
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Utilization of fluoroform for difluoromethylation in continuous flow: a concise synthesis of α-difluoromethyl-amino acids†
Manuel
Köckinger
a,
Tanja
Ciaglia
a,
Michael
Bersier
b,
Paul
Hanselmann
b,
Bernhard
Gutmann
 *ac and
C. Oliver
Kappe
*ac
*ac and
C. Oliver
Kappe
*ac
aInstitute of Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, 8010 Graz, Austria. E-mail: bernhard.gutmann@uni-graz.at; oliver.kappe@uni-graz.at
bMicroreactor Technology, Lonza AG, CH-3930 Visp, Switzerland
cResearch Center Pharmaceutical Engineering GmbH (RCPE), Inffeldgasse 13, 8010 Graz, Austria
First published on 30th October 2017
Abstract
Fluoroform (CHF3) can be considered as an ideal reagent for difluoromethylation reactions. However, due to the low reactivity of fluoroform, only very few applications have been reported so far. Herein we report a continuous flow difluoromethylation protocol on sp3 carbons employing fluoroform as a reagent. The protocol is applicable for the direct Cα-difluoromethylation of protected α-amino acids, and enables a highly atom efficient synthesis of the active pharmaceutical ingredient eflornithine.
The difluoromethyl group is found in an increasing array of pharmaceutical and agrochemical products.1 Not surprisingly, therefore, significant efforts have been devoted towards the development of novel protocols for the introduction of the CHF2-moiety into organic molecules.2 In addition to well-established methods based on the deoxyfluorination of aldehydes, various methods for direct difluoromethylation have recently become available.2 Among the cheapest and most versatile reagents for direct CHF2-transfer is chlorodifluoromethane (CHF2Cl, Freon 22). Chlorodifluoromethane is produced on a massive scale, particularly for the production of fluoropolymers, and it is available at a relatively low cost (Fig. 1). With sufficiently strong bases, CHF2Cl can be deprotonated. The so-formed carbanion, chlorodifluoromethanide (CF2Cl−), immediately loses chloride to generate an electrophilic singlet difluorocarbene (CF2Cl− → CF2 + Cl−). The short-lived difluorocarbene can then be trapped with a suitably reactive nucleophilic species to produce the difluoromethylated product (Fig. 1).2 This reaction has been shown to proceed successfully with a range of NH-, OH- and CH-acidic compounds.2 However, CHF2Cl is a strong ozone depleting gas and it is controlled under the Montreal Protocol. As a consequence, its production and usage has become increasingly limited and expensive. A plethora of alternative difluoromethane sources have been developed in recent years, including TMSCF2Br, (EtO)2POCF2Br, PhCOCF2Cl and CHF2OTf.2 Although these reagents cover the needs of chemists for difluoromethylation on a laboratory scale, their high cost, low atom economy and limited commercial availability prohibit their usage in an industrial setting.
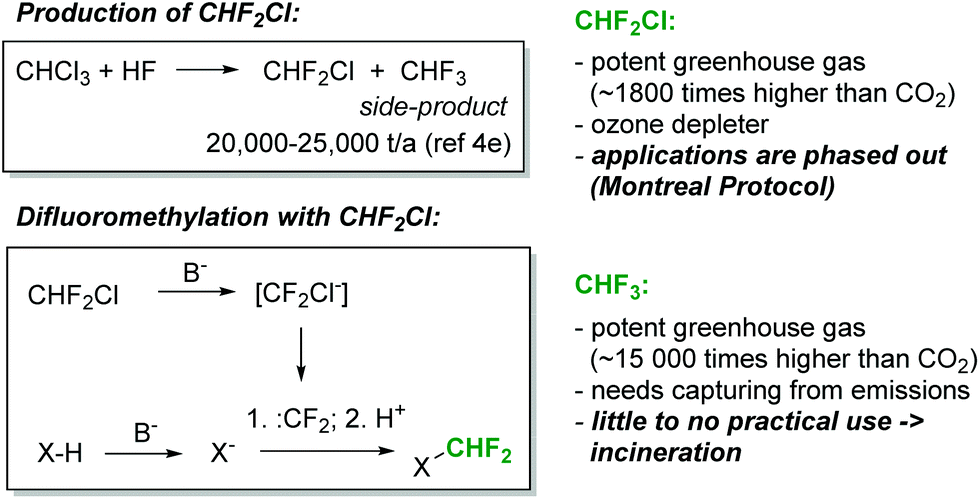 | ||
| Fig. 1 CHF3 is a large-volume by-product in the synthesis of CHF2Cl. | ||
The most attractive CF3- and CHF2-source is fluoroform (CHF3, Freon 23). Fluoroform is generated as a large-volume waste-product during the synthesis of chlorodifluoromethane (Fig. 1). It is a nontoxic and ozone-friendly gas with a boiling point of −82 °C. Since fluoroform has an extraordinary global warming potential (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 times higher than carbon dioxide over a 100-year period),3 its discharge into the environment is restricted by the Kyoto Protocol. As a consequence, fluoroform needs to be destroyed or, alternatively, captured and used as a feedstock for manufacturing. The latter option, though vastly preferable, is difficult to realize due to the extraordinarily low reactivity of fluoroform. Only very recently have the first synthetically relevant transformations involving fluoroform started to emerge.4 Most relevant for the present work is a