
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical approaches for the enhancement of 5-aminolevulinic acid-based photodynamic therapy and photodiagnosis
Kunal M.
Tewari†
and
Ian M.
Eggleston
 *
*
Department of Pharmacy and Pharmacology, University of Bath, Bath BA2 7AY, UK. E-mail: ie203@bath.ac.uk
First published on 8th October 2018
Abstract
The administration of 5-aminolevulinic acid (ALA) to generate enhanced intracellular levels of endogenous porphyrins is currently one of the most important approaches for photodynamic therapy (PDT) and photodiagnosis (PDD). Despite the great promise of ALA-based PDT, the physicochemical behaviour and chemical reactivity of ALA are problematic, and a variety of chemical approaches have been brought to bear to improve cellular delivery, enhance porphyrin production, and generate ALA prodrugs that have appropriate stability for convenient clinical use, as well as selectivity for cancerous tissues. While there has been considerable success, there are still a number of challenges to be addressed and opportunities to be exploited through application of chemical insight in this area.
 Kunal M. Tewari | Kunal Tewari received his PhD from University of Bath, Bath, UK in 2016 under the supervision of Dr Ian Eggleston. His research involved development of novel strategies for enhancement of delivery of 5-aminolevulinic acid (5-ALA) to specific cell types using targeted ALA dendrimeric prodrugs. He is currently working as a research associate at the University of Strathclyde and his current focus is centred around biomaterials science, especially the development of peptoid nanosheets. |
 Ian M. Eggleston | Ian Eggleston received his D.Phil. in Chemistry in 1990 from the University of Oxford, UK, and carried out postdoctoral work at the University of Wisconsin–Madison (USA), CNRS-INSERM Montpellier (France), and the University of Lausanne. In 1997 he joined the University of Dundee as Lecturer in Synthetic Organic Chemistry and moved to the University of Bath in 2006, where he is now Reader in Medicinal Chemistry. His research interests include the development of peptide-targeted agents for photodynamic therapy and light-triggered drug delivery, especially the synthesis of novel peptide-based prodrugs of 5-aminolevulinic acid. |
Introduction
In the last 40 years, 5-aminolevulinic acid (ALA) has attracted tremendous interest in the field of PDT and photobiology.1–5 Alongside the emergence of clinical applications and approvals for ALA-PDT for the treatment of cancerous and precancerous conditions, as well as in photodiagnosis (PDD) and fluorescence-guided surgery, it has become apparent that there is a real need for better ways to administer ALA and also to manipulate its bioconversion to protoporphrin IX (PpIX) via the heme cycle (Scheme 1).6,7 The aim of this review is to cast a focus on some of the chemical strategies that have been developed to improve the performance of ALA in photomedicine, and the opportunities that exist for future exploration by medicinal chemists.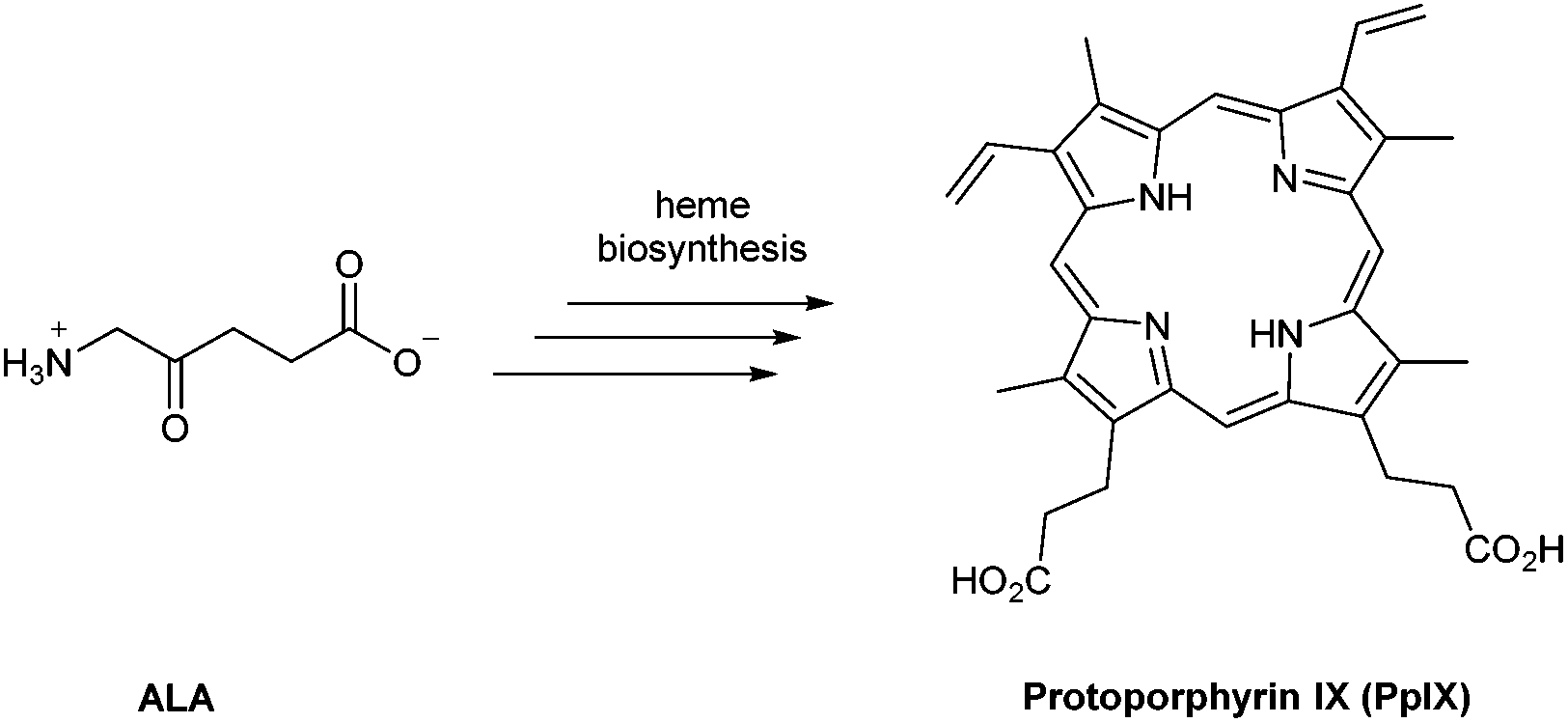 | ||
| Scheme 1 5-Aminolevulinic acid (ALA) and its bioconversion to protoporphyrin IX (PpIX). | ||
ALA is an early intermediate in the heme biosynthetic pathway and is not a photosensitiser itself (Scheme 1). ALA-PDT and related techniques exploit instead the downstream generation of the active sensitiser protoporphyin IX (PpIX), which is the penultimate species that is converted to heme. ALA can therefore be regarded as a prodrug for both PDT and photodiagnosis. The heme biosynthetic pathway is shown in detail in Scheme 2 and begins with a condensation reaction between glycine and succinyl-CoA in the mitochondrion to form ALA which becomes the source of C and N atoms for the construction of the heme macrocycle. This reaction is catalysed by ALA synthase (ALA-S), and the synthesized ALA then enters the cytosol where a condensation reaction between two units of ALA, mediated by ALA dehydratase (ALAD), yields the pyrrole derivative, porphobilinogen (PBG). Four units of PBG are combined to generate a linear tetrapyrrole intermediate under the influence of porphobilogen deaminase (PBG-D), followed by cyclisation by the enzyme uroporphyrinogen III cosynthase (UCS) to give the cyclic tetrapyrrole intermediate, uroporphyrinogen III. This intermediate now undergoes a decarboxylation of the four acetate side chains to methyl groups (catalysed by uroporphyrinogen decarboxylase, UGD) to give coproporphyrinogen III, which is then carried to the mitochondrion. Further oxidative steps catalysed by copropoprphyrinogen oxidase (CPO) and protoporphyrinogen oxidase (PPO) then lead to formation of PpIX. Insertion of Fe2+ into PpIX, mediated by ferrochelatase (FC), ultimately completes the heme cycle, and under normal physiological conditions, the whole pathway and thus the production of porphyrins is tightly controlled by negative feedback by heme itself which thus controls ALA synthesis. In a clinical setting, the administration of exogenous ALA or a suitable derivative bypasses this control mechanism and the rate-limiting step in the pathway becomes the final insertion of iron into PpIX by ferrochelatase. The net result is the temporary overproduction and accumulation of excess PpIX in cells which then renders them photosensitive and susceptible to PDT. The preferential accumulation of PpIX in rapidly dividing tumour cells relative to healthy cells not only provides ALA-PDT with an inherent selectivity relative to PDT with other photosensitisers, but also presents the means to accurately image the location of tumour tissue for PDD. From a medicinal chemistry perspective, the challenge is to develop ways to optimise the amount of ALA that is available to enter into the heme cycle and its subsequent conversion into PpIX within selected tissues. This can be tackled in one of two ways: either by manipulating a specific enzyme-catalysed reaction in the heme cycle with small molecules in order to maximise PpIX production from administered ALA, or by enhancing the delivery of ALA across biological barriers to the cells of interest by incorporating ALA into derivatives with more suitable physicochemical and targeting properties and from which ALA is then released (ALA prodrugs).
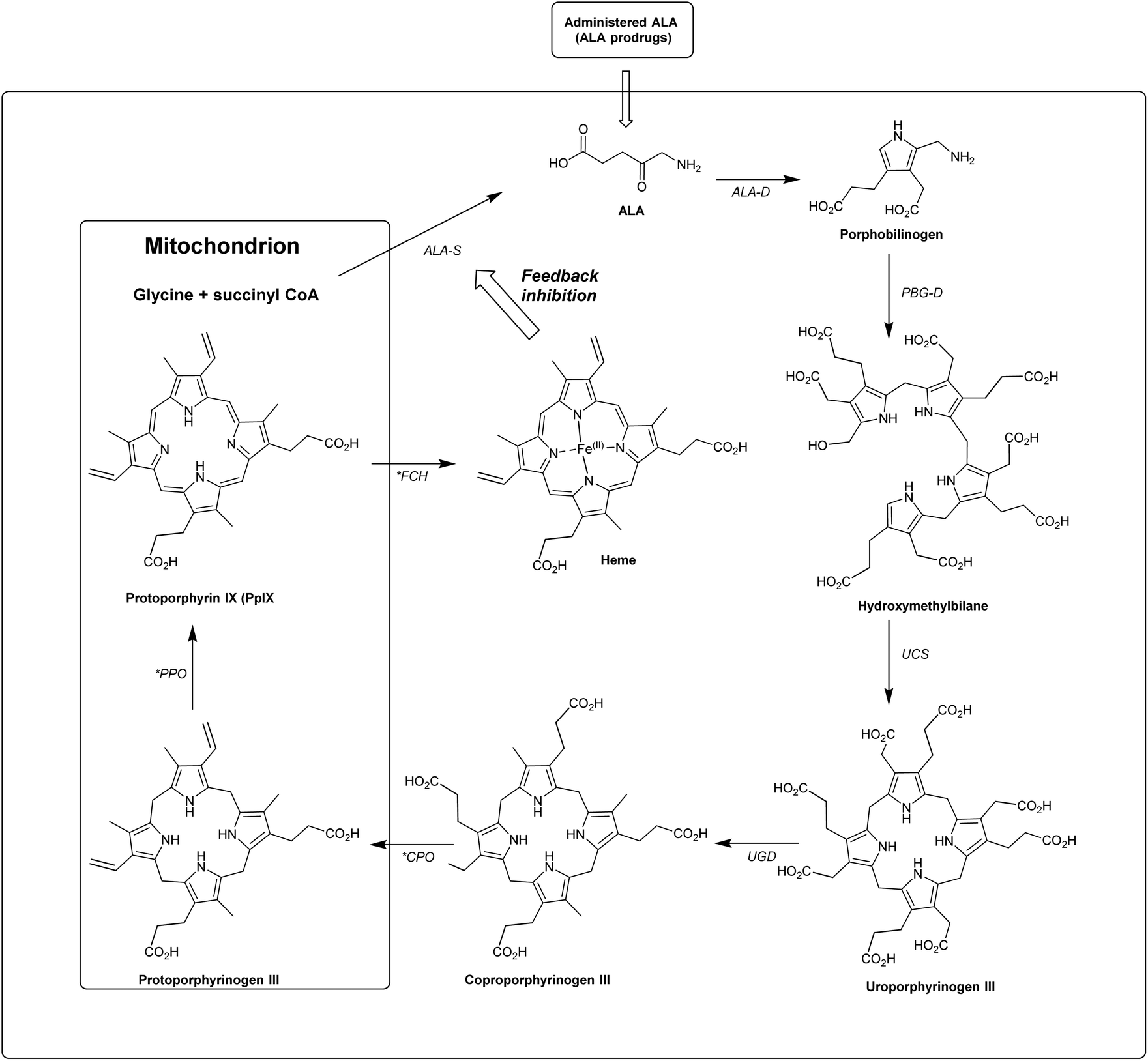 | ||
| Scheme 2 The heme biosynthetic pathway. 5-Aminolevulinic acid (ALA). Key steps that may be targeted chemically to maximise PpIX production are highlighted (*). Modified from Thunshelle et al.7 | ||
Drawbacks of ALA-PDT/PDD and strategies to improve its effectiveness
ALA is a zwitterion at physiological pH, and as such its hydrophilic nature severely impairs its passage through biological barriers to ultimately reach and enter the cells of interest.3 This is a significant challenge for topical application of ALA in dermatology where penetration through the lipophilic stratum corneum7,8 must be achieved, and has motivated many chemical and non-chemical approaches to overcome issues of low penetration and non-homogeneous distribution in the targeted tissues. The bioavailability of ALA itself is also limited when administered parenterally, and has been associated with significant adverse effects.3A variety of formulation-based chemical penetration enhancers have been investigated for topical ALA administration including, dimethyl sulfoxide (DMSO),9 1-[2-(decylthio)-ethyl]azacycpentan-2-one (HPE-101),10 glycerol monooleate,11 6-ketocholestanol (2% w/w)12 and oleic acid.13 Recently, nanoemulsion-based formulations of ALA such as BF-200 ALA14 have begun to show very promising clinical applications, particularly for the treatment of actinic keratoses (AK), and can not only enhance penetration of ALA through the strateum corneum, but also can improve the long term stability of the prodrug.15
This highlights a further challenge for both the clinical application of ALA and also strategies for chemical modification. At physiological or alkaline pH, ALA and simple ester derivatives (see below), are unstable with respect to self-condensation through formation of a dimeric Schiff base. Buffered solutions of ALA at physiological pH discolour even at low temperature due to decomposition of ALA, and thus stock solutions for clinical use would typically need to be freshly prepared.16 The instability of ALA under neutral and alkaline conditions has been investigated in some detail, and under alkaline conditions, it has been shown that ALA readily undergoes dimerization to form 2,5-dicarboxyethyl-3,6-dihydropyrazine (DHPY), which oxidises spontaneously to 2,5-dicarboxyethylpyrazine (PY, Scheme 3).17,18 Other transformations of the ALA dimer to form porphobilinogen (PBG) and pseudoporphobilinogen have also been proposed.18–20
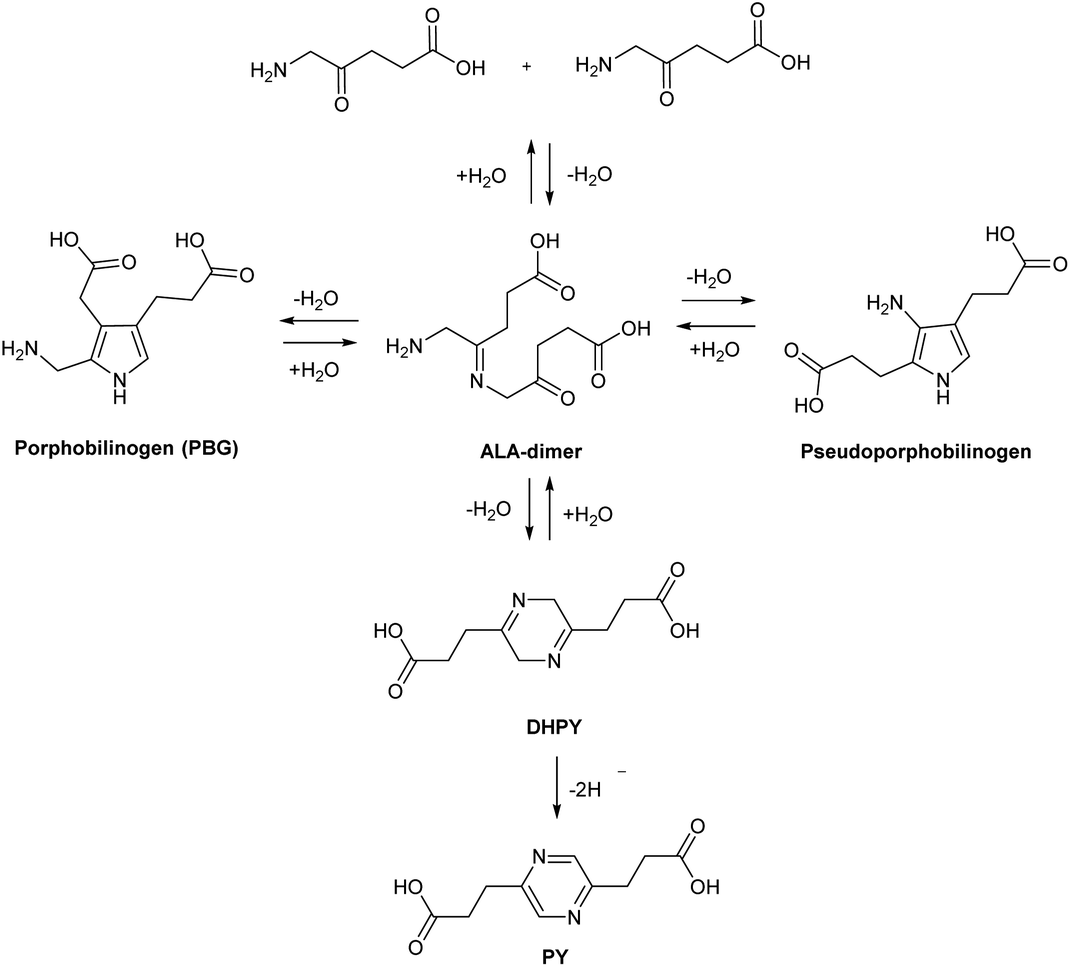 | ||
| Scheme 3 Instability of ALA at physiological pH and chemical conversion to non-heme precursors. | ||
In addition to the drawbacks associated with the physical properties of ALA and its chemical stability, a further problem that needs to be addressed for both topical and systemic ALA delivery is to ensure an adequate discrimination between normal and diseased (i.e. tumour) cells. In this case, the challenge for the chemist is to design ALA derivatives which are either selectively targeted towards particular cell types, or which release ALA only in the right biological context in the presence of a suitable enzyme activity. Many of the chemical strategies that have been devised to enhance ALA-induced PpIX production do indeed rely on this kind of prodrug approach,6 and this therefore forms a major part of the rest of this review.
