
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDouble enhancement of hydrogen storage capacity of Pd nanoparticles by 20 at% replacement with Ir; systematic control of hydrogen storage in Pd–M nanoparticles (M = Ir, Pt, Au)†
Hirokazu
Kobayashi
*ab,
Miho
Yamauchi
c,
Ryuichi
Ikeda
a,
Tomokazu
Yamamoto
de,
Syo
Matsumura
def and
Hiroshi
Kitagawa
 *afg
*afg
aDivision of Chemistry, Graduate School of Science, Kyoto University, Kitashirakawa-Oiwakecho, Sakyo-ku, Kyoto, 606-8502, Japan. E-mail: hkobayashi@kuchem.kyoto-u.ac.jp; kitagawa@kuchem.kyoto-u.ac.jp
bJST, PRESTO, 4-1-8 Honcho, Kawaguchi, Saitama, 332-0012, Japan
cInternational Institute for Carbon-Neutral Energy Research (I2CNER), Kyushu University, 744 Motooka, Nishi-ku, Fukuoka, 819-0395, Japan
dDepartment of Applied Quantum Physics and Nuclear Engineering, Graduate School of Engineering, Kyushu University, Motooka 744, Nishi-ku, Fukuoka, 819-0395, Japan
eThe Ultramicroscopy Research Center, Kyushu University, Motooka 744, Nishi-ku, Fukuoka, 819-0395, Japan
fInamori Frontier Research Center, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka, 819-0395, Japan
gInstitute for Integrated Cell-Material Sciences (iCeMS), Kyoto University, Yoshida, Sakyo-ku, Kyoto, 606-8501, Japan
First published on 24th May 2018
Abstract
We report on binary solid-solution nanoparticles (NPs) composed of Pd and Ir, which are not miscible at the equilibrium state of the bulk, for the first time, by means of a process of hydrogen absorption/desorption from core (Pd)/shell (Ir) NPs. Only 20 at% replacement with Ir atoms doubled the hydrogen-storage capability compared to Pd NPs, which are a representative hydrogen-storage material. Furthermore, the systematic control of hydrogen concentrations and the corresponding pressure in Pd and Pd–M NPs (M = Ir, Pt, Au) have been achieved based on the band filling control of Pd NPs.
Introduction
A wide range of materials including metallic alloys,1,2 metal complexes,3 carbon materials4 and metal–organic frameworks (MOFs)5 has been investigated as hydrogen storage materials. Among various kinds of hydrogen storage materials, hydrogen-storage metals or alloys provide one of the most promising ways to store hydrogen compactly and safely due to the stability of their hydrides. In particular, palladium is one of the representative materials for hydrogen storage. It can absorb about 1000 times its own volume of hydrogen at ambient pressure and temperature. Therefore, research on Pd hydride (Pd–H) has been intensively developed in various areas including hydrogen storage materials, purification filters, isotope separation membranes, and sensors.1,2,6–15 Many investigations on hydrogen storage have been conducted on the bulk system of Pd or Pd-based alloys in the last half century.Over recent years, metal nanoparticles (NPs) have received considerable interest as a new type of hydrogen-storage material16–31 in addition to catalytic, optical and magnetic materials. Especially, Pd or Pd-based alloy NPs have been investigated as a model to clarify the hydrogen-storage properties of nanosized metals.16–28 For example, it was shown that the amount of absorbed hydrogen and equilibrium pressure required to form Pd hydride decrease with decreasing particle size.16 For Pd–Au solid-solution NPs, 20 at% Au substitution of Pd drastically reduced more than half the amount of absorbed hydrogen.23 The decrease in the hydrogen concentrations of Pd–Au NPs is explained based on the filling of the 4d conduction band of Pd because of Au substitution, which serves as an electron donor to Pd. From our previous work, we expected that the replacement of Pd with Ir would enhance hydrogen-storage capacity because Ir may act as an electron acceptor to Pd as Ir (5d76s2) has two less electrons than Au (5d106s1). However, Pd and Ir do not mix in the bulk state, unlike the Pd–Au system;32 rather, the alloys segregate to form a domain structure.33
Here, we report binary solid solution NPs composed of Pd and Ir, which are not miscible at the equilibrium state of the bulk, for the first time, by means of a process of hydrogen absorption/desorption (PHAD) from core (Pd)/shell (Ir) NPs. Only 20 at% replacement of Pd with Ir atoms doubled the hydrogen-storage capability compared with pure Pd NPs, which are a representative hydrogen-storage material. Furthermore, the systematic control of hydrogen concentrations and the corresponding pressure in Pd and Pd–M NPs (M = Ir, Pt, Au) have been achieved based on the band-filling control of Pd NPs.
