
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The high-pressure and low-temperature structural behaviour of 2,2,2-trifluoroethanol†
S. A.
Barnett
 and
D. R.
Allan
*
and
D. R.
Allan
*
Diamond Light Source, Harwell Science and Innovation Campus, Chilton, OX11 0RZ, UK. E-mail: David.Allan@diamond.ac.uk
First published on 7th June 2019
Abstract
On cooling to 229 K at ambient pressure, 2,2,2-trifluoroethanol freezes to form a crystal structure characterised by hydrogen-bonded molecular chains in the orthorhombic space group Pca21 (form 1). On compression to 0.22 GPa at room temperature, however, liquid 2,2,2-trifluoroethanol crystallises in a structure adopting triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) symmetry (form 2). Although the form 1 and form 2 polymorphs are both characterised by the formation of hydrogen-bonded chains, the arrangement of the molecules within the chains are significantly different with the high-pressure form appearing to adopt a more strained configuration. The orientation of the molecules in form 2 is such that the hydrogen bonds in neighbouring chains are in close proximity with a bridging interchain O⋯O contact distance that is of the same order as the O⋯O distances found for the hydrogen bonds. The resulting close pairing of neighbouring catemeric chains produces molecular pillars in the structure of form 2.
symmetry (form 2). Although the form 1 and form 2 polymorphs are both characterised by the formation of hydrogen-bonded chains, the arrangement of the molecules within the chains are significantly different with the high-pressure form appearing to adopt a more strained configuration. The orientation of the molecules in form 2 is such that the hydrogen bonds in neighbouring chains are in close proximity with a bridging interchain O⋯O contact distance that is of the same order as the O⋯O distances found for the hydrogen bonds. The resulting close pairing of neighbouring catemeric chains produces molecular pillars in the structure of form 2.
Introduction
In the mono-alcohols (ROH), there is competition between the packing requirements of the relatively bulky R-group and the demand for the small hydroxyl groups to be sufficiently close for hydrogen bonding to occur.1 The packing motifs adopted for the smaller members of the mono-alcohol series are often relatively ‘thin’ where the crystal structures show that catemers are formed with the molecules arranged in an approximately coplanar alternating sequence about the central chain of hydrogen bonds. For mono-alcohol systems with bulkier R-groups, steric hindrance often prohibits the molecules from adopting this simple arrangement. If the R-group is particularly bulky then the molecules may no longer form hydrogen-bonded chains or catemers, but cyclic dimers, trimers, tetramers or hexamers can be created instead.As part of an ongoing programme to study the high-pressure and low-temperature structural behaviour of small molecule systems, we have been examining a range of mono-functional molecular materials. Among the first of these studies was the investigation of the structural behaviour of methanol and ethanol which are the simplest members of the linear alcohol series, H(CH2)nOH.2–5 For methanol, two orthorhombic crystalline phases are formed on cooling, while at pressures above about 4 GPa a triclinic P phase is formed. It is, however, possible to superpress the liquid phase and, if compression is suitably rapid, crystallisation is suppressed and the liquid becomes a pressure-induced glass. The extremely strained conformation of the hydrogen-bonded molecular chains in the high-pressure P crystal structure offers a partial explanation of why methanol is so difficult to crystallise at high pressure and so readily vitrifies.2 Ethanol, in contrast, almost invariably crystallises on compression at pressures above 3 GPa.3,4 Conversely, it is the low-temperature ethanol polymorph that exhibits the strained structure.5 Below the freezing temperature of ∼156 K, ethanol crystallises in the monoclinic Pc space group and its crystal structure is characterised by hydrogen-bonded molecular chains where pairs of molecules are linked together in trans and gauche conformers with the methyl groups of each molecule directed away from the centre of each pair. These molecular pairs alternate such that the molecules are arranged in a relatively strained sequence along the chain. In the high-pressure P21/c structure this strain is relieved by the orientations of the molecules about the hydrogen bonds adopting a simple alternating sequence.
