
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControl of spintronic and electronic properties of bimetallic and vacancy-ordered vanadium carbide MXenes via surface functionalization†
Shuo
Li
 a,
Junjie
He
b,
Petr
Nachtigall
a,
Lukáš
Grajciar
a and
Federico
Brivio
*a
a,
Junjie
He
b,
Petr
Nachtigall
a,
Lukáš
Grajciar
a and
Federico
Brivio
*a
aDepartment of Physical and Macromolecular Chemistry, Faculty of Science, Charles University in Prague, 128 43 Prague 2, Czech Republic. E-mail: briviof@natur.cuni.cz
bBremen Center for Computational Materials Science, University of Bremen, Am Fallturm 1, 28359 Bremen, Germany
First published on 13th November 2019
Abstract
MXenes are 2D transition metal carbides with high potential for overcoming limitations of conventional two-dimensional electronics. In this context, various MXenes have shown magnetic properties suitable for applications in spintronics, yet the number of MXenes reported so far is far smaller than their parental MAX phases. Therefore, we have studied the structural, electronic and magnetic properties of bimetallic and vacancy-ordered MXenes derived from a new (V2/3Zr1/3)2AlC MAX phase to assess whether MXene exfoliation would return stable magnetic materials. In particular, we have investigated the properties of pristine and surface-functionalized (V2/3Zr1/3)2CX2 bimetallic and (V2/3□1/3)2CX2 (where □ denotes the vacancies) vacancy-ordered MXenes (X = O, F and OH). Our density functional theory (DFT) calculations showed that modifying the MXene stoichiometry and/or MXene surface functionalization changes MXene properties. After testing all possible combinations of metallic motifs and functionalization, we identified (V2/3Zr1/3)2CX2, (V2/3□1/3)2CF2 and (V2/3□1/3)2C(OH)2 as stable structures. Among them, (V2/3Zr1/3)2CO2 MXene is predicted to be an FM intrinsic half-semiconductor with a remarkably high Curie temperature (TC) of 270 K. The (V2/3Zr1/3)2C(OH)2 MXene exhibits a rather low work function (WF) (1.37 eV) and is thus a promising candidate for ultra-low work function electron emitters. Conversely, the (V2/3□1/3)2CF2 MXene has a rather high WF and hence can be used as a hole injector for Schottky-barrier-free contact applications. Overall, our proof-of-concept study shows that theoretical predictions of MXene exfoliation and properties support further experimental research towards developing spintronics devices.
1 Introduction
2D materials are currently the subject of intense experimental and theoretical research for their extremely high aspect ratio, which accounts for their remarkable electronic and magnetic properties. Since graphene1 was first isolated, several other ultrathin 2D materials with the same honeycomb lattice have been described, such as boron nitride (h-BN),2 silicene,3 transition metal dichalcogenides,4,5 monolayer black phosphorus6 and MXenes,7 among others.8 MXenes are particularly appealing as candidates for spintronics, i.e., the manipulation of electronic spin for logic devices.9 However, the magnetic response required for spintronics applications is relatively rare in 2D materials.10 While magnetism can be introduced in various ways, e.g., via defects or dopants engineering or with external electric fields, our ability to control operational parameters for practical devices remains limited.11 For example, although phases with high spin-polarization can be achieved in transition metal dichalcogenides, controlling the distribution of dopants and defects is highly difficult.12–14 Accordingly, the design of new intrinsic 2D magnetic materials (which would not require such complicated engineering) would greatly improve their potential for spintronics applications. Several such new classes of compounds have emerged in recent years,15,16 including: half-metals,14,17,18 spin gapless semiconductors,19,20 bipolar magnetic semiconductors21 and half-semiconductors.22–24 Among them, half-semiconductors and half-metals have shown the most promising results regarding spin generation, injection, storage and detection,25,26 thus paving the way forward towards a new type of computer components.MXenes are 2D transition metal carbides (or nitrides) with the general formula M2C (where M is a transition metal) derived from the corresponding MAX phase structures by exfoliation upon chemical etching in aqueous hydrofluoric acid at room temperature.27,28 As a result of their synthesis, MXene surfaces are typically functionalized with groups such as O, F or OH.7 Considering their properties, MXenes have been proposed as materials suitable for applications such as transparent conductive films, electromagnetic interference absorption and shielding devices, electrocatalysts, lithium-ion batteries cathodes and supercapacitors.29–31 In particular, many MXenes have shown potential for spintronics since they are often half semiconductors or half metals.14,17,18 In the first case, the conduction band minimum (CBM) and the valence band maximum (VBM) are both fully polarized and both spin channels show a band gap. Conversely, only one channel shows a band gap, while the other is metallic in half metals. Several MXenes have also been predicted to be either half-semiconductors or half-metals, albeit with limited experimental evidence.14,17,18,32 Moreover, the electronic structures of MXenes can be fine-tuned by surface modification.33,34 Therefore, MXenes are candidates for other applications,35,36 such as emitter cathodes in light emitting diodes and field effect transistors.37,38
The development of MAX phases with bimetallic composition, in particular (Mo2/3Sc1/3)2AlC39–41 and (V2/3Zr1/3)2AlC,42 has recently opened new MXene research avenues. These bimetallic MXenes (often denoted metal-doped) with functionalized surfaces can be exfoliated by precisely controlling the thermodynamic conditions.39,40 In addition, a vacancy-ordered MXene (Mo2/3□1/3)2C (where □ denotes the missing transition metal atom) has been reported. Using first-principles calculations, H. Lind et al.43 have computationally shown how introducing different surface functional groups can be used to tune the band gap of (Mo2/3□1/3)2C.
