
Electrochemically tuned cobalt hydroxide carbonate with abundant grain boundaries for highly efficient electro-oxidation of hydrazine†
Xiaodong
Yan‡
 a,
Yuan
Liu‡
b,
Jinle
Lan
c,
Yunhua
Yu
c,
James
Murowchick
d,
Xiaoping
Yang
*c and
Zhonghua
Peng
*a
a,
Yuan
Liu‡
b,
Jinle
Lan
c,
Yunhua
Yu
c,
James
Murowchick
d,
Xiaoping
Yang
*c and
Zhonghua
Peng
*a
aDepartment of Chemistry, University of Missouri–Kansas City, Kansas City, Missouri 64110, USA. E-mail: PengZ@umkc.edu
bState Key Lab of New Ceramics and Fine Processing, School of Materials Science and Engineering, Tsinghua University, Beijing 100084, China
cState Key Laboratory of Organic–Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: yangxp@mail.buct.edu.cn
dDepartment of Geosciences, University of Missouri–Kansas City, Kansas City, Missouri 64110, USA
First published on 26th December 2017
Abstract
Grain boundaries bear many active sites for surface reactions (e.g. electrocatalysis and energy storage), and increasing the grain boundaries has become an important strategy to tune the number of surface active sites and the reaction kinetics. Herein, we show that a simple in situ electrochemical tuning method can be used to turn Co(OH)x(CO3)0.5(2−x) crystals into interconnected ultrafine nanoparticles with enriched grain boundaries. The grain boundaries offer numerous unparalleled active sites for highly efficient electro-oxidation of hydrazine. The electrochemically-tuned Co(OH)x(CO3)0.5(2−x) shows an onset potential (−1.12 V vs. Ag/AgCl) 180 mV smaller than that of the pristine Co(OH)x(CO3)0.5(2−x), and delivers a high current density of 62.4 mA cm−2 at −0.90 V vs. Ag/AgCl, which is 27 times higher than that of the pristine Co(OH)x(CO3)0.5(2−x). This work produces the most active non-metallic catalyst for hydrazine oxidation with catalytic activity comparable to the state-of-the-art catalysts. Moreover, our electrochemical tuning method is applicable to other materials (e.g. Co3O4, Co(OH)2 and Ni(OH)2).
1. Introduction
Fuel cells, batteries and supercapacitors are the three major electrochemical systems for energy storage and conversion, and potentially the mainstream energy supply systems for portable electronics and vehicles in future green and sustainable economies.1–8 Batteries, especially lithium-ion batteries, and supercapacitors have been successfully commercialized for decades, and are recently booming owing to their success in powering electric vehicles.9,10 However, the commercial batteries and supercapacitors suffer from low volumetric energy, limiting their lifetime per charge to days and even hours. Fuel cells, which directly convert chemical energy from fuels (methanol, hydrazine, et al.) to mechanical, electrical and/or thermal energy, are an ideal primary energy conversion device for uninterruptible power supply.1,11,12 Current fuels cells, however, rely on noble metal catalyst systems, whose high cost and scarcity greatly hampers the mass production and extensive applications of fuel cells.13 There is thus an urgent need for the development of low-cost, earth-abundant and highly-efficient catalyst materials for fuel cells.14–18Among various fuel cell technologies, direct hydrazine fuel cells, which were pioneered in the 1960s,19 have drawn increasing attention due to the ease in transportation and storage of the liquid hydrazine fuel, the high theoretical cell voltage (+1.61 V), the high energy and power density, and the pollution-free emissions (H2O and N2).11,12,20–24 The key component for fuel cells is the catalysts.14,16–18,25–34 So far, a variety of earth-abundant catalyst materials have been developed for hydrazine fuel cells.25–32 Among them, the first-row transition metal based nanomaterials (mainly Co, Ni and Cu) have attracted the most attention because of their relatively high earth abundance, low cost and decent catalytic activity.27–32 However, only metal nanocrystals have so far been shown to exhibit high catalytic performances. Their low-cost, easily-processable oxides and hydroxides are seldom reported as high-performing electrocatalysts towards hydrazine oxidation. The activity of an electrocatalyst depends strongly on its structure, composition, surface area, etc., and thus structure engineering as an effective tool in tuning the functional properties of the existing materials has spurred wide interest. For instance, recent researches show that structure-engineered metal/metal oxide heterostructures exhibit tunable catalytic activity towards water electrolysis,35–40 and present high activity towards hydrazine electro-oxidation.41 The enhanced performances of these metal/metal oxide heterostructures can be attributed, at least partially, to the increased interphase boundaries and/or grain boundaries.35–38,41 As indicated by numerous recent studies, grain boundaries that often include abundant defects greatly contribute to the active sites for surface electrochemical reactions. Aaronson et al. found that grain boundaries on platinum contributed significantly to its overall electrochemical behavior towards the Fe2+/Fe3+ redox reaction.42 Feng et al. reported that the CO2 electro-reduction activity of gold nanoparticles and the CO electro-reduction activity of copper nanoparticles were grain-boundary-dependent.43,44 Maillard and co-workers found that the active sites on the carbon-supported Pt nanoparticles towards CO monolayer oxidation was partially derived from the grain boundaries.45 Wang's group introduced a lithium electrochemical tuning method to controllably reduce zinc oxide into interconnected metal nanoparticles with enriched grain boundaries, resulting in a five-fold improvement towards CO2 reduction at an overpotential of −948 mV.46 Inspired by these studies, herein, we report that a simple in situ electrochemical tuning method can convert the Co(OH)x(CO3)0.5(2−x) crystals into interconnected ultrafine nanoparticles with abundant grain boundaries, turning an otherwise inactive catalyst into one of the most effective non-metallic catalysts for hydrazine oxidation. To the best of knowledge, this is the first report on the application of Co(OH)x(CO3)0.5(2−x) as a potential cost-effective yet highly efficient catalyst for hydrazine fuel cells. The abundant grain boundaries of the electrochemically tuned Co(OH)x(CO3)0.5(2−x) (ECT-Co(OH)x(CO3)0.5(2−x)) contribute to a high cathodic current density of 62.4 mA cm−2 at a potential of −0.90 V vs. Ag/AgCl, representing a 27-fold improvement compared to the pristine Co(OH)x(CO3)0.5(2−x). This opens a new avenue for endowing traditionally inactive materials with high activities towards electro-oxidation of hydrazine.