More recently, Ridout and Probert6 have studied the low-temperature and high-pressure structures of isopropyl alcohol (isopropanol, C3H8O) where they found that the more bulky R-group led to the two polymorphs, which both adopt the monoclinic P21/c space group, having very different packing motifs and densities. In the low-temperature phase, at 180 K, the molecules are arranged in one-dimensional helical hydrogen-bonded chains while the significantly denser high-pressure phase is characterised by the formation of isolated 8-membered rings, at 1.1 GPa. It appears, therefore, that the R-group in isopropanol exhibits ‘thin’ behaviour at low-temperature but its steric properties transform to ‘bulky’ behaviour at high-pressure. This behaviour is reminiscent of that of tertiary butyl alcohol (t-butanol, C4H10O) where the low-temperature triclinic, P, structure, phase-IV, is composed of hydrogen-bonded helical chains.7 The only other structure to be determined in the relatively complex phase diagram is that of the trigonal phase-II (P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ), which is found to be stable at both high-pressure (0.85 GPa) and low-temperature (220 K), and is characterised by hexameric rings that adopt the chair conformation. In contrast, for cyclobutanol (C4H7OH), both the low-temperature Aba2 structure and the high-pressure Pna21 structure are characterised by catemers.8 The R-group is not sufficiently bulky to drive the formation of rings but, instead, for the high-pressure phase the molecules are arranged in an alternating, and approximately coplanar, arrangement about the hydrogen bond, while in the low-temperature structure the molecules adopt a pseudo 3-fold helical arrangement. This behaviour strongly parallels the changes we have also previously observed between the low-temperature and high-pressure phases of phenol,9 and its halogenated derivatives 2-chlorophenol and 4-fluorophenol.10,11 Perhaps the most significant steric effects are observed in the phase-I crystal structures of both cyclopentanol12 and cyclohexanol13 (which are formed on either initial compression or initial cooling) where hydrogen bonding is hindered to such an extent that plastic phases are crystallised. In these phase-I structures the molecules have complete orientational disorder. For cyclohexanol, phase-I is cubic and hydrogen-bonded structures are formed on further cooling; the tetragonal phase-II (P
), which is found to be stable at both high-pressure (0.85 GPa) and low-temperature (220 K), and is characterised by hexameric rings that adopt the chair conformation. In contrast, for cyclobutanol (C4H7OH), both the low-temperature Aba2 structure and the high-pressure Pna21 structure are characterised by catemers.8 The R-group is not sufficiently bulky to drive the formation of rings but, instead, for the high-pressure phase the molecules are arranged in an alternating, and approximately coplanar, arrangement about the hydrogen bond, while in the low-temperature structure the molecules adopt a pseudo 3-fold helical arrangement. This behaviour strongly parallels the changes we have also previously observed between the low-temperature and high-pressure phases of phenol,9 and its halogenated derivatives 2-chlorophenol and 4-fluorophenol.10,11 Perhaps the most significant steric effects are observed in the phase-I crystal structures of both cyclopentanol12 and cyclohexanol13 (which are formed on either initial compression or initial cooling) where hydrogen bonding is hindered to such an extent that plastic phases are crystallised. In these phase-I structures the molecules have complete orientational disorder. For cyclohexanol, phase-I is cubic and hydrogen-bonded structures are formed on further cooling; the tetragonal phase-II (P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 21c) is composed of molecules in a tetrameric ring motif while the structures of phases III and III′ are monoclinic (Pc and P21/c respectively) with hydrogen-bonded 3-fold-helical molecular chains. For cyclopentanol, the plastic phase-I structure is hexagonal which, on compression, converts to an ordered structure identified as phase-IV (P21/c) where the molecules form pseudo 4-fold hydrogen-bonded molecular chains.
21c) is composed of molecules in a tetrameric ring motif while the structures of phases III and III′ are monoclinic (Pc and P21/c respectively) with hydrogen-bonded 3-fold-helical molecular chains. For cyclopentanol, the plastic phase-I structure is hexagonal which, on compression, converts to an ordered structure identified as phase-IV (P21/c) where the molecules form pseudo 4-fold hydrogen-bonded molecular chains.
