
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Robust open cellular porous polymer monoliths made from cured colloidal gels of latex particles†
Christopher T.
Desire
 a,
Andrea
Lotierzo
b,
R. Dario
Arrua
c,
Emily F.
Hilder
*c and
Stefan A. F.
Bon
*b
a,
Andrea
Lotierzo
b,
R. Dario
Arrua
c,
Emily F.
Hilder
*c and
Stefan A. F.
Bon
*b
aAustralian Centre for Research on Separation Science (ACROSS), School of Natural Sciences, University of Tasmania, Hobart, Australia
bDepartment of Chemistry, The University of Warwick, Coventry, CV4 7AL, UK. E-mail: S.Bon@warwick.ac.uk; Web: http://www.bonlab.info
cFuture Industries Institute, University of South Australia, Adelaide, Australia. E-mail: Emily.Hilder@unisa.edu.au; Tel: +(61) 883026292
First published on 4th May 2018
Abstract
The coagulation of oppositely charged latexes, prepared from the soap-free emulsion polymerisation of styrene using water as the reaction medium, resulted in the obtainment of colloidal gels that were porous in nature and held together by electrostatic interactions. Chemical crosslinking, involving the introduction of a water-soluble crosslinker, resulted in the obtainment of stronger chemical bonds between particles affording a rigid porous material known as a monolith. It was found that, in a simpler approach, these materials could be prepared using a single latex where the addition of ammonium persulfate both resulted in the formation of the colloidal gel and initiated the crosslinking process. The pore size of the resulting monoliths was predictable as this was observed to directly correlate to the particle diameter, with larger pores achieved using particles of increased size. All gels obtained in this work were highly mouldable and retained their shape, which allowed for a range of formats to be easily prepared without the requirement of a mould.
Introduction
Since their development in the 1990s1,2 polymer monoliths with an interconnected network of pores have attracted significant attention, in particular as materials for solid phase chemistry,3 as catalytic supports,4–8 metal chelating agents,9 tissue engineering scaffolds,10,11 controlled release devices,12 absorbents,13 chromatography,2,3,14–16 and for extraction and sample preparation.9,17–19 The polymer monoliths are characterised as a permanently rigid continuous piece of material with an open cellular porous structure, which allows fluid to flow through.2 When applied as stationary phases in separation science, they have several advantages over conventional formats such as packed-beds, owing to their high permeability, enhanced mass transfer as a result of convective flow, ease of miniaturisation, and the associated lower solvent/sample consumption. These properties allow for higher-throughput and greater process efficiency.14 In accordance polymer monoliths have been identified as greener alternatives to these other formats.4–7,10,14Polymer monoliths are most commonly prepared by free-radical polymerisation induced phase separation from a mixture of monomers, initiator, and solvent, the latter referred to as the porogen.3,20 During polymerisation the growing polymer chains undergo phase separation and precipitate from the mixture hereby forming the monolithic structure, which is often covalently crosslinked.21 The choice of porogen itself is more historical than based on a set of rigid scientific criteria; with most groups opting for previously published solvent mixtures.22 Note that the porogen in principle can also be a polymer, for example poly(ethylene glycol), which is not compatible with the polymer matrix formed upon polymerisation.23,24 A wide variety of monomers have been utilised for this approach, including acrylates,25,26 methacrylates,27,28 styrene/divinylbenzene29–31 and acrylamides.32,33
In particular poly(styrene)-based monoliths have been demonstrated to be green alternatives for catalysis,4–7 as absorbents,13 and for chromatography.14 However, the porogen utilised in their preparation, in most cases, consists of a mixture of toluene and dodecanol, and the monolith is often purified using THF.4–7,31 The use of toluene and THF is concerning from an industrial and environmental perspective, as both have been classed as problematic for implementation at the production scale, based on a set of safety, health and environmental criteria.34 It is therefore desirable to utilise greener alternatives. However replacement of the porogenic solvent is not a straightforward process, requiring re-optimisation of its composition and the polymerisation conditions, with no guarantee that suitable porous properties will be obtained.22,35 Water, in particular, is problematic for this approach, given the low water-solubility of styrene.
Other approaches, such as the use of emulsion templates, do allow for the use of water,13,36,37 however this typically requires the presence of relatively large amounts of surfactant, which introduces additional purification requirements and can be difficult to completely remove.10 In general, the toxicity and environmental impact of surfactant waste is unclear, requiring an in-depth investigation for individual cases.38–41 In addition, surfactants can also act as plasticisers for polymer-based materials reducing their mechanical properties.42,43 Ionic liquids have also been employed in the preparation of poly(styrene)-based monoliths,14 however much debate about their green credentials exist,44–48 in particular relating to their synthesis, environmental impact and intrinsic properties. Alternatives for the preparation of these materials should be explored to alleviate these concerns.
The use of particles as colloidal building blocks to fabricate porous materials has a range of attractive options. We previously demonstrated the fabrication of a gas sensor using a mixture of colloids.49 However, control of pore structure is tedious as one relies on ice crystal templating. A more straightforward route that caught our attention was to make use of colloidal gels, in particular those formed from the coagulation of oppositely charged particles.50–53 For example, Wang et al.51 prepared a porous network from the coagulation of oppositely charged poly(D,L-lactic-co-glycolic acid) (PLGA)-based nanoparticles, with potential for use as a tissue engineering scaffold. Colloidal gels themselves are soft supracolloidal materials with a characteristic yield stress, above which the assembled gel monolith breaks down into its colloidal components and flows, and this behaviour is useful for the preparation of injectable porous materials. For example, hydrogels prepared from the combination of oppositely charged dextran microspheres have been prepared and their potential as injectable and biodegradable tissue engineering scaffolds demonstrated.54–56
This approach could potentially be applied to the preparation of styrene-based porous materials as styrene-based particles of opposite charge can easily be prepared by soap-free emulsion polymerisation.57–60 Emulsion polymerisation is an attractive technique owing to its simplicity, low cost, high yield, and use of water as a non-toxic and environmentally friendly solvent. The use of water is also advantageous for its excellent heat dissipation during the course of polymerisation, and this technique has been widely utilised in industry for the preparation of large quantities of latex for surface coatings, such as paints and adhesives.61 The soap-free emulsion polymerisation approach is therefore particularly attractive due to the absence of surfactant,57–60 which in addition to the concerns raised above, can result in destabilisation of the latex upon removal.58,60
However, the poor mechanical rigidity of colloidal gels, due to the absence of covalent bonds between particles and their associated characteristic yield stress, makes physical handling and the application of pressure for flow-through applications, challenging. This is particularly problematic if the gel is intended for use as a stationary phase in liquid-based separation science. It would therefore be desirable to improve their rigidity, which can be achieved by introducing crosslinking points through chemical crosslinking.2,62 A similar process has previously been applied for poly(styrene)-based particles prepared from emulsion polymerisation for the preparation of macroporous materials.62,63 In this case the addition of salt (NaCl) to the swollen latex resulted in aggregation of the particles, which were then crosslinked using residual monomer. However, these particles were prepared using the surfactant sodium dodecyl sulfate.
Here we report the use of styrene-based polymer latexes, prepared from soap-free emulsion polymerisation, for the preparation of porous colloidal gels. To assemble the colloidal gel both the use of oppositely charged latexes (strategy 1) as well as the addition of electrolyte (strategy 2) to trigger flocculation are exploited. To reinforce the mechanical properties of the three dimensional colloidal network we covalently crosslink the particles together with a subsequent free radical polymerisation step. This “curing” process renders the originally soft colloidal gels into rigid porous monolithic materials as a greener alternative for the preparation of styrene-based polymer monoliths, with the use of water as an industrially and environmentally friendly solvent (low VOC), the absence of molecular surfactant (low SOC), and limited purification the main advantages. The synthetic strategies employed in this work are shown in Scheme 1.