Results and discussion
Synthesis and characterization of Pd–Ir NPs
Poly(N-vinyl-2-pyrrolidone) (PVP)-coated Pd NPs were first synthesized through a chemical reduction of PdCl2.34 An IrCl3 aqueous solution was slowly dropped onto the prepared Pd NPs having a mean diameter of 7.2 nm, and the mixture was stirred in the presence of H2 gas to form a core/shell structure (see Experimental details in the ESI†). The transmission electron microscope (TEM) image showed that the mean diameter of the NPs is 8.8 nm, and their size is larger than that of the Pd NPs used as a seed (Fig. S1†). The atomic ratio of Pd and Ir was determined by energy-dispersive X-ray (EDX) measurements, and the corresponding particles were characterized as Pd0.8Ir0.2.In order to investigate the structure of the as-prepared Pd–Ir NPs, we performed high angle annular dark field scanning transmission electron microscopy (HAADF-STEM) and EDX mapping. Fig. 1a shows the HAADF-STEM image of the as-prepared Pd–Ir NPs. Fig. 1b and c show the corresponding Ir-L and Pd-L STEM–EDX maps, respectively. Fig. 1d presents an overlay map of the Pd and Ir chemical distribution. These mapping data show that the obtained Pd/Ir NPs form a Pd core/Ir shell structure. The corresponding EDX line profiles of Pd and Ir clearly showed that the Pd–Ir NPs form a core/shell structure (Fig. 1e and f). We also confirmed the core/shell structure of the as-prepared Pd–Ir NPs from the powder X-ray diffraction (XRD) results (Fig. S2†).
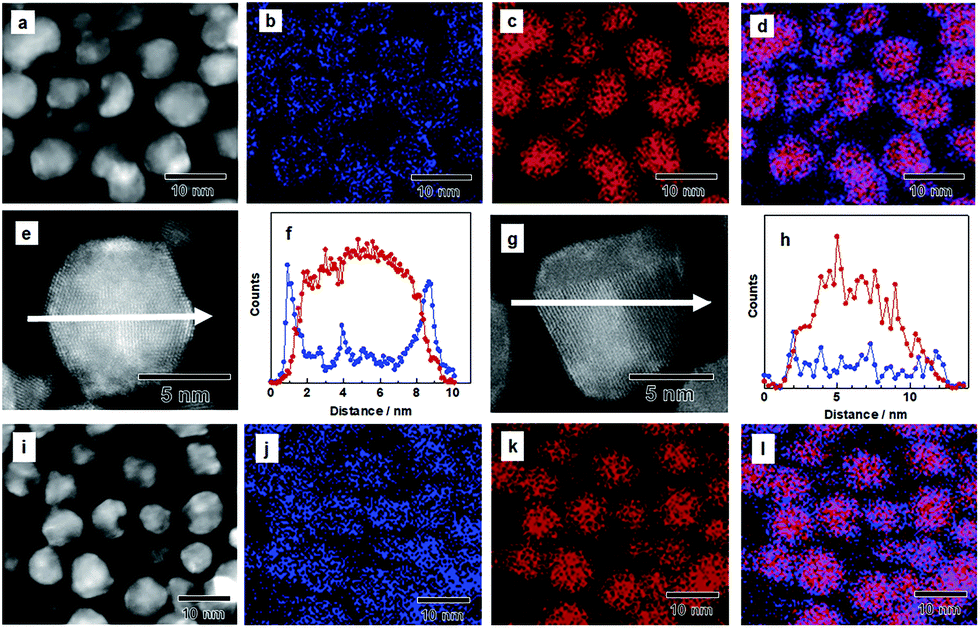 | ||
| Fig. 1 (a) HAADF-STEM image and EDX maps of (b) Ir and (c) Pd in Pd–Ir NPs before the PHAD. (d) Overlay image of (b) and (c) (blue, Ir; red, Pd). (f) Line-scanning profiles of Ir (blue) and Pd (red) across the as-prepared Pd/Ir core/shell NPs in the STEM image (e). (h) Line-scanning profiles of Ir (blue) and Pd (red) across Pd–Ir solid-solution NPs in the STEM image (g). (i) HAADF-STEM image and EDX maps of (j) Ir and (k) Pd in Pd–Ir solid-solution NPs after the PHAD. (l) Overlay image of (j) and (k) (blue, Ir; red, Pd). | ||
Then, we performed the PHAD at 373 K24,25 to obtain Pd–Ir solid-solution NPs (see Experimental details in the ESI†). Fig. 1i–l show the distribution of Pd and Ir elements and their superposition in Pd–Ir NPs after the PHAD. Fig. 1j indicates that Ir elements are randomly distributed in the Pd–Ir NPs after the PHAD, while those in the Pd–Ir NPs before the PHAD have doughnut shaped distributions due to the core/shell structure (Fig. 1b). The overlay map of Pd and Ir shown in Fig. 1l also supports the formation of a Pd–Ir solid-solution alloy. We further characterized the Pd–Ir NPs after the PHAD by EDX line scanning analysis. As shown in Fig. 1g and h, the EDX line profiles clearly demonstrated that Ir and Pd atoms are randomly distributed in the NPs. The distributions of Pd and Ir for Pd–Ir NPs after the PHAD have been investigated for several particles, and the same results were obtained (Fig. S3†). We also confirmed the occurrence of Pd–Ir alloying from the XRD patterns (Fig. S4†). From these results, it can be seen that the as-prepared core/shell Pd–Ir NPs successfully rearrange to Pd–Ir solid-solution NPs, in which Pd and Ir are atomically mixed by the PHAD at 373 K.
