CeO2 nanorods, synthesized by a one-pot hydrothermal method, exhibited good catalytic activity in hydroprocessing of rice straw biomass.276 Shape-controlled CeO2 nanorods preferentially expose a large fraction of reactive (110) and (100) crystal planes, which can provide a high concentration of redox active sites.269,307 Ni/CeO2 catalyst with a Ni/Ce molar ratio of 2/10 afforded 89% conversion of rice straw and 66.7% yield of bio-oil, which was attributed to homogeneous dispersion of Ni metal sites and abundant oxygen vacancies in cerium oxide.276 Biomass to MoO3 mass ratio played a pivotal role on the deoxygenation activity of MoO3/TiO2 and MoO3/ZrO2 catalysts (10 wt% MoO3 loading) for catalytic fast pyrolysis of pine biomass.56 Owing to adequate amounts of surface reduced Mo species (Mo3+ and Mo5+), the supported MoO3 catalysts converted approx. 27 mol% of the original carbon into hydrocarbons, mainly containing aromatics (7 C%), olefins (18 C%), and paraffins (2 C%), outperforming a bulk MoO3 catalyst.
4.5 Upgrading of carbohydrates and their derived compounds
In this section, the catalytic processing of polysaccharides (cellulose and hemicellulose), monosaccharides (glucose and xylose), and their derived compounds (alcohols, acids, esters, and furans) is discussed.Selective hydrogenation of monosaccharides, such as glucose into 1,2-PG and EG was also studied using a variety of nanoscale metal/metal oxide catalysts. It was pointed out that both Lewis acid and metal sites in Pd–WOx/Al2O3 catalysts are vital to achieve high yields of 1,2-PG from glucose.311 In contrast, Brønsted acid sites surprisingly have no contribution in the 1,2-PG production. The Pd–WOx(5 wt%)/Al2O3 catalyst, which has well-balanced Lewis acid and metal sites, afforded 92.2% glucose conversion and 60.8% 1,2-PG selectivity. A combined experimental and computational study was performed to understand the role of the support (MgO and ZnO) on the performance of Ni-based catalysts for the selective conversion of glucose into 1,2-PG and EG products.312 Remarkably, the MgO support promoted glucose conversion into fructose as well as the subsequent retro-aldol reaction of fructose at the C3–C4 position, resulting in high yields of 1,2-PG. The production of C3 polyols, including EG and 1,2-PG, from sorbitol was also studied over Cu/CaO–Al2O3 catalysts.313 By optimizing several parameters, such as Cu loading, catalyst pre-treatment, reaction temperature, and H2 pressure a combined selectivity of 84% to glycols and linear alcohols was obtained with Cu/CaO–Al2O3 catalyst. A possible reaction mechanism involving β-ketose formation on Cu–Ca2+ sites was proposed. The significance of metal–metal interactions in tailoring the efficiency of bimetallic Pd–Cu/ZrO2 catalysts was markedly noticed in sorbitol hydrogenolysis.314 A combined selectivity of 61.7% to EG, 1,2-PG, and glycerol was obtained at nearly 100% sorbitol conversion over Pd–Cu/ZrO2 (Cu/Pd = 5) catalyst, outperforming monometallic counterparts. Sugar alcohols (xylitol and arabitol) have plentiful applications in food, oral hygiene, and pharmaceutical industries. These sugars can be selectively synthesized via hydrogenolysis of xylose (monosaccharide) and hemicellulose (xylan, polysaccharide) over supported MP catalysts (M, metal = Pt & Ru and P, promoter = Sn, Ga, & Fe).315 Three supports, namely γ-Al2O3, SiO2–Al2O3, and carbon were used in this work. Among these catalysts, Pt(3.5)Sn(0.43)/γ-Al2O3 catalyst showed good activity with a 2.8 times higher yield of sugar alcohols from a polysaccharide compared to the corresponding monometallic Pt(3.5)/γ-Al2O3 catalyst. The improved activity of bimetallic catalysts is due to the existence of electron deficient Sn species, which promote polarization of the carbonyl groups, resulting in high yields of sugar alcohols. In addition, Pt(3.5)Sn(0.43)/γ-Al2O3 catalyst is efficiently reused for five cycles without considerable loss in catalytic activity. NiO addition has a great effect on the performance of Ru/TiO2 catalyst for the liquid phase xylose hydrogenation.316 Particularly, Ru(1%)/NiO(5%)–TiO2 catalyst showed excellent performance in terms of xylose conversion (99.9%) and xylitol selectivity (99.8%), outperforming Ru/TiO2, Ru/C, and RANEY® Ni catalysts.
The catalytic hydrogenation of levulinic acid (LA), a key platform chemical derived from carbohydrates, into γ-valerolactone (GVL) is one of the most crucial reactions in bio-refinery industry.317,318 Various nanostructured metal/metal oxide catalysts were developed for LA-to-GVL transformation. Ftouni et al.318 observed durable performance of Ru/ZrO2 catalyst towards GVL production even after multiple recycling tests. In contrast, Ru/TiO2 catalyst showed an abrupt deactivation after the first catalytic test, which was due to undesirable reduction of TiO2 in combination with partial coverage of Ru NPs on the support. The addition of water to the reaction mixture indicated a favorable effect in promoting the selective hydrogenation of LA to GVL product. Ru/ZrO2 catalyst exhibited outstanding stability in liquid phase LA hydrogenation in the presence of H2SO4 impurity compared to Ru/carbon catalyst due to preferential adsorption of sulfate species on the surface of ZrO2, preventing deactivation of Ru phase.319 The significance of Ru/ZrO2 catalyst in catalytic transfer hydrogenation of LA to GVL was also noticed.320 An equimolar mixture of formate/formic acid in water was used as hydrogen source. Doping with 0.1 wt% SiO2 was found to stabilize the tetragonal phase of ZrO2, resulting in improved catalyst stability in the liquid phase LA hydrogenation, even after four recycles. Ru precursor (RuCl3, RuNO(NO3)3, and Ru(NH3)6Cl3) used for the catalyst synthesis has a profound influence on the structure–activity performance of Ru/TiO2 (anatase) catalyst for the aqueous phase LA-to-GVL transformation.321 Best results in terms of turn-over frequency (TOF) and initial rate based on Ru intake were obtained over the catalysts synthesized using RuNO(NO3)3 and RuCl3 precursors, respectively. Alkali cations (Na+, K+, and Cs+) exhibited a substantial effect on the activity of Ru/Al2O3 catalyst for the liquid phase hydrogenation of LA to GVL.322 As the most polarizable cation, Cs+ strongly enhances the electron density on the Ru metal surface, which is essential for achieving higher reaction rates in LA hydrogenation.
Nanostructured bimetallic Au–Pd/TiO2 and Ru–Pd/TiO2 catalysts, prepared by a modified wet-impregnation method, were found to show improved GVL selectivity compared to the monometallic analogues, which was attributed to synergistic effect of nano-alloying.323 In addition, both supported nanoalloy catalysts showed exceptional stability in the liquid phase LA hydrogenation, even after three consecutive recycles. Al-Naji et al.324 studied LA hydrogenation under microwave-assisted, batch scale, and continuous flow conditions using Ni–Pt and Ni–Ru catalysts supported on ZrO2 and γ-Al2O3. Results revealed that Ni–Ru/γ-Al2O3 catalyst exhibits the highest LA conversion, due to the presence of high concentration of metallic Ru0 species on the surface of γ-Al2O3, together with its improved textural properties. However, Ni–Ru/γ-Al2O3 catalyst suffered rapid deactivation after four consecutive catalytic runs. This can be explained by a drastic loss in specific surface area (from 259 to 53 m2 g−1) and pore volume (0.7 to 0.2 cm3 g−1). The transformation of γ-Al2O3 to a soluble boehmite phase (AlOOH) was also a key reason for the deactivation of Ni–Ru/γ-Al2O3 catalyst. Effects of both metal oxides (ZrO2, Al2O3, Cr2O3, and BaO) and mixed oxides (Cr2O3–Al2O3 and BaO–Al2O3) on the activity of nanoscale Cu catalysts were investigated in the liquid phase hydrogenation of LA and its ester to GVL product.325 Among the supports studied, best catalytic results were obtained in the case of ZrO2 and Al2O3 supported Cu catalysts. In addition, Cu/ZrO2 catalyst can be reused for at least three cycles without much loss in catalytic activity. An efficient catalytic cascade strategy was developed for the synthesis of GVL, 1,4-pentanediol, and 2-methyl tetrahydrofuran from LA in water solvent using a Pd–Cu/ZrO2 catalyst.326 The addition of 1% Pd to 30% Cu/ZrO2 catalyst significantly improved its catalytic stability, revealing the importance of nano-alloying in catalytic biomass upgrading. Furthermore, Pd–Cu/ZrO2 catalyst exhibited good hydrothermal stability.
The catalytic vapor phase (or continuous-flow) hydrogenation of LA to GVL is a hot research topic because of its advantages (e.g., low consumption of catalysts, waste minimization, cost efficiency, improved product quality, and the integration of downstream processes) over batch scale catalytic processes. Varkolu et al.327 developed an efficient continuous-flow process for the selective hydrogenation of LA to GVL with formic acid as the hydrogen source. Among the catalysts tested, Ni/Al2O3 catalyst showed the best performance, which was attributed to high concentration of uniformly dispersed metallic Ni species on the surface of Al2O3 support. In another work, Varkolu et al.328 demonstrated the role of preparation method (impregnation, co-precipitation, deposition–precipitation, and citric acid assisted impregnation combustion) on the performance of Ni/SiO2 catalysts for LA hydrogenation. The authors found that Ni/SiO2 catalyst prepared by citric acid assisted method followed by calcination in inert gas flow displays good catalytic performance in LA-to-GVL transformation, due to the presence of optimum porosity, along with highly dispersed Ni NPs. The effect of support (Al2O3, SiO2, TiO2, ZSM-5 and SiO2–Al2O3) was evident on the structure–activity performance of Cu catalyst for the vapor phase hydrogenation of LA with formic acid as the hydrogen source.329 Owing to strong metal–support interactions, the Cu–SiO2 catalyst gave the highest LA conversion (56%) with 87% selectivity to GVL at 250 °C. The influence of both supports (SiO2, γ-Al2O3 and ZrO2)330 and the promoter (W)331 on the efficacy of Ni and Ni/TiO2 catalysts, respectively, was observed for the vapor phase hydrogenation of aqueous LA to GVL. Pyridine adsorption DRIFT studies were undertaken to understand the role of Brønsted- and Lewis-acid sites in tuning GVL selectivity over Ni based catalysts.
Selective hydrogenation of furans (furfural and 5-hydroxymethylfurfural) is of great importance since the resulting products have a myriad of applications in the chemical industry.332–334 Rogers et al.335 developed tandem site- and size-controlled Pd NPs immobilized on TiO2 for the hydrogenation of furfural to tetrahydrofurfuryl alcohol. The size of Pd NPs could be controlled by varying the temperature of the colloidal solution. However, no linear correlation was found between the Pd particle size and the product selectivity in furfural hydrogenation. Interestingly, a higher proportion of corner and edge sites, which are able to tune product selectivity in furfural hydrogenation, can be obtained by adjusting the solvent system (water–ethanol) of the colloid preparation. Both support nature (TiO2, carbon, MgO, Fe3O4, and Al2O3) and Pt addition had a notable effect on the efficiency of Pd catalysts for the liquid phase hydrogenation of furfual.336 The distribution of Pd–Pt nano-alloys on TiO2 surface is shown in Fig. 27. Among the supports used, TiO2-supported Pd catalyst exhibited the highest selectivity to tetrahydrofurfuryl alcohol. The activity of Pd/TiO2 catalyst (Pd = 0.47 wt%) could be improved by Pt addition (0.49 wt%), giving 95% yield of tetrahydrofurfuryl alcohol, which was attributed to synergistic Pd–Pt interactions stimulated by facile electron transfer effects (entry 5, Table 3). CuxNiy (x/y = 7:1, 3:1, 1:1, 1:3, 1:7) nano-alloys supported on Mg–Al–O mixed oxide were demonstrated to be efficient catalysts for the liquid phase furfural hydrogenation.337 The reduction temperature of the catalyst precursor played a crucial role in tuning the dispersion and composition of CuNi nanoalloys as well as the surface basicity of catalysts, which are vital for achieving improved product yields (99%) in furfural hydrogenation (entry 6, Table 3). By tuning the metal composition as well as changing the reaction solvent, the selectivity of the catalyst could be changed from tetrahydrofurfuryl alcohol to furfuryl alcohol. King et al.338 reported that Pd/TiO2 catalysts are highly selective towards 2-methylfuran product in the liquid phase furfural hydrogenation. However, Sn addition (1 wt%) to Pd/TiO2 catalyst led to a shift in product selectivity from 2-methylfuran to tetrahydrofurfuryl alcohol. This indicates the effect of Sn–Pd interactions in tuning product selectivity. An efficient catalytic process was developed for the direct conversion of furfural to cyclopentanone using CuZnAl mixed oxides.339 Effects of Cu/Zn molar ratio and the calcination temperature towards cyclopentanone production were investigated. Notably, the CuZnAl-0.5 (Cu/Zn = 0.5) catalyst calcined at 500 °C afforded about 62% yield of cyclopentanone (entry 7, Table 3). Moreover, CuZnAl-500–0.5 catalyst could be reused for at least five times without much loss in catalytic activity. The addition of Fe considerably enhanced the performance of Cu/γ-Al2O3 catalyst for the vapour phase hydrogenation of furfural.340 About 91% furfuryl alcohol yield was achieved over CuFe/γ-Al2O3 (Cu – 10 wt% and Fe – 10 wt%) catalyst, due to the presence of synergistic bimetallic particles and abundant oxygen vacancies in iron oxide (entry 8, Table 3). In recent work, Gupta et al.341 developed a feasible catalytic process for the room-temperature total hydrogenation of furans and furan/acetone aldol adducts over Ni–Pd nano-alloy catalysts. Promising catalytic results were obtained in the presence of Ni0.90Pd0.10 nano-alloy catalyst. The possibility of scaling-up this catalytic process was also demonstrated in this work. The significance of atomic ordering on the structure–activity properties of spherically shaped NixSny–Al2O3 catalysts was noticed for the vapor phase furfural hydrogenation.342 In addition, crystal structure and metal composition of NixSny–Al2O3 catalysts played a crucial role in furfural hydrogenation. Particularly, Ni3Sn2–Al2O3 catalyst showed the best performance with ∼65% furfural conversion and good selectivity (57–62%) to furfuryl alcohol at optimized reaction conditions. Doping of La into the ZrO2 led to the formation of active tetragonal phase zirconia with smaller particle size and high BET surface area.340 Besides, improved Co–ZrO2 interactions and abundant acid sites were found in Co/La–ZrO2, which are crucial for the efficient adsorption of carbonyl feedstock. As a result, 95% yield of furfural alcohol was obtained via aqueous phase furfural hydrogenation over Co/ZrLa0.2Ox catalyst at 40 °C, 2 MPa H2 in 10 h.
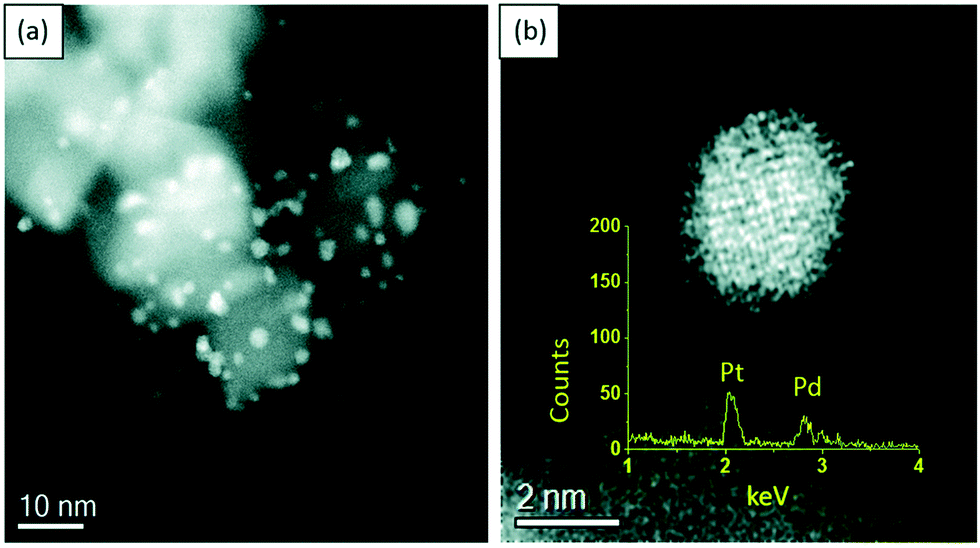 | ||
| Fig. 27 Scanning transmission electron microscopic images of Pd–Pt/TiO2 catalyst at different magnifications. The formation of random Pd–Pt nanoalloys is confirmed from the Z-contrast high-angle annular dark-field imaging and the X-ray energy-dispersive spectrum (inset image, b) taken from an individual particle. Reproduced with permission from ref. 336. Copyright (2018) Royal Society of Chemistry. | ||
The hydroprocessing of 5-hydroxymethylfurfural (HMF) can give 2,5-dimethylfuran (DMF), 2,5-bis(hydroxymethyl)furan (BHF), or 2,5-dimethyltetrahydrofuran (DMTHF), depending on the catalysts and the reaction conditions used. DMF is considered as a sustainable fuel additive and BHF is a key chemical for the production of polymers and fine chemicals. Table 4 presents the notable catalytic results obtained for the liquid phase hydrogenation of HMF over porous and nanoscale catalysts. By transforming Co3O4 using a controlled reduction step into a bi-functional Co–CoOx catalyst with well-balanced Co metal (hydrogenation site) and CoOx species (acid site), a high efficiency in the liquid phase hydrogenolysis of HMF to DMF (83.3% yield) was obtained (entry 2, Table 4).343 Cu–Zn nanoalloy with particle size <150 nm showed good catalytic activity in HMF hydrogenolysis with a 97% combined yield of DMF and DMTHF at optimized reaction conditions (entry 3, Table 4).344 This nanoalloy catalyst also converts 10 wt% HMF in cyclopentyl methyl ether solvent to DMF (90% yield). Kong et al.345 achieved improved reaction rates for HMF hydrogenolysis by adding Zn to a NiAl catalyst. The structure evolution of the NiZnAl catalyst is shown in Fig. 28A. The resulting NiZnAl mixed oxide catalyst provided a significantly enhanced DMF yield (93.6%) compared with that of NiAl catalyst (63.5%), which was attributed to electron modification and isolation of Ni atoms (entry 4, Table 4). Moreover, NiZnAl catalyst showed three times higher activity than that of NiAl catalyst in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/C–O hydrogenolysis over CO/C–C hydrogenation at similar HMF conversions (Fig. 28B). Ohyama et al.346 demonstrated that addition of Fe (up to 10 wt%) to Au/Al2O3 catalyst could improve its catalytic efficiency by 3–4 times in the hydrogenation of HMF to BHF, due to the formation of abundant surface-exposed Au nanoclusters (entry 5, Table 4). However, the activity of Au/FeOx/Al2O3 catalyst was decreased drastically with further increase of Fe content (greater than 10 wt%), because of the formation of inactive buried structures of Au nanoclusters in FeOx. Nickel oxide NPs supported on nanostructured tungsten oxide were demonstrated to be efficient for the aqueous phase conversion of HMF to DMF.347 With optimized parameters, such as Ni loading, reaction temperature, and H2 pressure, about 95% yield of DMF was achieved over 5% Ni/WO3 catalyst, due to the presence of highly dispersed Ni nanoparticles (entry 6, Table 4). Moreover, this catalyst could be successfully reused for at least five cycles without much loss in its activity and selectivity. Ample amounts of active Ni sites in 20 wt% Ni/LaFeO3 catalyst are crucial to achieve high yields of DMF (98.3%) from aqueous phase HMF hydroprocessing (entry 7, Table 4).348 Co-hydroprocessing of HMF and furfural was successfully investigated over mono- and bi-metallic Cu–Ni/TiO2 and Cu–Ni/Al2O3 catalysts.349 The Cu–Ni/TiO2 catalyst displayed 87.5% yield of DMF from HMF and 88.5% yield of methyl furan from furfural in a one-pot reaction (entry 8, Table 4). The selective segregation of Cu (enriched at the catalyst surface) and Ni sites (enriched at the TiO2 interface) played a crucial role on the activity and stability of the Cu–Ni/TiO2 catalyst.
O/C–O hydrogenolysis over CO/C–C hydrogenation at similar HMF conversions (Fig. 28B). Ohyama et al.346 demonstrated that addition of Fe (up to 10 wt%) to Au/Al2O3 catalyst could improve its catalytic efficiency by 3–4 times in the hydrogenation of HMF to BHF, due to the formation of abundant surface-exposed Au nanoclusters (entry 5, Table 4). However, the activity of Au/FeOx/Al2O3 catalyst was decreased drastically with further increase of Fe content (greater than 10 wt%), because of the formation of inactive buried structures of Au nanoclusters in FeOx. Nickel oxide NPs supported on nanostructured tungsten oxide were demonstrated to be efficient for the aqueous phase conversion of HMF to DMF.347 With optimized parameters, such as Ni loading, reaction temperature, and H2 pressure, about 95% yield of DMF was achieved over 5% Ni/WO3 catalyst, due to the presence of highly dispersed Ni nanoparticles (entry 6, Table 4). Moreover, this catalyst could be successfully reused for at least five cycles without much loss in its activity and selectivity. Ample amounts of active Ni sites in 20 wt% Ni/LaFeO3 catalyst are crucial to achieve high yields of DMF (98.3%) from aqueous phase HMF hydroprocessing (entry 7, Table 4).348 Co-hydroprocessing of HMF and furfural was successfully investigated over mono- and bi-metallic Cu–Ni/TiO2 and Cu–Ni/Al2O3 catalysts.349 The Cu–Ni/TiO2 catalyst displayed 87.5% yield of DMF from HMF and 88.5% yield of methyl furan from furfural in a one-pot reaction (entry 8, Table 4). The selective segregation of Cu (enriched at the catalyst surface) and Ni sites (enriched at the TiO2 interface) played a crucial role on the activity and stability of the Cu–Ni/TiO2 catalyst.
