
Towards sustainable manufacture of epichlorohydrin from glycerol using hydrotalcite-derived basic oxides†
Giacomo M.
Lari
,
Giorgio
Pastore
,
Cecilia
Mondelli
* and
Javier
Pérez-Ramírez
 *
*
Institute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, Vladimir-Prelog-Weg 1, 8093 Zurich, Switzerland. E-mail: cecilia.mondelli@chem.ethz.ch; jpr@chem.ethz.ch
First published on 16th October 2017
Abstract
Commercial two-step processes to convert glycerol into epichlorohydrin are more benign compared to the predominant industrial route starting from propene in terms of materials requirements and CO2 emissions. Still, the use of alkali hydroxides in stoichiometric amounts in the second reaction, i.e., the dehydrochlorination of the dichloropropanol intermediate, leads to the formation of large amounts of salt wastes, thus limiting the greenness of the technology. Here, we show for the first time that the latter transformation can be selectively conducted in the gas phase in the presence of a heterogeneous hydrotalcite-derived mixed oxide of Al and Mg. Upon reaction, the lamellar solid is rehydrated to a hydrotalcite-like compound, which can effectively activate the alcoholic group of dichloropropanol owing to its strong Brønsted basic character and moderately high surface area. In-depth characterisation of the porous, compositional, structural and acid/base properties demonstrates that the HCl formed during the reaction causes the progressive exchange of interlayer OH groups by Cl atoms, thus gradually diminishing the reactivity of the material. Facile calcination restores the original mixed oxide structure and is shown to enable three equivalent consecutive reaction runs. Since the HCl evolved along with water upon regeneration can be recycled in the first step of the process, i.e., glycerol hydrochlorination, our approach paves the way for a waste-free and more atom efficient biobased epichlorohydrin production process.
Introduction
The upgrading of vegetable oils, which substantially comprises their transesterification to produce biodiesel, has a low profitability compared to the conventional refining of petroleum to fuels and base chemicals.1 This is not merely due to the small difference between the price of the product and the cost of the feedstock, but relates to the fact that glycerol, the main by-product of the transesterification process, is obtained in greater quantities than the cosmetic, food and pharmaceutical industries are able to absorb and must, largely, be disposed of as waste.2 Thus, the economic and environmental footprint of a biodiesel plant would benefit from the establishment of an associated glycerol biorefinery, i.e. a facility where the triol is valorised into higher-value chemicals.3 Many conversion paths have been proposed to obtain compounds retaining the C3 backbone, such as lactic acid and lactates, diols, carbonates, ethers, esters and epichlorohydrin.4 The latter, which is conventionally produced starting from propene,5 has a world market of ca. 2 Mt per year and a very broad application range. Before the biodiesel production introduced burgeoning amounts of cheap glycerol in the market, this molecule was the key intermediate in the most relevant industrial route to the triol.6 Nowadays, epichlorohydrin is mainly used as a (co–)monomer in the preparation of epoxy resins and as an intermediate in the manufacture of other polymers (e.g., polyesters), explosives and commodities.7The processes proposed8 for the synthesis of this epoxide from glycerol are based on two reaction steps (Scheme 1) and some of them have been industrially applied up to a scale of 100 kt per year, as, for example, the Solvay's Epicerol process.9 Notably, this company highlighted the milder ecologic impact of their technology compared to the fossil fuel-based route in view of the inferior requirement for materials and CO2 emissions.8 The first transformation comprises the chlorination of liquid glycerol with HCl gas, which is bubbled through the reactor, in the presence of a catalyst, typically an organic carboxylic acid.10 After reaction, the mixture mostly contains various isomers of monochlorinated and dichlorinated species. Upon separation, the first class of molecules is recycled back to the hydrochlorination reactor along with unconverted glycerol, while the second is fed to a second reactor, typically a reactive-distillation unit, where an inorganic base is added to deprotonate the residual hydroxyl group. This induces ring closure, which forms the epoxide and is associated with the elimination of HCl. The catalyst applied in the first step can be recovered easily by distillation or filtration,9,10 depending on its physical state. In contrast, the conventional stoichiometric bases, like aqueous solutions of NaOH, KOH or Ca(OH)2,9 used in the second reaction lead to the formation of salt by-products. Furthermore, based on the nature of the alkaline compound, the salt can be soluble in the product feed, hence increasing the difficulty and the cost of its separation11 as well as posing serious corrosion issues to the units downstream of the reactor.12 Accordingly, to attain a greener glycerol-based production of epichlorohydrin further developments are required to meet to principles of waste reduction, atom economy and use of regenerable chemicals. Highly instrumental to this purpose would be the introduction of a heterogeneous reactant.13
 | ||
| Scheme 1 Preparation of epichlorohydrin from glycerol by hydrochlorination to dichloropropanols followed by hydroxide-mediated epoxide formation and undesired thermally-induced isomerisation of epichlorohydrin to chloroacetone. | ||
Based on the mechanism and the nature of the homogeneous alkaline compounds, solid bases such as alkali oxides,14 hydrotalcites15 or basic zeolites16 are expected to be most suited for the reaction. Nevertheless, it cannot be excluded that this transformation can proceed through different pathways, in analogy to the dehydrochlorination-oligomerisation of alkyl chlorides, which is mediated by acid sites.17 While a catalytic material is preferable, the identification of a suitable system appears as a challenging endeavor. Indeed, hydrochloric acid is generated, which, obviously, interacts with basic sites in a strong manner,18 likely leading to activity loss by poisoning. Focusing on a solid reactant, reusability would be a highly relevant parameter. This property has been demonstrated for basic metal oxides (CaO, MgO) applied to the dehydrohalogenation of alkyl halides, which were transformed into metal halides during the reaction but could be regenerated by calcination at 823 K.19
In this work, mixed oxides of magnesium and aluminum obtained by thermal activation of hydrotalcite-like compounds (HTlcs) are introduced as efficient solids for the gas-phase conversion of dichloropropanols to epichlorohydrin. The superiority of these basic solids is identified by evaluation of acid and basic compounds with variable strength. The selectivity towards epichlorohydrin is maximised by tuning the temperature as well as the concentration and strength of the basic sites through variation of the Mg/Al ratio and the activation conditions of the HTlc, which offers insights into structure-mechanism-activity relations. Since the testing of the best performer over prolonged reaction times evidences pronounced decrease in activity, the origins of this decay are unraveled and a facile strategy to restore the original level is demonstrated. Lastly, qualitative conclusions are drawn on the environmental and economic advantages of using a heterogeneous rather than a homogeneous base in a glycerol-to-epichlorohydrin process.
Experimental
Materials
γ-Al2O3 (Alfa Aesar, 99.997% metals basis), MgO (Strem Chemicals, 99.5%), a USY zeolite in protonic form with a bulk Si/Al ratio of 405 (Zeolyst, USY) and a Y zeolite in sodium form with a bulk Si/Al ratio of 2.6 (Zeolyst, CBV100) were used as received.Two Mg- and Al-based HTlcs with a nominal Mg/Al ratio of x = 2 and 4 (coded as HTx) were prepared by co-precipitation at pH 10. 500 cm3 of a x·0.25 M and 0.25 M aqueous solution of Mg(NO3)3·6H2O (Riedel-de Haen, >99%) and Al(NO3)3·9H2O (Sigma-Aldrich, >98%), respectively, were added dropwise to 600 cm3 of 2 M aqueous Na2CO3 (Sigma-Aldrich, >99.5%) at a rate of 3.5 cm3 min−1 while magnetically stirring (500 rpm) at ambient temperature. A 40 wt% NaOH (Sigma-Aldrich, 97%) solution was simultaneously added dropwise to keep the pH at 10. The obtained slurry was aged at 333 K for 6 h under stirring. After filtering and washing (3 times, 200 cm3 of deionised water each time), the precipitate was dried at 333 K for 18 h. Mixed metal oxides were obtained by calcination of the HTlcs at 673–973 K (ramp rate = 5 K min−1) for 5 h. These samples were labelled as HTx-cy, where y is the calcination temperature expressed in K.
