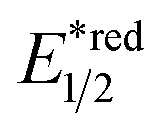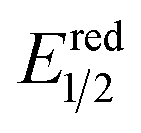DOI:
10.1039/C7SC00940B
(Edge Article)
Chem. Sci., 2017,
8, 4242-4249
Hybrid photocathode consisting of a CuGaO2 p-type semiconductor and a Ru(II)–Re(I) supramolecular photocatalyst: non-biased visible-light-driven CO2 reduction with water oxidation†
Received
1st March 2017
, Accepted 5th April 2017
First published on 6th April 2017
Abstract
A CuGaO2 p-type semiconductor electrode was successfully employed for constructing a new hybrid photocathode with a Ru(II)–Re(I) supramolecular photocatalyst (RuRe/CuGaO2). The RuRe/CuGaO2 photocathode displayed photoelectrochemical activity for the conversion of CO2 to CO in an aqueous electrolyte solution with a positive onset potential of +0.3 V vs. Ag/AgCl, which is 0.4 V more positive in comparison to a previously reported hybrid photocathode that used a NiO electrode instead of CuGaO2. A photoelectrochemical cell comprising this RuRe/CuGaO2 photocathode and a CoOx/TaON photoanode enabled the visible-light-driven catalytic reduction of CO2 using water as a reductant to give CO and O2 without applying any external bias. This is the first self-driven photoelectrochemical cell constructed with the molecular photocatalyst to achieve the reduction of CO2 by only using visible light as the energy source and water as a reductant.
Introduction
The use of artificial photosynthesis, particularly for the photochemical reduction of CO2, has gained much attention because it might provide solutions for overcoming both the problem of global warming and the shortage of fossil resources. Metal complex photocatalysts have been intensively developed for achieving the reduction of CO2 using the energy of visible light with a high selectivity for the products.1–3 In particular, supramolecular photocatalysts composed of both redox photosensitizer and catalyst units have exhibited high selectivity, durability, and efficiency for the reduction of CO2 under visible light conditions even in aqueous solutions,4,5 as well as in organic solvents.6–14 In these photocatalytic reactions, sacrificial reductants such as 1-benzyl-1,4-dihydronicotinamide,7 sodium ascorbate,4 and 2-(1,3-dimethyl-2,3-dihydro-1H-benzimidazol-2-yl)benzoic acid5 are necessary because of the low oxidizing power of the photosensitizer unit, which cannot sufficiently oxidize water, an abundant electron donor.
Hybrid photocatalysts and photoelectrodes composed of metal complexes and semiconductor materials with relatively high photochemical oxidizing power, in which the metal complexes act as catalysts alone in some cases15–24 but as photocatalysts in others,25–30 have recently been studied for the visible-light-driven photocatalytic reduction of CO2. In the cases where metal complexes are used as photocatalysts, hybrid photocatalysts with semiconductor particles can cope with both the high selectivity of the CO2 reduction and the strong oxidation power via the step-by-step excitation of both the redox photosensitizer unit of the metal complex photocatalyst and the semiconductor, so-called Z-scheme mechanism. However, visible-light-driven CO2 reduction accompanied by water oxidation has not been achieved yet using hybrids with these particle systems. We have previously reported a hybrid photocathode consisting of a Ru(II)–Re(I) supramolecular photocatalyst (RuRe) fixed on a NiO p-type semiconductor electrode for the photoelectrochemical reduction of CO2.31 In this system, the metal complex photocatalyst can drive the reduction of CO2 by the injection of electrons from an external electric circuit through the NiO semiconductor electrode without the addition of any sacrificial electron sources. This RuRe/NiO photoelectrochemical system was used to successfully achieve the catalytic reduction of CO2 using water as an electron source, combined with a CoOx/TaON n-type semiconductor photoanode for the oxidation of water.32 However, the performance of this photoelectrochemical cell was still insufficient, i.e., the combined photoelectrochemical cell needed the assistance of an external electrical bias (0.3 V) and a chemical bias (0.10 V) produced by a difference in pH, and the photoelectrochemical cell generated much lower yields of the reduction products (CO and H2) compared to the oxidation one (O2) from the viewpoint of electron balance.
One of the main reasons for these insufficiencies was the low activity of the NiO electrode. Although NiO has been widely used as an electrode material for p-type dye-sensitized photocathodes, the flat band position of NiO is relatively negative (+0.34 V vs. Ag/AgCl in a saturated aqueous solution of KCl at a pH of 7).33 This could cause a large loss of energy for passing an electron to the excited state of the Ru(II) photosensitizer unit (E1/2 = +0.51 V vs. Ag/AgCl)31 through the interface, leading to the requirement of a large external bias (totally 0.4 V) when this photocathode is used for constructing a photoelectrochemical cell combined with a photoanode. The consumption of electrons for reducing trivalent nickel ions (Ni3+) that originally existed in NiO could also be problematic because it lowers the Faraday efficiencies of the reduction reactions on the photocathode.31,32,34 Therefore, the development of new p-type semiconductor electrodes is necessary for the field of hybrid photoelectrochemical cells with supramolecular photocatalysts for reduction of CO2.