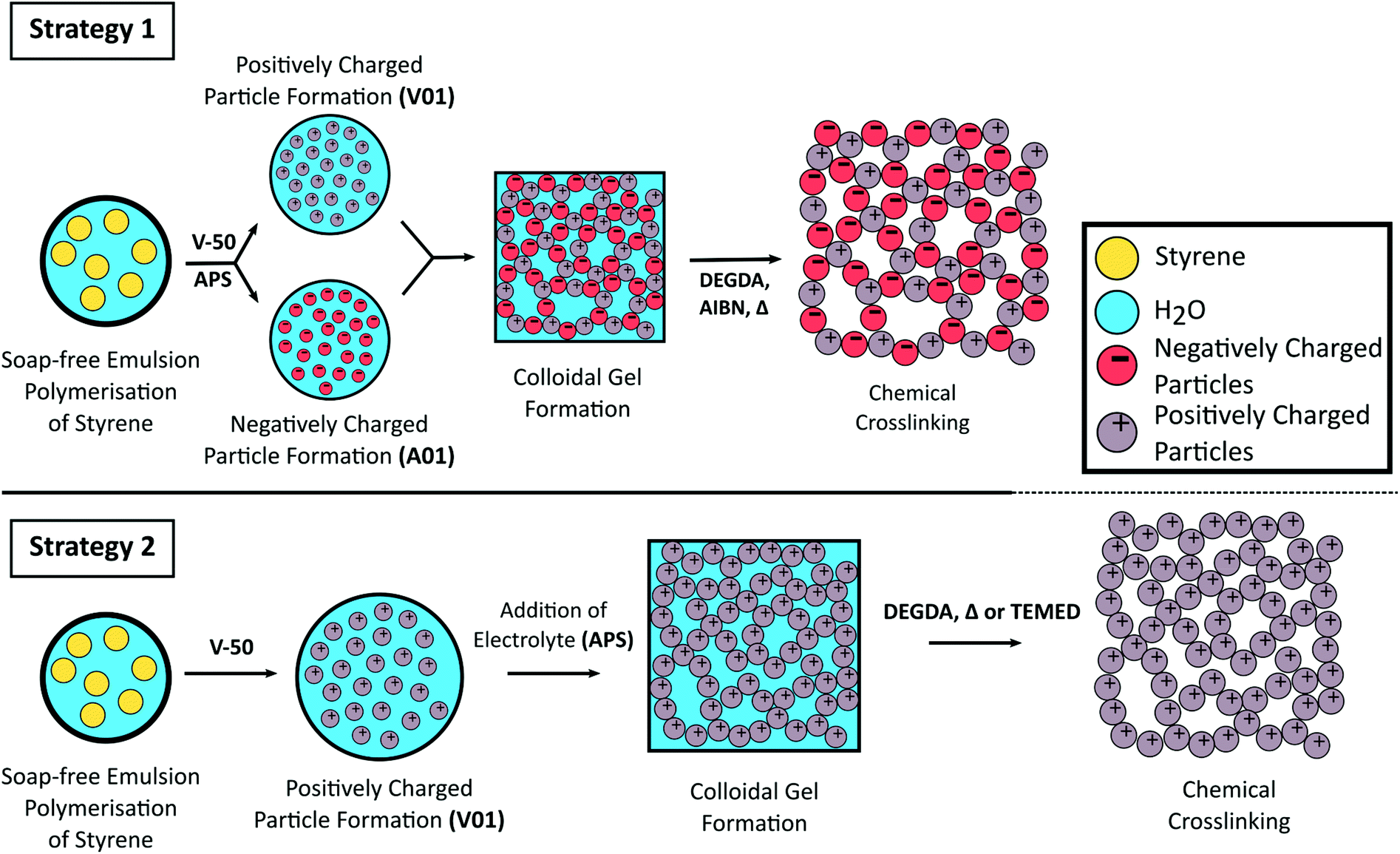 | ||
| Scheme 1 Schematic representation for the formation of crosslinked colloidal gels from oppositely charged latex particles prepared from the soap-free emulsion polymerisation of styrene using different initiators (Strategy 1) or from the addition of electrolyte to a cationic polymer latex (Strategy 2). | ||
Additionally, we show that some control of pore size can be gained upon varying the size of the polymer colloids used. Larger particles result in a corresponding increase in pore size, which is important for lower operating backpressures and enabling high-throughput when used for flow-through applications. Finally, the high mouldability intrinsic to these colloidal gels pre-curing allows for easy preparation of monolithic materials in a variety of macroscopic shapes.
Experimental section
Materials
Acetone (≥99%), ammonium persulfate (APS, 98%), di(ethylene glycol) diacrylate (DEGDA, 75%), dimethyl sulfoxide-d6 (DMSO-d6), divinylbenzene (DVB, 80%), styrene (Sty, ≥99%), 4-styrenesulfonic acid sodium salt, triethylamine (≥99%), N,N,N′,N′-tetramethylethylenediamine (TEMED, ≥99%), and 4-vinylbenzyl chloride (90%) were obtained from Sigma-Aldrich. Acetonitrile (ACN, ≥99.8%) and AR grade MeOH (≥99.6) were obtained from VWR. 2,2-Azobis(2-methylpropanimidamide) dihydrochloride (V-50, 98%) and hexadecane (≥98.5%) were obtained from Acros Organics. Azobisisobutyronitrile (AIBN, GPR) was obtained from BDH and was re-crystallised from MeOH. EtOH (>99%) was obtained from Chem-Supply. Sunflower oil (Woolworths essentials) was obtained from Woolworths Limited. Distilled H2O was utilised for all experiments. All chemicals used as received unless otherwise specified.Synthesis of the cationic co-monomer
The triethyl(4-vinylbenzyl)ammonium chloride (TEVBAC) cationic co-monomer was synthesised using the Menschutkin reaction between 4-vinylbenzyl chloride and triethylamine as reagents based on a method for the synthesis of trimethyl(vinylbenzyl)ammonium chloride (TMVBAC)57 as follows: 4-vinylbenzyl chloride (1.00 g, 6.55 × 10−3 mol) and triethylamine (1.99 g, 19.7 × 10−3 mol) were added to a 50 mL round bottom flask containing acetone (5 mL). This was sealed with a rubber septa and the contents shaken. The mixture was stored at room temperature in the dark during which crystallisation of the product occurred. The, white needle-like crystals were collected by Buchner filtration, and washing with aliquots of cold acetone. The crystals were stored under a dry nitrogen atmosphere because of their marked hydroscopic nature. The isolated product (40% yield‡) was characterised by 1H NMR (Fig. S1†) and 13C NMR (Fig. S2†). 1H-NMR (300 MHz, DMSO-d6) δ 1.10–1.53 (t, 9H, J = 6.9 Hz (CH3)3–CH2–N+), δ 3.02–3.42 (q, 6H, J = 7.2 Hz, CH3–(CH2)3–N+), δ 4.56 (s, 2H, Ar–CH+–N+), δ 5.27–5.65 (d, 1H, Jcis = 11.0 Hz, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–Ar), δ 5.83–6.05 (d, 1H, Jtrans = 17.6 Hz, CHC–Ar), δ 6.69–7.06 (dd, 1H, CH2CH–Ar), δ 7.48–7.66 (m, 4H, Ar). 13C-NMR (300 MHz, DMSO-d6) δ 8.1, δ 52.5, δ 59.8, δ 116.7, δ 127.0, δ 127.9, δ 133.4, δ 136.3, δ 139.2. In addition the crystal structure was determined (Fig. S3 and Tables S1–S3†). All of which were consistent with the formation of TEVBAC.
C–Ar), δ 5.83–6.05 (d, 1H, Jtrans = 17.6 Hz, CHC–Ar), δ 6.69–7.06 (dd, 1H, CH2CH–Ar), δ 7.48–7.66 (m, 4H, Ar). 13C-NMR (300 MHz, DMSO-d6) δ 8.1, δ 52.5, δ 59.8, δ 116.7, δ 127.0, δ 127.9, δ 133.4, δ 136.3, δ 139.2. In addition the crystal structure was determined (Fig. S3 and Tables S1–S3†). All of which were consistent with the formation of TEVBAC.
Synthesis of poly(styrene) latexes by soap-free emulsion polymerisation
A typical soap-free emulsion polymerisation process was adopted and is summarised as follows: styrene (9.9 g) was added to a continuous phase consisting of cationic (TEVBAC) or anionic (sodium styrene sulfonate) co-monomer (0.1 g) and H2O (90 g) in a 250 mL round bottom flask. A stirrer bar was added, the flask sealed with a rubber septa, and the contents purged with N2 gas for 20 min. The system was kept under N2 for the duration of the polymerisation with constant stirring. The reaction vessel was heated to 70 °C using an oil bath and after 15 min the initiator solution (0.01 g of either cationic (V-50) or anionic (ammonium persulfate) initiator dissolved in 1 mL of deoxygenated H2O) was injected through the septa. This was left at 70 °C for 12 h to reach near complete monomer conversion. The final solids content of the latexes were determined by gravimetry.Particles of considerably larger size were synthesised without ionic co-monomer under semi-batch conditions57 in a similar procedure to that above, except the continuous phase consisted of H2O (109.2 g) and MeOH (43.2 g), the initiator solution contained 0.3640 g of V-50 dissolved in 2 g of deoxygenated H2O. Styrene (18.2 g), which had been purged with N2, was added at a rate of 5 mL h−1 over a period of 4 h using a syringe pump. The MeOH was then removed by dialysing the latex against H2O for 1 week, replacing the water twice daily.