series of seminal publications from the laboratories of Mikami.5–7 Mikami and co-workers have shown that fluoroform can be utilized for the direct difluoromethylation of a variety of substrates utilizing strong lithium bases (lithium diisopropylamide (LDA) or lithium hexamethyldisilazide (LiHMDS)).5–7 In addition to Mikami's work, the difluoromethylation reactions of alkynes with CHF3 and tBuOK,8 as well as the difluoromethylation of phenols and malonates with aqueous KOH,9 have been described by Shibata and Dolbier, respectively. The mechanism of the difluoromethylation reaction is believed to resemble that of the corresponding reaction with CHF2Cl.8–10 An electrophilic singlet difluorocarbene is formed by the deprotonation of CHF3 and the subsequent rapid α-elimination of fluoride. Difluorocarbene then reacts with the anion of the substrate (Fig. 1).
800 times higher than carbon dioxide over a 100-year period),3 its discharge into the environment is restricted by the Kyoto Protocol. As a consequence, fluoroform needs to be destroyed or, alternatively, captured and used as a feedstock for manufacturing. The latter option, though vastly preferable, is difficult to realize due to the extraordinarily low reactivity of fluoroform. Only very recently have the first synthetically relevant transformations involving fluoroform started to emerge.4 Most relevant for the present work is a series of seminal publications from the laboratories of Mikami.5–7 Mikami and co-workers have shown that fluoroform can be utilized for the direct difluoromethylation of a variety of substrates utilizing strong lithium bases (lithium diisopropylamide (LDA) or lithium hexamethyldisilazide (LiHMDS)).5–7 In addition to Mikami's work, the difluoromethylation reactions of alkynes with CHF3 and tBuOK,8 as well as the difluoromethylation of phenols and malonates with aqueous KOH,9 have been described by Shibata and Dolbier, respectively. The mechanism of the difluoromethylation reaction is believed to resemble that of the corresponding reaction with CHF2Cl.8–10 An electrophilic singlet difluorocarbene is formed by the deprotonation of CHF3 and the subsequent rapid α-elimination of fluoride. Difluorocarbene then reacts with the anion of the substrate (Fig. 1).
The main objective of the present work was to establish a scalable, continuous flow synthesis route to Cα-difluoromethyl amino acids using fluoroform as the reagent (Scheme 1). Cα-Difluoromethyl amino acids are potent and selective irreversible inhibitors of their respective α-amino acid decarboxylases.11 Representatives of this class of compounds exhibit a broad spectrum of biological activities, such as antibacterial, antihypertensive, cancerostatic, and cytotoxic activities.11 Currently, only D,L-α-difluoromethylornithine (eflornithine), an inhibitor of ornithine decarboxylase, is in medical use (Scheme 1). Eflornithine has been explored as an anticancer agent, and it is in clinical use for the treatment of African sleeping sickness as well as of Pneumocystis carinii pneumonia, the most frequent opportunistic infection associated with acquired immunodeficiency syndrome (AIDS).11 It is on the World Health Organization's Model List of Essential Medicines. Two different strategies can be employed for the generation of Cα-difluoromethyl amino acids: (i) construction of the amino acids from fluorine-containing building blocks (e.g. α-difluoromethyl malonates),12,13 or (ii) direct substitution of the α-hydrogen of amino acids with a CHF2 moiety (Scheme 1).14 The direct Cα-difluoromethylation of Schiff base-protected α-amino acid methyl esters with CHF2Cl as the reagent has been demonstrated by Bey and others.14 The α-difluoromethylation of protected amino acids with CHF3 is currently not reported. Indeed, the protocol described herein is, to the best of our knowledge, the first example where fluoroform is utilized for the preparation of a pharmaceutical end-product.