2. Experimental section
2.1. Material synthesis
2.2. Property characterization
Morphologies of the samples were examined using scanning and transmission electron microscopy (SEM and TEM). The SEM images were collected using a field emission scanning electron microscope (Supra55, Carl Zeiss). The TEM analyses were conducted on a high-resolution transmission electron microscope (HRTEM, Tecnai G2 F30 S-TWIN) equipped with a scanning transmission electron microscope. The electron accelerating voltage for the transmission electron microscope was 200 kV. Structural analysis was performed with the collected Co(OH)x(CO3)0.5(2−x) powder on a wide-angle X-ray diffractometer (WAXD, D8 Advance, Bruker) using Cu Kα radiation (wavelength = 1.5418 Å). The elemental composition and chemical status of the electrodes were analyzed by using X-ray photoelectron spectroscopy (XPS, EscaLab 250, Thermo Fisher Scientific) with a monochromatic Al X-ray (hν = 1486.6 eV) as the excitation source.2.3. Electrochemical characterization.
Electrochemical measurements were carried out in a three-electrode system at room temperature. A Pt wire and an Ag/AgCl electrode were used as the counter and reference electrodes, respectively. Typically, 0.5 M N2H4 aqueous solution with 1.0 M KOH was used as the electrolyte. Linear sweep voltammetry was conducted at a scan rate of 5 mV s−1 to evaluate the catalytic performance of the working electrode. Long-term stability test was carried out by cyclic voltammetry in the voltage range from −1.2 to −0.9 V vs. Ag/AgCl with repeated scans at a scan rate of 20 mV s−1.To estimate the electrochemically active surface area (ECSA), following procedures were carried out. The electrochemically tuned electrode was first undergone three cycles of linear sweep voltammograms at a scan rate of 5 mV s−1. Subsequently, cyclic voltammograms were collected at varied scan rates of 5, 10, 20, 40, 60, and 80 mV s−1. The voltage window of the cyclic voltammograms was between −1.0 and −0.9 V vs. Ag/AgCl. The cyclic voltammograms from different electrodes were shown in Fig. S1 (ESI†). The double layer capacitance (Cdl) was estimated by plotting the Δj (Δj = ja − jc) against the scan rate, and the slope was twice of the Cdl. ja and jc are the anodic and cathodic current densities, respectively, from the voltammograms at the potential of −0.95 V vs. Ag/AgCl.
3. Results and discussion
Co(OH)x(CO3)0.5(2−x) was first grown on nickel foam substrates by hydrothermal reaction in the presence of 1.0 mmol Co(NO3)2 and 4.0 mmol urea in a mixture solution of water and ethanol. As shown in Fig. 1A and B, the Co(OH)x(CO3)0.5(2−x) film was uniformly grown on the surface of the nickel foam. On the surface of the Co(OH)x(CO3)0.5(2−x) film, thinly distributed nanosheets and nanowires were observed (Fig. 1B). TEM characterization of the nanosheets showed that the Co(OH)x(CO3)0.5(2−x) nanosheets were single crystals with continuous lattice fringes (Fig. 1C and D). The interplanar spacing of 0.442 nm is close to the (001) plane of Co(OH)(CO3)0.5 (JCPDS 48-0083), confirming the formation of the Co(OH)x(CO3)0.5(2−x). Note that the holes in the Co(OH)x(CO3)0.5(2−x) nanosheet in Fig. 1D were derived from the high-energy electron beam during the HRTEM characterization. The elevated local temperature by the high-energy electron beam decomposed part of the Co(OH)x(CO3)0.5(2−x) nanosheet. | ||
| Fig. 1 SEM (A, B, E and F), TEM (C and G) and HRTEM (D and H) images of Co(OH)x(CO3)0.5(2−x) before (A–D) and after (E–H) electrochemical tuning. | ||
ECT-Co(OH)x(CO3)0.5(2−x) was prepared by performing cyclic voltammetry cycles on the pristine Co(OH)x(CO3)0.5(2−x) electrode until the catalytic performance was stabilized. The morphology of the resulting ECT-Co(OH)x(CO3)0.5(2−x) was studied by SEM and TEM. The SEM images (Fig. 1E and F) of the ECT-Co(OH)x(CO3)0.5(2−x) showed similar feature to the pristine Co(OH)x(CO3)0.5(2−x). While the TEM image (Fig. 1G) of the ECT-Co(OH)x(CO3)0.5(2−x) nanosheets confirmed its well-maintained morphology, the HRTEM image taken from the same nanosheet showed that the ECT-Co(OH)x(CO3)0.5(2−x) nanosheets were composed of numerous ultrafine interconnected nanocrystals of sizes around 2–3 nm (Fig. 1H and Fig. S2, ESI†). This proves the high effectiveness of our electrochemical tuning method in transforming relatively large nanocrystals of Co(OH)x(CO3)0.5(2−x) into ultrafine nanocrystals with abundant grain boundaries. The spacing of 0.201 nm between adjacent planes (Fig. S2, ESI†) is indexed to the (050) plane of Co(OH)(CO3)0.5 (JCPDS 48-0083). XRD analysis was further used to probe the phase composition of the Co(OH)x(CO3)0.5(2−x) and ECT-Co(OH)x(CO3)0.5(2−x). The XRD patterns (Fig. 2A) of the Co(OH)x(CO3)0.5(2−x) and ECT-Co(OH)x(CO3)0.5(2−x) resemble each other and both match well with the XRD patterns of the Co(OH)x(CO3)0.5(2−x) reported in the literature.47–50 Most of the XRD peaks of ECT-Co(OH)x(CO3)0.5(2−x) are weaker, likely due to the ultra-small sizes of the nanocrystals in the ECT-Co(OH)x(CO3)0.5(2−x) and their decreased crystallinity caused by the abundant grain boundaries. On the other hand, diffraction signals at 2θ ≈ 14.4° and 24.0° in the XRD pattern of the ECT-Co(OH)x(CO3)0.5(2−x), which correspond to the (200) and (301) crystal planes of hexagonal cobalt hydroxide carbonate, respectively,49 are stronger, indicating that the percentage of those crystal planes increased after electrochemical tuning.