| Entry | Catalyst | Reaction conditions | Product | Catalytic activity | Key factors for the catalyst activity | Ref. | |
|---|---|---|---|---|---|---|---|
| Conv. (%) | Yield (%) | ||||||
| a DMF – 2,5-dimethylfuran. b DMTHF – 2,5-dimethyltetrahydrofuran. c BHF – 2,5-bis(hydroxymethyl)furan. | |||||||
| 1 | 2 wt% Ru/NaY zeolite | Molar ratio of HMF to metal is 200, 220 °C, 15 bar H2, THF, 1 h | DMFa | 100 | 78 | High dispersion of Ru NPs | 136 |
| 2 | Co3O4 | 0.5 g HMF, 0.1 g catalyst reduced at 400 °C, 170 °C, 12 h, 10 bar H2, 1,4-dioxane | DMF | 100 | 83.3 | Well-balanced metal (Co) and acid (CoOx) functionalities | 343 |
| 3 | Cu–Zn nanoalloy | 0.5 g HMF, 0.1 g CuZn, 220 °C, 18 h, 20 bar H2, cyclopentyl methyl ether | DMF + DMTHFb | 100 | 97 | Synergistic Cu–Zn interaction | 344 |
| 4 | NiZnAl mixed oxide | 1.5 g HMF, 0.15 g catalyst, 15 bar H2, 180 °C, 15 h, 1,4-dioxane | DMF | 100 | 93.6 | Electron modification and isolation of Ni atoms due to the formation of NiZn alloy | 345 |
| 5 | Au/FeOx/Al2O3 (Fe = 10%) | 0.1 M HMF (aq. 2 mL), 0.01 g catalyst, 80 °C, 30 bar H2, 2 h | BHFc | 96 | 96 | Ample amounts of surface exposed Au nanoclusters | 346 |
| 6 | 5 wt% Ni/WO3 | 3 mmol HMF, 0.05 g catalyst, H2O, 10 bar H2, 180 °C, 6 h | DMF | 99 | 95 | High dispersion of Ni nanoparticles | 347 |
| 7 | 20 wt% Ni/LaFeO3 catalyst | 1 mmol HMF, 0.1 g catalyst, n-tetradecane, 6 h, ethanol, 50 bar H2, 230 °C | DMF | 99 | 98.3 | Ample amounts of active metal sites | 348 |
| 8 | Cu (5 wt%)–Ni (5 wt%)/TiO2 | 0.5 g furfural loading, 0.25 g HMF loading, 0.3 g catalyst, 1,4-dioxane, 200 °C, 25 bar H2, 8 h | DMF | — | 87.5 | Selective segregation of Cu and Ni sites | 349 |
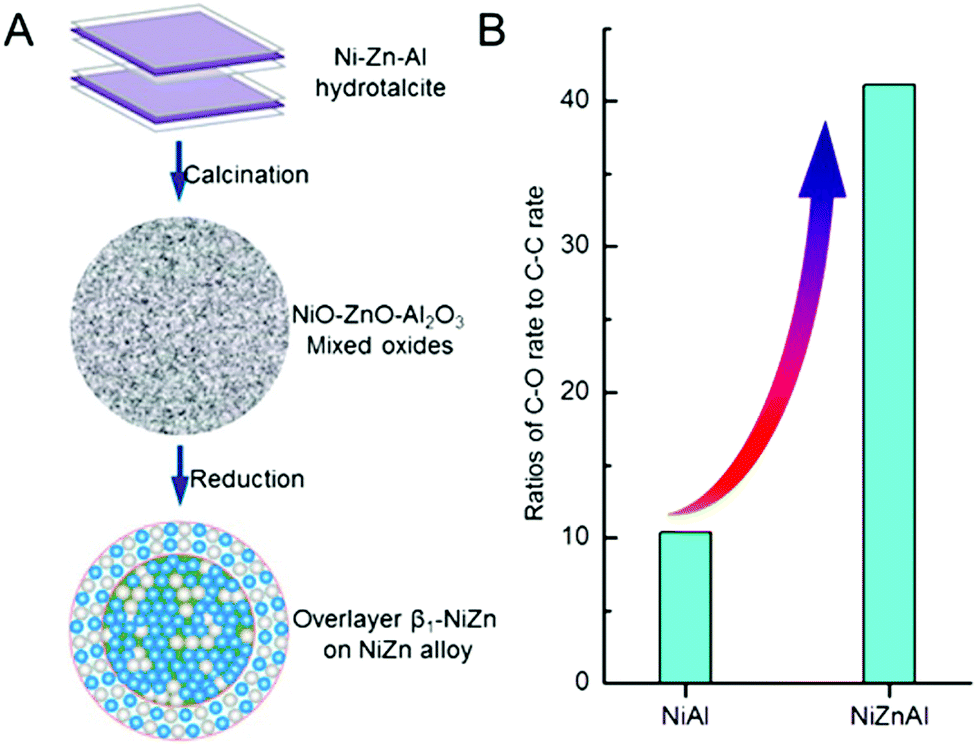 | ||
| Fig. 28 (A) Structure evolution of NiZnAl catalyst and (B) comparison of reaction rates for C–O vs. C–C bond scission over NiAl and NiZnAl catalysts at similar HMF conversions. Reproduced with permission from ref. 345. Copyright (2017) American Chemical Society. | ||
 | ||
| Fig. 29 Representative TEM images of 1% Au/TiO2 catalyst synthesized by sol-immobilization method and then calcined at (a) 250 °C, (b) 350 °C, (c) 450 °C and (d) 550 °C. Reproduced with permission from ref. 355. Copyright (2015) Royal Society of Chemistry. (e) Possible products from glucose oxidation. | ||
The Hutchings group has made significant advances towards glucose oxidation using metal oxide supported Au-based catalysts. They demonstrated that the selective, aerobic oxidation of glucose to gluconic acid can be carried out over supported Au–Pd catalysts without using a sacrificial base or pH control if a basic support (MgO) is used.354 Effects of both the preparation method and the post-synthesis treatment on the efficiency of 1% Au/TiO2 catalyst were also studied for base-free oxidation of glucose.355 The catalyst prepared by sol-immobilization method and calcined at 250 °C (Fig. 29a) showed the highest activity in terms of glucose conversion and gluconic acid selectivity, due to the presence of smaller sized Au nanoparticles. The particle size of Au increases with the increase of calcination temperature (Fig. 29). Moreover, the amount of stabilizing ligand used in the sol-immobilization method is a key parameter for improving the activity of Au/TiO2 catalysts. They also found that bimetallic Au–Pd/TiO2 and Au–Pt/TiO2catalysts display good activity in the oxidation of cellobiose (a disaccharide) to gluconic acid under base-free conditions.356 Among Al2O3, CeO2, and ZrO2, Au NPs supported on Al2O3 were found to be highly active for base-free aerobic oxidation of glucose.357 Using this catalyst, several reaction parameters, such as reaction time, temperature, and stirring rate were optimized to achieve improved gluconic acid yields. In case of non-Au based catalysts, for instance with Pt–Pd/TiO2 catalysts, glucaric acid is mainly obtained in glucose oxidation reaction, outperforming monometallic catalysts.358 However, Darrien et al.359 reported selective formation of glucaric acid in the presence of supported Au–Pt and Au–Pd catalysts. This indicates that the presence of Pt NPs is key for the formation of glucaric acid instead of gluconic acid, a commonly observed product in Au-catalyzed glucose oxidation reactions. On the other hand, Wojcieszak et al.360 noticed glucuronic acid production via base-free oxidation of various carbohydrates (glucose, fructose, and maltose) over Au/CeO2 catalyst. The addition of caesium led to improved activity of the Au/CeO2 catalyst. In contrast, base addition (K2CO3) to the reaction mixture resulted in the formation of various by-products, including gluconic acid.
The catalytic oxidation of biomass-derived furans, especially 5-hydroxymethylfurfural (HMF) gives a number of high-value chemicals, such as 2,5-diformylfuran (DFF), 5-hydroxymethyl-2-furancarboxylic acid (HMFCA), 5-formyl-2-furancarboxylic acid (FFCA), and 2,5-furandicarboxylic acid (FDCA), which can be used for the production of drugs, fungicides, and bio-polymers (Fig. 30d).361–363 Both noble and non-noble metal-based nanocatalysts were demonstrated to be efficient for liquid phase HMF oxidation, while product selectivity could be controlled by varying the catalyst compositions and reaction conditions, as shown in Table 5. Lei et al.251 pointed out the crucial role of the particle morphology and the preferentially exposed crystal facets on the catalytic performance of Pd nanocrystals for liquid phase HMF oxidation. Pd nano-octahedra enclosed by {111} facets were more efficient than Pd nanocubes enclosed by {100} facets in HMF oxidation, outperforming a commercial Pd/C catalyst. Pt NPs, stabilized by an ionic polymer, were also catalytically active in the liquid phase, base-free oxidation of HMF under mild conditions.362 To optimize the process towards FDCA production, thorough mechanistic and kinetic investigations were conducted. The aqueous phase oxidation of HMF to FDCA or HMFCA was carried out using TiO2 supported mono- and bi-metallic AuPd nanoalloys.364 An appropriate amount of polyvinylpyrrolidone was added during the catalyst synthesis to stabilize Au and Pd nanoparticles. Both Au and Pd NPs were able to convert HMF to HMFCA. Surprisingly, Pd NPs were inactive for further oxidation of HMFCA, while Au-containing catalysts showed good activity in the subsequent oxidation of HMFCA to FDCA. The combination of Au NPs and ordered mesoporous CeO2 was found to be promising for the liquid phase HMF oxidation (entry 3, Table 5).365 Controlled removal of silica template during CeO2 synthesis was vital to achieve abundant structural defect sites and enhanced catalytic properties, giving high yields of FDCA (92%). Pt NPs supported on ceria solid solution (Ce0.8Bi0.2O2−δ) efficiently catalyze the liquid phase HMF-to-FDCA transformation, outperforming a Pt/CeO2 catalyst (entry 4, Table 5).366 About 98% yield of FDCA was achieved within 30 min at room temperature. Moreover, this catalyst was reused for five times without much loss of FDCA selectivity, attributed to superior oxygen activation ability of Bi-doped ceria support. Comparable Au particle sizes were found on both TiO2 and CeO2 supports.367 However, Au/CeO2 catalyst showed a higher activity than the Au/TiO2 catalyst due to the existence of synergistic Au–CeO2 interface and abundant defects in the cerium oxide lattice. Nanostructured Ru/MnCo2O4 is a selective catalyst for the liquid phase oxidation of HMF towards DFF product.368 Both the Brønsted acidity of MnCo2O4 support and the Lewis acidity of Ru NPs were key to obtaining high yields of DFF (99%, entry 5, Table 5). The Ru/MnCo2O4 catalyst could also be reused for several consecutive cycles without much loss in catalytic activity.
 | ||
| Fig. 30 (a) SEM, (b) high-resolution TEM, and (c) TEM images of Ni–MnOx nanowires. The measured d-spacing of 0.51 nm corresponds to the (200) planes in the MnO2-type unit cell. Reproduced with permission from ref. 371. Copyright (2018) Royal Society of Chemistry. (d) Possible major products from oxidation of 5-hydroxymethylfurfural (HMF): 5-hydroxymethyl-2-furancarboxylic acid (HMFCA), 2,5-diformylfuran (DFF), 5-formyl-2-furancarboxylic acid (FFCA), and 2,5-furandicarboxylic acid (FDCA). | ||
| Entry | Catalyst | Reaction conditions | Product | Catalytic activity | Key factors for the catalyst activity | Ref. | |
|---|---|---|---|---|---|---|---|
| Conv. (%) | Yield (%) | ||||||
| a DFF – 2,5-diformylfuran. b FDCA – 2,5-furandicarboxylic acid. | |||||||
| 1 | 10V2O5@Cu–MOR (125) | 0.05 g HMF, 0.1 g catalyst, DMSO, 120 °C, 7 h, O2 balloon | DFFa | 100 | 91.5 | Strong interaction between the framework Cu species and the guest V sites | 53 |
| 2 | Co–Mn/nanospherical porous silica | 1 mmol HMF, 0.05 g catalyst, MeCN, 12 h, 120 °C, 7 mmol t-BuOOH | FDCAb | 100 | 72.4 | Strong metal–support interaction and high metal dispersion | 197 |
| 3 | 1.5 wt% Au/CeO2 | HMF/metal/NaOH molar ratio 1:0.01:4, 70 °C, 10 bar O2, 4 h |
FDCA | — | 92 | Strong Au–ceria interaction and ceria defects | 365 |
| 4 | 1 wt% Pt/Ce0.8Bi0.2O2−δ | Pt: HMF = 5 × 10−3 mol mol−1, 4 equiv. NaOH, 10 bar O2, 23 °C, 30 min | FDCA | 100 | 98 | High metal dispersion and oxygen vacancies of ceria | 366 |
| 5 | 1.8% Ru/MnCo2O4 | HMF/Ru ratio (50), 2 mmol HMF, toluene, 3 h, 130 °C, 10 bar O2 | DFF | 99 | 99 | Brønsted acidity of MnCo2O4 support and Lewis acidity of Ru NPs | 368 |
| 6 | MnOx–CeO2 (Mn/Ce = 6) | 0.5 mmol HMF, 0.1 g catalyst, H2O, 20 bar O2, 110 °C, 6 h, KHCO3/HMF = 2 | FDCA | 98 | 84 | High concentration of surface Mn4+ and Ce3+ sites | 369 |
| 7 | MnxFey (Mn/Fe = 3) | HMF:NaOH molar ratio (1:4), 1 mmol HMF, water, 8 bar O2, 0.05 g catalyst, 24 h, 90 °C |
FDCA | 93 | 30 | Synergy of Mn3+ and Mn4+ species and hematite phase | 370 |
| 8 | Ni-Doped MnOx nanowires | 0.126 g HMF, 0.336 g NaHCO3, 0.1 g catalyst, H2O, 100 °C, 8 bar O2, 28 h | FDCA | 100 | 94 | Ni3+O(–Mn4+)2 clusters | 371 |
| 9 | Mn–Co binary oxides (Mn/Co = 1/1) | 1 mmol HMF, 0.05 g catalyst, 140 °C, 10 g ethanol, 30 bar air, 2 h | DFF | 42.6 | 41.7 | High Mn4+/Mn3+ atomic ratio and exposed lattice oxygen species on the catalyst surface | 372 |
| 10 | Cu (4.8 mol%)–MnO2 | 0.252 g HMF, 0.1 g catalyst, ethanol, 3 bar O2, 140 °C, 5 h | DFF | 86 | 82.6 | Synergistic effect of Cu and MnO2 | 361 |
Several non-precious metal catalysts, especially manganese-based catalysts, were demonstrated to be efficient for the liquid phase HMF oxidation. For instance, MnOx–CeO2 mixed oxides, prepared by a co-precipitation method, exhibited good activity for the aerobic oxidation of HMF to FDCA using KHCO3 base.369 Especially, MnOx–CeO2 (Mn/Ce = 6) mixed oxide afforded high yields of FDCA (84%), which was attributed to the presence of ample amounts of surface Mn4+ and Ce3+ sites (entry 6, Table 5). A series of MnxFey mixed oxides, along with pristine metal oxides were studied for HMF-to-FDCA transformation in aqueous medium using NaOH base.370 Several reaction parameters, such as nature and amount of base, oxygen pressure, temperature, and catalyst composition were optimized for this reaction. The synergy of Mn3+ and Mn4+ species, coexisting with a hematite phase in MnxFey (Mn/Fe = 3) played a favorable role towards FDCA production (entry 7, Table 5). Yu et al.371 indicated the beneficial effect of metal doping in MnOx nanowires (M = Fe, Co, and Ni) for the aerobic oxidation of HMF in the presence of NaHCO3 base. Results revealed that M3+O(–Mn4+)2 clusters in the doped MnOx catalysts act as the active sites for HMF oxidation (entry 8, Table 5). Fig. 30 shows (a) SEM and (b, c) TEM images of Ni–MnOx nanowires. The activity order of the catalysts follows as MnOx < Fe–MnOx < Co–MnOx < Ni–MnOx, in line with order of their enhanced redox property and oxygen activation capacity. The promoting role of Mn4+/Mn3+ redox couple, along with surface exposed lattice oxygen species was distinctly observed in the Mn–Co binary oxide catalyzed HMF oxidation towards DFF product (entry 9, Table 5).372 A nanostructured Cu–MnO2 catalyst with rod morphology was found to be effective for the liquid phase HMF-to-DFF transformation (entry 10, Table 5).361 A 82.6% yield of DFF was obtained over Cu (4.8 mol%)–MnO2 catalyst, which can be reused for at least 5 times without much loss in catalytic activity. Nanocrystalline Li2CoMn3O8 spinel, developed by a gel pyrolysis method, exhibited good catalytic performance in HMF oxidation.373 About 80% isolated yield of FDCA was obtained in a gram scale reaction. The direct conversion of fructose to DFF via simultaneous dehydration and aerobic oxidation was also successfully carried out using 3D flower-like Ce–Mo micro/nano composite oxides.374 The catalytic activities of Ce10−xMoxOδ oxides were highly dependent on their composition, structure, and physicochemical properties. Especially, Ce9Mo1Oδ displayed the highest catalytic activity with 94% and 74% yields of DFF from HMF and fructose, respectively. Ni–Pd nanoalloys played a versatile catalytic role in the aerobic oxidation of various furans, including HMF, 2-furfuraldehyde, 2-furfuryl alcohol, 5-methyl-2-furfural and 5-methyl-2-furfuryl alcohol to the corresponding carboxylic acids in water.375 The addition of Ni not only led to enhanced catalytic activity of Pd NPs, but also to excellent stability (especially for Ni0.90Pd0.10), as the catalyst could be recycled up to ten times. This nanoalloy catalyst is also active for the direct transformation of fructose to furan carboxylic acids.
4.6 Valorization of lignin and its derived compounds
Catalytic hydrogenolysis is a widespread process for the efficient upgrading of lignin to high-value aromatics and fuel grade hydrocarbons. Zhang et al.382 revealed that NiRu catalyst containing 85% Ni and 15% Ru is highly active in the conversion of lignin to monomeric aromatic chemicals, outperforming monometallic Ni and Ru catalysts. The high performance of Ni85Ru15 catalyst was attributed to large amounts of surface-active atoms (compared with Ni), improved H2 and substrate activation (compared with Ni), and inhibition of benzene ring hydrogenation (compared with Ru). The catalytic hydrotreatment of Alcell lignin was performed using supported Ru (support = C, Al2O3, and TiO2), Pd (support = C and Al2O3), and a Cu/ZrO2 catalyst.383 Among them, Ru/TiO2 catalyst is highly active with a lignin oil (78 wt% on lignin intake) containing 9 wt% alkylphenols, 2.5 wt% aromatics, and 3.5 wt% catechols. Solvent has a profound effect on the hydrogenolysis of diphenyl ether and lignin carried out over a RANEY® Ni catalyst.384 Higher yields of cyclic alcohols and cyclic alkanes were obtained in the presence of methylcyclohexane solvent, whereas 2-propanol solvent yielded mainly cyclic alcohols, cyclic ketones, and unsaturated products. The hydrogenolysis of lignin in methanol, however, produced mostly phenols. Bimetallic Pd–Ni NPs supported on ZrO2 exhibited high selectivity in the cleavage of lignin β-O-4 linkages under hydrogen pressure conditions.385 The catalyst could be reused efficiently for ten and five times in the hydrogenolysis of a model compound and lignin, respectively, without much loss in catalytic activity and selectivity. The direct HDO of enzymatic lignin to C6–C9 cycloalkanes was successfully performed using a Ni catalyst supported on amorphous silica–alumina.386 About 80 wt% lignin conversion was found at 300 °C and 6 MPa H2. The synergy of acid and metal sites could be tailored to enhance the performance of Ni catalysts. Importantly, the presence of acid sites facilitates HDO of lignin-derived phenolic fragments to selectively obtain cyclic alkanes.
Interestingly, bulk MoO3 phase is also capable of catalyzing the vapor phase hydrodeoxygenation (HDO) of various lignin-derived oxygenates (phenol, m-cresol, anisole, guaiacol, and diphenyl ether) to benzene or toluene under H2 pressure conditions.390 MoO3 catalyst preferentially cleaved phenolic Ph–OMe bonds over the weaker aliphatic PhO–Me bonds during the HDO of lignin-derived compounds. Partial surface carburization of the catalyst, occurred during the reaction, played a crucial role in stabilizing the Mo5+ species on the MoO3 surface as well as in controlling the over-reduction of MoO3 to form an inactive MoO2 phase. In another work, MoO3/ZrO2 catalysts with varied MoO3 loadings (1 to 36 wt%) were tested for the vapor phase HDO of anisole at H2 pressures of ≤1 bar.391 The monolayer coverage of MoO3 on ZrO2 surface (optimized for 15 wt% MoO3) was crucial to obtain improved specific reaction rates in anisole HDO reaction. This is because the reduction of MoO3 crystallites to less reactive MoO2 is possible once the monolayer coverage is exceeded. Zhang et al.392 pointed out that oxygen vacancies present in MoO3 (Mo5+) exhibit a favourable effect in the selective cleavage of Ph–OH bonds during the liquid phase HDO of phenol to produce benzene (97% yield, entry 7, Table 2). The conversion of guaiacol to substituted alkylphenols was performed in supercritical ethanol without using additional hydrogen source.393 A total selectivity of 86% to alkylphenols at 99% guaiacol conversion was obtained at optimized reaction conditions. Characterization of used catalyst revealed that oxycarbohydride phase (MoOxCyHz) with Mo5+, formed under supercritical alcohol conditions, is the primary reason for improved conversion rates and high product selectivity.
Recently, much attention has been devoted towards understanding the role of metal oxide supports on the performance of metal/metal oxide catalysts for hydroprocessing of lignin-derived compounds. For instance, the catalytic efficiency of MoOx in liquid phase HDO of m-cresol to toluene (80–85%) was strongly dependent on the support nature (Al2O3, SBA-15, and SiO2).394 The activity order of investigated catalysts follows as MoOx/Al2O3 > MoOx/SBA-15 > MoOx/SiO2, which is in line with the reducibility of Mo species on the respective supports (entry 8, Table 2). Owing to high metal dispersion and strong metal–support interaction, the 10 wt% Co catalyst supported on TiO2 (compared to CeO2, ZrO2, Al2O3, and SiO2) showed the best HDO activity with >99.9% yield of propylcyclohexanols from lignin-derived phenols (entry 9, Table 2).395 Irrespective of the metal oxide support, all the catalysts selectively cleaved the Caryl–OCH3 bond in lignin-derived compounds, followed by hydrogenation of the aromatic ring when the –OCH3 group was present at the ortho-position. Interestingly, except for anatase TiO2-40 nm, the guaiacol conversion and the product selectivity did not significantly differ between the supported Au catalysts and the supports alone (anatase TiO2 in 45 μm, rutile TiO2, ZrO2, Al2O3, SiO2, and activated carbon).396 This indicates that Au NPs are barely active for the HDO of lignin-derived compounds. On the other hand, supported Pt based catalysts (TiO2, SiO2, Al2O3, and Nb2O5–Al2O3) displayed good activity in the vapour-phase HDO of diphenyl ether.397 The 0.40 wt% Pt/20Nb–Al2O3 catalyst afforded 100% diphenyl ether conversion and 99.2% cyclohexane selectivity due to the presence of abundant water-tolerant Lewis acid sites (entry 10, Table 2). Bimetallic NiCo/γ-Al2O3 catalysts were found to be promising in the aqueous phase HDO of guaiacol compared to supported monometallic Ni and Co catalysts.398 About 96% guaiacol conversion and 68% cyclohexanol selectivity were obtained over NiCo/γ-Al2O3 catalyst (Ni:Co = 1:1 with 20% total loading), which was ascribed to improved acidity and synergistic metal–support interaction (entry 11, Table 2). Liu et al.399 used density functional theory calculations to evaluate reaction mechanisms and product selectivity in HDO of guaiacol over Fe-alloyed (Ni, Pt) catalysts. Results revealed that catechol was the major product of guaiacol HDO reaction over both Fe-alloyed catalysts and was formed via dehydrogenation of methoxy (OCH3), followed by O–CH2 bond scission.
The Song group investigated the role of TiO2 addition on the structure–activity properties of Pd/SiO2 catalysts for the vapor phase HDO of guaiacol.400 Results demonstrated that TiO2 is essential to stabilize Pd particles as well as to activate the C–O bond in guaiacol, leading to higher conversion rates via enhanced deoxygenation activity. Consequently, Ti2.0Pd/SiO2 catalyst exhibited about 80% cyclohexane selectivity at full conversion of guaiacol (entry 12, Table 2). They also pointed out the crucial effect of TiO2 phase (anatase, rutile, and their mix, P25) on the performance of Pd catalysts for the vapor phase HDO of guaiacol.401 Interestingly, 5 wt% Pd catalyst supported on anatase TiO2 showed higher efficiency in guaiacol HDO to obtain cyclohexane due to the existence of partially reduced titanium species on the surface of anatase TiO2 that can efficiently cleave the C–O bond in guaiacol. The activity of 5 wt% Ru/TiO2 catalyst for the vapor phase HDO of guaiacol could be considerably promoted by ZrO2 addition.402 The authors propose that ZrO2 can hinder the migration of Ti3+ species on the surface of Ru particles and thus, more Ru active sites are available for the HDO of guaiacol to obtain benzene. Xu et al.403 also observed the favourable role of ZrO2 addition in improving the efficiency of 5 wt% Ru/La(OH)3 catalyst for the aqueous phase HDO of lignin-derived phenols. Zr and La can interact with each other to form a stable mixed (hydr)oxide, which effectively stabilizes active Ru particles. At optimized reaction conditions, 91.6% and 86.9% yields of cyclohexanol and alkyl cyclohexanols were obtained from guaiacol and alkyl phenols, respectively (entry 13, Table 2). Han et al.404 demonstrated the influence of shape-controlled TiO2 spheres in guaiacol hydrogenation. Promising results were obtained in terms of guaiacol conversion (100%) and cyclohexanol yield (84.2%) over Ru supported on mesoporous TiO2 hollow spheres embedded within SiO2 NPs. This remarkable catalytic activity is attributed to the unique spherical structure and improved acid properties. Thorough experimental and theoretical studies were performed to understand hydrogen-efficient HDO mechanism of phenol over Ru/TiO2 catalyst.405 Results indicated that the amphoteric nature of TiO2 facilitates the H2 heterolysis to generate an active site water molecule, promoting the direct substitution of phenolic OH with H to form benzene. Ambrusa et al.406 found that high dispersion of NiCu particles on TiO2 is the primary reason for achieving improved results in the liquid phase HDO of dibenzofuran to bicyclic hydrocarbons. Reaction temperature was also found to play a crucial role in tailoring product selectivity.