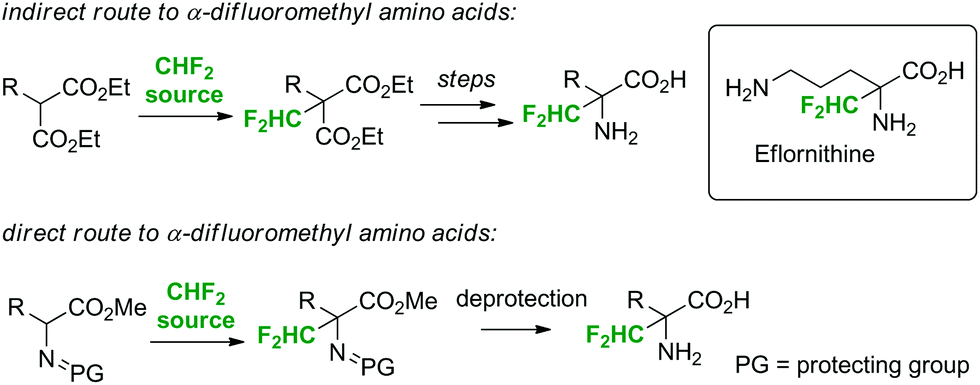 | ||
| Scheme 1 Synthesis of D,L-Cα-difluoromethyl amino acids. | ||
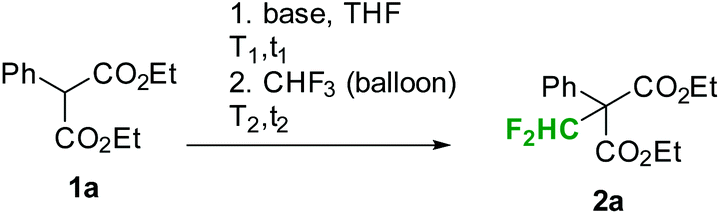
First preliminary batch experiments were performed with diethyl phenylmalonate (1a) as a model substrate (Table 1). For these reactions, malonate was dissolved in the desired solvent and a base was added. After stirring the mixture for a few minutes at the indicated temperatures (t1, T1 in Table 1), fluoroform was slowly passed through the reaction mixture under vigorous stirring (t2, T2 in Table 1). Using phenylmalonate 1a as a substrate, no product was formed with nBuLi, LDA or tBuOK as a base (entries 1 to 5 in Table 1). Furthermore, attempts to reproduce the results reported by Dolbier and co-workers failed in our hands.9 According to Dolbier's procedure, the phenylmalonate 1a is stirred in aqueous KOH for 30 min at room temperature. MeCN or dioxane is then added and fluoroform is bubbled through the solution.9 Our experiments under these reaction conditions did not yield any difluoromethylated product (entry 7 in Table 1). Small amounts of the desired product were formed using LiHMDS as a base (entry 6 in Table 1). Mikami and co-workers have already shown that fluoroform in combination with LiHMDS can be utilized for the direct difluoromethylation of cyclic amides, cyclic and open esters, and certain simple malonates.5 According to the authors, the reaction proceeds best with 2 equiv. of base and 5 equiv. of CHF3. Reaction times of 6 to 48 h at reaction temperatures of 0 to 25 °C were needed to provide the desired products in good yields.5
a
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Base (equiv.) | t 1 (min) | T 1 (°C) | t 2 (min) | T 2 (°C) | Convb (%) | Selb (%) | |
| a Reaction conditions: 0.5 mmol diethyl phenylmalonate 1a in 0.5 mL THF, base and CHF3 (balloon). b Analyzed by HPLC-UV/VIS at 215 nm. c Method B in ref. 9a. d 20% ethyl phenylacetate (by hydrolysis and decarboxylation). | |||||||
| 1 | tBuOK (3) | 5 | 25 | 60 | 25 | 28 | 0 |
| 2 | tBuOLi (3) | 5 | 25 | 60 | 25 | 4 | 0 |
| 3 | LDA (2) | 5 | 25 | 5 | 25 | 0 | 0 |
| 4 | nBuLi (2) | 5 | 25 | 5 | 25 | 0 | 0 |
| 5 | nBuLi (2) | 5 | −80 | 5 | −80 | 0 | 0 |
| 6 | LiHMDS (2) | 5 | 25 | 60 | 25 | 2 | 100 |
| 7c | KOH (15) | 30 | 25 | 120 | 25 | 32d | 0 |
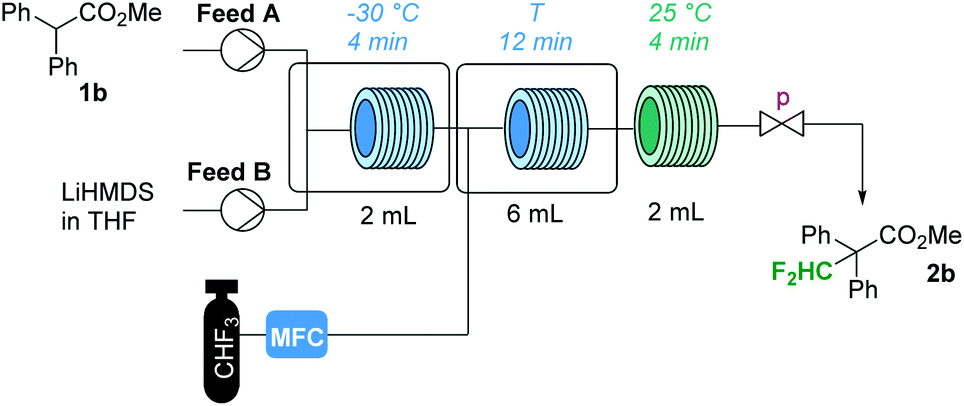