Alkyl ester prodrugs
The synthesis of ALA esters has so far proved to be the most effective and straightforward prodrug approach. The methyl ester of ALA (Metvix or MAL) 1a is indeed now a mainstay for the treatment of AK by both conventional and daylight PDT, as well as being clinically approved for the treatment of basal cell carcinomas (BCC). The hexyl ester 1f has been approved for fluorescence diagnosis in the bladder as Hexvix (now Cysview).7For these clinically approved examples, and also for many other esters of ALA, it has been shown that their enhanced lipophilicity compared to ALA itself to gives rise to better cellular permeability and thus quicker, more homogeneous, and more effective PpIX production in a variety of cell models. Many ALA ester prodrugs have been synthesized with linear or branched alkyl ester groups, and a selection of these are shown in Fig. 1.3 The preparation of such derivatives from the corresponding alcohols is straightforward and easily scaled-up to multi-gram quantities. Like ALA itself, simple esters have limited stability at neutral pH, although they may be stored in solution at pH 4 without significant degradation.21
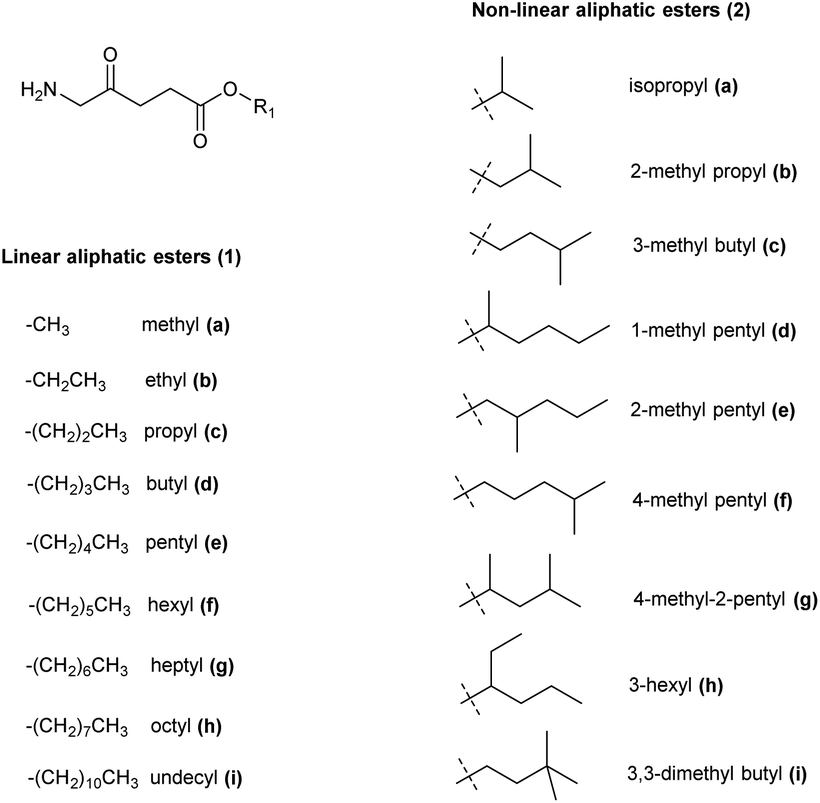 | ||
| Fig. 1 Aliphatic ester prodrugs of ALA 1a–i and 2a–i with a linear or branched alkyl component (reviewed by Fotinos et al.3). | ||
Peng et al. were the first to report a comparative study on the effectiveness of a series of ALA esters against ALA. This showed that the methyl, ethyl and propyl esters 1a–c produced more PpIX fluorescence in both murine and human tissues compared to free ALA.22 Similar observations were made by Kloek et al.23,24 and Gaullier et al.25 with straight-chain ALA esters up to the octyl ester 1h. Gaullier reported that for the lipophilic ALA esters 1f–h, PpIX production was 30–150 times more efficient than for free ALA, thus suggesting faster cellular uptake of the prodrugs and ALA-induced PpIX formation, with the maximum benefit being observed for the hexyl ester 1f. The improved performance of shorter chain esters compared to ALA upon topical administration to the skin is therefore more consistent with increased penetration through tissue towards the target cells, rather than enhanced cellular uptake as a result of a greater lipophilicity.25 Several studies have confirmed that simply changing the lipophilicity of aliphatic ester prodrugs of ALA is not the only factor that must be considered when seeking to optimise delivery and PpIX production. For example, Uehlinger et al.26 noted that the hexyl ester 1f produced an order of magnitude greater PpIX fluorescence than the corresponding cyclohexyl ester 3e (see Fig. 2) even though both the conjugates had similar log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values. More recently, DiVenosa et al.27 reported the evaluation of the undecyl ester 1i of ALA which accumulates rapidly in LM3 breast cancer cells to generate PpIX, by virtue of its high lipophilicity, but is much less effective when applied topically to the skin due to slow diffusion across the strateum corneum.
P values. More recently, DiVenosa et al.27 reported the evaluation of the undecyl ester 1i of ALA which accumulates rapidly in LM3 breast cancer cells to generate PpIX, by virtue of its high lipophilicity, but is much less effective when applied topically to the skin due to slow diffusion across the strateum corneum.
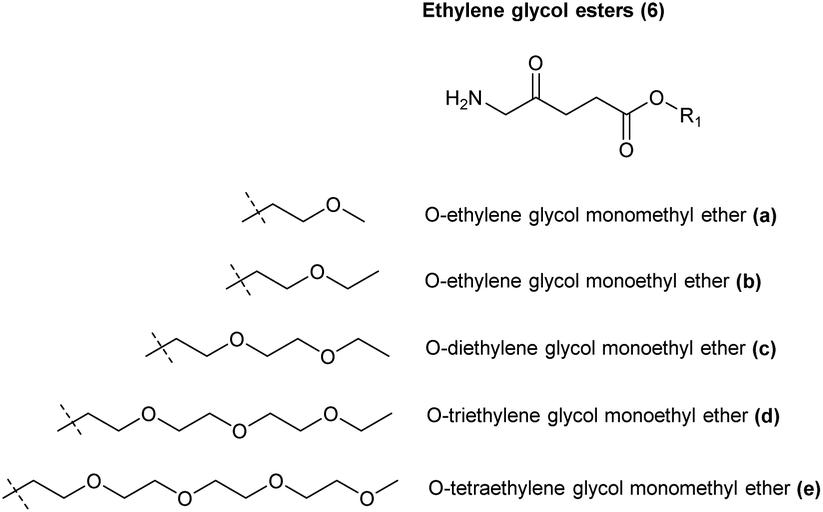 | ||
| Fig. 2 Ethylene glycol-based ester prodrugs of ALA 6a–f. | ||
The utility of ALA ester prodrugs of course depends critically upon the efficient release of ALA by cellular esterases upon internalisation. For most simple aliphatic esters, this is not problematic, however a comprehensive study by Whitaker et al. on the synthesis and in vitro biological evaluation of various linear and branched-ALA esters highlights the impact that ester structure may have on PpIX production from such prodrugs.28 Here, significantly lower PpIX production was observed in pancreatic cells for 2a, 2d, 2g and 2h compared to their linear counterparts which do not contain branching adjacent to the ester carbonyl group.
A range of ALA prodrugs have also been synthesized based on alicyclic or substituted benzyl esters which in some cases also display promising levels of PpIX compared to equimolar ALA (Fig. 3). Again, the need for a balance between lipophilicity and ease of ester hydrolysis is apparent among such compounds, and while they have not so far gained clinical approval, they provide further valuable indications to be inputted into the design of more elaborate ALA prodrug systems.
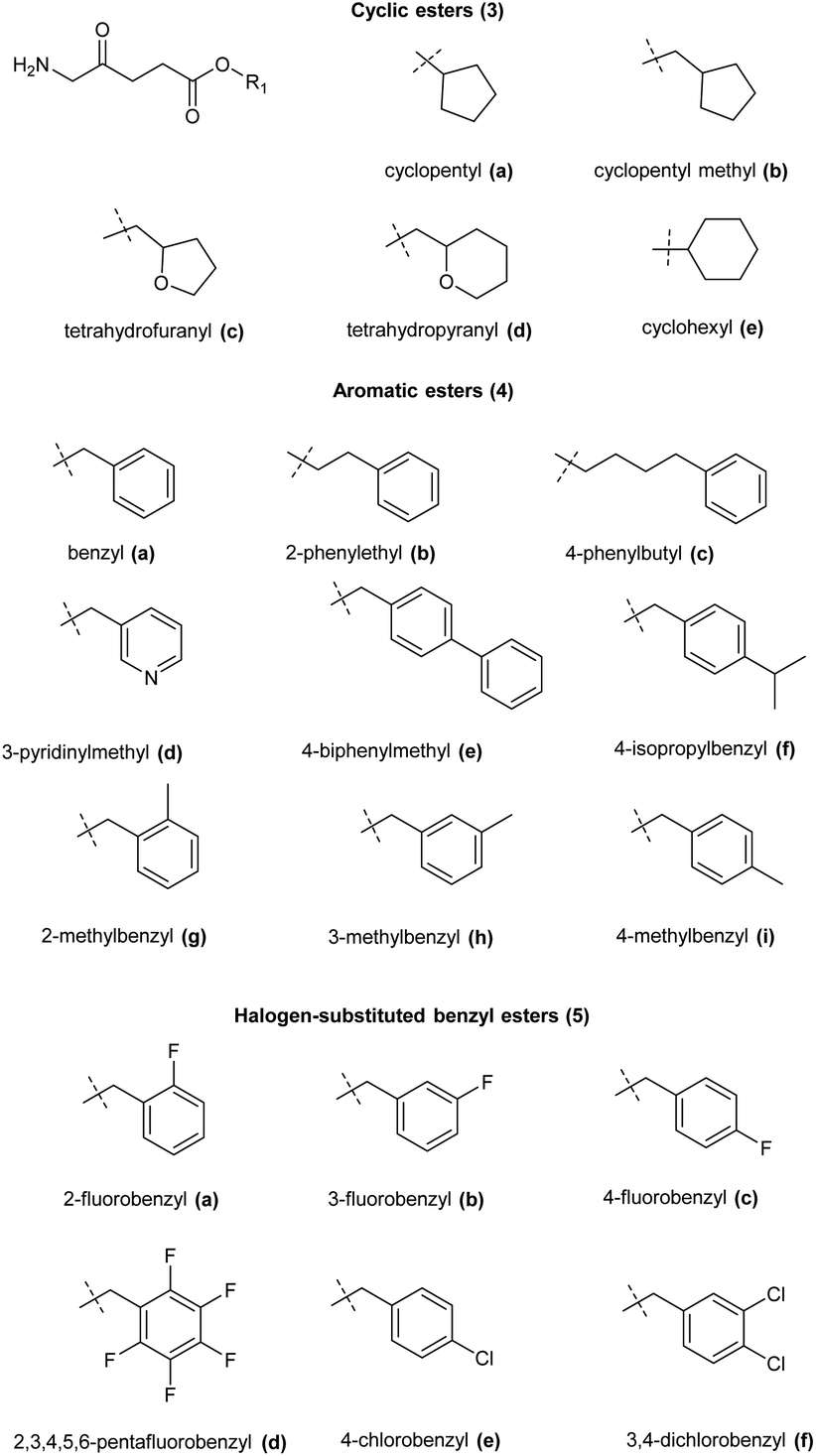 | ||
| Fig. 3 Cyclic ester prodrugs of ALA 3a–e and various substituted benzyl ester derivatives 4a–i and 5a–f. | ||
For example, Kloek et al. reported the synthesis and biological evaluation of tetrahydrofuranyl 3c and tetrahydropyranyl 3d esters of ALA which both showed enhanced PpIX fluorescence compared to ALA in vitro.23 Godal et al. reported the synthesis of the cyclohexyl ester of ALA 4e along with a variety of substituted benzylic and arylalkyl esters 4a–f of ALA, and biological evaluation in vitro/in vivo showed significantly enhanced PpIX fluorescence compared to ALA for aromatic esters, especially the benzyl and ring-alkylated benzyl esters 4a, 4b and 4g–i. In contrast, the cyclohexyl ester 4e produced the least PpIX fluorescence amongst all the prodrugs tested and was only as effective as ALA itself. Halogen-substituted benzyl esters 5a–f showed intermediate PpIX fluorescence, and amongst them the mono-halogenated esters showed higher PpIX fluorescence than the di- or penta-substituted esters.29
Although aliphatic ALA esters have been shown to induce higher PpIX production with an increase in carbon chain length, as already noted increasing ALA prodrug lipophilicity in this way can be counter-productive, bringing the limitations of slower tissue penetration as well as lower water solubility for esters which are derived from long-chain alcohols.27 In contrast, poly(ethylene glycol) derivatives, which have been used for a large number of pharmaceutical applications, potentially offer the advantage of both improved water and lipid solubility, as well as giving rise to a benign promoeity upon ester hydrolysis.30 To this end, Berger et al. reported the synthesis of ethylene glycol esters of ALA 6a–c (Fig. 4) and tested their biological efficacy in human and rat cell lines of carcinoma and endothelial origin. It was found that a high PpIX fluorescence compared to free ALA was observed in endothelial cells in comparison to tumour cells, especially for long-chain derivatives.31 Godal et al. also reported the synthesis of ethylene glycol esters 6d and 6e, but biological evaluation in vivo showed that a lower PpIX fluorescence was produced compared to the simple hexyl ester 1f.29
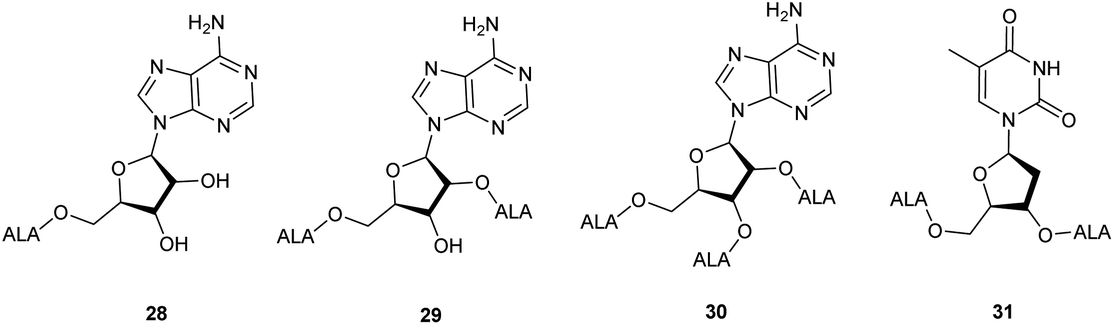 | ||
| Fig. 4 ALA-nucleoside bioconjugate prodrug 28–31 bearing up to three ALA units. | ||
As already noted, the use of ALA esters can result in increase PpIX production in various cell types, but with a greater enhancement in tumour cells. In this context, Brunner et al.32 observed that the simple hexyl ester 1f and benzyl ester 4a prodrugs which lack any specific targeting motif in their structures showed a significantly higher accumulation of PpIX in tumour cells compared to ALA, when applied at low concentrations and short incubation times. In a clinical setting, this means that appropriate PpIX levels for either PDT or PDD can be attained more rapidly and with less side effects. Preferential accumulation of ALA and its esters in tumour cells can also be correlated with the expression of specific uptake transporters33 and this has provided a rationale for the development of prodrugs that contain structural features that only enhance physical properties but can provide more effective targeted delivery.