Here, we report a novel hybrid photocathode (RuRe/CuGaO2) consisting of CuGaO2 as the p-type semiconductor electrode and RuRe as the photocatalyst for the reduction of CO2. CuGaO2 with a delafossite crystal structure is known to exhibit p-type semiconducting properties that are derived from native Cu+ vacancies.35 It has received attention as a transparent conducting oxide material36–38 and has also been studied as an alternative to NiO for dye-sensitized photocathodes due to both its high conductivity (10−1–10−2 S cm−1)38 and positive flat band potential, which was reported to be approximately 0.16 V more positive compared to that of NiO.39,40 Although CuGaO2 electrodes have been investigated as parts of dye-sensitized solar cells, to the best of our knowledge there are no reports on the application of CuGaO2 electrodes for photocatalytic reactions such as CO2 reduction and H2 evolution. The onset potential for the reduction of CO2 by the as-synthesized RuRe/CuGaO2 was revealed to be 0.4 V more positive in comparison to that of the RuRe/NiO electrode. A photoelectrochemical cell consisting of RuRe/CuGaO2 and a CoOx/TaON photoanode enabled the visible-light-driven catalytic reduction of CO2 using water as the reductant without applying any external bias.
Results and discussion
Preparation of the RuRe/CuGaO2 photocathode
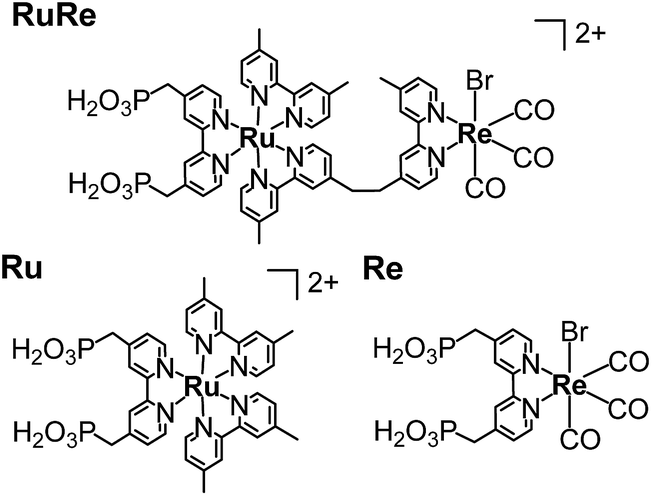
CuGaO2 powder was prepared using a solid-state reaction method. The XRD pattern of the synthesized powder revealed that the delafossite structure of the CuGaO2 was obtained with no obvious impurity phase (Fig. S1†). The UV-vis diffuse reflectance spectrum of the synthesized CuGaO2 powder indicated that the powder absorbs light at λ < 460 nm, which has been reported in previous research into CuGaO2 powder (Fig. S2†).41,42 The CuGaO2 electrodes were prepared by drop-casting a powder suspension onto FTO glass substrates following annealing with a N2 flow. SEM observations revealed that the polycrystalline CuGaO2 particles had rod-like shapes on the micron scale and the thickness of the stacked CuGaO2 particle layer of the electrode was approximately 15 μm (Fig. S3†). The dominant material at the solid–liquid interface of the electrode should be the deposited CuGaO2 particles and not the underlying flat FTO film. RuRe and its model complexes (Ru and Re) (see Chart 1) were synthesized in accordance with reported procedures.43 A CuGaO2 electrode was immersed in an acetonitrile solution containing the metal complex overnight to obtain hybridized photocathodes.