Encouraged by this result, we were keen to develop a continuous flow protocol for this reaction. Continuous processing techniques have had a significant impact on the development of more sustainable manufacturing routes for several pharmaceuticals.15–18 For gas–liquid reactions, several specific advantages exist: high pressure operation and fast gas–liquid mass transfer enhance the availability of the gaseous reagent in the liquid phase.16,17 In addition, the gaseous reagent can be dosed into the liquid phase with precise stoichiometry using mass flow controllers.16,17 Our initial flow setup consisted of two continuous syringe pumps to introduce (i) a 0.5 M solution of substrate in THF (Feed A, Table 2), and (ii) a commercial solution of LiHMDS (Feed B, Table 2). The two feeds were mixed in a Y-shaped connector in a cooling bath. The substrate is deprotonated in a 2 mL residence loop (reactor 1), before the mixture is combined with fluoroform in a second Y-shaped connector in a second cooling bath. The flow rate of the fluoroform stream was controlled using a calibrated mass flow controller (MFC). The combined mixture then went through a second cooled residence loop (reactor 2) and left the flow system through a third residence loop at room temperature (reactor 3) and an adjustable back pressure regulator. With pressures of ∼5 bar and temperatures below ∼25 °C for reactor 2, fluoroform dissolved completely in the liquid feed. At higher temperatures at this pressure, distinct gas–liquid segments were formed. The processed reaction mixture was finally collected in a quench solution of aqueous HCl/Et2O and the organic phase was analyzed by GC-FID and 19F-NMR spectroscopy. Methyl diphenylacetate 1b was used as the model substrate for the initial optimization. With flow rates of 300 μL min−1 for Feed A, 200 μL min−1 for Feed B and 8.3 mL min−1 for fluoroform, a stoichiometry of 1:1.3:2.5 for substrate/LiHMDS/CHF3 and a total residence time of ∼20 min was obtained. The flow reactions clearly revealed that the conversion increased with a decrease in temperature and an increase in pressure (Table 2). Also the selectivity increased slightly with decreasing temperatures (Table 2). As already observed by Mikami and co-workers, the best results are obtained with 2 equiv. of base (see Table S1 in the ESI†). Also, for fluoroform, 2 to 3 equiv. were identified as the ideal amount.