In the Pd–Ir system, various phase-segregated structures have been reported, including core/shell35–40 and alloy.41–43 In contrast, the solid-solution NPs, in which Pd and Ir atoms are homogeneously mixed, have rarely been reported44,45 Only a few reports suggest that the binary solid-solution NPs may originate from the fact that Pd and Ir are not atomically mixed and are segregated to form a domain structure below 1673 K.33 Our results in this report demonstrate the first example of solid-solution NPs in which Pd and Ir are atomically mixed by means of the PHAD. Furthermore, the mildness of the PHAD can prevent the size growth caused by the coalescence of each NP. The mean diameters of the NPs were maintained before/after the PHAD (Fig. S1†). The effective method allows us to develop various kinds of solid-solution NPs by precisely controlling the size and compositional ratio of initial core/shell NPs.
It is known that hydrogen molecules invade inside metal or alloy lattices as hydrogen atoms and generate defect structures with superabundant vacancies, promoting atomic diffusion and structural change of alloys.46–48 In this study, the vacancies or lattice defects over the entire structure produced by the PHAD are considered to play an important role in the atomic rearrangements from the Pd–Ir core/shell structure to the solid-solution one.
Hydrogen storage properties of Pd–Ir NPs
To investigate the states of 2H absorbed inside Pd–Ir solid-solution NPs, solid-state 2H NMR spectra were measured. In the spectrum of Pd–Ir NPs, a sharp signal around 0 ppm and a broad signal at a higher field were observed (Fig. 2a). Considering that a single sharp line was obtained at 3.4 ppm for 2H2 gas (Fig. 2c), the sharp component in the Pd–Ir spectrum is attributed to deuterium atoms on the surface of particles which exchanged with free deuterium gas (2H2) or deuterium atoms (2H) inside the NPs, and the broad component is attributed to 2H absorbed inside the particles.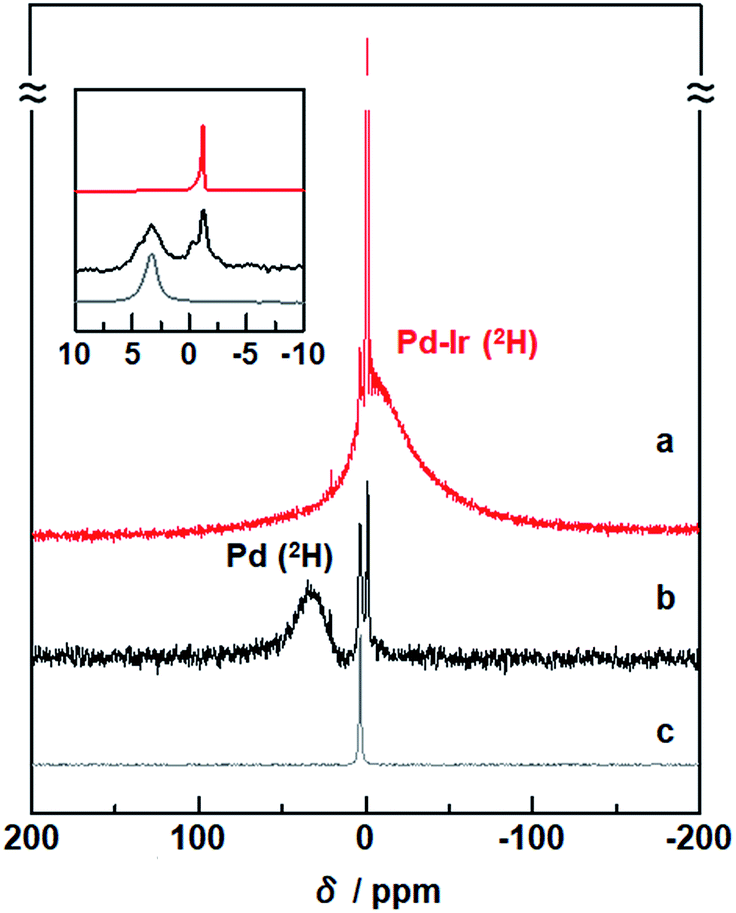 | ||