Biomolecule conjugates and polyvalent ALA ester derivatives
The esterification of ALA with various vitamins, monosaccharides and nucleoside derivatives has been explored both with a view to tailoring the physical properties of ALA and enhancing the transport of ALA into cells. For example, the attempted synthesis of novel ester bioconjugates of ALA with vitamin E (or α-tocopherol) 8 or vitamin D312 has been reported by Vallinayagam et al.34 as shown in Scheme 4. Typically, more complex ester prodrugs of ALA such as the desired conjugates 11 and 15 are prepared using a N-alkoxycarbonyl-protected ALA derivative such as Boc-ALA 9 or the ALA surrogate, azidolevulinic acid 10, with activation of the carboxylic acid function using Steglich-type esterification conditions with a carbodiimide reagent (DCC or EDC) and DMAP as catalyst. Both 9 and 10 are unable to undergo competing Schiff base chemistry during the key coupling step. For the molecules in Scheme 4, although vitamin E conjugate 11 was successfully prepared, the vitamin D conjugate 15 was unstable to final deprotection chemistry. The original rationale for the preparation of ester derivatives of ALA with these hydrophobic vitamins was to potentially facilitate the transport of ALA into cancer cells through the ease of incorporation of such bioconjugates into cell membranes.35 In parallel, the enhancement of ALA-induced PpIX production by vitamin D has also been studied;36 the observed enhancement of photodynamic cell death by vitamin D suggests that there may be potential for developing multifunctional ALA prodrugs of this type that can activate more than one cell-death pathway (see below).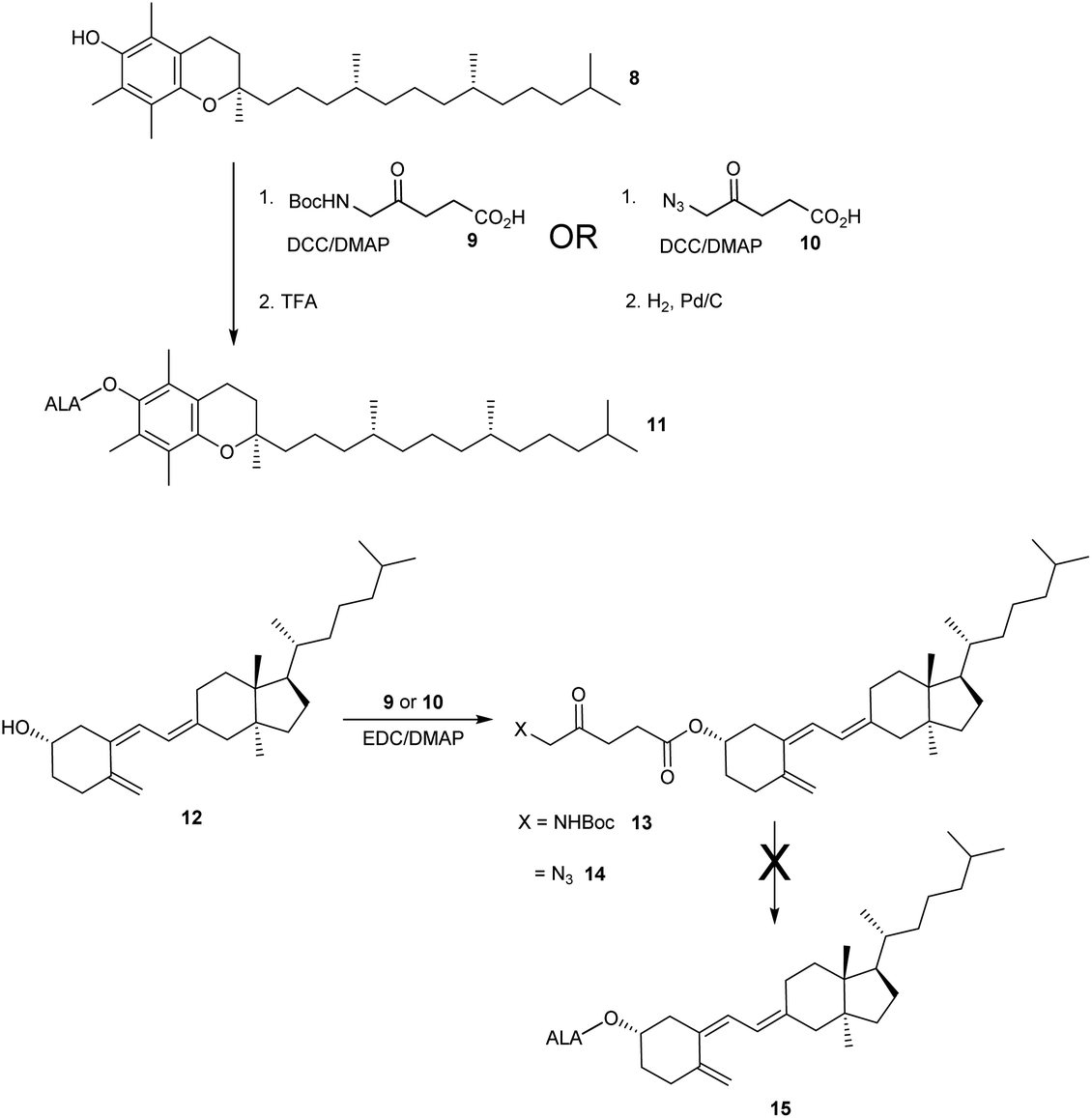 | ||
| Scheme 4 Synthesis of ester bioconjugate prodrugs of ALA. | ||
The targeting of various photosensitisers with sugars has been very widely investigated as a means of enhancing selective accumulation in cancerous vs. normal cells. To this end, Vallinayagam et al.37 reported the esterification of ALA with several simple monosaccharides. The presence of multiple hydroxyl functions in the ester component here presents an additional challenge for the chemist and required the development of distinct synthetic strategies to produce ALA conjugates selectively monoacylated on the 1-hydroxyl (anomeric) or 6-hydroxyl groups. As shown in Scheme 5, regioselective acylation of glucose 16 at the 6-position was achieved in moderate yield by enzyme-catalysed transesterification with the Boc-ALA derivative 17 using subtilisin, followed by acidic deprotection of the ALA component in 18 to give the desired conjugate 19. Similar regioselective acylations could also be achieved with galactose 20 and mannose 24 and using porcine pancreatic lipase, again in reasonable yields. In order to effect esterification directly on the anomeric hydroxyl of these sugars, a multi-step orthogonal protection scheme was needed, using benzyl protection of the other hydroxyl groups, as outlined in Scheme 6 for the preparation of the galactose conjugate 23. A simpler, enzyme-mediated approach to 1-functionalised derivatives could however be achieved without additional hydroxyl protection by first derivatising the anomeric hydroxyl with an ethylene glycol spacer, as illustrated for the mannose derivative 27 (Scheme 7).
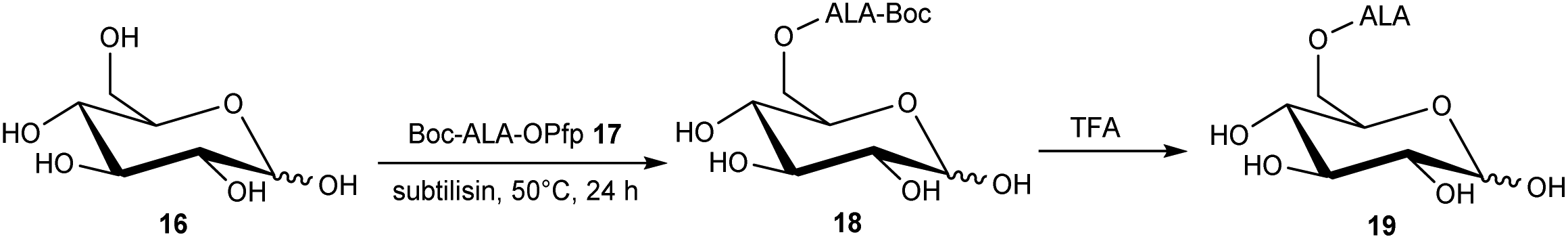 | ||
| Scheme 5 Enzyme-mediated syntheses of a regioselectively 6′-O-acylated ALA-glucose bioconjugate prodrug 19. | ||
 | ||
| Scheme 6 Chemical synthesis of regioselectively acylated 1′-O-ALA-galactose bioconjugate prodrug 23via an orthogonal protecting group strategy. | ||
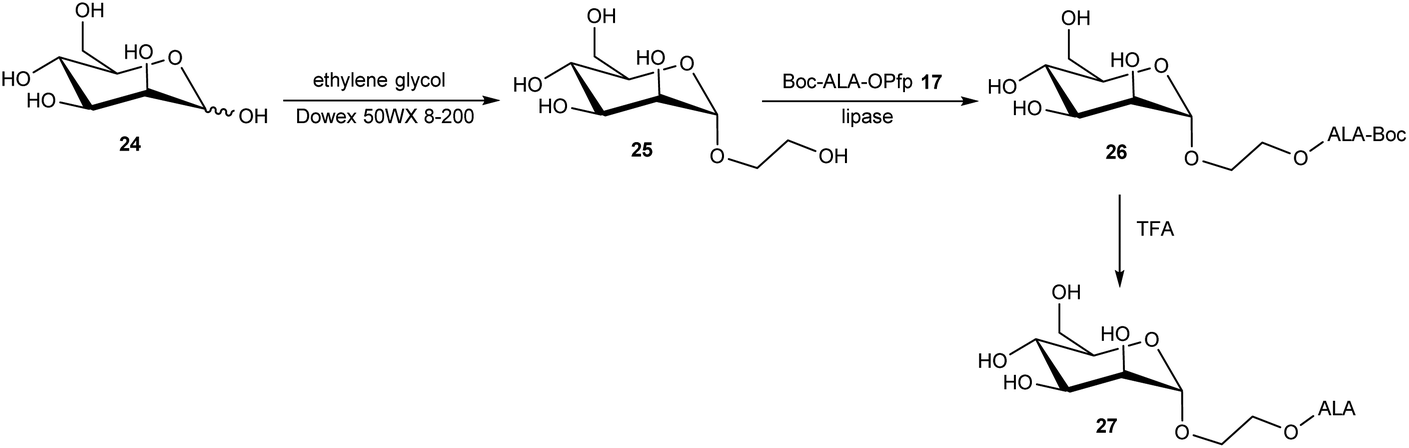 | ||
| Scheme 7 Enzyme-mediated regioselective synthesis of ALA-bioconjugate prodrug 27via introduction of a spacer unit at the anomeric position of the sugar. | ||
Evaluation of these prodrugs in human colon, breast and lung carcinoma cells and angiogenic endothelial cells revealed that PpIX production was similar to ALA itself, whereas in normal fibroblasts PpIX production from the prodrugs was very low. Surprisingly, there was little difference in PpIX production between the 1- or 6-acylated ALA esters such as 19 and 23, and the insertion of a spacer as in 27 tended to decrease PpIX production. Furthermore, competition experiments with the parent sugars 16, 20, and 24, or their 1-OMe derivatives had no effect on PpIX production, suggesting that such prodrugs do not appear to produce any cell-selectivity associated with sugar-mediated uptake mechanisms. It was concluded that prodrugs of this type may nonetheless be valuable for topical delivery of ALA to tumour cells with the reduced PpIX production in normal cells serving to spare them upon ALA-PDT. Moreover, the production of a harmless, alcohol component upon prodrug cleavage should again be a potential advantage for these or other prodrugs based upon natural sugars.
ALA esters with potential targeting properties have also been prepared by esterification of the hydroxyl functions of nucleoside derivatives.38 A feature of these prodrugs, as with other sugar-based derivatives, is the potential to produce derivatives with an increased payload of ALA compared to conventional esters, which is of interest since 8 copies of ALA must be delivered to the cell for bioconversion to one molecule of PpIX. Once again, prodrugs containing a single ALA unit, demand a chemical approach that allows for selective esterification of primary and secondary hydroxyl functions of the nucleoside component. To this end, an ALA prodrug of adenosine 28, selectively monoacylated at the primary 5′-hydroxyl was prepared in modest yield directly from the parent nucleosides by Gurba et al. again utilising enzyme-mediated transesterification.38 Attempted peracylation of the ribose component of adenosine using standard carbodiimide chemistry (DCC/DMAP) was less efficient and led to a mixture of di- and triacylated products from which di- and tri-ALA ester prodrugs 29 and 30 were subsequently obtained. In contrast, the deoxyribose unit of thymidine could be converted to the expected diester in excellent yield under similar conditions, providing prodrug 31 bearing two ALA units. Interestingly, evaluation of 28–31 in various cancer cell lines (human colon (SW480), breast (MCF7), lung (A549), ovarian (A2780), cervix (HeLa)) revealed that PpIX production did not clearly correlate with the number of copies of ALA present. Indeed, the adenosine conjugates 28–30 showed poor PpIX production compared to ALA in all the cell lines tested, suggesting that they may not be substrates for relevant adenosine receptors. Encouragingly, the thymidine derivative 31 did produce more PpIX (approximately 1.5-fold) than equimolar ALA in most cell lines, which supports the idea that it may be possible to develop ALA prodrugs that carry multiple copies of ALA and which can be targeted to specific tumour cells.
Cyclodextrins are well-known nanocarriers of biological origin that present multiple hydroxyl functions for attachment of drug entities such as ALA. In a preliminary investigation by Aggelidou et al.,39 beta-cyclodextrin which disposes seven primary hydroxyl functions, could be esterified with up to three ALA units via the azido derivative 10, using DCC/DMAP chemistry. The water-soluble ALA prodrug 32 that was obtained (Fig. 5) was found to effectively produce PpIX in MCF7 cells in comparison to equimolar ALA, which suggests that such compounds may have significant potential for the delivery of enhanced payloads of ALA, if effective functionalisation of all primary hydroxyls can be achieved.
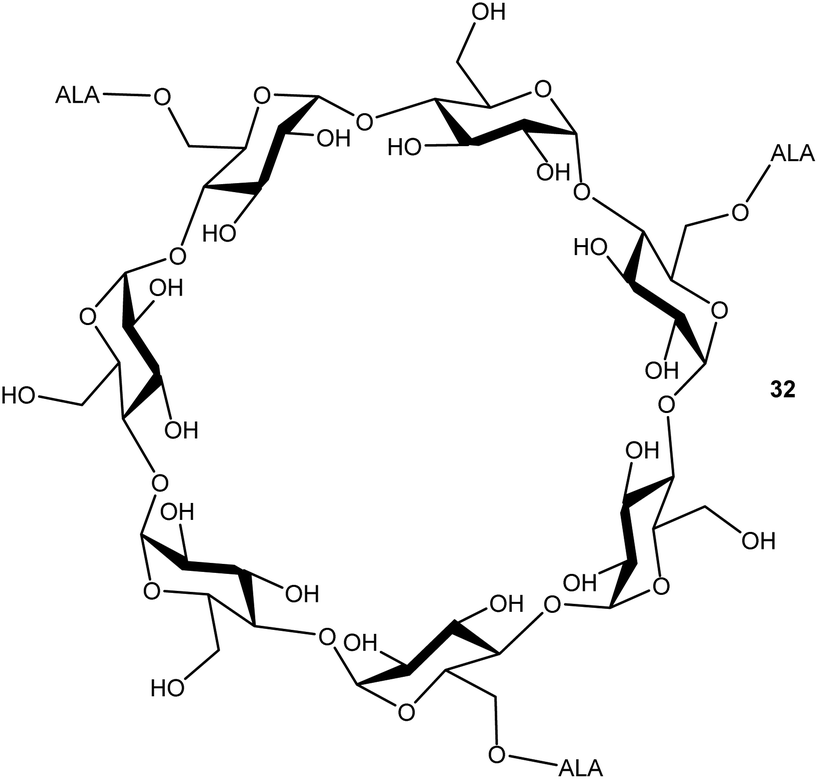 | ||
| Fig. 5 Cyclodextrin-based trivalent ALA prodrug ester 32. The conjugate obtained presents three ALA units which are presumably randomly distributed over the upper (primary hydroxyl) face of the scaffold. | ||
The use of other polyvalent drug carriers such as dendrimers have shown significant promise for the delivery of high payloads of photosensitisers in PDT. This approach has been explored in some depth as a means of overcoming the poor systemic bioavailability of ALA in both PDT and PDD. Battah et al. in 2001 first reported the synthesis of a series of novel ALA-containing dendrimers bearing 6 to 18 ALA residues in which ALA residues are attached to the periphery of the dendrimer structure via ester linkages.40 First-generation dendrimers 34–36 with 6 or 9 ALA units were prepared by attachment of a tris(Boc-ALA) derivative 33 to a di- or tripodal aromatic or tripodal aliphatic core via amide bond formation (Fig. 6). Second generation dendrimers 37 and 38 with 18 ALA units were then prepared by amplification of the building block (dendron) 33 and coupling to the tripodal aromatic cores used previously thus providing derivatives with an increased distance between the dendrimer core and the ALA units. This convergent growth approach has the advantage of ensuring the formation of prodrugs of defined size and loading, which is more challenging to achieve via a divergent strategy starting from a multifunctional core unit (see above), and potentially provides a route to ALA dendrimers of sufficient size to result in enhanced permeability and retention in tumour tissue. Evaluation of first generation dendrimeric ester 35 and second generation derivative 37 in the in the keratinocyte cell line, PAM 212 at a concentration of 0.1 mM, revealed a significantly enhanced level of PpIX fluorescence after 4 h incubation compared to equimolar ALA. Although no direct correlation was observed between the number of ALA units and PpIX fluorescence, 37 appeared to have a greater than two-fold efficacy over 35, with a correspondingly higher phototoxicity upon irradiation of cells. No dark toxicity was observed for either dendrimer at this concentration.