|
|
||||
|---|---|---|---|---|
| T (°C) | p (bar) | Convb (%) | Selb (%) | |
|
a Reaction conditions: Feed A: 0.5 M diethyl phenylmalonate 1b in THF; Feed B: 1 M LiHMDS in THF; with flow rates for Feed A/Feed B/CHF3 = 0.30:0.20:8.3 mL min−1, respectively, the following conditions were obtained: LiHMDS (1.33 equiv.); CHF3 (2.5 equiv.).
b Analyzed by GC-FID. For a detailed description of the experiments, see the ESI.
|
||||
| 1 | 40 | 5 | 63 | 81 |
| 2 | 25 | 5 | 65 | 80 |
| 3 | −10 | 5 | 82 | 88 |
| 4 | −15 | 10 | 86 | 91 |
| 5 | −15 | 12 | 92 | 91 |
The general reaction conditions were suitable for a variety of substrates (Fig. 2). Malonates with sterically benign alkyl groups in the α-position performed particularly well (1d to 1f in Fig. 2), while the phenyl derivative 1a resulted in low conversion. The reaction was remarkably clean, with the unreacted substrate and tris(trimethylsilyl)amine being the only contaminants in the crude mixture after washing with water. The analytically pure compounds were isolated by preparative thin-layer chromatography (TLC). The yield of diethyl methylmalonate 2e was the same as that previously reported for the batch protocol, even though the reaction time for the present procedure was significantly shorter (20 min vs. 20 h for the batch procedure).5 The yield of product 2c was lower than that reported for the batch procedure (39% vs. 78% according to 19F-NMR spectroscopy).5 The other difluoromethylated compounds prepared in this work have not been previously reported.
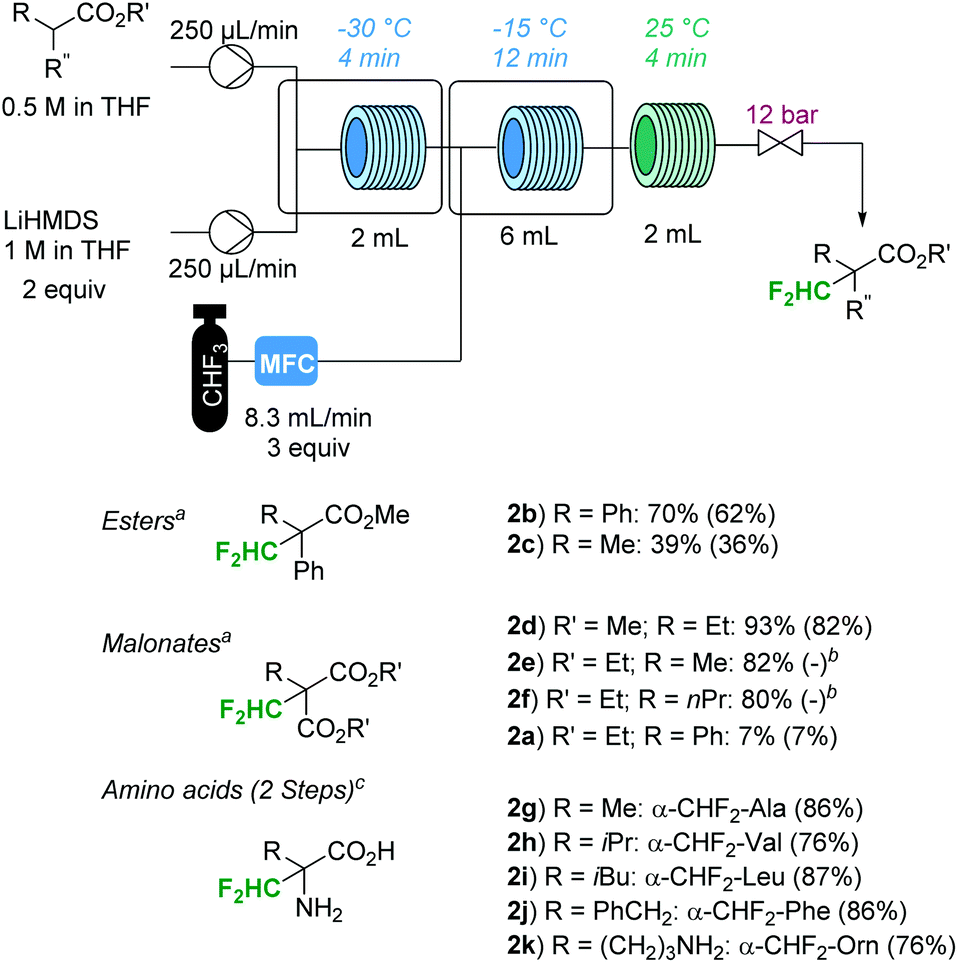 | ||
| Fig. 2 Continuous flow Cα-difluoromethylation with fluoroform. a19F-NMR yields (trifluorotoluene as an internal standard); isolated yields (TLC) in parentheses. bIsolation was not attempted. cIsolated yields after 2 steps (Cα-difluoromethylation and hydrolysis of the N-benzylidene- and the O-methyl-protecting groups). For experimental details, see the ESI.† | ||
As mentioned in the introduction, the synthesized α-difluoromethyl malonates can be converted to the respective α-difluoromethyl amino acids in a multi-step transformation (Scheme 1).12,13 However, since a direct difluoromethylation of natural α-amino acids has clear advantages, reactions with Schiff base-protected α-amino acid methyl esters were investigated.14 The N-benzylidene-protected α-amino acid methyl esters were readily available from the parent amino esters using literature procedures (for experimental details, see the ESI†).14 Gratifyingly, subjecting the N-benzylidene-protected α-amino acid methyl esters 1g to 1k to the general reaction conditions resulted in >95% conversion to the desired product. Indeed, the reaction was remarkably clean with only one CHF2-moiety (two doublets of doublets) detectable in the 19F-NMR spectra of the crude reaction mixtures. Difluoromethylation on the imine carbon, a side reaction typically encountered in reactions with CHF2Cl as a