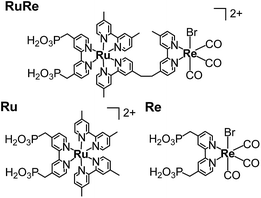 |
| | Chart 1 Structures and abbreviations of the Ru(II)–Re(I) supramolecular photocatalyst (RuRe) and its model mononuclear metal complexes used as the photosensitizer unit (Ru) and the catalyst unit (Re). | |
Photoelectrochemical properties of the RuRe/CuGaO2 photocathode
The photoelectrochemical properties of the synthesized RuRe/CuGaO2 electrode were investigated under irradiation at λex > 460 nm, which can be selectively absorbed by the Ru photosensitizer unit of RuRe, in an aqueous solution containing NaHCO3 (50 mM) saturated with CO2, which was used as the supporting electrolyte. Fig. 1(a) shows the current–potential curves of the RuRe/CuGaO2 electrode under continuous visible-light irradiation (λex > 460 nm) and under dark conditions. This clearly indicates that the RuRe/CuGaO2 electrode generated a cathodic photocurrent under irradiation. The difference in the observed current between the irradiation and dark conditions indicated that the cathodic photoresponse of the RuRe/CuGaO2 electrode started at approximately +0.3 V vs. Ag/AgCl (equivalent to +0.9 V vs. RHE). It should be noted that this onset potential of the photocurrent was approximately 0.4 V more positive than that of RuRe/NiO (ca. −0.1 V, Fig. 1(b)).31 A pristine CuGaO2 electrode showed a slight cathodic photoresponse (ca. −1 μA cm−2) under irradiation at λex > 460 nm (Fig. S4†), which was almost negligible compared to the photocurrent from the RuRe/CuGaO2 electrode, as shown in Fig. 1(a). The anodic current obtained from ca. +0.2 V vs. Ag/AgCl in the dark was derived from the self-oxidation of the CuGaO2 surface42 but not from the redox reaction of the immobilized RuRe.
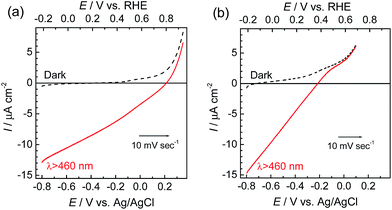 |
| | Fig. 1 Current–potential curves of RuRe/CuGaO2 (a) and RuRe/NiO31 (b) electrodes under continuous visible-light irradiation (λex > 460 nm) and under dark conditions. A 50 mM aqueous solution of NaHCO3 (pH 6.6) saturated with CO2 was used as the electrolyte. | |
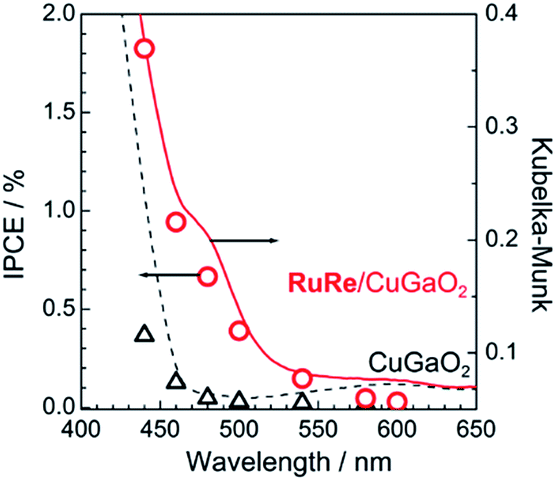
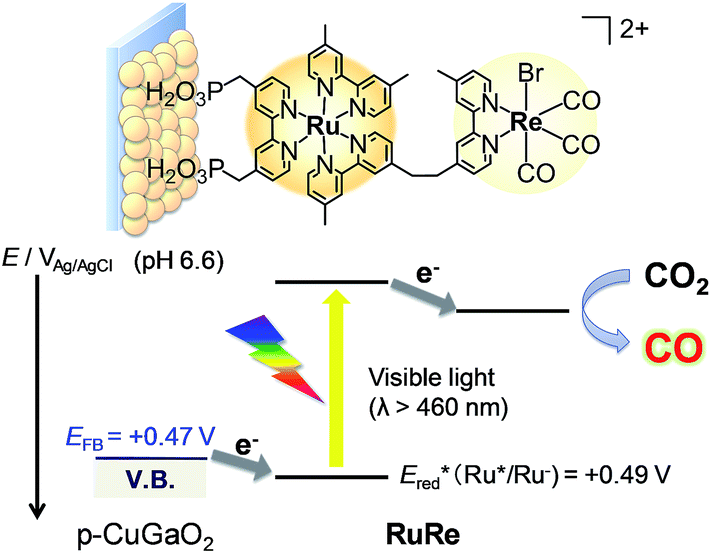
Fig. 2 shows the dependence of the IPCE of the RuRe/CuGaO2 electrode and a bare CuGaO2 electrode without RuRe on the wavelength of light used for irradiation when the applied potential was −0.3 V vs. Ag/AgCl. The diffuse reflectance spectra of these electrodes are also shown in Fig. 2. The RuRe/CuGaO2 electrode displayed light absorption up to 600 nm, which was derived from the Ru photosensitizer unit of the hybridized RuRe. In the case of the RuRe/CuGaO2 electrode, the dependence agreed well with the absorption spectrum of the electrode, whereas the bare CuGaO2 electrode exhibited almost no photoresponse under irradiation at λex > 460 nm. The flat band potential of the CuGaO2 electrode in the reaction solution was estimated using electrochemical impedance spectroscopy (Fig. S5†) to be +0.47 V, which was more negative than the reduction potential of the excited state of RuRe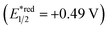 .44 These results obviously indicate that the photocurrent was induced by the injection of the electrons from the CuGaO2 electrode into the excited Ru photosensitizer unit of RuRe, as shown in Fig. 3. To conclude this section, we successfully synthesized the new hybrid photocathode (RuRe/CuGaO2) for the reduction of CO2 with an onset potential that was approximately 0.4 V more positive in comparison to that reported for the RuRe/NiO photocathode.