An alkaline-activated USY zeolite with basic character (USY-AT) was synthesised by adding 3.3 g of a high-silica USY zeolite (Si/Al = 405, Zeolyst) to 100 cm3 of a 0.1 M methanolic (Sigma-Aldrich, 99.8%) solution of NaOH. After stirring for 10 min at room temperature, the solid was filtered and washed with methanol (3 times, 100 cm3 each time).20
An alkali-activated Y zeolite (Y-AT) was prepared in two steps. Firstly, a zeolite Y (Zeolyst, CBV100) was dealuminated at 373 K in a 0.15 M solution of H4EDTA (Fluka, >99%, 15 cm3 per gram of zeolite) for 24 h, recovered by filtration, washed with deionised water (3 times, 30 cm3 per gram of zeolite) and dried at 338 K for 16 h. Thereafter, the solid obtained was treated in 0.05–0.2 M aqueous NaOH (30 cm3 per gram of zeolite) at 338 K for 30 min, using an Easymax 102 setup (Mettler Toledo). After quenching in an ice bath and filtering, the zeolite was washed with deionised water (3 times, 30 cm3 per gram of zeolite) and dried at 338 K for 16 h.21
MgO/USY was prepared by dry impregnation. 4 cm3 of a 0.25 M aqueous solution of Mg(NO3)2·6H2O (Fluka, 99%) were added dropwise to 4 g of the USY zeolite at room temperature. The sample was dried at 338 K overnight and calcined at 823 K (5 K min−1) for 5 h.18
Characterisation
The elemental composition of the materials was determined by X-ray fluorescence spectroscopy (XRF) using an Orbis Micro XRF instrument equipped with a Rh source operated at 35 kV and 500 mA. N2 sorption at 77 K was conducted using a Micromeritics TriStar analyser. Prior to the measurements, the solids were degassed at 573 K under vacuum for 3 h. Powder X-ray diffraction (XRD) was performed using a PANalytical X'Pert PRO-MPD diffractometer with Ni-filtered Cu Kα radiation (λ = 0.1541 nm), acquiring data in the 10–60° 2θ range with an angular step size of 0.05° and a counting time of 2 s per step. Al magic angle spinning nuclear magnetic resonance (27Al MAS NMR) spectroscopy was conducted in a Bruker Avance 700 spectrometer operated at 182.4 MHz using 4 mm ZrO2 rotors spun at 10 kHz. Spectra were acquired accumulating 512 scans using a pulse length of 1 ms, a recycle delay of 1 s and solid (NH4)Al(SO4)2 as a reference (δ = 0.00 ppm). Temperature-programmed desorption of carbon dioxide (CO2-TPD) and temperature programmed oxidation with O2 (TPO) were carried out using a Micromeritics Autochem II chemisorption analyser coupled with a MKS Cirrus 2 quadrupole mass spectrometer. For the first measurements, the samples (0.05 g) were treated in a He flow (20 cm3 min−1) at 373 K for 2 h. Afterwards, CO2 (50 pulses, 1 cm3 each pulse) carried by He (10 cm3 min−1) was adsorbed at 323 K, followed by He purging at the same temperature for 1 h. CO2 desorption was performed using a He flow of 10 cm3 min−1 and monitored in the 323–973 K range (10 K min−1). The concentration of basic sites (CB), expressed as mmol of CO2 per gram, was obtained by multiplying the area of the desorption curve by a calibration factor obtained from the decomposition of known amounts of Na2CO3 (Sigma-Aldrich, >99.5%) and dividing it by the mass of the sample. Upon TPO, the samples (0.05 g) were treated in a He flow (20 cm3 min−1) at 373 K for 2 h. Thereafter, the temperature was lowered to 323 K and the flow composition and rate modified to 10 vol% O2 in He and 10 cm3 min−1, respectively. The sample was heated up to 973 K (10 K min−1) while monitoring the gases evolved. The chlorine and the carbon contents of the used solids were determined by elemental analysis using a LECO CHN-9000 instrument. Transmission electron micrographs (TEM) were acquired using a FEI Talos F200A instrument operated at 200 kV.Testing
The gas-phase dehydrochlorination (Aldrich-Fine Chemicals, 98%) was studied at ambient pressure using a continuous-flow fixed-bed reactor setup (Microactivity Reference, PID Eng&Tech) comprising: (i) a syringe pump for the admission of the 10 wt% aqueous solution of the reactant, (ii) a mass flow controller for feeding N2 (PanGas, 99.99%), (iii) a tubular quartz reactor (12 mm internal diameter) equipped with a frit to support the heterogeneous bed and a thermocouple to monitor its temperature and heated in an oven and (iv) a condenser kept at 273 K. 0.1–0.5 g of material (0.2–0.5 mm particle size, 3–12 mm bed thickness) were loaded into the reactor and the system was heated to the desired temperature (403–673 K) under a N2 flow of 100 cm3 min−1. Thereafter, an aqueous dichloropropanol solution (5–10 wt%) was admitted for 0.5–3 h. In order to verify the theoretical possibility to vaporise dichloropropanol under the reaction conditions, the physical state of the dichloropropanol–water mixture was calculated for solute concentrations ranging from 0 to 100 wt% and between 273–473 K based on the UNIFAC model and using the RGibbs function in Aspen Plus V8.6.Liquid samples were collected from the condenser every 15 min, which were analysed using an HP 6890 gas chromatograph equipped with an HP-5 capillary column and a flame ionisation detector. Quantification was achieved based on the absolute peak areas. Calibration curves were measured in the 0.5–10, 0.5–7 and 0.5–3 wt% ranges for 1,3-dichloro-2-propanol, epichlorohydrin and chloroacetone, respectively. The calibration response factor of unknown compounds was assumed to be the same as that of epichlorohydrin.
The conversion of the substrate was calculated as the number of moles of substrate reacted divided by number of moles of substrate fed and the selectivity towards the product i as the number of moles of product formed divided by the total number of moles of products, according to the following equations, where the subscripts 0/1 refer to the reactor inlet/outlet mixture:

| Si = ni,1/(ndichloropropanol,0 − ndichloropropanol,1). |