reagent, was not observed.14b Due to the instability of the Schiff base to hydrolysis, isolation of the intermediate products was not attempted. Instead, the N-benzylidene- and the O-methyl-protecting groups were directly removed by heating the crude product in 6 N HCl in a microwave batch autoclave (150 °C for 45 min). The released benzaldehyde was removed by extracting with ether and the aqueous phase was concentrated. After recrystallization from MeOH/EtOH, the Cα-difluoromethyl amino acids were obtained as their monohydrochloride salts (dihydrochloride salt for product 2k). As expected, [α]20D measurements of product 2h revealed that the chirality of the substrate was lost. The yields were above 70% for all tested amino acids over the two reaction steps, i.e. difluoromethylation and deprotection (Fig. 2). Importantly, eflornithine (2g) was isolated in 76% yield after the two reaction steps. The yield of eflornithine for the present chromatography-free method is significantly higher than that previously reported for the less desirable process based on chlorodifluoromethane (37% to 40%).14d,e
It should be noted that the process described herein consumes only two of the three introduced equivalents of fluoroform. For large scale applications, the separation of fluoroform from the processed solution and recycling needs to be considered.19 Further optimization of the difluoromethylation process, in particular with regard to stoichiometry and reaction time, is ongoing in our laboratories and will be reported in due course.
Conclusions
A gas–liquid continuous flow difluoromethylation protocol employing fluoroform as a reagent was reported. Fluoroform, a by-product of Teflon manufacture with little current synthetic value, is the most attractive reagent for difluoromethylation reactions. The continuous flow process allows this reaction to be performed within reaction times of 20 min with 2 equiv. of base and 3 equiv. of fluoroform. Importantly, the protocol allows the direct Cα-difluoromethylation of protected α-amino acids. These compounds are highly selective and potent inhibitors of pyridoxal phosphate-dependent decarboxylases. The starting materials are conveniently derived from the commercially available α-amino acid methyl esters, and the final products are obtained in excellent purities and yields after simple hydrolysis and precipitation. The developed process appears to be especially appealing for industrial applications, where atom economy, sustainability, reagent cost and reagent availability are important factors.Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) Fluorine-containing Amino Acids, Synthesis and Properties, ed. V. P. Kukhar and V. A. Soloshonok, Wiley & Sons, Chichester, 1995 Search PubMed; (b) Organofluorine Compounds in Medicinal Chemistry & Biomedical Applications, ed. R. Filler, Y. Kobayashi and L. M. Yagupolskii, Elsevier, Amsterdam, 1993 Search PubMed.