.44 These results obviously indicate that the photocurrent was induced by the injection of the electrons from the CuGaO2 electrode into the excited Ru photosensitizer unit of RuRe, as shown in Fig. 3. To conclude this section, we successfully synthesized the new hybrid photocathode (RuRe/CuGaO2) for the reduction of CO2 with an onset potential that was approximately 0.4 V more positive in comparison to that reported for the RuRe/NiO photocathode.
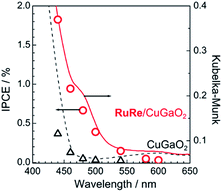 |
| | Fig. 2 Dependence on wavelength of the incident-photon-to-current efficiencies of the RuRe/CuGaO2 (red circles) and bare CuGaO2 (black triangles) electrodes at −0.3 V vs. Ag/AgCl. An aqueous solution containing 50 mM NaHCO3 saturated with CO2 was used as the electrolyte (pH 6.6). The diffuse reflectance spectra of these electrodes are also shown: RuRe/CuGaO2 (red solid line) and CuGaO2 (black broken line). | |
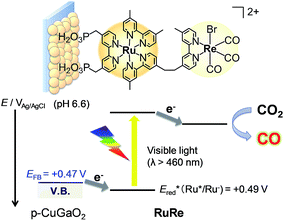 |
| | Fig. 3 Reaction scheme of the reduction of CO2 by the RuRe/CuGaO2 hybrid photocathode. | |
Photoelectrochemical reduction of CO2 using the RuRe/CuGaO2 photocathode
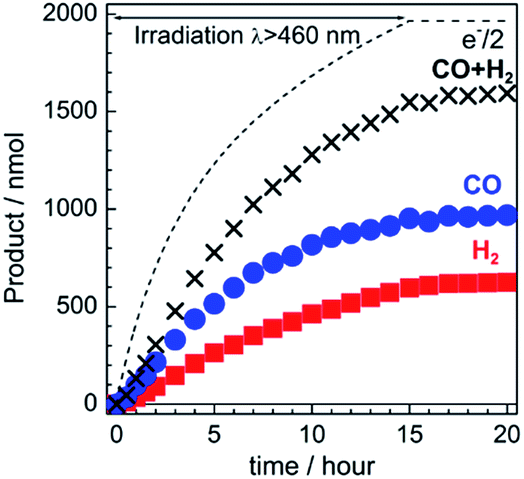
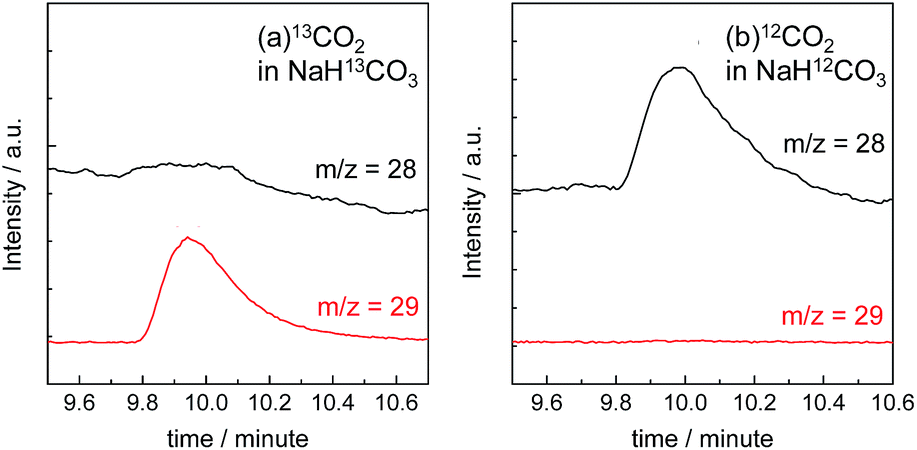
The gas products were analyzed during the continuous visible-light irradiation (λex > 460 nm) of the RuRe/CuGaO2 photocathode in an aqueous solution saturated with CO2 in the presence of an applied potential. Fig. 4 shows the time courses of both the produced CO and H2, and a half amount of electrons passing at an applied potential of −0.3 V vs. Ag/AgCl. After irradiating for 15 h, 966 nmol of CO and 622 nmol of H2 were detected as the reduction products when the turnover number for the formation of CO (TONCO), which was based on the RuRe deposited on the electrode, was 125, and the total faradaic efficiency for the production of the reduced products (CO + H2) was 81%. In the irradiated solution, HCOOH was not detected (the detection limit of the capillary electrophoresis system used was 2 μmol). An isotope tracer experiment using 13CO2 and NaH13CO3 was also conducted to confirm the source of the carbon for the produced CO. GC-MS analysis using NaH13CO3 as the electrolyte under a 13CO2 atmosphere revealed that 13CO was the main product of the reaction with a very small proportion of 12CO, where the 13C content was 99% in both 13CO2 and NaH13CO3 (Fig. 5(a)). It is worth noting that the Re unit of RuRe has three 12CO ligands, and the exchange of the 12CO ligands with 13CO proceeded in the photocatalytic 13CO2 reduction reactions in homogeneous systems. On the other hand, only 12CO was detected in the gas phase of the reaction using ordinary CO2 and NaHCO3 (Fig. 5(b)). These results clearly indicate that the source of carbon for CO was CO2.