The carbon balance was calculated as the ratio between the number of moles of products and the number of moles of glycerol fed and was always higher than 90%. The experimental error, determined by three repetitions of selected runs, was within 5%, which allowed excluding the occurrence of bypass phenomena in the experiments applying the thinner beds.
Results and discussion
Evaluation of solid acids and bases
Since the identification of a suitable solid for the dehydrochlorination of dichloropropanol to epichlorohydrin is here tackled for the first time, a diverse set of materials was selected for assessement. In analogy with the stoichiometric homogeneous reactants commonly used in the established liquid-phase processes, part of the compounds comprised solid bases of different nature and strength. Specifically, they included earth-alkali oxides (MgO), alkali-activated zeolites (USY-AT)20 and HTlc in as-synthesised form, i.e., layered double hydroxides with formula [Mg2+nAl3+m(OH)2(n+m)]m+[CO32+]m/2·H2O (here with n = 4 and m = 2, HT2), or in calcined state, i.e., Mg–Al mixed oxides (here calcined at 823 K, HT2-c823). These materials have a strong Lewis-, strong Brønsted-, weak Brønsted- and strong Lewis-basic character, respectively. γ-Al2O3 and a zeolite Y in its sodium form with a bulk nominal Si/Al ratio of 2.6 were chosen as representative strong and weak Lewis acids, in this order. Strong Brønsted acidity was featured in an ultra-stable Y zeolite in protonic form with a bulk nominal Si/Al ratio of 405 (H-USY). Finally, to assess the relevance of the co-presence of acidity and basicity, 1.1 wt% of MgO was supported on the H-USY zeolite (MgO/USY) and the zeolite Y was modified by an alkaline treatment in 0.15 M NaOH (Y-AT). The porous and acid/base properties of these solids are in line with those reported in the literature for analogous materials (Table 1 and Table S1 in the ESI†).15,18,20,21 All of the systems were tested in a continuous-flow fixed-bed reactor kept at 473 K by feeding an aqueous dichloropropanol solution (5 wt%) at a weight hourly space velocity (WHSV) of 6 h−1 along with an inert carrier for 0.5 h (Fig. 1 and Table S2†). Solids possessing Brønsted and Lewis acidity generally displayed very low activity (<1% dichloropropanol conversion) and selectivity towards epichlorohydrin (<15%). This behaviour was rationalised by the capability of acid sites to catalyse dehydration and oligomerisation reactions, leading to fouling,22 which was evidenced by the black colour assumed by the white powders upon the test and confirmed by CHN analysis (up to 8 wt% C, Table S1†). In addition to the generation of carbonaceous species, chloroacetone was formed as a by-product with a selectivity up to 25%. The poor performance of solid acids improved through the incorporation of basic sites. In fact, the dichloropropanol conversion increased from 0.5 and 1.0% to 2.2 and 3.8% over Y-AT and MgO/USY, respectively, and the epichlorohydrin selectivity raised to ca. 50% over both solids. However, coke was still produced in a significant amount in these two cases (4–5 wt% C), due to the marked acidity retained by the zeolites upon their modification. Superior epichlorohydrin yields were attained over purely basic solids, except for USY-AT. Indeed, MgO, the HTlc and the mixed Mg–Al oxide displayed a conversion of 5, 6 and 8% and an epichlorohydrin selectivity of ca. 30, 70 and 80%, respectively. Notably, coke formation was limited over these materials (<3 wt% C).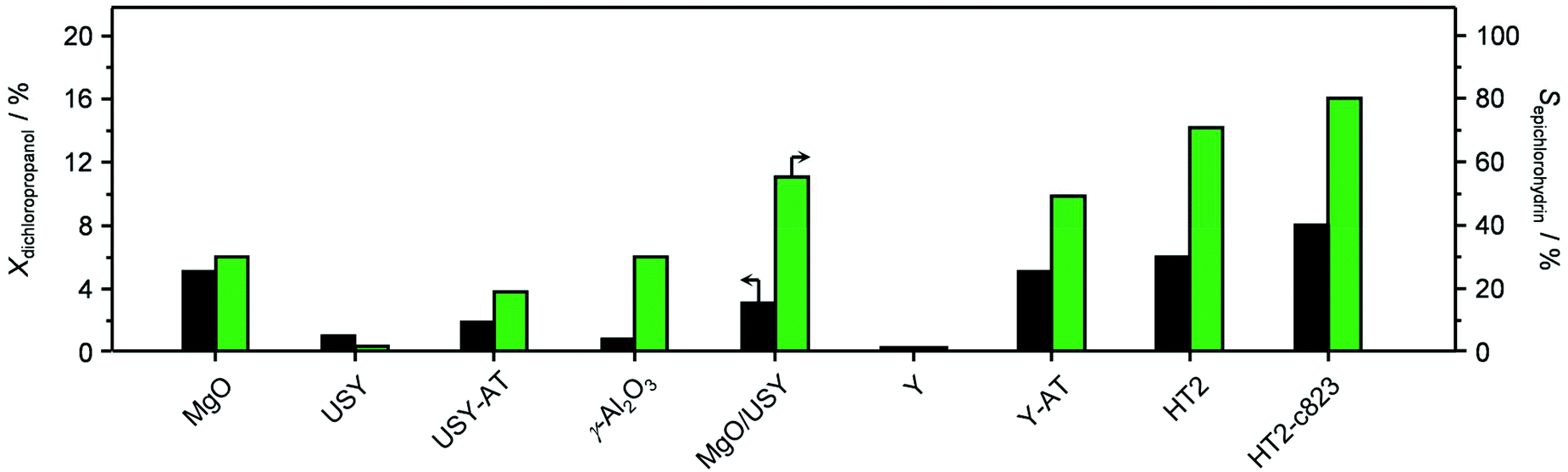 | ||
| Fig. 1 Dichloropropanol conversion and epichlorohydrin selectivity in the presence of different materials. T = 473 K, P = 1 bar, t = 0.5 h and WHSV = 6 h−1. | ||
| Sample | Molar Mg/Ala (—) | Crystalline phase(s)b |
V
pore![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (cm3 g−1) c (cm3 g−1) |
S
BETd (m2 g−1) |
C
Be (μmol g−1) |
T
desf (K) |
T
decf (K) |
Cg (wt%) |
|---|---|---|---|---|---|---|---|---|
| a XRF. b XRD. HT = hydrotalcite, MMO = mixed metal oxide. c Volume of N2 adsorbed at p/p0 = 0.98. d Total surface area, BET method. e Concentration of basic sites, CO2-TPD. f Temperature corresponding to the maximum of the desorption (des) and decomposition (dec) peaks in the CO2-TPD profiles. g Carbon content of the used materials, elemental analysis. | ||||||||
| HT2 | 1.97 | HT | 0.47 | 81 | 85 | 404 | 623 | 0.9 |
| HT4 | 3.52 | HT | 0.42 | 65 | 89 | 395 | 595 | 0.4 |
| HT2-c823 | 1.82 | MMO | 0.58 | 197 | 600 | 532 | — | 1.0 |
| HT4-c673 | 3.31 | HT + MMO | 0.45 | 148 | 412 | 473 | 650 | 0.3 |
| HT4-c823 | 3.50 | MMO | 0.60 | 223 | 482 | 573 | — | 1.0 |
| HT4-c973 | 3.52 | MMO | 0.58 | 203 | 355 | 536 | — | 1.1 |
Design of Mg–Al mixed oxides
Based on its remarkable performance, the Mg–Al oxide was selected for further development. Since such compound was obtained from a hydrotalcite-like precursor, it was conceived to vary its properties by altering the composition of the starting double layered hydroxide. Thus, an additional sample was prepared with a Mg/Al ratio of 4 (HT4). A ratio lower than 2 was excluded, since for this value it is not feasible to attain a solid with pure hydrotalcite structure, while a ratio higher than 4 was irrelevant, since it leads to materials with limited ion-exchange properties.23 The actual metal ratio in the as-synthesised samples was close to the nominal value and, in all cases, the surface area was comprised between 70–80 m2 g−1, in agreement with previous literature (Table 1). The XRD patterns of the solids show the typical reflections of a rhombohedral (3R) layered double hydroxide structure (Fig. 2a), which are shifted to lower 2θ values due to the expansion of the unit cell size caused by the higher Mg content.24 TEM confirmed the platelet-like structure of the crystals (Fig. 3a), which exhibit extensive intergrowth and are oriented in a random fashion. All of the 27Al MAS NMR spectra