| Fig. 2 Solid-state 2H NMR spectra of (a) Pd–Ir NPs after the PHAD, (b) Pd NPs,24 and (c) 2H2 gas as a reference. The measurements were performed at 86.7 kPa of 2H2 gas at 303 K. | ||
In the spectra of Pd, and Pd–Au and Pd–Pt solid-solution NPs, the broad signals of 2H absorbed inside the lattices were observed at 26.9, 17.5, and 8.3 ppm, respectively (Fig. S5†).23,24 A broad line, originating from 2H inside the Pd–Ir lattice, is observed at a higher field (−16.5 ppm) (Fig. 2a).
Pd hydride shows the lowest field shift of 2H atoms, compared with other alloy hydrides. The d band in Pd is almost filled after hydride formation. As a result, the correlation of the d spin in Pd is weakened, and the Knight shift mainly affects the NMR shift of 2H in Pd hydride (Fig. 2b).49 On the other hand, for the Pd–Ir NPs, the observed signal is remarkably shifted upfield (Fig. 2a). The NMR shift can be interpreted by the polarization of the s electrons of 2H arising from d spin paramagnetism of the Pd–Ir hydride. Because the d band of Pd–Ir has many holes, spin correlation between the conduction electrons of the PdIr hydride gives rise to the polarized spin of the 2H s electrons in the PdIr hydride. The intrinsic chemical shifts suggest that the electron density around the 2H nuclei depends on the change of d-band filling associated with mixing d orbitals of different metals, and the higher-field shift of Pd–Ir NPs indicates the formation of the atomic-level Pd–Ir alloy.
Hydrogen pressure–composition (PC) isotherms of Pd–Ir solid-solution NPs were measured at 303 K to quantify how much hydrogen was absorbed by alloying of Pd and Ir. As shown in Fig. 3a, Pd NPs have a hydrogen-absorption capacity of 0.24 H/Pd24 at 101.3 kPa. Considering that Ir monometallic NPs have a poor hydrogen-absorption capacity of 0.18 at 101.3 kPa,29 the significantly enhanced hydrogen absorption (0.48 H/Pd0.8–Ir0.2) in Pd–Ir NPs being twice that in Pd NPs is attributed to the synergetic effect of Pd and Ir at the atomic level.