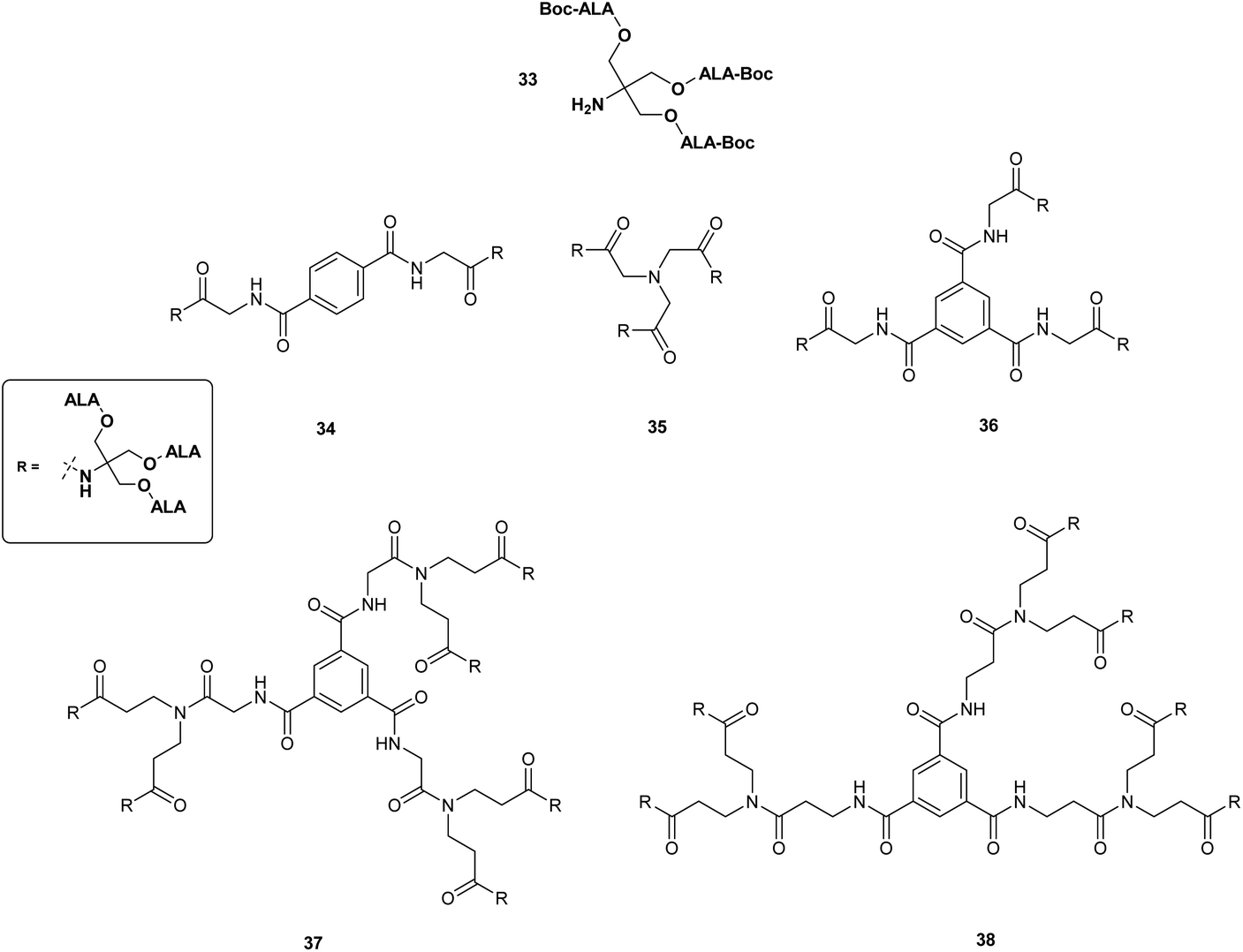 | ||
| Fig. 6 First and second generation dendrimeric ALA prodrug esters 34–38. | ||
A further expanded second generation dendrimer 39 (Fig. 7) was subsequently prepared by MacRobert and coworkers and evaluated in detail in both PAM 212 keratinocyte and A431 human epidermoid cancer cell lines.41 This again confirmed the ability of such dendrimers to efficiently pass through cell membranes and release ALA intracellularly, with porphyrin production again being significantly higher than for equimolar ALA in both cell lines at low concentrations (0.01–0.1 mM). At higher prodrug concentrations, porphyrin production was significantly lower, possibly due to intermolecular Schiff base formation at physiological pH in culture medium as observed for most ALA ester derivatives with a free amino function.
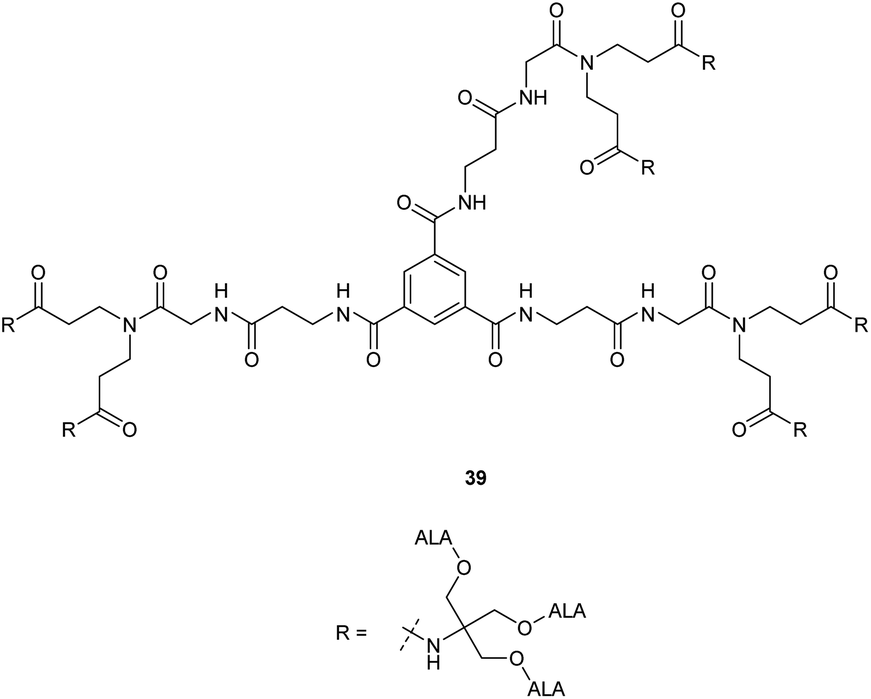 | ||
| Fig. 7 Expanded second generation dendrimeric ALA prodrug ester 39 presenting 18 ALA units. | ||
Studies with low temperature incubation and endocytosis inhibitors suggested that cellular uptake of 39 is primarily via micropinocytosis and thus the improved performance of second generation derivatives 37 and 39 over first generation derivative 34 may reflect increased uptake of the larger dendrimers by endocytic processes. Uptake of 39 by endocytosis may also result in stabilisation against imine formation between ALA units due to protonation of their amino functions upon incorporation within acidic lysosomes. A further very important finding for 39 is that there is a marked difference in the kinetics of PpIX accumulation compared to ALA which ultimately may have significant importance for the application of these prodrugs, particularly for PDD. PpIX production for 39 was found to be sustained for up to 24 h, at which point PpIX levels from ALA itself were significantly lower, consistent with the metabolism of porphyrin to heme within a few hours of incubation. The slower kinetics for 39 was proposed to result from gradual release of ALA by ester hydrolysis from the internalised prodrug which in turn is likely to be related to the steric accessibility of the multiple ALA units to intracellular esterases. It was indeed noted that the second generation dendrimer 37 which incorporates a shorter spacer structure between the ALA units and the prodrug core performed rather less effectively than 38 suggesting that the ALA ester units here are slightly less accessible. Complementary studies by the same researchers42 on three different ALA dendrons 40–42 each with three ALA units (Fig. 8) further support the importance of steric effects in determining the rate of ALA release from these systems and hence PpIX synthesis. Dendron derivative 41 for instance which incorporates a longer, propyl linker to the quaternary carbon, was found to be the most effective, with the ALA ester units here again being relatively more accessible for enzyme-mediated hydrolysis. 41 was also studied for in vitro efficacy in LM3 lung carcinoma cells and was found to produce similar porphyrin levels compared to free ALA.43 It was also found to be effective in vivo when administered either topically or systemically to tumour-bearing mice with higher peak porphyrin levels being observed compared to the hexyl and methyl esters of ALA, although it was inferior to ALA itself. This may be due to retention of 41 within the stratum corneum at the application site when applied topically, as this triester conjugate is fairly lipophilic compared to ALA, which highlights again the fine balance that needs to be achieved in the design of ALA prodrugs with optimal physicochemical properties for both cell uptake and tissue penetration.
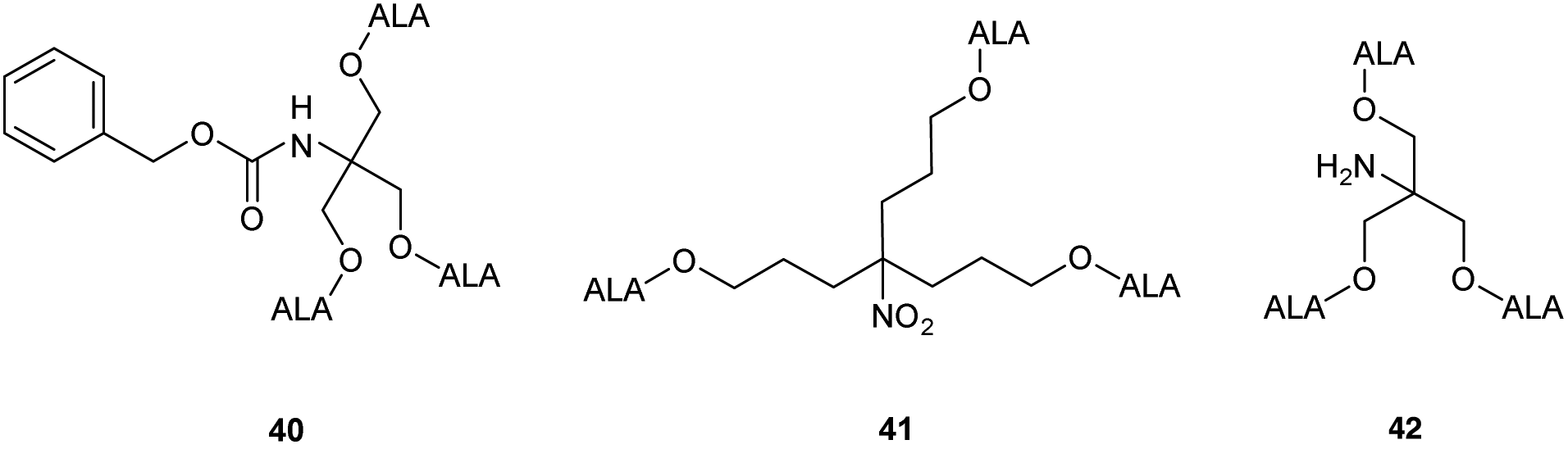 | ||
| Fig. 8 ALA dendron ester derivatives with varying structural features that may lead to altered rates of ALA release upon cleavage by esterases. | ||
The sustained and enhanced PpIX production observed with the 18 ALA dendron 39 in in vitro studies was also confirmed by in vivo evaluation in a murine tumour model. Here, 39 showed a sustained release of PpIX for over 24 h, and the basal values were not reached until 48 h post-intraperitoneal administration of 39.44 In contrast, PpIX production for ALA was observed for only 3–4 h. While these results indicate that the kinetics of porphyrin production is a function of the rate of ester hydrolysis in 39, it is also of significance that the integrated porphyrin accumulation for 39 and ALA at equivalent molar ratios was observed to comparable for the 48 h time course. This demonstrates that the release of the ALA unit from 39 was essentially complete and the total porphyrin production is comparable – an essential feature to be achieved for the design of an effective polyvalent ALA prodrug derivative.
Based on these promising features, François et al.45 evaluated 39 for fluorescence diagnosis of bladder cancer, wherein a sustained release of PpIX was observed even after 24 h post-administration of the prodrug intravesically. A significantly lower dose of the prodrug was required to produce the same effect in comparison to free ALA (0.7 vs. 180 mM) and ALA hexyl ester 1f (0.7 vs. 8 mM). Significant tumour specificity vs. muscle and normal urothelium was also observed as compared to 1f making it a potentially viable tool for use in fluorescence-guided cystoscopy.
Most recently Rodriguez et al.46 have investigated ALA dendrimers 43 (six ALA units) and 44 (nine ALA units) (Fig. 9) for PDT in mammary LM3 carcinoma cells. In vitro studies showed enhanced PpIX production (ca. four times) for both 43 and 44 at a lower concentration as compared to free ALA. The presence of the extended propyl spacer in the ester units of these derivatives would be expected to facilitate ALA release and PpIX production compared to previously reported compounds. Topical application of 43 and 44 on the skin overlying a subcutaneous LM3 implanted tumour showed lower amounts of PpIX in distant skin as compared to free ALA, which induced similar profiles of PpIX in distant skin and skin overlying tumour. This suggests a promising use of these dendrimeric ALA prodrugs in superficial cancer models. A possible application in vascular PDT was also identified for 43 and 44 by assessing their selectivity towards macrophages over endothelial cells. Evaluation in Raw 264.7 macrophages and HMEC-1 microvasculature cells showed enhanced porphyrin synthesis by 44 (6 fold) and 43 (4.6 fold) in macrophages as compared to endothelial cells. Free ALA in contrast had only a slight selectivity towards macrophages (1.7 fold).