- For recent reviews, see: (a) C. Ni and J. Hu, Synthesis, 2014, 842–863 Search PubMed; (b) D. E. Yerien, S. Barata-Vallejo and A. Postigo, Chem. – Eur. J., 2017, 23, 14676–14701 CrossRef CAS PubMed.
- P. Forster, V. Ramaswamy, P. Artaxo, T. Berntsen, R. Betts, D. W. Fahey, J. Haywood, J. Lean, D. C. Lowe, G. Myhre, J. Nganga, R. Prinn, G. Raga, M. Schulz and R. Van Dorland, in Changes in atmospheric constituents and in radiative forcing. In Climate change 2007: The Physical Science Base, Fourth Assessment Report of the Intergovernmental Panel on Climate Change, ed. S. Soloman, D. Qin, M. Manning, Z. Chen, M. Marquis, K. B. Averyt, M. Tignor and H. L. Miller, Cambridge University Press, Cambridge, 2007 Search PubMed.
- For selected publications, see: (a) T. Shono, M. Ishifune, T. Okada and S. Kashimura, J. Org. Chem., 1991, 56, 2–4 CrossRef CAS; (b) B. Folléas, I. Marek, J.-F. Normant and L. Saint-Jalmes, Tetrahedron, 2000, 56, 275–283 CrossRef; (c) J. Russell and N. Roques, Tetrahedron, 1998, 54, 13771–13782 CrossRef CAS; (d) A. Zanardi, M. A. Novikov, E. Martin, J. Benet-Buchholz and V. V. Grushin, J. Am. Chem. Soc., 2011, 133, 20901–20913 CrossRef CAS PubMed; (e) P. Novák, A. Lishchynskyi and V. V. Grushin, J. Am. Chem. Soc., 2012, 134, 16167–16170 CrossRef PubMed; (f) G. K. S. Prakash, P. V. Jog, P. T. D. Batamack and G. A. Olah, Science, 2012, 338, 1324–1327 CrossRef CAS PubMed.
- T. Iida, R. Hashimoto, K. Aikawa, S. Ito and K. Mikami, Angew. Chem., Int. Ed., 2012, 51, 9535–9538 CrossRef CAS PubMed.
- K. Aikawa, K. Maruyama, K. Honda and K. Mikami, Org. Lett., 2015, 17, 4882–4885 CrossRef CAS PubMed.
- K. Aikawa, K. Maruyama, J. Nitta, R. Hashimoto and K. Mikami, Org. Lett., 2016, 18, 3354–3357 CrossRef CAS PubMed.
- S. Okusu, E. Tokunaga and N. Shibata, Org. Lett., 2015, 17, 3802–3805 CrossRef CAS PubMed.
- (a) C. S. Thomoson and W. R. Dolbier, J. Org. Chem., 2013, 78, 8904–8908 CrossRef CAS PubMed; (b) C. S. Thomoson, L. Wang and W. R. Dolbier, J. Fluorine Chem., 2014, 168, 34–39 CrossRef CAS.
- For an alternative mechanistic hypothesis suggested by Mikami and co-workers, see ref. 5.
- For a review on the synthesis of Cα-difluoromethyl amino acids, see: (a) X.-L. Qiu, W.-D. Meng and F.-L. Qing, Tetrahedron, 2004, 60, 6711–6745 CrossRef CAS; (b) R. Smits, C. D. Cadicamo, K. Burger and B. Koksch, Chem. Soc. Rev., 2008, 37, 1727–1739 RSC.
- B. Gutmann, P. Hanselmann, M. Bersier, D. Roberge and C. O. Kappe, J. Flow Chem., 2017, 7, 46–51 CrossRef.