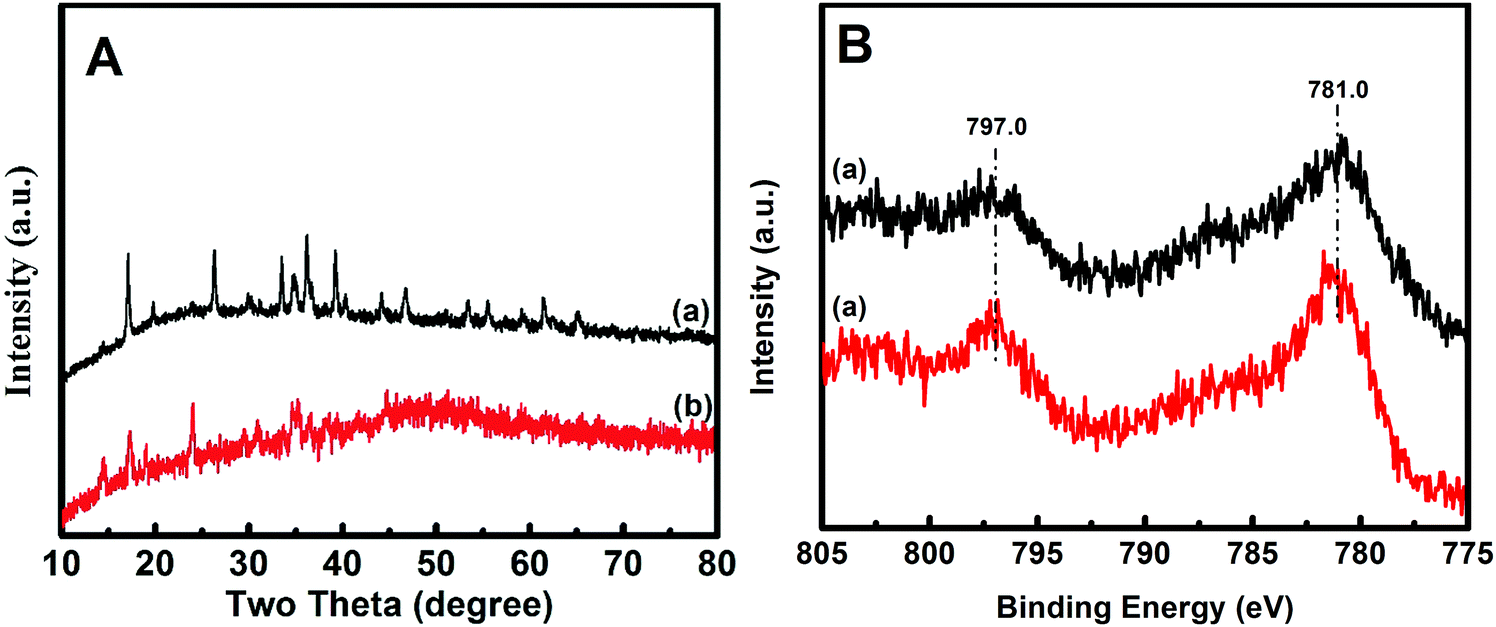 | ||
| Fig. 2 XRD patterns (A) and Co 2p XPS spectra (B) of the Co(OH)x(CO3)0.5(2−x) (a) and ECT-Co(OH)x(CO3)0.5(2−x) (b). | ||
XPS analysis was conducted to survey the surface chemical states and elemental composition of the samples. All XPS spectra were calibrated with the C 1s peak (284.6 eV). The Co(OH)x(CO3)0.5(2−x) and ECT-Co(OH)x(CO3)0.5(2−x) presented very similar XPS survey spectra (Fig. S3, ESI†) with signals from cobalt, oxygen and carbon elements.35Fig. 2B displays the Co 2p core-level XPS spectra of the Co(OH)x(CO3)0.5(2−x) and ECT-Co(OH)x(CO3)0.5(2−x). The similar Co 2p XPS spectra suggest similar surface chemical states of Co in the Co(OH)x(CO3)0.5(2−x) and ECT-Co(OH)x(CO3)0.5(2−x). The Co 2p3/2 peak at 781.0 eV and the Co 2p1/2 peak at 797.0 eV were assigned to the Co2+ species.51,52 Note that the Co 2p peaks at 781.0 and 797.0 eV were intensified after electrochemical tuning, indicative of the increased surface Co2+ sites. This could be to the consequence of the abundant grain boundaries in the ECT-Co(OH)x(CO3)0.5(2−x).