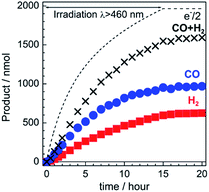 |
| | Fig. 4 Time courses of the chemical products (CO and H2) and a half amount of electrons passing through the RuRe/CuGaO2 photocathode under continuous visible-light irradiation (λex > 460 nm) at −0.3 V vs. Ag/AgCl. A 50 mM aqueous solution of NaHCO3 (pH 6.6) that was saturated with CO2 was used as the electrolyte. | |
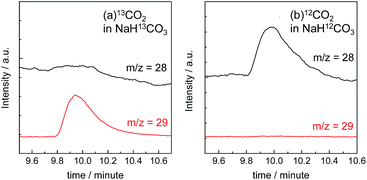 |
| | Fig. 5 GC-MS chromatograms of the gas products in the reaction chamber after irradiation (the detected m/z = 28 for 12CO and 29 for 13CO). The RuRe/CuGaO2 photocathode was irradiated at λex > 460 nm in an aqueous solution containing 50 mM sodium bicarbonate (pH 6.6) at −0.7 V vs. Ag/AgCl for 5 h using 13CO2 and NaH13CO3 (a) and ordinary CO2 and NaHCO3 (b). | |
Table 1 shows the results of the photoelectrochemical reactions that were conducted under various conditions. The RuRe/CuGaO2 electrode could produce CO as the main product with H2 under visible-light irradiation at an applied potential from −0.1 V to −0.7 V vs. Ag/AgCl (Table 1, entries 1–3). The TONCO of the RuRe/CuGaO2 electrode was more than twice the reported value of the RuRe/NiO electrode (32; entry 10), even at more positive potentials, i.e., −0.3 V and −0.1 V. Carbon monoxide was not produced under an Ar atmosphere even when the same photocathode was used, whereas H2 was produced (entry 4). The reactions using the bare CuGaO2 electrode or electrodes with only one of the model mononuclear complexes, i.e., Ru or Re, did not lead to the formation of any CO (entries 5, 6, 7 and 8). In these cases, almost negligible or small amounts of H2 were produced during the irradiation. Therefore, the CuGaO2 electrode, the RuRe supramolecular photocatalyst, and a CO2 atmosphere may all be required for the photoelectrochemical reduction of CO2. We note that an electrode that was loaded with both the model complexes (Re and Ru) at random produced a much smaller amount of CO (entry 9), which indicates that the molecular design of RuRe, which enables the sufficiently efficient transfer of electrons from the reduced photosensitizer unit to the catalyst unit, is quite important for developing photocathodes for the reduction of CO2.
Table 1 Photoelectrochemical reactions using various electrodes under various conditionsa
| Entry |
Sample |
Complex/nmol |
Potentialb/V |
Products |
F
red
/% |
| CO/nmol (TONCO) |
H2/nmol |
|
The electrode was irradiated at λex > 460 nm using a 300 W Xe lamp for 15 h. A 50 mM aqueous solution of NaHCO3 was used as the electrolyte.
Versus Ag/AgCl (sat. KCl).
Under an Ar atmosphere.
The electrode was prepared via immersion in solutions of Re and Ru in sequence.
Data from ref. 32 (irradiated for 12 h).