4.7 Valorization of lipids
The catalytic valorization of lipids (oils & fats) and their model compounds to produce biodiesel is discussed in this section.407,408 Several Mo- and W-based mixed oxides were found to show good catalytic activities for biodiesel synthesis via liquid phase transesterification of triglycerides (oils) or esterification of fatty acids with simple alcohols (mainly methanol). Xie et al.409 for instance, indicated that 30% WO3/SnO2 can be used as solid acid catalyst for the transesterification of soybean oil (79% conversion) with methanol to produce biodiesel. This catalyst can be reused up to four cycles without much loss in catalytic activity. However, there was a drastic decrease in biodiesel yield (57.8%) after 5th recycle, due to the blockage of catalyst active sites by adsorbed intermediates/byproducts. In another work, a mixed oxide Al2O3–ZrO2 was used as support for WOx catalyst, which displayed about 90% conversion of soybean oil at 250 °C that could be maintained up to 100 h of time on stream.410 In contrast, very low oil conversions were obtained in the presence of sulfated tin oxide (SO42−/SnO2) and sulfated zirconia (SO42−/ZrO2) catalysts. According to López et al.,411 tungstated zirconia was the most suitable catalyst for both the transesterification of triglycerides and the esterification of carboxylic acids to produce biodiesel. This catalyst was more active than the titania–zirconia for esterification and its activity could be easily regenerated than the sulfated zirconia by simply re-calcining the catalyst in air. Although sulfated zirconia is catalytically active for biodiesel synthesis, a huge loss of sulfur was observed during reaction. Waste cooking oil, containing high amounts of free fatty acids, was used for the synthesis of biodiesel over MoO3/ZrO2, WO3/ZrO2, WO3/ZrO2–Al2O3, and MoO3/SiO2 catalysts.412 Among them, MoO3/SiO2 was found to be the most promising catalyst, achieving 79% biodiesel yield at optimized reaction conditions. Komintarachat et al.413 studied the role of different supports, namely Al2O3, SiO2, SnO2, and ZnO on the performance of WOx catalysts for the transesterification of waste cooking oil (15% free fatty acid content). The activity of supported WOx catalysts, based on biodiesel yields, was in the following order: Al2O3 > SiO2 > SnO2 > ZnO. The high activity of WOx/Al2O3 catalyst can be explained by larger BET surface area (260 m2 g−1) and greater pore volume (0.95 cm3 g−1) of Al2O3 support, since bulky triglyceride molecules can efficiently adsorb/desorb through larger pores. Guldhe et al.414 pointed out the potential catalytic role of WO3/ZrO2 solid acid in the conversion of S. obliquus lipids, giving 94.6% biodiesel yield, which is comparable to that obtained in the presence of a liquid acid catalyst (e.g., H2SO4) and higher than that of an enzyme catalyst (immobilized Pseudomonas fluorescence lipase). The properties of biodiesel obtained in this work can comply with the specifications set by American and European biodiesel standards. Compared to WO3 based catalyst, MoO3/ZrO2–TiO2 is effective for the esterification of oleic acid with methanol to biodiesel.415 This is due to well-balanced acid and specific surface area properties in MoO3/ZrO2–TiO2 catalyst. For the same reaction, a pellet-type WO3/ZrO2 catalyst provided a 70% steady state conversion of oleic acid for 140 h.416 The oxidation state of tungsten species played a crucial role in the activity of WO3/ZrO2 catalyst for biodiesel synthesis. dos Santos et al.417 investigated the effect of tungsten loadings in WOx/ZrO2 catalyst for the esterification of palmitic acid. Polymeric tungsten species dispersed on tetragonal ZrO2 can provide abundant Brønsted acidic sites, which are key active sites for palmitic acid esterification. Notably, the 15 wt% WOx/ZrO2 catalyst exhibited a high resistant to leaching, and hence excellent reusability for several recycles without much loss in catalytic activity.Various CaTiO3, CaMnO3, CaZrO3 and Ca2Fe2O5 perovskites, prepared by milling and then calcination, were tested for biodiesel synthesis via methanolysis of sunflower oil.418 Best catalytic results (biodiesel production) were obtained over two-phase CaO/CaTiO3 or CaO/Ca2Fe2O5 catalysts at 165 °C, which was attributed to the presence of isolated CaO active phase. As a result, a number of CaO-based oxides were developed for biodiesel synthesis.419–422 Sr–Zr mixed oxides were also active in one-pot esterification and transesterification of waste cooking oil with ethanol.423 Owing to well-balanced acid–base properties, mixed oxide with a 2:1 atomic ratio of Sr–Zr, followed by calcination at 650 °C, displayed the highest catalytic performance towards biodiesel production. In another work, Mg was added to SrO to improve its catalytic activity for biodiesel production.424 Super strong basic sites in Sr–Mg mixed oxides were formed via a partial solid-state reaction induced by thermal treatment at 600°C. These novel basic sites together with the original basic sites from SrO played a beneficial role in transesterification reaction, achieving about 96% biodiesel yield at optimized reaction conditions.
4.8 Glycerol upgrading
Nanostructured metal/metal oxide based catalysts were found to be highly effective for the conversion of glycerol to a number of valuable chemicals, such as acetins, acetals, glyceric acids, glyceraldehyde, lactic acid, 1,2-propanediol, 1,3-propanediol, dihydroxyacetone, acrylic acid, acrylonitrile, acetol, allyl alcohol, 1-propanol, propylene, etc.425,426 Glycerol acetins, produced via acetylation of glycerol with acetic acid or acetic anhydride, have found useful applications as antiknock and fuel additives. Various mixed oxides, namely TiO2–ZrO2, WOx/TiO2–ZrO2 and MoOx/TiO2–ZrO2, along with ZrO2 were examined for the acetylation of glycerol with acetic acid in liquid phase.427 Several reaction parameters, such as catalyst loading, temperature, time, and molar ratio of acetic acid to glycerol were optimized. Owing to rational textural and acid properties, MoOx/TiO2–ZrO2 catalyst afforded full conversion (100%) of glycerol with a high selectivity to triacetin. Glycerol acetals and ketals, important chemicals as fuel-additives, bases, scents, and flavors, are synthesized via liquid phase condensation of glycerol with aldehydes and ketones over ZrO2428 and SnO2 based solid acid catalysts.429 High concentration of acid and redox sites is crucial for this reaction, and hence improved product yields were obtained with Mo6+ doped SnO2 solid acid.429 The catalytic significance of various mixed oxides, such as CeO2–ZrO2,430 promoted TiO2–ZrO2,431 and SnO2 based catalysts432,433 has been also markedly noted in glycerol acetylation and acetalization reactions.Glycerol oxidation is a sequential reaction that follows multiple pathways and gives various high-value chemicals (Fig. 31).425,434 The major challenges associated with the catalytic oxidation of glycerol are (i) catalyst stability, (ii) product selectivity, and (iii) using alkaline-free conditions.426 A series of nanostructured CuCo catalysts, prepared by a co-precipitation method, were studied for the aqueous phase oxidation of glycerol under basic conditions.435 Both the Cu/Co ratio and the post-treatment are the foremost factors to achieve improved product yields (glyceric, glycolic, and formic acid) in glycerol oxidation over nanoscale CuCo catalysts. The catalytic efficiency of isolated Au clusters (from 1 to 5 atoms) dispersed on TiO2 in the oxidation of glycerol using a NaOH solution can be enhanced by tuning the synthesis conditions of Au NPs, such as temperature and solvent.436 The synthesis of Au NPs in H2O at 1 °C resulted in a higher activity in glycerol oxidation compared with other Au/TiO2 catalysts reported in the literature. Au loading and Au particle size are also important for the Au/TiO2 catalyzed glycerol oxidation reaction.437 Results suggested that low Au loadings favor the formation of smaller Au NPs with narrow size distribution, leading to improved catalytic efficiency as smaller Au particles act as much stronger adsorption sites for glycerol. Ag NPs supported on Al2O3 were found to be effective for the liquid phase oxidation of glycerol to glycolic acid.438 About 85% glycerol conversion and 57% selectivity to glycolic acid were achieved over a 3.6 wt% Ag loaded catalyst. Díaz et al.439 reported kinetic models to evaluate the deactivation of Al2O3 supported Au, Pt and Ag NPs for glycerol oxidation. Notably, Pt/Al2O3 and Au/Al2O3 favored glyceric acid production, while glycolic acid was the major product over Ag/Al2O3 catalyst. Among the catalysts tested, Pt/Al2O3 was the least stable catalyst. The catalytic efficiency of nanoscale Ag/CeO2 catalyst is considerably improved for glycerol oxidation after the addition of Au, Pd or Pt.440 Mainly, the 5% Pt containing Ag95–Pt5/CeO2 catalyst showed the best performance with 54% glycerol conversion and 51% glycolic acid selectivity. Interestingly, further increase of the Pt amount (up to 50%) led to a noticeable shift in product selectivity from glycolic acid to glyceric acid. A synergistic effect of Au–Pd nano-alloys supported on ceria–zirconia was observed for the glycerol oxidation and the activity of the catalysts increased with the increase of Au:Pd ratio.441 The oxidation pretreatment of 2.2AuPd/Ce0.62Zr0.38O2 catalyst at 700 °C led to an improved performance for the oxidation of glycerol, attributed to the homogenous dispersion of Au–Pd nano-alloys. Recent results indicated that the introduction of Cu and Ag modifies the Au electronic state in Au/ZnO catalyst, enriched by strong metal–metal interactions.442 The AuCu/ZnO catalyst having metallic Au with the lowest negative charge, was found to be the most active catalyst for glycerol oxidation using molecular oxygen. The utilization of supported bimetallic NPs based on Au, Pd, and Pt resulted in enhanced catalytic activities in glycerol oxidation due to the synergistic metal–metal and metal–support interactions.426 The use of basic metal oxides (e.g., MgO) as supports for Au-based catalysts seemed to be promising route for base-free oxidation of glycerol.
 | ||
| Fig. 31 Possible major products from glycerol oxidation reaction. | ||
Lactic acid, a platform chemical for the production of biodegradable fibers, polylactic acid esters, and other bio-based chemicals, is commercially produced (∼95% of world production) via enzyme-catalyzed fermentation of carbohydrates (glucose and sucrose).443,444 However, this process is slow and involves high cost of enzymes, large amounts of salt waste generation and tedious workup separation procedures. The direct transformation of glycerol to lactic acid via glycerol oxidation to glyceraldehyde has been investigated as an alternative to the fermentation route. Three types of Cu based catalysts, such as Cu/SiO2, CuO/Al2O3, and bulk Cu2O with a base promoter (NaOH) were studied for the synthesis of lactic acid from glycerol in liquid phase without using reducing and oxidizing agents.443 Both CuO/Al2O3 and bulk Cu2O catalysts showed higher glycerol conversions (∼95%) compared to that of Cu/SiO2 catalyst (75%) at nearly identical lactic acid selectivity (75–80%). CuO/ZrO2 catalyst was also found to be effective for direct conversion of glycerol to lactic acid in aqueous solution.444 Cu loading (∼30%) was optimized to attain abundant metallic Cu species in CuO/ZrO2 catalyst, which could promote both dehydrogenation and hydrogenolysis of glycerol towards lactic acid production (94.6% yield). Palacio et al.445 demonstrated that the preparation method of nanostructured Co3O4/CeO2 catalyst could strongly affect both glycerol conversion and lactic acid selectivity. Compared to deposition–precipitation synthesized catalyst, improved results i.e. 79.8% lactic acid selectivity and 85.7% glycerol conversion were obtained over the impregnated Co3O4/CeO2 catalyst, which contains uniformly dispersed crystalline Co3O4 NPs in synergistic interaction with the CeO2 support. Cyclic dimer of lactic acid (i.e. lactide) is a key intermediate formed during the production of polylactic acid.446 Lactide is currently produced by a two-step procedure, involving synthesis of a pre-polymer via water removal from concentrated lactic acid solution, followed by thermal depolymerization of pre-polymer chain through intramolecular esterification so called “backbiting”. Alternatively, the Sels group developed a novel, efficient one-pot method for the synthesis of lactide from methyl lactate using TiO2/SiO2 catalysts.447 Lactide selectivity was as high as 90% at 42% methyl lactate conversion over 5% TiO2/SiO2 catalyst at 220 °C, while bulk TiO2 (both rutile and anatase) was almost inactive for lactide formation. A strong correlation between specific catalytic activity of TiO2 and its electronic/geometric features was observed. A higher number of weak Ti–O–Si bonds and low coordination number of Ti sites in TiO2/SiO2 composite are essential for improved methyl lactate-to-lactide transformation.
The gas phase dehydration of glycerol to acrolein (an important chemical for the chemical and agricultural industries) was studied using W–Nb–O complex mixed oxides, along with WO3/Nb2O5 and WO3/ZrO2 solid acids.448 The structure of W–Nb–O strongly affects the product distribution in glycerol dehydration. Both W–Nb–O and WO3/ZrO2 catalysts displayed a lower deactivation rate than a zeolite catalyst. Calcination temperature (varied in the range of 110–700 °C) played a central role in tuning the structure–activity properties of Ta2O5 for the gas phase dehydration of glycerol.449 Particularly, Ta2O5 catalyst calcined at 350 °C exhibited the best performance in terms of glycerol consumption rate and acrolein selectivity, which was attributed to the hydrated amorphous state and the strong acidic properties. Foo et al.450 investigated the role of Lewis and Brønsted acidic sites in the dehydration of glycerol over niobia and Na+-exchanged niobia catalysts. Results revealed that Lewis acidic sites favored the formation of hydroxyacetone, while acrolein selectivity is improved in the presence of more Brønsted acid sites than the Lewis acid sites. Effects of coke formation on the product distribution was examined in the gas phase dehydration of glycerol over WO3/TiO2 catalyst.451 Surprisingly, selectivity to acrolein increased as coke formed on the catalyst during the 14 h reaction time. The coke was suggested to cover specific active sites (weak and medium acidic sites), responsible for undesirable side reactions, resulting in improved glycerol-to-acrolein conversion, which is typically catalyzed by strong acidic sites.
Acrylic acid and its esters are very important chemicals for the manufacture of polymers, plastics, and synthetic rubbers.452 The Ueda group developed several metal oxide-based catalysts for the conversion of glycerol (or acrolein) to acrylic acid.453–456 In 2017, Sun et al.98 provided a critical review on glycerol-to-acrylic acid conversion using a variety of solid catalysts, including metal oxides.
Owing to synergistic redox properties, metal oxide supported transition metals (e.g., Ru, Rh, Pt, Pd, Cu, Co, and Ni) are attractive catalysts for the hydrogenolysis of glycerol to obtain 1,2-propanediol (1,2-PDO) or 1,3-propanediol (1,3-PDO). Both 1,2-PDO and 1,3-PDO have widespread applications in pharmaceutical, antifreeze additives, cosmetics, and polymer industries.457Table 6 summarizes the most important catalytic results obtained in glycerol hydrogenolysis over porous and nanoscale catalysts. For instance, the aqueous phase hydrogenolysis of glycerol to 1,3-PDO was studied over a Pt/WOx catalyst.458 Owing to larger specific surface area and abundant oxygen vacancies, the mesoporous WOx support improved the dispersion of isolated Pt atoms. A very high space–time yield (3.78 gg Pt−1 h−1) towards 1,3-PDO was obtained with this catalyst, which was attributed to synergistic Pt–WOx interface and specificity of bond formation between glycerol and WOx. Addition of Au to Pt/WOx catalyst led to improved glycerol conversion (81.4%) with a 51.6% selectivity to 1,3-PDO.459 The in situ generated Brønsted acidic sites and spatially separated pairs of H+ and H− function were the key factors for the efficient conversion of glycerol to 1,3-PDO (entry 3, Table 6). The promoting effect of non-noble Ni addition on the performance of Ir/γ-Al2O3 catalyst was also noticed for glycerol hydrogenolysis.460 Results indicated that the calcination step was crucial to obtain strong Ir–Ni interaction and consequently a significant increase in glycerol conversion and 1,2-PDO selectivity. Sun et al.461 observed a considerable leaching of Zn from PdZn alloys, resulting in rapid catalyst deactivation in the liquid phase hydrogenolysis of glycerol. Surprisingly, the physical mixture of Pd/m-ZrO2 and ZnO inhibited the catalyst deactivation in glycerol hydrogenolysis, due to the in situ formation of stabilized PdZn alloy layers on Pd surfaces.
| Entry | Catalyst | Reaction conditions | Product | Catalytic activity | Key factors for the catalyst activity | Ref. | |
|---|---|---|---|---|---|---|---|
| Conv. (%) | Yield (%) | ||||||
| a 1,3-PDO – 1,3-propanediol. b 1,2-PDO – 1,2-propanediol. | |||||||
| 1 | 2%Pt–5%Cu/H-mordenite | Vapor phase, 210 °C, H2 flow rate of 80 mL min−1, glycerol concentration of 10 wt% | 1,3-PDOa | 90 | 52.6 | Synergistic bimetallic phase-support interaction, Brønsted acidity of H-mordenite and its multiple pore system | 143 |
| 2 | 5 wt% Cu/SBA-15 | Vapor phase, 0.4 g catalyst, 220 °C, H2 flow rate of 30 mL min−1, WHSV of 1.03 h−1, 10 h | 1,2-PDOb | 90 | 75.6 | Homogeneous Cu dispersion and catalyst acidity | 216 |
| 3 | Au–Pt/WOx (2 wt% Pt and 0.1 wt% Au) | Liquid phase, 0.6 g catalyst, aq. solution of glycerol (12 g, 5 wt%), 140 °C, 10 bar H2, 12 h | 1,3-PDO | 81.4 | 42 | In situ generated Brønsted acid sites and spatially separated pairs of H+ and H− function | 459 |
| 4 | 20%Cu–10%ZrO2–MgO | Liquid phase, 0.6 g catalyst, 20 wt% of glycerol, 40 bar H2, 8 h, 180 °C | 1,2-PDO | 62 | 60 | High concentration of available Cu sites and moderate basicity of the catalyst | 462 |
| 5 | Cu–Al2O3–ZrO2 (20 wt% Zr) | Vapor phase, 20 wt% glycerol ethanol solution, 27.8 mL h−1 liquid flow rate, 100 mL min−1 H2 flow rate, H2:glycerol = 95:1 (mole ratio), 4 g catalyst, WHSV = 1.21 h−1 230 °C, 35 bar operating pressure, data acquisition after steady operation for 3 h |
1,2-PDO | 97 | 92.4 | High dispersion of Cu particles and synergistic Cu–Zr interaction | 463 |
| 6 | CuO–MgO–CeO2 (Cu/Ce/Mg atomic ratio = 1/3/5) | Liquid phase, 1 g catalyst, 50 g of 20 wt% aqueous glycerol solution, 200 °C, 60 bar H2 pressure, 10 h | 1,2-PDO | 51.8 | 50.4 | High surface area, ample amounts of acid sites, abundant oxygen vacancies, and improved reducibility | 457 |
| 7 | ZnPd/ZnO–3Al reduced at 350 °C | Liquid phase, 0.1 g catalyst, 230 °C, 30 bar H2, 5 mL of 20 wt% glycerol aqueous solution, 6 h | 1,2-PDO | 70 | 64.8 | Smaller sized ZnPd particles, improved reduction of ZnO, and abundant oxygen vacancies | 468 |
Multicomponent Cu–ZrO2–MgO catalysts, prepared by a co-precipitation method with various Cu and ZrO2 loadings, were found to be effective for the liquid phase hydrogenolysis of glycerol.462 The efficacy of the catalysts is highly dependent on the concentration of available Cu sites and moderate catalyst basicity. About 97% selectivity to 1,2-PDO and 62% glycerol conversion were obtained over 20%Cu–10%ZrO2–MgO catalyst (entry 4, Table 6). Cai et al.463 examined both bare Cu–Al2O3 and ZrO2-promoted Cu–Al2O3 catalysts for glycerol-to-1,2-PDO transformation in vapour phase. Compared to Cu–Al2O3 catalyst, ZrO2-promoted Cu–Al2O3 catalyst showed durable activity for glycerol hydrogenolysis, which was attributed to high dispersion of Cu particles and synergistic Cu–Zr interaction (entry 5, Table 6). Mallesham et al.457 demonstrated the beneficial effect of Ce on the catalytic performance of Cu–Mg mixed oxide for the liquid phase hydrogenolysis of glycerol to 1,2-PDO (entry 6, Table 6). Results revealed that the addition of Ce to Cu–Mg–O leads to improved specific surface area, enriched redox properties, and ample amounts of acid–base sites, and consequently high yields of 1,2-PDO over CuO–MgO–CeO2 catalyst (Cu/Ce/Mg atomic ratio = 1/3/5). Leaching of Zn species and Cu sintering were the primary reasons for the deactivation of bimetallic CuZn catalysts in glycerol hydrogenolysis.464 The significance of support nature and metal addition in improving catalyst stability and product selectivity in glycerol hydrogenolysis is discussed in recent articles.465–469
4.9 Terpenes upgrading
Terpenes are volatile unsaturated hydrocarbons found in many plants, such as pine, citrus, conifers, peppermint, spearmint, caraway, and dill. Some important examples of monoterpenes (e.g., α-pinene, myrtenol, carvone, p-cymene, and citral) are shown in Fig. 32. The catalytic oxidation of terpenes gives a number of value-added terpenoid products, which can be used as flavors, fragrances, spices and cosmetics. Costa et al.470 used ammonia hydroxide to stabilize Au hydroxides on MgO support, enabling the formation of highly dispersed Au NPs, which are indispensable to achieve higher yields of carbonylic monoterpenoids from the aerobic oxidation of bio-derived monoterpenic alcohols. Addition of Cu, Co or Ru has a considerable effect on the efficacy of nanoscale Au/TiO2 catalyst for α-pinene oxidation.471 The bimetallic AuCu/TiO2 catalyst displayed the best performance in terms of α-pinene conversion and verbenone product selectivity due to synergistic metal–metal and metal–support interactions. Mn–Fe–O mixed oxide is a highly selective catalyst in the aerobic oxidation of p-cymene towards terephthalic acid, an important chemical for the production of poly(ethylene terephthalate) (PET).472p-Cymene can be produced from terpenes or eucalyptol. About 51% yield of terephthalic acid was obtained over Mn–Fe–O catalyst with molecular oxygen as the oxidant, outperforming Mn2O3, Fe2O3, CoFe2O4, NiFe2O4, and V/Al2O3 catalysts. | ||
| Fig. 32 Some important examples of monoterpenes. | ||
The selective hydrogenation of terpenes is an important process for the production of valuable fine chemicals.473 The direct synthesis of naturanol from α-pinene oxide, involving isomerization of α-pinene oxide to campholenic aldehyde in liquid phase, followed by its hydrogenation to naturanol, was performed using bimetallic Pt–Sn/SiO2 catalysts.474 Oxidized Sn species acted as active sites for isomerization step, while Sn/Pt ratio affected both the hydrogenation of α-pinene oxide and the naturanol selectivity. The conversion of carvone and selectivity to carvomenthone achieved in Al2O3 supported metal-catalyzed carvone hydrogenation reaction followed the order: Pd > Rh > Ru and Rh > Pd > Ru, respectively.475 Interestingly, supercritical CO2 as the reaction medium showed a promoting effect in carvone hydrogenation. Ekou et al.476 indicated that catalyst preparation method and activation pre-treatment of Rh/TiO2 and Pt/TiO2 catalysts were the foremost factors for selective hydrogenation of citral into unsaturated alcohols. Results suggested that TiO(2−x) species play a vital role in the activation of CO groups of citral to obtain desirable products. A comprehensive review was published on the selective hydrogenation of citral over metal-based solid catalysts.473 In recent years, several nanoscale bimetallic477 and supported metal nanocatalysts478,479 were also investigated for citral hydrogenation.