General procedure for the preparation of colloidal gels
The latexes were initially concentrated under reduced pressure (to overall solids content of ∼30 wt%, the value determined gravimetrically). The higher solids latexes were then diluted with H2O to obtain the desired solid content (in the range 5–25 wt%) needed for the colloidal gel formation experiments.Strategy 1: A mixture consisting of oppositely charged latexes was then prepared in a glass vial by mixing equal amounts of the positively charged latex and the negatively charged latex. Flocculation was promoted by mild sonication using an Elma Elmasonic P sonicator bath (80 kHz, 5 min, 100% power). The gel was left to sett at room temperature for at least 2 h prior to characterisation. Inversion of the vial was performed to evaluate the cohesiveness of the gel,50,64,65 with photographs taken after 20 min equilibration time.
Preparation of crosslinked colloidal gels
A series of colloidal gels were prepared at 20 wt% as described above, except one latex was diluted with different amounts of DEGDA (containing 1 wt% AIBN w.r.t latex solids) in the range 10–30 wt% (w.r.t. total solid content of the resulting gel) and the amount of H2O added was adjusted accordingly. For example, to prepare 1 g of gel from 30 wt% latexes with 20 wt% DEGDA, 0.33 g of A01 was diluted with 0.04 g of DEGDA and 0.13 g H2O, while 0.33 g of V01 was diluted with 0.17 g of H2O before combining. After equilibration (20 min) these colloidal gels were placed in an oil bath at 60 °C for 24 h. Neither the latexes nor the resulting gels were degassed prior to curing. The resulting materials were then washed in H2O with gentle agitation and characterised once the washings remained visually clear.A series of crosslinked colloidal gels were also obtained using a single latex using strategy 2. Here, a cationic polymer latex was diluted with H2O and various amounts of DEGDA in the range 15–65 wt% (w.r.t. solids). However instead of AIBN, APS was added at a concentration of 1 wt% (w.r.t. solids) using a 0.04 mg mL−1 solution of APS, to promote coagulation before equilibration and curing. The amount of H2O added to the latex was varied so that upon addition of DEGDA and APS solution, an overall latex concentration of 20 wt% was obtained. The colloidal gels were then cured thermally, or by the addition of TEMED at room temperature.
Macroscopic shape variation of the polymer monoliths
Colloidal gels (2 g each) were prepared in 10 mL glass vials using the approaches described above, where DEGDA was incorporated at 30 wt% (w.r.t. solids). Cylindrical formats were obtained simply by using glass vials as the mould and curing. The resulting materials were removed by carefully smashing the glass vials. Flat sheets were prepared by sandwiching the gel between two glass slides (76 mm × 26 mm, 1.0–1.2 mm thick, Academy Science Limited) and curing. Removal of the top slide resulted in the obtainment of a continuous flat sheet. Other formats (such as a pyramid) were also prepared by moulding the gel using a spatula into the desired shape and then curing. The resulting materials were all gently washed with H2O using a wash bottle, air-dried, and then photographed. The polymer disks for porosity measurements were prepared by crosslinking 1 g of the colloidal gels using either 20 or 30 wt% DEGDA (w.r.t. solids) in 4 mL glass vials. After curing the vials were carefully smashed and the resulting disks were removed and rinsed with H2O. These were then dried in a vacuum oven for 1 week prior to analysis.Characterisation
Nuclear magnetic resonance (NMR) spectra were recorded in DMSO-d6 on a Bruker Advance III HD operating at 300 MHz at room temperature. NMR data was exported and redrawn using Origin® 8.5 (Northampton, MA, USA). Crystal structures were determined by mounting suitable crystals on a glass fiber with Fomblin oil®, which were then placed on an Xcalibur Gemini diffractometer with a CCD area detector. Crystals were kept at 150 K during the data collection66 and the structure was solved using Olex267 with the ShelXS66 structure solution program using Direct Methods and refined with the ShelXL68 refinement package using least squares minimisation.Particle size and particle size distributions were measured by dynamic light scattering (DLS) using a Malvern Instruments Zetasizer (Nano-ZS) using dilute latex samples. Zeta potentials were also measured using this instrument using dilute latex samples. SEM micrographs were obtained using a Zeiss Supra™ 55VP field emission scanning electron microscope, with secondary electron detection, operating in high vacuum mode with an acceleration voltage of 15 kV. Samples were first dispersed in H2O and evaporated onto silicon wafers attached to aluminium stubs, before being sputter-coated with carbon using an Emitech K950X sputter-coater or gold coated using a Polaron Range sputter coater. The average pore size was estimated by measuring the diameter of 500 pores. Histograms were obtained from these data sets using 22 bins, where the bin width was calculated by dividing the range of values by the number of bins. Theoretical normal distributions were also obtained based on the mean and standard deviation over a range of ±3 standard deviations using 200 points.
The specific surface area was obtained with the Brunauer–Emmett–Teller method69 using a Micromeritics Tristar II 2020 automated nitrogen sorption-desorption instrument. Prior to analysis, all samples were dried in a Micromeritics SmartPrep at 60 °C for 48 h. This was performed using ∼200 mg of sample and in triplicate. Mercury intrusion porosimetry (MIP) was performed on selected samples using a Micromeritics AutoPore IV 9505 porosimeter. Penetrometers with a stem volume of 0.4120 mL and a bulb volume of 3 mL were used. Intrusion pressure was started at 1.5 psi and was increased to a final value of 33, 000 psi. The average pore diameter was calculated using the range 30–4000 nm. This was performed in triplicate using ∼200 mg of sample.
The porosity was estimated by immersing dry polymer disks in a variety of solvents for 24 h. At least three disks for each sample were immersed and their mass and dimensions were recorded both prior and after immersion. At least one of the disks for each sample was immersed in the solvents for only 30 min. The porosity in the wet state (φw) can then be calculated from the following equation:
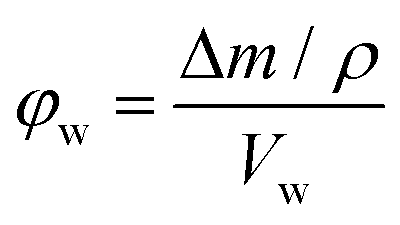 | (1) |
Results and discussion
The preparation of oppositely charged latexes
Two latexes of opposite charge were prepared from the soap-free emulsion polymerisation of styrene using APS and V-50 as water-soluble initiators, and were denoted as A01 and V01, respectively. These initiators provide a surface charge to the latex through their fragmentation, with a negative charge provided by APS70 and a positive charge provided by V-50,57,71 thus promoting latex stability through electrostatic stabilisation.70 Ionic co-monomers of similar charge, sodium styrene sulfonate in the case of APS and TEVBAC in the case of V-50 (1 wt% for each system), were also included to enhance the stability of these latexes.In the synthesis of both A01 and V01 near complete monomer conversion was achieved. The polymer latexes had characteristic low particle size dispersities, and possessed an average particle diameter in the order of 100 nm (Table 1), with A01 having an average diameter of 80 ± 10 nm by SEM and 109 ± 1 nm by DLS, while V01 had an average diameter of 130 ± 20 nm by SEM and 172.5 ± 0.4 by DLS. This was consistent with previous reports.57,70,72,73 In addition, for both A01 and V01 the sign of their zeta potentials were consistent with their expected charge (Table 1). Both latexes also appeared stable with no obvious sign of coagulation, and their SEM images (Fig. 1) showed no evidence of secondary nucleation, which is the generation of a new smaller batch of particles. The reason for the difference in average particle diameters as determined by SEM and DLS is related to the way that DLS works. DLS measurements tend to be an overestimate, as (1) the scattering intensity is more pronounced for larger particle sizes (the intensity is proportional to the sixth power of particle diameter) and (2) it measures the hydrodynamic size which means that the extent of the electrostatic double layer needs to be taken into account in the data interpretation.
 | ||
| Fig. 1 SEM images of latexes taken from dilute samples (A) A01, (B) V01 and (C) V02. Scale bar is 200 nm. | ||
| Samplea | Z-Aveb/nm | Average PDIc | Z /nm | wt%e | Conversionf/% | ζ /mV |
|---|---|---|---|---|---|---|
| a The following nomenclature is used, samples prepared with APS start with an A, while those prepared with V-50 start with a V. b Average particle diameter determined from DLS measurements using dilute samples. c Average polydispersity index obtained from the DLS measurements. d Average particle diameter measured directly from SEM images of dilute samples with at least 300 particles measured. e Average wt% determined from gravimetry after synthesis. f Conversion determined from mass of monomer added and the mass of latex obtained. g Average zeta potential determined from dilute latex samples. h Estimate of conversion before dialysis. | ||||||
| A01 | 109 ± 1 | 0.010 ± 0.009 | 80 ± 10 | 9.79 ± 0.03 | 92 | −53 ± 3 |
| V01 | 172.5 ± 0.4 | 0.02 ± 0.02 | 130 ± 20 | 9.2 ± 0.3 | 93 | 34 ± 1 |
| V02 | 560 ± 10 | 0.05 ± 0.04 | 470 ± 50 | 9.4 ± 0.2 | 93h | 41.2 ± 0.5 |
No purification of these latexes was performed in order to keep the synthetic strategy as simple as possible, and to demonstrate the versatility of this approach. It should also be noted that large quantities of latex can easily be prepared using this methodology,57,70,72,73 with the size of the reactor or flask the main limiting factor, which is an important consideration for preparation at the production scale.