The electrocatalytic performances towards hydrazine oxidation over the ECT-Co(OH)x(CO3)0.5(2−x) and pristine Co(OH)x(CO3)0.5(2−x) electrodes were evaluated in a typical three-electrode system with a mixture solution of 0.5 M N2H4 and 1.0 M KOH as the electrolyte. Fig. 3 displays the polarization curves of the ECT-Co(OH)x(CO3)0.5(2−x) and pristine Co(OH)x(CO3)0.5(2−x) electrodes. The pristine Co(OH)x(CO3)0.5(2−x) was found to be inactive towards electro-oxidation of hydrazine at potentials below −0.90 V vs. Ag/AgCl. As shown in Fig. 3A, the current density of the pristine Co(OH)x(CO3)0.5(2−x) electrode only reached ∼2.2 mA cm−2 at a relatively high potential of −0.90 V vs. Ag/AgCl. Excitingly, the ECT-Co(OH)x(CO3)0.5(2−x) electrode reached a markedly high current density of ∼62.4 mA cm−2 at the same potential, which represented a 27-fold improvement after electrochemical tuning. On the other hand, the onset potential, defined as the potential where the current density reaches 1.0 mA cm−2, was found to be −1.12 vs. Ag/AgCl for the ECT-Co(OH)x(CO3)0.5(2−x) electrode. This is a dramatic decrease of 180 mV as compared to that of the pristine Co(OH)x(CO3)0.5(2−x) electrode, confirming the high activity of the ECT-Co(OH)x(CO3)0.5(2−x) electrode owing to the numerous active sites primarily derived from the enriched grain boundaries. The onset potential of the ECT-Co(OH)x(CO3)0.5(2−x) electrode is among the lowest reported in the literature (Table 1). More importantly, our preparation method is simple and does not involve complex processes and harsh conditions used in most of the other synthesis methods. In addition, our method uses inexpensive source materials, and is easily scalable.
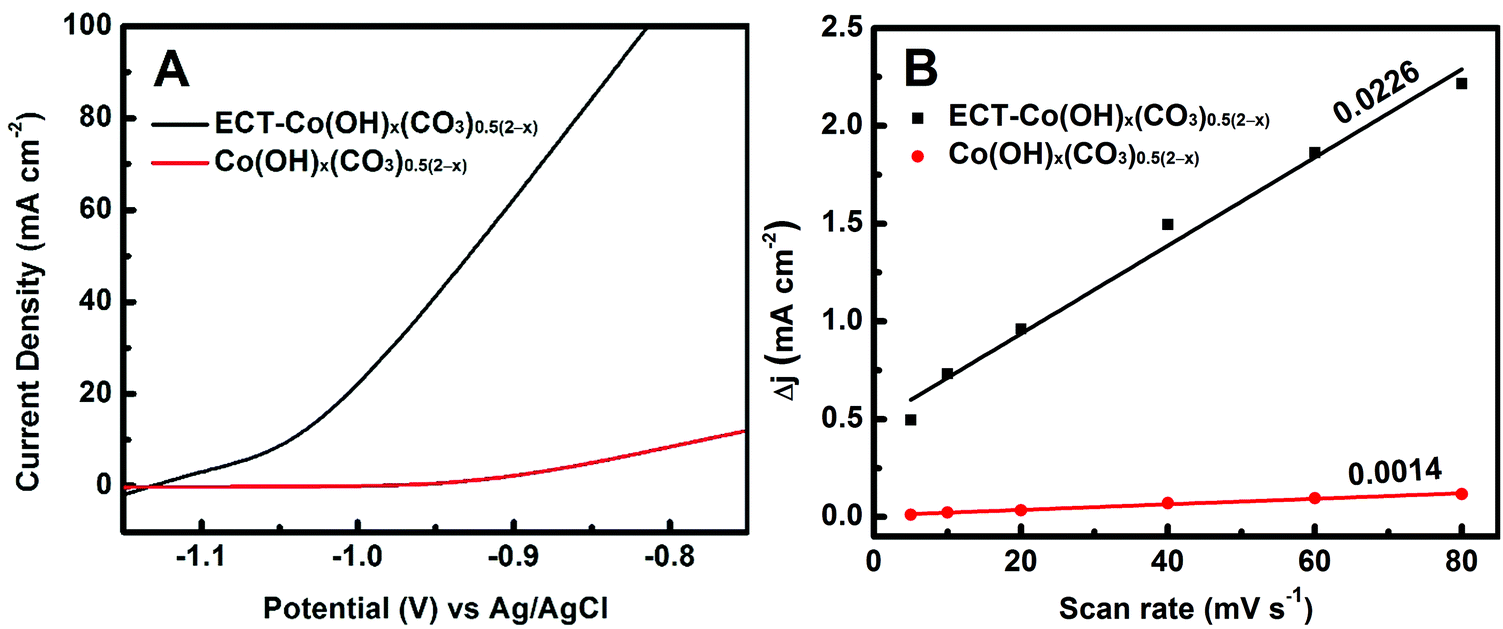 | ||
| Fig. 3 (A) Polarization curves of the ECT-Co(OH)x(CO3)0.5(2−x) and pristine Co(OH)x(CO3)0.5(2−x) electrodes. (B) Charging current density difference Δj (ja − jc) plotted against scan rate. | ||
| Sample | Electrolyte | v (mV s−1) | Con. of N2H4 (M) | Onset potentiala (V) | Ref. |
|---|---|---|---|---|---|
| a All the potentials are relative to Ag/AgCl. | |||||
| Co/carbon fiber cloth | 1.0 M KOH | 10 | 0.02 | −1.10 | 27 |
| Cu nanowires on Cu foil | 3.0 M NaOH | 25 | 1.0 | −0.82 | 29 |
| Ni60Co40 alloy | 1.0 M KOH | 20 | 0.1 | −1.17 | 31 |
| Ni nanoflowers on Ni foam | 3.0 M KOH | 1 | 0.5 | −1.05 | 53 |
| Nanostructured Cu | 9.0 M KOH | 20 | 2.1 | −0.96 | 54 |
| Cu film on Cu Foil | 3.0 M NaOH | 5 | 1.0 | −0.76 | 55 |
| NiS2 nanosheets on Ti mesh | 1.0 M KOH | 5 | 0.5 | −0.983 | 56 |
| FeP nanosheets on Ni foam | 1.0 M KOH | 5 | 0.5 | −1.023 | 57 |
| Ni2P on Ni foam | 1.0 M KOH | 5 | 0.5 | −1.12 | 58 |
| ECT-Co(OH)x(CO3)0.5(2−x) | 1.0 M KOH | 5 | 0.5 | −1.12 | This work |
It is suspected that the improved catalytic performance is correlated to the increased grain boundaries in the ECT-modified electrode. While it is impossible to quantitatively measure the grain boundaries, the ECSA may be measured and to some extent reflect the quantity of the grain boundaries, since ECSA is proportional to the number of surface active sites. ECSA was estimated from the Cdl, as Cdl was linearly proportional to the effective active surface area.59,60 The profile of the charging current density difference Δj (ja − jc) versus scan rate is shown in Fig. 3B. The Cdl values of Co(OH)x(CO3)0.5(2−x) before and after electrochemical tuning were found to