- (a) P. Bey, F. Gerhart, V. V. Dorsselaer and C. Danzin, J. Med. Chem., 1983, 26, 1551–1556 CrossRef CAS PubMed; (b) T. Tsushima, K. Kawada, S. Iahihara, N. Uchida, O. Shiratori, J. Higaki and M. Hirata, Tetrahedron, 1988, 44, 5375–5387 CrossRef CAS; (c) D. Schirlin, F. Gerhart, J. M. Hornsperger, M. Hamon, J. Wagner and M. J. Jungs, J. Med. Chem., 1988, 31, 30–36 CrossRef CAS PubMed; (d) S. N. Osipov, A. S. Golubev, N. Sewald and K. Burger, Tetrahedron Lett., 1979, 38, 5965–5966 CrossRef; (e) T. Tsushima and K. Kawada, Tetrahedron Lett., 1985, 26, 2445–2448 CrossRef CAS; (f) P. Bey and D. Schirlin, Tetrahedron Lett., 1978, 52, 5225–5228 CrossRef; (g) S. N. Osipov, A. S. Golubev, N. Sewald, T. Michel, A. F. Kolomiets, A. V. Fokin and K. Burger, J. Org. Chem., 1996, 61, 7521–7528 CrossRef CAS PubMed; (h) I. I. Gerus, A. A. Kolomeitsev, M. I. Kolycheva and V. P. Kukhar, J. Fluorine Chem., 2000, 105, 31–33 CrossRef CAS.
- (a) P. Bey and J. P. Vevert, Tetrahedron Lett., 1978, 14, 1215–1218 CrossRef; (b) P. Bey, J. B. Ducep and D. Schirlin, Tetrahedron Lett., 1984, 25, 5657–5660 CrossRef CAS; (c) P. Bey, J.-P. Vevert, V. V. Dorsselaer and M. Kolb, J. Org. Chem., 1979, 44, 2732–2742 CrossRef CAS; (d) M. Seki, M. Suzuki and K. Matsumoto, Biosci., Biotechnol., Biochem., 1993, 57, 1024–1025 CrossRef CAS; (e) P. Bey and M. Jung (Merrell Toraude et Compagnie), Method of treating benign prostatic hypertrophy, US4330559, 1982 Search PubMed; (f) P. Bey and M. Jung (Merrell Toraude et Compagnie), 2-(Difluoromethyl)-2,5-diaminopentanoic acid, US4413141, 1983 Search PubMed.
- D. Dallinger and C. O. Kappe, Curr. Opin. Green Sust. Chem., 2017, 7, 6–12 CrossRef and references cited therein.
- (a) B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed; (b) M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC; (c) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- (a) H. P. Gemoets, Y. Su, M. Shang, V. Hessel, R. Luque and T. Noël, Chem. Soc. Rev., 2016, 45, 83–117 RSC; (b) C. J. Mallia and I. R. Baxendale, Org. Process Res. Dev., 2016, 20, 327–360 CrossRef CAS.
- For a recent protocol for introducing CF2 using photochemistry in a continuous flow, see: X.-J. Wei, W. Boon, V. Hessel and T. Noël, ACS Catal., 2017, 7, 7136–7140 CrossRef CAS PubMed.
- Methods for continuous flow gas–liquid separation are well developed. For previous reports on continuous inline gas removal, see: (a) P. Koos, U. Gross, A. Polyzos, M. O'Brien, I. Baxendale and S. V. Ley, Org. Biomol. Chem., 2011, 9, 6903–6908 RSC; (b) M. O'Brien, N. Taylor, A. Polyzos, I. R. Baxendale and S. V. Ley, Chem. Sci., 2011, 2, 1250–1257 RSC; (c) K. Skowerski, S. J. Czarnocki and P. Knapkiewicz, ChemSusChem, 2014, 7, 536–542 CrossRef CAS PubMed; (d) M. J. Fink, M. Schön, F. Rudroff, M. Schnurch and M. D. Mihovilovic, ChemCatChem, 2013, 5, 724–727 CrossRef CAS; (e) D. Cantillo, B. Wolf, R. Goetz and C. O. Kappe, Org. Process Res. Dev., 2017, 21, 125–132 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, further optimization data and characterization of all compounds. See DOI: 10.1039/c7gc02913f |
| This journal is © The Royal Society of Chemistry 2018 |