Formation of colloidal gels
The possibility of preparing colloidal gels was explored by combining A01 and V01 at equal weight percent at a variety of concentrations in the range 5 to 25 wt%. Since these latexes were originally synthesised at ∼10 wt%, in order to obtain latexes of different concentration, both latexes were concentrated under reduced pressure to ∼30 wt% and then diluted. It is possible to prepare latexes with higher concentrations using the soap-free emulsion polymerisation approach,71 in a more energy efficient process, however alterations in the monomer concentration during synthesis is known to influence both the particle size and the number of particles present in the resulting latex.71,72 This was avoided so that any differences between these gels could be attributed solely to the particle concentration. No significant changes in the properties of these latexes (particle size, particle size distribution, and the sign of their zeta potential) were observed upon concentration (Table S4†). To promote gel formation the vials containing the mixture of the latexes were sonicated mildly to ensure the same energy input in all cases, as this is likely to be variable when shaking these vials by hand.Upon inspection, clumps were observed, rather than a continuous gel, for the lower concentrations of 5 and 10 wt%, and these exhibited significant flow upon inversion (Fig. 2).
 | ||
| Fig. 2 Photographs of colloidal gels obtained by combining A01 and V01 at equal weight percent. (A) Taken with vials upright 2 h after preparation, (B) taken 20 min after inversion. Particle concentration: (a) 5 wt%; (b) 10 wt%; (c) 15 wt%; (d) 20 wt%; (c) 25 wt%. Also shown are individual latex solutions at 25 wt%: (f) A01 and (g) V01. (C) Upright, (D) taken 20 min after inversion. | ||
This is a result of the particles being too distant from each other, due to the high water content.55 Cohesive gels were only obtained for particle concentrations greater than 10 wt%, with 15, 20 and 25 wt% resulting in a single plug of material, which exhibited greater resistance to flow (Fig. 2). Physical manipulation of these gels revealed that more viscous structures were obtained for latex concentrations of 20 and 25 wt%, when compared to 15 wt%. In comparison, individual latexes exhibited a high degree of flow (Fig. 2). The cohesive nature of these gels is therefore predominately provided by the electrostatic interactions between the oppositely charged particles, although it does also depend on van der Waals interactions and steric hindrance.51,74 An increase in the number of particles per unit volume therefore results in an increase in the number of these interactions, with increases in particle concentration corresponding to gels with greater viscosity and elastic moduli.50 In addition, these gels appeared to be highly mouldable and capable of retaining their shape, which makes them excellent candidates for polymer monolith precursors, as the ability to easily prepare a variety of formats is one of the advantages polymer monoliths posses over conventional formats such as packed-beds.75
SEM analysis (Fig. 3) revealed that the materials prepared at particle concentrations of 15, 20 and 25 wt%, as well as the clumps obtained at 5 and 10 wt% (Fig. S4†), possessed a porous morphology. No significant difference in morphology was observed with the particle concentrations utilised, and their porous nature appeared to be the result of interstitial space between closely packed particles in a cluster, coupled with the presence of voids, presumably resulting from multiple clusters intersecting imperfectly. This resulted in an average pore size of 100 ± 50 nm for the particle concentration of 20 wt%. This is in comparison to the large cellular domains sometimes observed with other systems.51–53 This porous structure did not appear to be related to the removal of H2O during the imaging process, as individual latexes, which were dried and then imaged, appeared to be more densely packed with a higher degree of order, and no particle clusters were observed (Fig. 4). However it is important to consider that these SEM images may not be representative of the structure of the colloidal gel in solution, as any shrinkage, as a result of their non-rigid nature, could have altered their morphology. Regardless, the particle arrangement observed resulted in pore sizes in the order of the particle dimensions, with pores less than 300 nm present.
 | ||
| Fig. 3 SEM images of colloidal gels obtained by combining A01 and V01 at equal weight percent. Particle concentration: (A) 15 wt%; (B) 20 wt%; (C) 25 wt%. Scale bar is 250 nm. | ||
 | ||
| Fig. 4 SEM images comparing (A) the colloidal gel obtained from the combination of A01 and V01 at 20 wt%, with individual dried latexes at 20 wt%: (B) A01 and (C) V01. Scale bar is 250 nm. | ||
Formation of crosslinked polymer monoliths from colloidal gels
Chemical crosslinking was employed to improve the rigidity of the colloidal gels made at 20 wt% overall solids. This concentration of polymer colloids was chosen as it was sufficiently cohesive (Fig. 2) while still maintaining a high water content and therefore porosity. A chemical crosslinking step was utilised to improve the mechanical properties of polymer monoliths, while also restricting the degree to which the network can shrink or swell in different solvent environments.2,76 The increase in mechanical properties is due to the introduction of covalent bonds, during the chemical crosslinking process, and these are stronger than the particle interactions responsible for the cohesive nature of the gels.55 We chose to reinforce our colloidal gels with 10–30 wt% crosslinkable monomer, to be utilised in the curing step to prepare the polymer monoliths.Initial experiments focused on the incorporation of DVB, which is commonly used in the preparation of poly(styrene)-based monoliths.15,31,75,77 However, this resulted in significant immediate coagulation of the individual latexes, even for DVB contents as low as 10 wt% (w.r.t. solids) (Table S5†). We therefore changed DVB for a less hydrophobic crosslinker. As such DEGDA was incorporated in the range of 10–30 wt% (w.r.t. solids). An additional benefit is that DEGDA, being an acrylate, polymerises markedly faster than divinylbenzene which can be of benefit as it reduces the overall curing time. The crosslinking monomer has the ability to swell the polymer particles and potentially upon colloidal gel formation lead to capillary bridging between particles. The relatively low water solubility of DEGDA should encourage the polymerisation to occur onto/in the colloidal gel network rather than gelling of the aqueous phase, which would reduce the porosity and permeability of the resulting material. The incorporation of DEGDA in this range did not appear to compromise the stability of the individual latexes, nor the ability to obtain cohesive gels.
The resulting gels were therefore cured thermally using AIBN as initiator by dissolving this initially in the DEGDA crosslinker. When 10 wt% DEGDA (w.r.t. total solid content of the gel) was utilised, this resulted in a material post-curing with the same consistency as the original colloidal gel before its reinforcement through polymerisation. However when DEGDA concentrations of 15 wt% and above were utilised rigid polymer monoliths were obtained. Washing these materials with H2O or MeOH did not appear to compromise their integrity and the washings remained clear, suggesting the latex particles were incorporated into the continuous network.
SEM analysis (Fig. 5) revealed that the material prepared with 15 wt% DEGDA possessed a very similar morphology to that of the non-crosslinked colloidal gels (Fig. 3). However, closer inspection revealed there were regions where multiple particles were fused together, with what appeared to be a smooth polymer coating. This coating is consistent with previous reports, where DEGDA was used to encapsulate calcium carbonate particles.78 As the DEGDA content was increased this fused morphology became more predominant. This is clearer at higher magnification (Fig. S5†). Thicker coatings were present for 25 wt%, however this material appeared more heterogeneous, with large variations in thickness of the coating observed.
 | ||
| Fig. 5 SEM images of crosslinked colloidal gels obtained by combining 20 wt% of A01 and V01 and cured with different concentrations of DEGDA. DEGDA concentration (w.r.t. total solid content of the gel): (A) 15 wt%; (B) 20 wt%; (C) 25 wt%; (D) 30 wt%. Scale bar is 500 nm. | ||
In terms of the porous morphology, the presence of this polymer coating resulted in a reduction in the interstitial space between the particles in a single cluster, however this did not appear to compromise the voids present between adjacent clusters, with an average void size of 140 ± 80 nm by SEM and 80 ± 10 nm by MIP for the material prepared with 20 wt% DEGDA. This was not statistically different to that of the original colloidal gels, however the slightly higher value obtained by SEM could be related to reduced shrinkage upon drying, associated with increased crosslinking density.2,76 The polymer monoliths made by this supracolloidal approach possessed a specific surface area of 38 ± 4 m2 g−1, slightly higher than the surface areas typically achieved with conventional polymer monoliths, which are often below 10 m2 g−1.20,79 The material also exhibited a type II isotherm (Fig. S13†), consistent with the obtainment of a macroporous material. As expected, the thickest polymer coatings were achieved for 30 wt% DEGDA, and this did appear to compromise its porous nature. As such the use of 20 wt% DEGDA appeared to be optimal for these materials, as this resulted in a reasonably homogenous porous material with good rigidity.