The Simakova group developed a potential catalytic route for the direct amination of myrtenol with aniline using nanoscale Au catalysts supported on different metal oxides (e.g., MgO, ZrO2, CeO2, Al2O3, and La2O3).480 The resulting terpene amines are very useful for the production of drugs (for neurological diseases). A correlation between the acid–base properties of support and the performance of Au catalysts was established in this study. Au/ZrO2 catalyst afforded about 52% selectivity to secondary amines of myrtenol at nearly complete conversion. They also examined the role of hydrogen addition and the reaction temperature towards product selectivity in myrtenol amination over Au/ZrO2 catalyst.481 The addition of H2 (1 bar) at 100 °C reaction temperature led to complete conversion of myrtenol with 68% amine yield, preserving CC bond in the initial myrtenol structure. On the other hand, the reduction of both CC and CN bonds was possible with the formation of two diastereomers (around 93% yield) when hydrogen was added at 180 °C reaction temperature. Conversion rates and product selectivity in myrtenol amination over nanosized Au-based catalysts were strongly dependent on catalyst pre-activation482 as well as on the addition of alcohols and formic acid.483
4.10 Bioethanol conversion
A promising strategy for the conversion of bio-ethanol into isobutene was developed using nanosized ZnxZryOz catalysts.484,485 A high selectivity of isobutene up to 83% at full conversion of bioethanol was obtained at optimized reaction conditions. Selective deposition of ZnO on ZrO2 led to optimization of ZrO2 acidity and provided the required basicity, favoring the ethanol-to-isobutene transformation. Optimization of acid sites played a prominent role in controlling undesired reactions of bio-ethanol dehydration and acetone polymerization/coking, resulting in high yields of isobutene. It was suggested that basic sites could catalyze ethanol dehydrogenation and aldol-condensation reactions to yield acetone, while Brønsted acidic sites promote acetone-to-isobutene reaction pathway. Scandium doped indium oxide was found to be active for bio-ethanol upgrading. However, propylene was obtained as the main product up to 60% yields in the presence of water and hydrogen, outperforming zeolite-based catalysts (propylene yield is up to 20–30%).486,4875. Importance of water in liquid phase biomass valorization
Biomass conversion reactions are mostly carried out in liquid phase, rather than in gas phase normally used in petrochemical industry, because of low thermal stability of biomass feedstocks originating from highly oxygenated species.488–490 On the other hand, effective utilization of bulky/viscous biomass molecules, facile optimization of process variables, improved product selectivity, and high atom economy are the persuasive factors to carry out biomass conversions in liquid phase. Therefore, liquid phase biomass processing is of great importance for the future biorefineries. Various solvents, including water, ionic liquids, bio-based solvents, and conventional organic solvents have been used for biomass valorization. Water is particularly a desirable solvent because of its high abundance, ecofriendly nature, and ability to solubilize biomass-derived oxygenated products.490 In addition, water can serve as a reactant, promoter, or as a catalytic active phase for biomass processing (Fig. 33).491–493 Biomass naturally contains huge moisture contents (30–60%, depending on the source) because water is one of the key reagents for photosynthesis process to produce biomass (Fig. 2). It is obvious that liquid phase biomass processing can result in the generation of significant amounts of water, along with bio-derived chemicals. Moreover, water is a byproduct in the dehydration reaction of various biomass-derived compounds, such as glucose, fructose, xylose, and glycerol. By using water as reaction medium, the process to separate in situ generated water formed during biomass conversion, can be simplified by creating a bi-phasic system in which water would be retained in the polar phase, while most bio-derived organic chemicals will reside in the apolar phase. Consequently, energy-intensive steps required for the purification of bio-based chemicals can be avoided, leading to improved product yields with high quality and reduced process costs. | ||
| Fig. 33 Importance of water in liquid phase biomass valorization and its impact on catalyst stability. | ||
Water is an important reactant for the hydrolysis of various biomass molecules, including lignocellulose, cellulose, hemicellulose, cellobiose, and triglycerides to produce valuable platform molecules.493–495 Water is also essential for the production of levulinic acid from carbohydrates and HMF. The application of hydrothermal conditions, normally >100 °C and 10 bar, is indispensable for the efficient fractionation of biomass into platform molecules or building blocks.284 Sometimes, supercritical conditions (374 °C and 221 bar for water) are required to enhance mass transfer properties and to increase reaction rates during biomass processing.492 Under these sub- and super-critical conditions, water can provide hydronium ions (H3O+) and hydroxyl species (OH−), which have very important roles in biomass conversion. For instance, hydronium ions can act as acidic sites for the acid-catalyzed reactions, especially for the hydrolysis of cellulose to glucose.37,492,496 On the other hand, hydroxyl species can act as the source of oxygen for biomass oxidation reactions, such as HMF oxidation to 2,5-furandicarboxylic acid497 and galacturonic acid oxidation to galactaric acid.498 Recently, Meng et al.499 pointed out the promoting role of water, which can suppress the side reactions of HMF, in the decarbonylation of HMF towards furfuryl alcohol synthesis.
6. Hydrothermal stability and deactivation of nanoporous catalysts during aqueous phase biomass valorization
Although water plays an important role in liquid phase biomass processing, there are two major concerns of the catalysts that must be taken into consideration when using water as solvent namely, (i) hydrothermal stability and (ii) catalyst deactivation.489,500,501 These problems are mainly due to the inevitable utilization of harsh hydrothermal reaction conditions, which may degrade catalyst structure and consequently their catalytic performance. The deactivation of a solid catalyst can be determined in terms of undesirable structural changes, active phase leaching, loss of surface area, and blockage of pores/active sites (Fig. 33). In hot liquid water conditions (>150 °C), desilication or dealumination of zeolites often occurs via hydrolysis of Si–O–Si and Si–O–Al bonds, respectively, leading to severe degradation of zeolite frameworks.8,488,502 Gardner et al.502 found that solvated Al3+ ions leached from ZSM-5 zeolite were the active sites for the aqueous phase glucose-to-fructose isomerization (reaction conditions: 170 °C and 30 min). Leaching of Si or Al species into the reaction mixture was drastically induced in the presence of inorganic metal salts, which are naturally present in biomass feedstocks. Since the catalyst active sites (Al3+) were leached into the reaction solution, the ZSM-5 catalyst loses its recycling efficiency for glucose isomerization. Leaching of heterocations from zeolite frameworks was also observed during aqueous phase biomass conversions.489,503,504 Incorporating heterocations into the zeolite frameworks can stimulate the distortions in bond angles and bond lengths, due to the disparity in host–guest properties. It is therefore obvious that incorporated heterocations can be more rapidly leached compared to constituent atoms (Si or Al) of the zeolites, under the hydrothermal conditions.47 For instance, leaching of both extra-framework Ti and active framework Ti species from a titanium silicalite zeolite was found during furfural oxidation reaction in aqueous H2O2, and thus, rapid catalyst deactivation was observed.503 The ordered structure of mesoporous silica and its surface area can also be affected adversely at elevated hydrothermal conditions (≥200 °C).502,505 About 90% loss in the surface area of a mesoporous silica was observed at 200 °C for 10 h hydrothermal treatment due to the degradation of ordered mesoporous structure elicited by the hydrolysis of silica in hot liquid water.Leaching of metal nanoparticles (e.g., Zn) and acidic species (e.g., –SO3H) is possible during aqueous phase reactions due to the solvation and chelating effects. For instance, Zn leaching from PdZn461 and CuZn464 nanoalloys was found to be the main reason for catalyst deactivation in glycerol hydrogenolysis reaction. Biomass contains several oxygen, nitrogen or sulfur functionalities in the form of sugars or acids, which can form complexes with metal components of the catalysts, accelerating the leaching and the aggregation of metal NPs.489 Under hydrothermal conditions (>200 °C and autogenous pressure), basic metal oxides (e.g., MgO and γ-Al2O3) can form hydroxides with increased solubility in water that can lead to severe metal leaching and drastically reduced catalyst reusability.506,507 Sulfur leaching via hydrolysis of sulfate groups is the primary reason for the deactivation of SO3H-functionalized porous catalysts in aqueous phase biomass conversions.508,509 The application of harsh reaction conditions can induce the leaching of metal salts (potassium, calcium and magnesium salts), present in biomass feedstocks, into the reaction solution.510 These metal ions can replace the protons on the solid acids and can ultimately reduce the concentration of active sites needed for the acid-catalyzed reactions, such as dehydration of carbohydrates to HMF and hydrolysis of cellulose to glucose.
Blockage of pores/active sites by insoluble reaction intermediates/byproducts,423,504 incorporated metals/catalyst precursors55,200 or by solvent molecules511 is another key factor for the deactivation of porous and nanostructured catalysts during aqueous phase biomass conversions. Pore blockage in microporous zeolites and mesoporous silica catalysts can adversely affect their specific surface area as well as the accessibility of active sites (usually present inside the pores). Compared to mesoporous silica catalysts (2–50 nm pore size), zeolites are more prone to suffer pore blockage because of their microporous nature (<2 nm pore size). It was found that insoluble oligomeric byproducts can accumulate predominantly on the outer regions of Sn-beta zeolite and can drastically affect the mass transport properties during retro-aldolization and isomerization of glucose.504 Incorporation of larger sized metals, such as Ce into the zeolites should be avoided because they can block the micropores, resulting in low conversion rates as observed for the reaction of tert-butanol with 3,4-dihydro-2H-pyran over Ce/MCM-22 zeolite.55 Interestingly, the concentration of acid sites in Ce/MCM-22 was not affected by hot water treatment at 160 °C for 48 h. Pore blockage and surface area loss are also possible in mesoporous silica materials if the impregnated catalyst precursors contain very large complex species, like isopolytungstates.200 In addition to the accumulation of reaction byproducts, accumulation of water molecules on Brønsted acid sites (present in Ag/zeolite catalyst) was also noticed, impeding their accessibility for the reactant molecules, leading to low conversion rates in an alkene oxidation reaction.511 The polar character of basic metal oxides (e.g., MgO) is the key factor for the accumulation of undesirable nucleophiles (e.g., water and oligomers), which can block the active sites and accelerate catalyst deactivation as observed in aldol condensation of biomass-derived cyclopentanone.506 Moreover, soluble byproducts obtained during biomass processing or impurities present in biomass can cause fouling or poisoning of catalyst active sites.512 All the drawbacks emphasized in this section have become a barrier for the practical application of porous and nanostructured catalysts in aqueous phase biomass conversions. Therefore, the development of efficient, durable, and water-tolerant catalysts is essential for sustainable biomass valorization.
Over the last ten years, many useful strategies have been developed to enhance the hydrothermal stability and catalytic performance of porous and nanostructured catalysts for a variety of aqueous phase chemical transformations, including biomass valorization. Improving the hydrophobicity of nanoporous catalysts is one of the most promising strategies to enhance their hydrothermal stability. Moreover, it can enrich the catalyst wettability towards bio-derived organic chemicals. As a result, the direct contact of water molecules with zeolite can be inhibited as shown in Fig. 34.490 Zeolite frameworks are typically composed of siloxane bonds, but they can also contain hydrophilic silanol (SiOH) groups, which originate from framework siloxy (i.e., SiO−) defects formed during hydrothermal synthesis of zeolites.513–515 These SiOH species are known to be the main active sites for the hydrolysis of zeolite frameworks in hot liquid water conditions (>150 °C and autogenous pressure). Hence, the formation of SiOH defects should be prevented to improve hydrothermal stability of the zeolites, which can be done by using alternative charge-balancing anions (i.e., F−). However, this process involves the utilization of environmentally unfriendly media, such as NaF, NH4F, and HF.516 In contrast, the functionalization of zeolites by silylation approach has attracted much attention as this strategy allows the anchoring of hydrophobic organosilane groups on the external/internal surfaces of the zeolites.113,490,513,517 This hydrophobization route using organosilanes can impede the interactions of water molecules with zeolites, thus stabilizing the framework against harsh hydrothermal conditions (Fig. 34).490 Alternatively, Grand et al.518 used a tungsten incorporation method to inhibit the formation of silanol defects in nanosized MFI-type zeolites. This approach greatly enhanced the structure stability of the zeolites because of the formation of W–O–Si bridges, which are more stable than Si–O–Si bonds.
 | ||
| Fig. 34 The behavior of hydrophobized zeolites, prepared by silylation approach, in aqueous and organic phases. Reproduced with permission from ref. 490. Copyright (2012) American Chemical Society. | ||
A structural reconstruction strategy was also found to improve hydrophobicity of nanosized Sn-beta zeolites.519 This process involves dealumination using HNO3 treatment, followed by degradation at hydrothermal conditions (140 °C for 45 min) and finally, self-assembly at 140 °C for 1 h. The resulting zeolites showed high catalytic performance in aqueous phase redox reactions, which was attributed to their outstanding hydrophobicity. The selective anchoring of metal organic framework (MOF) nanospheres on a hierarchical zeolite (FAU) assisted to improve its hydrophobic nature, due to the surface functionalization of the zeolite.520 The degree of surface functionalization and hydrophobicity can be easily tuned by optimizing the amount of deposited MOF and the number of deposition cycles. Zeolite-encapsulated metal nanoparticles may contain contaminated water because of the hydrothermal conditions used for their synthesis.145 To circumvent this problem, a solvent-free method was used to synthesize AuPd@zeolite catalysts, which exhibited good hydrothermal stability for the aerobic oxidation of bioethanol due to the absence of contaminated water in the catalyst structure.
Zeolitization, silylation, carbon coating, surface modification using functional molecules (e.g., oleic acid), and incorporating heteroatoms (e.g., Co, Ce, or Al) are commonly used methods to improve hydrothermal stability of mesoporous silica materials.507,521–523 Especially, carbon coating is of great research interest because carbon is hydrophobic, which can efficiently prevent the mesoporous silica from hydrolytic attack under hydrothermal conditions. This approach involves the deposition of carbon thin films, obtained by controlled pyrolysis of carbohydrates (e.g., sucrose or glucose), on mesoporous SiO2. Fig. 35 shows the coating of carbon on the pore walls of SBA-15 material.505 With metal catalysts supported on carbon-coated mesoporous silica, improved hydrothermal stability and high catalytic performance in aqueous phase biomass conversions were obtained compared with the uncoated analogues.505,524,525 Pyrolysis of carbon precursor at lower temperature (300 °C rather than 600 °C) is vital to achieve better metal dispersion and improved hydrophobicity for Pd/carbon-coated SBA-15 catalyst.525 This is because low-temperature pyrolyzed carbon overlayer (300 °C) contains higher amounts of oxygen-containing functional groups, which can stabilize Pd NPs during aqueous phase furfural hydrogenation. The formation of surface hydroxyl groups, responsible for the hydrolysis of Si–O–Si bonds in mesoporous silica, was partially hindered by doping a metal into the SBA-15 material.521 As a result, the metal-doped SBA-15 materials (metal = Co, Ce, and Al) provided higher hydrothermal stability compared with the undoped material. Oleic acid functionalization strategy was also found to be promising to enhance the hydrophobicity of mesoporous silica due to the formation of strong bonds between carbonyl group of oleic acid with silanol group on mesoporous silica surface.523 However, a considerable reduction in specific surface area, pore volume, and pore size was observed because of the adsorption of oleic acid molecules on the inner pore surface of mesoporous silica.
 | ||
| Fig. 35 HRTEM images (a and c) and elemental carbon maps (b and d) of calcined SBA-15 and 10 wt% carbon-coated SBA-15 material. Reproduced with permission from ref. 505. Copyright (2012) John Wiley and Sons. | ||
The coordination of a metal with another metal or metal oxide is an attractive strategy, not only to achieve enhanced structure–activity properties, but also to improve hydrothermal stability of nanostructured metal/metal oxide catalysts. For instance, mixed oxide supported metal particles (e.g., Ru/Zr–W–O)310 and metal oxide supported bimetallic particles (e.g., Pd–Cu/ZrO2)326 were found to be highly active for cellulose hydrogenolysis and levulinic acid hydrogenation in aqueous phase, respectively, compared to the pristine counterparts, which was attributed to strong metal–metal and metal–support interactions. Such synergistic interactions were also observed in unsupported mixed oxides, namely SiO2–Al2O3,269 and La2O3–ZrO2.526,527 These mixed oxides afforded improved hydrothermal stability and high catalytic performance for the conversion of biomass-derived molecules (e.g., cellobiose, inulin, and acetic acid) to value-added chemicals. Carbon coating, as has been demonstrated for mesoporous silica, also plays a key role in improving hydrothermal stability of metal oxides (e.g., γ-Al2O3).507,528,529 The structural, acid, and redox properties of metal/coated-metal oxide catalysts (e.g., Pd/coated-γ-Al2O3) can be controlled by the degree of carbon coating.528 In contrast to the coating of γ-Al2O3 support by carbon layer, Pham et al.507 developed a carbon-overcoated strategy, which can protect the whole structure of metal/metal oxide catalysts (e.g., Cu/γ-Al2O3 catalyst) against hydrolytic attack, without showing detrimental effects on their catalytic activity. As a result, carbon-overcoated Cu/γ-Al2O3 catalyst presented high hydrothermal stability for the aqueous phase hydrogenation of furfural to furfuryl alcohol compared with the uncoated catalyst. Moreover, the carbon in Cu/γ-Al2O3 can be functionalized, providing additional active sites for biomass valorization. On the other hand, O'Neill et al.530 proposed an oxide (alumina) overcoating method for the stabilization of Cu/γ-Al2O3 catalyst. This approach greatly prevented Cu sintering and leaching during aqueous phase furfural hydrogenation. It must be noted that γ-Al2O3 can convert to soluble boehmite phase (AlOOH) at high hydrothermal conditions (>200 °C) and can result in the degradation of alumina coating.507 Alkyl phosphonate coating was also found to improve hydrothermal stability of γ-Al2O3, attributed to strong Al2O3–POx bonds, which can inhibit the formation of hydrated boehmite phase. As a result, the specific surface area of functionalized γ-Al2O3 was unaffected over a 20 h hydrothermal treatment at 200 °C and autogenous pressure.531 In recent work, Ngo et al.506 reported a facile strategy to enhance hydrophobicity of basic metal oxides (e.g., MgO) by functionalizing with octadecyltrichlorosilane. The resulting functionalized MgO catalyst exhibited improved hydrothermal stability in the aqueous phase aldol condensation of biomass-derived cyclopentanone, outperforming conventional MgO-based catalysts. In another recent work, the hydrophobicity of Pd/γ-Al2O3 catalysts, enhanced by the deposition of organophosphonic acids, played a pivotal role to achieve higher product yields (p-cresol) in hydrodeoxygenation of biomass-derived vanillin.532
The development of hierarchically porous zeolite- and silica-based catalysts is the utmost potential route to improve mass transport properties during aqueous phase biomass conversions. In this review (Sections 2.4 and 3.3), we have summarized several strategies developed for the synthesis of hierarchical porous catalysts, while discussing the ensuing catalytic effects in the relevant biomass conversions. The deposition of insoluble heavy byproducts on the surface and pores of the catalysts can be prevented by using hydrophobic solvent mixtures (e.g., ethanol/water and GVL/water) instead of pure water.504,533,534 If the catalyst still contains the deposited insoluble byproducts, a simple calcination step under air atmosphere at the requisite temperature can be used to remove them from the catalyst surface.503
7. Conclusions and future prospects
During the last ten years, significant research efforts have been made towards tailoring the porosity and the nanoscale properties of solid catalysts, aiming to develop state-of-the-art catalysts with improved properties for a large spectrum of industrially important catalytic applications. The most promising porous and nanosized materials that have paved the way for exciting scientific discoveries in heterogeneous catalysis are microporous zeolites, mesoporous silicas, and nanoscale metals/metal oxides. Although several important applications of these fascinating catalysts have been reported in the recent literature, their catalytic role in valorizing biomass into useful fine chemicals and biofuels has particularly attracted paramount attention in view of a more sustainable economy and a carbon-neutral society.In this review, a wide variety of potential catalytic strategies for the valorization of both edible (lipids and starch) and non-edible (lignocellulose) biomass substrates as well as their derived compounds have been demonstrated over microporous zeolites, mesoporous silicas, and nanoscale metals/metal oxides. Despite the nature of biomass feedstock, hydroprocessing is indeed the most appealing route for efficient biomass upgrading.535 The resulting products can strongly resemble the petro-based products. Since hydroprocessing often involves multiple reaction steps, the development of versatile solid catalysts with adequate amounts of necessary active sites is essential for achieving improved reaction rates. Catalytic fast pyrolysis (CFP) is another attractive process to convert lignocellulose biomass into transportation fuels in a single-step process. Low quality of pyrolytic bio-oil, uncontrolled product selectivity, and fast catalyst deactivation due to coke formation are the major constraints in CFP technology. Hence, the design of efficient and stable solid catalysts that are able to deal with coke as well as with various decomposition intermediates formed during CFP processing, represents a potential solution to tackle the above concerns.536 Catalytic oxidation is one of the widely used processes in biorefinery research to produce a variety of key platform chemicals for polymer, food, and pharmaceutical industries. The catalytic oxidation of 5-hydroxymethylfurfural (HMF) to 2,5-furandicarboxylic acid (FDCA) is of great importance since FDCA is a promising substitute for petroleum-derived terephthalic acid in the manufacture of poly(ethylene terephthalate) polymer (PET). However, the design of milder and greener reaction methodologies as well as efficient solid catalysts for HMF-to-FDCA transformation remains a great challenge. Catalytic transesterification is another important reaction for valorizing edible biomass, especially lipids (oils & fats) to biodiesel fuel.537 In addition, hydrolysis, isomerization, dehydration, esterification, decarbonylation, etherification, and many other processes were also effective in biomass valorization to useful fuels and fine chemicals.
One of the most interesting and challenging subjects in biorefinery is the efficient valorization of lignin, since it is the only scalable and renewable feedstock for the production of aromatic chemicals. In this context, reductive fractionation of lignocellulose using solid catalysts has attracted much attention as this strategy provides two valuable separated fractions: (i) lignin oil containing high-value phenolic compounds and (ii) valorizable solid carbohydrate pulp. The obtained lignin-derived molecules can be upgraded to polymer precursors or fuel-grade chemicals using hydroprocessing reactions. As well, carbohydrate pulp can be used for the production of paper, platform molecules, or biofuels. Over the last five years, notable findings on this novel “lignin-first” biorefinery concept were reported. However, there are still several hurdles that needs to be addressed in this process, including the use of mild reaction conditions, simplifying reaction methodology, as well as improving catalyst selectivity and its reusability.
This review has provided a large number of approaches to tailor unique catalytic properties of microporous zeolites, mesoporous silicas, and nanoscale metals/metal oxides. Doping of heteroatoms into the zeolite structures, developing hierarchical zeolites with wide-ranging pore dimensions, and introducing new active sites by modifying synthesis conditions are some of the vital strategies demonstrated to improve zeolites’ properties for efficient biomass valorization. Poor hydrothermal stability in the presence of inorganic salts is one of major disadvantages of zeolites for their applications in biomass upgrading. For example, the presence of NaCl promotes the hydrolysis of Si–O–Al bonds of ZSM-5 zeolite, resulting in the leaching of active Al3+ species into the reaction mixture that can catalyze the dehydration reaction of glucose through a homogeneous catalytic process, which is associated with catalyst recovery and reusability concerns.502 Similar types of modification routes used for the zeolites, such as heteroatom doping, immobilizing acid–base and redox functionalities, multimodal pore structures, and tailoring pore morphologies have been also suggested for mesoporous silica catalysts. Owing to large pore geometries, mesoporous silica catalysts can facilitate efficient diffusion of (bulky) bio-based molecules towards catalytically active sites, thus overcoming mass transport problems often encountered with microporous zeolites. However, mesoporous silica contains negligible amounts of acid sites, which are indispensable for a number of biomass upgrading reactions, such as hydrolysis, hydrodeoxygenation, dehydration, and esterification. Although immobilization of acid functional species (e.g., –SO3H) into the mesoporous silica framework has been attempted, leaching of these acid groups is inevitable during biomass upgrading, due to the application of elevated temperature/pressure conditions as well as the presence of large amounts of water in biomass feedstocks. Pristine mesoporous silica catalysts also suffer from low hydrothermal stability because of their small wall thickness and low degree of cross-linked silicate units.