In this context, we computationally investigated the MAX phase (V2/3Zr1/3)2AlC and the properties of the corresponding bimetallic (V2/3Zr1/3)2C MXene and related vacancy-ordered (V2/3□1/3)2C MXene. Our aim is to determine whether suitable surface functionalization affects the electronic and magnetic properties of both (V2/3Zr1/3)2CX2 and (V2/3□1/3)2CX2 (X = O, F and OH) MXenes based on first-principles calculations.
2 Methods
Density functional theory (DFT) was used to identify structures, electronic and magnetic properties of MXenes along with ab initio molecular dynamics (AIMD) and Monte Carlo methods for calculating the kinetic stability and Curie temperature of pristine and functionalized MXenes. DFT calculations were performed using the Vienna ab initio simulation package (VASP)44,45 based on the PAW method. The wave-function was converged to a threshold of 10−5 eV with a plane-wave energy cut-off set to 500 eV and a k-point mesh of 15 × 15 × 1 following the Monkhorst–Pack method for 2D structures. All the structures were optimized to converge the interatomic forces below a threshold of 0.01 eV Å−1 using the generalized gradient approximation (GGA) PBE46 exchange–correlation functional; the dispersion forces have been described using the DFT-D3 method.47 The MXenes unit cell was obtained from the corresponding MAX phase bulk structure by cutting the M2C slab perpendicular to the [001] direction and adding a vacuum region of 15 Å. To improve the generally underestimated band gaps by GGA methods,18 the band structure and projected density of states (PDOS) were obtained with the hybrid HSE0648 functional. The work function (WF) was derived from the energy difference between the Fermi level and the vacuum level.34Most MXenes have the high-symmetry P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 1m space group.14,31,49 In our case, the symmetry of bimetallic and vacancy-ordered MXenes is lower than that of the V2C MXene due to the presence of Zr atoms (or vacancy). To accurately assess the band structure and phonon dispersion of MXenes, we expanded the definition of symmetry points of the hexagonal Brillouin zone (see Fig. S1, ESI†), thus breaking the degeneracy of M and K points and naming the now inequivalent points as M1, K1, M2, K2, M3, K3, as previously reported for (Mo2/3□1/3)2CX2.43 The energy difference at symmetry points is significant and therefore cannot be disregarded. All band structures are in the ESI† (Fig. S9 and S10).
1m space group.14,31,49 In our case, the symmetry of bimetallic and vacancy-ordered MXenes is lower than that of the V2C MXene due to the presence of Zr atoms (or vacancy). To accurately assess the band structure and phonon dispersion of MXenes, we expanded the definition of symmetry points of the hexagonal Brillouin zone (see Fig. S1, ESI†), thus breaking the degeneracy of M and K points and naming the now inequivalent points as M1, K1, M2, K2, M3, K3, as previously reported for (Mo2/3□1/3)2CX2.43 The energy difference at symmetry points is significant and therefore cannot be disregarded. All band structures are in the ESI† (Fig. S9 and S10).
The vibrational properties within the harmonic approximation are entirely defined within the dynamical matrix (Hessian matrix) calculated at the density functional perturbation theory (DFPT)50 level as implemented in VASP. The post-processing and analysis has thus been performed using the software PhonoPy.51 The convergence standards have been increased to 10−7 eV and to 10−6 eV Å−1 for the wave-function and interatomic forces, respectively. The other parameters have been kept consistent with the geometry optimization.