display a signal centred at 10 ppm, indicating the octahedral (Oh) symmetry of the Al centres (Fig. 2b).15a The basicity of the HTlcs was probed by CO2-TPD (Fig. 2c). For all samples, the curves evidence a low-intensity peak with maximum at ca. 400 K, indicating the presence of few (<80 mmol g−1) weak basic sites, which is accompanied by a second, very pronounced signal peaking between 600–700 K, which was produced by the decomposition of interlayer carbonates. The area of this second contribution is larger for HT2 than for HT4, in line with the greater Al substitution and, thus, capacity of accommodating interlayer anions. After calcination at 823 K, the two samples possessed the same Mg/Al ratios as the corresponding starting HTlcs (Table 1) and ca. 2.5–3.5 times higher surface area (150–220 m2 g−1). Their diffractograms almost exclusively comprise two relatively broad reflections at 43 and 62°2θ (Fig. 2d), which indicate the transformation of the double layered hydroxide into a mixed oxide retaining the layered structure (Scheme 2).15 Platelet-like crystals were visualised by TEM (Fig. 3b), which also highlighted an increased disorder, i.e., an augmentation of intercrystalline mesoporosity. The latter is in line with the higher surface area (Table 1). 27Al MAS NMR spectroscopy revealed that about one third of the Al atoms changed their environment upon calcination attaining a tetrahedral geometry (Td) (Fig. 2e), which is generally quite distorted considering the broad shape of the resonance at 80 ppm, and that, based on the appearance of a peak at 18 ppm, about half of the remaining octahedral sites became highly distorted (C3v). The density of basic centres of the calcined samples underwent a ca. 8-fold increase (Fig. 2f and Table 1) and was maximal for HT2-c823. Still, although preserving sites of mild and moderate strength, HT4-c823 possessed the strongest centres (Tdes > 773 K).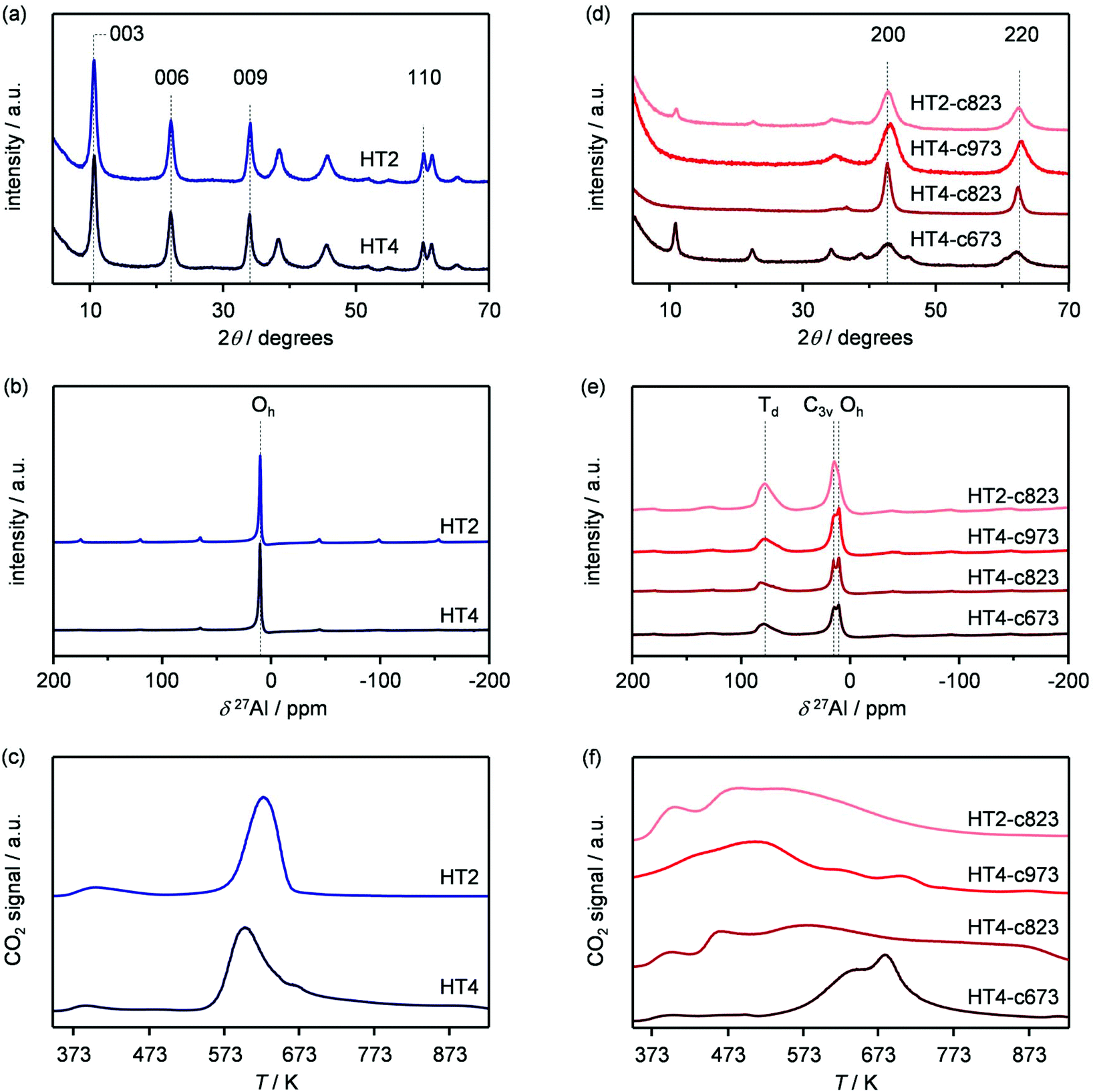 | ||
| Fig. 2 (a, d) XRD, (b, e) 27Al MAS NMR and (c, f) CO2-TPD profiles of the (a–c) as-synthesised HTx and (d–f) calcined HTx-cy materials. JCPDS 22-700 and 87-0653 comprise the reference diffractograms used for the assignment of the HTlc and the mixed metal oxide phases, respectively. | ||
 | ||
| Fig. 3 TEM images of the (a) HT4 precursor and of the calcined HT4-c823 material in its (b) fresh, (c) used and (d) regenerated forms. | ||
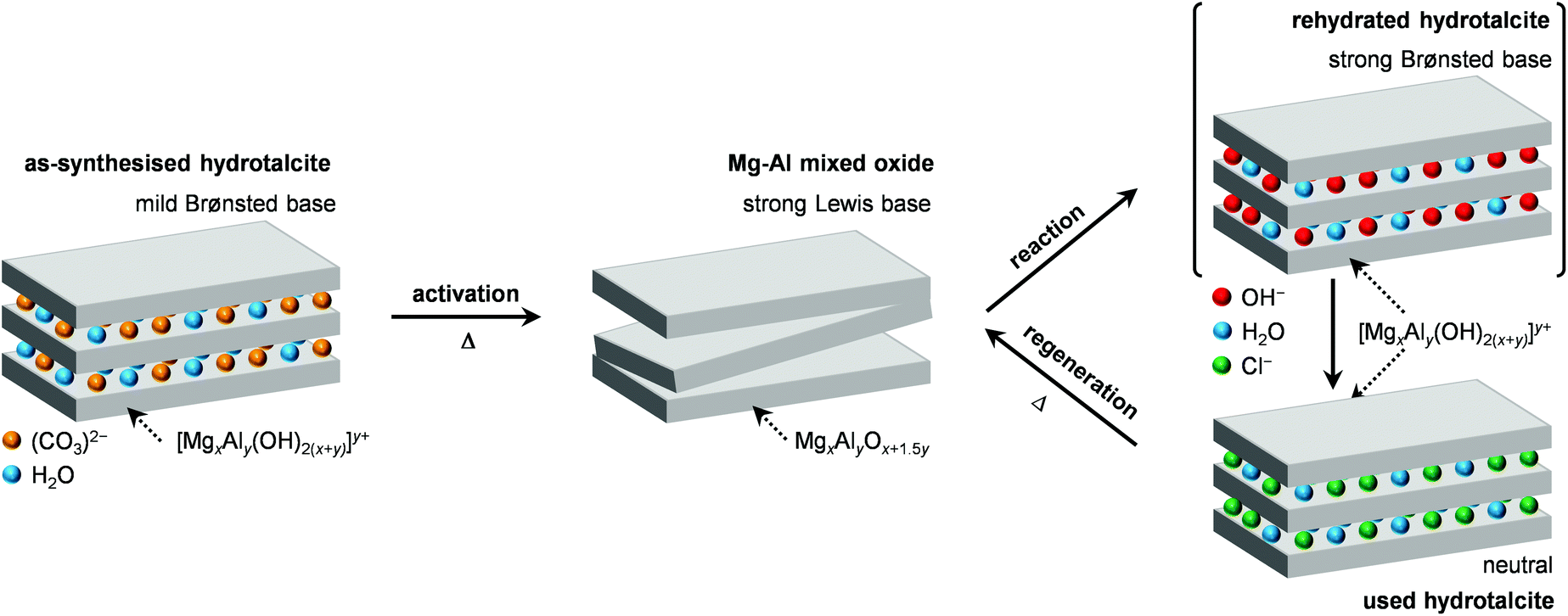 | ||
| Scheme 2 Generation of the Mg–Al mixed oxide by calcination of a HTlc, its modification upon use in the dichloropropanol-to-epichlorohydrin reaction and its subsequent regeneration by calcination. | ||
To modify the characteristics of the mixed oxide, we also considered the thermal treatment of the starting HTlcs, tuning its temperature. Thus, HT4 was additionally calcined at 673 and 973 K. The use of milder conditions led to an only partial decomposition of the hydrotalcite to the mixed oxide, as demonstrated by the surface area, which was intermediate between that of the precursor and that of the mixed oxide obtained upon calcination at 823 K, by the detection of unconverted precursor by XRD (Fig. 2d) and by the coexistence of octahedral and distorted octa- and tetrahedral species according to 27Al MAS NMR spectroscopy (Fig. 2e). After calcination at 973 K, only a mixed oxide phase was evidenced in the diffractogram, which featured sharper peaks than those for the other two samples, suggesting a moderate increase of the crystallite size due to sintering at high temperatures.
The two new sets of samples were tested in the dehydrochlorination of dichloropropanol under the same conditions applied in the initial evaluation of materials except for the temperature, which was decreased to 423 K with the purpose of boosting the selectivity towards epichlorohydrin. Since the boiling point of dichloropropanol is 450 K, a phase diagram for the dichloropropanol–water mixture was calculated to verify that the reactant would be present as a vapour at this temperature and at 403 K, which is the mildest condition applied in the testing in this study. For the concentration employed, the plot (Fig. S1 in the ESI†) suggests that this is fulfilled, since both mixture components are in the gas phase above 373 K. With respect to the HTlcs, HT4 showed an only slightly lower conversion and epichlorohydrin selectivity than the newly evaluated HT2 (5 versus 6% and 70 versus 75%, respectively, Fig. 4a and Table S2†). A noticeable boost in activity and selectivity was observed over all calcined solids, which were maximised over HT4-c823 (20% dichloropropanol conversion, 90% epichlorohydrin selectivity). It should be noted that these two parameters are linked, i.e., a higher selectivity is achieved at a higher conversion level. This was explained by the fact that the concentration of chloroacetone, the main by-product, was equal in all tests, suggesting that its rate of formation is not influenced by the nature of the solid and is likely generated through a thermally-driven pathway. By analogy with the decomposition of other epoxides,25 it was hypothesised that this by-product arises from the thermal isomerisation of epichlorohydrin. Experiments conducted heating the latter at 673 K confirmed the formation of the ketonic product with a yield of 78%. Minor quantities of coke were found on all the solids after reaction (Table 1). Since no dependence of these amounts on acid/base or textural properties was observed, thermal mechanisms are likely responsible for its formation. Comparison of the dichloropropanol conversion over the various mixed oxides with the type of their basic sites did not unveil any correlation, while their surface area appeared as a better descriptor for activity (Fig. 4b), hinting at the active-site accessibility, rather than its nature, as the most relevant parameter.