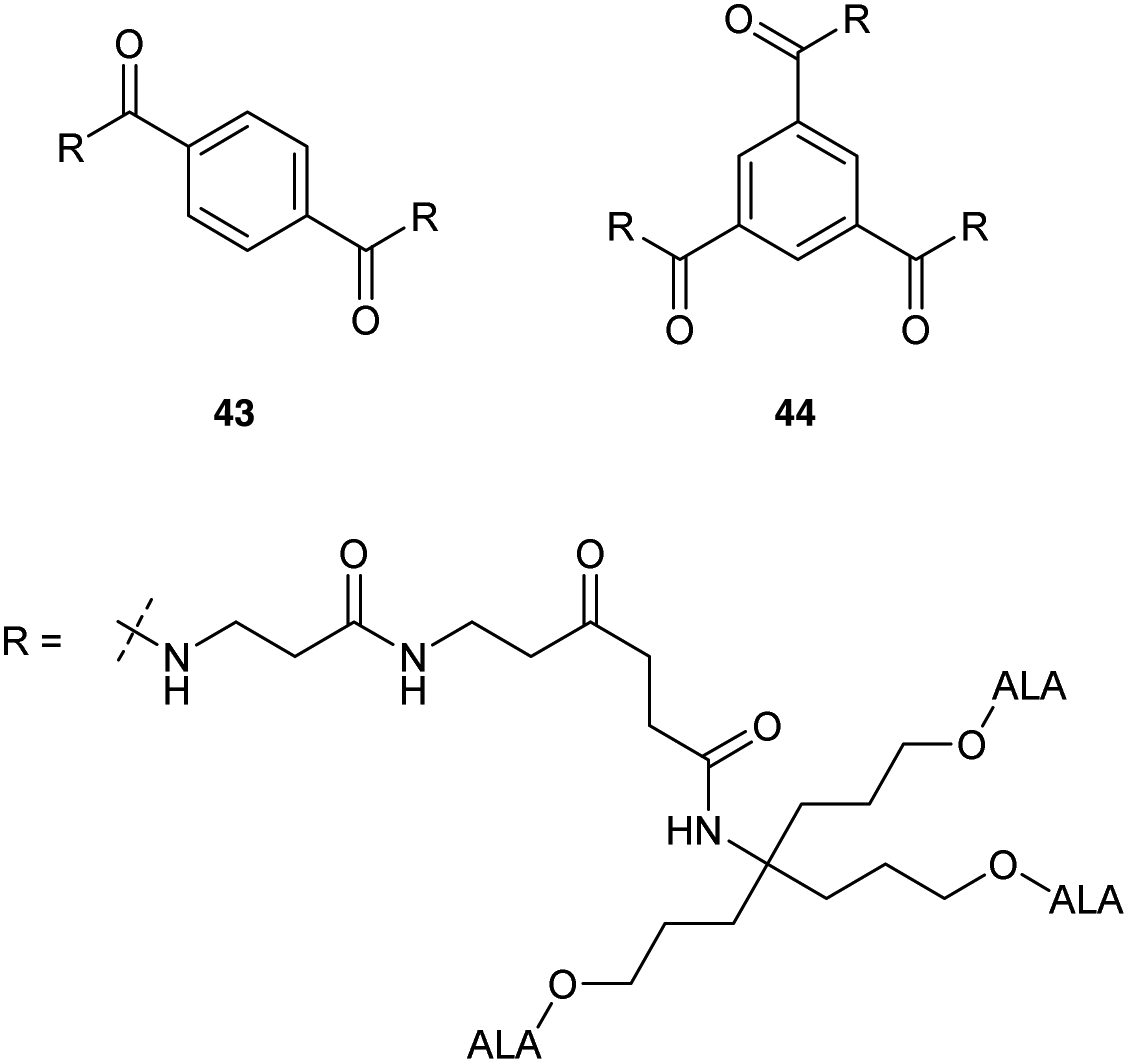 | ||
| Fig. 9 ALA dendron derivatives with a more sterically accessible ester components. | ||
Miscellaneous ester prodrugs
With the growing recognition of the clinical possibilities offered by some of the ester derivatives described above, chemists have been challenged to design more novel ALA prodrugs with bespoke properties for PDT, as well as to come up with new methods for their synthesis. A few brief examples can be highlighted here. Pavani et al.47 have utilised a hydroxy-functionalised zinc phthalocyanine derivative as a template for attachment of four ALA units, thereby producing a system for intracellular ALA delivery with the potential to provide two synergistic photosensitisers. The phthalocyanine unit here provides a functional promoiety, which is a feature that has been deliberately designed into acyloxyalkyl ester prodrugs of ALA by Rephaeli and coworkers48 as a means to deliver a potential histone deacetylase inhibitor (e.g. butyric acid) alongside ALA to provide an additional non-light induced cell killing mechanism. Gola et al.49 have recently reported the use of the multicomponent Passerini reaction to produce structurally diverse esters of ALA instead of applying a classic acylation approach. By combining a protected ALA derivative with various isocyanides and formaldehyde, various novel ALA esters with tailored lipophilicity could be prepared in good yield. Some of these compounds gave significantly higher levels of porphyrin production than ALA in more than one cell line, and the most promising derivative had comparable PDT efficiency to the hexyl ester 1f in LM2 mammary adenocarcinoma cells, at a concentration where ALA itself produced no cell death. Lastly, as has already been noted, the release of ALA from various ester prodrug derivatives may be strongly influenced by the structure of the pro-moiety, as well as different expression levels of esterases in normal compared to tumour cells. Soares et al.50 have demonstrated that ALA may be released from lipophilic coumarin ester derivatives by application of an external light source. This removes the dependence of ALA release upon steric effects in an enzyme-mediated hydrolysis reaction, and could provide an interesting way of triggering ALA release on demand in a time and spatio-dependent fashion.Modulators of the heme pathway: iron chelators
As has already been noted, targeting key steps in the heme cycle with appropriate small molecules is a potential chemical strategy by which the conversion of administered ALA to PpIX can be optimised. Moreover, it has been observed that differences in the accumulation of PpIX in healthy cells and malignant cells are due to differences in the activity of enzymes in the haem biosynthetic pathway such as porphobilinogen deaminase and ferrochelatase.51 Molecules such as 2-allyl-2-isopropylacetamide are known to stimulate ALA synthase and ALA dehydratase activities,52 while molecules that inhibit protoporphyrinogen oxidase53 or upregulate coproporphyrinogen oxidase54 have been identified which show positive results in ALA-PDT. While this highlights the potential for targeting various steps in the heme cycle in order to enhance intracellular PpIX production most attention has been paid to targeting the insertion of ferrous iron (Fe2+) into the PpIX core, which is catalysed by ferrochelatase in the last step of heme biosynthesis. This forms the basis of a therapy combining iron chelators with ALA PDT where cells are thus deprived of Fe2+ in the last step of heme synthesis, leading to higher levels of PpIX.55,56Diverse iron chelators such as EDTA, desferrioxamine (DFO, 45), and dexrazoxane all shown potential in preclinical studies for increasing PpIX levels for PDT upon ALA administration.7 EDTA is however, a non-selective chelator which binds other biologically relevant metal ions as well as Fe2+. More significantly, it has a markedly lower affinity for iron compared to DFO. DFO 45 (Fig. 10) is a hexadentate siderophore which has been shown to have antiproliferative activities against various tumour cell lines,57 and has been extensively studied for use in the treatment of iron overload disorders.58 However, due to its hydrophilic nature it is unsuitable for topical application, and since prolonged exposure to strong iron chelators such as DFO 45 may lead to iron starvation within healthy tissue,59 its suitability for use in ALA-PDT or PDD is somewhat uncertain.60,61
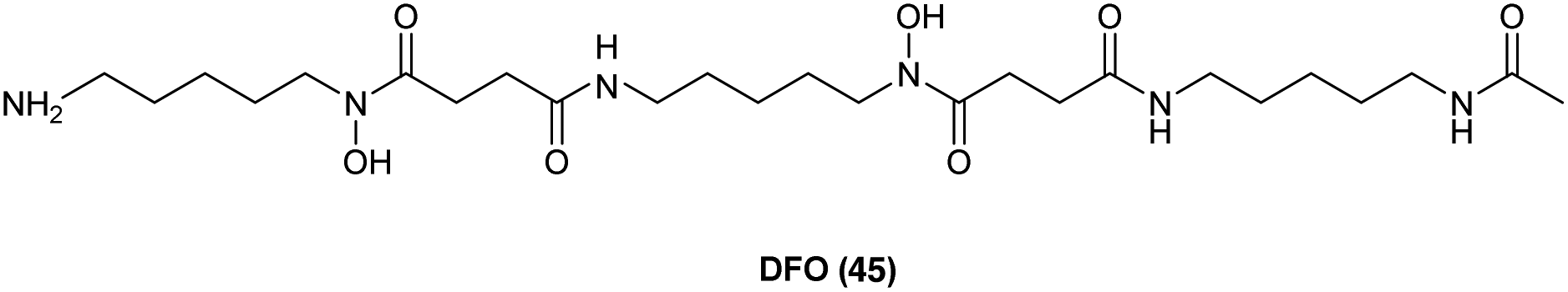 | ||
| Fig. 10 Desferrioxamine (DFO) 45. A potent iron chelator that has been studied for enhancement of PpIX production in ALA-PDT. | ||
One compound that has shown particular promise for use in ALA-PDT is 1,2-diethyl-3-hydroxypyridin-4-one, more commonly known as CP94 46 (Fig. 11). The low molecular weight and greater lipophilicity of CP94 compared to both DFO and EDTA, make it particularly effective for chelating intracellular iron. CP94 has high affinity for Fe3+ and as such the chemical basis for its application to enhance ALA-PDT lies in its ability of promote the oxidation of Fe2+ to Fe3+ at low concentration by molecular oxygen.62 Following the initial observation of the efficacy of combined ALA and CP94 treatment,63 many other studies have demonstrated the effectiveness of ALA and CP94 both in vitro and in vivo for PDT, as well as potential applications in fluorescence-guided surgery.64,65 For example, Blake et al.66 investigated the combination of CP94 with ALA and MAL 1a in human glioma cells, and observed that CP94 significantly enhanced PpIX accumulation and subsequent PDT cytotoxicity. This combination may therefore be useful both in photodiagnosis and /or PpIX-induced PDT treatment of glioblastoma, where prognosis is typically poor. The combination of ALA or MAL and CP94 has also been shown to be safe and effective in clinical pilot studies for the topical PDT of nodular basal cell carcinomas.67,68
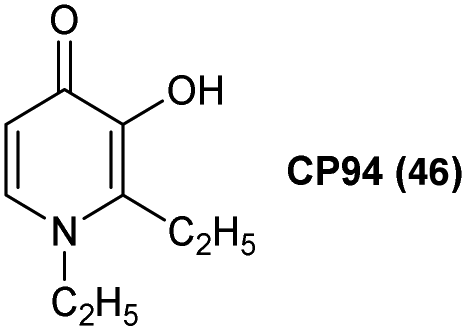 | ||
| Fig. 11 Structure of CP94. A clinically validated iron chelator for ALA-PDT. | ||
The difference in pharmacological properties between ALA and a lipophilic iron chelator such as CP94, or indeed any modulator of heme synthesis, is a potential challenge that must be addressed upon coadministration. Zhou and coworkers62 have sought to address this problem via the innovative approach of incorporating the structure of hydroxypyridone-(HPO)-type iron chelator into the ester moiety of an ALA prodrug. Conjugates of this type (Fig. 12) are more lipophilic than ALA itself and as predicted were able to efficiently deliver and release both ALA and HPO simultaneously into MCF-7 and MCF-7R cells with a corresponding enhancement in PpIX levels compared to ALA alone, or separate coadministration of ALA and HPOs. The best performing derivatives embodied a relatively long, flexible aliphatic linker in the ester unit between ALA and the bulkier HPO function, suggesting effective uptake by passive diffusion as well as the absence of any steric effects on cleavage by cellular esterases. These compounds (n = 6–12) were found to produce higher levels of PpIX fluorescence than the benchmark ALA hexyl ester.
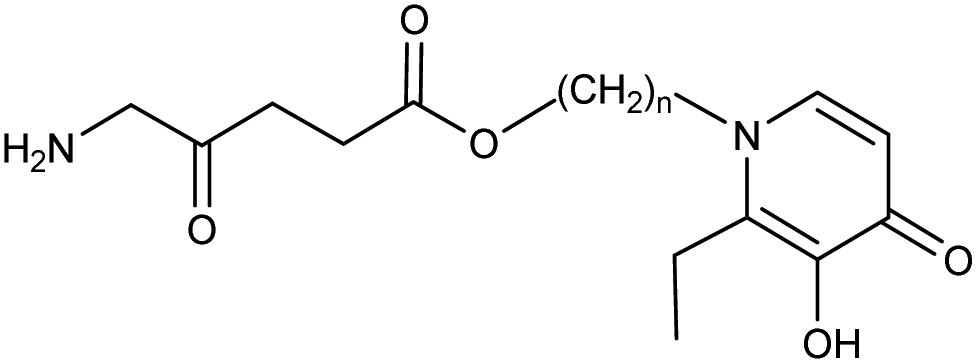 | ||
| Fig. 12 General structure of ALA-HPO conjugates. | ||
A very recent report by Anayo et al.69 has also revealed that a related combined ALA prodrug and iron chelator, AP2-18, is able to outperform the coadministration of ALA or MAL and CP94. The authors suggest that this novel patented ALA prodrug has the potential to address the challenge of delivering greater PpIX accumulation to the base of thicker nodular basal cell carcinoma (nBCC) tumours that are typically encountered clinically, and so may be a valuable new tool for dermatological PDT.
Studies by Zhou et al. have also revealed the potential for linking a HPO to the N-terminus of an ALA derivative,70 while Zhu et al. have described ALA-HPO ester conjugates where the N-terminus of ALA is acylated (see following section).71 Once again, examples of such conjugates have been shown to be superior to ALA itself for PpIX production in a variety of tumour cell lines, with levels of intracellular porphyrin fluorescence correlating well with cellular photoxicity upon irradiation.
Finally, Mrozek-Wilczkiewicz et al.72 have reported the concept of enhancing ALA-PDT using thiosemicarbazone (TSC) iron chelators, which have been previously developed as stand-alone anticancer agents (Fig. 13). Some of the compounds synthesised show exceptional (sub-nanomolar) dark cytotoxicity in HCT 116 colorectal carcinoma cells with a wild-type (+/+) or deleted (−/−) tumour suppressor TP53 gene. Although some synergistic effects on photocytotoxicity were observed with X and Y when used in combination with ALA-PDT, the sub-nanomolar concentrations of chelators used do not appear to be consistent with an enhancement of PpIX levels through simple sequestration of iron. A possible explanation for the effectiveness of the ALA-PDT-TSC combination may therefore be rather the result of two direct cytotoxic effects; one stemming from the PDT effect of the PpIX generated, the other due to ROS produced from the TSC-iron complexes via the Fenton reaction. This suggests an interesting new chemical approach for improved ALA-PDT that might be exploited further through the synthesis of novel combined ALA-TSC prodrugs.
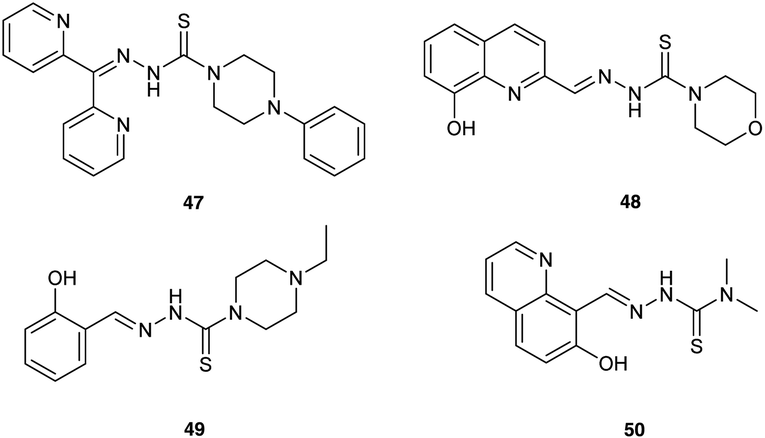 | ||
| Fig. 13 Novel thiosemicarbazones studied for synergistic iron chelation and ALA-PDT. | ||
N-Acylated ALA derivatives and ALA peptide prodrugs
A very obvious way to address the chemical stability issues that are associated with ALA and clinically used ester derivatives is to mask the primary amino function by the formation of an amide derivative. While this is attractive in principle, the effectiveness of such ALA prodrugs then depends upon the expression of a suitable protease activity in the targeted cells to liberate ALA. Indeed, while a number of simple N-acylated ALA derivatives have been reported, such as the formyl or acetyl derivatives 51 and 52 (Fig. 14), which are stable at physiological pH and also more lipophilic than ALA, in vitro studies have in general revealed very low PpIX production, consistent with inefficient release of ALA.3,73 | ||
| Fig. 14 Structures of formyl and acetyl ALA derivatives. | ||
In response to the challenges posed by simple N-acylated ALA derivatives, Babic et al.74 have developed ALA prodrugs in which the amino group of ALA is masked either by conversion to a phosphonamido group, or a novel amide where the pro-moiety contains a phosphate linkage that provides a trigger for cleavage and ALA release (Scheme 8). Both the compounds 53 and 54 were found to be chemically stable at acidic, basic, and physiological pH, however they were efficiently cleaved in vitro by alkaline phosphatase to liberate the hexyl ester of ALA. The activation of 53 and 54 is clearly structure-dependent, with complete conversion of 54 occurring in around 60 min, approximately ten times faster than for the phosphonamido derivative 53. This provides the possibility to fine-tune the rate of ALA release from such compounds in vivo. To this end, activation by alkaline phosphatases is very attractive since they are ubiquitous enzymes that are present in all tissues and overexpressed in various tumours. This not only suggests a possible application for these prodrugs for selective PDT, but also PDD. Both compounds were shown to produce higher levels of PpIX as well as reduced toxicity compared to 1f in U87MG glioblastoma cells over a 24 h time course, notwithstanding the slower rate of activation of 53. Their clinical potential was further validated in vivo in a U87MG glioblastoma spheroid tumour model in chick embryos. Additional studies75 with these prodrugs in a panel of different cancer cell lines revealed the level of PpIX production varied significantly, as might be expected based on known levels of phosphatase expression. As well as U87MG, the prodrugs performed best in PC3 and MCF7 cells, which points to their potential for future clinical development for applications in glioblastoma, prostate and breast cancer respectively.
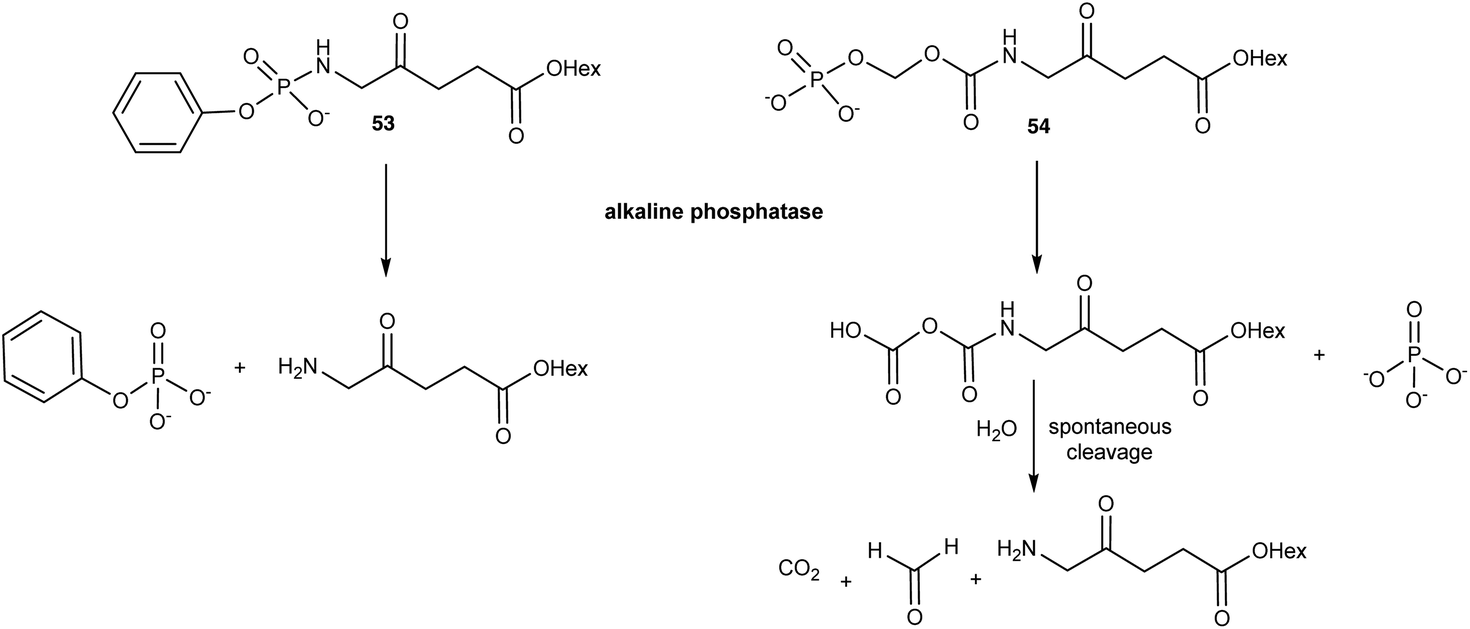 | ||
| Scheme 8 Bioconversion of phosphatase-sensitive ALA prodrugs 53 and 54 to release the hexyl ester of ALA 1f. | ||
In a further application of this approach, Babic and coworkers76 have reported a prodrug of ALA 55 in which the amino function is masked by an acyl group that is susceptible to cleavage induced by β-glucuronidase activity. Such ALA prodrugs could again represent promising tools for tumour detection as elevated expression of β-glucuronidase activity has been associated with various cancer types, including lung, breast and ovarian cancers. In this case, cleavage of the glucuronyl unit from the N-acyl promoiety of 55 triggers the release of the hexyl ester of ALA via a self-immolative 1,6-elimination process (Scheme 9). Preliminary biological evaluation showed that this prodrug produced similar levels of fluorescence as ALA-Hex in breast cancer MCF7 cell, but with much lower dark toxicity.