To evaluate the stability of functionalized (V2/3Zr1/3)2CX2 MXenes, the formation energy (Eform) of the unit cell for each M–X bond is calculated as:
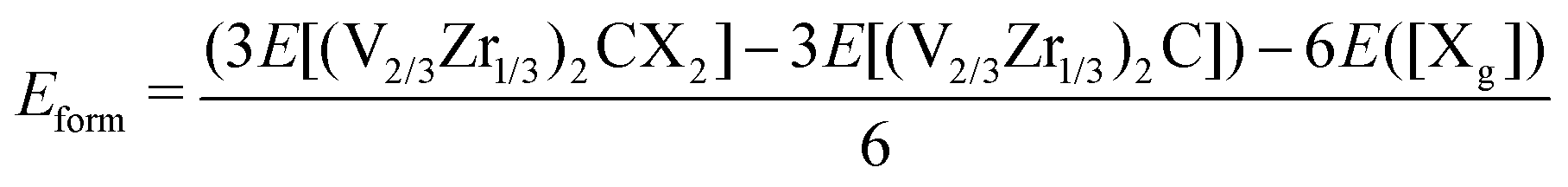 | (1) |
For a quick assessment of structural stability, we considered a set of ab initio molecular dynamics (AIMD), as implemented in VASP. These simulations have been completed using the Nosé algorithm52 in the NVT ensemble at room temperature (300 K) for 9 ps. We used a scalar, collinear magnetic model, and we considered the ferromagnetic (FM) configuration and the three possible antiferromagnetic (AFM) states to calculate the preferred magnetic ground state structures of (V2/3Zr1/3)2CX2 system (Fig. S2, ESI†). To comprehensively assess the magnetic behavior of these MXenes, we have constructed an Ising model (see Fig. S3, ESI†) using exchange coupling parameters derived from DFT simulations. This allowed us to calculate the Curie temperature (TC) and specific heats (CV) (see ESI†) by Monte Carlo simulations (see Section 3.2.3) performed with the open-source software ALPS.53
3 Result and discussion
3.1 Structural analysis
MXene structures derive from corresponding bulk MAX phases, as shown in Fig. 1. The pristine (V2/3Zr1/3)2C MXene slab is formed (upon removal of the Al layer) by three hexagonal layers stacked on top of each other, and the layer of C atoms is sandwiched between layers composed of V and Zr atoms (Fig. 1b). The Zr atoms are all aligned along the short diagonal of the ab-plane. Functionalized MXenes are obtained by surface termination (F, OH, or O termination), while vacancy-ordered MXenes are obtained by removing Zr atoms. The structural details (e.g., lattice constant, bond lengths, etc.) are outlined in Table S1 (ESI†).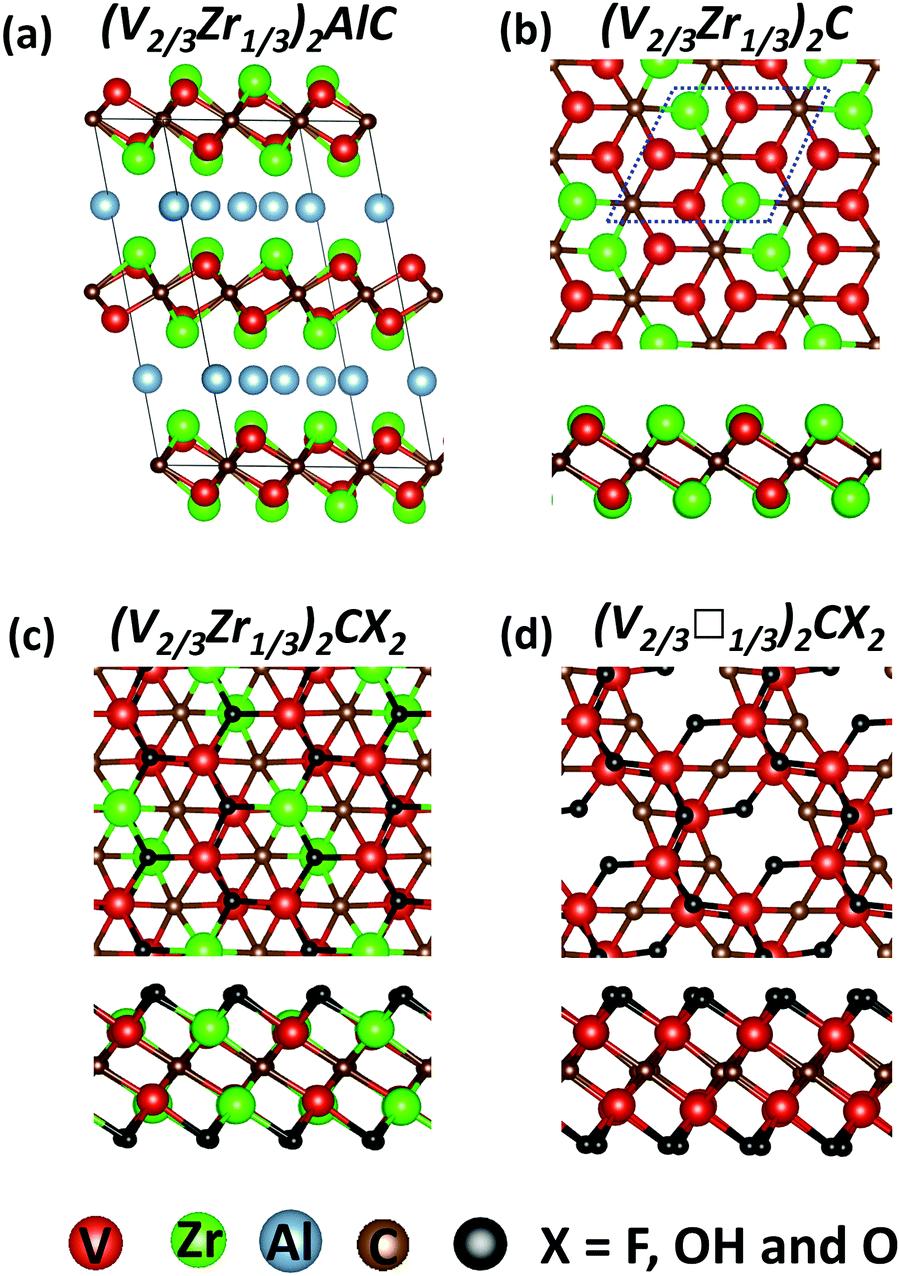 | ||
| Fig. 1 Structures of parent MAX phase (V2/3Zr1/3)2AlC (a), pristine bimetallic MXene (V2/3Zr1/3)2C (b), functionalized bimetallic MXene (V2/3Zr1/3)2CX2 (c) and vacancy-ordered (V2/3□1/3)2CX2 (X = F, OH and O) MXene (d). The unit cell of MXenes is marked by a blue dotted line. | ||
Our calculations suggest that the bimetallic (V2/3Zr1/3)2C MXene can be obtained from a parent (V2/3Zr1/3)2AlC MAX phase. First, the calculated exfoliation energy of (V2/3Zr1/3)2CAl is very similar to that of V2AlC54 (2.59 J m−2versus 2.53 J m−2, respectively, as shown in Fig. S4, ESI†). Second, the absence of soft modes at the Γ-point, as shown by the dispersion of the vibrational modes, ensures the dynamical stability of the structure, which is further supported by AIMD calculations performed at 300 K for (V2/3Zr1/3)2C (Fig. S5, ESI†). All these results indicate that bimetallic MXene is kinetically stable and can be obtained experimentally, similarly to V2C.55
The stability and other properties of MXenes depend on surface functionalization, which in turn depends on the exfoliation process. Experimental investigation of related V2C MXenes56 has shown that different conditions (i.e. solvent, O2 partial pressure, etc.) during the exfoliation process, lead to different surface decorations.7,27,57 In analogy to the study by Harris et al., we selected the following surface functional groups: O, F and OH. The functional groups are positioned above the hollow site formed by three neighbouring C mirroring the positions of the metals on the opposite layer. Such structures are systematically more stable than structures with functional groups sitting on top of the C atoms (Fig. S6, ESI†).