F
red: Faradaic efficiency for reduction reaction.
n.d.: not detected.
|
| 1 |
RuRe/CuGaO2 |
7.9 |
−0.7 |
668(85) |
387 |
68 |
| 2 |
RuRe/CuGaO2 |
7.7 |
−0.3 |
966(125) |
622 |
81 |
| 3 |
RuRe/CuGaO2 |
9.3 |
−0.1 |
781(84) |
494 |
72 |
| 4c |
RuRe/CuGaO2 |
8.3 |
−0.3 |
n.d.g |
346 |
52 |
| 5 |
CuGaO2 |
— |
−0.7 |
n.d.g |
25 |
7 |
| 6 |
CuGaO2 |
— |
−0.3 |
n.d.g |
Trace |
— |
| 7 |
Ru/CuGaO2 |
9.6 |
−0.3 |
n.d.g |
64 |
20 |
| 8 |
Re/CuGaO2 |
14.2 |
−0.3 |
n.d.g |
25 |
13 |
| 9d |
(Ru,Re)/CuGaO2 |
Re: 11.1, Ru: 4.4 |
−0.3 |
35(3, based on Re) |
31 |
34 |
| 10e |
RuRe/NiO32 |
11.2 |
−0.7 |
361(32) |
36 |
64 |
As was described above, the production of H2 was detected using the RuRe/CuGaO2 photocathode in reactions under both Ar and CO2 atmospheres, whereas only a small amount of H2 was produced in the case of RuRe/NiO. It has been reported that the decomposition products of both the Ru photosensitizer unit in systems using RuRe supramolecular photocatalysts and Ru in a mixed system of Ru and Re, i.e., a complex of the form [RuII(N⁁N)2(solvent)2]n+ (N⁁N = diimine ligand), were gradually produced during the photocatalytic reactions, and these acted as catalysts for both the evolution of H2 and the formation of HCOOH from CO2.5,45 However, the Ru/CuGaO2 photocathode produced a smaller amount of H2 than that of the RuRe/CuGaO2 photocathode. Therefore, the generation of H2 might result not only from the decomposition products of RuRe but also from the coexistence of photoexcited RuRe on the CuGaO2 surface. Because it has been reported that CuGaO2 worked as a H2-evolving photocathode under UV light irradiation (CuGaO2 itself can absorb UV light),42 back electron transfer from photoexcited RuRe to the CuGaO2 surface, which could act as an active site of H2 evolution, might induce the production of H2. Detailed studies of the reaction mechanism of the production of H2 and its suppression are in progress in our laboratory.
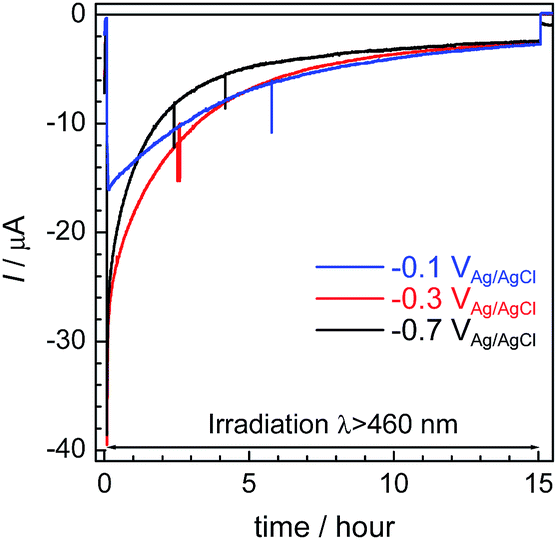
Fig. 6 shows the time courses of the photocurrent using the RuRe/CuGaO2 photocathode at the potentials of −0.1 V, −0.3 V, and −0.7 V vs. Ag/AgCl under irradiation at λex > 460 nm, which correspond to entries 1–3 in Table 1. In all of the cases, a photocurrent was observed even after irradiation for 15 h. However, the photocurrent decreased in the first stage of the photoelectrochemical reaction, and the rates of formation of CO and H2 also became slower in accordance with the decline in the photocurrent (Fig. 4 for the case at −0.3 V; Fig. S6† for the other cases). To confirm the reasons for the decline in the photocurrent and the rates of product formation, an estimation of the amount of “electrochemically active” RuRe species on the CuGaO2 surface was conducted using cyclic voltammetry in an acetonitrile solution (Fig. S7 and Table S1†) according to reported procedures, where the areas of the oxidation peaks of RuII/RuIII were used for the estimation.32 This indicated that about 80% of the RuRe lost its electrochemical activity after reacting for 15 h, which was probably due to (photo)desorption and/or photodecomposition of RuRe and could account for the decline in the photocurrent and the photocatalytic activity. In addition, the difference between the peak potentials of the redox reaction of RuII/RuIII was greater after the reaction (Fig. S7†), which indicates that the ohmic resistance of the electrode increased. Because the XPS measurements of the electrodes showed no obvious change in the electronic states of the Cu and Ga on the surface of the CuGaO2 particles during the reaction (Fig. S8†), the increase in the internal resistance of the CuGaO2 electrode might not be caused by the collapse of the GuGaO2 surface. An increase in inter-particle resistance probably proceeded in the electrode owing to the gradual deterioration of physical contact among the GuGaO2 particles. This might induce the deactivation of the photocathode. We note that the peaks of Ru and Re in the XPS measurements were too small to be identified, possibly due to the low loading density of RuRe on CuGaO2.