To examine the effect of increasing the steric hindrance of the R-group in ethanol we have determined the low-temperature and high-pressure structures of 2,2,2-trifluoroethanol (where the hydrogen atoms on the methyl group have been replaced with fluorine) with the expectation that there would be a significant modification of the conformation of any, likely, hydrogen-bonded molecular chains. Although fluorine has a high electronegativity14 and would be anticipated to have correspondingly high electron withdrawing properties (with the electronegative fluorine reducing the electron donating power of the ethyl group), the replacement of the terminal methyl group C–H bonds in ethanol by C–F bonds will have only a secondary effect on the hydrogen bond due to the influence of the moderating methylene (–CH2–) bridge. Therefore, we expect that steric affects will have a more direct influence on the differences between the crystal structures observed in ethanol and trifluoroethanol.
Herein, we report the crystal structure of the high-pressure phase of 2,2,2-trifluoroethanol (CF3CH2OH) along with the crystal structure of its low-temperature phase which, to the best of our knowledge, have not been reported previously.
Experimental
2,2,2-Trifluoroethanol (CF3CH2OH) is a clear, colourless liquid with a melting point of 229 K and a boiling point of 347 K. A sample of 99.5+% purity was used, as received from Sigma-Aldrich, for all subsequent experiments.For the low-temperature study a cryostream15 was used to cool the sample, which was contained within a capillary mounted on the diffractometer. The temperature was then cycled near the freezing point until the original powder was transformed into a small number of crystals.
For the high-pressure study the sample was loaded into a Merrill–Bassett diamond-anvil cell, equipped with Boehler–Almax cut diamonds and their matched tungsten carbide backing seats.16 A 200 μm thick tungsten gasket, that had been pre-indented to a thickness of ∼100 μm, was used with a 200 μm hole drilled through it to act as a pressure chamber. A ruby was added for pressure calibration via the ruby fluorescence method.17 The 2,2,2-trifluoroethanol sample was compressed until solid and the temperature carefully cycled (using a heat gun) so that the polycrystalline material was partially melted each time until only a single crystallite remained. This crystallite then grew in size so as to fill the gasket hole on subsequent cooling of the cell. The sample pressure at ambient temperature was found to be 0.22 GPa, as measured using the ruby fluorescence method.
X-ray diffraction data were collected in Experiments Hutch 2 (EH2) of Beamline I19, at Diamond Light Source,18 using the Newport kappa-geometry 4-circle diffractometer fitted with a Dectris Pilatus 300 K pixel-array photon-counting detector. Both datasets were collected using a wavelength of λ = 0.4895 Å, with a step size and exposure time of 0.2° and 0.4 seconds respectively. The low-temperature data collection was carried out with a single 360° φ scan, in order to minimise the contribution of other crystals, at a cryostream temperature of 229 K. The collected frames were integrated using XIA219 on one of the major crystallites, and the data were corrected for absorption effects using AIMLESS,20 an empirical method. The high pressure dataset was collected using a series of ω scans to give the optimal completeness when using a diamond anvil cell. The data were integrated with the program CrysAlisPro21 which incorporates routines that omit regions of the detector shaded by the diamond-anvil cell from integration. Both structures were solved by dual-space methods22 and refined by least-squares refinement on all unique measured F2 values.23 For both forms, the alkyl hydrogen atoms were placed geometrically and refined using a riding model, whereas the hydroxyl hydrogen atoms were located from the difference map and refined freely. The data collection, integration and refinement statistics for both the low-temperature and high-pressure data sets are presented in Table 1.