Formation of porous materials using a single latex
Curing of the colloidal gels was performed using AIBN as thermal initiator, as we noticed that the addition of APS to the individual latexes resulted in their coagulation, with small clumps initially observed which became larger over time. This occurred due to the increase in ionic strength, which depresses the electrostatic double-layer, allowing for greater contact between the particles.58,60,71,80,81 If APS was present at 1 wt% (w.r.t solids), or higher, full coagulation of the latex was observed over a period of 2 h for V01. While coagulation of A01 and V01 prevented their combination, SEM analysis (Fig. S6A†) revealed that the gel obtained for V01 also possessed a porous structure similar to the previous colloidal gels obtained (Fig. 3). This potentially allows for the preparation of rigid porous materials using only one latex, where the initiator not only promotes the crosslinking process, but also the formation of the colloidal gel itself. The addition of salt to particle suspensions has previously been used to induce their aggregation allowing for the obtainment of macroporous materials.62,82,83 However, the ability to use the thermal initiator, which itself is a salt, to initiate this process further simplifies this process. DEGDA was therefore included in the range 15–65 wt% (w.r.t. solids) before the addition of APS. Again the presence of DEGDA did not compromise the ability to obtain cohesive gels and thermal curing resulted in the obtainment of rigid cylinders in all cases.SEM analysis (Fig. 6) revealed similar results to that obtained above with 15 wt% DEGDA resembling that of the non-crosslinked colloidal gels, and as the DEGDA content was increased a fused morphology became more predominant up to 30 wt% DEGDA. The average pore sizes for these polymer monoliths was also similar with a value of 170 ± 60 nm by SEM and 150 ± 20 nm by MIP in the case of 30 wt% DEGDA. This material possessed a specific surface area of 25 ± 3 m2 g−1, which was consistent with a slightly higher average pore size. Coagulation, prior to curing, appeared to be a requirement for the obtainment of these porous materials, as simply curing the latex, using AIBN instead of APS, resulted in a non-porous material consisting of particles trapped within bulk polymer (Fig. S7†). Increasing the DEGDA content above 30 wt% resulted in significantly thicker coatings, and in contrast to the smooth coatings obtained previously, a cauliflower type morphology was observed. This was present for both 40 and 65 wt% DEGDA. It is likely that the increased DEGDA content is simply resulting in the formation of uneven polymer layers, alternatively this could be resulting in the formation of a secondary batch of particles, which are fusing with the existing particle network, however this is less likely. Regardless, the thicker coatings significantly reduced the porous nature of these materials and it is clear that the preparation of porous materials is possible using this approach. In addition to offering a simpler process for the preparation of these porous materials the use of APS as initiator also has an additional advantage, that is it can be coupled with TEMED to allow for rapid polymerisation of these materials at room temperature84–86 (Fig. S9†).
 | ||
| Fig. 6 SEM images of crosslinked colloidal gels obtained from the addition of APS at 1 wt% (w.r.t. solids) to 20 wt% V01 and cured with different concentrations of DEGDA. DEGDA concentration (w.r.t. solids): (A) 15 wt%; (B) 20 wt%; (C) 25 wt%; (D) 30 wt%; (E) 40 wt%; (F) 65 wt%. Scale bar is 500 nm. | ||
Given the pore sizes observed appear to be in the order of the particle dimensions, the use of larger particles would be expected to result in the production of larger pores, which is an important consideration for obtaining materials with greater permeability and lower resistance to mass transfer.14,85,87 A positively charged latex with larger particle diameter was therefore synthesised using V-50 as initiator and a continuous phase consisting of a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture of H2O and MeOH as outlined by Bon et al.57 This was synthesised without co-monomer, and these particles were denoted as V02 (Table 1). The resulting latex possessed an average particle diameter of 470 ± 50 nm by SEM and 560 ± 10 nm by DLS and a positive zeta potential. This was consistent with previous reports where an ionic co-monomer was absent.57,70,72 In addition, no secondary nucleation was apparent (Fig. 1). Dialysis of this latex against H2O was performed to remove this co-solvent, allowing for fair comparisons to the materials prepared with H2O only. The addition of APS at 1 wt% (w.r.t. solids) to 20 wt% V02 also resulted in full coagulation of this latex and this gel was porous in nature (Fig. S6B†). Thermally curing these gels with DEGDA concentrations in the range 20–65 wt% (w.r.t. solids) produced rigid cylinders and SEM analysis (Fig. 7) revealed similar trends to those above, with 20 wt% DEGDA resembling that of the colloidal gel, while DEGDA contents of 30 and 40 wt% resulted in predominately fused structures with thicker coatings. Fig. S8† clearly demonstrates that this fused morphology is directly related to the presence of the crosslinker, and not as a result of coagulation, or drying of these latexes. In terms of the porous morphology, larger pore sizes were observed compared to the materials prepared with the smaller V01 particles (Fig. 6), with an average pore size of 0.8 ± 0.6 μm by SEM and 0.64 ± 0.07 μm by MIP for the material prepared with 30 wt% DEGDA. This increase in pore size was also supported by a significantly lower specific surface area of 5.6 ± 0.2 m2 g−1. Voids of this size are important for applications requiring high permeability such as chromatography,88 as flow-through catalytic reactors,5 or for the transport of nutrients in tissue engineering.89 Lower magnification images (Fig. S10†) demonstrate the porous morphologies of these materials more clearly. In the case of 65 wt% DEGDA a material with cauliflower type morphology was again observed with, as a result of the thicker coating, significantly reduced void size.
:2 mixture of H2O and MeOH as outlined by Bon et al.57 This was synthesised without co-monomer, and these particles were denoted as V02 (Table 1). The resulting latex possessed an average particle diameter of 470 ± 50 nm by SEM and 560 ± 10 nm by DLS and a positive zeta potential. This was consistent with previous reports where an ionic co-monomer was absent.57,70,72 In addition, no secondary nucleation was apparent (Fig. 1). Dialysis of this latex against H2O was performed to remove this co-solvent, allowing for fair comparisons to the materials prepared with H2O only. The addition of APS at 1 wt% (w.r.t. solids) to 20 wt% V02 also resulted in full coagulation of this latex and this gel was porous in nature (Fig. S6B†). Thermally curing these gels with DEGDA concentrations in the range 20–65 wt% (w.r.t. solids) produced rigid cylinders and SEM analysis (Fig. 7) revealed similar trends to those above, with 20 wt% DEGDA resembling that of the colloidal gel, while DEGDA contents of 30 and 40 wt% resulted in predominately fused structures with thicker coatings. Fig. S8† clearly demonstrates that this fused morphology is directly related to the presence of the crosslinker, and not as a result of coagulation, or drying of these latexes. In terms of the porous morphology, larger pore sizes were observed compared to the materials prepared with the smaller V01 particles (Fig. 6), with an average pore size of 0.8 ± 0.6 μm by SEM and 0.64 ± 0.07 μm by MIP for the material prepared with 30 wt% DEGDA. This increase in pore size was also supported by a significantly lower specific surface area of 5.6 ± 0.2 m2 g−1. Voids of this size are important for applications requiring high permeability such as chromatography,88 as flow-through catalytic reactors,5 or for the transport of nutrients in tissue engineering.89 Lower magnification images (Fig. S10†) demonstrate the porous morphologies of these materials more clearly. In the case of 65 wt% DEGDA a material with cauliflower type morphology was again observed with, as a result of the thicker coating, significantly reduced void size.