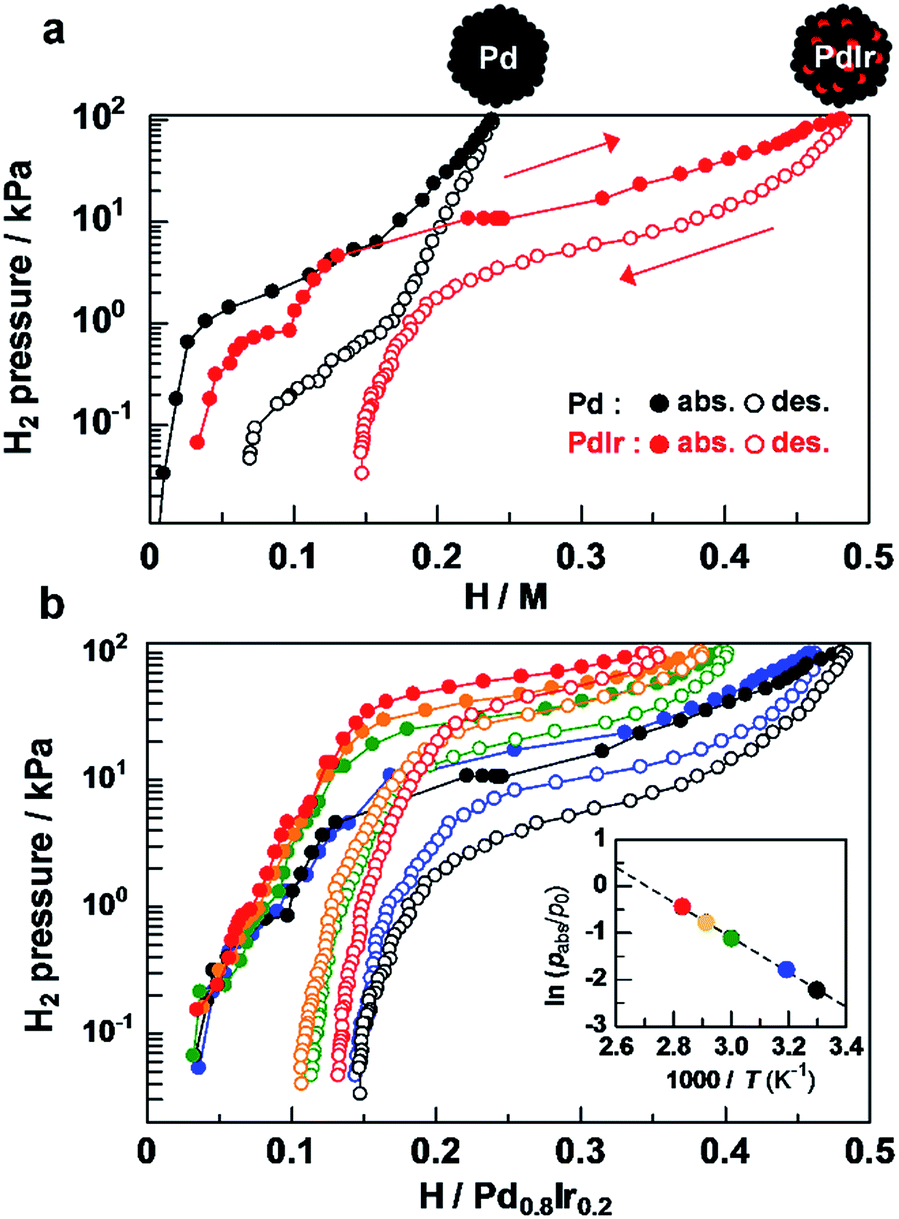 | ||
| Fig. 3 (a) PC isotherms of Pd NPs and Pd0.8–Ir0.2 solid solution NPs at 303 K. (b) Temperature dependency of PC isotherms of Pd–Ir NPs (black: 303 K, blue: 313 K, green: 333 K, orange: 343 K, and red: 353 K) and the van't Hoff plot of the absorption process (inset). | ||
The difference in hydrogen-storage properties together with the metal species in the Pd-based bimetallic NPs is discussed (Fig. S6†). The total amounts of hydrogen absorbed at 101.3 kPa were 0.24, 0.11, 0.33, and 0.48 H/M for Pd,24 Pd0.8–Au0.2,23 Pd0.79–Pt0.21,24 and Pd0.8–Ir0.2 NPs, respectively, which demonstrate that hydrogen concentrations are strongly affected by metal species in Pd-based bimetallic NPs. Interestingly, the 20 at% replacement of Pd with Ir provides the largest amount of hydrogen in Pd and Pd–M bimetallic NPs (M = Ir, Pt, Au). The total wt% including PVP which is used as a protecting polymer and has no hydrogen storage ability was estimated to be 0.12, 0.05, 0.14 and 0.18 wt% for Pd, Pd0.8–Au0.2, Pd0.79–Pt0.21, and Pd0.8–Ir0.2 NPs, respectively. To the best of our knowledge, Pd–Ir NPs absorb a larger amount of hydrogen at 303 K and 1 atm, compared with physisorption based materials such as MOFs,5 carbon based materials4,50–52 and zeolites53 (<0.1 wt% at 0.1 MPa, 298 K).
It is well known that particle size affects hydrogen storage properties in cases of Pd,16 Rh30 or Ir29 NPs. However, the mean diameters of Pd, Pd–Pt, Pd–Ir and Pd–Au NPs used in this study were 7.2 ± 1.4 nm, 8.1 ± 0.9 nm, 8.8 ± 1.7 nm and 8.4 ± 1.6 nm, respectively, and the size dependency of hydrogen storage properties is negligible. Furthermore, the crystal sizes of these nanoparticles were similar. The PHAD method allows us to properly compare the hydrogen storage properties of solid solution NPs with the same size and same compositional ratio. Therefore, the change in the electronic states of Pd associated with solid solution alloying is a dominant factor for hydrogen storage properties of Pd-based solid-solution NPs.