| Form 1 | Form 2 | ||
|---|---|---|---|
| Chemical formula | C2H3F3O | C2H3F3O | |
| Molecular weight | 100.04 | 100.04 | |
| Temperature (K)/pressure (GPa) | 229/0 | 295/0.22 | |
| Crystal system | Orthorhombic | Triclinic | |
| Space group | Pca21 |
P |
|
| Unit cell | a (Å) | 8.6040(7) | 4.9159(7) |
| b (Å) | 10.0578(8) | 8.8894(18) | |
| c (Å) | 9.1461(8) | 9.226(3) | |
| α (°) | 90 | 111.71(2) | |
| β (°) | 90 | 103.03(2) | |
| γ (°) | 90 | 90.970(10) | |
| V (Å3) | 791.48(11) | 362.68(15) | |
| Z/Z′ | 8/2 | 4/2 | |
| Density (g cm−3) | 1.679 | 1.832 | |
| Flack/extinction coefficient | −0.7(10)/0.099(10) | —/— | |
| Reflections collected | 9973 | 2016 | |
| Unique reflections/Rint | 2767/0.0518 | 637/0.0290 | |
| R 1 [I > 2σ] | 0.0331 | 0.0478 | |
| wR2 [all data] | 0.1058 | 0.1430 | |
| GoF on F2 | 0.875 | 1.088 | |
| Largest diff. peak and hole (e A−3) | 0.157/−0.161 | 0.142/−0.118 | |
Results and discussion
Form 1 of 2,2,2-trifluoroethanol (TFE) was found to crystallise at a temperature of 229 K, at ambient pressure, in the orthorhombic space group Pca21 with 2 molecules in the asymmetric unit, see Fig. 1a. At a fairly modest pressure of 0.22 GPa, at room temperature, TFE crystallises as form 2 adopting the triclinic P space group, also with Z′ = 2, see Fig. 1b.
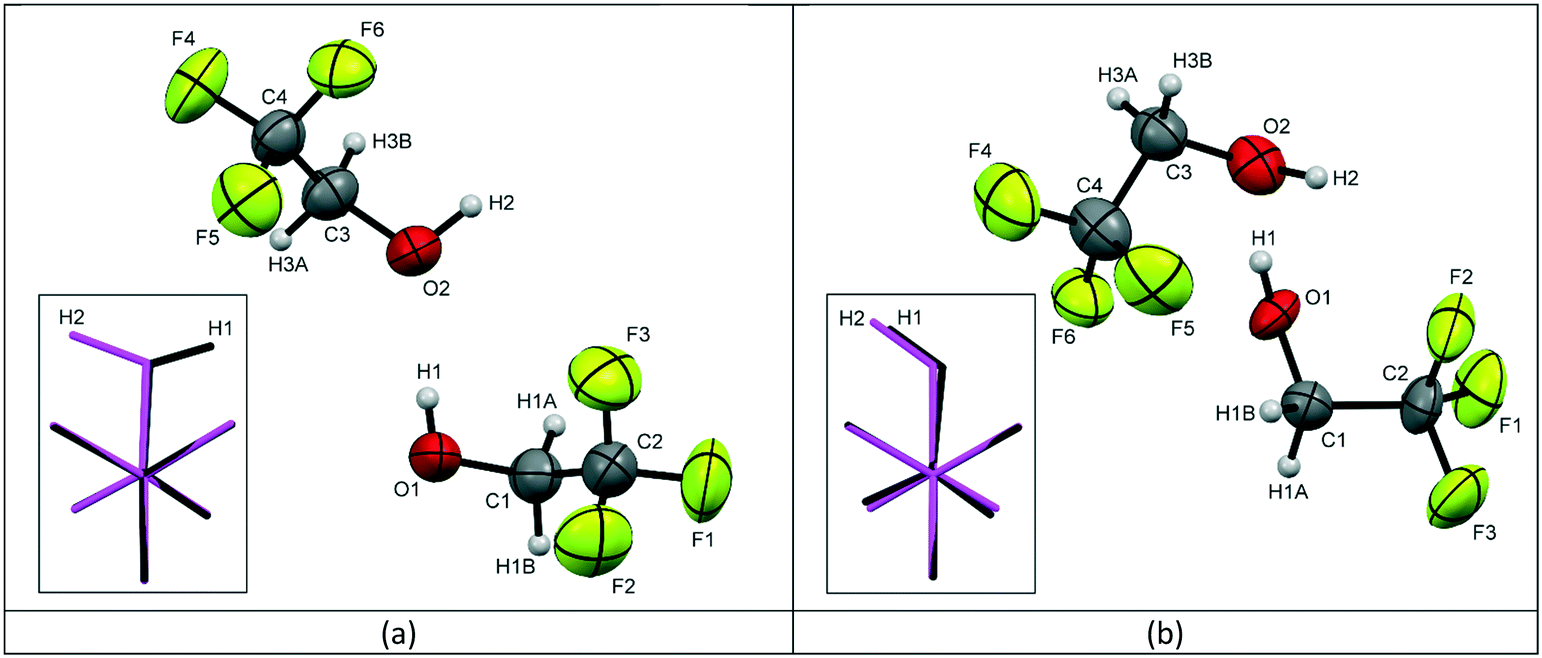 | ||
| Fig. 1 The labelling schemes used in the crystal structures of TFE for (a) the low temperature form 1 and (b) the high pressure form 2, with thermal ellipsoids drawn at the 50% probability level. The inserted images show the molecular overlay‡ of the two independent molecules in the asymmetric unit with molecule 1 (O1, C1, C2, F1, F2, F3) shown in black and molecule 2 (O2, C3, C4, F4, F5, F6) shown in pink (C – dark grey, H – light grey, O – red, F – green). | ||