Nanoscale metals/metal oxides are the most widely used solid catalysts for biomass valorization because of their excellent redox properties and unusual structural defects, along with nanosize-dependent catalytic properties. Tailoring the particle size and morphology of metallic materials at nanoscale range (1–100 nm in at least one dimension) could generate remarkable changes in their structural and electronic properties that could be largely different from the corresponding bulk metal catalysts. Nano-alloys, nanosized metal oxides, metal oxide supported single metal atom catalysts, and shape-controlled metal oxide supported metal NPs are some of the novel, appealing metal-based nanocatalysts discussed in this review in view of their unusual catalytic performances in biomass valorization. However, difficulties in the separation and recovery of nanoscale metal catalysts from reaction mixtures may limit their applications in biomass valorization. A feasible solution to tackle this issue is the development of composite nanocatalysts with magnetic properties. For instance, the addition of magnetic Fe3O4 to metal nanocatalysts can provide new nanocomposite catalysts with magnetic properties. The resulting catalysts can be efficiently recovered from the reaction mixtures using external magnetic field without affecting their activity and selectivity.538 In this review, several useful strategies have been summarized to improve the hydrothermal stability and to inhibit the deactivation of porous and nanostructured catalysts during aqueous phase biomass conversions. However, more stimulating research must be undertaken towards the development of active, multifunctional solid catalysts for the large-scale biomass valorization to produce high yields of fuels and chemicals. Based on the above-mentioned challenges and opportunities, we believe that this comprehensive review may provide useful insights for the design of advanced porous and nanoscale solid catalysts with versatile characteristics (Fig. 36) to support a viable biorefinery.
 | ||
| Fig. 36 Key characteristics of porous and nanoscale catalysts for achieving improved structure–activity performance in biomass valorization. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement number-747968 (project acronym: CATLIGCAR). E. P. acknowledges the Research Foundation of Flanders (FWO Vlaanderen). E. V. M. and B. F. S. thank the Industrial Research Fund of KU Leuven (IOF, grant ZKC8139) and VLAIO (Catalisti cluster SBO project SPICY). V. I. P. kindly acknowledges UEFISCDI for financial support (projects PN-III-P4-ID-PCE-2016-0146 and 32PCCDI-2018). We also acknowledge the financial support from VLAIO/Catalisti (ARBOREF), the Research Foundation Flanders (FWO) (WOG and BioFact Excellence of Science project), and the INTERREG BIO-HArT project which is established by a contribution of the European Interreg V Flanders-The Netherlands program that stimulates innovation, sustainable energy, a healthy environment and the labor market by means of cross-border projects.References
- R. Zhang, N. Liu, Z. Lei and B. Chen, Chem. Rev., 2016, 116, 3658–3721 CrossRef CAS PubMed.
- L. T. Mika, E. Cséfalvay and Á. Németh, Chem. Rev., 2018, 118, 505–613 CrossRef CAS PubMed.
- A. F. Lee, J. A. Bennett, J. C. Manayil and K. Wilson, Chem. Soc. Rev., 2014, 43, 7887–7916 RSC.
- R. Lødeng, C. Ranga, T. Rajkhowa, V. I. Alexiadis, H. Bjørkan, S. Chytil, I. H. Svenum, J. Walmsley and J. W. Thybaut, Biomass Convers. Biorefin., 2017, 7, 343–359 CrossRef.
- A. Deneyer, E. Peeters, T. Renders, S. Van den Bosch, N. Van Oeckel, T. Ennaert, T. Szarvas, T. I. Korányi, M. Dusselier and B. F. Sels, Nat. Energy, 2018, 3, 969–977 CrossRef CAS.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- Z. Zhang, J. Song and B. Han, Chem. Rev., 2017, 117, 6834–6880 CrossRef CAS PubMed.
- T. Ennaert, J. Van Aelst, J. Dijkmans, R. De Clercq, W. Schutyser, M. Dusselier, D. Verboekend and B. F. Sels, Chem. Soc. Rev., 2016, 45, 584–611 RSC.
- Q. Liu, S. C. Chmely and N. Abdoulmoumine, Energy Fuels, 2017, 31, 3525–3536 CrossRef CAS.
- A. K. Chandel, B. C. M. Gonçalves, J. L. Strap and S. S. da Silva, Crit. Rev. Biotechnol., 2015, 35, 281–293 CrossRef PubMed.
- A. K. Chandel, V. K. Garlapati, A. K. Singh, F. A. F. Antunes and S. S. da Silva, Bioresour. Technol., 2018, 264, 370–381 CrossRef CAS PubMed.
- G. Sahu, N. K. Gupta, A. Kotha, S. Saha, S. Datta, P. Chavan, N. Kumari and P. Dutta, ChemBioEng Rev., 2018, 5, 231–252 CrossRef CAS.
- G. Pathak, D. Das, K. Rajkumari and L. Rokhum, Green Chem., 2018, 20, 2365–2373 RSC.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- W. Schutyser, S. den Bosch, T. Renders, T. De Boe, S.-F. Koelewijn, A. Dewaele, T. Ennaert, O. Verkinderen, B. Goderis, C. M. Courtin and B. F. Sels, Green Chem., 2015, 17, 5035–5045 RSC.
- T. Renders, W. Schutyser, S. den Bosch, S.-F. Koelewijn, T. Vangeel, C. M. Courtin and B. F. Sels, ACS Catal., 2016, 6, 2055–2066 CrossRef CAS.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- P. Sudarsanam, R. Zhong, S. Van den Bosch, S. M. Coman, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2018, 47, 8349–8402 RSC.
- R. De Clercq, M. Dusselier and B. F. Sels, Green Chem., 2017, 19, 5012–5040 RSC.
- D. Wang and D. Astruc, Chem. Soc. Rev., 2017, 46, 816–854 RSC.
- S. Mintova, M. Jaber and V. Valtchev, Chem. Soc. Rev., 2015, 44, 7207–7233 RSC.
- J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, ChemSusChem, 2009, 2, 18–45 CrossRef CAS PubMed.
- Y. Wang, S. De and N. Yan, Chem. Commun., 2016, 52, 6210–6224 RSC.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- L. D. Rampino and F. F. Nord, J. Am. Chem. Soc., 1941, 63, 2745–3268 CrossRef CAS.
- K. E. Kavanagh and F. F. Nord, J. Am. Chem. Soc., 1943, 65, 2121–2125 CrossRef CAS.
- G. Parravano, J. Catal., 1970, 18, 320–328 CrossRef CAS.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301–309 CrossRef CAS.
- J. Liang, Z. Liang, R. Zou and Y. Zhao, Adv. Mater., 2017, 29, 1701139 CrossRef PubMed.
- D. P. Serrano, J. M. Escola and P. Pizarro, Chem. Soc. Rev., 2013, 42, 4004–4035 RSC.
- D. Verboekend, N. Nuttens, R. Locus, J. Van Aelst, P. Verolme, J. C. Groen, J. Perez-Ramirez and B. F. Sels, Chem. Soc. Rev., 2016, 45, 3331–3352 RSC.
- C. T. Kresge and W. J. Roth, Chem. Soc. Rev., 2013, 42, 3663–3670 RSC.
- L.-B. Sun, X.-Q. Liu and H.-C. Zhou, Chem. Soc. Rev., 2015, 44, 5092–5147 RSC.
- L. Wang and F.-S. Xiao, Green Chem., 2015, 17, 24–39 RSC.
- X.-Y. Yang, A. Leonard, A. Lemaire, G. Tian and B.-L. Su, Chem. Commun., 2011, 47, 2763–2786 RSC.
- M. H. Amin, P. Sudarsanam, M. R. Field, J. Patel and S. K. Bhargava, Langmuir, 2017, 33, 10632–10644 CrossRef CAS PubMed.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- C. Gerardin, J. Reboul, M. Bonne and B. Lebeau, Chem. Soc. Rev., 2013, 42, 4217–4255 RSC.
- S. Abate, K. Barbera, G. Centi, P. Lanzafame and S. Perathoner, Catal. Sci. Technol., 2016, 6, 2485–2501 RSC.
- M. Xu, C. Mukarakate, D. J. Robichaud, M. R. Nimlos, R. M. Richards and B. G. Trewyn, Top. Catal., 2016, 59, 73–85 CrossRef CAS.
- C. Perego, A. Bosetti, M. Ricci and R. Millini, Energy Fuels, 2017, 31, 7721–7733 CrossRef CAS.
- G. Busca, Microporous Mesoporous Mater., 2017, 254, 3–16 CrossRef CAS.
- P. Ferrini, J. Dijkmans, R. De Clercq, S. Van de Vyver, M. Dusselier, P. A. Jacobs and B. F. Sels, Coord. Chem. Rev., 2017, 343, 220–255 CrossRef CAS.
- H. Hernando, J. Fermoso, C. Ochoa-Hernández, M. Opanasenko, P. Pizarro, J. M. Coronado, J. Čejka and D. P. Serrano, Catal. Today, 2018, 304, 30–38 CrossRef CAS.
- X. Querol, N. Moreno, J. C. Umaña, A. Alastuey, E. Hernández, A. López-Soler and F. Plana, Int. J. Coal Geol., 2002, 50, 413–423 CrossRef CAS.
- S. De, S. Dutta and B. Saha, Catal. Sci. Technol., 2016, 6, 7364–7385 RSC.
- H. Y. Luo, J. D. Lewis and Y. Román-Leshkov, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 663–692 CrossRef CAS PubMed.
- J. Jae, G. A. Tompsett, A. J. Foster, K. D. Hammond, S. M. Auerbach, R. F. Lobo and G. W. Huber, J. Catal., 2011, 279, 257–268 CrossRef CAS.
- Y. Wei, T. E. Parmentier, K. P. de Jong and J. Zecevic, Chem. Soc. Rev., 2015, 44, 7234–7261 RSC.
- B. Smit and T. L. M. Maesen, Nature, 2008, 451, 671–678 CrossRef CAS PubMed.
- H. Li, S. Yang, A. Riisager, A. Pandey, R. S. Sangwan, S. Saravanamurugan and R. Luque, Green Chem., 2016, 18, 5701–5735 RSC.
- T. C. Keller, E. G. Rodrigues and J. Pérez-Ramírez, ChemSusChem, 2014, 7, 1729–1738 CrossRef CAS PubMed.
- W. Zhang, J. Xie, W. Hou, Y. Liu, Y. Zhou and J. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 23122–23132 CrossRef CAS PubMed.
- A. Herbst and C. Janiak, CrystEngComm, 2017, 19, 4092–4117 RSC.
- B. Gil, W. J. Roth, J. Grzybek, A. Korzeniowska, Z. Olejniczak, M. Eliáš, M. Opanasenko and J. Čejka, Catal. Today, 2018, 304, 22–29 CrossRef CAS.
- K. Murugappan, C. Mukarakate, S. Budhi, M. Shetty, M. R. Nimlos and Y. Roman-Leshkov, Green Chem., 2016, 18, 5548–5557 RSC.
- V. Paasikallio, K. Kalogiannis, A. Lappas, J. Lehto and J. Lehtonen, Energy Technol., 2017, 5, 94–103 CrossRef CAS.
- P. K. W. Likun, H. Zhang, T. Vitidsant, P. Reubroycharoen and R. Xiao, Energy Fuels, 2017, 31, 6120–6131 CrossRef CAS.
- H. Zhang, P. K. W. Likun and R. Xiao, Sci. Total Environ, 2018, 618, 151–156 CrossRef CAS PubMed.
- P. Sirous Rezaei, H. Shafaghat and W. M. A. W. Daud, RSC Adv., 2015, 5, 65408–65414 RSC.
- B. M. E. Chagas, C. Dorado, M. J. Serapiglia, C. A. Mullen, A. A. Boateng, M. A. F. Melo and C. H. Ataíde, Fuel, 2016, 179, 124–134 CrossRef CAS.
- Y.-K. Park, M. L. Yoo, S. H. Jin and S. H. Park, Renewable Energy, 2015, 79, 20–27 CrossRef CAS.
- Z. Wang, R. Ma, W. Lin and W. Song, Int. J. Green Energy, 2016, 13, 853–858 CrossRef.
- Y. Zhang, P. Chen and H. Lou, J. Energy Chem., 2016, 25, 427–433 CrossRef.
- C. A. Mullen and A. A. Boateng, ACS Sustainable Chem. Eng., 2015, 3, 1623–1631 CrossRef CAS.
- E. L. Schultz, C. A. Mullen and A. A. Boateng, Energy Technol., 2017, 5, 196–204 CrossRef CAS.
- L. Mao, Y. Li and Z. C. Zhang, Front. Chem. Sci. Eng., 2018, 12, 50–58 CrossRef CAS.
- K. Soongprasit, V. Sricharoenchaikul and D. Atong, J. Anal. Appl. Pyrolysis, 2017, 124, 696–703 CrossRef CAS.
- G. Kabir, A. T. M. Din and B. H. Hameed, Bioresour. Technol., 2018, 249, 42–48 CrossRef CAS PubMed.
- Z. Ma, A. Ghosh, N. Asthana and J. van Bokhoven, ChemCatChem, 2017, 9, 954–961 CrossRef CAS.
- J. R. Christianson, S. Caratzoulas and D. G. Vlachos, ACS Catal., 2015, 5, 5256–5263 CrossRef CAS.
- G. Zhang, P. Feng, W. Zhang, H. Liu, C. Wang, H. Ma, D. Wang and Z. Tian, Microporous Mesoporous Mater., 2017, 247, 158–165 CrossRef CAS.
- M. Moreno-Recio, J. Santamaría-González and P. Maireles-Torres, Chem. Eng. J., 2016, 303, 22–30 CrossRef CAS.
- R. Otomo, T. Yokoi and T. Tatsumi, ChemCatChem, 2015, 7, 4180–4187 CAS.
- L. Li, J. Ding, J.-G. Jiang, Z. Zhu and P. Wu, Chin. J. Catal., 2015, 36, 820–828 CrossRef CAS.
- T. D. Swift, H. Nguyen, Z. Erdman, J. S. Kruger, V. Nikolakis and D. G. Vlachos, J. Catal., 2016, 333, 149–161 CrossRef CAS.
- T.-Y. Cheng, P.-Y. Chao, Y.-H. Huang, C.-C. Li, H.-Y. Hsu, Y.-S. Chao and T.-C. Tsai, Microporous Mesoporous Mater., 2016, 233, 148–153 CrossRef CAS.
- X. Xiang, J. Cui, G. Ding, H. Zheng, Y. Zhu and Y. Li, ACS Sustainable Chem. Eng., 2016, 4, 4506–4510 CrossRef CAS.
- L. Bing, Z. Zhang and K. Deng, Ind. Eng. Chem. Res., 2012, 51, 15331–15336 CrossRef CAS.
- I. Delidovich, K. Leonhard and R. Palkovits, Energy Environ. Sci., 2014, 7, 2803–2830 RSC.
- H. Li, S. Saravanamurugan, S. Yang and A. Riisager, Green Chem., 2016, 18, 726–734 RSC.
- F. Yang, S. Zhang, Z. C. Zhang, J. Mao, S. Li, J. Yin and J. Zhou, Catal. Sci. Technol., 2015, 5, 4602–4612 RSC.
- J. Cui, J. Tan, T. Deng, X. Cui, Y. Zhu and Y. Li, Green Chem., 2016, 18, 1619–1624 CAS.
- N. K. Gupta, A. Fukuoka and K. Nakajima, ACS Catal., 2017, 7, 2430–2436 CrossRef CAS.
- X. Zhao, G. Yu, J. Li, Y. Feng, L. Zhang, Y. Peng, Y. Tang and L. Wang, ACS Sustainable Chem. Eng., 2018, 6, 4105–4114 CrossRef CAS.
- H. P. Winoto, B. S. Ahn and J. Jae, J. Ind. Eng. Chem., 2016, 40, 62–71 CrossRef CAS.
- E. Y. C. Jorge, T. de, M. Lima, C. G. S. Lima, L. Marchini, W. N. Castelblanco, D. G. Rivera, E. A. Urquieta-Gonzalez, R. S. Varma and M. W. Paixao, Green Chem., 2017, 19, 3856–3868 RSC.
- Q. Yao, L. Xu, Z. Han and Y. Zhang, Chem. Eng. J., 2015, 280, 74–81 CrossRef CAS.
- M. A. Mellmer, J. M. R. Gallo, D. M. Alonso and J. A. Dumesic, ACS Catal., 2015, 5, 3354–3359 CAS.
- E. Mahmoud, J. Yu, R. J. Gorte and R. F. Lobo, ACS Catal., 2015, 5, 6946–6955 CrossRef CAS.
- M. M. Antunes, S. Lima, P. Neves, A. L. Magalhães, E. Fazio, F. Neri, M. T. Pereira, A. F. Silva, C. M. Silva, S. M. Rocha, M. Pillinger, A. Urakawa and A. A. Valente, Appl. Catal., B, 2016, 182, 485–503 CrossRef CAS.
- P. Charoenwiangnuea, T. Maihom, P. Kongpracha, J. Sirijaraensre and J. Limtrakul, RSC Adv., 2016, 6, 105888–105894 CAS.
- Y. Wang, P. Zhang, G. Huang, Q. Yuan, Y. Guan and P. Wu, ACS Sustainable Chem. Eng., 2017, 5, 6645–6653 CrossRef CAS.
- M. E. Sad, L. F. G. Peña, C. L. Padró and C. R. Apesteguía, Catal. Today, 2018, 302, 203–209 CrossRef CAS.
- P. M. Veiga, A. C. L. Gomes, C. O. Veloso and C. A. Henriques, Appl. Catal., A, 2017, 548, 2–15 CrossRef CAS.
- G. M. Lari, P. Y. Dapsens, D. Scholz, S. Mitchell, C. Mondelli and J. Perez-Ramirez, Green Chem., 2016, 18, 1249–1260 RSC.
- J. Dijkmans, M. Dusselier, D. Gabriëls, K. Houthoofd, P. C. M. M. Magusin, S. Huang, Y. Pontikes, M. Trekels, A. Vantomme, L. Giebeler, S. Oswald and B. F. Sels, ACS Catal., 2015, 5, 928–940 CrossRef CAS.
- D. Sun, Y. Yamada, S. Sato and W. Ueda, Green Chem., 2017, 19, 3186–3213 RSC.
- A. S. de Oliveira, S. J. S. Vasconcelos, J. R. de Sousa, F. F. de Sousa, J. M. Filho and A. C. Oliveira, Chem. Eng. J., 2011, 168, 765–774 CrossRef CAS.
- L. Huang, F. Qin, Z. Huang, Y. Zhuang, J. Ma, H. Xu and W. Shen, Ind. Eng. Chem. Res., 2016, 55, 7318–7327 CrossRef CAS.
- H. Zhang, Z. Hu, L. Huang, H. Zhang, K. Song, L. Wang, Z. Shi, J. Ma, Y. Zhuang, W. Shen, Y. Zhang, H. Xu and Y. Tang, ACS Catal., 2015, 5, 2548–2558 CrossRef CAS.
- R. Beerthuis, L. Huang, N. R. Shiju, G. Rothenberg, W. Shen and H. Xu, ChemCatChem, 2018, 10, 211–221 CrossRef CAS PubMed.
- Y. P. Wijaya, I. Kristianto, H. Lee and J. Jae, Fuel, 2016, 182, 588–596 CrossRef CAS.
- C.-C. Chang, H. Je Cho, J. Yu, R. J. Gorte, J. Gulbinski, P. Dauenhauer and W. Fan, Green Chem., 2016, 18, 1368–1376 RSC.
- I. F. Teixeira, B. T. W. Lo, P. Kostetskyy, M. Stamatakis, L. Ye, C. C. Tang, G. Mpourmpakis and S. C. E. Tsang, Angew. Chem., Int. Ed., 2016, 55, 13061–13066 CrossRef CAS PubMed.
- P. Ferrini, S. F. Koelewijn, J. Van Aelst, N. Nuttens and B. F. Sels, ChemSusChem, 2017, 10, 2249–2257 CrossRef CAS PubMed.
- Y. Liao, R. Zhong, E. Makshina, M. d’Halluin, Y. van Limbergen, D. Verboekend and B. F. Sels, ACS Catal., 2018, 8, 7861–7878 CrossRef CAS.
- Y. Liao, M. d’Halluin, E. Makshina, D. Verboekend and B. F. Sels, Appl. Catal., B, 2018, 234, 117–129 CrossRef CAS.
- G. Afreen, T. Patra and S. Upadhyayula, Mol. Catal., 2017, 441, 122–133 CrossRef CAS.
- M. Heuchel, F. Reinhardt, N. Merdanoğlu, E. Klemm and Y. Traa, Microporous Mesoporous Mater., 2017, 254, 59–68 CrossRef CAS.
- S. Ai, M. Zheng, Y. Jiang, X. Yang, X. Li, J. Pang, J. Sebastian, W. Li, A. Wang, X. Wang and T. Zhang, AIChE J., 2017, 63, 4032–4042 CrossRef CAS.
- C. P. Nash, A. Ramanathan, D. A. Ruddy, M. Behl, E. Gjersing, M. Griffin, H. Zhu, B. Subramaniam, J. A. Schaidle and J. E. Hensley, Appl. Catal., A, 2016, 510, 110–124 CrossRef CAS.
- S. Xu, H. Sheng, T. Ye, D. Hu and S. Liao, Catal. Commun., 2016, 78, 75–79 CrossRef CAS.
- C. K. Narula, Z. Li, E. M. Casbeer, R. A. Geiger, M. Moses-Debusk, M. Keller, M. V. Buchanan and B. H. Davison, Sci. Rep., 2015, 5, 16039 CrossRef CAS PubMed.
- M. A. Sanhoob, O. Muraza, E. N. Shafei, T. Yokoi and K.-H. Choi, Appl. Catal., B, 2017, 210, 432–443 CAS.
- X. Wang, X. Hu, C. Song, K. W. Lux, M. Namazian and T. Imam, Ind. Eng. Chem. Res., 2017, 56, 12046–12055 CrossRef CAS.
- L. Zhang, T. N. Pham, J. Faria and D. E. Resasco, Appl. Catal., A, 2015, 504, 119–129 CrossRef CAS.
- Z. Song, A. Takahashi, N. Mimura and T. Fujitani, Catal. Lett., 2009, 131, 364–369 CrossRef CAS.
- W. Xia, A. Takahashi, I. Nakamura, H. Shimada and T. Fujitani, J. Mol. Catal. A: Chem., 2010, 328, 114–118 CrossRef CAS.
- C. Duan, X. Zhang, R. Zhou, Y. Hua, J. Chen and L. Zhang, Catal. Lett., 2011, 141, 1821–1827 CrossRef CAS.
- F. Xue, C. Miao, Y. Yue, W. Hua and Z. Gao, Green Chem., 2017, 19, 5582–5590 RSC.
- E. V. Makshina, M. Dusselier, W. Janssens, J. Degreve, P. A. Jacobs and B. F. Sels, Chem. Soc. Rev., 2014, 43, 7917–7953 RSC.
- E. V. Makshina, W. Janssens, B. F. Sels and P. A. Jacobs, Catal. Today, 2012, 198, 338–344 CrossRef CAS.
- V. L. Sushkevich and I. I. Ivanova, ChemSusChem, 2016, 9, 2216–2225 CrossRef CAS PubMed.
- P. S. Rezaei, H. Shafaghat and W. M. A. W. Daud, Green Chem., 2016, 18, 1684–1693 RSC.
- L. Sun, X. Zhang, L. Chen, B. Zhao, S. Yang and X. Xie, J. Anal. Appl. Pyrolysis, 2016, 121, 342–346 CrossRef CAS.
- S. Cheng, L. Wei, X. Zhao, E. Kadis and J. Julson, Energy Technol., 2016, 4, 706–713 CrossRef CAS.
- M. M. Yung, A. K. Starace, C. Mukarakate, A. M. Crow, M. A. Leshnov and K. A. Magrini, Energy Fuels, 2016, 30, 5259–5268 CrossRef CAS.
- S. Ogo, Y. Okuno, H. Sekine, S. Manabe, T. Yabe, A. Onda and Y. Sekine, ChemistrySelect, 2017, 2, 6201–6205 CrossRef CAS.
- N. A. S. Ramli and N. A. S. Amin, Appl. Catal., B, 2015, 163, 487–498 CrossRef CAS.
- N. A. S. Ramli and N. A. S. Amin, Energy Convers. Manage., 2015, 95, 10–19 CrossRef CAS.
- W. Zhang, T. Meng, J. Tang, W. Zhuang, Y. Zhou and J. Wang, ACS Sustainable Chem. Eng., 2017, 5, 10029–10037 CrossRef CAS.
- Q. Zhang, T. Wang, J. Tan, Q. Zhang, Y. Li and L. Ma, Int. J. Green Energy, 2016, 13, 767–773 CrossRef CAS.
- C.-B. Chen, M.-Y. Chen, B. Zada, Y.-J. Ma, L. Yan, Q. Xu, W. Li, Q.-X. Guo and Y. Fu, RSC Adv., 2016, 6, 112477–112485 RSC.