The formation energies of (V2/3Zr1/3)2CX2-functionalized MXenes calculated from eqn (1) are large, −5.21, −7.55 and −4.48 eV for X = F, OH and O, respectively, thus highlighting the formation of strong chemical bonds on MXene surfaces (V or Zr atoms). This is also supported by AIMD simulation at 300 K (Fig. S7, ESI†). However, when removing Zr, the (V2/3□1/3)2C and (V2/3□1/3)2CO2 structures are subjected to large interatomic forces and heavily distorted during just a short AIMD simulation. We also found that only (V2/3□1/3)2CF2 and (V2/3□1/3)2C(OH)2 are kinetically stable, based on both AIMD and phonon spectra calculations (Fig. S8, ESI†). Thus, the interaction between metals and surface functional groups is weaker in vacancy-ordered MXenes than in bimetallic MXenes.
3.2 Magnetic and electronic properties
 | ||
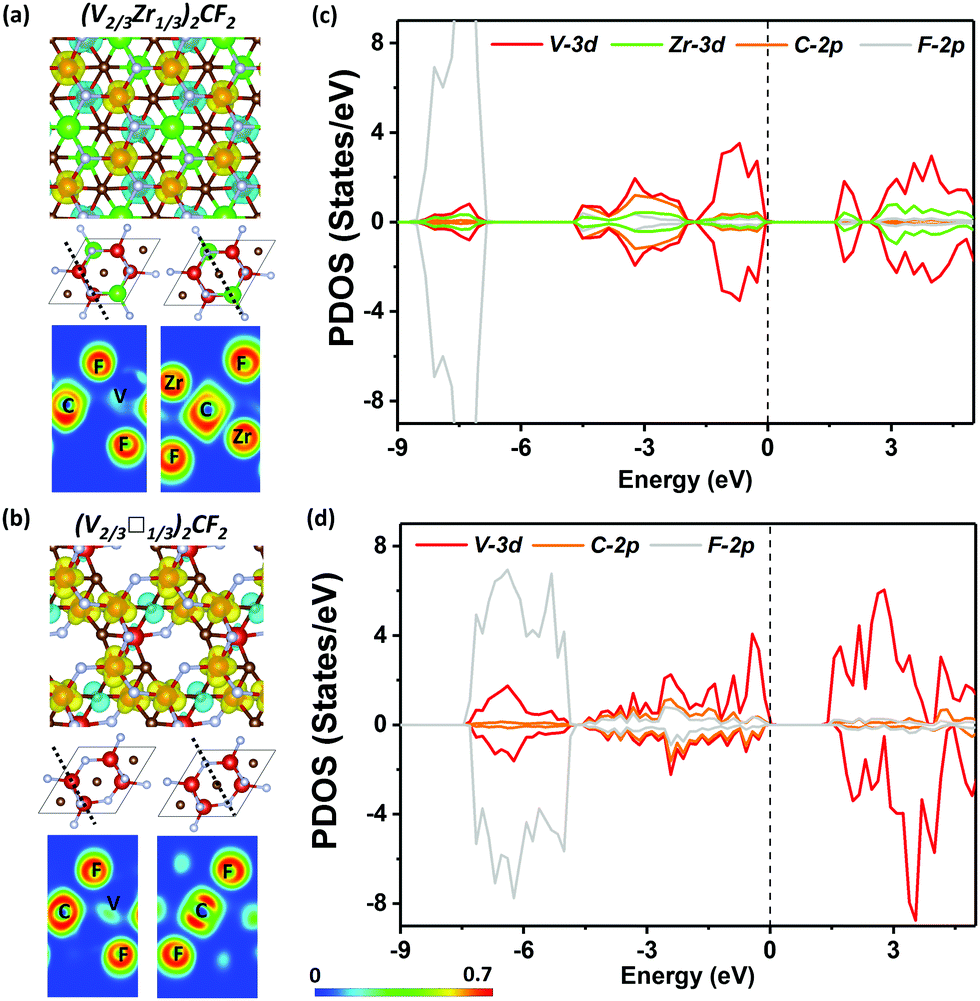
| Fig. 2 Spin polarized charge densities and electron localization function (ELF) maps (perpendicular to [001] direction) are shown for (V2/3Zr1/3)2CF2 (a) and (V2/3□1/3)2F2 (b), where spin up and spin down densities are shown in yellow and light blue, respectively. The units of color scale is “probability”. Project density of states (PDOS) of (V2/3Zr1/3)2CF2 (c) and (V2/3□1/3)2F2 (d). Fermi level (black dotted line) is set to zero. | ||
Electronic local function (ELF) maps show that (V2/3Zr1/3)2C has distinct characteristics of electrons localized on V atoms, which leads to the super-exchange mechanism. The band structure of (V2/3Zr1/3)2C shows a semiconducting character with a band gap of 0.78 eV (Fig. S9, ESI†), and PDOS shown in Fig. 2c are in line with ELF. The states near CBM and VBM have main contributions from the V 3d orbitals, while the contributions from Zr 3d orbitals and C 2p orbitals are insignificant.