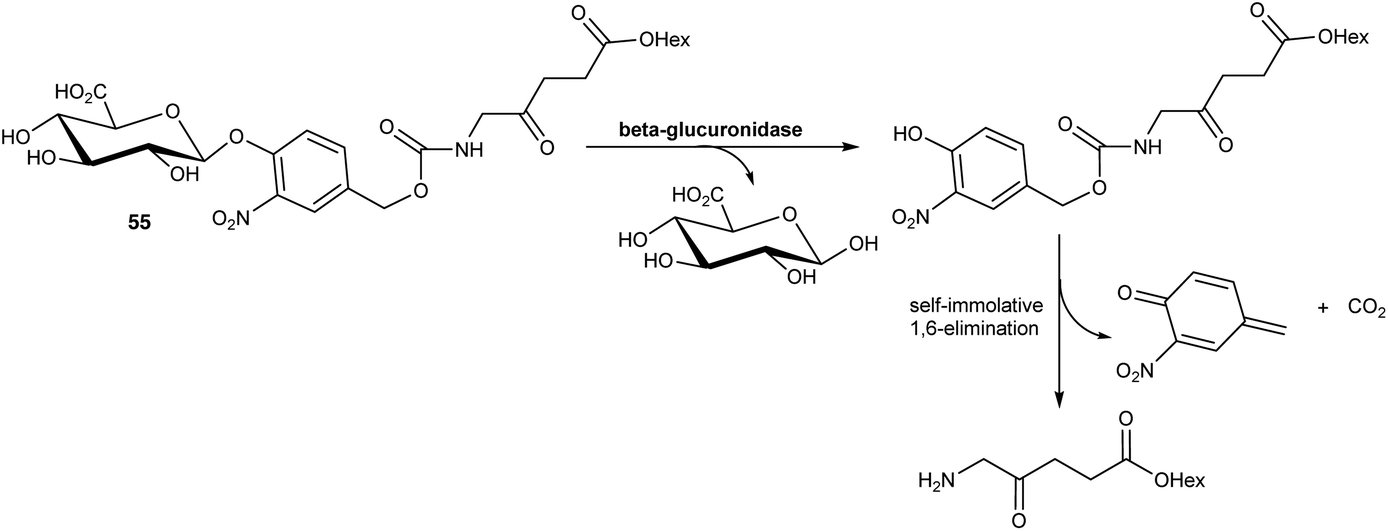 | ||
| Scheme 9 Cleavage of ALA prodrug 55 by β-glucuronidase activity. | ||
Peptide derivatives of ALA have also been synthesized to provide prodrugs with improved physicochemical properties relative to ALA itself. This approach also provides the possibility of achieving selective ALA release in particular cell lines which express proteases for which the prodrug is a potential substrate. Berger et al. investigated two types of ALA peptide prodrugs (Fig. 15) and evaluated them for their ability to produce PpIX.31,77 The first approach involved synthesizing acidic, basic and neutral ALA dipeptide derivatives 56a–e as potential substrates for cellular aminopeptidases APA, APB or APN/M.31 Biological evaluation in endothelial and carcinoma cell lines A549, HCEC and EC219 showed that prodrugs containing a neutral amino acid sidechain such as 56c induced the highest PpIX production, however a basic amino acid derivative (56d) or acidic derivative (56e) did not show any favourable results (minimal PpIX production). The second approach involved the synthesis and evaluation of pseudo tri- and tetrapeptide derivatives 57a–f (Gly-Gly and Gly-Pro) of ALA as potential substrates for cellular di/tripeptidases or peptide transporters at the cell surface, to target human endothelial cells or carcinoma cells in cancer.77 In this case, studies in human endothelial (HCEC) and human lung carcinoma cells (A549) showed the peptide derivatives of ALA to be substrates for Gly/Pro-specific cellular transport systems and enzymatic activities. The dipeptide derivatives were also less cytotoxic in the absence of light compared to the dipeptide derivatives 56a–e.
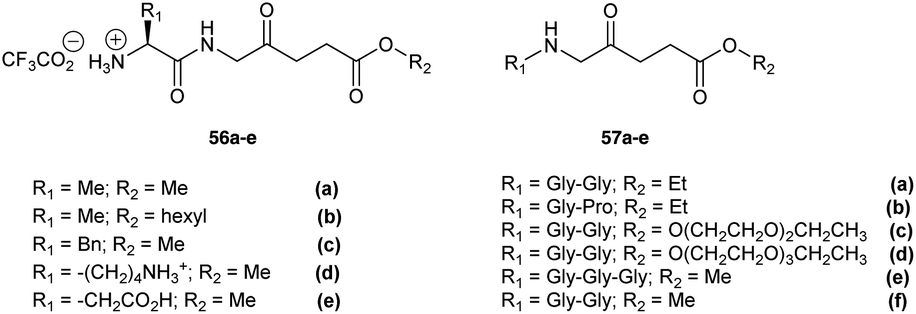 | ||
| Fig. 15 Peptide prodrugs of ALA prepared by Berger et al.31,77 | ||
A significant feature in the synthetic approaches used by Berger et al. was that an excess of the activated amino acid components or ALA derivative was used for the formation of the critical amino acid-ALA peptide bond, presumably to compensate for the instability of ALA under the basic coupling conditions. Rogers et al. developed an alternative approach by employing only one equivalent each of an acylated amino acid succinimido ester and ALA·HCl.78 The instability of ALA under basic conditions was avoided by slow neutralisation of ALA·HCl in the presence of the active esters (Scheme 6). The intermediate Boc-protected peptides were also used to synthesise the corresponding N-acetyl derivatives, thereby avoiding any potential epimerisation in the coupling reaction (Scheme 10).
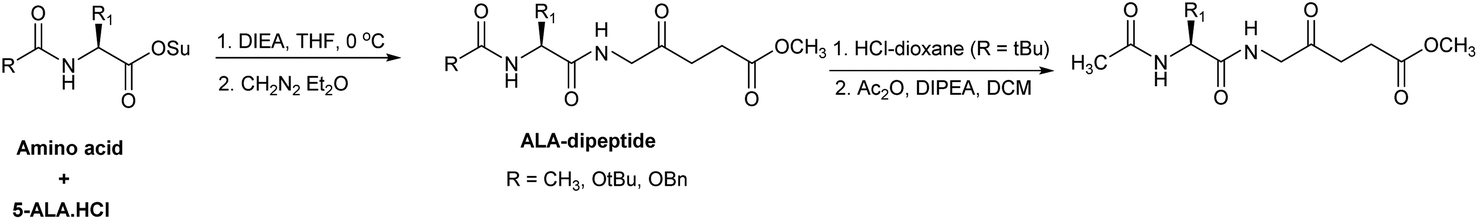 | ||
| Scheme 10 Synthesis of ALA peptide derivatives by Rogers et al. avoiding self-condensation of ALA during the coupling step. | ||
Simple ALA peptide prodrugs of the general structure shown above embody a number of desirable features for improved topical and systemic ALA administration. As has already been noted, acylation of the amino function of ALA provides chemical stability, while esterification of the carboxyl function increases lipophilicity for enhanced membrane permeability. Compounds of this type can be termed “double prodrugs of ALA”, a term recently coined by Babic and coworkers.76 There is also considerable scope to tailor the physicochemical properties of the prodrugs through careful choice of the amino acid residue to provide compounds that are overall more lipophilic than ALA, yet are still water soluble. Unlike other peptide prodrugs reported by Berger et al.,31,77 and also the phosphatase-sensitive prodrugs of Babic,74 subject to the choice of a suitable amino acid residue the compounds are not charged at physiological pH.
Bourré et al.79 described the biological evaluation of the dipeptide prodrugs 55 and 56 with the ALA amino group blocked with either Ac-L-Phe or Ac-D-Phe respectively, as well as the corresponding L-amino acid containing derivative 57 with a free carboxylic acid function (Fig. 16). Fluorescence spectroscopy revealed both 55 and 57 to be efficiently taken up in transformed PAM212 keratinocytes and generated greater levels of PpIX compared to equimolar ALA, although as anticipated better results were obtained with the neutral prodrug 58 (methyl ester) compared to 60 (free carboxy terminus). Significantly, no PpIX production was observed with the D-amino acid containing prodrug 59 consistent with the lack of a suitable intracellular enzyme with D-peptidase activity. All three dipeptide derivatives were stable in solution at physiological pH, in contrast to the corresponding dipeptides with a free N-terminus, presumably due to intermolecular Schiff base formation between the terminal amino acid amino function and the carbonyl function of an ALA residue, or possibly intramolecular cyclisation to generate a reactive six-membered Schiff base intermediate. PDT studies with both 58 and 60 in cells were in agreement with the results obtained from fluorescence pharmacokinetics, and the cell survival was significantly reduced in comparison to equimolar doses of ALA. 58 was also evaluated in a pig skin explant model in comparison to ALA and was found to produce higher porphyrin fluorescence by a factor of three demonstrating that 58 was capable of being transported into the skin and metabolised into PpIX in an ex vivo setting.
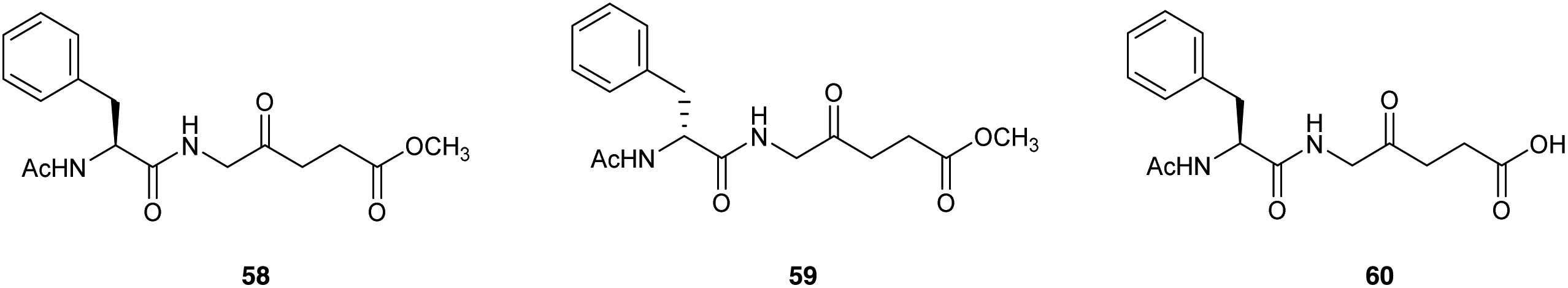 | ||
| Fig. 16 Phenylalanine-derived ALA dipeptide derivatives 58–60. | ||
Eggleston and coworkers80 subsequently reported a series of 27 dipeptide prodrugs of ALA with the same general structure Ac-Xaa-ALA-OR (i.e. analogues of 58 and 59) where Xaa was an L- or D-α-amino acid providing a varying level of lipophilicity and water solubility to the prodrug. The method of Rogers et al.78 was simplified by simply slowly neutralising HCl·ALA-Me or other ALA esters in the presence of the appropriate Boc or Fmoc-protected amino acid active esters, to give an intermediate dipeptide ester which could then be converted to the required acetyl derivatives in excellent yield and on a gram scale. A significant observation from this study was that there was no clear correlation as might have been expected between the lipophilicity of the prodrugs and the level of PpIX production in PAM212 keratinocytes. Quantification of the amount of ALA internalised using an improved fluorescence-based method81 revealed that in fact all the ALA prodrugs were taken up efficiently compared to ALA itself, by a combination of passive and active transport mechanisms, but predictably only the L-amino acid derivatives produced PpIX. This clearly demonstrated not only potential for designing peptide prodrugs of ALA which are cleaved by a disease-specific/cell-line specific enzyme activity, but also the importance of being able to independently assess PpIX production and ALA uptake when attempting to rationally design ALA prodrugs. In this study, further variation of the ester component of the lead L-Phe derivative to fine-tune lipophilicity also led to the identification of a dipeptide double prodrug whose photocytotoxicity was comparable to that of the hexyl ester of ALA 1f at concentrations where ALA itself was completely ineffective.
In a further study by the same researchers,82 it was shown that compounds of this general structure which contain an L-amino acid are in fact substrates for the enzyme acylpeptide hydrolase (APEH), a member of the prolyl oligopeptidase family of serine proteases, and for which D-amino acid derivatives are not substrates, as is also the case for compounds where the terminal acetyl function is replaced by a benzyloxycarbonyl group.79 This corroborated the earlier finding that PpIX production is also limited for these compounds in cell lines with a low expression of this enzyme such as Caco-2 and AF549.80 Further in vivo studies with these compounds has provided further evidence for their potential use in topical treatment of basal cell carcinomas and their effectiveness upon systemic administration compared to ALA or its hexyl ester 1f.83,84
Some similar dipeptide-based double prodrugs of ALA have recently been reported by Chen and coworkers,85 again exploring the variation of the ester function. This theme has also been investigated in a somewhat different way by Zhu et al.,71 where two of the lead dipeptide motifs identified by Eggleston and coworkers78 were esterified with an HPO iron chelator. Zhou et al.70 on the other hand, have modified the same lead dipeptides by acylation of the N-terminal amino acid residue with a suitably functionalised HPO. In both cases, codelivery of ALA and an iron chelator in the form of a dipeptide prodrug could be shown to be more effective than either ALA alone, or coadministration of ALA with CP94 as a stand-alone chelator.
Several studies have reported the synthesis and biological evaluation of more complex ALA peptide prodrugs which have been designed to provide improved cellular delivery and also highly selective ALA release. For example, Dixon et al. reported the synthesis of a 9-residue ALA-containing cell-penetrating peptide (CPP) conjugate 61 using Fmoc-solid phase chemistry (SPPS).86 The synthesized peptide was based on a heptapeptide fragment of the cell penetrating peptide, penetratin (Fig. 17), and in this proof of concept study for CPP-mediated ALA delivery, it was possible to demonstrate effective uptake in PAM212 cells and intracellular conversion of the peptide to PpIX. This was the first example of the synthesis of an ALA-containing peptide by SPPS, and incorporation of the ALA residue was achieved by attachment of the preformed dipeptide, Fmoc-Leu-ALA-OH, thereby avoiding the potentially problematic acylation of ALA itself upon the solid phase. Use of a hindered 2-chlorotrityl resin support also helped prevent any potential intramolecular Schiff base formation upon acylation of the Leu-ALA dipeptide unit.
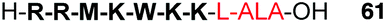 | ||
| Fig. 17 A cell-penetrating peptide-ALA conjugate based on the penetratin CPP sequence.86 | ||
Abd-Elgaliel et al.87 have adopted the same chemical strategy for the SPPS of a cathepsin E-cleavable ALA peptide prodrug 62 (Fig. 18). Biological evaluation showed that ALA was selectively liberated in cathepsin E-positive pancreatic tumour cells, but not in normal pancreatic tissue, thus allowing delivery of selective photodynamic therapy (PDT) to cancerous tissues.
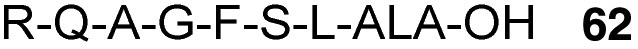 | ||
| Fig. 18 A cathepsin E-activatable ALA peptide derivative.87 | ||
Other peptide-based approaches for ALA delivery have also focused on the tagging of the amino function of ALA with an oligopeptide carrier. One notable example is the use of oligo(L-His) peptides which may be employed to provide tailored lipophilicity and water solubility to an attached cargo, and at physiological pH the oligo-His motif is able to act as a proton receptor and facilitate entry into tumour cells. In a study by Johnson et al.,88 a series of pH-sensitive ALA-oligo(L-His) prodrugs 63 were prepared by ring opening polymerization of 1-benzyl-N-carboxy-L-histidine anhydride initiated by the amino function of ALA itself. The activity of these prodrugs was assessed in the human colon cancer HCT116 cell line revealing promising phototoxicity and selectivity compared to ALA, especially for the conjugate with 15 L-His residues (Fig. 19).