be 0.0007 and 0.0113 F, respectively. This indicates that the ECSA of the ECT-Co(OH)x(CO3)0.5(2−x) electrode is 16.1 times as high as that of the pristine Co(OH)x(CO3)0.5(2−x) electrode. It is believed that the large ECSA of the ECT-Co(OH)x(CO3)0.5(2−x) leads to its dramatically improved catalytic performance, while the large ECSA is undoubtedly derived from the abundant grain boundaries. Further, normalized current density was calculated by dividing the current density with the relative ECSA (The relative ECSA of the pristine Co(OH)x(CO3)0.5(2−x) was set as 1). The normalized current density of the ECT-Co(OH)x(CO3)0.5(2−x) electrode (3.9 mA cm−2) was about 1.8 times that of the pristine Co(OH)x(CO3)0.5(2−x) electrode (2.2 mA cm−2) at the potential of −0.9 V vs. Ag/AgCl. This suggests that the active grain boundary sites are superior to the regular active sites on the surface of the pristine Co(OH)x(CO3)0.5(2−x). This can be explained as follows. Adsorption of OH− and N2H4 onto the surface of the catalyst is reported to be the rate-determining processes (N2H4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ads + OH−ads → N2H3ads + H2O + e−),61,62 and the adsorption of OH− onto the surface Co sites herein is the first step towards hydrazine oxidation.61 The rich grain boundaries exposed more accessible Co sites as evidenced by the XPS, where many defective Co sites may exist, thus enhancing the OH− adsorption and contributing to the greatly decreased overpotential necessary for the electro-oxidation of hydrazine.61,63
ads + OH−ads → N2H3ads + H2O + e−),61,62 and the adsorption of OH− onto the surface Co sites herein is the first step towards hydrazine oxidation.61 The rich grain boundaries exposed more accessible Co sites as evidenced by the XPS, where many defective Co sites may exist, thus enhancing the OH− adsorption and contributing to the greatly decreased overpotential necessary for the electro-oxidation of hydrazine.61,63
The effect of mass loadings of Co(OH)x(CO3)0.5(2−x) on the catalytic performance was evaluated. As shown in Fig. 4A, a mass loading of about 1.0 mg cm−2 appears to be optimal: lower loading led to poorer performance while higher loading did not further improve the catalytic performance. We also investigated the effect of the concentration of the hydrazine on the performance (Fig. 4B). As was expected, no redox reaction was detected without N2H4. At a low N2H4 concentration of 0.05 M, the reaction was highly diffusion-controlled, evidenced by the limited current increase with potential. This is consistent with the literature.26–28 At high N2H4 concentrations of ≥0.1 M, the current density increased linearly with potential when the potential was over −1.0 V vs. Ag/AgCl, and the catalytic performance was slightly but continuously improved with increasing the concentration of N2H4 from 0.2 to 1.0 M.
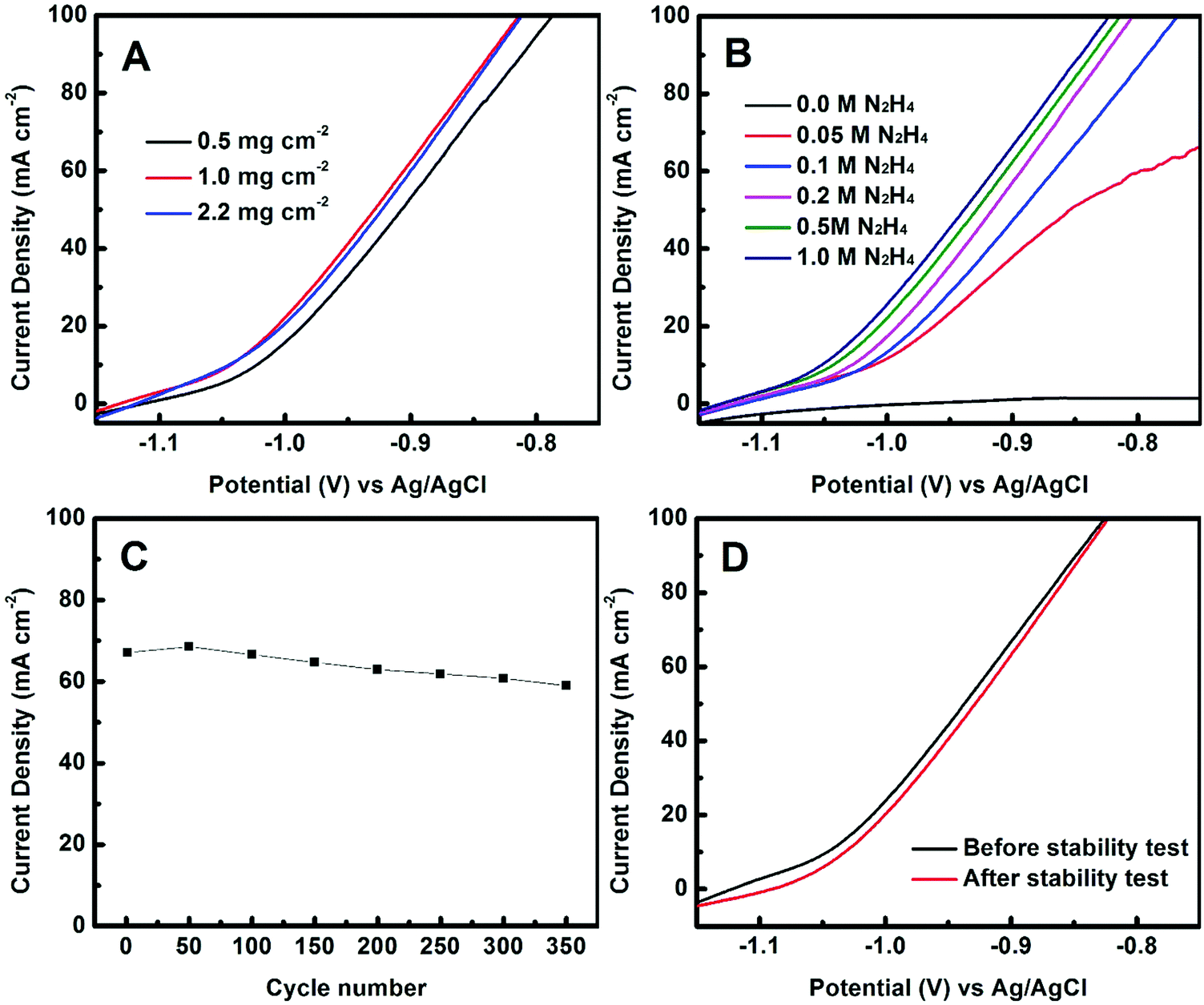 | ||
| Fig. 4 (A) Polarization curves of the ECT-Co(OH)x(CO3)0.5(2−x) with different mass loading. (B) Polarization curves of the ECT-Co(OH)x(CO3)0.5(2−x) with varied concentrations of N2H4. (C) Cycling stability of the ECT-Co(OH)x(CO3)0.5(2−x) electrode: current density at the potential of −0.90 V vs. Ag/AgCl against cycle number. (D) Polarization curves of the ECT-Co(OH)x(CO3)0.5(2−x) electrode before and after the stability test. | ||