 | ||
| Fig. 7 SEM images of crosslinked colloidal gels obtained from the addition of APS at 1 wt% (w.r.t. solids) to 20 wt% V02 and cured with different concentrations of DEGDA. DEGDA concentration (w.r.t. solids): (A) 20 wt%; (B) 30 wt%; (C) 40 wt%; (D) 65 wt%. Scale bar is 500 nm. | ||
Provided the DEGDA content was below that required for the onset of this morphology, the pore sizes obtained for these materials appear to be directly correlated to particle size, with voids present in the order of the particle dimensions. This is clear when comparing the pore size distributions obtained from the 20 wt% gels by SEM (Fig. 8) and the pore size distributions obtained from the cross-linked materials from MIP (Fig. S14†). This potentially allows the porous properties of these materials to be easily predicted, allowing for the preparation of materials specifically designed for particular applications, without an extensive optimisation process, as is the case when using a new porogenic solvent or monomer system.22 For example, materials with small pore sizes and higher surface areas are useful for bulk catalysis, adsorbents, and for gas storage, whereas larger pore sizes are important for applications such as flow-through reactors, biochromatography and tissue engineering. In addition, the particle size and particle size distribution can easily be varied in the soap-free emulsion polymerisation approach, through changes in a variety of parameters, which include the reaction temperature, monomer concentration, initiator and co-monomer concentration, and the ionic strength.61
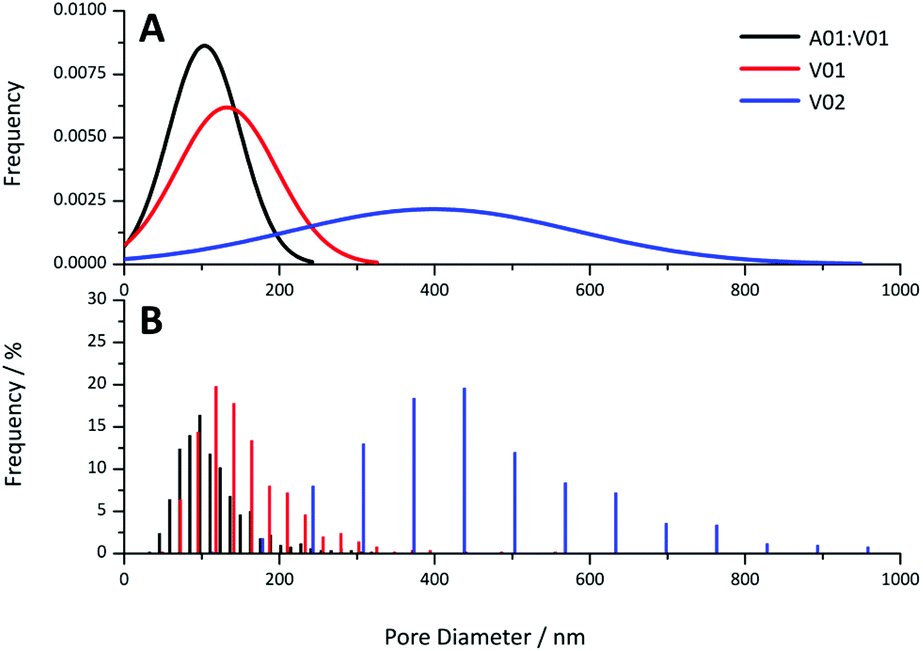 | ||
| Fig. 8 (A) Theoretical normal distribution and (B) histograms obtained for pore diameter of the A01:V01, V01 and V02 gels at 20 wt% obtained from the SEM images. | ||
Preparation of crosslinked colloidal gels with different shapes
All gels prepared in this work, including those prepared using a single latex, were highly mouldable, which potentially allows for the preparation of these materials in a variety of formats. The use of vials has already been demonstrated to result in the formation of rigid cylinders (Fig. 9A), however a range of other formats can also easily be prepared as demonstrated in Fig. 9 for the colloidal gels obtained using V02. A flat sheet (Fig. 9B) was prepared simply by sandwiching the gel between two glass slides, while a pyramid (Fig. 9C) was prepared by moulding the gel with a spatula into the desired shape on a glass slide. This was possible as these gels are capable of maintaining their shape in the absence of an external force, and both became rigid after curing. The other gels used in this work could also easily be prepared in these formats (for example the materials obtained using V01 is shown in Fig. S11†). | ||
| Fig. 9 Photographs of rigid porous materials obtained in a variety of formats from the addition of APS to V02 with 30 wt% DEGDA (w.r.t. solids). (A) Cylinder; (B) flat sheet; (C) & (D) pyramid. Scale bar is 5 mm. | ||
This demonstrates that a wide variety of formats can readily be prepared, which is important for enabling their use in a wide variety of applications. For example, pumping the gels into column housing could enable their use for chromatography,14 or as catalytic supports,4–7 while the flat sheet format could be useful for the manufacture of plates for thin-layer chromatography (TLC).16 The freestanding nature of these gels is particularly advantageous as it provides the possibility of preparing these materials without a mould (Fig. 9C & D), which is not possible when using a porogenic solvent.22
Solvent behaviour
In order to access the suitability of these materials for different applications, polymer disks prepared from the combination of A01 and V01 or using V01 and V02 only were immersed in solvents of varying polarity. For the gels obtained from A01 and V01 20 wt% DEGDA (w.r.t. solids) was utilised in the crosslinking process, while 30 wt% DEGDA (w.r.t. solids) was utilised for the V01 and V02 gels. The solvents investigated included Milli-Q H2O, MeOH, EtOH, ACN, hexadecane and sunflower oil. The porosity values calculated for these disks by immersion in these solvents are shown in Table 2, as well as the theoretical porosity, which was calculated from the H2O content, assuming full conversion and incorporation of the crosslinker into the resulting material.| Sample | [DEGDA]a/wt% | φ w/% (H2O) | φ w/% (MeOH) | φ w/% (EtOH) | φ w/% (ACN) | φ w/% (hexadecane) | φ w/% (sunflower oil) | φ T/% |
|---|---|---|---|---|---|---|---|---|
| a Concentration of DEGDA used w.r.t. solids. φw signifies the porosity, while φT indicates the theoretical porosity. | ||||||||
| A01:V01 | 20 | 50 ± 10 | 75 ± 3 | 69 ± 6 | 72 ± 7 | 67 ± 4 | 77 ± 4 | 76 |
| V01 | 30 | 67 ± 7 | 69 ± 7 | 70 ± 8 | 68 ± 7 | 74 ± 1 | 76 ± 4 | 74 |
| V02 | 30 | 72 ± 6 | 71 ± 5 | 73 ± 4 | 79 ± 2 | 67 ± 4 | 77 ± 5 | 74 |
In most cases the values obtained were in agreement with the theoretical porosities, which were 76% and 74% when 20 and 30 wt% DEGDA were utilised for crosslinking, respectively. These values are higher than that of conventional polymer monoliths, which are often prepared with a porosity of 60%, but similar to that of poly(HIPE)s which have porosities in excess of 74%. The change in volume observed for these disks was also negligible for all solvents (Table S6†), excluding acetonitrile, suggesting these values were reflective of the porosity in the dry state. Acetonitrile is a relatively good solvent for linear poly(styrene), so in this case it is possible that some linear chains from the original latex particles may hypothetically leach out. To avoid this the original latexes could have been lightly crosslinked with 0.5–1 wt% (w.r.t. styrene) of divinylbenzene in their formulation. Given these disks were simply immersed in these solvents, without any applied pressure, this also suggested that the liquid was being drawn into the pores of the polymer disk by capillary action, rather than the swelling of the polymer. This was also observed to occur rapidly with negligible change in mass of the disks after 30 min of immersion, even when sunflower oil was utilised as the solvent.
This behaviour is particularly important for several applications and suggests the potential for these materials to be utilised in TLC or for extraction, where greener solvents such as ethanol or even aqueous solutions could be utilised. The uptake of H2O was of particular interest given the strong hydrophobic character typically associated with poly(styrene) monoliths.90 It appeared as though the inclusion of DEGDA resulted in an increase in the hydrophilicity of the material allowing for H2O uptake by capillary action, which is typically not possible for poly(styrene)-based monoliths. In fact the amount of DEGDA present appeared to directly correlate to the amount of H2O absorbed, with the disks prepared with 20 wt% DEGDA absorbing significantly lower amounts of H2O by mass (Table S7 & Fig. S12†), resulting in a lower than expected porosity of 50 ± 10%. This is in comparison to the disks prepared with 30 wt% DEGDA, which had porosities consistent with those obtained using the other solvents (Table 2). For all other solvents the amount absorbed correlated to the pore volume of these disks, with the mass of solvent entering the disks ranging from 110 to 220% by mass relative to the mass of the dry disks (Table S7 & Fig. S12†), thus resulting in porosity values of ∼70% (Table 2).
The inclusion of DEGDA has therefore resulted in the ability of these disks to absorb solvents of varying polarities ranging from H2O to hexadecane. In addition, no incompatibility with these solvents was observed with minimal swelling/shrinkage of these disks as a result of the crosslinking process utilised. These disks did however swell to a small degree in acetonitrile, with a change in volume of ∼20% compared to the original volume observed (Table S6†). However, no shrinkage or swelling was observed for the other solvents.