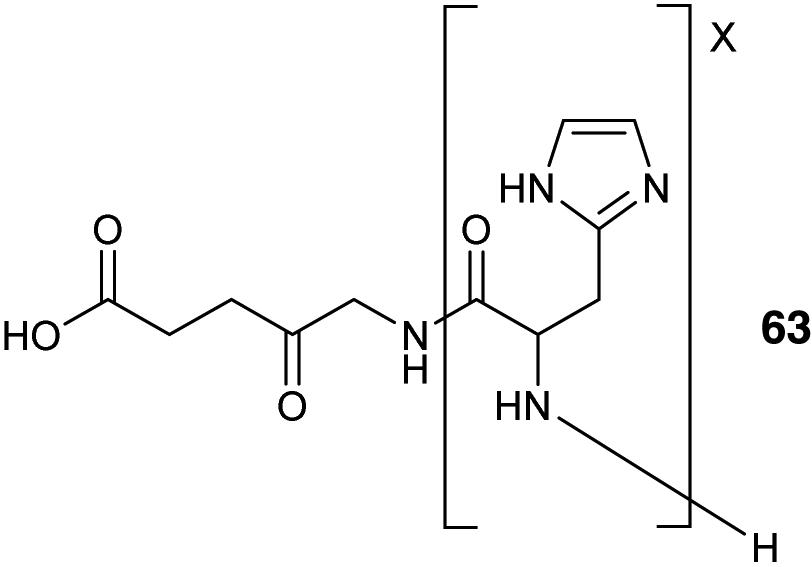 | ||
| Fig. 19 General structure of ALA-oligo(L-His) prodrugs 63.88 | ||
Finally, the synthesis of a novel folic acid-targeted ALA conjugate has been recently described by Guaragna et al.89 Targeting of folate receptors has been frequently exploited for the delivery of anticancer drugs, and in this example the methyl ester of ALA was conjugated to folic acid via a peptide spacer consisting of four consecutive β-Ala units linked to the β-carboxylic acid function of the Asp residue of folic acid. The spacer was included to ensure adequate water solubility and cellular stability of the final conjugate 64, and the whole carrier unit was prepared via SPPS on Rink amide resin, with an α-linked Asp residue at the C-terminus. This then left the β-acid function of the Asp residue free for acylation of the amino function of ALA (see Fig. 20). No biological and stability studies have yet been reported for this interesting targeted ALA derivative which once more illustrates the possibility of linking larger synthetic peptides to ALA.
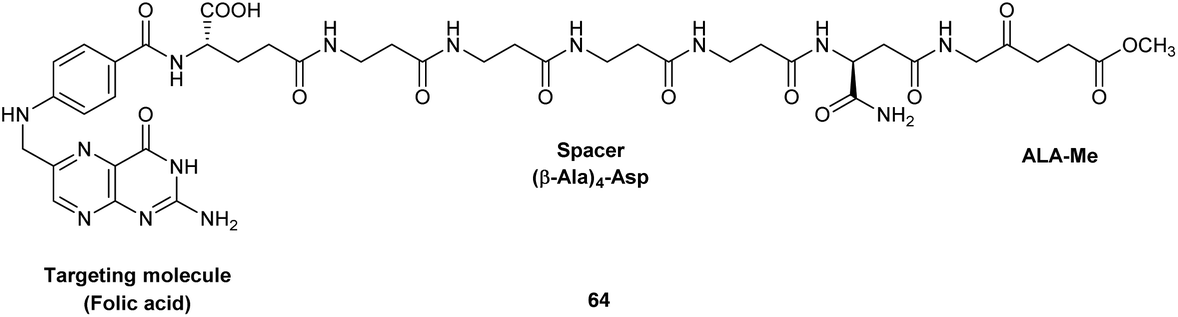 | ||
| Fig. 20 A potential folic acid-targeted ALA prodrug.89 | ||
Applications of nanotechnology and nanochemistry to the design of ALA prodrugs
The development of ALA dendrimers have already been described in detail as a promising strategy for the systemic delivery of a clinically useful payload of ALA for either PDT or PDD. In recent years, a growing number of other strategies have been explored that draw upon the knowledge gained from the synthesis and biological properties of simpler ALA esters and peptide derivatives to connect the challenge of ALA delivery to the field of nanotechnology.7 Notable examples include the encapsulation of ALA into polymeric90 or gold nanoparticles,91 and most recently the preparation of ALA-squalene nanoassemblies via esterification of ALA itself with squalene alcohol.92 Self-assembly of ALA squalene esters in water produces monodisperse systems with average diameter 70 nm, and a high ALA drug loading of 26%. These nanoassemblies show very promising PpIX production in human prostate PC3 and U87MG glioblastoma cells which is superior to that of the hexyl ester of ALA suggesting that they have great potential for PDD and tumour-selective PDT.Several examples of peptide-targeted ALA nanosystems have also been recently reported that provide for both efficient targeting and context-dependent (i.e. pH-selective) ALA release.93,94
Summary
Since the discovery of the clinical potential of ALA-mediated PDT considerable progress has been made in developing simple ALA derivatives that have improved bioavailability and stability compared to ALA itself, and also in identifying compounds that may be administered alongside ALA to maximise the production of PpIX in PDT and PDD applications. The methyl ester of ALA is now a mainstay for topical ALA-PDT in the clinic, while the hexyl ester is widely accepted for use in PDD. Challenges that remain to be solved are the development of effective platforms for systemic delivery of ALA, and also precise tumour targeting, particularly for applications in diagnosis and surgery. There are many opportunities for chemists to tackle these issues, drawing on the lessons from the wealth of knowledge already gathered about ALA and its derivatives and the application of modern methods of synthetic chemistry.Conflicts of interest
There are no conflicts to declare.References
- J. C. Kennedy, R. H. Pottier and D. C. Pross, Photodynamic therapy with endogenous protoporphyrin IX: basic principles and present clinical experience, J. Photochem. Photobiol., B, 1990, 6, 143–148 CrossRef CAS.
- Q. Peng, T. Warloe, K. Berg, J. Moan, M. Kongshaug, K. E. Giercksky and J. M. Nesland, 5-Aminolevulinic acid-based photodynamic therapy, Cancer, 1997, 79, 2282–2308 CrossRef CAS PubMed.
- N. Fotinos, M. A. Campo, F. Popowycz, R. Gurny and N. Lange, 5-Aminolevulinic acid derivatives in photomedicine: characteristics, application and perspectives, Photochem. Photobiol., 2006, 82, 994–1015 CrossRef CAS PubMed.
- B. Krammer and K. Plaetzer, ALA and its clinical impact: from bench to bedside, Photochem. Photobiol. Sci., 2008, 7, 283–289 RSC.
- F. Harris and L. Pierpoint, Photodynamic therapy based on 5-aminolevulinic acid and its use as an antimicrobial agent, Med. Res. Rev., 2012, 32, 1292–1327 CrossRef CAS PubMed.
- M. Wachowska, A. Muchowicz, M. Firczuk, M. Gabrysiak, M. Winiarska, M. Wanczyk, K. Bojarczuk and J. Golab, Aminolevulinic Acid (ALA) as a prodrug in photodynamic therapy of cancer, Molecules, 2011, 16, 4140–4164 CrossRef CAS.
- C. Thunshelle, R. Yin, Q. Chen and M. R. Hamblin, Current advances in 5-aminolevulinic acid mediated photodynamic therapy, Curr. Dermatol. Rep., 2016, 5, 179–190 CrossRef PubMed.
- D. I. J. Morrow, P. A. McCarron, A. D. Woolfson, P. Juzenas, A. Juzienne, V. Iani, J. Moan and R. F. Donnelly, Influence of penetration enhancers on topical delivery of 5-aminolevulinic acid from bioadhesive patches, J. Pharm. Pharmacol., 2010, 62, 906–913 CrossRef PubMed.
- Z. Malik, G. Kostenich, L. Roitman, B. Ehrenberg and A. Orenstein, Topical application of 5-aminolevulinic acid, DMSO and EDTA - protoporphyrin IX accumulation in skin and tumors of mice, J. Photochem. Photobiol., B, 1995, 28, 213–218 CrossRef CAS.
- J. T. H. M. van den Akker, V. Iani, W. M. Star, H. J. C. M. Sterenborg and J. Moan, Topical application of 5-amino levulinic acid hexyl ester and 5-aminolevulinic acid to normal nude mouse skin: Differences in protoporphyrin IX fluorescence kinetics and the role of the stratum corneum, Photochem. Photobiol., 2000, 72, 681–689 CrossRef PubMed.
- R. Steluti, F. S. DeRosa, J. Collett, A. C. Tedesco and M. V. L. B. Bentley, Topical glycerol monooleate/propylene glycol formulations enhance 5-aminolevulinic acid in vitro skin delivery and in vivo protophorphyrin IX accumulation in hairless mouse skin, Eur. J. Pharm. Biopharm. Sci., 2005, 60, 439–444 CrossRef CAS PubMed.
- B. G. Auner, E. Petzenhauser and C. Valenta, Influence of 6-ketocholestanol on skin permeation of 5-aminolevulinic acid and evaluation of chemical stability, J. Pharm. Sci., 2004, 93, 2780–2787 CrossRef CAS PubMed.
- M. B. R. Pierre, E. Ricci Jr., A. C. Tedesco and M. V. L. B. Bentley, Oleic acid as optimizer of the skin delivery of 5-aminolevulinic acid in photodynamic therapy, Pharm. Res., 2006, 23, 360–366 CrossRef CAS PubMed.
- L. Schmitz, B. Novak, A. K. Hoek, H. Luebbert and T. Dirschka, Epidermal penetration and protoporphyrin IX formation of two different 5-aminolevulinic acid formulations in ex vivo human skin, Photodiagn. Photodyn. Ther., 2016, 14, 40–46 CrossRef CAS PubMed.
- U. Reinhold, A review of BF-200 ALA for the photodynamic tretament of mild-to-moderate actinic keratosis, Future Oncol., 2017, 13, 2413–2428 CrossRef CAS PubMed.
- Ø. B. Gadmar, J. Moan, E. Scheie, L.-W. Ma and Q. Peng, The stability of 5-aminolevulinic acid in solution, J. Photochem. Photobiol., B, 2002, 67, 187–193 CrossRef.
- M. Novo, G. Huttmann and H. Diddens, Chemical instability of 5-aminolevulinic acid used in the fluorescence diagnosis of bladder tumours, J. Photochem. Photobiol., B, 1996, 34, 143–148 CrossRef CAS.
- A. Bunke, O. Zerbe, H. Schmid, G. Burmeister, H. P. Merkle and B. Gander, Degradation mechanism and stability of 5-aminolevulinic acid, J. Pharm. Sci., 2000, 89, 1335–1341 CrossRef CAS PubMed.
- B. Franck and H. Stratmann, Condensation products of the porphyrin precursor, 5-aminolevulinic acid, Heterocycles, 1981, 15, 919–923 CrossRef CAS.
- J. J. Scott, Synthesis of crystallisable porphobilinogen, Biochem. J., 1956, 62, P6–P6 Search PubMed.
- M. Kaliszewski, M. Kwasny, A. Juzeniene, P. Juzenas, A. Graczyk, L.-W. Ma, V. Iani, P. Mikolajewska and J. Moan, Formation of protoporphyrin IX from carboxylic-and amino-derivatives of 5-aminolevulinic acid, J. Photochem. Photobiol., B, 2007, 87, 67–72 CrossRef CAS PubMed.
- Q. Peng, J. Moan, T. Warloe, V. Iani, H. B. Steen, A. Bjørseth and J. M. Nesland, Build-up of esterified aminolevulinic-acid-derivative-induced porphyrin fluorescence in normal mouse skin, J. Photochem. Photobiol., B, 1996, 34, 95–96 CrossRef CAS.
- J. Kloek and G. M. J. B. van Henegouwen, Prodrugs for 5-aminolevulinic acid for photodynamic therapy, Photochem. Photobiol., 1996, 64, 994–1000 CrossRef CAS PubMed.
- J. Kloek, W. Akkermans and G. M. J. B. van Henegouwen, Derivatives of 5-aminolevulinic acid for photodynamic therapy: enzymatic conversion into protoporphyrin, Photochem. Photobiol., 1998, 67, 150–154 CrossRef CAS PubMed.
- J. M. Gaullier, K. Berg, Q. Peng, H. Anholt, P. K. Selbo, L. W. Ma and J. Moan, Use of 5-aminolevulinic acid esters to improve photodynamic therapy on cells in culture, Cancer Res., 1997, 57, 1481–1486 CAS.
- P. Uehlinger, M. Zellweger, G. Wagnières, L. Juillerat-Jeanneret, H. van den Bergh and N. Lange, 5-Aminolevulinic acid and its derivatives: physical chemical properties and protoporphyrin IX formation in cultured cells, J. Photochem. Photobiol., B, 2000, 54, 72–80 CrossRef CAS.
- G. DiVenosa, L. Hermida, H. Fukuda, M. V. Defain, L. Rodriguez, L. Mannone, A. MacRobert, A. Casas and A. Batlle, Comparison of liposomal formulations of ALA undecanoyl ester for its use in photodynamic therapy, J. Photochem. Photobiol., B., 2009, 96, 152–158 CrossRef CAS PubMed.
- C. J. Whitaker, S. H. Battah, M. J. Forsyth, C. Edwards, R. W. Boyle and E. K. Matthews, Photosensitization of pancreatic tumour cells by delta-aminolaevulinic acid esters, Anti-Cancer Drug Des., 2000, 15, 161–170 CAS.
- A. Godal, N. O. Nilsen, J. Klaveness, J. E. Branden, J. M. Nesland and Q. Peng, New derivatives of 5-aminolevulinic acid for photodynamic therapy: Chemical synthesis and porphyrin production in in vitro and in vivo biological systems, J. Environ. Pathol., Toxicol. Oncol., 2006, 25, 109–126 CrossRef CAS.
- R. F. V. Lopez, N. Lange, R. Guy and M. V. L. B. Bentley, Photodynamic therapy of skin cancer: controlled drug delivery of 5-ALA and its esters, Adv. Drug Delivery Rev., 2004, 56, 77–94 CrossRef CAS PubMed.
- Y. Berger, A. Greppi, O. Siri, R. Neier and L. Juillerat-Jeanneret, Ethylene glycol and amino acid derivatives of 5-aminolevulinic acid as new photosensitizing precursors of protoporphyrin IX in cells, J. Med. Chem., 2000, 43, 4738–4746 CrossRef CAS PubMed.
- H. Brunner, F. Hausmann, R. Krieg, E. Endlicher, J. Schölmerich, R. Knuechel and H. Messmann, The effects of 5-aminolevulinic acid esters on protoporphyrin IX production in human adenocarcinoma cell lines, Photochem. Photobiol., 2001, 74, 721–725 CrossRef CAS PubMed.
- T. Teshigawara, M. Mizuno, I. Ishii, Y. Kitajima, F. Utsumi, J. Sakata, H. Kajiyama, K. Shibata, M. Ishizuka and F. Kikkawa, Novel potential photodynamic therapy strategy using 5-Aminolevulinic acid for ovarian clear-cell carcinoma, Photodiagn. Photodyn. Ther., 2018, 21, 121–127 CrossRef CAS PubMed.
- R. Vallinayagam, J. Weber and R. Neier, Novel bioconjugates of aminolevulinic acid with vitamins, Org. Lett., 2008, 10, 4453–4455 CrossRef CAS PubMed.
- C. Yang, T. Wu, Y. Qi and Z. Zhang, Recent advances in the application of vitamin E TPGS for drug delivery, Theranostics, 2018, 8, 464–485 CrossRef PubMed.
- N. Sato, B. W. Moore, S. Keevey, J. A. Drazba, T. Hasn and E. V. Maytin, Vitamin D enhances ALA-induced protoporphyrin IX production and photodynamic cell death in 3-D organotypic cultures of keratinocytes, J. Invest. Dermatol., 2006, 127, 925–934 CrossRef PubMed.
- R. Vallinayagam, F. Schmitt, J. Barge, G. Wagnieres, V. Wenger, R. Neier and L. Juillerat-Jeanneret, Glycoside esters of 5-aminolevulinic acid for photodynamic therapy of cancer, Bioconjugate Chem., 2008, 19, 821–839 CrossRef CAS PubMed.
- P. Gurba, R. Vallinayagam, F. Schmitt, J. Furrer, L. Juillerat-Jeanneret and R. Neier, Novel bioconjugates of aminolevulinic acid with nucleosides, Synthesis, 2008, 3957–3962 CAS.
- C. Aggelidou, T. A. Theodossiou, A. R. Goncalves, M. Lampropoulu and K. Yannakopoulou, A versatile δ-aminolevulinic acid (ALA)-cyclodextrin bimodal conjugate-prodrug for PDT applications with the help of intracellular chemistry, Beilstein J. Org. Chem., 2014, 10, 2414–2420 CrossRef PubMed.
- S. H. Battah, C.-E. Chee, H. Nakanishi, S. Gerscher, A. J. MacRobert and C. Edwards, Synthesis and biological studies of 5-aminolevulinic acid-containing dendrimers for photodynamic therapy, Bioconjugate Chem., 2001, 12, 980–988 CrossRef CAS PubMed.