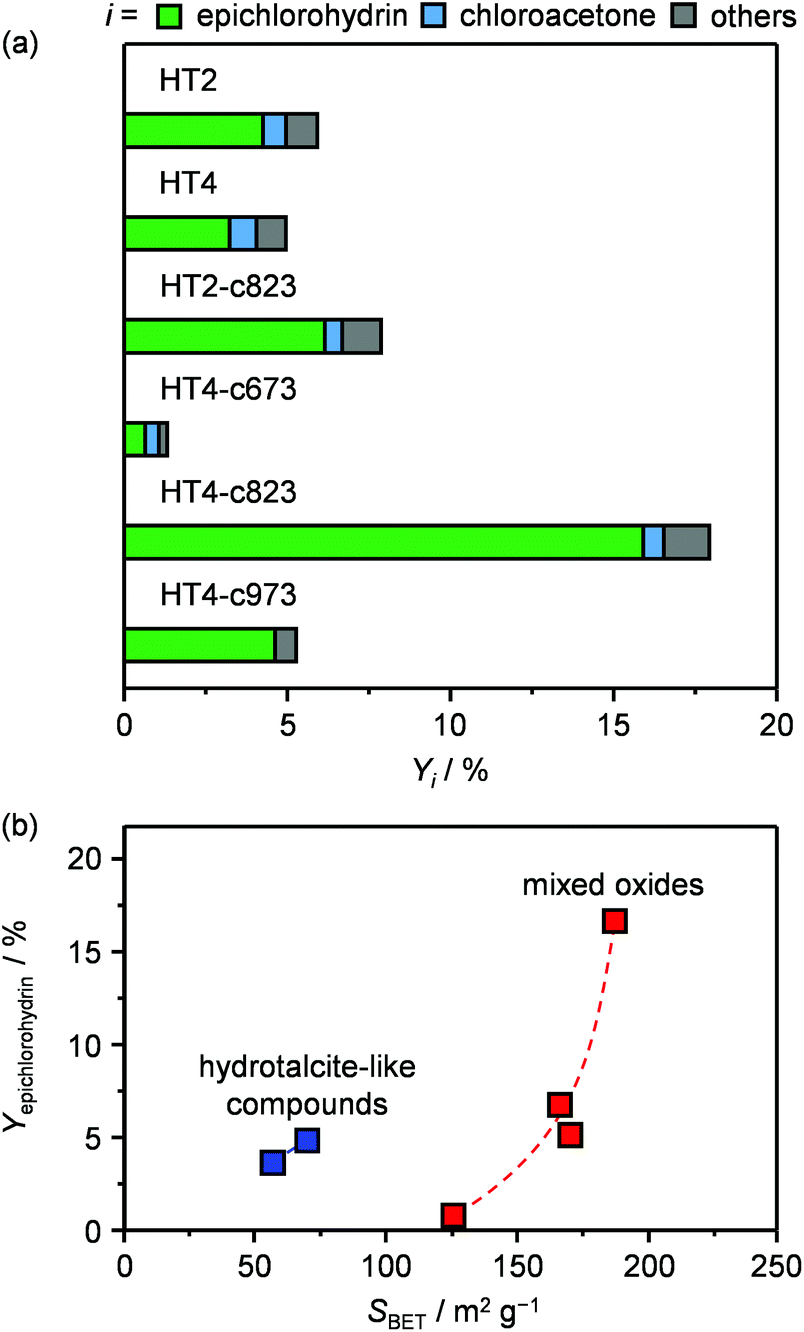 | ||
| Fig. 4 (a) Yield of epichlorohydrin, chloroacetone and other by-products upon conversion of dichloropropanol in the presence of HTx and HTx-cy materials. (b) Epichlorohydrin yield as a function of their specific surface area. T = 423 K, P = 1 bar, t = 0.5 h and WHSV = 6 h−1. | ||
Optimisation of the reaction conditions
In view of the superior epichlorohydrin yield obtained in the presence of HT4-c823, the influence of the reaction parameters on the performance was investigated using this sample. In the 403–673 K range, the dichloropropanol conversion increased monotonously with the temperature (Fig. 5a). The only exception was observed at 403 K, since an unexpectedly high conversion was observed. This was explained by the occurrence of capillary condensation in the micropores (vide infra) of the material and to the consequently altered diffusion and reaction kinetics. In contrast, the selectivity towards the desired product, epichlorohydrin, was comprised between 80 and 100% below 423 K and rapidly decreased between 423–573 K, approaching 0% at the highest temperature. In line with these results, the reactant was thermally decomposed in blank tests in an amount that corresponds to only 2 and up to 20% in the 403–423 K and 423–673 K ranges, respectively (Fig. S2†). The selectivity trend for chloroacetone was exactly opposite to that of epichlorohydrin, i.e., the formation of the ketonic compound was negligible at low temperatures and almost exclusive at high temperatures. These results indicate that a temperature of 423 K, as applied in the evaluation of Mg–Al-based solids, was optimal to maximise the selectivity towards the desired product.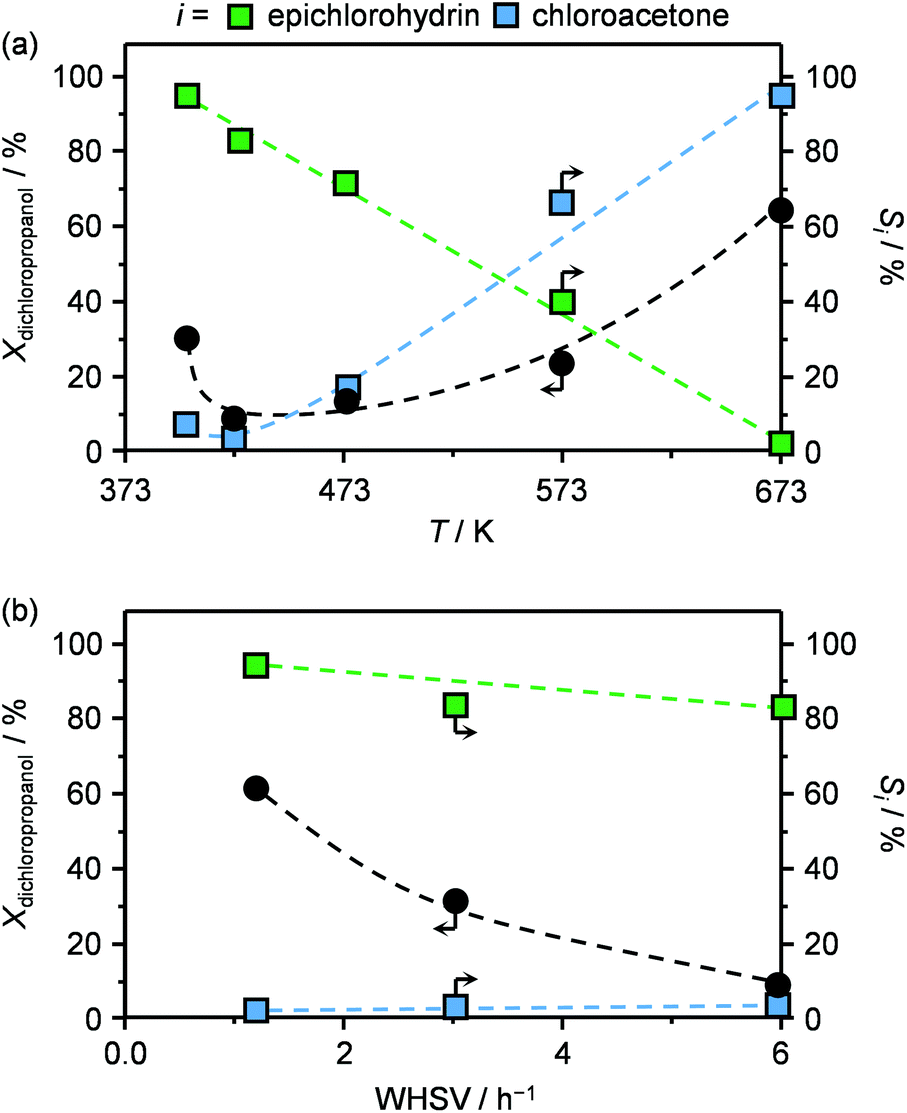 | ||
| Fig. 5 Dichloropropanol conversion and selectivity towards epichlorohydrin and chloroacetone in the presence of HT4-c823 as a function of (a) temperature and (b) weight hourly space velocity. T = 423 K, P = 1 bar, t = 0.5 h and WHSV = 6 h−1. | ||
An increase of the space velocity through the application of a lower mass of solid base led to a pronounced decrease in the conversion and a slight drop of the selectivity towards epichlorohydrin (Fig. 5b). The latter observation is in agreement with the fact that, since the residence time in the heated section of the reactor was unaltered, the thermally-driven generation of by-products was not affected.
The best performing material, HT4-c823, was tested over a prolonged reaction time under the best conditions (423 K and 1.2 h−1). The initially high (ca. 60%) dichloropropanol conversion was found to rapidly decrease to 15% during the first hour (Fig. 6a). Similarly, the high epichlorohydrin selectivity observed in the first stage of the test diminished, in line with the fact that chloroacetone and coke were generated at a constant yield.