- H. Xia, J. Zhang, X. Yan, S. Xu and L. Yang, J. Fuel Chem. Technol., 2015, 43, 575–580 CrossRef CAS.
- A. S. Nagpure, N. Lucas and S. V. Chilukuri, ACS Sustainable Chem. Eng., 2015, 3, 2909–2916 CrossRef CAS.
- C. Wang, Z. Liu, L. Wang, X. Dong, J. Zhang, G. Wang, S. Han, X. Meng, A. Zheng and F.-S. Xiao, ACS Catal., 2018, 8, 474–481 CrossRef CAS.
- M. Hellinger, H. W. P. Carvalho, S. Baier, D. Wang, W. Kleist and J.-D. Grunwaldt, Appl. Catal., A, 2015, 490, 181–192 CrossRef CAS.
- L. Wang, F. Yuan, L. Liu, N. Mominou, S. Li, C. Li and W. Wang, J. Ind. Eng. Chem., 2015, 21, 494–499 CrossRef CAS.
- X. Xu, E. Jiang, Z. Li and Y. Sun, Fuel, 2018, 221, 440–446 CrossRef CAS.
- P. S. Rezaei, H. Shafaghat and W. M. A. W. Daud, RSC Adv., 2015, 5, 51278–51285 RSC.
- F. Wang, X. Kang, M. Zhou, X. Yang, L. Gao and G. Xiao, Appl. Catal., A, 2017, 539, 80–89 CrossRef CAS.
- S. S. Priya, P. Bhanuchander, V. P. Kumar, S. K. Bhargava and K. V. R. Chary, Ind. Eng. Chem. Res., 2016, 55, 4461–4472 CrossRef CAS.
- T. Hanaoka, T. Miyazawa, K. Shimura and S. Hirata, Chem. Eng. J., 2015, 263, 178–185 CrossRef CAS.
- J. Zhang, L. Wang, L. Zhu, Q. Wu, C. Chen, X. Wang, Y. Ji, X. Meng and F. Xiao, ChemSusChem, 2015, 8, 2867–2871 CrossRef CAS PubMed.
- H. Chen, Y. Dai, X. Jia, H. Yu and Y. Yang, Green Chem., 2016, 18, 3048–3056 RSC.
- A. Wang, D. Austin, A. Karmakar, G. M. Bernard, V. K. Michaelis, M. M. Yung, H. Zeng and H. Song, ACS Catal., 2017, 7, 3681–3692 CrossRef CAS.
- A. Witsuthammakul and T. Sooknoi, Catal. Sci. Technol., 2016, 6, 1737–1745 RSC.
- L. Y. Jia, M. Raad, S. Hamieh, J. Toufaily, T. Hamieh, M. M. Bettahar, G. Mauviel, M. Tarrighi, L. Pinard and A. Dufour, Green Chem., 2017, 19, 5442–5459 RSC.
- T. C. Hoff, D. W. Gardner, R. Thilakaratne, J. Proano-Aviles, R. C. Brown and J.-P. Tessonnier, Appl. Catal., A, 2017, 529, 68–78 CrossRef CAS.
- D. P. Gamliel, H. J. Cho, W. Fan and J. A. Valla, Appl. Catal., A, 2016, 522, 109–119 CrossRef CAS.
- M. Asadieraghi and W. M. A. W. Daud, Energy Convers. Manage., 2015, 101, 151–163 CrossRef CAS.
- S. C. Oh, T. Nguyendo, Y. He, A. Filie, Y. Wu, D. T. Tran, I. C. Lee and D. Liu, Catal. Sci. Technol., 2017, 7, 1153–1166 RSC.
- H. Li, Z. Fang, J. Luo and S. Yang, Appl. Catal., B, 2017, 200, 182–191 CrossRef CAS.
- J. Zhang, L. Wang, G. Wang, F. Chen, J. Zhu, C. Wang, C. Bian, S. Pan and F.-S. Xiao, ACS Sustainable Chem. Eng., 2017, 5, 3123–3131 CrossRef CAS.
- S. Song, L. Di, G. Wu, W. Dai, N. Guan and L. Li, Appl. Catal., B, 2017, 205, 393–403 CrossRef CAS.
- G. M. Lari, B. Puértolas, M. S. Frei, C. Mondelli and J. Pérez-Ramírez, ChemCatChem, 2016, 8, 1507–1514 CrossRef CAS.
- M. M. Antunes, P. Neves, A. Fernandes, S. Lima, A. F. Silva, M. F. Ribeiro, C. M. Silva, M. Pillinger and A. A. Valente, Catal. Sci. Technol., 2016, 6, 7812–7829 RSC.
- J. Xing, L. Song, C. Zhang, M. Zhou, L. Yue and X. Li, Catal. Today, 2015, 258, 90–95 CrossRef CAS.
- L. Wang, J. Zhang, X. Yi, A. Zheng, F. Deng, C. Chen, Y. Ji, F. Liu, X. Meng and F.-S. Xiao, ACS Catal., 2015, 5, 2727–2734 CrossRef CAS.
- Y. Zhang, C. Xia, M. Lin, Q. Duan, B. Zhu, X. Peng, B. Wang, S. Yuan, Y. Liu and X. Shu, Catal. Commun., 2017, 101, 1–4 CrossRef CAS.
- M. Popova, H. Lazarova, Y. Kalvachev, T. Todorova, Á. Szegedi, P. Shestakova, G. Mali, V. D. B. C. Dasireddy and B. Likozar, Catal. Commun., 2017, 100, 10–14 CrossRef CAS.
- Y. Deng, J. Wei, Z. Sun and D. Zhao, Chem. Soc. Rev., 2013, 42, 4054–4070 RSC.
- W. Li, Q. Yue, Y. Deng and D. Zhao, Adv. Mater., 2013, 25, 5129–5152 CrossRef CAS PubMed.
- N. Linares, A. M. Silvestre-Albero, E. Serrano, J. Silvestre-Albero and J. Garcia-Martinez, Chem. Soc. Rev., 2014, 43, 7681–7717 RSC.
- Y. Shi, Y. Wan and D. Zhao, Chem. Soc. Rev., 2011, 40, 3854–3878 RSC.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 CrossRef CAS PubMed.
- X. Yan, S. Komarneni and Z. Yan, J. Colloid Interface Sci., 2013, 390, 217–224 CrossRef CAS PubMed.
- P. Akhter, M. Hussain, G. Saracco and N. Russo, Nanoscale Res. Lett., 2014, 9, 158 CrossRef PubMed.
- S. S. Kim, W. Zhang and T. J. Pinnavaia, Science, 1998, 282, 1302–1305 CrossRef CAS.
- S. Kerkhofs, T. Willhammar, H. Van Den Noortgate, C. E. A. Kirschhock, E. Breynaert, G. Van Tendeloo, S. Bals and J. A. Martens, Chem. Mater., 2015, 27, 5161–5169 CrossRef CAS PubMed.
- R. Ciriminna, A. Fidalgo, V. Pandarus, F. Béland, L. M. Ilharco and M. Pagliaro, Chem. Rev., 2013, 113, 6592–6620 CrossRef CAS PubMed.
- J. Dhainaut, J.-P. Dacquin, A. F. Lee and K. Wilson, Green Chem., 2010, 12, 296–303 RSC.
- Y. Yang, C. Ochoa-Hernández, P. Pizarro, V. A. de la Peña O'shea, J. M. Coronado and D. P. Serrano, Appl. Catal., B, 2016, 197, 206–213 CrossRef CAS.
- S. B. A. Hamid, M. M. Ambursa, P. Sudarsanam, L. H. Voon and S. K. Bhargava, Catal. Commun., 2017, 94, 18–22 CrossRef.
- J. Andas, F. Adam and I. A. Rahman, Appl. Surf. Sci., 2014, 315, 154–162 CrossRef CAS.
- N. Linares, E. Serrano, M. Rico, A. Mariana Balu, E. Losada, R. Luque and J. Garcia-Martinez, Chem. Commun., 2011, 47, 9024–9035 RSC.
- X. Li, Q. Xia, V. C. Nguyen, K. Peng, X. Liu, N. Essayem and Y. Wang, Catal. Sci. Technol., 2016, 6, 7586–7596 RSC.
- N. A. Brunelli, K. Venkatasubbaiah and C. W. Jones, Chem. Mater., 2012, 24, 2433–2442 CrossRef CAS.
- A. Romero, E. Alonso, Á. Sastre and A. Nieto-Márquez, Microporous Mesoporous Mater., 2016, 224, 1–8 CrossRef CAS.
- L. Yang, X. Yang, E. Tian and H. Lin, ChemSusChem, 2015, 9, 36–41 CrossRef PubMed.
- L. Yang, X. Yang, E. Tian, V. Vattipalli, W. Fan and H. Lin, J. Catal., 2016, 333, 207–216 CrossRef CAS.
- L. Zhen, F. Gang, P. Chunyan, L. Wang, C. Ping, L. Hui and Z. Xiaoming, Chin. J. Catal., 2012, 33, 1696–1705 Search PubMed.
- J. Pang, M. Zheng, X. Li, L. Song, R. Sun, J. Sebastian, A. Wang, J. Wang, X. Wang and T. Zhang, ChemistrySelect, 2017, 2, 309–314 CrossRef CAS.
- M. Moreno-Recio, I. Jiménez-Morales, P. L. Arias, J. Santamaría-González and P. Maireles-Torres, ChemistrySelect, 2017, 2, 2444–2451 CrossRef CAS.
- V. B. F. Custodis, S. A. Karakoulia, K. S. Triantafyllidis and J. A. van Bokhoven, ChemSusChem, 2016, 9, 1134–1145 CrossRef CAS PubMed.
- S. Jeenpadiphat, C. Kasiban and D. N. Tungasmita, J. Chem. Technol. Biotechnol., 2016, 91, 2519–2528 CrossRef CAS.
- T. Mochizuki, D. Atong, S.-Y. Chen, M. Toba and Y. Yoshimura, Catal. Commun., 2013, 36, 1–4 CrossRef CAS.
- Y. P. Wijaya, D. J. Suh and J. Jae, Catal. Commun., 2015, 70, 12–16 CrossRef CAS.
- M. Di Serio, R. Turco, P. Pernice, A. Aronne, F. Sannino and E. Santacesaria, Catal. Today, 2012, 192, 112–116 CrossRef CAS.
- H. Teruki, K. Kouji and I. Masakazu, Chem. Lett., 2011, 40, 614–616 CrossRef.
- L. Xu, W. Wei, H. Li and H. Li, ACS Catal., 2014, 4, 251–258 CrossRef CAS.
- Y. Zhang, Y. Chen, Y. Shen, Y. Yan, J. Pan, W. Shi and L. Yu, ChemPlusChem, 2016, 81, 108–118 CrossRef CAS.
- Z. Xiao, Y. Fan, Y. Cheng, Q. Zhang, Q. Ge, R. Sha, J. Ji and J. Mao, Fuel, 2018, 215, 406–416 CrossRef CAS.
- Y. Kuwahara, Y. Magatani and H. Yamashita, Catal. Today, 2015, 258, 262–269 CrossRef CAS.
- P. Demma Carà, R. Ciriminna, N. R. Shiju, G. Rothenberg and M. Pagliaro, ChemSusChem, 2014, 7, 835–840 CrossRef PubMed.
- F. Yang, Y. Ding, J. Tang, S. Zhou, B. Wang and Y. Kong, Mol. Catal., 2017, 435, 144–155 CrossRef.
- W. Wei, Y. Zhao, S. Peng, H. Zhang, Y. Bian, H. Li and H. Li, ACS Appl. Mater. Interfaces, 2014, 6, 20851–20859 CrossRef CAS PubMed.
- J. Chen, F. Lu, J. Zhang, W. Yu, F. Wang, J. Gao and J. Xu, ChemCatChem, 2013, 5, 2822–2826 CrossRef CAS.
- M. Kaur, R. Malhotra and A. Ali, Renewable Energy, 2018, 116, 109–119 CrossRef CAS.
- N. Godard, A. Vivian, L. Fusaro, L. Cannavicci, C. Aprile and D. P. Debecker, ChemCatChem, 2211, 9, 2211–2218 CrossRef.
- Y. Cao, J. Wang, M. Kang and Y. Zhu, J. Mol. Catal. A: Chem., 2014, 381, 46–53 CrossRef CAS.
- Z. Xiao, J. Mao, J. Ji, R. Sha, Y. Fan and C. Xing, J. Fuel Chem. Technol., 2017, 45, 641–650 CrossRef CAS.
- Z. Xiao, Q. Ge, C. Xing, C. Jiang, S. Fang, J. Ji and J. Mao, J. Energy Chem., 2016, 25, 434–444 CrossRef.
- J. Grams, N. Potrzebowska, J. Goscianska, B. Michalkiewicz and A. M. Ruppert, Int. J. Hydrogen Energy, 2016, 41, 8656–8667 CrossRef CAS.
- A. Romero, A. Nieto-Márquez and E. Alonso, Appl. Catal., A, 2017, 529, 49–59 CrossRef CAS.
- C. García-Sancho, I. Sádaba, R. Moreno-Tost, J. Mérida-Robles, J. Santamaría-González, M. López-Granados and P. Maireles-Torres, ChemSusChem, 2013, 6, 635–642 CrossRef PubMed.
- S. Srivastava, P. Mohanty, J. K. Parikh, A. K. Dalai, S. S. Amritphale and A. K. Khare, Chin. J. Catal., 2015, 36, 933–942 CrossRef CAS.
- S. Li, Y. Wang, L. Gao, Y. Wu, X. Yang, P. Sheng and G. Xiao, Microporous Mesoporous Mater., 2018, 262, 154–165 CrossRef CAS.
- C. P. Jiménez-Gómez, J. A. Cecilia, R. Moreno-Tost and P. Maireles-Torres, ChemSusChem, 2017, 10, 1448–1459 CrossRef PubMed.
- Z. Huang, W. Pan, H. Zhou, F. Qin, H. Xu and W. Shen, ChemSusChem, 2013, 6, 1063–1069 CrossRef CAS PubMed.
- C. Tian, X. Zhu, S. Chai, Z. Wu, A. Binder, S. Brown, L. Li, H. Luo, Y. Guo and S. Dai, ChemSusChem, 2014, 7, 1703–1709 CrossRef CAS PubMed.
- Y. Kuwahara, W. Kaburagi, Y. Osada, T. Fujitani and H. Yamashita, Catal. Today, 2017, 281, 418–428 CrossRef CAS.
- R. Sheikh, G. N. Shao, Z. Khan, N. Abbas, H. Kim and Y. Park, Asia-Pac. J. Chem. Eng., 2015, 10, 339–346 CrossRef CAS.
- A. Carrero, A. J. Vizcaíno, J. A. Calles and L. García-Moreno, J. Energy Chem., 2017, 26, 42–48 CrossRef.
- M. Harisekhar, V. P. Kumar, S. S. Priya and K. V. R. Chary, J. Chem. Technol. Biotechnol., 2015, 90, 1906–1917 CrossRef CAS.
- V. Shanmugam, R. Zapf, S. Neuberg, V. Hessel and G. Kolb, Appl. Catal., B, 2017, 203, 859–869 CrossRef CAS.
- H.-J. Chae, T.-W. Kim, Y.-K. Moon, H.-K. Kim, K.-E. Jeong, C.-U. Kim and S.-Y. Jeong, Appl. Catal., B, 2014, 150–151, 596–604 CrossRef CAS.
- J. L. Cheong, Y. Shao, S. J. R. Tan, X. Li, Y. Zhang and S. S. Lee, ACS Sustainable Chem. Eng., 2016, 4, 4887–4894 CrossRef CAS.
- W. Janssens, E. V. Makshina, P. Vanelderen, F. De Clippel, K. Houthoofd, S. Kerkhofs, J. A. Martens, P. A. Jacobs and B. F. Sels, ChemSusChem, 2015, 8, 994–1008 CrossRef CAS PubMed.
- G. V. Mamontov, M. V. Grabchenko, V. I. Sobolev, V. I. Zaikovskii and O. V. Vodyankina, Appl. Catal., A, 2016, 528, 161–167 CrossRef CAS.
- F. G. Baddour, C. P. Nash, J. A. Schaidle and D. A. Ruddy, Angew. Chem., Int. Ed., 2016, 55, 9026–9029 CrossRef CAS PubMed.
- D. Duraczyńska, A. Michalik-Zym, B. D. Napruszewska, R. Dula, R. P. Socha, L. Lityńska-Dobrzyńska, A. Gaweł, K. Bahranowski and E. M. Serwicka, ChemistrySelect, 2016, 1, 2148–2155 CrossRef.
- R. N. Olcese, G. Lardier, M. Bettahar, J. Ghanbaja, S. Fontana, V. Carré, F. Aubriet, D. Petitjean and A. Dufour, ChemSusChem, 2013, 6, 1490–1499 CrossRef CAS PubMed.
- Y. Yang, G. Lv, L. Deng, B. Lu, J. Li, J. Zhang, J. Shi and S. Du, Microporous Mesoporous Mater., 2017, 250, 47–54 CrossRef CAS.
- T. M. Sankaranarayanan, A. Berenguer, C. Ochoa-Hernández, I. Moreno, P. Jana, J. M. Coronado, D. P. Serrano and P. Pizarro, Catal. Today, 2015, 243, 163–172 CrossRef CAS.
- N. T. T. Tran, Y. Uemura, S. Chowdhury and A. Ramli, Appl. Catal., A, 2016, 512, 93–100 CrossRef CAS.
- M. M. Ambursa, P. Sudarsanam, L. H. Voon, S. B. A. Hamid and S. K. Bhargava, Fuel Process. Technol., 2017, 162, 87–97 CrossRef CAS.
- N. T. T. Tran, Y. Uemura and A. Ramli, Procedia Eng., 2016, 148, 1252–1258 CrossRef CAS.
- X. Zhang, W. Tang, Q. Zhang, T. Wang and L. Ma, Appl. Energy, 2018, 227, 73–79 CrossRef CAS.
- H. Huang, C. A. Denard, R. Alamillo, A. J. Crisci, Y. Miao, J. A. Dumesic, S. L. Scott and H. Zhao, ACS Catal., 2014, 4, 2165–2168 CrossRef CAS.
- Y. Huang, P.-Y. Chao, T.-Y. Cheng, Y. Ho, C.-T. Lin, H.-Y. Hsu, J.-J. Wong and T.-C. Tsai, Chem. Eng. J., 2016, 283, 778–788 CrossRef CAS.
- A. Najafi Chermahini, H. Hafizi, N. Andisheh, M. Saraji and A. Shahvar, Res. Chem. Intermed., 2017, 43, 5507–5521 CrossRef CAS.
- P. A. Russo, M. M. Antunes, P. Neves, P. V. Wiper, E. Fazio, F. Neri, F. Barreca, L. Mafra, M. Pillinger, N. Pinna and A. A. Valente, Green Chem., 2014, 16, 4292–4305 RSC.
- J. C. Manayil, V. C. dos Santos, F. C. Jentoft, M. Granollers Mesa, A. F. Lee and K. Wilson, ChemCatChem, 2017, 9, 2231–2238 CrossRef CAS.
- L. Sherry and J. A. Sullivan, Catal. Today, 2011, 175, 471–476 CrossRef CAS.
- D. N. Durgasri, T. Vinodkumar, P. Sudarsanam and B. M. Reddy, Catal. Lett., 2014, 144, 971–979 CrossRef CAS.
- P. Sudarsanam, K. Kuntaiah and B. M. Reddy, New J. Chem., 2014, 38, 5991–6001 RSC.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- R. C. R. Santos, D. M. V. Braga, A. N. Pinheiro, E. R. Leite, V. N. Freire, E. Longhinotti and A. Valentini, Catal. Sci. Technol., 2016, 6, 4986–5002 RSC.
- I. E. Wachs and K. Routray, ACS Catal., 2012, 2, 1235–1246 CrossRef CAS.
- F. Yang, D. Deng, X. Pan, Q. Fu and X. Bao, Natl. Sci. Rev., 2015, 2, 183–201 CrossRef CAS.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- M. Okumura, T. Fujitani, J. Huang and T. Ishida, ACS Catal., 2015, 5, 4699–4707 CrossRef CAS.
- P. Sudarsanam, B. Mallesham, P. S. Reddy, D. Großmann, W. Grünert and B. M. Reddy, Appl. Catal., B, 2014, 144, 900–908 CrossRef CAS.
- P. Sudarsanam, B. Mallesham, A. Rangaswamy, B. G. Rao, S. K. Bhargava and B. M. Reddy, J. Mol. Catal. A: Chem., 2016, 412, 47–55 CrossRef CAS.
- P. Sudarsanam, P. R. Selvakannan, S. K. Soni, S. K. Bhargava and B. M. Reddy, RSC Adv., 2014, 4, 43460–43469 RSC.
- P. Sudarsanam, B. Mallesham, D. N. Durgasri and B. M. Reddy, J. Ind. Eng. Chem., 2014, 20, 3115–3121 CrossRef CAS.
- C. Marsden, E. Taarning, D. Hansen, L. Johansen, S. K. Klitgaard, K. Egeblad and C. H. Christensen, Green Chem., 2008, 10, 168–170 RSC.
- Y. Li and W. Shen, Chem. Soc. Rev., 2014, 43, 1543–1574 RSC.
- D. Lei, K. Yu, M.-R. Li, Y. Wang, Q. Wang, T. Liu, P. Liu, L.-L. Lou, G. Wang and S. Liu, ACS Catal., 2017, 7, 421–432 CrossRef CAS.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099–8139 RSC.
- P. Deka, B. J. Borah, H. Saikia and P. Bharali, Chem. Rec., 2018, 18, 1–13 CrossRef.
- A. Holewinski, H. Xin, E. Nikolla and S. Linic, Curr. Opin. Chem. Eng., 2013, 2, 312–319 CrossRef.
- S. Navalón and H. García, Nanomaterials, 2016, 6, 123 CrossRef PubMed.
- Z. Zhang and G. W. Huber, Chem. Soc. Rev., 2018, 47, 1351–1390 RSC.
- L. Yang, J. Su, X. Yang and H. Lin, in Reaction Pathways and Mechanisms in Thermocatalytic Biomass Conversion I., ed. M. Schlaf and Z. C. Zhang, Springer Singapore, 2016, pp. 171–202 Search PubMed.
- B. Hammer and J. K. Nørskov, Impact of Surface Science on Catalysis, Academic Press, 2000, vol. 45, pp. 71–129 Search PubMed.
- J. K. Nørskov, F. Studt, F. Abild-Pedersen and T. Bligaard, Fundamental Concepts in Heterogeneous Catalysis. J, Wiley Online Library, 2014 Search PubMed.
- K. Sun, M. Kohyama, S. Tanaka and S. Takeda, J. Phys. Chem. A, 2012, 116, 9568–9573 CrossRef CAS PubMed.
- Y. Xu and M. Mavrikakis, J. Phys. Chem. B, 2003, 107, 9298–9307 CrossRef CAS.
- R. K. Rai, D. Tyagi, K. Gupta and S. K. Singh, Catal. Sci. Technol., 2016, 6, 3341–3361 RSC.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- M. B. Gawande, R. K. Pandey and R. V. Jayaram, Catal. Sci. Technol., 2012, 2, 1113–1125 RSC.
- A. S. Burange and M. B. Gawande, Encyclopedia of Inorganic and Bioinorganic Chemistry, 2016, pp. 1–19 Search PubMed.
- J. C. Védrine, Catalysts, 2017, 7, 341 CrossRef.
- B. Govinda Rao, P. Sudarsanam, A. Rangaswamy and B. M. Reddy, Catal. Lett., 2015, 145, 1436–1445 CrossRef CAS.
- L. Katta, P. Sudarsanam, B. Mallesham and B. M. Reddy, Catal. Sci. Technol., 2012, 2, 995–1004 RSC.
- P. Sudarsanam, B. Hillary, M. H. Amin, N. Rockstroh, U. Bentrup, A. Brückner and S. K. Bhargava, Langmuir, 2018, 34, 2663–2673 CrossRef CAS PubMed.
- J. Wang, X. Zhang, Q. Sun, S. Chan and H. Su, Catal. Commun., 2015, 61, 57–61 CrossRef CAS.
- M. Watanabe, Y. Aizawa, T. Iida, R. Nishimura and H. Inomata, Appl. Catal., A, 2005, 295, 150–156 CrossRef CAS.
- G. Pacchioni, Catal. Lett., 2015, 145, 80–94 CrossRef CAS.