The catalyst stability of the ECT-Co(OH)x(CO3)0.5(2−x) electrode was evaluated by cyclic voltammetry measurements. As shown in Fig. 4C, the current density at the potential of −0.90 V vs. Ag/AgCl maintained 89% of its initial value after 350 cycles. Polarization curves collected before and after the stability test showed that the catalytic performance only decreased slightly after 350 cycles (Fig. 4D). For instance, the degradation in current density at −0.90 V vs. Ag/AgCl from the polarization curves before and after the stability test was calculated to be 5.4%. These results show that the ECT-Co(OH)x(CO3)0.5(2−x) catalyst has good stability. The SEM image of the ECT-Co(OH)x(CO3)0.5(2−x) electrode after 350 cycles was collected and is shown in Fig. S4 (ESI†).
To find out whether the electrochemical tuning method can be applied to other materials, the Co3O4, Co(OH)2 and Ni(OH)2 electrodes were subjected to the same electrochemical tuning process. As shown in Fig. S5–S7 (ESI†) and Fig. 5, compared with their corresponding pristine electrodes, improved catalytic performances were observed for all electrochemically tuned electrodes. The extent of improvement, however, is different for different materials. The Co3O4 showed the least amount of improvement while cobalt hydroxide carbonate presented the largest increase and the highest catalytic performance after the electrochemical tuning. The current density of the ECT-Co(OH)x(CO3)0.5(2−x) electrode doubled that of the second best one (the ECT-Co(OH)2 electrode). This suggest that the intrinsic structure of the cobalt hydroxide carbonate plays a critical role in its restructuring during the electrochemical tuning process.
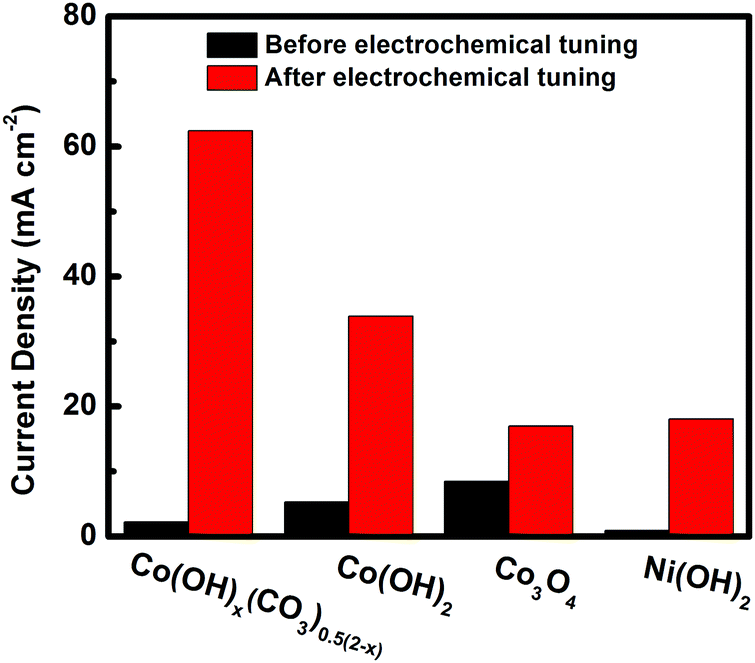 | ||
| Fig. 5 Current density at the potential of −0.90 V vs. Ag/AgCl plotted against catalyst before and after electrochemical tuning. | ||
4. Conclusions
In summary, a simple in situ electrochemical tuning method has been shown to be able to convert an inactive cobalt hydroxide carbonate into a highly efficient catalyst for electro-oxidation of hydrazine. Structurally, the electrochemical tuning turns single crystals into interconnected ultrafine crystalline particles of a few nanometers, creating abundance grain boundaries. As a catalyst for electro-oxidation of hydrazine, the electrochemically tuned Co(OH)x(CO3)0.5(2−x) showed a performance comparable to the state-of-the-art catalysts, delivering a low onset potential (−1.12 V vs. Ag/AgCl), a high current density (62.4 mA cm−2 at −0.9 V vs. Ag/AgCl) and good long-term stability. The active grain boundary sites are found to be superior to the regular surface-active sites towards catalytic oxidation of hydrazine due to the unique structure of the gain boundary regions. This study provides new insights in developing low-cost, advanced electrocatalysts for hydrazine fuel cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. Z. acknowledges the support from the National Science Foundation (DMR1308577). X. Y. thanks the funds provided by the University of Missouri-Kansas City, School of Graduate Studies.References
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4269 CrossRef CAS PubMed.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- J. Wang, H. Tang, L. Zhang, H. Ren, R. Yu, Q. Jin, J. Qi, D. Mao, M. Yang, Y. Wang, P. Liu, Y. Zhang, Y. Wen, L. Gu, G. Ma, Z. Su, Z. Tang, H. Zhao and D. Wang, Nat. Energy, 2016, 1, 16050 CrossRef CAS.