For form 1, the two molecules are conformationally very similar, with the molecular overlay‡ of the two independent molecules in the asymmetric unit calculated to be 0.0096. The overlay, see Fig. 1a inset, shows that the F–C–C–O torsion angles for the two molecules are comparable (O1–C1–C2–F1 (black) = −176.08(17)°; O2–C3–C4–F4 (pink) = −177.91(18)°) and highlights that the fundamental difference between the two molecules is the opposing orientations of the hydroxyl hydrogens. The four remaining F–C–C–O torsion angles for the two molecules average 59.7°, with a variance of 12.0. The structure is characterised by the formation of hydrogen-bonded molecular chains, with a relatively unstrained arrangement similar to that of the monoclinic Pc phase of ethanol,5 see Fig. 2a. The chains are aligned parallel with the crystallographic c-axis (Fig. 2b) and these chains, in turn, are arranged in layers stacked orthogonally to the a-axis, see Fig. 2c.
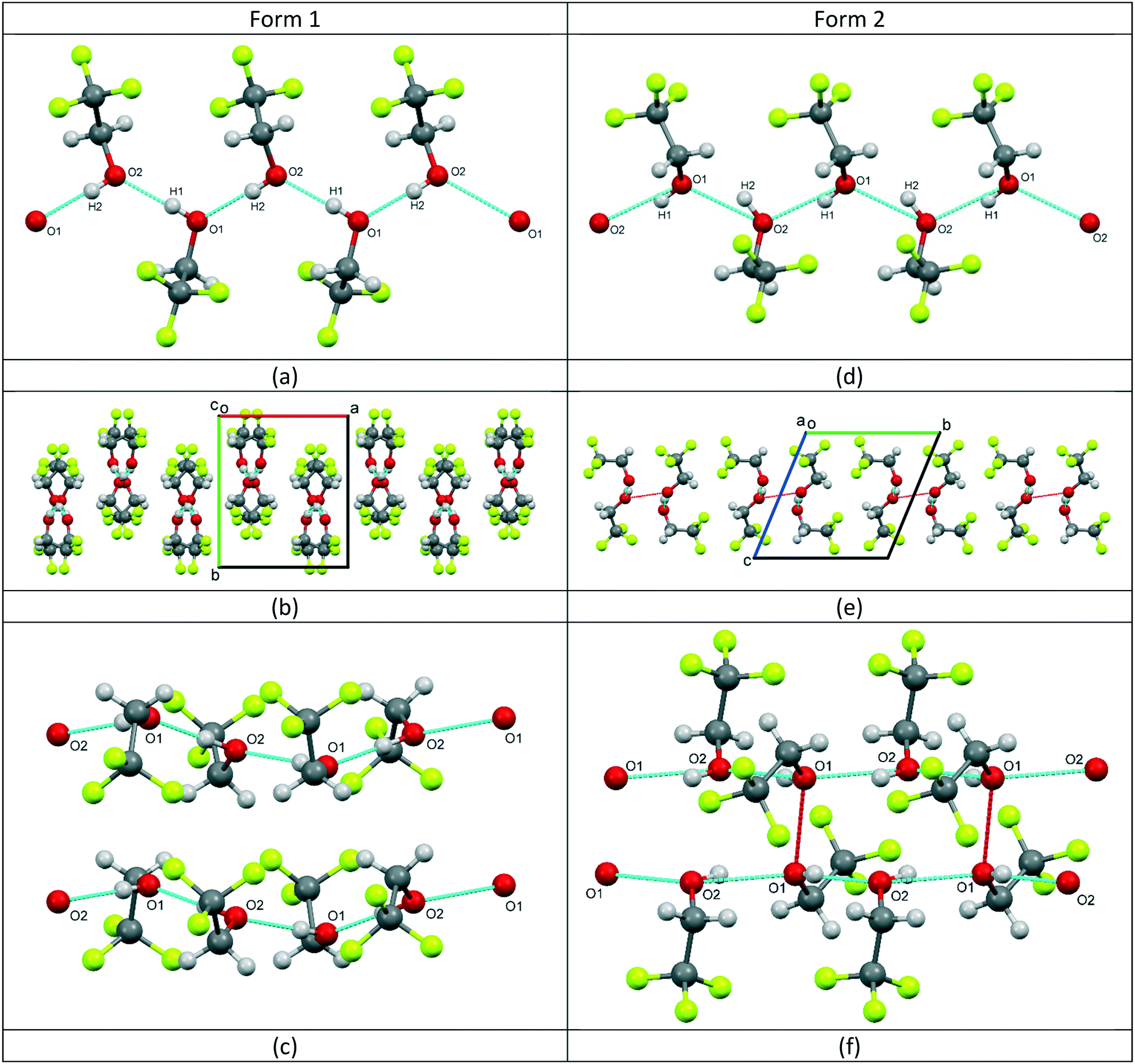 | ||
| Fig. 2 The structure of TFE form 1 showing (a) the hydrogen-bonded chain formed along the c-axis, (b) stacking of the hydrogen-bonded chains and (c) the layers formed orthogonal to the a-axis; the structure of TFE form 2 showing (d) the hydrogen-bonded chain formed along the a-axis, (e) the orthogonal nature of the trifluoro groups within the hydrogen-bonded chain and the exposed hydrogen bond and (f) the double chain pillars. The dashed lines show the hydrogen bonds in light blue and the short O⋯O contacts in red. | ||