To explore the origin of the enhanced hydrogen storage capability by atomic-level Pd–Ir alloying, the temperature dependence of the plateau pressure in the absorption isotherms was investigated at H/Pd0.8–Ir0.2 = 0.25 (Fig. 3b). From the van't Hoff plots of H2 shown in the Fig. 3b inset, the enthalpy change was estimated to be −31.0 kJ (H2 mol)−1 for Pd–Ir NPs, while those of Pd–Au, Pd, and Pd–Pt were estimated to be −3.5, −18.6, and −25.0 kJ (H2 mol)−1, respectively (Fig. S7–S10†). These results demonstrate that the Pd–Ir alloy provides a favorable environment to form stable hydrides, which is also supported by the results of recent theoretical calculations.54 These specific enthalpy and entropy changes of bimetallic NPs allow us to precisely control the plateau pressure, which is the operating pressure for absorbing/desorbing hydrogen by changing the metal species doped into Pd (Fig. S11†). In addition, the particle size and high hydrogen storage capacity of Pd–Ir solid solution NPs were retained after the measurements of temperature-dependent PC isotherms (Fig. S12†).
The hydrogen storage concentration of bulk Pd is known to correlate with the number of 4d holes in the conduction band at the Fermi level,55 and the capability increases with increasing the number of 4d band holes. Pd–Rh bulk56–60 and its nanoalloys have been reported for hydrogen absorption based on the band-filling effect.26 In the Pd–Rh system, the charge transfer to Rh causes an increase in the number of 4d-band holes of Pd–Rh alloys; therefore Pd–Rh alloys can absorb a larger amount of hydrogen than Pd. Considering that Rh and Ir elements are located in the same group of the periodic table, the greatly enhanced hydrogen capability in Pd–Ir NPs may originate from band filling of Pd NPs. Interestingly, the hydrogen storage capacity of the obtained Pd–Ir NPs (0.48 H/M) was significantly larger than that of Pd–Rh NPs (0.29 H/M).26 In contrast, the decrease in the hydrogen storage capability of Pd–Au NPs is based on the filling of the Pd 4d conduction band by Au substitution, which acts as an electron donor to Pd.23 These results demonstrate that the hydrogen concentration in Pd-based bimetallic NPs is controlled by selecting metal species doped in Pd NPs.
Conclusions
In summary, by means of the PHAD of Pd/Ir core/shell NPs, we first synthesized binary Pd–Ir solid-solution NPs in which Pd and Ir are atomically mixed. The obtained Pd–Ir NPs have double the hydrogen-storage capability of Pd NPs which are a representative hydrogen-storage material. Moreover, the hydrogen-storage capacity in Pd-based solid-solution alloy NPs is controlled by the metal species substituted in Pd NPs due to the band-filling change of Pd 4d. Because Pd and Ir are well known as important catalysts in a wide range of industries, we expect that the Pd–Ir solid-solution alloy NPs are useful as efficient catalysts for various reactions. We hope that the results in this study will contribute to not only a design for the development of highly concentrated hydrogen-storage nanomaterials, but also effective catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Core Research for Evolutional Science and Technology (CREST), Japan Science and Technology Agency (JST). STEM observations were performed as part of a program conducted by the Advanced Characterization Nanotechnology Platform sponsored by the MEXT, Japan.Notes and references
- F. A. Lewis, The Palladium Hydrogen System, Academic Press, London, 1967 Search PubMed.
- Hydrogen in Metals II, ed. G. Alefeld, J. Völkl, Springer, Berlin, Heidelberg, 1978 Search PubMed.
- S. Orimo, Y. Nakamori, J. R. Eliseo, A. Züttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111–4132 CrossRef PubMed.
- W.-C. Xu, K. Takahashi, Y. Matsuo, Y. Hattori, M. Kumagai, S. Ishiyama, K. Kaneko and S. Iijima, Int. J. Hydrogen Energy, 2007, 32, 2504–2512 CrossRef.
- M. P. Suh, H. J. Park, T. K. Prasad and D. W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef PubMed.
- A. J. Maeland and T. R. P. Gibb, J. Phys. Chem., 1961, 65, 1270 CrossRef.
- J. W. Simons and T. B. Flanagan, J. Phys. Chem., 1965, 69, 3581 CrossRef.
- E. F. W. Seymour, R. M. Cotts and W. D. Williams, Phys. Rev. Lett., 1975, 35, 165 CrossRef.
- S. Yun, J. H. Ko and S. T. Oyama, J. Membr. Sci., 2011, 369, 482 CrossRef.