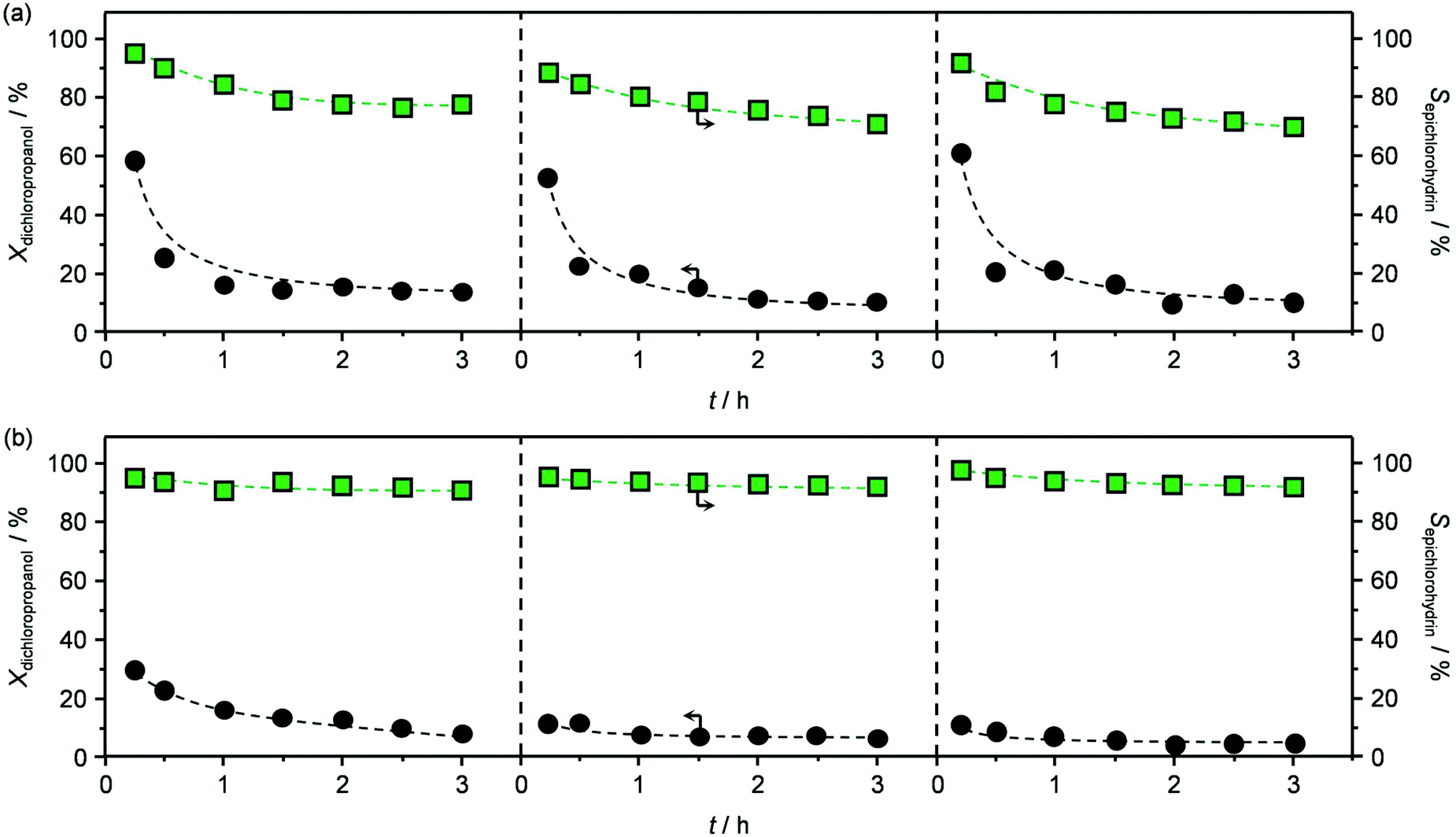 | ||
| Fig. 6 Dichloropropanol conversion and selectivity towards epichlorohydrin in three consecutive reaction cycles with intermediate regeneration of HT4-c823at (a) T = 423 K, P = 1 bar and WHSV = 1.2 h−1 and (b) T = 403 K, P = 1 bar and WHSV = 6 h−1. | ||
Characterisation of the used solid
In order to gain insights into the fate of the solid in the reaction, the used HT4-c823 was characterised thoroughly. Elemental analysis indicated a high chlorine content (10.7 wt%, Table 2), which was comparable to the amount effectively released upon formation of the epichlorohydrin and chloroacetone products, which confirms the exclusive participation of the mixed oxide as a solid reactant in the conversion of dichloropropanol. From a structural viewpoint, a portion of the material re-assumed the hydrotalcite structure that characterised the precursor (Fig. 7a). It is well known that upon contacting layered double hydroxide-derived oxides with liquid water or with its vapour at 303–423 K the material converts back into a HTlc featuring hydroxide instead of carbonate anions.26 Due to the smaller size of OH−, the interlayer spacing is lower than for the original material and the reflections in the XRD pattern appear at slightly higher 2θ. The presence of water in the reaction, the comparable conditions and the shift in peak position (ca. 1.0 2θ) in the diffractogram are consistent with the hydration of the mixed oxide. Still, in view of the fact that HT2-c823 acquires chlorine upon use, no metal chlorides are formed and the chlorine anion has a comparable size to OH−, Cl− species shall occupy interlayer positions. Actually, it is supposed that the mixed oxide is firstly converted into a rehydrated hydrotalcite, in which the OH− species serve as basic sites for the reaction with dichloropropanol and are progressively substituted by Cl− ions (Scheme 2). In analogy to these considerations, it is speculated that carbonate species might have been at least partly replaced by hydroxide species in HT2 and HT4 upon use and that their inferior conversion level compared to mixed oxides is likely due to the milder basicity of residual carbonates and their lower surface area (Fig. 4 and Table 1). In line with the structural changes, the surface area of HT4-c823 after reaction diminished to an intermediate value between those determined for HT4 and HT4-c823 in fresh forms (Tables 1 and 2). TEM analysis evidenced that the layered organisation of the solid was retained, although the morphology of the platelets became less defined (Fig. 3c). In agreement with the conversion of part of the aluminium sites from a tetrahedral or distorted octahedral into an octahedral geometry, the signal at 18 ppm in the 27Al MAS NMR spectrum exhibited a comparatively higher intensity (Fig. 7b). The CO2-TPD profile indicated a small amount of residual basic sites (Fig. 7c). This corroborates the predominance of neutral Cl− anion over basic OH− species in the interlayer spaces (Scheme 2) and hints that the residual mixed oxide phase is located in the interior of the particles. The difficulty of the reactant to reach the core of the material explains the gradual loss in activity. Indeed, at the beginning of the reaction, dichloropropanol can easily access the basic sites present at the external surface, while, at later stages, it has to diffuse into the pores to find unreacted sites. If the reactant freely diffused in the solid, the conversion level would stay at 100% and the reaction would last 1.15 h over HT2-c823 under the conditions applied.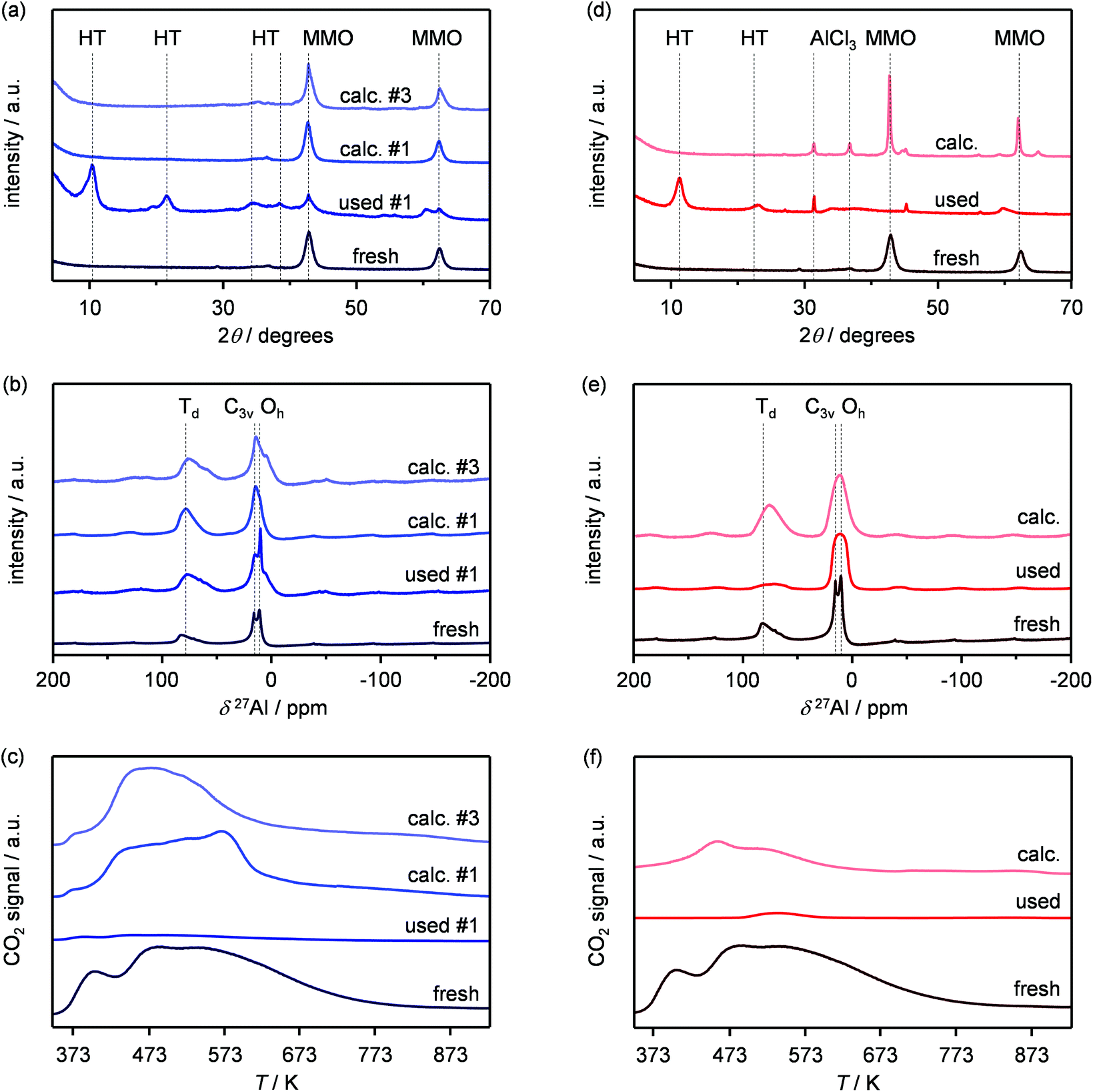 | ||
| Fig. 7 (a) XRD, (b) 27Al MAS NMR and (c) CO2-TPD profiles of the HT4-c823 material used at 423 K. (d) XRD, (e) 27Al MAS NMR and (f) CO2-TPD profiles of the HT4-c823 material used at 403 K. JCPDS 22-700, 87-0653 and 01-1133 comprise the reference diffractograms used for the assignment of the HTlc, the mixed metal oxide and AlCl3 phases, respectively. | ||
| HT4-c823 sample | Molar Mg/Ala (—) | Crystalline phase(s)b |
V
porec (cm3 g−1) |
S
BETd (m2 g−1) |
C
Be (μmol g−1) |
T
desf (K) |
Clg (wt%) | Cg (wt%) |
|---|---|---|---|---|---|---|---|---|
| a XRF. b XRD. HT = hydrotalcite, MMO = mixed metal oxide. c Volume of N2 adsorbed at p/p0 = 0.98. d Total surface area, BET method. e Concentration of basic sites, CO2-TPD. f Temperature corresponding to the maximum of the desorption (des) and decomposition (dec) peaks in the CO2-TPD profiles. g Carbon content of the used materials, elemental analysis. h T = 423 K, P = 1 bar and WHSV = 6 h−1. i T = 398 K, P = 1 bar and WHSV = 6 h−1. | ||||||||
| Fresh | 3.50 | MMO | 0.60 | 223 | 482 | 573 | — | — |
| Used #1h | 3.25 | HT + MMO | 0.47 | 125 | 6 | — | 10.7 | 0.9 |
| Calcined #1 | 3.41 | MMO | 0.51 | 205 | 425 | 540 | 0.9 | 0.0 |
| Used #2h | 3.38 | HT + MMO | 0.42 | 110 | 8 | — | 10.0 | 1.1 |
| Calcined #2 | 3.45 | MMO | 0.52 | 199 | 409 | 545 | 1.1 | 0.0 |
| Used #3h | 3.32 | HT + MMO | 0.44 | 112 | 5 | — | 11.3 | 0.9 |
| Calcined #3 | 3.38 | MMO | 0.50 | 195 | 397 | 500 | 0.8 | 0.0 |
| Usedi | 1.81 | HT + MMO + others | 0.41 | 89 | 2 | — | 12.1 | 0.6 |
| Calcined | 1.75 | MMO + others | 0.47 | 145 | 227 | 473 | 6.2 | 0.0 |
Regeneration studies
In order to recover the original mixed oxide structure, the used sample was subjected to calcination at 823 K, which was effective in reducing the chlorine content to only less than 1 wt%. Based on the chemical behaviour of metal chlorides,19 it is put forward that the elimination of chlorine from the HTlc generated in the reaction could follow two main mechanisms involving molecular oxygen provided in the treatment or water released by the solid upon heating: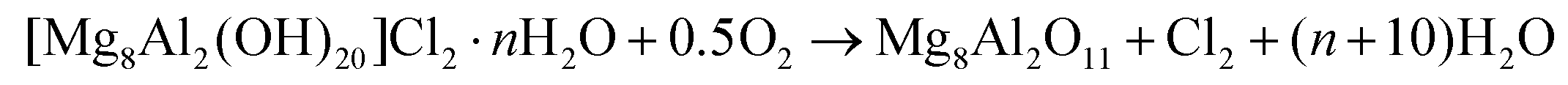
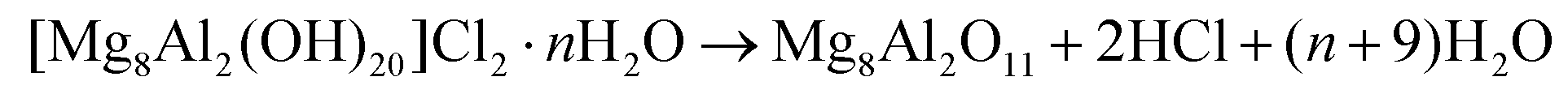