The magnetic properties of (V2/3Zr1/3)2CO2 are qualitatively different. The presence of surface oxygens changes the magnetic ordering, resulting in FM ground state with ΔE = EAFM − EFM of 33.59 meV per unit cell with respect to the most stable AFM state. The total magnetic moment of (V2/3Zr1/3)2CO2 is 4 μB per unit cell. d-Electrons of V atoms can induce the spin-polarization of the neighbouring C atoms via double-exchange mechanism59 (Fig. 2b). The presence of oxygens on the MXene surface also leads to charge transfer towards the surface (as shown by Bader charge analysis,60 Table S1, ESI†), and this affects the position of the V 3d in the spin-down channel (see PDOS, Fig. 2d), which is shifted down in energy and no longer participates in VBM. However, surface oxygens have no effect on the spin-up channel. This is reflected on the band structure, which exhibits distinct half-semiconductor features. The two half-semiconducting gaps are 0.53 and 1.85 eV for spin-up and spin-down channels (Fig. S9, ESI†), respectively. The difference of the band edge energy between the two spin channels (ΔECBM = ECBM(down) − ECBM(up) and ΔEVBM = EVBM(down) − EVBM(up)) shows the typical half-semiconducting character (see Table S2, ESI† for complete set of characteristics).
Both (V2/3Zr1/3)2CF2 and (V2/3Zr1/3)2C(OH)2 MXenes exhibits similar magnetic properties, and only the results regarding the former are discussed here (see Fig. S10, ESI† for further details on the latter). The (V2/3Zr1/3)2CF2 MXene exhibits an AFM magnetic character, similarly to pristine (V2/3Zr1/3)2C. The AFM magnetic configuration is remarkably stable, showing ΔE = −841.08 meV. Such analogous value has already been reported for V2CF2.61 The analysis of spin-polarized densities and ELF suggests that (V2/3Zr1/3)2C(OH)2 MXene shows an analogous behaviour to the pristine MXene, i.e., super-exchange mechanism. The presence of F surface atoms widens the band gap due to electron density localization around the halide centre, in analogy to the O-terminated surface. However, the FM state is not stabilized in (V2/3Zr1/3)2CF2 since both V 3d spin channels are equally shifted (Fig. 3a and c).
 | ||
| Fig. 3 (a and b) The inserted background figure shows spin-polarized charge densities on (V2/3Zr1/3)2CF2 and (V2/3□1/3)2CF2, where spin up and spin down densities are shown in yellow and light blue, respectively. The sections (dot lines) of electron localization function (ELF) maps are perpendicular to (001) direction. The units of color scale is “probability”. (c) and (d) The project density of states (PDOS) of (V2/3Zr1/3)2CF2 and (V2/3□1/3)2CF2. Fermi level is set up to zero with the black dotted line. | ||
The FM ordering results from the effect of V d-electrons, which induce spin-polarization of neighbouring C atoms via a double-exchange mechanism, as shown by the ELF map in Fig. 3b. The band structure shows a half-semiconducting behaviour (Fig. S9, ESI†), which originates from the spin-polarization of CBM observed in (V2/3□1/3)2CF2. We can also observe a smaller polarization on VBM, due to the slight shift in the PDOS of the V 3d orbitals in the spin-down channel. The full set of electronic properties are outlined in Table S2 (ESI†). The polarization of the band edges can be explained by PDOS, wherein, in addition to the V 3d orbital, C and F orbitals also significantly contribute to the VBM (3d). We also considered the (V2/3□1/3)2C(OH)2, which behaves similarly to (V2/3□1/3)2CF2; however, due to the lower electronegativity of OH, the effect of spin polarization is weaker and hence ΔE is smaller.