- H. Li, A. Caravella and H. Y. Xu, J. Mater. Chem. A, 2016, 4, 14069 Search PubMed.
- W. Luo and D. F. Cowgill, J. Phys. Chem. C, 2013, 117, 13861 Search PubMed.
- G. Sicking, J. Less-Common Met., 1984, 101, 169 CrossRef.
- F. Leardini, J. F. Fernandez, J. Bodega and C. Sanchez, J. Phys. Chem. Solids, 2008, 69, 116 CrossRef.
- C. Christofides and A. Mandelis, J. Appl. Phys., 1990, 68, R1 CrossRef.
- J. RaviPrakash, A. H. McDaniel, M. Horna, L. Pilione, P. Sunal, R. Messier, R. T. McGrath and F. K. Schweighardt, Sens. Actuators, B, 2007, 120, 439 CrossRef.
- M. Yamauchi, R. Ikeda, H. Kitagawa and M. Takata, J. Phys. Chem. C, 2008, 112, 3294 Search PubMed.
- H. Kobayashi, M. Yamauchi, H. Kitagawa, Y. Kubota, K. Kato and M. Takata, J. Am. Chem. Soc., 2008, 130, 1828 CrossRef PubMed.
- M. Yamauchi, H. Kobayashi and H. Kitagawa, ChemPhysChem, 2009, 10, 2566 CrossRef PubMed.
- A. Pundt, C. Sachs, M. Winter, M. T. Reetz, D. Fritsch and R. Kirchheim, J. Alloys Compd., 1999, 293–295, 480 CrossRef.
- C. Sachs, A. Pundt and R. Kirchheim, Phys. Rev. B, 2001, 64, 075408 CrossRef.
- M. Yamauchi and H. Kitagawa, Synth. Met., 2005, 153, 353 CrossRef.
- G. Li, H. Kobayashi, S. Dekura, R. Ikeda, Y. Kubota, K. Kato, M. Takata, T. Yamamoto, S. Matsumura and H. Kitagawa, J. Am. Chem. Soc., 2014, 136, 10222 CrossRef PubMed.
- H. Kobayashi, M. Yamauchi, R. Ikeda and H. Kitagawa, Chem. Commun., 2009, 4806 RSC.
- H. Kobayashi, M. Yamauchi, H. Kitagawa, Y. Kubota, K. Kato and M. Takata, J. Am. Chem. Soc., 2010, 132, 5576 CrossRef PubMed.
- H. Kobayashi, K. Kusada and H. Kitagawa, Acc. Chem. Res., 2015, 48, 1551 CrossRef PubMed.
- H. Kobayashi, H. Morita, M. Yamauchi, R. Ikeda, H. Kitagawa, Y. Kubota, K. Kato and M. Takata, J. Am. Chem. Soc., 2012, 134, 12390 CrossRef PubMed.
- H. Kobayashi, M. Yamauchi, H. Kitagawa, Y. Kubota, K. Kato and M. Takata, J. Am. Chem. Soc., 2008, 130, 1818 CrossRef PubMed.
- K. Kusada, H. Kobayashi, R. Ikeda, Y. Kubota, M. Takata, S. Toh, T. Yamamoto, S. Matsumura, N. Sumi, K. Sato, K. Nagaoka and H. Kitagawa, J. Am. Chem. Soc., 2014, 136, 1864 CrossRef PubMed.
- H. Kobayashi, M. Yamauchi and H. Kitagawa, J. Am. Chem. Soc., 2012, 134, 6893 CrossRef PubMed.
- H. Kobayashi, H. Morita, M. Yamauchi, R. Ikeda, H. Kitagawa, Y. Kubota, K. Kato and M. Takata, J. Am. Chem. Soc., 2011, 133, 11034 CrossRef PubMed.
- Y. Isobe, M. Yamauchi, R. Ikeda and H. Kitagawa, Synth. Met., 2003, 135–136, 757 CrossRef.
- H. Okamoto and T. B. Massalski, Bull. Alloy Phase Diagrams, 1985, 6, 229 CrossRef.
- S. N. Tripathi, S. R. Bharadwaj and M. S. Chandrasekharaiah, J. Phase Equilib., 1991, 12, 603 CrossRef.
- T. Teranishi and M. Miyake, Chem. Mater., 1998, 10, 594 CrossRef.
- H. Ye, K. Yang, J. Tao, Y. Liu, Q. Zhang, S. Habibi, Z. Nie and X. Xia, ACS Nano, 2017, 11, 2052 CrossRef PubMed.