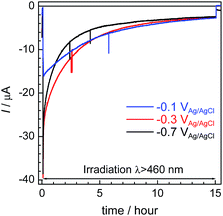 |
| | Fig. 6 Time courses of the photocurrent using the RuRe/CuGaO2 photocathode at various potentials (corresponding to entries 1–3 in Table 1) under irradiation at λex > 460 nm. | |
Photoelectrochemical reduction of CO2 using a hybrid electrochemical cell with a Z-scheme configuration
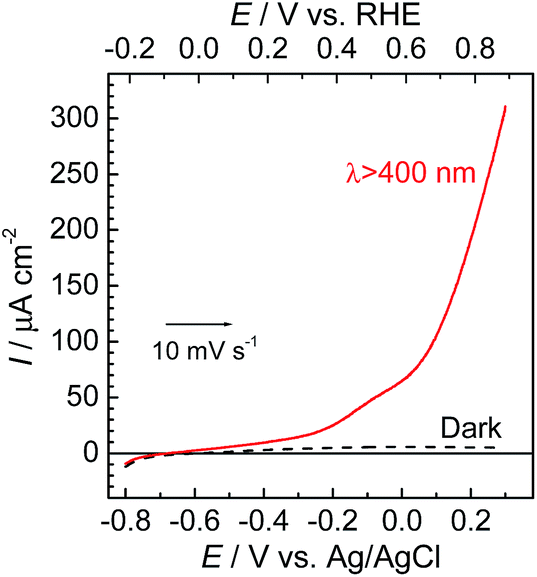
To couple the photoelectrochemical reduction of CO2 to the oxidation of water, the CoOx/TaON photoanode, which has been reported to be an efficient photoanode for the water oxidation reaction, was examined as the counter photoanode in a full photoelectrochemical cell using the RuRe/CuGaO2 hybrid photocathode. The CoOx/TaON electrode was prepared according to a previously reported method.46Fig. 7 shows the current–potential curve of the CoOx/TaON photoanode in a 50 mM aqueous solution of NaHCO3 with a pH of 6.6 that was purged with CO2, which is the same electrolyte as was used for the photocathode. An anodic photocurrent was clearly observed, as in the cases of the aqueous solutions of Na2SO4 (pH 8) and NaHCO3 (pH 8.3), that were both purged with Ar.46 In addition, the evolution of O2 under visible-light irradiation in a 50 mM aqueous solution of NaHCO3 purged with CO2 was confirmed by the analysis of the gas products at an applied potential of +0.2 V vs. Ag/AgCl, and its total faradaic efficiency was 93% after irradiating for 60 min (Fig. S9†).
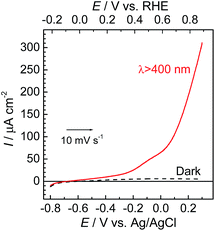 |
| | Fig. 7 Current–potential curves of the CoOx/TaON photoanode under continuous visible-light irradiation (λex > 400 nm) and under dark conditions. A 50 mM aqueous solution of NaHCO3 (pH 6.6) saturated with CO2 was used as the electrolyte. | |
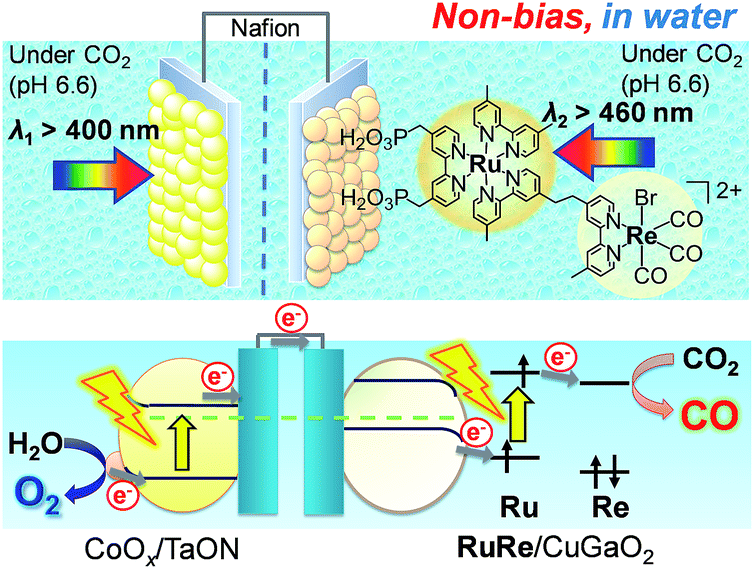
Fig. 8 shows a schematic representation of the hybrid photoelectrochemical cell consisting of the RuRe/CuGaO2 photocathode and the CoOx/TaON photoanode in a Z-scheme configuration. Two chambers containing the electrolyte solution purged with CO2 were separated by a Nafion membrane, and one photoelectrode was installed in each chamber. Visible light with λex > 460 nm and λex > 400 nm was used to irradiate the RuRe/CuGaO2 photocathode and the CoOx/TaON photoanode, respectively. It is worth nothing that the photoelectrochemical reaction was conducted under short-circuit conditions with the same electrolyte for both electrodes, i.e., there was no external electrical or chemical bias between the electrodes. In addition, a higher intensity of irradiated light was utilized for the photocathode (4.5 × 1017 photon cm−2 s−1 in the wavelength range 460–600 nm) than that for the photoanode (4.1 × 1016 photon cm−2 s−1 in the wavelength range 400–600 nm) to achieve sufficient photoexcitation of the loaded RuRe on the photocathode.