- X. Xiao, H. Bergstrom, R. Saenger, B. Johnson, R. Sun and A. Peterson, Catal. Sci. Technol., 2018, 8, 1819–1827 RSC.
- S. Tosoni, H.-Y. T. Chen, A. Ruiz Puigdollers and G. Pacchioni, Philos. Trans. R. Soc., A, 2018, 376, 20170056 CrossRef PubMed.
- T. Prasomsri, T. Nimmanwudipong and Y. Roman-Leshkov, Energy Environ. Sci., 2013, 6, 1732–1738 RSC.
- D. Chen, Q. Ma, L. Wei, N. Li, Q. Shen, W. Tian, J. Zhou and J. Long, J. Anal. Appl. Pyrolysis, 2018, 130, 169–180 CrossRef CAS.
- S. M. Schimming, O. D. LaMont, M. König, A. K. Rogers, A. D. D’Amico, M. M. Yung and C. Sievers, ChemSusChem, 2015, 8, 2073–2083 CrossRef CAS PubMed.
- K. A. Rogers and Y. Zheng, ChemSusChem, 2016, 9, 1750–1772 CrossRef CAS PubMed.
- C. A. Gaertner, J. C. Serrano-Ruiz, D. J. Braden and J. A. Dumesic, Ind. Eng. Chem. Res., 2010, 49, 6027–6033 CrossRef CAS.
- P. Sudarsanam, L. Katta, G. Thrimurthulu and B. M. Reddy, J. Ind. Eng. Chem., 2013, 19, 1517–1524 CrossRef CAS.
- L. Katta, P. Sudarsanam, G. Thrimurthulu and B. M. Reddy, Appl. Catal., B, 2010, 101, 101–108 CrossRef CAS.
- J.-O. Shim, D.-W. Jeong, W.-J. Jang, K.-W. Jeon, B.-H. Jeon, S. Y. Cho, H.-S. Roh, J.-G. Na, C. H. Ko, Y.-K. Oh and S. S. Han, Renewable Energy, 2014, 65, 36–40 CrossRef CAS.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed.
- H. Xiong, H. N. Pham and A. K. Datye, Green Chem., 2014, 16, 4627–4643 RSC.
- A. G. Chakinala, J. K. Chinthaginjala, K. Seshan, W. P. M. van Swaaij, S. R. A. Kersten and D. W. F. Brilman, Catal. Today, 2012, 195, 83–92 CrossRef CAS.
- M. W. Hahn, J. R. Copeland, A. H. van Pelt and C. Sievers, ChemSusChem, 2013, 6, 2304–2315 CrossRef CAS PubMed.
- B. C. Gates, M. Flytzani-Stephanopoulos, D. A. Dixon and A. Katz, Catal. Sci. Technol., 2017, 7, 4259–4275 RSC.
- A. Fukuoka and P. L. Dhepe, Chem. Rec., 2009, 9, 224–235 CrossRef CAS PubMed.
- K. Tomishige, Y. Nakagawa and M. Tamura, Green Chem., 2017, 19, 2876–2924 RSC.
- H. Saikia, R. Duarah, P. Sudarsanam, S. K. Bhargava and P. Bharali, ChemistrySelect, 2017, 2, 2123–2130 CrossRef CAS.
- S. J. Tauster, Acc. Chem. Res., 1987, 20, 389–394 CrossRef CAS.
- M. Kim and H. Lee, ACS Sustainable Chem. Eng., 2017, 5, 11371–11376 CrossRef CAS.
- X. Wang, G. Zhao, H. Zou, Y. Cao, Y. Zhang, R. Zhang, F. Zhang and M. Xian, Green Chem., 2011, 13, 2690–2695 RSC.
- B. Hillary, P. Sudarsanam, M. H. Amin and S. K. Bhargava, Langmuir, 2017, 33, 1743–1750 CrossRef CAS PubMed.
- S. Putla, M. H. Amin, B. M. Reddy, A. Nafady, K. A. Al Farhan and S. K. Bhargava, ACS Appl. Mater. Interfaces, 2015, 7, 16525–16535 CrossRef CAS PubMed.
- P. Sudarsanam, B. Hillary, B. Mallesham, B. G. Rao, M. H. Amin, A. Nafady, A. M. Alsalme, B. M. Reddy and S. K. Bhargava, Langmuir, 2016, 32, 2208–2215 CrossRef CAS PubMed.
- P. Sudarsanam, B. Hillary, D. K. Deepa, M. H. Amin, B. Mallesham, B. M. Reddy and S. K. Bhargava, Catal. Sci. Technol., 2015, 5, 3496–3500 RSC.
- P. Sudarsanam and B. Reddy, in Nanotechnology Commercialization, ed. T. Tsuzuki, Pan Stanford Publishing, Singapore, 2013, pp. 73–138 Search PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- W. Schutyser, T. Renders, S. den Bosch, S.-F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- S. den Bosch, T. Renders, S. Kennis, S.-F. Koelewijn, G. den Bossche, T. Vangeel, A. Deneyer, D. Depuydt, C. M. Courtin, J. M. Thevelein, W. Schutyser and B. F. Sels, Green Chem., 2017, 19, 3313–3326 RSC.
- P. Ferrini and R. Rinaldi, Angew. Chem., Int. Ed., 2014, 53, 8634–8639 CrossRef CAS PubMed.
- Y.-L. Wang, J.-F. Zheng, S. Yan, Y.-Y. Ye, S.-R. Li, D. Wang and Y.-Q. Liu, Energy Fuels, 2017, 31, 2907–2913 CrossRef CAS.
- A. P. Tathod and P. L. Dhepe, Bioresour. Technol., 2015, 178, 36–44 CrossRef CAS PubMed.
- T. He, Z. Jiang, P. Wu, J. Yi, J. Li and C. Hu, Sci. Rep., 2016, 6, 38623 CrossRef PubMed.
- R. Li, B. Li, X. Kai and T. Yang, Fuel Process. Technol., 2017, 167, 363–370 CrossRef CAS.
- P. Sudarsanam, B. Hillary, M. H. Amin, S. B. A. Hamid and S. K. Bhargava, Appl. Catal., B, 2016, 185, 213–224 CrossRef CAS.
- M. Zheng, J. Pang, R. Sun, A. Wang and T. Zhang, ACS Catal., 2017, 7, 1939–1954 CrossRef CAS.
- Z. Tai, J. Zhang, A. Wang, J. Pang, M. Zheng and T. Zhang, ChemSusChem, 2013, 6, 652–658 CrossRef CAS PubMed.
- M. Lucas, K. Fabičovicová and P. Claus, ChemCatChem, 2018, 10, 612–618 CrossRef CAS.
- C. Liu, C. Zhang, S. Sun, K. Liu, S. Hao, J. Xu, Y. Zhu and Y. Li, ACS Catal., 2015, 5, 4612–4623 CrossRef CAS.
- Z. Tan, L. Shi, Y. Zan, G. Miao, S. Li, L. Kong, S. Li and Y. Sun, Appl. Catal., A, 2018, 560, 28–36 CrossRef CAS.
- X. Jin, J. Shen, W. Yan, M. Zhao, P. S. Thapa, B. Subramaniam and R. V. Chaudhari, ACS Catal., 2015, 5, 6545–6558 CrossRef CAS.
- Y. Jia and H. Liu, Chin. J. Catal., 2015, 36, 1552–1559 CrossRef CAS.
- A. P. Tathod and P. L. Dhepe, Green Chem., 2014, 16, 4944–4954 RSC.
- M. Yadav, D. K. Mishra and J.-S. Hwang, Appl. Catal., A, 2012, 425–426, 110–116 CrossRef CAS.
- L. Yan, Q. Yao and Y. Fu, Green Chem., 2017, 19, 5527–5547 RSC.
- J. Ftouni, A. Muñoz-Murillo, A. Goryachev, J. P. Hofmann, E. J. M. Hensen, L. Lu, C. J. Kiely, P. C. A. Bruijnincx and B. M. Weckhuysen, ACS Catal., 2016, 6, 5462–5472 CrossRef CAS.
- J. Ftouni, H. C. Genuino, A. Muñoz-Murillo, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemSusChem, 2017, 10, 2891–2896 CrossRef CAS PubMed.
- Y. Gao, H. Zhang, A. Han, J. Wang, H. Tan, E. Tok, S. Jaenicke and G. Chuah, ChemistrySelect, 2018, 3, 1343–1351 CrossRef CAS.
- A. S. Piskun, J. Ftouni, Z. Tang, B. M. Weckhuysen, P. C. A. Bruijnincx and H. J. Heeres, Appl. Catal., A, 2018, 549, 197–206 CrossRef CAS.
- S. Cao, J. R. Monnier and J. R. Regalbuto, J. Catal., 2017, 347, 72–78 CrossRef CAS.
- W. Luo, M. Sankar, A. M. Beale, Q. He, C. J. Kiely, P. C. A. Bruijnincx and B. M. Weckhuysen, Nat. Commun., 2015, 6, 6540 CrossRef PubMed.
- M. Al-Naji, A. Yepez, A. M. Balu, A. A. Romero, Z. Chen, N. Wilde, H. Li, K. Shih, R. Gläser and R. Luqueb, J. Mol. Catal. A: Chem., 2016, 417, 145–152 CrossRef CAS.
- A. M. Hengne and C. V. Rode, Green Chem., 2012, 14, 1064–1072 RSC.
- S. C. Patankar and G. D. Yadav, ACS Sustainable Chem. Eng., 2015, 3, 2619–2630 CrossRef CAS.
- M. Varkolu, V. Velpula, D. R. Burri and S. R. R. Kamaraju, New J. Chem., 2016, 40, 3261–3267 RSC.
- M. Varkolu, D. R. Burri, S. R. R. Kamaraju, S. B. Jonnalagadda and W. E. van Zyl, Chem. Eng. Technol., 2017, 40, 719–726 CrossRef CAS.
- S. Lomate, A. Sultana and T. Fujitani, Catal. Lett., 2018, 148, 348–358 CrossRef CAS.
- V. V. Kumar, G. Naresh, M. Sudhakar, C. Anjaneyulu, S. K. Bhargava, J. Tardio, V. K. Reddy, A. H. Padmasri and A. Venugopal, RSC Adv., 2016, 6, 9872–9879 RSC.
- V. V. Kumar, G. Naresh, S. Deepa, P. G. Bhavani, M. Nagaraju, M. Sudhakar, K. V. R. Chary, J. Tardio, S. K. Bhargava and A. Venugopal, Appl. Catal., A, 2017, 531, 169–176 CrossRef CAS.
- K. Gupta, R. K. Rai and S. K. Singh, ChemCatChem, 2018, 10, 2326–2349 CrossRef CAS.
- X. Li, P. Jia and T. Wang, ACS Catal., 2016, 6, 7621–7640 CrossRef CAS.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sadaba and M. Lopez Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC.
- S. M. Rogers, C. R. A. Catlow, C. E. Chan-Thaw, A. Chutia, N. Jian, R. E. Palmer, M. Perdjon, A. Thetford, N. Dimitratos, A. Villa and P. P. Wells, ACS Catal., 2017, 7, 2266–2274 CrossRef CAS.
- R. Albilali, M. Douthwaite, Q. He and S. H. Taylor, Catal. Sci. Technol., 2018, 8, 252–267 RSC.
- J. Wu, G. Gao, J. Li, P. Sun, X. Long and F. Li, Appl. Catal., B, 2017, 203, 227–236 CrossRef CAS.
- G. M. King, S. Iqbal, P. J. Miedziak, G. L. Brett, S. A. Kondrat, B. R. Yeo, X. Liu, J. K. Edwards, D. J. Morgan, D. K. Knight and G. J. Hutchings, ChemCatChem, 2015, 7, 2122–2129 CrossRef CAS.
- J. Guo, G. Xu, Z. Han, Y. Zhang, Y. Fu and Q. Guo, ACS Sustainable Chem. Eng., 2014, 2, 2259–2266 CrossRef CAS.
- M. Manikandan, A. K. Venugopal, A. S. Nagpure, S. Chilukuri and T. Raja, RSC Adv., 2016, 6, 3888–3898 RSC.
- K. Gupta and S. K. Singh, ACS Sustainable Chem. Eng., 2018, 6, 4793–4800 CrossRef CAS.
- V. S. Marakatti, N. Arora, S. Rai, S. C. Sarma and S. C. Peter, ACS Sustainable Chem. Eng., 2018, 6, 7325–7338 CrossRef CAS.
- D. Li, Q. Liu, C. Zhu, H. Wang, C. Cui, C. Wang and L. Ma, J. Energy Chem., 2019, 30, 34–41 CrossRef.
- G. Bottari, A. J. Kumalaputri, K. K. Krawczyk, B. L. Feringa, H. J. Heeres and K. Barta, ChemSusChem, 2015, 8, 1323–1327 CrossRef CAS PubMed.
- X. Kong, Y. Zhu, H. Zheng, Y. Zhu and Z. Fang, ACS Sustainable Chem. Eng., 2017, 5, 11280–11289 CrossRef CAS.
- J. Ohyama, Y. Hayashi, K. Ueda, Y. Yamamoto, S. Arai and A. Satsuma, J. Phys. Chem. C, 2016, 120, 15129–15136 CrossRef CAS.
- N. Siddiqui, A. S. Roy, R. Goyal, R. Khatun, C. Pendem, A. N. Chokkapu, A. Bordoloi and R. Bal, Sustainable Energy Fuels, 2018, 2, 191–198 RSC.
- M.-Y. Chen, C.-B. Chen, B. Zada and Y. Fu, Green Chem., 2016, 18, 3858–3866 RSC.
- B. Seemala, C. M. Cai, C. E. Wyman and P. Christopher, ACS Catal., 2017, 7, 4070–4082 CrossRef CAS.
- H. Zhang, M. Okumura and N. Toshima, J. Phys. Chem. C, 2011, 115, 14883–14891 CrossRef CAS.
- N. Toshima and H. Zhang, Macromol. Symp., 2012, 317–318, 149–159 CrossRef CAS.
- H. Zhang and N. Toshima, J. Colloid Interface Sci., 2013, 394, 166–176 CrossRef CAS PubMed.
- H. Zhang, L. Wang, L. Lu and N. Toshima, Sci. Rep., 2016, 6, 30752 CrossRef CAS PubMed.
- P. J. Miedziak, H. Alshammari, S. A. Kondrat, T. J. Clarke, T. E. Davies, M. Morad, D. J. Morgan, D. J. Willock, D. W. Knight, S. H. Taylor and G. J. Hutchings, Green Chem., 2014, 16, 3132–3141 RSC.
- Y. Cao, X. Liu, S. Iqbal, P. J. Miedziak, J. K. Edwards, R. D. Armstrong, D. J. Morgan, J. Wang and G. J. Hutchings, Catal. Sci. Technol., 2016, 6, 107–117 RSC.
- Y. Cao, S. Iqbal, P. J. Miedziak, D. R. Jones, D. J. Morgan, X. Liu, J. Wang and G. J. Hutchings, J. Chem. Technol. Biotechnol., 2017, 92, 2246–2253 CrossRef CAS.
- C. Megías-Sayago, S. Ivanova, C. López-Cartes, M. A. Centeno and J. A. Odriozola, Catal. Today, 2017, 279, 148–154 CrossRef.
- X. Jin, M. Zhao, M. Vora, J. Shen, C. Zeng, W. Yan, P. S. Thapa, B. Subramaniam and R. V. Chaudhari, Ind. Eng. Chem. Res., 2016, 55, 2932–2945 CrossRef CAS.
- E. Derrien, M. Mounguengui-Diallo, N. Perret, P. Marion, C. Pinel and M. Besson, Ind. Eng. Chem. Res., 2017, 56, 13175–13189 CrossRef CAS.
- R. Wojcieszak, I. M. Cuccovia, M. A. Silva and L. M. Rossi, J. Mol. Catal. A: Chem., 2016, 422, 35–42 CrossRef CAS.
- X. Tong, L. Yu, H. Chen, X. Zhuang, S. Liao and H. Cui, Catal. Commun., 2017, 90, 91–94 CrossRef CAS.
- S. Siankevich, G. Savoglidis, Z. Fei, G. Laurenczy, D. T. L. Alexander, N. Yan and P. J. Dyson, J. Catal., 2014, 315, 67–74 CrossRef CAS.
- N. K. Gupta, S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2011, 13, 824–827 RSC.
- A. Lolli, S. Albonetti, L. Utili, R. Amadori, F. Ospitali, C. Lucarelli and F. Cavani, Appl. Catal., A, 2015, 504, 408–419 CrossRef CAS.
- A. Lolli, R. Amadori, C. Lucarelli, M. G. Cutrufello, E. Rombi, F. Cavani and S. Albonetti, Microporous Mesoporous Mater., 2016, 226, 466–475 CrossRef CAS.
- Z. Miao, T. Wu, J. Li, T. Yi, Y. Zhang and X. Yang, RSC Adv., 2015, 5, 19823–19829 RSC.
- S. Albonetti, A. Lolli, V. Morandi, A. Migliori, C. Lucarelli and F. Cavani, Appl. Catal., B, 2015, 163, 520–530 CrossRef CAS.
- D. K. Mishra, J. K. Cho and Y. J. Kim, J. Ind. Eng. Chem., 2018, 60, 513–519 CrossRef CAS.
- X. Han, C. Li, X. Liu, Q. Xia and Y. Wang, Green Chem., 2017, 19, 996–1004 RSC.
- F. Neaţu, R. S. Marin, M. Florea, N. Petrea, O. D. Pavel and V. I. Pârvulescu, Appl. Catal., B, 2016, 180, 751–757 CrossRef.
- K. Yu, Y. Liu, D. Lei, Y. Jiang, Y. Wang, Y. Feng, L.-L. Lou, S. Liu and W. Zhou, Catal. Sci. Technol., 2018, 8, 2299–2303 RSC.
- Z. Gui, S. Saravanamurugan, W. Cao, L. Schill, L. Chen, Z. Qi and A. Riisager, ChemistrySelect, 2017, 2, 6632–6639 CrossRef CAS.
- A. Jain, S. C. Jonnalagadda, K. V. Ramanujachary and A. Mugweru, Catal. Commun., 2015, 58, 179–182 CrossRef CAS.
- Z. Yang, W. Qi, R. Su and Z. He, ACS Sustainable Chem. Eng., 2017, 5, 4179–4187 CrossRef CAS.
- K. Gupta, R. K. Rai, A. D. Dwivedi and S. K. Singh, ChemCatChem, 2016, 9, 2760–2767 CrossRef.
- J. Zakzeski and B. M. Weckhuysen, ChemSusChem, 2011, 4, 369–378 CrossRef CAS PubMed.
- C. G. Boeriu, F. I. Fiţigău, R. J. A. Gosselink, A. E. Frissen, J. Stoutjesdijk and F. Peter, Ind. Crops Prod., 2014, 62, 481–490 CrossRef CAS.
- X. Huang, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ChemSusChem, 2014, 7, 2276–2288 CrossRef CAS PubMed.
- X. Huang, C. Atay, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ACS Catal., 2015, 5, 7359–7370 CrossRef CAS.
- T. I. Korányi, X. Huang, A. E. Coumans and E. J. M. Hensen, ACS Sustainable Chem. Eng., 2017, 5, 3535–3543 CrossRef PubMed.
- M. Chen, X. Ma, R. Ma, Z. Wen, F. Yan, K. Cui, H. Chen and Y. Li, Ind. Eng. Chem. Res., 2017, 56, 14025–14033 CrossRef CAS.
- J. Zhang, J. Teo, X. Chen, H. Asakura, T. Tanaka, K. Teramura and N. Yan, ACS Catal., 2014, 4, 1574–1583 CrossRef CAS.
- A. Kloekhorst and H. J. Heeres, ACS Sustainable Chem. Eng., 2015, 3, 1905–1914 CrossRef CAS.
- X. Wang and R. Rinaldi, ChemSusChem, 2012, 5, 1455–1466 CrossRef CAS PubMed.
- J. Zhang, Y. Cai, G. Lu and C. Cai, Green Chem., 2016, 18, 6229–6235 RSC.
- J. Kong, B. Li and C. Zhao, RSC Adv., 2016, 6, 71940–71951 RSC.
- W. Schutyser, G. Van den Bossche, A. Raaffels, S. Van den Bosch, S.-F. Koelewijn, T. Renders and B. F. Sels, ACS Sustainable Chem. Eng., 2016, 4, 5336–5346 CrossRef CAS.
- W. Schutyser, S. den Bosch, J. Dijkmans, S. Turner, M. Meledina, G. Van Tendeloo, D. P. Debecker and B. F. Sels, ChemSusChem, 2015, 8, 1805–1818 CrossRef CAS PubMed.
- J. Yi, Y. Luo, T. He, Z. Jiang, J. Li and C. Hu, Catalysts, 2016, 6, 12 CrossRef.
- T. Prasomsri, M. Shetty, K. Murugappan and Y. Roman-Leshkov, Energy Environ. Sci., 2014, 7, 2660–2669 RSC.
- M. Shetty, K. Murugappan, W. H. Green and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2017, 5, 5293–5301 CrossRef CAS.
- X. Zhang, J. Tang, Q. Zhang, Q. Liu, Y. Li, L. Chen, C. Wang and L. Ma, Catal. Today, 2019, 319, 41–47 CrossRef CAS.
- K. Cui, L. Yang, Z. Ma, F. Yan, K. Wu, Y. Sang, H. Chen and Y. Li, Appl. Catal., B, 2017, 219, 592–602 CrossRef CAS.
- V. O. O. Gonçalves, C. Ciotonea, S. Arrii-Clacens, N. Guignard, C. Roudaut, J. Rousseau, J.-M. Clacens, S. Royer and F. Richard, Appl. Catal., B, 2017, 214, 57–66 CrossRef.
- X. Liu, W. Jia, G. Xu, Y. Zhang and Y. Fu, ACS Sustainable Chem. Eng., 2017, 5, 8594–8601 CrossRef CAS.
- J. Mao, J. Zhou, Z. Xia, Z. Wang, Z. Xu, W. Xu, P. Yan, K. Liu, X. Guo and Z. C. Zhang, ACS Catal., 2017, 7, 695–705 CrossRef CAS.
- W. Guan, X. Chen, S. Jin, C. Li, C.-W. Tsang and C. Liang, Ind. Eng. Chem. Res., 2017, 56, 14034–14042 CrossRef CAS.
- M. Zhou, J. Ye, P. Liu, J. Xu and J. Jiang, ACS Sustainable Chem. Eng., 2017, 5, 8824–8835 CrossRef CAS.
- X. Liu, W. An, Y. Wang, C. H. Turner and D. E. Resasco, Catal. Sci. Technol., 2018, 8, 2146–2158 RSC.
- M. Lu, J. Zhu, M. Li, Y. Shan, M. He and C. Song, Energy Fuels, 2016, 30, 6671–6676 CrossRef.
- M. Lu, H. Du, B. Wei, J. Zhu, M. Li, Y. Shan and C. Song, Energy Fuels, 2017, 31, 10858–10865 CrossRef CAS.
- M. Lu, H. Du, B. Wei, J. Zhu, M. Li, Y. Shan, J. Shen and C. Song, Ind. Eng. Chem. Res., 2017, 56, 12070–12079 CrossRef CAS.
- G.-Y. Xu, J.-H. Guo, Y.-C. Qu, Y. Zhang, Y. Fu and Q.-X. Guo, Green Chem., 2016, 18, 5510–5517 RSC.
- B. Han, Z. Bao, T. Liu, H. Zhou, G. Zhuang, X. Zhong, S. Deng and J. Wang, ChemistrySelect, 2017, 2, 9599–9606 CrossRef CAS.
- R. C. Nelson, B. Baek, P. Ruiz, B. Goundie, A. Brooks, M. C. Wheeler, B. G. Frederick, L. C. Grabow and R. N. Austin, ACS Catal., 2015, 5, 6509–6523 CrossRef CAS.
- M. M. Ambursa, T. H. Ali, H. V. Lee, P. Sudarsanam, S. K. Bhargava and S. B. A. Hamid, Fuel, 2016, 180, 767–776 CrossRef CAS.
- F. Su and Y. Guo, Green Chem., 2014, 16, 2934–2957 RSC.
- A. M. Ruhul, M. A. Kalam, H. H. Masjuki, I. M. R. Fattah, S. S. Reham and M. M. Rashed, RSC Adv., 2015, 5, 101023–101044 RSC.
- W. Xie and T. Wang, Fuel Process. Technol., 2013, 109, 150–155 CrossRef CAS.