In the first case, the chloride ion is oxidised to elemental chlorine, while along the second path HCl is produced through an acid–base reaction. Since only HCl was detected by MS analysis upon TPO of the used material (Fig. S3†), the second route was predominant. XRD confirmed the disappearance of the hydrotalcite-like phase and the restoration of the mixed oxide (Fig. 7a). Accordingly, the surface area increased back to an only slightly lower value than for the fresh HT4-c823 sample (Table 2) and 27Al MAS NMR indicated that the proportion between aluminium sites with tetrahedral and octahedral geometry was similar to that of the original material (Fig. 7b). Moreover, CO2-TPD evidenced that the solid regained basicity (Fig. 7c). Notably, the concentration of basic sites of the regenerated oxide is almost equal to that of the starting material when relating the values to their surface areas (Table 2). In addition, the mass and Mg/Al ratio of the calcined sample were equivalent to the data measured for the fresh solid, considering the amount of chlorine that remained in the material, indicating that metals leaching was negligible.
Testing of the calcined sample produced analogous results to those obtained in the first run (Fig. 6a), except for the slightly inferior initial activity, which is in line with the moderately lower surface area and basicity and the small amount of residual chorine contained. Remarkably, calcination of the re-used material was equally effective in regenerating the mixed oxide as the treatment conducted after the first reaction (Table 2) and, accordingly, the reaction data collected in the third test were comparable to those of the second run (Fig. 6a).
Finally, since the epichlorohydrin selectivity was 15% higher at 403 K, HT4-c823 was assessed also at this 20 K lower temperature. Under these conditions, the dichloropropanol conversion progressively decreased reaching a similar level to that attained at the end of the run at 423 K (Fig. 6b). In contrast, the selectivity towards epichlorohydrin remained almost unperturbed at ca. 95%. This is rationalised by the minor occurrence of the thermal decomposition of the product to chloroacetone at 403 K. Analysis of the used material uncovered substantial differences with respect to the sample retrieved after the reaction at 423 K. In this case, the mixed oxide was fully converted into a hydrotalcite-like phase as well as AlCl3 (Fig. 7d). Thus, Cl− ions formed upon reaction not only exchanged OH− species at interlayer positions but also generated a metal salt. 27Al MAS NMR spectroscopy only detected hexacoordinated Al sites (Fig. 7e), in line with the octahedral geometry of this metal in the hydrotalcite and in AlCl3, and CO2-TPD indicated a full depletion on the basicity (Fig. 7f), as expected from the neutral nature of OH-free hydrotalcite and the metal chloride. The distinct alterations underwent by the sample at 403 K are likely due to the pronounced condensation observed on the solid reactant at this temperature, suggesting that the transformation took place in a liquid phase. Although the equilibrium diagram indicates that a pure vapour phase should be present, capillary condensation might have been induced by the surface rugosity and/or the liquid mixture might not have fully vaporised due to insufficient heat transfer rates during the short residence time in the heated zone of the reactor. The first hypothesis is supported by the application of the Kelvin equation.
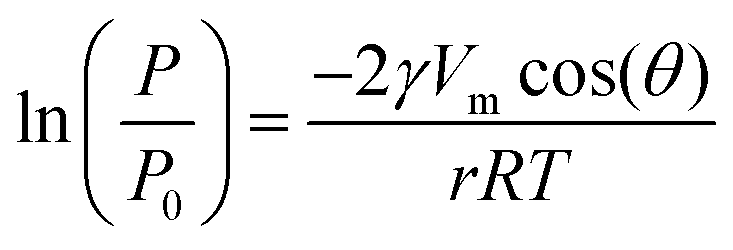
When using the values typical of pure water in view of the diluted nature of the system (i.e., a surface tension γ of 6 × 10−2 N m−1, a molar volume Vm of 1.8 × 10−5 m3 mol−1, a contact angle θ of 0 degrees and a condensation pressure P0 of 2.2 bar), a liquid phase is expected to form in pores with a diameter r smaller than ca. 6 nm, which are responsible for a significant fraction of the porosity of HT4-c823 (Fig. S4†). The presence of water greatly facilitates ion migration within the solid, which helped the formation of more thermodynamically-stable phases, i.e. aluminium chloride.27 Calcination of this material converted the hydrotalcite phase into the mixed oxide, but the latter produced very sharp reflections, which is indicative of a severe increase of the particle size. XRD also evidenced that AlCl3 remained in the material, in line with its decomposition temperature in excess of 900 K.27 These findings are in agreement with the only partial restoration of the surface area and the still high Cl content in the sample (Table 2). The basicity of the calcined solid was strongly inferior to that of the fresh mixed oxide in view of the enrichment in Mg of the system. In line with the characterisation data, the initial activity of the calcined solid was remarkably lower than that of the freshly prepared mixed oxide. A subsequent regeneration-reaction cycle showed a further deterioration of the performance. Accordingly, operation of the mixed oxide in the gas phase is a prerequisite to attain stable reaction-regeneration cycles. For this purpose, the well-known memory effect of layered double hydroxides is another crucial factor, since it enables the interconversion between hydrotalcite and mixed oxide forms with minimal modification of their basic and textural properties.15 From a practical viewpoint, a continuous production of epichlorohydrin can be realised operating two reactors simultaneously, one dedicated to the desired transformation and one to the regeneration of the solid base.
Overall, the use of a solid base in the gas phase in place of homogeneous alkaline solutions allows the compliance of the glycerol-to-epichlorohydrin process with three main green chemistry principles. Firstly, the generation of a salt byproduct is avoided, simplifying downstream purification and reducing waste treatment efforts. Secondly, the solid reactant is regenerable, reducing the material needs. Thirdly, the HCl evolved upon its regeneration could be easily recycled in the dehalogenation of glycerol to dichloropropanols (Scheme 3), maximising the atom economy and the cost-related performance of the overall glycerol-to-epichlorohydrin route. Additionally, it is worth noting that the solid base shares an equivalent non-toxic nature to the homogeneous counterpart.