- L. Zheng, X. Niu, J. Zhao, T. Liu, Y. Liu and Y. Yang, J. Nanosci. Nanotechnol., 2016, 16, 5984 CrossRef PubMed.
- X. Xia, L. F. Cosme, J. Tao, H. C. Peng, G. Niu, Y. Zhu and Y. Xia, J. Am. Chem. Soc., 2014, 136, 10878 CrossRef PubMed.
- X. Xia, J. Zhang, N. Lu, M. J. Kim, K. Ghale, Y. Xu, E. McKenzie, J. Liu and H. Ye, ACS Nano, 2015, 9, 9994 CrossRef PubMed.
- H. Ziaeiazad, C. X. Yin, J. Shen, Y. Hu, D. Karpuzov and N. Semagina, J. Catal., 2013, 300, 113 CrossRef.
- J. Chen, Y. Li, Z. Gao, G. Wang, J. Tian, C. Jiang, S. Zhu and R. Wang, Electrochem. Commun., 2013, 37, 24 CrossRef.
- L. Piccolo, S. Nassreddine, M. Aouine, C. Ulhaq and C. Geantet, J. Catal., 2012, 292, 173 CrossRef.
- M. H. M. T. Assumpção, S. G. da Silva, R. F. B. De Souza, G. S. Buzzo, E. V. Spinacé, M. C. Santos, A. O. Neto and J. C. M. Silva, J. Power Sources, 2014, 268, 129 CrossRef.
- T. Yang, Y. Ma, Q. Huang, G. Cao, S. Wan, N. Li, H. Zhao, X. Sun and F. Yin, Electrochem. Commun., 2015, 59, 95 CrossRef.
- J. Bao, M. Dou, H. Liu, F. Wang, J. Liu, Z. Li and J. Ji, ACS Appl. Mater. Interfaces, 2015, 28, 15223 Search PubMed.
- J. Chen, Y. Li, S. Liu, G. Wang, J. Tian, C. Jiang, S. Zhu and R. Wang, Appl. Surf. Sci., 2013, 287, 457 CrossRef.
- N. Fukumuro, M. Yokota, S. Yae, H. Matsuda and Y. Fukai, J. Alloys Compd., 2013, 580, S55 CrossRef.
- N. Mukaibo, Y. Shimizu, Y. Fukai and T. Hiroi, Mater. Trans., 2008, 49, 2815 CrossRef.
- E. Hayashi, Y. Kurokawa and Y. Fukai, Phys. Rev. Lett., 1998, 80, 5588 CrossRef.
- P. Brill, J. Voitlander and B. Bunsenges, Phys. Chem., 1973, 77, 1097 Search PubMed.
- W. Zhao, V. Fierro, C. Zlotea, M. T. Izquierdo, C. C. César, M. Latroche and A. Celzard, Int. J. Hydrogen Energy, 2012, 37, 5072–5080 CrossRef.
- S. Schaefer, V. Fierro, M. T. Izquierdo and A. Celzard, Int. J. Hydrogen Energy, 2016, 41, 12146–12156 CrossRef.
- S. Schaefer, V. Fierro, A. Szczurek, M. T. Izquierdo and A. Celzard, Int. J. Hydrogen Energy, 2016, 41, 17442–17452 CrossRef.
- J. Dong, X. Wang, H. Xu, Q. Zhao and J. Li, Int. J. Hydrogen Energy, 2007, 32, 4998–5004 CrossRef.
- T. Yayama, T. Ishimoto and M. Koyama, J. Alloys Compd., 2015, 653, 444 CrossRef.
- D. A. Papaconstantopoulos, B. M. Klein, E. N. Economou and L. L. Boyer, Phys. Rev. B, 1978, 17, 141 CrossRef.
- H. Noh, W. Luo and T. B. Flanagan, J. Alloys Compd., 1993, 196, 7 CrossRef.
- H. Noh, T. B. Flanagan, B. Cerundolo and A. Craft, Scr. Metall. Mater., 1991, 25, 225 CrossRef.
- H. Noh, J. D. Clewley, T. B. Flanagan and A. P. Craft, J. Alloys Compd., 1996, 240, 235 CrossRef.
- H. Noh, J. D. Clewley, T. B. Flanagan and A. P. Craft, Int. J. Hydrogen Energy, 2000, 25, 853 CrossRef.
- B. Baranowski, S. Majchrzak and T. B. Flanagan, J. Phys. Chem., 1973, 77, 35 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01460d |
| This journal is © The Royal Society of Chemistry 2018 |