Conclusions
Mechanically robust and solvent resistant polymer monoliths were made using colloidal gels which were reinforced through a post-polymerisation step using diacylates as crosslinker. The colloidal building blocks for the gels were poly(styrene) latexes synthesised by soap-free emulsion polymerisation. This waterborne and low-SOC approach potentially offers a greener alternative in comparison to the use of a porogenic solvent or an emulsion template, with the use of only H2O and/or MeOH as solvents, the absence of surfactant, and minimal purification the main advantages. Chemical crosslinking was employed through the introduction of DEGDA.The phase separation of the crosslinker during curing resulted in the presence of a polymer coating, with increases in the DEGDA content resulting in thicker coatings and ultimately a predominately fused morphology. It is expected that the presence of this coating would have modified the surface chemistry, and may offer an alternative method for surface functionalisation through the incorporation of additional water-soluble crosslinkers. This is a focus of future work. Initial experiments focused on the preparation of these materials using two latexes, however it was found that similar materials could be obtained through the addition of APS to a single latex, where it both promoted the formation of the colloidal gel and initiated the crosslinking process. The use of APS as initiator also allowed for the rapid curing of these materials at room temperature through the addition of TEMED.
In conjunction to the greener advantages, this approach also offered some unique advantages over conventional synthetic strategies. For example, the pore size of these materials was found to be in the order of the particle dimensions, with the use of larger particles resulting in materials with larger pore size. Given particles of different size can easily be prepared using the soap-free emulsion polymerisation approach this offers the ability to easily prepare materials with desired porous properties for particular applications. Additionally, the high mouldability of all gels prepared in this work afforded the possibility to prepare these materials in a variety of formats and more importantly without the use of a mould. This approach is therefore expected to be applicable for the preparation of polymer monoliths for a wide variety of applications, including but not limited to, tissue engineering, catalysis, chromatography, extraction, sample preparation, and as absorbents. In particular these monoliths were found to posses relatively high porosities and were capable of rapidly absorbing solvents of varying polarity by capillary action, which suggested their applicability for TLC and extraction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Australian Research Council's Discovery funding scheme (DP130101471). C. T. D was the recipient of both an Endeavour Fellowship provided by the Australian Government and an Australian Government Research Training Program Scholarship. We gratefully acknowledge Guy Clarkson for assistance with the obtainment of crystal structures (Department of Chemistry, University of Warwick).Notes and references
- S. Hjertén, J.-L. Liao and R. Zhang, J. Chromatogr., A, 1989, 473, 273–275 CrossRef.
- F. Svec and J. M. J. Fréchet, Anal. Chem., 1992, 64, 820–822 CrossRef CAS.
- C. Viklund, F. Svec and J. M. J. Fréchet, Chem. Mater., 1996, 8, 744–750 CrossRef CAS.
- M. I. Burguete, A. Cornejo, E. García-Verdugo, J. García, M. J. Gil, S. V. Luis, V. Martínez-Merino, J. A. Mayoral and M. Sokolova, Green Chem., 2007, 9, 1091–1096 RSC.
- B. Altava, M. I. Burguete, E. García-Verdugo, S. V. Luis and M. J. Vicent, Green Chem., 2006, 8, 717–726 RSC.
- V. Chiroli, M. Benaglia, A. Puglisi, R. Porta, R. P. Jumde and A. Mandoli, Green Chem., 2014, 16, 2798–2806 RSC.
- O. Bortolini, A. Cavazzini, P. Dambruoso, P. P. Giovannini, L. Caciolli, A. Massi, S. Pacifico and D. Ragno, Green Chem., 2013, 15, 2981–2992 RSC.
- S. Xie, F. Svec and J. M. J. Fréchet, Biotechnol. Bioeng., 1999, 62, 30–35 CrossRef CAS PubMed.
- H. Wang, H. Zhang, Y. Lv, F. Svec and T. Tan, J. Chromatogr., A, 2014, 1343, 128–134 CrossRef CAS PubMed.
- S. Zhang, W. Luo, W. Yan and B. Tan, Green Chem., 2014, 16, 4408–4416 RSC.
- E. M. Christenson, W. Soofi, J. L. Holm, N. R. Cameron and A. G. Mikos, Biomacromolecules, 2007, 8, 3806–3814 CrossRef CAS PubMed.
- H. Zhang and A. I. Cooper, Adv. Mater., 2007, 19, 2439–2444 CrossRef CAS.
- P. Jing, X. Fang, J. Yan, J. Guo and Y. Fang, J. Mater. Chem. A, 2013, 1, 10135–10141 CAS.
- Y.-H. Shih, B. Singco, W.-L. Liu, C.-H. Hsu and H.-Y. Huang, Green Chem., 2011, 13, 296–299 RSC.
- F. Svec and Y. Lv, Anal. Chem., 2015, 87, 250–273 CrossRef CAS PubMed.
- D. Yin, Y. Guan, H. Gu, Y. Jia and Q. Zhang, RSC Adv., 2017, 7, 7303–7309 RSC.
- E. Candish, H. J. Wirth, A. A. Gooley, R. A. Shellie and E. F. Hilder, J. Chromatogr., A, 2015, 1410, 9–18 CrossRef CAS PubMed.
- J. Krenkova and F. Foret, J. Sep. Sci., 2011, 34, 2106–2112 CAS.
- E. Candish, A. Khodabandeh, M. Gaborieau, T. Rodemann, R. A. Shellie, A. A. Gooley and E. F. Hilder, Anal. Bioanal. Chem., 2017, 409, 2189–2199 CrossRef CAS PubMed.
- F. Svec and J. M. J. Fréchet, Chem. Mater., 1995, 7, 707–715 CrossRef CAS.
- C. Viklund, E. Pontén, B. Glad and K. Irgum, Chem. Mater., 1997, 9, 463–471 CrossRef CAS.
- F. Svec, J. Chromatogr., A, 2010, 1217, 902–924 CrossRef CAS PubMed.
- C. T. Desire, R. D. Arrua, M. Talebi, N. A. Lacher and E. F. Hilder, J. Sep. Sci., 2013, 36, 2782–2792 CrossRef CAS PubMed.
- R. D. Arrua, D. Serrano, G. Pastrana, M. Strumia and C. I. A. Igarzabal, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6616–6623 CrossRef CAS.
- M. Bedair and Z. El Rassi, J. Chromatogr., A, 2003, 1013, 35–45 CrossRef CAS PubMed.
- B. Gu, J. M. Armenta and M. L. Lee, J. Chromatogr., A, 2005, 1079, 382–391 CrossRef CAS PubMed.
- X. Huang, Q. Wang, H. Yan, Y. Huang and B. Huang, J. Chromatogr., A, 2005, 1062, 183–188 CrossRef CAS PubMed.
- E. C. Peters, M. Petro, F. Svec and J. M. J. Fréchet, Anal. Chem., 1998, 70, 2296–2302 CrossRef CAS PubMed.
- W. Jin, H. Fu, X. Huang, H. Xiao and H. Zou, Electrophoresis, 2003, 24, 3172–3180 CrossRef CAS PubMed.
- H. Oberacher, A. Premstaller and C. G. Huber, J. Chromatogr., A, 2004, 1030, 201–208 CrossRef CAS PubMed.
- Q. C. Wang, F. Svec and J. M. J. Fréchet, J. Chromatogr., A, 1994, 669, 230–235 CrossRef CAS PubMed.
- K. Zhang, C. Yan, J. Yang, Z. Zhang, Q. Wang and R. Gao, J. Sep. Sci., 2005, 28, 217–224 CrossRef CAS PubMed.
- D. Hoegger and R. Freitag, J. Chromatogr., A, 2001, 914, 211–222 CrossRef CAS PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- B. P. Santora, M. R. Gagné, K. G. Moloy and N. S. Radu, Macromolecules, 2001, 34, 658–661 CrossRef CAS.
- S. D. Kimmins and N. R. Cameron, Adv. Funct. Mater., 2011, 21, 211–225 CrossRef CAS.
- N. R. Cameron and D. C. Sherrington, Adv. Polym. Sci., 1996, 126, 162–214 CrossRef.
- T. Cserháti, E. Forgács and G. Oros, Environ. Int., 2002, 28, 337–348 CrossRef.
- J. Guilbot, S. Kerverdo, A. Milius, R. Escola and F. Pomrehn, Green Chem., 2013, 15, 3337–3354 RSC.
- M. C. Morán, A. Pinazo, L. Pérez, P. Clapés, M. Angelet, M. T. García, M. P. Vinardell and M. R. Infante, Green Chem., 2004, 6, 233–240 RSC.
- P. Verdia, H. Q. N. Gunaratne, T. Y. Goh, J. Jacquemin and M. Blesic, Green Chem., 2016, 18, 1234–1239 RSC.
