
A self-assembled nanotube supported by halogen bonding interactions†
Stuart R.
Kennedy
 a,
Mawgan U.
Main
b,
Colin R.
Pulham
*a,
Irene
Ling
*c and
Scott J.
Dalgarno
*b
a,
Mawgan U.
Main
b,
Colin R.
Pulham
*a,
Irene
Ling
*c and
Scott J.
Dalgarno
*b
aSchool of Chemistry, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: C.R.Pulham@ed.ac.uk
bInstitute of Chemical Sciences, Heriot-Watt University, William Perkin Building, Riccarton, Edinburgh, Scotland EH14 4AS, UK. E-mail: S.J.Dalgarno@hw.ac.uk
cSchool of Science, Monash University Malaysia, Jalan Lagoon Selatan, Bandar Sunway, 46150 Selangor, Malaysia. E-mail: ireneling@monash.edu
First published on 3rd January 2019
Abstract
Di-propoxycalix[4]arene is known to self assemble into a nanotube in the solid state. The introduction of two bromine atoms at distal positions across the upper-rim of the calixarene framework modulates nanotube packing, a feature supported by complementary halogen bonding interactions.
Controlled self-assembly remains a fundamental challenge for the supramolecular chemist, especially when one considers that many types of intermolecular interaction can play a role in directing the final outcome. Calix[4]arenes (C[4]s) are cyclic polyphenols, many of which adopt cone or pinched-cone conformations depending on the degree of functionalisation (e.g. alkylation) at the lower-rim.1 C[4]s have played a pivotal role in the development of supramolecular chemistry, and much has been learned from detailed single crystal X-ray diffraction (SCXRD) studies; an excellent example of this is the emergence and understanding of porosity in the seemingly non-porous sublimate of p-tBu-calix[4]arene.2 In the vast majority of cases, including the aforementioned example, SCXRD studies have revealed bi-layer arrangements in which the molecules alternate in an up-down anti-parallel manner. This is due to the formation of many non-covalent interactions (e.g. CH⋯π and π-stacking) between the constituent molecules within these highly favourable arrangements (Fig. 1A).3
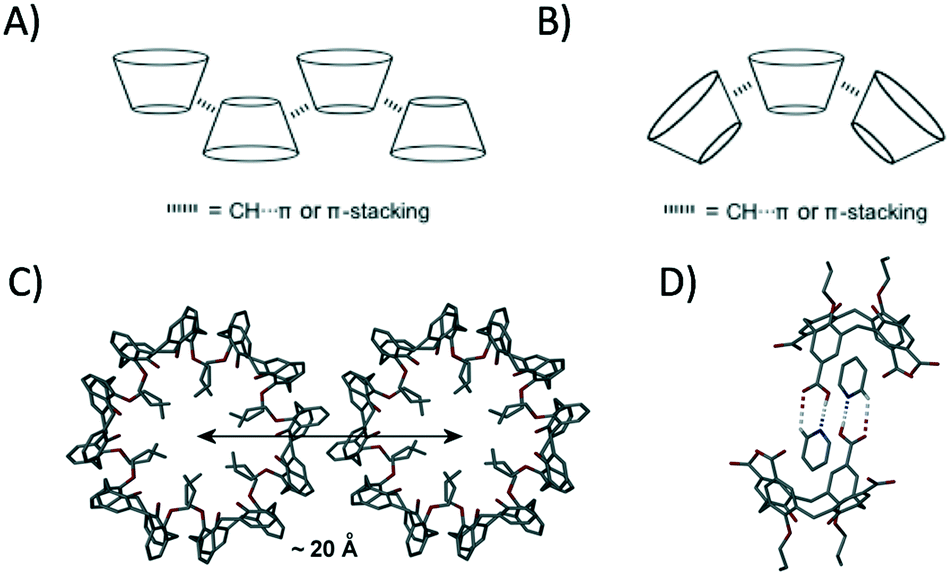 | ||
| Fig. 1 A) Schematic of anti-parallel bilayer C[4] packing. B) Schematic of parallel C[4] packing with induced curvature. C) Expanded structure of 1 showing nanotube spacing and interdigitation.5 D) Heterosynthon formation between the p-carboxylato analogue of 1 upon crystallisation from pyridine.6 Dashed lines represent hydrogen bonding interactions. H atoms omitted for clarity in C and D except for those involved in H-bonding interactions. Colour code: C – grey, O – red, N – blue, H – white. Figures not to scale. | ||
Although this is the case, there are a small number of reported examples in which cone-shaped C[4]s buck the trend, packing in an alternative parallel fashion to (typically) afford spherical or tubular assemblies; parallel packing invokes curvature in any prevailing structure because of the cone shape of the constituent building blocks (Fig. 1B).4 Di-propoxycalix[4]arene, 1, is a molecule that has been shown to pack as a triply helical nanotube in the solid state (Fig. 1C).5 The central core of the nanotube is compact, and the lower-rim propoxy chains (that point inwards) are arranged such that there is only a very small channel present. Neighbouring nanotubes pack through ‘cog-like’ interdigitation, with symmetry equivalent (s.e.) molecules self-including as shown in Fig. 1C; this interdigitation is stabilised by complementary π-stacking and CH⋯π interactions within the cavities of s.e. of 1. In previous work we showed that it was possible to modulate the packing of this nanotube motif through the introduction of carboxylic acids at the upper-rim of 1.6 Subsequent crystallisation from pyridine had the effect of increasing inter-tubule spacing by from ∼20 Å to ∼27 Å due to the formation of complementary host–guest heterosynthons between neighbouring nanotubes (Fig. 1D).
Halogen bonding has emerged as an excellent addition to the toolbox of interactions that the supramolecular chemist can exploit when seeking to control or influence self-assembly and/or host–guest chemistry.7 With this in mind, we have begun to investigate how this type of interaction may affect the assembly behaviour of C[4] building blocks, in particular those that exhibit preferences to form tubular assemblies. In this contribution we report our initial findings concerning the effect of halogen introduction at the upper-rim of the framework in 1, notably the ability to modulate nanotube packing through the formation of complementary halogen bonding interactions. We also present a concise summary of these building blocks, some of which exhibit a tendency to undergo parallel packing; this is with a view to outlining the potential to utilise a range of intermolecular interactions (including halogen bonding) to drive or control the formation of such challenging assemblies.
Upper-rim halogenation of the C[4] framework can depend on a number of different factors, an example being the presence or absence of directing groups at the lower-rim that subsequently influence functionalisation. Given that the nanotube assembly of 1 was our target for modulation, we synthesised readily accessible halogenated derivatives 2–4 as shown in Fig. 2.8–12 Crystallisation of 2 and 3 was undertaken in a range of common laboratory solvents. Single crystals were obtained in just one case, that being compound 2 from acetone, although they were not found to be a solvate (vide infra). In stark contrast, compound 4 was found to be poorly soluble, and as such it was not possible to obtain single crystals to monitor any effect of upper-rim iodination.
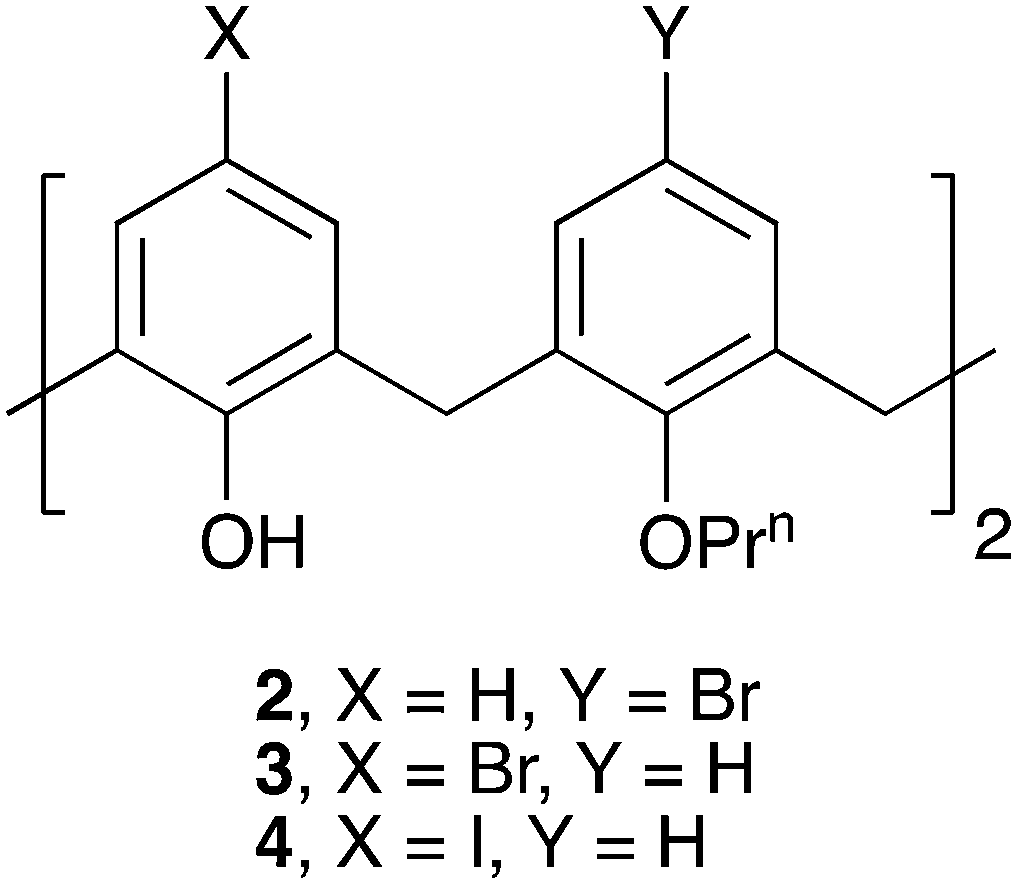 | ||
| Fig. 2 Upper-rim halogenated derivatives synthesised and used in the present study. | ||
Colourless single crystals of 2 that were suitable for diffraction studies were obtained upon dissolution in acetone and standing over a number of weeks.‡ The crystals were found to be of trigonal symmetry and structure solution was carried out in the space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) . The asymmetric unit comprises one molecule of 2, and symmetry expansion at the upper-rim reveals that s.e. molecules self-include in a cog-like manner (Fig. 3A) akin to that of 1 (Fig. 1C), despite the fact that two H atoms at the upper-rim have been replaced with bromines. Comparison of the self-included dimers in 1 and 2 reveals that the distance between centroids generated between the lower-rim oxygens has increased from 8.85 Å to 10.03 Å respectively as a direct result of halogenation coupled with self-inclusion.5 Further expansion of the structure shows a concomitant increase in the intertubule spacing, moving from ∼20 Å to ∼23 Å (compare Fig. 1C and 3B). This represents approximately half of the modulation achieved via the introduction of carboxyl groups to the framework of 1 as outlined above.6
. The asymmetric unit comprises one molecule of 2, and symmetry expansion at the upper-rim reveals that s.e. molecules self-include in a cog-like manner (Fig. 3A) akin to that of 1 (Fig. 1C), despite the fact that two H atoms at the upper-rim have been replaced with bromines. Comparison of the self-included dimers in 1 and 2 reveals that the distance between centroids generated between the lower-rim oxygens has increased from 8.85 Å to 10.03 Å respectively as a direct result of halogenation coupled with self-inclusion.5 Further expansion of the structure shows a concomitant increase in the intertubule spacing, moving from ∼20 Å to ∼23 Å (compare Fig. 1C and 3B). This represents approximately half of the modulation achieved via the introduction of carboxyl groups to the framework of 1 as outlined above.6
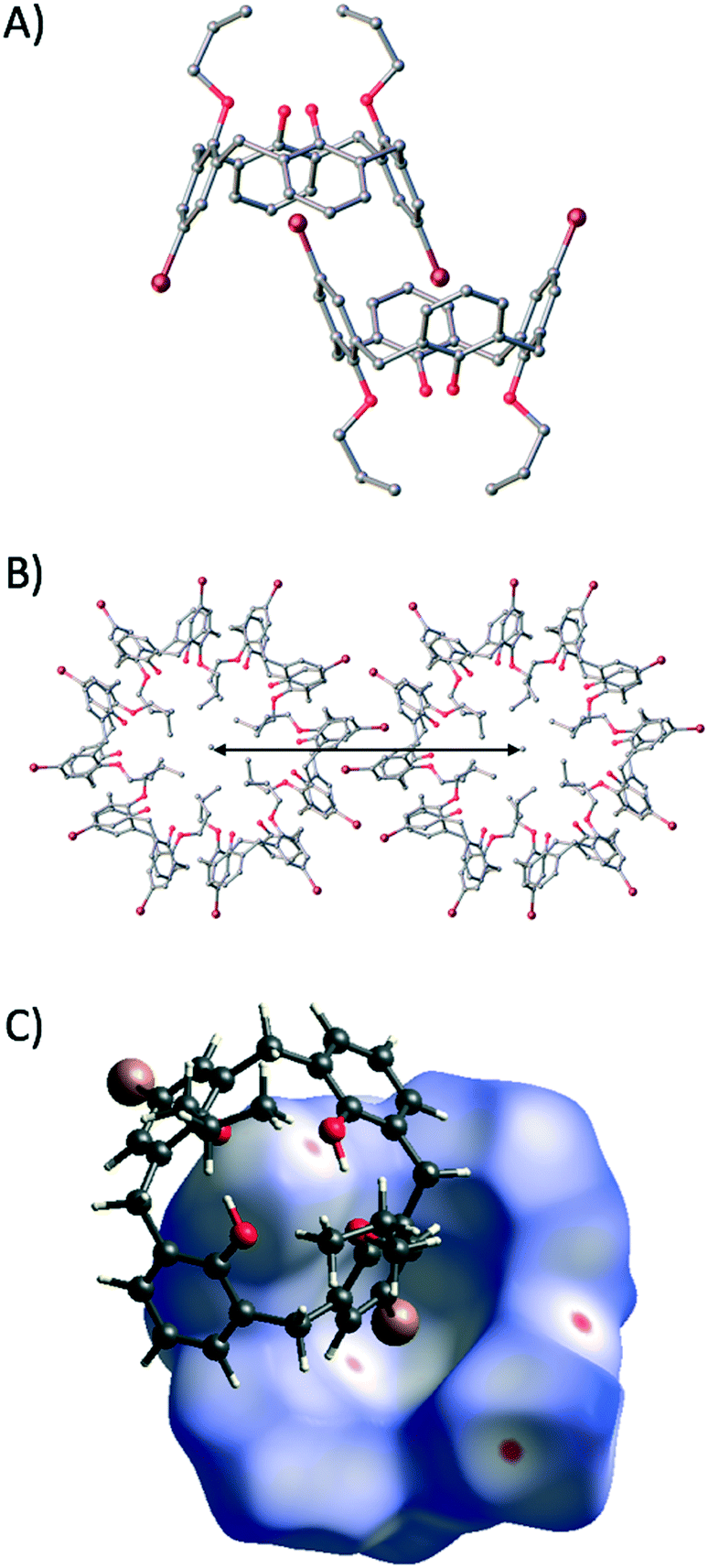 | ||
| Fig. 3 A) Self-included dimer found in the single crystal X-ray structure of 2. B) Cog-like interdigitation of nanotubes of 2 showing the intertubule spacing of ∼23 Å. C) Picture from the Hirshfeld analysis showing the Br⋯arene interaction found in the self-included dimer as a red spot on the cavity interior surface. H atoms omitted for clarity in A and B. Colour code: C – grey, O – red, H – white, Br – maroon. Figures not to scale. | ||
Given the versatility observed for this nanotube system, we thought it pertinent to survey the Cambridge Structural Database (CSD) with a view to establishing how rare these combined features are with respect to assembly. A search of the CSD13 for all structures containing lower-rim di-alkoxy C[4]s reveals that dimeric or cog-like self-inclusion is a relatively common assembly motif (41 hits).5,14,15 This self-inclusion phenomenon is also found to be tolerant towards the presence or a variety of upper-rim groups, as long as they are able to form complementary host–guest interactions with the cavity of a s.e. molecule.
Hirshfeld analysis16 of 2 (see ESI†) reveals a crystallographically unique Br⋯π interaction as shown by the red spot on the cavity interior surface in Fig. 3C; this occurs with a Br⋯C distance of 3.486 Å. Examination of the regions between the nanotubes reveals chains of Ar–H⋯Br interactions but little evidence of Br⋯Br interactions. Given that there were no directly relevant literature examples for structural comparison with 2, we expanded our search criteria accordingly as outlined below.
A search for C[4]s that are doubly substituted at the upper-rim with distally positioned halogens returned just 11 hits, 5 of which deviate from the cone conformation and thus are not useful for comparison. Four of the six remaining hits (CSD codes GUDMEY, QADTUL, WAZZOO and XIGLEE) are lower-rim tetra-substituted C[4]s,17 meaning that they necessarily adopt pinched cone conformations in the solid state, and as a consequence of this do not have cavities occupied by either guest molecules or s.e. C[4]s. One of the two remaining structures (KEHTEX) is a heterobimetallic cluster in which a lithium ion and ligated THF occupy the C[4] cavity,18 leaving just one relevant hit (XIGLAA) in which there is a di-halogenated C[4] with an open cavity suitable for forming host–guest interactions.17d Inspection of this structure reveals that the upper-rim substitution pattern is analogous to that of compound 3, as bromination has been performed para- to the lower-rim hydroxyl groups rather than the alkoxy groups; the C[4] in XIGLAA differs from 3 in that it possesses lower-rim benzyloxy rather than propoxy groups. Symmetry expansion of the ASU in XIGLAA reveals self-inclusion (Fig. S1†) in an analogous manner to that found in 1, and not involving Br⋯π interactions. Considering the structure of 2, the self-inclusion in XIGLAA may be a direct result of alternative halogenation positions, but this will require broader investigation.
Expansion of the search criteria to include C[4]s halogenated at all four upper-rim positions returned a total of 17 hits.13 Similar elimination of non-cone conformers reduced this to 10 hits, 8 of which have the C[4] in a pinched-cone conformation due to lower-rim tetra-substitution (CSD codes FAJFAX, FOQDAR, HACYIU, HACYOA, KARNIB01, NIGPOI, QADTOF and QUWLOK).17b,19 The 2 remaining hits, (CIPQEY20 and QEVBEZ21) both contain 5,11,17,23-tetrabromo-25,27-dimethoxy-26,28-dihydroxycalix[4]arene. The cavity of the C[4] in CIPQEY is occupied by a different guest molecule so can be eliminated for the purposes of comparison with 2. Symmetry expansion of the ASU in QEVBEZ does generate a self-included dimer and, interestingly, Hirshfeld analysis16 reveals that there are no short Br⋯π interactions (Fig. S2†) as is the case for 2. This is surprising, and appears to be due to slight shifting of the C[4]s such that they are aligned in a more symmetric manner (i.e. less offset within the confines of the cavity).
Further expansion to include calix[4]arene and a halobenzene fragment returned 11 hits,13 none of which are relevant or useful for comparison with the halogen interactions present in the structure of 2. Finally, expansion of criteria to include other halogenated guests, as well as other structurally related hosts, returned a number of hits reported in a study by Diederich and co-workers, all of which relate to the elegant encapsulation of monohalo- and (±)-trans-1,2-dihalocyclohexanes in enantiopure alleno-acetylnic cages.22 Although markedly different with respect to host structure, these complexes are useful for comparison with the structure of 2, as halogen⋯π interactions occur to varying extents depending on the particular guest being encapsulated. In these examples, a Br⋯π distance of 3.6 Å is recorded for a well confined guest in the resorcinarene-based cavitand host, and the average interaction distance can be correlated to polarizability of the halogen.
The shorter Br⋯π distance observed in 2 may be due to a series of factors that likely includes the propensity of 1 to pack in a parallel manner/in nanotubes. That said, a wide-ranging study of halogenated C[4] derivatives, if one can overcome solubility/synthetic issues, would provide great insight into halogen polarizability effects with respect to self-assembly with these multi-component systems.
Conclusions
To conclude, we have reported the modulation of a self-assembled C[4] nanotube through the formation of halogen bonding interactions. A detailed survey of the CSD reveals that there is relatively little structural information available for useful comparison, suggesting that much is yet to be unearthed with respect to halogen bonding and its use in the controlled self-assembly of C[4]-based building blocks. This is particularly interesting as one may potentially utilise the C[4] cavity in concert with synthetic alteration at various regions of the molecular framework in order to promote the construction of targeted assemblies. For example, the ability to control or drive parallel packing of C[4]s in order to reliably form nanotube or spherical assemblies is an extremely challenging goal, but one that may be achieved once an enhanced understanding of how specific interactions within the C[4] cavity guide assembly protocols. Future work will focus on expanding our library of halogenated C[4] derivatives, addressing solubility issues, and using halogen bonding interactions to promote the formation of systems that deviate from anti-parallel bi-layer packing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Royal Society Newton Mobility Fund for supporting this work under grant reference NI170151. We would also like to thank Dr Gary Nichol for assistance with collection of single crystal diffraction data.Notes and references
- For example of synthetic alteration and subsequent conformational control over the calix[4]arene framework see: C. D. Gutsche, Calixarenes 2001, Kluwer Academic Publishers, Dordrecht, 2001, ch. 1 Search PubMed; I. Thondorf, A. Shivanyuk and V. Böhmer, Calixarenes 2001, Kluwer Academic Publishers, Dordrecht, 2001, ch. 1 Search PubMed.
- S. J. Dalgarno, P. K. Thallapally, L. J. Barbour and J. L. Atwood, Chem. Soc. Rev., 2007, 36, 236 RSC , and referneces therein.
- For a relevant review of calix[4]arene bi-layer formation see: J. L. Atwood, L. J. Barbour, M. J. Hardie and C. L. Raston, Coord. Chem. Rev., 2001, 222, 3 CrossRef CAS.
- For example see: G. W. Orr, L. J. Barbour and J. L. Atwood, Science, 1999, 285, 1049 CrossRef CAS PubMed; I. Ling, H. Kumari, M. Mirzamani, A. N. Sobolev, C. J. Garvey, J. L. Atwood and C. L. Raston, Chem. Commun., 2018, 54, 10824 RSC.
- L. G. Kuz'mina, G. G. Sadikov, J. A. K. Howard, E. A. Shokova and V. V. Kovalev, Kristallografiya, 2003, 48, 272 CrossRef CAS; S. Pakhomova, J. Ondracek, M. Vindys and I. Stibor, Z. Kristallogr. - New Cryst. Struct., 1997, 212, 459 Search PubMed.
- S. Kennedy and S. J. Dalgarno, Chem. Commun., 2009, 5275 RSC; S. Kennedy, P. Cholewa, R. D. McIntosh and S. J. Dalgarno, CrystEngComm, 2013, 15, 1520 RSC.
- For examples of recent reviews please see: A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514 CrossRef CAS PubMed; G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef PubMed; K. Rissanen, Chem. Soc. Rev., 2017, 46, 2638 RSC.
- Unfortunately it is not possible to isolate the entire series of analogues due to synthetic challenges, in particular the chloro derivatives. This would have been ideal for a full system analysis, but is unavoidable.
- To our knowledge it is not possible to isolate fully upper-rim halogenated analogues of 1.
- For the synthesis of 2 see: O. Hudecek, P. Curinova, J. Budka and P. Lhoták, Tetrahedron, 2011, 67, 5213 CrossRef CAS.
- For the synthesis of 3 see: V. Stastny, P. Lhoták, V. Michlová, I. Stibor and J. Sykora, Tetrahedron, 2002, 67, 7207 CrossRef.
- For the synthesis of 4 see: B. Klenke and W. Friedrichsen, J. Chem. Soc., Perkin Trans. 1, 1998, 3377 RSC.
- Correct as of 16/10/18, CSD version 5.39, 2018.
- (a) M. Duta, Z. Asfari, A. Hagege, P. Thuery and M. Leroy, Supramol. Chem., 2004, 16, 205 CrossRef CAS; (b) A. N. Lazar, N. Dupont, A. Navaza and A. W. Coleman, Chem. Commun., 2006, 1076 RSC; (c) K. No, H. J. Lee, K. M. Park, S. S. Lee, K. H. Noh, S. K. Kim, J. Y. Lee, J. S. Kim and J. Heterocyclic, Chem, 2004, 41, 211 CAS; (d) S. Banthia and A. Samanta, Org. Biomol. Chem., 2005, 3, 1428 RSC; (e) B. Bensenane, Z. Asfari, C. Platas-Iglesias, D. Esteban-Gómez, F. Djafri, M. Elhabiri and L. J. Charbonnière, Dalton Trans., 2016, 45, 15211 RSC; (f) Z.-G. Luo, Y. Zhao, C. Ma, L. Cao, S.-H. Ai, J.-S. Hu and X.-M. Xu, Jiegou Huaxue, 2014, 33, 1117 CAS; (g) P. J. A. Kenis, E. G. Kerver, B. H. M. Snellink-Ruel, G. J. van Hummel, S. Harkema, M. C. Flipse, R. H. Woudenberg, J. F. J. Engbersen and D. N. Reinhoudt, Eur. J. Org. Chem., 1998, 1089 CrossRef CAS; (h) P. Seigle-Ferrand, S. B. Sdira, C. Felix, R. Lamartine, C. Bavoux, B. Fenet, F. Bayard and F. Vocanson, Mater. Sci. Eng., C, 2006, 26, 181 CrossRef CAS; (i) P. G. Jones and M. Freytag, CSD Commun., 2010 Search PubMed; (j) S. Patra, R. Gunupuru, R. Lo, E. Suresh, B. Ganguly and P. Paul, New J. Chem., 2012, 36, 988 RSC; (k) K. Takenaka, Y. Obora and Y. Tsuji, Inorg. Chim. Acta, 2004, 357, 3895 CrossRef CAS; (l) F. Vita, M. Vorti, G. Orlandini, V. Zanichelli, C. Massera, F. Ugozzoli, A. Arduini and A. Secchi, CrystEngComm, 2016, 18, 5017 RSC; (m) M. Duta, Z. Asfari and P. Thuery, CSD Commun., 2006 Search PubMed; (n) H. Halouani, I. Dumazet-Bonnamour, M. Perrin and R. Lamartine, J. Org. Chem., 2004, 69, 6521 CrossRef CAS PubMed; (o) C. Redshaw, O. Rowe, D. L. Hughes, A.-M. Fuller, I. A. Ibarra and S. M. Humphrey, Dalton Trans., 2013, 42, 1983 RSC; (p) X. Zeng, X. Han, L. Chen, Q. Li, F. Xu, X. He and Z.-Z. Zhang, Tetrahedron Lett., 2002, 43, 131 CrossRef CAS; (q) X. Zeng, L. Weng, L. Chen, F. Xu, Q. Li, X. Leng, X. He and Z.-Z. Zhang, Tetrahedron, 2002, 58, 2647 CrossRef CAS; (r) Z. Bo, Y.-Z. Li, X.-F. Lu and G.-Y. Lu, J. Chem. Crystallogr., 2005, 35, 281 CrossRef CAS; Z. Bo, C.-Z. Zhang, G.-Y. Lu and F. Liu, Chin. J. Chem., 2006, 24, 124 Search PubMed; (s) H. M. Chawla, S. P. Singh and S. Upreti, Tetrahedron, 2006, 62, 9758 CrossRef CAS; (t) K. Stein and R. Schnorr, CSD Commun, 2015 Search PubMed; (u) P. Thuery, M. Lance, M. Nierlich, N. Reynier, V. Lamare, J.-F. Dozol, M. Saadioui, Z. Asfari and J. Vicens, An. Quim., 1997, 93, 324 CAS; (v) M. Fehlinger and W. Abraham, J. Inclusion Phenom. Macrocyclic Chem., 2007, 58, 263 CrossRef CAS; (w) P. Rashatasakhon, A. Jaiyu, R. Rojanathanes, N. Muangsin, N. Chaichit and M. Sukwattanasinitt, J. Mol. Struct., 2010, 963, 22 CrossRef CAS; (x) S. K. Kim, S. H. Kim, H. J. Kim, S. H. Lee, S. W. Lee, J. Ko, R. A. Bartsch and J. S. Kim, Inorg. Chem., 2005, 44, 786 Search PubMed; (y) D. Maity, M. Bhatt, M. L. Desai, E. Suresh, M. K. Si, V. P. Boricha, B. Ganguly and P. Paul, Supramol. Chem., 2017, 29, 600 CrossRef CAS; (z) S. Ullmann, R. Schnorr, C. Laube, B. Abel and B. Kersting, Dalton Trans., 2018, 47, 5801 RSC.
- (a) S. Ullmann, R. Schnorr, M. Handke, C. Laube, B. Abel, J. Matysik, M. Findeisen, R. Rüger, T. Heine and B. Kersting, Chem. – Eur. J., 2017, 23, 3824 CrossRef CAS PubMed; (b) A. Guelzim, S. Khrifi, F. Baert, M. Saadioui, Z. Asfari and J. Vicens, Cryst. Struct. Commun., 1958, 199, 53 Search PubMed; (c) Z.-Y. Xie, N.-T. Hou, Y.-Z. Zhu, H.-B. Song and J.-Y. Zheng, Chem. Lett., 2008, 37, 478 CrossRef CAS; (d) A. McConnell, C. Serpell and P. D. Beer, New J. Chem., 2012, 36, 102 RSC; (e) H. M. Chawla, S. P. Singh, S. N. Sahu and S. Upreti, Tetrahedron, 2006, 62, 7854 CrossRef CAS; (f) J. P. Ward, J. M. White and C. G. Young, Tetrahedron, 2013, 69, 8824 CrossRef CAS; (g) V. Ramakrishna, S. Patra, E. Suresh, A. K. Bhatt, P. A. Bhatt, A. Hussain and P. Paul, Inorg. Chem. Commun., 2012, 22, 85 CrossRef CAS; (h) A. M. Reichwein, W. Verboom, S. Harkema, A. L. Spek and D. N. Reinhoudt, J. Chem. Soc., Perkin Trans. 2, 1994, 1167 RSC; (i) N. Dupont, A. N. Lazar, F. Perret, O. Danylyuk, K. Suwinska, A. Navaza and A. W. Coleman, CrystEngComm, 2008, 10, 975 RSC; (j) H. Halouani, I. Dumazet-Bonnamour, C. Duchamp, C. Bavoux, N. Ehlinger, M. Perrin and R. Lamartine, Eur. J. Org. Chem., 2002, 4202 CrossRef CAS; (k) P. Kuhn, D. Semeril, C. Jeunesse, D. Matt, P. J. Lutz, R. Louis and M. Neuburger, Dalton Trans., 2006, 3647 RSC; (l) S. J. Coles, C. W. Hall and M. B. Hursthouse, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, o29 CrossRef; (m) F. Perret, A. N. Lazar, O. Shkurenko, K. Suwinska, N. Dupont, A. Navaza and A. W. Coleman, CrystEngComm, 2006, 8, 890 RSC.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627 CrossRef CAS PubMed; M. A. Spackman and P. G. Byrom, Chem. Phys. Lett., 1997, 267, 215 CrossRef.
- (a) L. Grubert, H. Henning and W. Abraham, Tetrahedron, 2009, 65, 5936 CrossRef CAS; (b) D. R. Evans, M. Huang, J. C. Fettinger and T. L. Williams, Inorg. Chem., 2002, 41, 5986 CrossRef CAS; (c) J. Han, F.-L. Wang, Y.-X. Liu, F.-Y. Zhang, J.-B. Meng and Z.-J. He, ChemPlusChem, 2012, 77, 196 CrossRef CAS; (d) K. Takenaka, Y. Obora, L.-H. Jiang and Y. Tsuji, Organometallics, 2002, 21, 1158 CrossRef CAS.
- L. Liu, L. N. Zakharov, J. A. Golen, A. L. Rheingold, W. H. Watson and T. A. Hanna, Inorg. Chem., 2006, 45, 4247 CrossRef CAS PubMed.
- L. J. Barbour, G. W. Orr and J. L. Atwood, Chem. Commun., 1998, 1901 RSC; M. Osipov, Q. Chu, S. J. Geib, D. P. Curran and S. G. Weber, Beilstein J. Org. Chem., 2008, 4, 36 Search PubMed; S. E. Matthews, V. Felix, M. G. B. Drew and P. D. Beer, Org. Biomol. Chem., 2003, 1, 1232 RSC; P. G. Jones and M. Freytag, CSD Communication, 2010 RSC; L. J. Barbour, G. W. Orr and J. L. Atwood, Chem. Commun., 1997, 1439 RSC.
- F. Maharaj, D. C. Craig, M. L. Scudder, R. Bishop and N. Kumar, J. Inclusion Phenom. Macrocyclic Chem., 2007, 59, 17 CrossRef CAS.
- Y.-H. Luo, X.-J. Hu, J.-K. Liu, H. Zhang, Y. Li, H.-J. Yang and R.-J. Want, Z. Kristallogr. - New Cryst. Struct., 2006, 221, 327 CAS.
- C. Gropp, T. Husch, N. Trapp, M. Reiher and F. Diederich, J. Am. Chem. Soc., 2017, 139, 12190 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the Hirshfeld analyses, additional figures to support discussion and the crystallographic information file (CIF) for 2. CCDC 1875214. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce01824c |
‡ Compounds 2–4 were synthesised according to literature procedures.10–12 Crystallisation of 2: compound 2 was dissolved in acetone and diffraction quality single crystals grew upon standing over a number of weeks. Crystal data for compound 2 (CCDC 1875214): C34H34Br2O4, M = 666.43 g mol−1, trigonal, space group R, a = 37.3853(7) Å, c = 11.2109(2) Å, V = 13569.8(6) Å3, Z = 18, T = 120.01(10) K, μ(Mo Kα) = 2.725 mm−1, Dcalc = 1.468 g cm−3, 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 040 reflections measured (5.766° ≤ 2Θ ≤ 59.31°), 7953 unique (Rint = 0.0668, Rsigma = 0.0602) which were used in all calculations. The final R1 was 0.0523 (I > 2σ(I)) and wR2 was 0.1164 (all data). 040 reflections measured (5.766° ≤ 2Θ ≤ 59.31°), 7953 unique (Rint = 0.0668, Rsigma = 0.0602) which were used in all calculations. The final R1 was 0.0523 (I > 2σ(I)) and wR2 was 0.1164 (all data). |
| This journal is © The Royal Society of Chemistry 2019 |