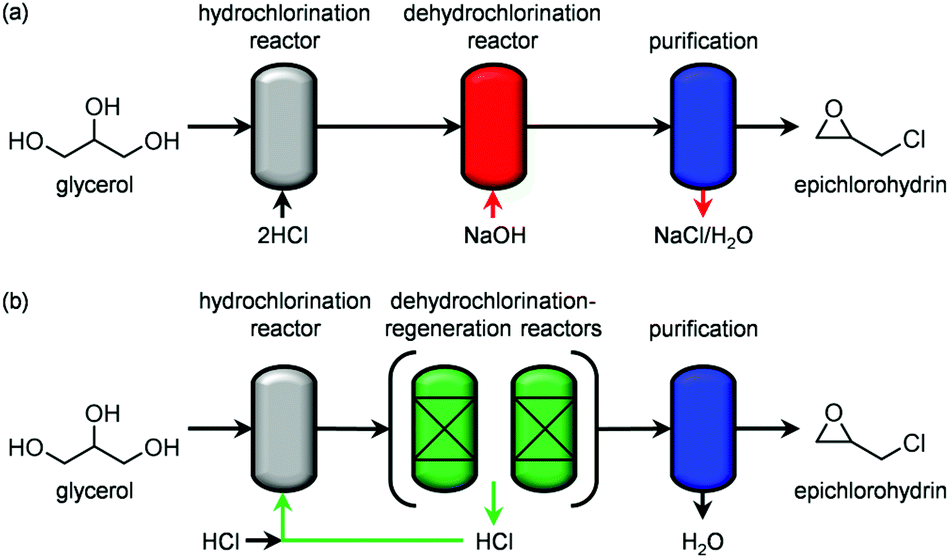 | ||
| Scheme 3 Simplified flow sheet for (a) the conventional glycerol-to-epichlorohydrin process and (b) the process herein introduced based on HTlc-derived oxides for the second reaction, showing the reaction-regeneration units as well as the chlorine recycling loop. | ||
Conclusions
In this study, a solid and recyclable base is introduced as alternative to alkali hydroxides for the dehydrochlorination of glycerol-derived dichloropropanol to epichlorohydrin. Upon testing of compounds with variable acid/base properties, Lewis-basic mixed oxides obtained from hydrotalcite-like materials emerged as the best-performing solids. Their selectivity pattern was found to be strongly influenced by temperature, since total selectivity towards epichlorohydrin was achieved below 423 K, while the product thermally decomposed to chloroacetone under harsher conditions. Optimisation of the space velocity enabled epichlorohydrin yields as high as 60%. Thorough characterisation enabled the elucidation of the reaction mechanism. The latter involves the initial formation of the actual active phase from the lamellar mixed oxide, a rehydrated hydrotalcite, and the subsequent progressive replacement of the hydroxide ions at the interlayer positions by chlorine ions. Simple calcination appeared effective in restoring a mixed oxide phase with equivalent porous, structural and basic properties and, thus, reactivity to the starting solid after a first and second reuse. Since this treatment releases HCl, a closed chlorine loop within the whole glycerol-to-epichlorohydrin process was envisaged when reutilising hydrochloric acid for the dehalogenation of the triol (Scheme 3). Based on the absence of salt by-products and the enhancement of the atom economy through HCl recycling, the new approach shown in this work holds potential toward a greener preparation of epichlorohydrin from renewable resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swiss National Science Foundation (Project Number 200020-159760). Dr S. Mitchell is thanked for the TEM analyses and the microscopy centre of ETH Zurich (ScopeM) is acknowledged for providing access to its facilities.References
- (a) Y. Wang and C. Wu, Energy Econ., 2012, 34, 2167–2181 CrossRef; (b) E. L. Schultz, D. T. de Souza and M. C. T. Damaso, Chem. Biol. Technol. Agric., 2014, 1, 1–7 CrossRef.
- J. R. M. Almeida, L. C. L. Fávaro and B. F. Quirino, Biotechnol. Biofuels, 2012, 5, 48 CrossRef CAS PubMed.
- M. J. Haas, A. J. McAloon, W. C. Yee and T. A. Foglia, Bioresour. Technol., 2006, 97, 671–678 CrossRef CAS PubMed.
- M. Pagliaro, R. Cirimmina, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS PubMed.
- K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, Wiley-VCH, Weinheim, 2008, pp. 267–312 Search PubMed.
- K. B. Cofer, US Pat, 2838574, 1958 Search PubMed.
- (a) E. Ahad, US Pat, 4891438, 1990 Search PubMed; (b) A. Harada, M. Furue and S.-I. Nozakura, Polym. J., 1981, 13, 777–781 CrossRef CAS; (c) A. R. Spurr, J. Ultrastruct. Res., 1969, 1, 31–43 CrossRef.
- W. J. Kruper, D. J. Schreck, K. L. Kearns, R. D. Varjian, M. E. Jones, R. M. Campbell, B. D. Hook, J. R. Briggs and J. G. Hippler, US Pat, 80888957, 2012 Search PubMed.
- (a) P. Gilbeau, US. Pat, 755725, 2009 Search PubMed; (b) P. Gilbeau, US Pat, 20120199786, 2012 Search PubMed.
- E. Santacesaria, R. Tesser, M. Di Serio, L. Casale and D. Verde, Ind. Eng. Chem. Res., 2010, 49, 964–970 CrossRef CAS.
- E. L. Ligero and T. M. K. Ravagnani, Chem. Eng. Process., 2003, 42, 543–552 CrossRef CAS.
- S. C. Okoro, M. Montgomery, F. J. Frandsen and K. Pantleon, Energy Fuels, 2014, 28, 6447–6458 CrossRef CAS.
- (a) P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS PubMed; (b) C. Len and R. Luque, Sustainable Chem. Processes, 2014, 2, 1–10 CrossRef.
- (a) R. Luque, L. Herrero-Davila, J. M. Campelo, J. H. Clark, J. M. Hidalgo, D. Luna, J. M. Marinas and A. A. Romero, Energy Environ. Sci., 2008, 1, 542–548 RSC; (b) J. M. Montero, M. A. Isaacs, A. F. Lee, J. M. Lynam and K. Wilson, Surf. Sci., 2016, 646, 170–178 CrossRef CAS.
- (a) S. Abelló, F. Medina, D. Tichit, J. Pérez-Ramírez, J. C. Groen, J. E. Sueiras, P. Salagre and Y. Cesteros, Chem. – Eur. J., 2005, 11, 728–739 CrossRef PubMed; (b) G. M. Lari, A. B. L. de Moura, L. Weimann, S. Mitchell, C. Mondelli and J. Pérez-Ramírez, J. Mater. Chem. A, 2017, 5, 16200–16211 RSC; (c) S. M. de Rezende, C. A. Franchini, M. Laura Dieuzeide, A. M. Duarte de Farias, N. Amadeo and M. A. Fraga, Chem. Eng. J., 2015, 272, 108–118 CrossRef CAS.
- D. Verboekend, T. C. Keller, S. Mitchell and J. Pérez-Ramírez, Adv. Funct. Mater., 2013, 23, 1923–1934 CrossRef CAS.
- M. Ibáñez, M. Gamero, J. Ruiz-Martínez, B. M. Weckhuysen, A. T. Aguayo, J. Bilbao and P. Castaño, Catal. Sci. Technol., 2016, 6, 296–306 Search PubMed.
- B. Puértolas, T. C. Keller, S. Mitchell and J. Pérez-Ramírez, Appl. Catal., B, 2016, 184, 77–86 CrossRef.
- (a) G. Huang, G. Lu, J. Wang and J. Yu, J. Anal. Appl. Pyrolysis, 2011, 91, 159–164 CrossRef; (b) N. C. Christensen, US Pat, 2413292, 1946 Search PubMed.
- G. M. Lari, K. Desai, C. Mondelli and J. Pérez-Ramírez, Catal. Sci. Technol., 2016, 6, 2706–2714 CAS.
- G. M. Lari, B. Puértolas, M. S. Frei, C. Mondelli and J. Pérez-Ramírez, ChemCatChem, 2016, 8, 1507–1514 CrossRef CAS.
- A. Aho, N. Kumar, K. Eränen, T. Salmi, M. Hupa and D. Y. Murzin, Process Saf. Environ. Prot., 2007, 85, 473–480 CrossRef CAS.
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 273–301 CrossRef.
- D. G. Cantrell, L. J. Gillie, A. F. Lee and K. Wilson, Appl. Catal., A, 2005, 287, 183–190 CrossRef CAS.
- R. R. Baldwin, A. Keen and R. W. Walker, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 435–456 RSC.
- J. Pérez-Ramírez, S. Abelló and N. M. van der Pers, Chem. – Eur. J., 2007, 13, 870–878 CrossRef PubMed.
- L. Catoire and M. T. Swihart, J. Electrochem. Soc., 2002, 149, 261–267 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Characterisation data of fresh and used materials, simulated phase diagram of dichloropropanol–water mixture, blank tests at the reaction temperatures studied and pore size distribution of the best performer. See DOI: 10.1039/c7gc02610b |
| This journal is © The Royal Society of Chemistry 2018 |