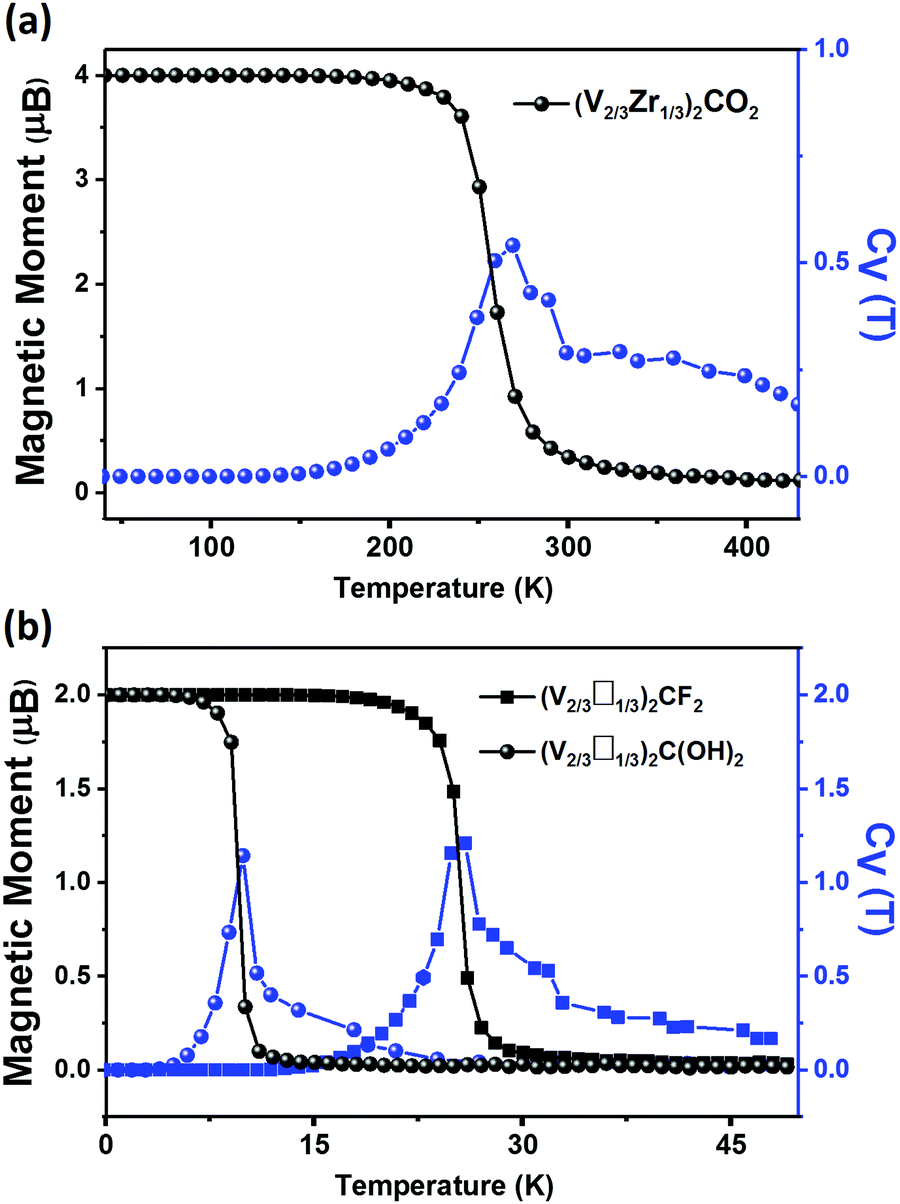 | ||
| Fig. 4 Variation of the total magnetic momentum (per unit cell) of (a) (V2/3Zr1/3)2CO2 (b) (V2/3□1/3)2CF2 and (V2/3□1/3)2C(OH)2 with respect to the temperature. Corresponding specific heats CV are shown in blue. | ||
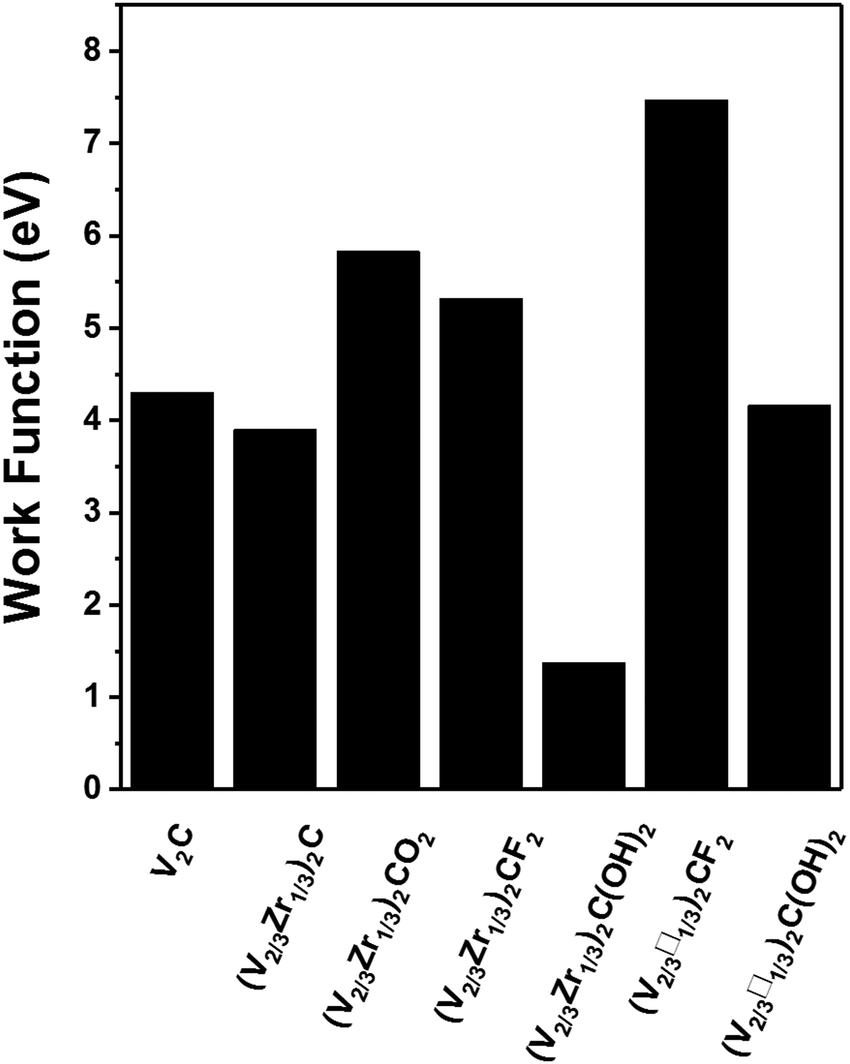 | ||
| Fig. 5 Work functions of V2C, pristine (V2/3Zr1/3)2C, bimetallic (V2/3Zr1/3)2CX2 and vacancy-ordered (V2/3□1/3)2CX2 MXenes. | ||
4 Conclusions
Our results show that modifying the stoichiometry and/or surface functionalization of MXenes changes their properties qualitatively. Therefore they are excellent candidates for applications in spintronics because their electric and magnetic properties can be tuned for specific purposes. In this study, we identified (V2/3Zr1/3)2CX2, (V2/3□1/3)2CF2 and (V2/3□1/3)2C(OH)2 as stable candidates. Among them, the (V2/3Zr1/3)2CO2 and (V2/3□1/3)2CF2 and (V2/3□1/3)2C(OH)2 are half-semiconductor materials. The predicted Curie temperature for (V2/3Zr1/3)2CO2 (270 K) is higher than that of the experimentally reported 2D CrI3 crystals (45 K).16 Hence, of the MXenes tested in this study, (V2/3Zr1/3)2CO2 is the best candidate for spintronic applications.The functional groups of MXenes can radically change the composition of their frontier orbitals, thereby affecting their work function. In particular, we found that (V2/3Zr1/3)2C(OH)2 MXene can be used as an ultra-low work function electron emitter and its work function (1.37 eV) is lower than that of Sc2C(OH)2 MXene (1.6 eV), as reported by Khazaei et al. Conversely, the (V2/3□1/3)2CF2 MXene has a rather high WF of 7.47 eV, which is thus higher than that of the Pt metal.33 Thanks to these properties, (V2/3□1/3)2CF2 MXenes can be used for holes injection in applications requiring Schottky-barrier-free contacts. Therefore, (V2/3Zr1/3)2C(OH)2 and vacancy-ordered (V2/3□1/3)2CF2 MXenes are also promising candidates for electronic devices.
Overall, the results presented in this study establish a new family of MXenes with intrinsic magnetism, which makes them ideal candidates for both spintronic and electronic applications in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A support from OP VVV “Excellent Research Teams”, project CUCAM, is also acknowledged. S. L. acknowledges the support from GAUK project (Grant No. 792218). F. B. is supported by the “International Mobility of Researchers at Charles University” (CZ.02.2.69/0.0/0.0/16_027/0008495). Thanks Carlos V. Melo for his precious contribution of writing guidance. The crystallographic structures and inputs to reproduce this work are available on gitlab at the address: https://gitlab.com/praguelab/shuo2019_work1 and can be cited using the following DOI: 10.5281/zenodo.3476885.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- Y. Lin, T. V. Williams and J. W. Connell, J. Phys. Chem. Lett., 2009, 1, 277–283 CrossRef.
- B. Lalmi, H. Oughaddou, H. Enriquez, A. Kara, S. Vizzini, B. Ealet and B. Aufray, Appl. Phys. Lett., 2010, 97, 223109 CrossRef.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263 CrossRef.
- M. Chhowalla, Z. Liu and H. Zhang, Chem. Soc. Rev., 2015, 44, 2584–2586 RSC.
- H. Liu, Y. Du, Y. Deng and D. Y. Peide, Chem. Soc. Rev., 2015, 44, 2732–2743 RSC.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed.
- H. Zhang, ACS Nano, 2015, 9, 9451–9469 CrossRef CAS PubMed.
- I. Žutić, J. Fabian and S. D. Sarma, Rev. Mod. Phys., 2004, 76, 323 CrossRef.
- A. Fert, N. Reyren and V. Cros, Nat. Rev. Mater., 2017, 2, 17031 CrossRef CAS.
- A. C. Neto, F. Guinea, N. M. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109 CrossRef.
- Z. Lin, A. McCreary, N. Briggs, S. Subramanian, K. Zhang, Y. Sun, X. Li, N. J. Borys, H. Yuan and S. K. Fullerton-Shirey, et al. , 2D Mater., 2016, 3, 042001 CrossRef.
- E. Torun, H. Sahin, S. Singh and F. Peeters, Appl. Phys. Lett., 2015, 106, 192404 CrossRef.
- C. Si, J. Zhou and Z. Sun, ACS Appl. Mater. Interfaces, 2015, 7, 17510–17515 CrossRef CAS PubMed.
- C. Gong, L. Li, Z. Li, H. Ji, A. Stern, Y. Xia, T. Cao, W. Bao, C. Wang and Y. Wang, et al. , Nature, 2017, 546, 265 CrossRef CAS PubMed.
- B. Huang, G. Clark, E. Navarro-Moratalla, D. R. Klein, R. Cheng, K. L. Seyler, D. Zhong, E. Schmidgall, M. A. McGuire and D. H. Cobden, et al. , Nature, 2017, 546, 270 CrossRef CAS PubMed.
- G. Gao, G. Ding, J. Li, K. Yao, M. Wu and M. Qian, Nanoscale, 2016, 8, 8986–8994 RSC.
- J. He, P. Lyu and P. Nachtigall, J. Mater. Chem. C, 2016, 4, 11143–11149 RSC.
- S. Ouardi, G. H. Fecher, C. Felser and J. Kübler, Phys. Rev. Lett., 2013, 110, 100401 CrossRef PubMed.