- A. N. Ghebremeskel, C. Vemavarapu and M. Lodaya, Int. J. Pharm., 2007, 328, 119–129 CrossRef CAS PubMed.
- S. Kovačič, N. B. Matsko, K. Jerabek, P. Krajnc and C. Slugovc, J. Mater. Chem. A, 2013, 1, 487–490 Search PubMed.
- A. Jordan and N. Gathergood, Chem. Soc. Rev., 2015, 44, 8200–8237 RSC.
- D. Coleman and N. Gathergood, Chem. Soc. Rev., 2010, 39, 600–637 RSC.
- M. Smiglak, W. M. Reichert, J. D. Holbrey, J. S. Wilkes, L. Sun, J. S. Thrasher, K. Kirichenko, S. Singh, A. R. Katritzky and R. D. Rogers, Chem. Commun., 2006, 2554–2556 RSC.
- M. J. Earle, J. M. Esperanca, M. A. Gilea, J. N. Lopes, L. P. Rebelo, J. W. Magee, K. R. Seddon and J. A. Widegren, Nature, 2006, 439, 831–834 CrossRef CAS PubMed.
- T. Welton, Green Chem., 2011, 13, 225 RSC.
- C. A. L. Colard, R. A. Cave, N. Grossiord, J. A. Covington and S. A. F. Bon, Adv. Mater., 2009, 21, 2894–2898 CrossRef CAS.
- H. Wang, M. B. Hansen, D. W. Lowik, J. C. van Hest, Y. Li, J. A. Jansen and S. C. Leeuwenburgh, Adv. Mater., 2011, 23, H119–H124 CrossRef CAS PubMed.
- Q. Wang, L. Wang, M. S. Detamore and C. Berkland, Adv. Mater., 2008, 20, 236–239 CrossRef CAS.
- Q. Wang, S. Jamal, M. S. Detamore and C. Berkland, J. Biomed. Mater. Res., Part A, 2011, 96, 520–527 CrossRef PubMed.
- Q. Wang, Z. Gu, S. Jamal, M. S. Detamore and C. Berkland, Tissue Eng., Part A, 2013, 19, 2586–2593 CrossRef CAS PubMed.
- S. R. Van Tomme, C. F. van Nostrum, S. C. de Smedt and W. E. Hennink, Biomaterials, 2006, 27, 4141–4148 CrossRef CAS PubMed.
- S. R. V. Tomme, M. J. v. Steenbergen, S. C. D. Smedt, C. F. v. Nostrum and W. E. Hennink, Biomaterials, 2005, 26, 2129–2135 CrossRef PubMed.
- S. R. V. Tomme, C. F. v. Nostrum, M. Dijkstra, S. C. D. Smedt and W. E. Hennink, Eur. J. Pharm. Biopharm., 2008, 70, 522–530 CrossRef PubMed.
- S. A. F. Bon, H. V. Beek, P. Piet and A. L. German, J. Appl. Polym. Sci., 1995, 58, 19–29 CrossRef CAS.
- Y. Chonde and I. M. Krieger, J. Appl. Polym. Sci., 1981, 26, 1819–1827 CrossRef CAS.
- J. R. McCracken and A. Datyner, J. Appl. Polym. Sci., 1974, 18, 3365–3372 CrossRef CAS.
- J. W. Goodwin, J. Hearn, C. C. Ho and R. H. Ottewill, Br. Polym. J., 1973, 5, 341–362 CrossRef.
- S. C. Thickett and R. G. Gilbert, Polymer, 2007, 48, 6965–6991 CrossRef CAS.
- N. Marti, F. Quattrini, A. Butté and M. Morbidelli, Macromol. Mater. Eng., 2005, 290, 221–229 CrossRef CAS.
- A. Cingolani, D. Cuccato, G. Storti and M. Morbidelli, Macromol. Mater. Eng., 2018, 303, 1700417 CrossRef.
- L. Yu, Z. Zhang, H. Zhang and J. Ding, Biomacromolecules, 2009, 10, 547–1553 CrossRef PubMed.
- L. Yu, G. Chang, H. Zhang and J. Ding, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1122–1133 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- J. H. Kim, M. Chainey, M. S. El-Aasser and J. W. Vanderhoff, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 171–183 CrossRef CAS.
- L.-J. Liu and I. M. Krieger, J. Polym. Sci., Part A: Polym. Chem., 1981, 19, 3013–3026 CrossRef CAS.
- M. S.-D. Juang and I. M. Krieger, J. Polym. Sci., Part A: Polym. Chem., 1976, 14, 2089–2107 CrossRef CAS.
- S. R. Turner, R. A. Weiss and R. D. Lundberg, J. Polym. Sci., Part A: Polym. Chem., 1985, 23, 535–548 CrossRef CAS.
- V. Tohver, A. Chan, O. Sakurada and J. e. A. Lewis, Langmuir, 2001, 17, 8414–8421 CrossRef CAS.
- R. D. Arrua, T. J. Causon and E. F. Hilder, Analyst, 2012, 137, 5179–5189 RSC.
- F. Svec and J. M. J. Fréchet, Ind. Eng. Chem. Res., 1999, 38, 34–48 CrossRef CAS.
- Q. C. Wang, F. Svec and J. M. J. Fréchet, Anal. Chem., 1993, 65, 2243–2248 CrossRef CAS PubMed.
- H. McKenzie, Doctor of Philosophy, University of Warwick, 2014 Search PubMed.
- X. Huang, S. Zhang, G. A. Schultz and J. Henion, Anal. Chem., 2002, 74, 2336–2344 CrossRef CAS PubMed.
- Z. Liu and H. Xiao, Polymer, 2000, 41, 7023–7031 CrossRef CAS.
- K. Kang, C. Y. Kan, Y. Du and D. S. Liu, J. Appl. Polym. Sci., 2004, 92, 433–438 CrossRef CAS.
- V. Mittal, N. B. Matsko, A. Butté and M. Morbidelli, Macromol. Mater. Eng., 2008, 2, 215–221 CAS.
- M. Bechtle, A. Butte, G. Storti and M. Morbidelli, J. Chromatogr., A, 2010, 1217, 4675–4681 CrossRef CAS PubMed.
- R. D. Arrua, A. Nordborg, P. R. Haddad and E. F. Hilder, J. Chromatogr., A, 2013, 1273, 26–33 CrossRef PubMed.
- R. D. Arrua, P. R. Haddad and E. F. Hilder, J. Chromatogr., A, 2013, 1311, 121–126 CrossRef CAS PubMed.
- A. Khodabandeh, R. Dario Arrua, C. T. Desire, T. Rodemann, S. A. F. Bon, S. C. Thickett and E. F. Hilder, Polym. Chem., 2016, 7, 1803–1812 RSC.
- F. Svec, J. Sep. Sci., 2004, 27, 1419–1430 CrossRef CAS PubMed.
- F. Svec, J. Sep. Sci., 2004, 27, 747–766 CrossRef CAS PubMed.
- F. M. Plieva, I. Y. Galaev and B. Mattiasson, J. Sep. Sci., 2007, 30, 1657–1671 CrossRef CAS PubMed.
- Z. Jiang, N. W. Smith and Z. Liu, J. Chromatogr., A, 2011, 1218, 2350–2361 CrossRef CAS PubMed.
- A. L. Barbarini, D. A. Estenoz and D. M. Martino, J. Appl. Polym. Sci., 2015, 132, 41947 CrossRef.
- P. Zarras and O. Vogl, J. Macromol. Sci., Part A: Pure Appl.Chem., 2000, 37, 817–840 CrossRef.
- Y. Dong, J. Yin, J. Yuan and X. Zhao, Polymer, 2016, 97, 408–417 CrossRef CAS.
- G. Leone, A. Boglia, F. Bertini, M. Canetti and G. Ricci, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4473–4483 CrossRef CAS.
- Y. Tominaga, T. Kubo, K. Sueyoshi, K. Hosoya and K. Otsuka, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3153–3158 CrossRef CAS.
- H.-K. Kim, M.-S. Lee, S.-Y. Lee, Y.-W. Choi, N.-J. Jeong and C.-S. Kim, J. Mater. Chem. A, 2015, 3, 16302–16306 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8gc01055b |
| ‡ The synthetic conditions were not optimised as part of this work. Higher yielding synthetic routes are also available for the preparation of the TEVBAC monomer,91–93 however they are more complicated. Alternatively, TMVBAC, which can also stabilize latexes prepared with V-50,57 is currently readily available for purchase from the Sigma-Aldrich catalogue.94–96 |
| This journal is © The Royal Society of Chemistry 2018 |