The two independent molecules in the asymmetric unit of the high pressure form of TFE (form 2) have the same hydroxyl hydrogen orientation, but the overlay match‡ is much poorer with an RMS of 0.0199. Fig. 1b inset also clearly demonstrates that the conformation of molecule 1 (black) is offset from a perfect staggered arrangement with an O1–C1–C2–F3 torsion angle of −174.6(4)° compared to 179.5(5)° for O2–C3–C4–F4 (molecule 2, pink). This is exemplified by the greater variance (16.6) for the spread of the F–C–C–O torsion angles, although the average is the same as for form 1 (59.7°). Like form 1, form 2 is also characterised by the formation of hydrogen-bonded chains, see Fig. 2d. However, the arrangement of the trifluoro groups with respect to the plane of hydrogen bonds is quite different to that of the low-temperature phase as they are aligned approximately orthogonally to one another rather than parallel, compare Fig. 2b and e. This arrangement of the trifluoro groups means the hydrogen bond is exposed and, as can be seen in Fig. 2e, a bridging O⋯O contact of 2.892(7) Å is made with the neighbouring hydrogen-bonded chain. This distance is of the same order as the O⋯O distances found for the hydrogen bonds in both structures, see Table 2. The resulting close pairing of neighbouring catemeric chains produces pillars in the structure, aligned along the crystallographic a-axis, see Fig. 2f.
| D–H⋯A | D–H/Å | H⋯A/Å | D⋯A/Å | D–H⋯A/° |
|---|---|---|---|---|
| Symmetry codes: i = 3/2 − x, y, z − 1/2; ii = x − 1, y, z. | ||||
| Form 1 | ||||
| O1–H1⋯O2 | 0.77(3) | 1.93(3) | 2.696(2) | 178(3) |
| O2–H2⋯O1i | 0.94(3) | 1.77(3) | 2.716(2) | 179(3) |
| Form 2 | ||||
| O1–H1⋯O2 | 0.81(10) | 1.99(7) | 2.762(7) | 158(10) |
| O2–H2⋯O1ii | 0.92(14) | 1.94(7) | 2.714(6) | 140(12) |
| O2–H2⋯F2 | 0.92(14) | 2.68(16) | 3.251(11) | 121(7) |
It is notable that both hydrogen bonds in form 2 exhibit a large deviation from linearity (158° and 140°, although the large uncertainties in these angles mean they are poorly defined), whereas, the angles formed by the hydrogen bonds in the low temperature form 1 are 178° and 179°. It is possible that the weak halogen bond to F2, formed by O2, could be responsible for the significant deviation observed in form 2.
In ethanol, the low temperature phase is unusual as it consists of pairs of molecules which alternate about the central axis of the hydrogen bonds. The high pressure phase is more closely related to forms I and II of TFE, as individual molecules alternate about the hydrogen-bonded chain. However, in both phases of ethanol the torsion angles are similar, and within the range expected for a staggered conformation, and these are comparable with the torsion angles found for both forms of TFE, thereby indicating that the molecular conformation is not strongly influenced by the molecular packing.
The unusual orientation of the TFE molecules within the hydrogen-bonded chains, combined with the non-linear hydrogen bonds, indicates that the structure is probably highly strained. This suggests that there is potential for the structure to undergo a phase transition on increasing the pressure and that the exposed oxygen atoms, oriented back-to-back between adjacent chains, could be in a favourable position to allow cross-linking hydrogen bonds to be formed.
Conclusions
2,2,2-Trifluoroethanol, a liquid at ambient conditions, has been compressed at room temperature, and cooled at ambient pressure, in order to induce crystallisation. TFE has been shown to be polymorphic with two novel crystal structures determined, both characterised by hydrogen-bonded chains. This behaviour is broadly reminiscent of the high-pressure and low-temperature behaviour of ethanol. The closest structural similarities are between the form 1 TFE structure and the structure of the high-pressure phase of ethanol, which both exhibit a simple alternating arrangement of molecules about the hydrogen bond within each chain. The fluoridation of the methyl group in ethanol, however, is not sufficient to make the R-group bulky enough for the closure of the molecular chains into hydrogen bonded rings.All of the O–H⋯O distances are well within the range of standard lengths for hydrogen-bonding in alcohols;2–13i.e., the electronegativity of the fluorine atoms appears to have a very limited effect on the overall structure and the structural differences observed, compared with those of ethanol, are likely to be dominated by steric effects.
As the orientation of the molecular chains in form 2 is such that the hydrogen bonds in neighbouring chains are in close proximity, with a bridging interchain O⋯O contact distance (2.892 Å) that is of the same order as the O⋯O distances found for the hydrogen bonds (∼2.72 Å), it could be expected that this region of the structure would become unstable under further compression. This instability could potentially lead to either bond formation or a further phase transition. We are currently studying TFE to higher pressures with both single-crystal X-ray diffraction and time-of-flight neutron powder-diffraction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr. Tamas Javorfi for supplying the sample and Dr. Craig Bull for helpful comments and suggestions. They are also grateful to Diamond Light Source Limited for the provision of beamtime on I19.Notes and references
- C. P. Brock and L. L. Duncan, Chem. Mater., 1994, 6, 1307 CrossRef CAS.