- X. Wang, Phys. Rev. Lett., 2008, 100, 156404 CrossRef CAS PubMed.
- X. Li, X. Wu, Z. Li, J. Yang and J. Hou, Nanoscale, 2012, 4, 5680–5685 RSC.
- W.-B. Zhang, Q. Qu, P. Zhu and C.-H. Lam, J. Mater. Chem. C, 2015, 3, 12457–12468 RSC.
- J. Liu, Q. Sun, Y. Kawazoe and P. Jena, Phys. Chem. Chem. Phys., 2016, 18, 8777–8784 RSC.
- M. Kan, S. Adhikari and Q. Sun, Phys. Chem. Chem. Phys., 2014, 16, 4990–4994 RSC.
- X. Li and J. Yang, Natl. Sci. Rev., 2016, 3, 365–381 CrossRef CAS.
- D. Zhong, Sci. Adv., 2017, 3, e1603113 CrossRef PubMed.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322–1331 CrossRef CAS PubMed.
- V. M. H. Ng, H. Huang, K. Zhou, P. S. Lee, W. Que, J. Z. Xu and L. B. Kong, J. Mater. Chem. A, 2017, 5, 3039–3068 RSC.
- J.-C. Lei, X. Zhang and Z. Zhou, Front. Phys., 2015, 10, 276–286 CrossRef.
- J. He, G. Ding, C. Zhong, S. Li, D. Li and G. Zhang, Nanoscale, 2019, 11, 356–364 RSC.
- A. S. Ingason, M. Dahlqvist and J. Rosén, J. Phys.: Condens. Matter, 2016, 28, 433003 CrossRef CAS PubMed.
- Y. Liu, H. Xiao and W. A. Goddard III, J. Am. Chem. Soc., 2016, 138, 15853–15856 CrossRef CAS PubMed.
- M. Khazaei, M. Arai, T. Sasaki, A. Ranjbar, Y. Liang and S. Yunoki, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 075411 CrossRef.
- M. Khazaei, A. Ranjbar, M. Ghorbani-Asl, M. Arai, T. Sasaki, Y. Liang and S. Yunoki, Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 93, 205125 CrossRef.
- B.-C. Min, K. Motohashi, C. Lodder and R. Jansen, Nat. Mater., 2006, 5, 817 CrossRef CAS PubMed.
- Y. Ando, Y. Gohda and S. Tsuneyuki, Surf. Sci., 2012, 606, 1501–1506 CrossRef CAS.
- J. Freeouf and J. Woodall, Appl. Phys. Lett., 1981, 39, 727–729 CrossRef CAS.
- Q. Tao, M. Dahlqvist, J. Lu, S. Kota, R. Meshkian, J. Halim, J. Palisaitis, L. Hultman, M. W. Barsoum and P. O. Persson, et al. , Nat. Commun., 2017, 8, 14949 CrossRef.
- A. Thore and J. Rosén, Phys. Chem. Chem. Phys., 2017, 19, 21595–21603 RSC.
- M. Khazaei, V. Wang, C. Sevik, A. Ranjbar, M. Arai and S. Yunoki, Phys. Rev. Mater., 2018, 2, 074002 CrossRef CAS.
- M. Dahlqvist, J. Lu, R. Meshkian, Q. Tao, L. Hultman and J. Rosen, Sci. Adv., 2017, 3, e1700642 CrossRef PubMed.
- H. Lind, J. Halim, S. Simak and J. Rosén, Phys. Rev. Mater., 2017, 1, 044002 CrossRef.
- G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- N. C. Frey, A. Bandyopadhyay, H. Kumar, B. Anasori, Y. Gogotsi and V. B. Shenoy, ACS Nano, 2019, 13, 2831–2839 CrossRef CAS PubMed.
- S. Baroni, S. De Gironcoli, A. Dal Corso and P. Giannozzi, Rev. Mod. Phys., 2001, 73, 515 CrossRef CAS.
- A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- A. F. Albuquerque, F. Alet, P. Corboz, P. Dayal, A. Feiguin, S. Fuchs, L. Gamper, E. Gull, S. Gürtler and A. Honecker, et al. , J. Magn. Magn. Mater., 2007, 310, 1187–1193 CrossRef CAS.
- Z. Guo, J. Zhou and Z. Sun, J. Mater. Chem. A, 2017, 5, 23530–23535 RSC.
- F. Liu, J. Zhou, S. Wang, B. Wang, C. Shen, L. Wang, Q. Hu, Q. Huang and A. Zhou, J. Electrochem. Soc., 2017, 164, A709–A713 CrossRef CAS.
- K. J. Harris, M. Bugnet, M. Naguib, M. W. Barsoum and G. R. Goward, J. Phys. Chem. C, 2015, 119, 13713–13720 CrossRef CAS.
- M. Khazaei, M. Arai, T. Sasaki, C.-Y. Chung, N. S. Venkataramanan, M. Estili, Y. Sakka and Y. Kawazoe, Adv. Funct. Mater., 2013, 23, 2185–2192 CrossRef CAS.
- J. Kanamori, J. Phys. Chem. Solids, 1959, 10, 87–98 CrossRef CAS.
- P.-G. De Gennes, Phys. Rev., 1960, 118, 141 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- J. Hu, B. Xu, C. Ouyang, S. A. Yang and Y. Yao, J. Phys. Chem. C, 2014, 118, 24274–24281 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp05638f |
| This journal is © the Owner Societies 2019 |