- S. Furuta, H. Matsuhashi and K. Arata, Catal. Commun., 2004, 5, 721–723 CrossRef CAS.
- D. E. López, J. G. Goodwin, D. A. Bruce and S. Furuta, Appl. Catal., A, 2008, 339, 76–83 CrossRef.
- K. Jacobson, R. Gopinath, L. C. Meher and A. K. Dalai, Appl. Catal., B, 2008, 85, 86–91 CrossRef CAS.
- C. Komintarachat and S. Chuepeng, Ind. Eng. Chem. Res., 2009, 48, 9350–9353 CrossRef CAS.
- A. Guldhe, P. Singh, F. A. Ansari, B. Singh and F. Bux, Fuel, 2017, 187, 180–188 CrossRef CAS.
- M. Veillette, A. Giroir-Fendler, N. Faucheux and M. Heitz, Chem. Eng. J., 2017, 308, 101–109 CrossRef CAS.
- Y.-M. Park, D.-W. Lee, D.-K. Kim, J.-S. Lee and K.-Y. Lee, Catal. Today, 2008, 131, 238–243 CrossRef CAS.
- V. C. dos Santos, K. Wilson, A. F. Lee and S. Nakagaki, Appl. Catal., B, 2015, 162, 75–84 CrossRef CAS.
- Ž. Kesić, I. Lukić, M. Zdujić, Č. Jovalekić, V. Veljković and D. Skala, Fuel Process. Technol., 2016, 143, 162–168 CrossRef.
- M. Kouzu, A. Fujimori, T. Suzuki, K. Koshi and H. Moriyasu, Fuel Process. Technol., 2017, 165, 94–101 CrossRef CAS.
- N. Kaur and A. Ali, RSC Adv., 2015, 5, 13285–13295 RSC.
- G. Moradi, M. Mohadesi, R. Rezaei and R. Moradi, Can. J. Chem. Eng., 2015, 93, 1531–1538 CrossRef CAS.
- R. G. Prado, G. D. Almeida, A. R. de Oliveira, P. M. T. G. de Souza, C. C. Cardoso, V. R.-L. Constantino, F. G. Pinto, J. Tronto and V. M. D. Pasa, Energy Fuels, 2016, 30, 6662–6670 CrossRef.
- N. Kaur and A. Ali, RSC Adv., 2014, 4, 43671–43681 RSC.
- K. Faungnawakij, B. Yoosuk, S. Namuangruk, P. Krasae, N. Viriya-empikul and B. Puttasawat, ChemCatChem, 2012, 4, 209–216 CrossRef CAS.
- B. Katryniok, H. Kimura, E. Skrzynska, J.-S. Girardon, P. Fongarland, M. Capron, R. Ducoulombier, N. Mimura, S. Paul and F. Dumeignil, Green Chem., 2011, 13, 1960–1979 RSC.
- A. Villa, N. Dimitratos, C. E. Chan-Thaw, C. Hammond, L. Prati and G. J. Hutchings, Acc. Chem. Res., 2015, 48, 1403–1412 CrossRef CAS.
- P. S. Reddy, P. Sudarsanam, G. Raju and B. M. Reddy, Catal. Commun., 2010, 11, 1224–1228 CrossRef CAS.
- P. S. Reddy, P. Sudarsanam, B. Mallesham, G. Raju and B. M. Reddy, J. Ind. Eng. Chem., 2011, 17, 377–381 CrossRef CAS.
- B. Mallesham, P. Sudarsanam, G. Raju and B. M. Reddy, Green Chem., 2013, 15, 478–489 RSC.
- P. S. Reddy, P. Sudarsanam, G. Raju and B. M. Reddy, J. Ind. Eng. Chem., 2012, 18, 648–654 CrossRef CAS.
- P. Sudarsanam, B. Mallesham, A. N. Prasad, P. S. Reddy and B. M. Reddy, Fuel Process. Technol., 2013, 106, 539–545 CrossRef CAS.
- B. Mallesham, P. Sudarsanam and B. M. Reddy, Catal. Sci. Technol., 2014, 4, 803–813 RSC.
- B. Mallesham, P. Sudarsanam and B. M. Reddy, Ind. Eng. Chem. Res., 2014, 53, 18775–18785 CrossRef CAS.
- S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17–45 RSC.
- G. Dodekatos and H. Tüysüz, ChemCatChem, 2017, 9, 610–619 CrossRef CAS.
- S. M. Rogers, C. R. A. Catlow, C. E. Chan-Thaw, D. Gianolio, E. K. Gibson, A. L. Gould, N. Jian, A. J. Logsdail, R. E. Palmer, L. Prati, N. Dimitratos, A. Villa and P. P. Wells, ACS Catal., 2015, 5, 4377–4384 CrossRef CAS.
- C. D’Agostino, G. Brett, G. Divitini, C. Ducati, G. J. Hutchings, M. D. Mantle and L. F. Gladden, ACS Catal., 2017, 7, 4235–4241 CrossRef.
- E. Skrzynska, S. Zaid, A. Addad, J.-S. Girardon, M. Capron and F. Dumeignil, Catal. Sci. Technol., 2016, 6, 3182–3196 RSC.
- J. A. Díaz, E. Skrzyńska, S. Zaid, J. Girardon, M. Capron, F. Dumeignil and P. Fongarland, J. Chem. Technol. Biotechnol., 2017, 92, 2267–2275 CrossRef.
- S. Zaid, E. Skrzyńska, A. Addad, S. Nandi, L. Jalowiecki-Duhamel, J.-S. Girardon, M. Capron and F. Dumeignil, Top. Catal., 2017, 60, 1072–1081 CrossRef CAS.
- C. M. Olmos, L. E. Chinchilla, E. G. Rodrigues, J. J. Delgado, A. B. Hungría, G. Blanco, M. F. R. Pereira, J. J. M. Órfão, J. J. Calvino and X. Chen, Appl. Catal., B, 2016, 197, 222–235 CrossRef CAS.
- I. Kaskow, P. Decyk and I. Sobczak, Appl. Surf. Sci., 2018, 444, 197–207 CrossRef CAS.
- D. Roy, B. Subramaniam and R. V. Chaudhari, ACS Catal., 2011, 1, 548–551 CrossRef CAS.
- G.-Y. Yang, Y.-H. Ke, H.-F. Ren, C.-L. Liu, R.-Z. Yang and W.-S. Dong, Chem. Eng. J., 2016, 283, 759–767 CrossRef CAS.
- R. Palacio, S. Torres, D. Lopez and D. Hernandez, Catal. Today, 2018, 302, 196–202 CrossRef CAS.
- M. Dusselier, P. Van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 RSC.
- R. De Clercq, M. Dusselier, E. Makshina and B. F. Sels, Angew. Chem., Int. Ed., 2018, 57, 3074–3078 CrossRef CAS PubMed.
- K. Omata, S. Izumi, T. Murayama and W. Ueda, Catal. Today, 2013, 201, 7–11 CrossRef CAS.
- L.-Z. Tao, B. Yan, Y. Liang and B.-Q. Xu, Green Chem., 2013, 15, 696–705 RSC.
- G. S. Foo, D. Wei, D. S. Sholl and C. Sievers, ACS Catal., 2014, 4, 3180–3192 CrossRef CAS.
- M. Dalil, D. Carnevali, M. Edake, A. Auroux, J.-L. Dubois and G. S. Patience, J. Mol. Catal. A: Chem., 2016, 421, 146–155 CrossRef CAS.
- R. Beerthuis, G. Rothenberg and N. R. Shiju, Green Chem., 2015, 17, 1341–1361 RSC.
- K. Omata, K. Matsumoto, T. Murayama and W. Ueda, Catal. Today, 2016, 259, 205–212 CrossRef CAS.
- F. Wang, J. Xu, J. Dubois and W. Ueda, ChemSusChem, 2010, 3, 1383–1389 CrossRef CAS PubMed.
- C. Chen, N. Kosuke, T. Murayama and W. Ueda, ChemCatChem, 2013, 5, 2869–2873 CrossRef CAS.
- K. Omata, K. Matsumoto, T. Murayama and W. Ueda, Chem. Lett., 2014, 43, 435–437 CrossRef CAS.
- B. Mallesham, P. Sudarsanam, B. V. S. Reddy and B. M. Reddy, Appl. Catal., B, 2016, 181, 47–57 CrossRef CAS.
- J. Wang, X. Zhao, N. Lei, L. Li, L. Zhang, S. Xu, S. Miao, X. Pan, A. Wang and T. Zhang, ChemSusChem, 2016, 9, 784–790 CrossRef CAS PubMed.
- X. Zhao, J. Wang, M. Yang, N. Lei, L. Li, B. Hou, S. Miao, X. Pan, A. Wang and T. Zhang, ChemSusChem, 2017, 10, 819–824 CrossRef CAS PubMed.
- A. J. Pamphile-Adrián, P. P. Florez-Rodriguez, M. H. M. Pires, G. Perez and F. B. Passos, Catal. Today, 2017, 289, 302–308 CrossRef.
- Q. Sun, S. Wang and H. Liu, ACS Catal., 2017, 7, 4265–4275 CrossRef CAS.
- V. Rekha, N. Raju, C. Sumana, S. Paul Douglas and N. Lingaiah, Catal. Lett., 2016, 146, 1487–1496 CrossRef CAS.
- F. Cai, W. Zhu and G. Xiao, Catal. Sci. Technol., 2016, 6, 4889–4900 RSC.
- D. Durán-Martín, M. L. Granados, J. L. G. Fierro, C. Pinel and R. Mariscal, Top. Catal., 2017, 60, 1062–1071 CrossRef.
- S. García-Fernández, I. Gandarias, Y. Tejido-Núñez, J. Requies and P. L. Arias, ChemCatChem, 2017, 9, 4508–4519 CrossRef.
- M. Gu, Z. Shen, L. Yang, B. Peng, W. Dong, W. Zhang and Y. Zhang, Ind. Eng. Chem. Res., 2017, 56, 13572–13581 CrossRef CAS.
- X. Li, B. Zhang, Q. Wu, C. Zhang, Y. Yu, Y. Li, W. Lin, H. Cheng and F. Zhao, J. Catal., 2016, 337, 284–292 CrossRef CAS.
- X. Li, Q. Wu, B. Zhang, C. Zhang, W. Lin, H. Cheng and F. Zhao, Catal. Today, 2018, 302, 210–216 CrossRef CAS.
- I. C. Freitas, R. L. Manfro and M. M. V. M. Souza, Appl. Catal., B, 2018, 220, 31–41 CrossRef CAS.
- V. V. Costa, M. Estrada, Y. Demidova, I. Prosvirin, V. Kriventsov, R. F. Cotta, S. Fuentes, A. Simakov and E. V. Gusevskaya, J. Catal., 2012, 292, 148–156 CrossRef CAS.
- S. Ajaikumar, J. Ahlkvist, W. Larsson, A. Shchukarev, A.-R. Leino, K. Kordas and J.-P. Mikkola, Appl. Catal., A, 2011, 392, 11–18 CrossRef CAS.
- F. Neaţu, G. Culică, M. Florea, V. I. Parvulescu and F. Cavani, ChemSusChem, 2016, 9, 3102–3112 CrossRef PubMed.
- A. Stolle, T. Gallert, C. Schmoger and B. Ondruschka, RSC Adv., 2013, 3, 2112–2153 RSC.
- G. Neri, G. Rizzo, A. S. Aricò, C. Crisafulli, L. De Luca, A. Donato, M. G. Musolino and R. Pietropaolo, Appl. Catal., A, 2007, 325, 15–24 CrossRef CAS.
- C. I. Melo, R. Bogel-Lukasik, M. G. da Silva and E. Bogel-Lukasik, Green Chem., 2011, 13, 2825–2830 RSC.
- T. Ekou, L. Ekou, A. Vicente, G. Lafaye, S. Pronier, C. Especel and P. Marécot, J. Mol. Catal. A: Chem., 2011, 337, 82–88 CrossRef CAS.
- J. Zhou, Y. Yang, C. Li, S. Zhang, Y. Chen, S. Shi and M. Wei, J. Mater. Chem. A, 2016, 4, 12825–12832 RSC.
- L. Yang, Z. Jiang, G. Fan and F. Li, Catal. Sci. Technol., 2014, 4, 1123–1131 RSC.
- W. Li, G. Fan, L. Yang and F. Li, Catal. Sci. Technol., 2016, 6, 2337–2348 RSC.
- Y. S. Demidova, I. L. Simakova, M. Estrada, S. Beloshapkin, E. V. Suslov, D. V. Korchagina, K. P. Volcho, N. F. Salakhutdinov, A. V. Simakov and D. Y. Murzin, Appl. Catal., A, 2013, 464–465, 348–356 CrossRef CAS.
- Y. S. Demidova, E. V. Suslov, I. L. Simakova, E. S. Mozhajcev, D. V. Korchagina, K. P. Volcho, N. F. Salakhutdinov, A. Simakov and D. Y. Murzin, J. Mol. Catal. A: Chem., 2017, 426, 60–67 CrossRef CAS.
- I. L. Simakova, Y. S. Demidova, M. Estrada, S. Beloshapkin, E. V. Suslov, K. P. Volcho, N. F. Salakhutdinov, D. Y. Murzin and A. Simakov, Catal. Today, 2017, 279, 63–70 CrossRef CAS.
- Y. S. Demidova, E. V. Suslov, I. L. Simakova, K. P. Volcho, E. Smolentseva, N. F. Salakhutdinov, A. Simakov and D. Y. Murzin, Mol. Catal., 2017, 433, 414–419 CrossRef CAS.
- J. Sun, K. Zhu, F. Gao, C. Wang, J. Liu, C. H. F. Peden and Y. Wang, J. Am. Chem. Soc., 2011, 133, 11096–11099 CrossRef CAS PubMed.
- C. Liu, J. Sun, C. Smith and Y. Wang, Appl. Catal., A, 2013, 467, 91–97 CrossRef CAS.
- S. Mizuno, M. Kurosawa, M. Tanaka and M. Iwamoto, Chem. Lett., 2012, 41, 892–894 CrossRef CAS.
- M. Iwamoto, S. Mizuno and M. Tanaka, Chem. – Eur. J., 2013, 19, 7214–7220 CrossRef CAS PubMed.
- T. Ennaert, J. Geboers, E. Gobechiya, C. M. Courtin, M. Kurttepeli, K. Houthoofd, C. E. A. Kirschhock, P. C. M. M. Magusin, S. Bals, P. A. Jacobs and B. F. Sels, ACS Catal., 2015, 5, 754–768 CrossRef CAS.
- I. Sádaba, M. López Granados, A. Riisager and E. Taarning, Green Chem., 2015, 17, 4133–4145 RSC.
- P. A. Zapata, J. Faria, M. P. Ruiz, R. E. Jentoft and D. E. Resasco, J. Am. Chem. Soc., 2012, 134, 8570–8578 CrossRef CAS PubMed.
- A. Kruse and N. Dahmen, J. Supercrit. Fluids, 2015, 96, 36–45 CrossRef CAS.
- K. Wu, Y. Wu, Y. Chen, H. Chen, J. Wang and M. Yang, ChemSusChem, 2016, 9, 1355–1385 CrossRef CAS PubMed.
- L. T. Mika, E. Cséfalvay and I. T. Horváth, Catal. Today, 2015, 247, 33–46 CrossRef CAS.
- L. Vilcocq, É. Rebmann, Y. W. Cheah and P. Fongarland, ACS Sustainable Chem. Eng., 2018, 6, 5555–5565 CrossRef CAS.
- L. Negahdar, I. Delidovich and R. Palkovits, Appl. Catal., B, 2016, 184, 285–298 CrossRef CAS.
- V. C. Nguyen, N. Q. Bui, M. Eternot, T. T. H. Vu, P. Fongarland and N. Essayem, Mol. Catal., 2018, 458, 171–179 CrossRef CAS.
- S. E. Davis, B. N. Zope and R. J. Davis, Green Chem., 2012, 14, 143–147 RSC.
- R. K. Pazhavelikkakath Purushothaman, F. van der Klis, A. E. Frissen, J. van Haveren, A. Mayoral, A. van der Bent and D. S. van Es, Green Chem., 2018, 20, 2763–2774 RSC.
- Q. Meng, D. Cao, G. Zhao, C. Qiu, X. Liu, X. Wen, Y. Zhu and Y. Li, Appl. Catal., B, 2017, 212, 15–22 CrossRef CAS.
- F. Liu, K. Huang, A. Zheng, F.-S. Xiao and S. Dai, ACS Catal., 2018, 8, 372–391 CrossRef CAS.
- J. N. Jocz, P. E. Savage and L. T. Thompson, Ind. Eng. Chem. Res., 2018, 57, 8655–8663 CrossRef CAS.
- D. W. Gardner, J. Huo, T. C. Hoff, R. L. Johnson, B. H. Shanks and J.-P. Tessonnier, ACS Catal., 2015, 5, 4418–4422 CrossRef CAS.
- A. C. Alba-Rubio, J. L. G. Fierro, L. León-Reina, R. Mariscal, J. A. Dumesic and M. L. Granados, Appl. Catal., B, 2017, 202, 269–280 CrossRef CAS.
- W. N. P. van der Graaff, C. H. L. Tempelman, F. C. Hendriks, J. Ruiz-Martinez, S. Bals, B. M. Weckhuysen, E. A. Pidko and E. J. M. Hensen, Appl. Catal., A, 2018, 564, 113–122 CrossRef CAS.
- H. N. Pham, A. E. Anderson, R. L. Johnson, K. Schmidt-Rohr and A. K. Datye, Angew. Chem., Int. Ed., 2012, 51, 13163–13167 CrossRef CAS PubMed.
- D. T. Ngo, T. Sooknoi and D. E. Resasco, Appl. Catal., B, 2018, 237, 835–843 CrossRef CAS.
- H. N. Pham, A. E. Anderson, R. L. Johnson, T. J. Schwartz, B. J. O’Neill, P. Duan, K. Schmidt-Rohr, J. A. Dumesic and A. K. Datye, ACS Catal., 2015, 5, 4546–4555 CrossRef CAS.
- M. Karaki, A. Karout, J. Toufaily, F. Rataboul, N. Essayem and B. Lebeau, J. Catal., 2013, 305, 204–216 CrossRef CAS.
- R. F. Perez, S. J. Canhaci, L. E. P. Borges and M. A. Fraga, Catal. Today, 2017, 289, 273–279 CrossRef CAS.
- S. Kang, J. Fu and G. Zhang, Renewable Sustainable Energy Rev., 2018, 94, 340–362 CrossRef CAS.
- H. Yang, C. Ma, X. Zhang, Y. Li, J. Cheng and Z. Hao, ACS Catal., 2018, 8, 1248–1258 CrossRef CAS.
- C. Sievers, Y. Noda, L. Qi, E. M. Albuquerque, R. M. Rioux and S. L. Scott, ACS Catal., 2016, 6, 8286–8307 CrossRef CAS.
- S. Prodinger, M. A. Derewinski, A. Vjunov, S. D. Burton, I. Arslan and J. A. Lercher, J. Am. Chem. Soc., 2016, 138, 4408–4415 CrossRef CAS PubMed.
- J. S. Bates and R. Gounder, J. Catal., 2018, 365, 213–226 CrossRef CAS.
- M. J. Cordon, J. W. Harris, J. C. Vega-Vila, J. S. Bates, S. Kaur, M. Gupta, M. E. Witzke, E. C. Wegener, J. T. Miller, D. W. Flaherty, D. D. Hibbitts and R. Gounder, J. Am. Chem. Soc., 2018, 140, 14244–14266 CrossRef CAS PubMed.
- Z. Jin, L. Wang, Q. Hu, L. Zhang, S. Xu, X. Dong, X. Gao, R. Ma, X. Meng and F.-S. Xiao, ACS Catal., 2018, 8, 5250–5254 CrossRef CAS.
- H.-T. Vu, F. M. Harth and N. Wilde, Front. Chem., 2018, 6, 143 CrossRef PubMed.
- J. Grand, S. N. Talapaneni, A. Vicente, C. Fernandez, E. Dib, H. A. Aleksandrov, G. N. Vayssilov, R. Retoux, P. Boullay, J.-P. Gilson, V. Valtchev and S. Mintova, Nat. Mater., 2017, 16, 1010–1015 CrossRef CAS PubMed.
- Z. Zhu, H. Xu, J. Jiang, H. Wu and P. Wu, Chem. Commun., 2017, 53, 12516–12519 RSC.
- D. Suttipat, W. Wannapakdee, T. Yutthalekha, S. Ittisanronnachai, T. Ungpittagul, K. Phomphrai, S. Bureekaew and C. Wattanakit, ACS Appl. Mater. Interfaces, 2018, 10, 16358–16366 CrossRef CAS PubMed.
- Y. Tang, Y. Chen, Y. Wu, M. Zheng, C. Zhang, M. Yang and G. Cao, Microporous Mesoporous Mater., 2016, 224, 420–425 CrossRef CAS.
- R. A. Mayanovic, H. Yan, A. D. Brandt, Z. Wang, M. Mandal, K. Landskron and W. A. Bassett, Microporous Mesoporous Mater., 2014, 195, 161–166 CrossRef CAS.
- J. Choi, Y. Han, S. Park, J. Park and H. Kim, J. Nanomater., 2014, 2014, 580347 Search PubMed.
- M. N. Gatti, F. Pompeo, G. F. Santori and N. N. Nichio, Catal. Today, 2017, 296, 26–34 CrossRef CAS.
- J. Huo, R. L. Johnson, P. Duan, H. N. Pham, D. Mendivelso-Perez, E. A. Smith, A. K. Datye, K. Schmidt-Rohr and B. H. Shanks, Catal. Sci. Technol., 2018, 8, 1151–1160 RSC.
- J. A. Lopez-Ruiz, A. R. Cooper, G. Li and K. O. Albrecht, ACS Catal., 2017, 7, 6400–6412 CrossRef CAS.
- A. Aronne, M. Di Serio, R. Vitiello, N. J. Clayden, L. Minieri, C. Imparato, A. Piccolo, P. Pernice, P. Carniti and A. Gervasini, J. Phys. Chem. C, 2017, 121, 17378–17389 CrossRef CAS.
- J. Oh, T. W. Kim, K. Jeong, J. H. Park and Y.-W. Suh, ChemCatChem, 2018, 10, 3892–3900 CrossRef CAS.
- H. Xiong, T. J. Schwartz, N. I. Andersen, J. A. Dumesic and A. K. Datye, Angew. Chem., Int. Ed., 2015, 54, 7939–7943 CrossRef CAS PubMed.
- B. J. O’Neill, D. H. K. Jackson, A. J. Crisci, C. A. Farberow, F. Shi, A. C. Alba-Rubio, J. Lu, P. J. Dietrich, X. Gu, C. L. Marshall, P. C. Stair, J. W. Elam, J. T. Miller, F. H. Ribeiro, P. M. Voyles, J. Greeley, M. Mavrikakis, S. L. Scott, T. F. Kuech and J. A. Dumesic, Angew. Chem., Int. Ed., 2013, 52, 13808–13812 CrossRef PubMed.
- T. Van Cleve, D. Underhill, M. Veiga Rodrigues, C. Sievers and J. W. Medlin, Langmuir, 2018, 34, 3619–3625 CrossRef CAS PubMed.
- P. Hao, D. K. Schwartz and J. W. Medlin, ACS Catal., 2018, 11165–11173 CrossRef CAS.
- Y. Rodenas, R. Mariscal, J. L. G. Fierro, D. Martín Alonso, J. A. Dumesic and M. López Granados, Green Chem., 2018, 20, 2845–2856 RSC.
- W. N. P. van der Graaff, C. H. L. Tempelman, G. Li, B. Mezari, N. Kosinov, E. A. Pidko and E. J. M. Hensen, ChemSusChem, 2016, 9, 3145–3149 CrossRef CAS PubMed.
- S. Gillet, M. Aguedo, L. Petitjean, A. R. C. Morais, A. M. da Costa Lopes, R. M. Lukasik and P. T. Anastas, Green Chem., 2017, 19, 4200–4233 RSC.
- C. Liu, H. Wang, A. M. Karim, J. Sun and Y. Wang, Chem. Soc. Rev., 2014, 43, 7594–7623 RSC.
- I. B. Laskar, K. Rajkumari, R. Gupta, S. Chatterjee, B. Paul and L. Rokhum, RSC Adv., 2018, 8, 20131–20142 RSC.
- B. Liu and Z. Zhang, ACS Catal., 2016, 6, 326–338 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |