
Dual-functional fluorescent molecular rotor for endoplasmic reticulum microviscosity imaging during reticulophagy†
Ying
He
a,
Jinwoo
Shin
b,
Wanjun
Gong
a,
Pintu
Das
a,
Jinghan
Qu
a,
Zhigang
Yang
 *a,
Wufan
Liu
a,
Chulhun
Kang
c,
Junle
Qu
*a and
Jong Seung
Kim
*b
*a,
Wufan
Liu
a,
Chulhun
Kang
c,
Junle
Qu
*a and
Jong Seung
Kim
*b
aKey Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Province, College of Optoelectronic Engineering, Shenzhen University, Shenzhen 518060, China. E-mail: zhgyang@szu.edu.cn; jlqu@szu.edu.cn
bDepartment of Chemistry, Korea University, Seoul 02841, Korea. E-mail: jongskim@korea.ac.kr
cGraduate School of East-West Medical Science, Kyung Hee University, Yongin 446-701, Korea
First published on 30th January 2019
Abstract
The microviscosity change associated with reticulophagy is an important component for studying endoplasmic reticulum (ER) stress disorders. Here, a BODIPY-arsenicate conjugate 1-based fluorescent molecular rotor was designed to covalently bind vicinal dithiol-containing proteins in the ER, exhibiting a bifunction of reticulophagy initiation and microviscosity evaluation. Therefore, we could quantify the local viscosity changes during reticulophagy based on the fluorescence lifetime changes of probe 1.
Autophagy is a highly conserved cellular process in eukaryotes that is critical for cell survival and maintenance. In particular, autophagy can catabolize cytoplasmic organelles, proteins, and macromolecules for removing defective structures (e.g., misfolded proteins), recycling components, or energy production.1 Autophagy of the endoplasmic reticulum (ER), termed reticulophagy, is a form of selective autophagy that is linked to the unfolded protein response for maintaining cell homeostasis.2 Reticulophagy largely contributes to the protein quality control process in the ER; however, the accumulation of unfolded or misfolded proteins in the ER can lead to ER stress,3–5 resulting in several pathophysiological processes and severe diseases such as tumorigenesis,6 neurodegenerative diseases,7 and diabetes.8,9 Therefore, gaining a comprehensive understanding of the reticulophagy process could provide a promising approach for the diagnosis and therapy of these diseases.10
Biological processes of protein interactions, mobility, and folding that frequently occur in the ER are largely determined by local viscosity at a reasonable speed.11–15 During ER stress, the large amount of unfolded or misfolded proteins accumulating in the ER leads to marked changes in viscosity.16 Cellular viscosity plays a very important role in governing diffusion-mediated processes; accordingly, abnormal changes in viscosity have been directly linked to several diseases and pathologies.13 In reticulophagy, the ER is partially engulfed by the lysosome to form an autophagosome. Since the local environmental conditions, including viscosity and pH, in the autophagosomes would be markedly different from those of the ER, focusing on the changes in local viscosity may contribute to gaining a better understanding of the autophagy process.
Fluorescence lifetime imaging (FLIM) is an ideal tool to quantitatively study the cellular environment, and has been widely used to study autophagic processes.17–19 Recently, various molecular rotors have been reported to sense and quantify local viscosity by FLIM in living cells15,20–22 or in other complex environments.23–25 In our previous work, we developed a probe with two modalities of fluorescent ratiometry and lifetime imaging to measure intracellular viscosity, including in the mitochondria and ER.15,20 Arsenicals such as 1,3,2-dithiarsenolane are able to selectively bind to vicinal dithiol-containing proteins (VDPs),26 and various fluorogenic probes have been developed to image VDPs in cancer cells or mitochondria.27–33 Such probes can also be used to label nascent proteins with vicinal dithiols in the ER, which would inhibit the folding or lead to the misfolding of these VDPs to subsequently initiate reticulophagy.27 However, few studies have investigated the viscosity changes occurring around nascent proteins during reticulophagy. Therefore, fluorescent probes capable of specifically imaging the distribution of VDPs in the ER and quantifying the concurrent local viscosity variations during reticulophagy have attracted a great deal of attention.34
In this context, we have investigated the viscosity variations of the ER during reticulophagy with a dual-functional molecular rotor (probe 1), which can be used as an ER stress initiator by labeling VDPs and simultaneously acting as a microviscosity sensor. The fluorescent probe was composed of a boron-dipyrromethene (BODIPY)-based fluorescent rotor and an moiety (Scheme 1). The probe can sense local viscosity, which is then visualized by FLIM microscopy, and its arsenicate moiety can covalently bind to nascent proteins with vicinal dithiols to prohibit their folding or induce their misfolding, resulting in ER stress and ultimately reticulophagy. Concurrently, through FLIM imaging, the BODIPY molecular rotor can quantify the consequent viscosity changes around the VDPs in the ER of live cells.
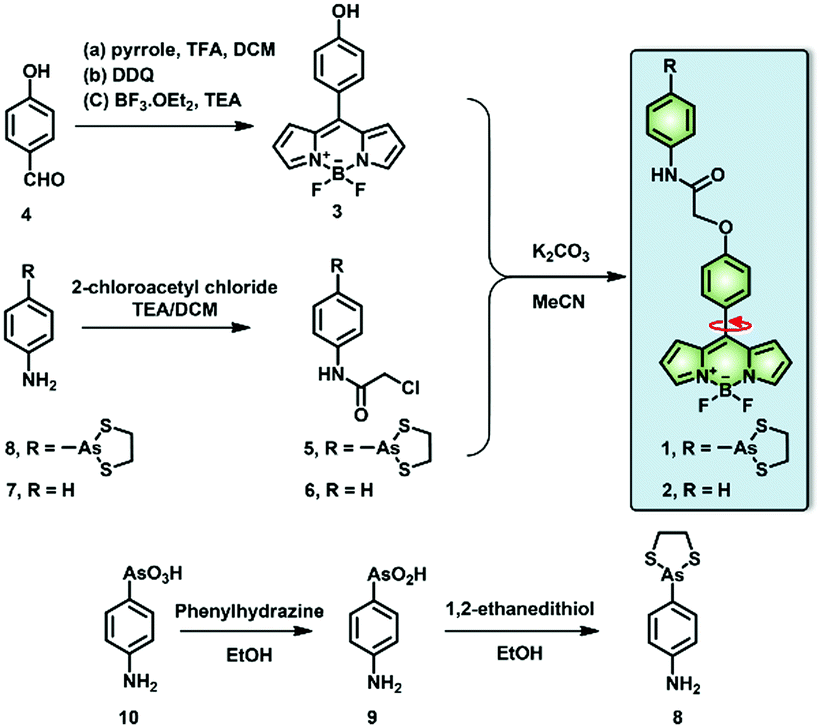 | ||
| Scheme 1 Synthetic scheme of the target molecular rotors (1, 2). | ||
First, all compounds were prepared according to the procedures detailed in our previous study,15 and were well characterized by 1H, 13C NMR and mass spectrometry (Fig. S13–S26, ESI†). Second, to validate the use of probe 1 in determining local viscosity, we first examined the fluorescence response of probe 1 towards arbitrary changes in viscosity. The fluorescent spectra and fluorescence decay of probe 1 were measured in binary solvent systems of water–glycerol (Fig. 1) and ethanol–glycerol (Fig. S1 and S2, ESI†), respectively. The fluorescence intensity of probe 1 (516 nm) increased markedly with the increase of solvent viscosity at room temperature (25 °C) [Fig. 1(a)]. With the change from water (0.8 cP) to glycerol (95%, ca. 767 cP), the fluorescence intensity linearly increased by approximately 20-fold (R2 = 0.99) [Fig. 1(b)]. The fluorescence lifetime (τ) of probe 1 also increased with an increase in the viscosity of the solution [Fig. 1(c)] from approximately 0.5 ns in water to approximately 4.6 ns in 99% glycerol, showing a strong exponential relationship (R2 = 0.99) [Fig. 1(d)]. This increase of fluorescence lifetime can be interpreted as gradual suppression of the free rotation around the C–C bond between BODIPY and the phenyl unit by increasing solvent viscosity, resulting in a decrease in energy consumption in the form of non-radiation.15 To verify the stability of probe 1, we examined the fluorescence intensity changes under continuous light exposure [Fig. S3(a), ESI†]. To eliminate the metal-effect and pH-effect, we examined the fluorescence response of probe 1 to different biologically abundant metal ions and different pH solutions [Fig. S3(b and c), ESI†].
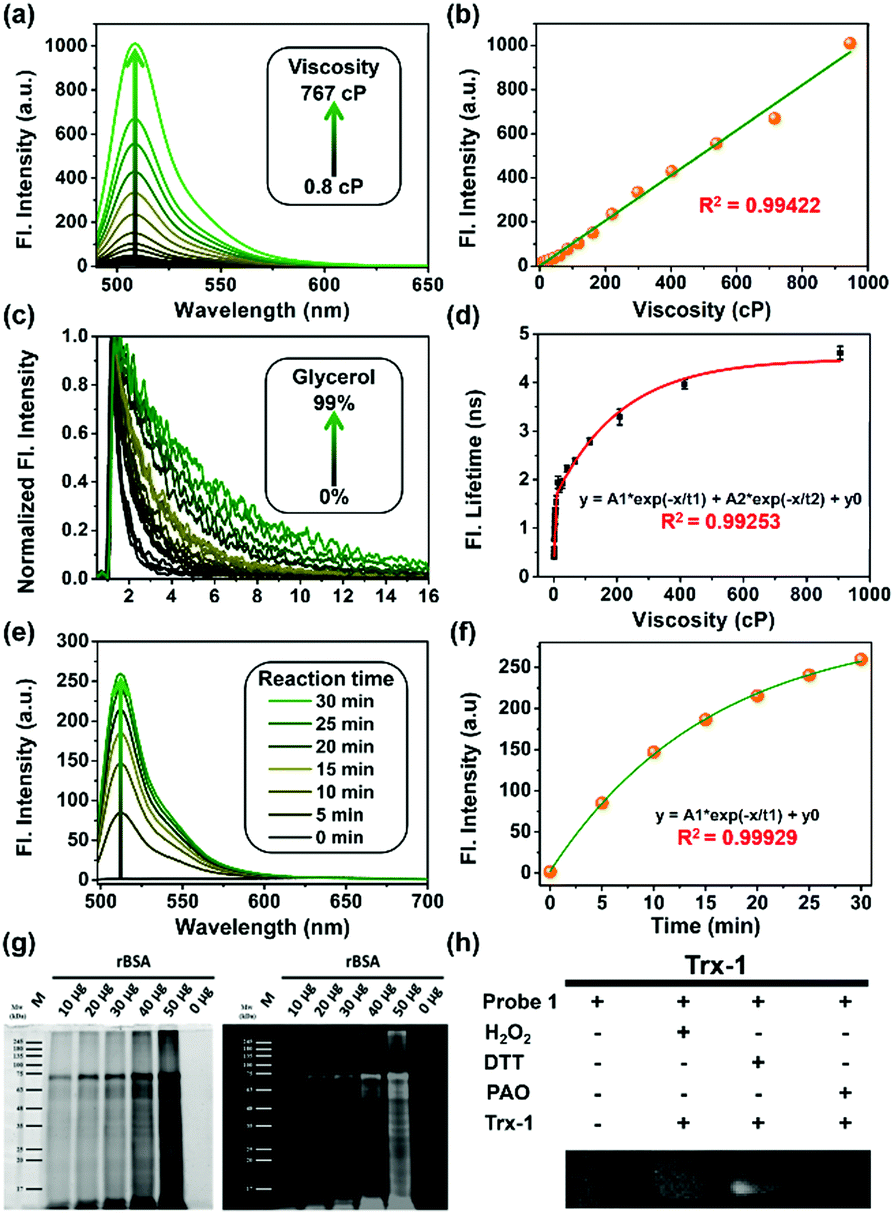 | ||
| Fig. 1 (a) Fluorescence changes and (c) lifetime decay of probe 1 (2 μM) with the variation of viscosity in a water–glycerol binary solution. (b) Linear relationship between fluorescence intensity and viscosity in mixed solvents. (d) Exponential relationship between fluorescence lifetime and solution viscosity excited at 488 nm, collected at 500–530 nm, using a TCSPC commercial DCS-120 FLIM system (Becker & Hickl, Germany). (e) Fluorescent changes in probe 1 (1 μM) upon reaction with BSA. (f) Fluorescence intensity changes of probe 1vs. reaction time. (g) SDS-PAGE results showing that probe 1 covalently bound to different amounts of reduced BSA. (h) Reduced thioredoxin-1 (rTrx-1) in the presence of different reagents; DTT was used to reduce Trx-1, H2O2 was used to oxidize Trx-1, and 4-aminophenylarsenic (PAO) is a popular specific ligand for protein vicinal dithiols. | ||
Next, to verify the reactivity of probe 1 towards VDPs, it was incubated with various proteins. Bovine serum albumin (BSA) and thioredoxin-1 (Trx-1) were first treated with dithiothreitol (DTT) to obtain reduced forms of the proteins (rBSA and rTrx-1, respectively). Then, rBSA or rTrx-1 was incubated with probe 1 (1 μM) at 37 °C through vortex mixing. As shown in Fig. 1, the fluorescence of probe 1 increased with prolonged incubation time [Fig. 1(e and f)], and was saturated upon incubation for approximately 30 min, demonstrating the binding between probe 1 and the VDPs. This interaction was further confirmed by SDS-PAGE analysis [Fig. 1(g)]. The protein bands with molecular weight of ∼66 kDa, observed from both Coomassie Brilliant Blue (CBB) staining and fluorescent imaging, became wider with increasing amounts of rBSA [Fig. 1(g)]. Furthermore, different forms of Trx-1 were used to examine the binding selectivity of probe 1 towards vicinal dithiols. Trx-1 was treated with H2O2, DTT, and 4-aminophenylarsenic, respectively, and then incubated with probe 1. The SDS-PAGE results showed that only dithiols (rTrx-1) could be selectively labeled by the arsenic moiety [Fig. 1(h)]. The reaction of fluorescent probes 1 and 2 (1 μM) with rBSA are shown in Fig. S4 (ESI†). Furthermore, we confirmed that probe 1 could covalently bind the target proteins in vitro (Fig. S5–S7, S9 and S10, ESI†).
In contrast to probe 1, the reference compound 2, which does not possess the As-moiety, could not form covalent bonds with VDPs (Fig. S5, S6 and S8–S10, ESI†). These results suggest that probe 1 was specific for VDPs in the nascent proteins in the ER, which could result in misfolded or unfolded VDPs in the ER, leading to ER stress.
Co-localization experiments were carried out to examine the sub-organelle localization of probe 1 inside live HeLa cells. Commercially available Mito-, Lyso- and ER-trackers were incubated separately for 5 min with probe 1 (1 μM) in HeLa cells, and then confocal images were taken using a Leica SP8 microscope with separate excitation and collecting windows. As shown in the merged images in Fig. 2, probe 1 and ER-Tracker showed the best overlapping fluorescence, in a punctuated pattern, implying that probe 1 predominantly localizes in the ER [Fig. 2(c), Pearson's coefficient r = 0.98]. Probe 1 was also found to partly co-localize with the Mito- and Lyso-trackers, albeit with faint intensities [Fig. 2(f) and (i), r = 0.63 and 0.62, respectively]. Probe 1 was further incubated with HeLa cells and then treated with Lyso-Tracker. Initially, probe 1 and Lyso-Tracker were found to be distinctly localized at the ER and lysosome, respectively. However, after 1 h of incubation, some green fluorescence (probe 1) was also observed in the organelles labeled with Lyso-Tracker, indicating that the ER gradually fused with the lysosome upon the interaction of probe 1 with the ER (Fig. S11 and S12, ESI†). As the ER fused with the lysosome, the Pearson's coefficient increased from 0.62 to 0.82 (Fig. S11, ESI†), demonstrating that probe 1 can induce reticulophagy.
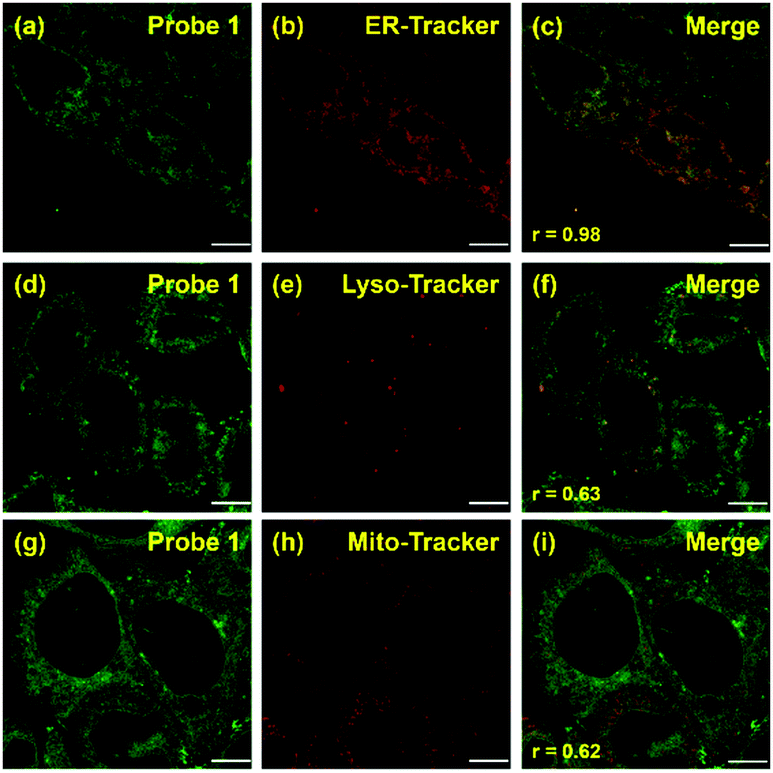 | ||
| Fig. 2 Fluorescent imaging of probe 1 in HeLa cells. The co-localization experiment was carried out by co-incubating probe 1 [1 μM, (a), (d), and (g); green] with ER-, Lyso-, and Mito-Trackers [1 μM, (b), (e), and (h), respectively; red]. Merged images [(c), (f), and (i)] were obtained by overlapping (a), (d), and (g) with (b), (e), and (h), respectively. Scale bar = 10 μm (Pearson's coefficient is specified as r [(c), (f), and (i)]). | ||
 | ||
| Fig. 3 Immunofluorescence imaging of LC3B staining in HeLa cells. Panels (a–c) represent the HeLa cells incubated without probe 1, and panels (d–f) represent the HeLa cells incubated with probe 1 (1 μM) for 1 h. (a–d) Excited at 647 nm, collected at 660–730 nm using a Leica SP8 confocal microscope system; scale bar = 10 μm. Alexa Fluor 647-conjugated goat to rabbit IgG was used as the secondary antibody (Abcam, catalog no. ab150079). | ||
To further verify that probe 1 can induce cell reticulophagy, we used LC3B antibody [Fig. 3(a)]. LC3B is a mammalian homolog of the autophagy-related protein Atg8 in yeast, which is the most commonly used tool in reticulophagy research.35–38 After probe 1 was incubated with HeLa cells for 1 h, the LC3B was observed to aggregate in the cytoplasm, indicating the occurrence of autophagy [Fig. 3(a and d)]. The interaction between the ER and lysosomes was further investigated through FLIM, and the changes in microviscosity were measured during the initiation of ER–lysosome fusion. Probe 1 (1 μM) was incubated with HeLa cells for either 5 min or 1 h, and then images were obtained using the Leica TCS SP8 confocal microscope. As shown in Fig. 4(c and d), the average lifetime was found to increase from 2.36 ns in the ER to 2.74 ns, with a corresponding increase in the viscosity from 66 to 107 cP. As reticulophagy of the cells was induced, the lifetime in the ER increased from 2.37 to 2.61 ns, with a corresponding increase in the viscosity from 67 to 92 cP. In the ER–lysosome-fused globules such as autolysosomes, the lifetime was 3.3 ns with a corresponding viscosity of 188 cP [Fig. 4(d)]. The viscosity was calculated as the exponential relationship between fluorescence lifetime vs. viscosity (Fig. 1), demonstrating that the viscosity in the ER increased during the fusion with the lysosome in the course of reticulophagy. Our results therefore suggest that HeLa cells could release the ER stress by initiating the fusion of the ER with lysosomes to digest the unfolded (or misfolded) VDPs, which in turn increased the internal viscosity. These findings further confirmed that probe 1 specifically enables measurements of viscosity changes associated with reticulophagy.
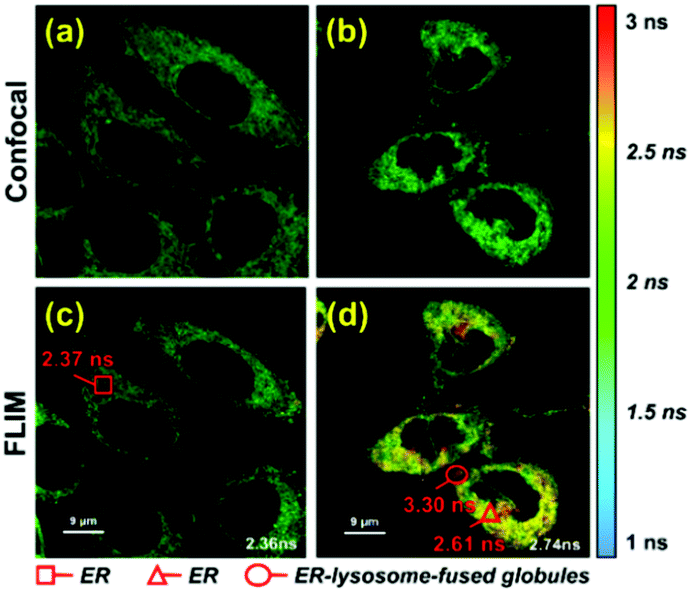 | ||
| Fig. 4 Fluorescent imaging of probe 1 in HeLa cells with different incubation times. (a) Fluorescent imaging and (c) fluorescence lifetime imaging (FLIM) of probe 1 (1 μM) incubated with HeLa cells for 5 min; and (b) fluorescent imaging and (d) FLIM of probe 1 (1 μM) co-cultured with HeLa cells for 1 h. Cells were excited at 488 nm, and measurements were collected at 500–530 nm for FLIM imaging. | ||
In conclusion, we developed a microenvironment-sensitive, dual-functional fluorescent probe 1, which is able to induce intracellular reticulophagy while also allowing for quantification of the local viscosity changes of the ER in live cells, thereby providing a quantitative method to monitor the dynamic autophagic processes in live cells. The bifunctional probe was composed of a molecular rotor (BODIPY) and a phenylarsenicate moiety, which can covalently bind to VDPs to initiate ER stress and to sense the viscosity changes with good exponential responses. Moreover, using probe 1, we explored the dynamic changes of the microenvironment in sub-organelles during the formation of autolysosomes, which exhibited the great potential of the probe for monitoring viscosity changes of the ER, and thereafter visualizing biological events of significance. Furthermore, probe 1 was able to specifically label VDPs in the nascent proteins in the ER, which could result in ER stress because of the accumulation of misfolded or unfolded VDPs. The viscosity increased in HeLa cells, and the ER stress was released by fusion of the ER with lysosomes to digest the unfolded (or misfolded) VDPs. Therefore, probe 1 could be a promising tool for the study of ER autophagy.
This work has been partially supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (CRI project no. 2018R1A3B1052702, JSK) and the National Natural Science Foundation of China (61525503/61875131/61620106016/61835009/81727804); the Basic Research Program of China (2015CB352005); Guangdong Natural Science Foundation Innovation Team (2014A030312008); and Shenzhen Basic Research Project (JCYJ20170818100931714/JCYJ20150930104948169).
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. C. Kimmelman and E. White, Cell Metab., 2017, 25, 1037–1043 CrossRef CAS PubMed.
- S. Bernales, S. Schuck and P. Walter, Autophagy, 2007, 3, 285–287 CrossRef PubMed.
- H. O. Rashid, R. K. Yadav, H. R. Kim and H. J. Chae, Autophagy, 2015, 11, 1956–1977 CrossRef CAS PubMed.
- W. S. Lee, W. H. Yoo and H. J. Chae, Curr. Mol. Med., 2015, 15, 735–745 CrossRef CAS PubMed.
- D. Molino, A. C. Nascimbeni, F. Giordano, P. Codogno and E. Morel, Commun. Integr. Biol., 2017, 10, e1401699 CrossRef PubMed.
- G. Ramadori, G. Konstantinidou, N. Venkateswaran, T. Biscotti, L. Morlock, M. Galie, N. S. Williams, M. Luchetti, A. Santinelli, P. P. Scaglioni and R. Coppari, Cell Metab., 2015, 21, 117–125 CrossRef CAS PubMed.
- T. Yamanaka and N. Nukina, Front. Neurosci., 2018, 12, 91 CrossRef PubMed.
- E. Bachar-Wikstrom, J. D. Wikstrom, Y. Ariav, B. Tirosh, N. Kaiser, E. Cerasi and G. Leibowitz, Diabetes, 2013, 62, 1227–1237 CrossRef CAS PubMed.
- O. Nadiv, M. Shinitzky, H. Manu, D. Hecht, C. T. Roberts, Jr., D. LeRoith and Y. Zick, Biochem. J., 1994, 298(Pt 2), 443–450 CrossRef CAS PubMed.
- L. Li, J. Xu, L. Chen and Z. Jiang, Acta Biochim. Biophys. Sin., 2016, 48, 774–776 CrossRef PubMed.
- M. K. Kuimova, Chimia, 2012, 66, 159–165 CrossRef CAS PubMed.
- M. K. Kuimova, S. W. Botchway, A. W. Parker, M. Balaz, H. A. Collins, H. L. Anderson, K. Suhling and P. R. Ogilby, Nat. Chem., 2009, 1, 69–73 CrossRef CAS PubMed.
- K. Luby-Phelps, Int. Rev. Cytol., 2000, 192, 189–221 CAS.
- C. W. Lai, D. E. Aronson and E. L. Snapp, Mol. Biol. Cell, 2010, 21, 1909–1921 CrossRef CAS PubMed.
- Z. Yang, Y. He, J. H. Lee, W. S. Chae, W. X. Ren, J. H. Lee, C. Kang and J. S. Kim, Chem. Commun., 2014, 50, 11672–11675 RSC.
- C. Gitler, B. Zarmi and E. Kalef, Anal. Biochem., 1997, 252, 48–55 CrossRef CAS PubMed.
- A. Loukil, M. Zonca, C. Rebouissou, V. Baldin, O. Coux, M. Biard-Piechaczyk, J. M. Blanchard and M. Peter, J. Cell Sci., 2014, 127, 2145–2150 CrossRef CAS PubMed.
- E. J. Feeney, S. Austin, Y. H. Chien, H. Mandel, B. Schoser, S. Prater, W. L. Hwu, E. Ralston, P. S. Kishnani and N. Raben, Acta Neuropathol. Commun., 2014, 2, 2 CrossRef PubMed.
- L. Hou, P. Ning, Y. Feng, Y. Ding, L. Bai, L. Li, H. Yu and X. Meng, Anal. Chem., 2018, 90, 7122–7126 CrossRef CAS PubMed.
- Z. Yang, Y. He, J. H. Lee, N. Park, M. Suh, W. S. Chae, J. Cao, X. Peng, H. Jung, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 9181–9185 CrossRef CAS.
- M. Ren, B. Deng, K. Zhou, X. Kong, J. Y. Wang and W. Lin, Anal. Chem., 2017, 89, 552–555 CrossRef CAS.
- K. Zhou, M. G. Ren, B. B. Deng and W. Y. Lin, New J. Chem., 2017, 41, 11507–11511 RSC.
- L. E. Shimolina, M. A. Izquierdo, I. Lopez-Duarte, J. A. Bull, M. V. Shirmanova, L. G. Klapshina, E. V. Zagaynova and M. K. Kuimova, Sci. Rep., 2017, 7, 41097 CrossRef CAS.
- M. V. Shirmanova, L. E. Shimolina, M. M. Lukina, E. V. Zagaynova and M. K. Kuimova, Adv. Exp. Med. Biol., 2017, 1035, 143–153 CrossRef CAS.
- R. Mercade-Prieto, L. Rodriguez-Rivera and X. D. Chen, Photochem. Photobiol. Sci., 2017, 16, 1727–1734 RSC.
- S. Lee, S. M. Kim and R. T. Lee, Antioxid. Redox Signaling, 2013, 18, 1165–1207 CrossRef CAS.
- K. Nakahira, S. M. Cloonan, K. Mizumura, A. M. Choi and S. W. Ryter, Antioxid. Redox Signaling, 2014, 20, 474–494 CrossRef CAS.
- X. Zhang, F. Yang, J. Y. Shim, K. L. Kirk, D. E. Anderson and X. Chen, Cancer Lett., 2007, 255, 95–106 CrossRef CAS.
- C. Huang, Q. Yin, W. Zhu, Y. Yang, X. Wang, X. Qian and Y. Xu, Angew. Chem., Int. Ed. Engl., 2011, 50, 7551–7556 CrossRef CAS.
- S. Shen, X. F. Li, W. R. Cullen, M. Weinfeld and X. C. Le, Chem. Rev., 2013, 113, 7769–7792 CrossRef CAS.
- C. Huang, T. Jia, M. Tang, Q. Yin, W. Zhu, C. Zhang, Y. Yang, N. Jia, Y. Xu and X. Qian, J. Am. Chem. Soc., 2014, 136, 14237–14244 CrossRef CAS.
- Y. Wang, Y. Zhong, Q. Wang, X. F. Yang, Z. Li and H. Li, Anal. Chem., 2016, 88, 10237–10244 CrossRef CAS.
- F. Liu, H. J. Liu, X. J. Liu, W. Chen, F. Wang, R. Q. Yu and J. H. Jiang, Anal. Chem., 2017, 89, 11203–11207 CrossRef CAS.
- H. Lee, Z. Yang, Y. Wi, T. W. Kim, P. Verwilst, Y. H. Lee, G. I. Han, C. Kang and J. S. Kim, Bioconjugate Chem., 2015, 26, 2474–2480 CrossRef CAS.
- Y. Wang, Y. Li, F. Wei and Y. Duan, Trends Biotechnol., 2017, 35, 1181–1193 CrossRef CAS.
- J. Wu, Y. Dang, W. Su, C. Liu, H. Ma, Y. Shan, Y. Pei, B. Wan, J. Guo and L. Yu, Biochem. Biophys. Res. Commun., 2006, 339, 437–442 CrossRef CAS.
- S. Mai, B. Muster, J. Bereiter-Hahn and M. Jendrach, Autophagy, 2012, 8, 47–62 CrossRef CAS.
- J. Huang, W. Pan, D. Ou, W. Dai, Y. Lin, Y. Chen and X. Chen, J. Cardiovasc. Pharmacol., 2015, 66, 576–583 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc00300b |
| This journal is © The Royal Society of Chemistry 2019 |