- S. Battah, S. Balaratnam, A. Casas, S. O'Neill, C. Edwards, A. Batlle, P. Dobbin and A. J. MacRobert, Macromolecular delivery of 5-aminolaevulinic acid for photodynamic therapy using dendrimer conjugates, Mol. Cancer Ther., 2007, 6, 876–885 CrossRef CAS PubMed.
- S. Battah, S. O'Neill, C. Edwards, S. Balaratnam, P. Dobbin and A. J. MacRobert, Enhanced porphyrin accumulation using dendritic derivatives of 5-aminolaevulinic acid for photodynamic therapy: an in vitro study, Int. J. Biochem. Cell Biol., 2006, 38, 1382–1392 CrossRef CAS PubMed.
- G. M. Di Venosa, A. G. Casas, S. Battah, P. Dobbin, H. Fukuda, A. J. MacRobert and A. Batlle, Investigation of a novel dendritic derivative of 5-aminolaevulinic acid for photodynamic therapy, Int. J. Biochem. Cell Biol., 2006, 38, 82–91 CrossRef CAS PubMed.
- A. Casas, S. Battah, G. Di Venosa, P. Dobbin, L. Rodriguez, H. Fukuda, A. Batlle and A. J. MacRobert, Sustained and efficient porphyrin generation in vivo using dendrimer conjugates of 5-ALA for photodynamic therapy, J. Controlled Release, 2009, 135, 136–143 CrossRef CAS PubMed.
- A. François, S. Battah, A. J. MacRobert, L. Bezdetnaya, F. Guillemin and M.-A. D'Hallewin, Fluorescence diagnosis of bladder cancer: a novel in vivo approach using 5-aminolevulinic acid (ALA) dendrimers, BJU Int., 2012, 110, E1155–E1162 CrossRef PubMed.
- L. Rodriguez, P. Vallecorsa, S. Battah, G. Di Venosa, G. Calvo, L. Mamone, D. Sáenz, M. C. Gonzalez, A. Batlle, A. J. MacRobert and A. Casas, Aminolevulinic acid dendrimers in photodynamic treatment of cancer and atheromatous disease, Photochem. Photobiol. Sci., 2015, 14, 1617–1627 RSC.
- C. Pavani, C. M. L. Francisco, N. R. S. Gobo, K. T. de Oliveira and M. S. Baptista, Improved photodynamic activity of a dual phthalocyanine-ALA photosensitiser, New J. Chem., 2016, 40, 9666–9671 RSC.
- G. Berkovitch, D. Doron, A. Nudelmanm, Z. Malik and A. Rephaeli, Novel multifunctional acyloxyalkyl ester prodrugs of 5-aminolevulinic acid display improved anticancer activity independent and dependent on photoactivation, J. Med. Chem., 2008, 51, 7356–7369 CrossRef CAS PubMed.
- G. F. Gola, G. M. Di Venosa, D. A. Saenz, G. H. Calvo, G. M. Cabrera, A. G. Casas and J. A. Ramirez, Synthesis of chemically diverse esters of 5-aminolevulinic acid for photodynamic therapy via the multicomponent Passerini reaction, RSC Adv., 2016, 6, 89492–89498 RSC.
- A. M. S. Soares, G. Hungerford, M. S. T. Goncalves and S. P. G. Costa, Light triggering of 5-aminolevulinic acid from fused coumarin ester cages, New J. Chem., 2017, 41, 2997–3005 RSC.
- X. Yang, W. H. Li, P. Palasubernian, K. A. Myers, C. G. Wang and B. Chen, Effects of silencing heme biosynthesis enzymes on 5-aminolevulinic acid-mediated protoporphyrin IX fluoresence and photodynamic therapy, Photochem. Photobiol., 2015, 91, 923–930 CrossRef CAS PubMed.
- A. H. Laftah, R. J. Simpson, T. J. Peters and K. B. Raja, The effect of haem biosynthesis inhibitors and inducers on intestinal iron absorption and liver haem biosynthetic enzyme activities, Toxicol. Appl. Pharmacol., 2008, 229, 273–280 CrossRef CAS PubMed.
- V. H. Fingar, T. J. Wieman, K. S. McMahon, P. S. Haydon, B. P. Halling, D. A. Yuhas and J. W. Winkleman, Photodynamic therapy using a protoporphyrinogen oxidase inhibitor, Cancer Res., 1997, 57, 4551–4556 CAS.
- D. F. Yang, J. H. Chen, C. P. Chiang, Z. Huang, J. W. Lee, C. J. Liu, J. L. Chang and Y. C. Hsu, Improve efficacy of topical ALA-PDT by calcipotriol through up-regulation of coproporphyrinogen oxidase, Photodiagn. Photodyn. Ther., 2014, 11, 331–341 CrossRef CAS PubMed.
- S. Filian, H. Hönigsmann and B. Ortel, Photodynamic therapy of epithelial skin tumours using delta-aminolaevulinic acid and desferrioxamine, Br. J. Dermatol., 1995, 133, 282–288 CrossRef.
- K. Berg, H. Anholt, O. Bech and J. Moan, The influence of iron chelators on the accumulation of protoporphyrin IX in 5-aminolaevulinic acid-treated cells, Br. J. Cancer, 1996, 74, 688–697 CrossRef CAS PubMed.
- J. L. Buss, F. M. Torti and S. V. Torti, The role of iron chelation in cancer therapy, Curr. Med. Chem., 2003, 10, 1021–1034 CrossRef CAS PubMed.
- Y. Ma, T. Zhou, X. Kong and R. C. Hider, Chelating agents for the treatment of systemic iron overload, Curr. Med. Chem., 2012, 19, 2816–2827 CrossRef CAS PubMed.
- P. T. Doulias, S. Christoforidis, U. T. Brunk and D. Galaris, Endosomal and lysosomal effects of desferrioxamine: Protection of HeLa cells from hydrogen peroxide-induced DNA damage and induction of cell-cycle arrest, Free Radical Biol. Med., 2003, 35, 719–728 CrossRef CAS PubMed.
- K. Chudry, R. C. Brooke, W. Farrar and L. E. Rhodes, The effect of an iron chelating agent on protoporphyrin IX levels and phototoxicity in topical 5-aminolaevulinic acid photodynamic therapy, Br. J. Dermatol., 2003, 149, 124–130 CrossRef.
- A. L. R. Ribeiro de Souza, K. Marra, J. Gunn, K. S. Samkoe, S. C. Kanick, S. C. Davis, M. S. Chapman, E. V. Maytin, T. Hasan and B. W. Pogue, Comparing desferrioxamine and light fractionation enhancement of ALA-PpIX photodynamic therapy in skin cancer, Br. J. Cancer, 2016, 115, 805–813 CrossRef PubMed.
- S. Battah, R. C. Hider, A. J. MacRobert, P. S. Dobbin and T. Zhou, Hydroxypyridine and 5-aminolaevulinic acid conjugates for photodynamic therapy, J. Med. Chem., 2017, 60, 3498–3510 CrossRef CAS PubMed.
- O. Bech, D. Phillips, J. Moan and A. J. MacRobert, A hydroxypyridone (CP94) enhances the protoporphyrin IX formation in 5-aminolaevulinic acid treated cells, J. Photochem. Photobiol., B, 1997, 41, 136–144 CrossRef CAS.
- A. Curnow, B. W. McIlroy, M. J. Postle-Hacon, J. B. Porter, A. J. MacRobert and S. G. Bown, Enhancement of 5-aminolaevulinic acid-induced photodynamic therapy in normal rat colon using hydroxypyridinone iron-chelating agents, Br. J. Cancer, 1998, 78, 1278–1282 CrossRef CAS PubMed.
- E. Blake, J. Allen and A. Curnow, An in vitro comparison of the effects of the iron-chelating agents CP94 and dexrazoxanem on protoporphyrin IX accumulation for photodynamic therapy and/or fluorescence guided resection, Photochem. Photobiol., 2011, 87, 1419–1426 CrossRef CAS PubMed.
- E. Blake and A. Curnow, The hydroxypyridinone iron chelator CP94 can enhance PpIX-induced PDT of cultured human glioma cells, Photochem. Photobiol., 2010, 86, 1154–1160 CrossRef CAS PubMed.
- A. Pye, S. Campbell and A. Curnow, Enhancement of methyl-aminolevulinate photodynamic therapy by iron chelation with CP94: an in vitro investigation and clinical dose-escalating safety study for the treatment of nodular basal cell carcinoma, J. Cancer Res. Clin. Oncol., 2008, 134, 841–849 CrossRef CAS PubMed.
- S. Campbell, C. Morton, R. Alayaha, S. Horton, A. Pye and A. Curnow, Clinical investigation of the novel iron chelating agent, CP94 to enhance topical photodynamic therapy of nodular basal cell carcinoma, Br. J. Dermatol., 2008, 159, 387–393 CrossRef CAS PubMed.
- L. Anayo, A. Magnussen, A. Perry, M. Wood and A. Curnow, Am experimental investigation of a novel iron chelating protoporphyrin IX prodrug for the enhancement of photodynamic therapy, Lasers Surg. Med., 2018, 50, 552–565 CrossRef PubMed.
- T. Zhou, L.-L. Shao, C.-f. Zhu, R. C. Hider, B. J. Reeder, A. Jabeen, A. J. MacRobert, G. Ren and X. Liang, Design and synthesisof 5-aminolaevulinic acid/3-hydroxypyridone conjugates for photodynamic therapy: enhancementof protoporphyrin IX production and photo-toxicity in tumor cells, MedChemComm, 2016, 7, 1190–1196 RSC.
- C.-F. Zhu, S. Battah, X. Kong, B. J. Reeder, R. C. Hider and T. Zhou, Design, synthesis and biological evaluation of 5-aminolaevulinicacid/3-hydroxypyridone conjugates as potential photodynamic therapeutical agents, Bioorg. Med. Chem. Lett., 2015, 25, 558–561 CrossRef CAS PubMed.
- A. Mrozek-Wilczkiewicz, M. Serda, R. Musiol, G. Malecki, A. Szurko, A. Muchowicz, J. Golab, A. Ratuszna and J. Polanski, Iron chelators in photodynamic therapy revisisted: synergistic effect by novel highly active thiosemicarbazones, ACS Med. Chem. Lett., 2014, 5, 336–339 CrossRef CAS PubMed.
- A. Casas, A. M. D. Batlle, A. R. Butler, D. Robertson, E. H. Brown, A. MacRobert and P. A. Riley, Comparative effect of ALA derivatives on protoporphyrin IX production in human and rat skin organ cultures, Br. J. Cancer, 1999, 80, 1525–1532 CrossRef CAS PubMed.
- A. Babic, V. Herceg, I. Ateb, E. Allémann and N. Lange, Tunable phosphatase-sensitive prodrugs of 5-aminolevulinic acid for tumor fluorescence photodetection, J. Controlled Release, 2016, 235, 155–164 CrossRef CAS PubMed.
- V. Herceg, N. Lange, E. Allémann and A. Babic, Activity of phosphatase-sensitive 5-aminolevulinic acid prodrugs in cancer cell lines, J. Photochem. Photobiol., B, 2017, 171, 34–42 CrossRef CAS PubMed.
- V. Herceg, S. Adriouach, K. Janikowska, E. Allémann and A. Babic, Design, synthesis and in vitro evaluationn of β-glucuronidase-sensitive prodrug of 5-aminolevulinic acid for photodiagnosis of breast cancer cell, Bioorg. Chem., 2018, 78, 372–380 CrossRef CAS PubMed.
- Y. Berger, L. Ingrassia, R. Neier and L. Juillerat-Jeanneret, Evaluation of dipeptide-derivatives of 5-aminolevulinic acid as precursors for photosensitizers in photodynamic therapy, Bioorg. Med. Chem., 2003, 11, 1343–1351 CrossRef CAS PubMed.
- L. M.-A. Rogers, P. G. McGivern, A. R. Butler, A. J. MacRobert and I. M. Eggleston, An efficient synthesis of 5-aminolaevulinic acid (ALA)-containing peptides for use in photodynamic therapy, Tetrahedron, 2005, 61, 6951–6958 CrossRef CAS.
- L. Bourré, F. Giuntini, I. M. Eggleston, M. Wilson and A. J. MacRobert, 5-aminolaevulinic acid peptide prodrugs enhance photosensitization for photodynamic therapy, Mol. Cancer Ther., 2008, 7, 1720–1729 CrossRef PubMed.
- F. Giuntini, L. Bourré, A. J. MacRobert, M. Wilson and I. M. Eggleston, Improved peptide prodrugs of 5-ALA for PDT: rationalization of cellular accumulation and protoporphyrin IX production by direct determination of cellular prodrug uptake and prodrug metabolization, J. Med. Chem., 2009, 52, 4026–4037 CrossRef CAS PubMed.
- F. Giuntini, L. Bourré, A. J. MacRobert, M. Wilson and I. M. Eggleston, Quantitative determination of 5-aminolaevulinic acid and its esters in cell lysates by HPLC-fluorescence, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 875, 562–566 CrossRef CAS PubMed.
- L. Bourré, F. Giuntini, I. M. Eggleston, M. Wilson and A. J. MacRobert, Protoporphyrin IX enhancement by 5-aminolaevulinic acid peptide derivatives and the effect of RNA silencing on intracellular metabolism, Br. J. Cancer, 2009, 100, 723–731 CrossRef PubMed.
- G. Di Venosa, P. Vallecorsa, F. Giuntini, L. Mamone, A. Batlle, S. Vanzuli, A. Juarranz, A. J. MacRobert, I. M. Eggleston and A. Casas, The use of dipeptide derivatives of 5-aminolaevulinic acid promotes their entry to tumor cells and improves tumor selectivity of photodynamic Therapy, Mol. Cancer Ther., 2015, 14, 440–451 CrossRef CAS PubMed.
- C. Perotti, A. Casas, H. Fukuda, P. Sacca and A. Batlle, ALA and ALA hexyl ester induction of porphyrins after their systemic administration to tumour bearing mice, Br. J. Cancer, 2002, 23, 790–795 CrossRef PubMed.
- W. Zhu, Y. H. Gao, C.-H. Song, Z.-B. Lu, N. T. Namulinda, Y.-P. Han, Y.-J. Yan, L.-X. Wang and Z.-L. Chen, Synthesis and evaluation of new 5-aminolevulinic acid derivatives as prodrugs of protoporphyrin for photodynamic therapy, Photochem. Photobiol. Sci., 2017, 16, 1623–1630 RSC.
- M. J. Dixon, L. Bourré, A. J. MacRobert and I. M. Eggleston, Novel prodrug approach to photodynamic therapy: Fmoc solid-phase synthesis of a cell permeable peptide incorporating 5-aminolaevulinic acid, Bioorg. Med. Chem. Lett., 2007, 17, 4518–4522 CrossRef CAS PubMed.
- W. R. Abd-Elgaliel, Z. Cruz-Monserrate, H. Wang, C. D. Logsdon and C.-H. Tung, Pancreatic cancer-associated Cathepsin E as a drug activator, J. Controlled Release, 2013, 167, 221–227 CrossRef CAS PubMed.
- R. P. Johnson, C.-W. Chung, Y.-I. Jeong, D. H. Kang, H. Suh and I. Kim, Poly(L-histidine)-tagged 5-aminolevulinic acid prodrugs: new photosensitizing precursors of protoporphyrin IX for photodynamic colon cancer therapy, Int. J. Nanomed., 2012, 7, 2497–2512 CAS.
- A. Guaragna, G. N. Roviello, S. D'Errico, C. Paolella, G. Palumbo and D. D'Alonzo, Solid phase synthesis of a novel folate-conjugated 5-aminolevulinic acid methyl ester based photosensitizer for selective photodynamic therapy, Tetrahedron Lett., 2015, 56, 775–778 CrossRef CAS.
- L. Shi, X. Wang, F. Zhao, H. Luan, Q. Tu, Z. Huang and H. Wang, In vitro evaluationof 5-aminolevulinic acid (ALA) loaded PLGA nanoparticles, Int. J. Nanomed., 2013, 8, 2669–2676 CrossRef PubMed.
- K. D. Goncalves, K. de Oliveira, D. P. Viera and L. C. Courrol, Synthesis and characterisation of aminolevulinic acid gold nanoparticles: photo and sonosensitizer agent for artherosclerosis, J. Lumin., 2018, 197, 317–323 CrossRef CAS.
- A. Babic, V. Herceg, E. Bastien, H.-P. Lassalle, L. Bezdetnaya and N. Lange, 5-Aminolevulinic acid-squalene nanoasemblies for tumor photodetection and therapy: in vitro studies, Nanoscale Res. Lett., 2018, 13, 1–9 CrossRef CAS PubMed.
- J. N. Wu, H. J. Han, Q. Jin, Z. Li and J. Ji, Design and proof of programmed 5-aminolevulinic acid prodrug nanocarriers for targeted photodynamic cancer therapy, ACS Appl. Mater. Interfaces, 2017, 9, 14596–14605 CrossRef CAS PubMed.
- J. N. Wu, Y. Lin, H. Li, Q. Jin and J. Ji, Zwitterionic stealth peptide-capped 5-aminolevulinic acid prodrug nanoparticles for targeted photodynamic therapy, J. Colloid Interface Sci., 2017, 485, 251–259 CrossRef CAS PubMed.
Footnote |
| † Current address: Department of Pure and Applied Chemistry, Technology and Innovation Centre, 99 George Street, Glasgow G1 1RD, UK. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2018 |