- D. R. Allan, S. J. Clark, M. J. P. Brugmans, G. J. Ackland and W. L. Vos, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, R11809 CrossRef CAS (and references therein).
- D. R. Allan and S. J. Clark, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 6328 CrossRef CAS.
- C. L. Bull, S. A. Barnett, D. R. Allan and W. G. Marshall, High Pressure Res., 2019, 39(1), 179 CrossRef CAS.
- P.-G. Jönsson, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 232 CrossRef.
- J. Ridout and M. R. Probert, CrystEngComm, 2014, 16, 7397 RSC.
- P. A. McGregor, D. R. Allan, S. Parsons and S. J. Clark, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 599 CrossRef PubMed.
- P. A. McGregor, D. R. Allan, S. Parsons and C. R. Pulham, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 449 CrossRef PubMed.
- D. R. Allan, S. J. Clark, A. Dawson, P. A. McGregor and S. Parsons, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 1018 CrossRef PubMed.
- I. D. H. Oswald, D. R. Allan, W. D. S. Motherwell and S. Parsons, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 69 CrossRef PubMed.
- I. D. H. Oswald, D. R. Allan, G. M. Day, W. D. S. Motherwell and S. Parsons, Cryst. Growth Des., 2005, 5, 1055 CrossRef CAS.
- S. A. Moggach, D. R. Allan, P. Lozano-Casal and S. Parsons, J. Synchrotron Radiat., 2005, 12, 590 CrossRef CAS PubMed.
- R. M. Ibberson, S. Parsons, D. R. Allan and A. M. T. Bell, Acta Crystallogr., Sect. B: Struct. Sci., 2008, 64, 573 CrossRef CAS PubMed.
- M. Heger, T. Scharge and M. A. Suhm, Phys. Chem. Chem. Phys., 2013, 15, 16065 RSC.
- J. Cosier and A. M. Glazer, J. Appl. Crystallogr., 1986, 19, 105 CrossRef CAS.
- S. A. Moggach, D. R. Allan, S. Parsons and J. E. Warren, J. Appl. Crystallogr., 2008, 41, 249 CrossRef CAS.
- H.-K. Mao, J. Xu and P. M. Bell, J. Geophys. Res.: Solid Earth, 1986, 91, 4673 CrossRef CAS.
- H. Nowell, S. A. Barnett, K. E. Christensen, S. J. Teat and D. R. Allan, J. Synchrotron Radiat., 2012, 19, 435 CrossRef CAS PubMed.
- P. Evans, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 72 CrossRef CAS PubMed; P. R. Evans and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1204 CrossRef PubMed; M. D. Winn, et al. , Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235 CrossRef PubMed; G. Winter, J. Appl. Crystallogr., 2010, 43, 186 CrossRef; G. Winter, et al. , Acta Crystallogr., Sect. D: Struct. Biol., 2018, 74, 85 CrossRef PubMed.
- CCP4 7.0.062: AIMLESS (version 0.7.2), 27/05/18.
- Rigaku Oxford Diffraction, CrysAlisPro, Rigaku Europe, Unit B6, Chaucer Business Park, Watery Lane, Kemsing Sevenoaks, Kent TN15 6QY, United Kingdom, 2017 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453 CrossRef CAS.
Footnotes |
| † CCDC Form 1 1906644, form 2 1906643. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ce00485h |
| ‡ The molecular overlay RMS (root mean square) value was calculated using the “molecule overlay” feature of Mercury24 for the two independent molecules in the asymmetric unit. Just the carbon and fluorine atoms were matched up for the calculation in order to emphasise the differences in the orientation of the hydroxyl groups. |
| This journal is © The Royal Society of Chemistry 2019 |