- J. Zhang, H. Ren, J. Wang, J. Qi, R. Yu, D. Wang and Y. Liu, J. Mater. Chem. A, 2016, 4, 17673–17677 CAS.
- H. Ren, R. Yu, J. Wang, Q. Jin, M. Yang, D. Mao, D. Kisailus, H. Zhao and D. Wang, Nano Lett., 2014, 14, 6679–6684 CrossRef CAS PubMed.
- J. Wang, H. Tang, H. Ren, R. Yu, J. Qi, D. Mao, H. Zhao and D. Wang, Adv. Sci., 2014, 1, 1400011 CrossRef PubMed.
- J. Qi, X. Lai, J. Wang, H. Tang, H. Ren, Y. Yang, Q. Jin, L. Zhang, R. Yu, G. Ma, Z. Su, H. Zhao and D. Wang, Chem. Soc. Rev., 2015, 44, 6749–6773 RSC.
- L. Lu, X. Han, J. Li, J. Hua and M. Ouyang, J. Power Sources, 2013, 226, 272–288 CrossRef CAS.
- A. F. Burke, Proc. IEEE, 2007, 95, 806–820 CrossRef.
- B. C. Ong, S. K. Kamarudin and S. Basri, Int. J. Hydrogen Energy, 2017, 42, 10142–10157 CrossRef CAS.
- N. V. Rees and R. G. Compton, Energy Environ. Sci., 2011, 4, 1255–1260 CAS.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- H. Yin, H. Tang, D. Wang, Y. Gao and Z. Tang, ACS Nano, 2012, 6, 8288–8297 CrossRef CAS PubMed.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS PubMed.
- H. Tang, H. Yin, J. Wang, N. Yang, D. Wang and Z. Tang, Angew. Chem., Int. Ed., 2013, 52, 5585–5589 CrossRef CAS PubMed.
- S. Zhao, H. Yin, L. Du, L. He, K. Zhao, L. Chang, G. Yin, H. Zhao, S. Liu and Z. Tang, ACS Nano, 2014, 8, 12660–12668 CrossRef CAS PubMed.
- J. Sun, H. Yin, P. Liu, Y. Wang, X. Yao, Z. Tang and H. Zhao, Chem. Sci., 2016, 7, 5640–5646 RSC.
- G. E. Evans and K. V. Kordesch, Science, 1967, 158, 1148–1152 CAS.
- A. Serov, M. Padilla, A. J. Roy, P. Atanassov, T. Sakamoto, K. Asazawa and H. Tanaka, Angew. Chem., 2014, 126, 10504–10507 CrossRef.
- Y. Meng, X. Zou, X. Huang, A. Goswami, Z. Liu and T. Asefa, Adv. Mater., 2014, 26, 6510–6516 CrossRef CAS PubMed.
- T. Asset, A. Roy, T. Sakamoto, M. Padilla, I. Matanovic, K. Artyushkova, A. Serov, F. Maillard, M. Chatenet, K. Asazawa, H. Tanaka and P. Atanassov, Electrochim. Acta, 2016, 215, 420–426 CrossRef CAS.
- G. Feng, Y. Kuang, P. Li, N. Han, M. Sun, G. Zhang and X. Sun, Adv. Sci., 2017, 4, 1600179 CrossRef PubMed.
- J. Jeong, M. Choun and J. Lee, Angew. Chem., Int. Ed., 2017, 56, 13513–13516 CrossRef CAS PubMed.
- A. Serov and C. Kwak, Appl. Catal., B, 2010, 98, 1–9 CrossRef CAS.
- R. Liu, K. Ye, Y. Gao, W. Zhang, G. Wang and D. Cao, Electrochim. Acta, 2015, 186, 239–244 CrossRef CAS.
- R. Liu, X. Jiang, F. Guo, N. Shi, J. Yin, G. Wang and D. Cao, Electrochim. Acta, 2013, 94, 214–218 CrossRef CAS.
- Y. Ma, H. Wang, J. Key, S. Ji, W. Lv and R. Wang, J. Power Sources, 2015, 300, 344–350 CrossRef CAS.
- J. Huang, S. Zhao, W. Chen, Y. Zhou, X. Yang, Y. Zhu and C. Li, Nanoscale, 2016, 8, 5810–5814 RSC.
- T.-Y. Jeon, M. Watanabe and K. Miyatake, ACS Appl. Mater. Interfaces, 2014, 6, 18445–18449 CAS.
- J. Sanabria-Chinchilla, K. Asazawa, T. Sakamoto, K. Yamada, H. Tanaka and P. Strasser, J. Am. Chem. Soc., 2011, 133, 5425–5431 CrossRef CAS PubMed.
- S. R. Hosseini, S. Ghasemi and M. Kamali-Rousta, J. Power Sources, 2017, 343, 467–476 CrossRef CAS.
- X. Zhang, H. Yin, J. Wang, L. Chang, Y. Gao, W. Liu and Z. Tang, Nanoscale, 2013, 5, 8392–8397 RSC.
- J. Wang, J. Gong, Y. Xiong, J. Yang, Y. Gao, Y. Liu, X. Lu and Z. Tang, Chem. Commun., 2011, 47, 6894–6896 RSC.
- X. Yan, L. Tian, M. He and X. Chen, Nano Lett., 2015, 15, 6015–6021 CrossRef CAS PubMed.
- X. Yan, K. Li, L. Lyu, F. Song, J. He, D. Niu, L. Liu, X. Hu and X. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 3208–3214 CAS.
- X. Yan, L. Tian, K. Li, S. Atkins, H. Zhao, J. Murowchick, L. Liu and X. Chen, Adv. Mater. Interfaces, 2016, 3, 1600368 CrossRef.
- X. Yan, L. Tian and X. Chen, J. Power Sources, 2015, 300, 336–343 CrossRef CAS.
- H. Yin, S. Zhao, K. Zhao, A. Muqsit, H. Tang, L. Chang, H. Zhao, Y. Gao and Z. Tang, Nat. Commun., 2015, 6, 6430 CrossRef CAS PubMed.
- S. Zhao, Y. Wang, J. Dong, C.-T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- X. Yan, Y. Liu, J. Lan, Y. Yu, J. Murowchick, X. Yang and Z. Peng, Mater. Chem. Front., 2018, 2, 96–101 RSC.
- B. D. B. Aaronson, C.-H. Chen, H. Li, M. T. M. Koper, S. C. S. Lai and P. R. Unwin, J. Am. Chem. Soc., 2013, 135, 3873–3880 CrossRef CAS PubMed.
- X. Feng, K. Jiang, S. Fan and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4606–4609 CrossRef CAS PubMed.
- X. Feng, K. Jiang, S. Fan and M. W. Kanan, ACS Cent. Sci., 2016, 2, 169–174 CrossRef CAS PubMed.
- F. Maillard, S. Schreier, M. Hanzlik, E. R. Savinova, S. Weinkauf and U. Stimming, Phys. Chem. Chem. Phys., 2005, 7, 385–393 RSC.
- K. Jiang, H. Wang, W.-B. Cai and H. Wang, ACS Nano, 2017, 11, 6451–6458 CrossRef CAS PubMed.
- R. Xu and H. C. Zeng, J. Phys. Chem. B, 2003, 107, 12643–12649 CrossRef CAS.
- D. Ghosh, M. Mandal and C. K. Das, Langmuir, 2015, 31, 7835–7843 CrossRef CAS PubMed.
- S. L. Wang, L. Q. Qian, H. Xu, G. L. Lü, W. J. Dong and W. H. Tang, J. Alloys Compd., 2009, 476, 739–743 CrossRef CAS.
- T. M. Masikhwa, J. K. Dangbegnon, A. Bello, M. J. Madito, D. Momodu and N. Manyala, J. Phys. Chem. Solids, 2016, 88, 60–67 CrossRef CAS.
- V. M. Lebarbier, A. M. Karim, M. H. Engelhard, Y. Wu, B.-Q. Xu, E. J. Petersen, A. K. Datye and Y. Wang, ChemSusChem, 2011, 4, 1679–1684 CrossRef CAS PubMed.
- H. Tang, H. Yin, J. Wang, N. Yang, D. Wang and Z. Tang, Angew. Chem., Int. Ed., 2013, 52, 5585–5589 CrossRef CAS PubMed.
- G. Feng, Y. Kuang, Y. Li and X. Sun, Nano Res., 2015, 8, 3365–3371 CrossRef CAS.
- E. Granot, B. Filanovsky, I. Presman, I. Kuras and F. Patolsky, J. Power Sources, 2012, 204, 116–121 CrossRef CAS.
- Z. Lu, M. Sun, T. Xu, Y. Li, W. Xu, Z. Chang, Y. Ding, X. Sun and L. Jiang, Adv. Mater., 2015, 27, 2361–2366 CrossRef CAS PubMed.
- J. Wang, X. Ma, T. Liu, D. Liu, S. Hao, G. Du, R. Rong, A. M. Asiri and X. Sun, Mater. Today Energy, 2017, 3, 9–14 CrossRef.
- L. Zhang, D. Liu, S. Hao, L. Xie, F. Qu, G. Du, A. M. Asiri and X. Sun, ChemistrySelect, 2017, 2, 3401–3407 CrossRef CAS.
- C. Tang, R. Zhang, W. Lu, Z. Wang, D. Liu, S. Hao, G. Du, A. M. Asiri and X. Sun, Angew. Chem., 2017, 129, 860–864 CrossRef.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef CAS PubMed.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CAS.
- T. Sakamoto, H. Kishi, S. Yamaguchi, D. Matsumura, K. Tamura, A. Hori, Y. Horiuchi, A. Serov, K. Artyushkova, P. Atanassov and H. Tanaka, J. Electrochem. Soc., 2016, 163, H951–H957 CrossRef CAS.
- M. Petek and S. Bruchenstein, J. Electroanal. Chem. Interfacial Electrochem., 1973, 47, 329–333 CrossRef CAS.
- M. D. García Azorero, M. L. Marcos and J. González velasco, Electrochim. Acta, 1994, 39, 1909–1914 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00511c |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2018 |