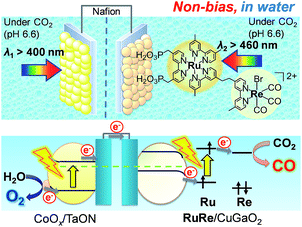 |
| | Fig. 8 Schematic image of the hybrid photoelectrochemical cell in a Z-scheme configuration. | |
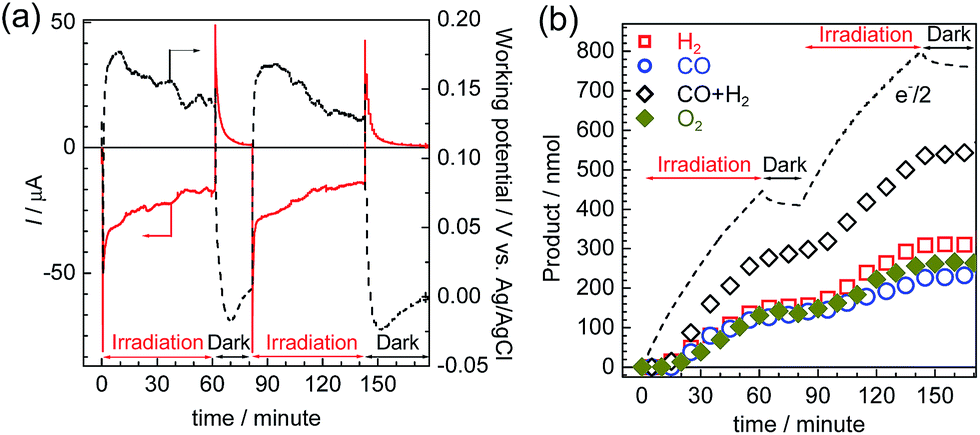
Photoelectrolysis using the constructed photoelectrochemical cell was conducted with intermittent visible-light irradiation for a total of 2 h. Fig. 9 shows the time courses of the photocurrent and electrode potentials (a) and the chemical products (b). The generation of a photocurrent and a corresponding increase in the products were clearly observed under irradiation, and the working potential of the electrodes was confirmed to be around +0.15 V vs. Ag/AgCl, at which point both of the photoelectrodes could generate a photocurrent under irradiation via the respective half reactions. The products of the reduction (CO and H2) and oxidation (O2) were detected in the cathode and anode chambers, respectively (Fig. S10†). A total of 232 nmol of CO was produced, and the TONCO reached 22 based on the amount of immobilized RuRe before the irradiation. The total faradaic efficiency of the cathodic reaction, including the evolution of H2 (311 nmol), was 72%, whereas that of the anodic reaction was 70% (266 nmol O2). Notably, an electron balance between the reduction products (CO and H2) and the oxidation product (O2) was almost acquired in this photoelectrochemical reaction, while in the case of the corresponding system using the NiO electrode instead of CuGaO2, there were much less reduction products than the produced O2.32 The slight decrease in the half amount of electrons when the light was cut in Fig. 9(b) means that a reverse current flowed, which was possibly derived from the discharge of the electrical double layers of the electrodes. When only the cathode or the anode was irradiated, the photocurrent was relatively small in comparison to that when both of the electrodes were under irradiation (Fig. S11†), which suggests that photoexcitation of both of the electrodes is required for the subsequent progress of the photochemical reduction of CO2 with water as the reductant. On the basis of these data, we conclude that this hybrid photoelectrochemical cell can drive both the reduction of CO2 and the oxidation of water by the stepwise photoexcitation of RuRe and TaON, i.e., via Z-scheme-type electron transfer, which gives CO and O2 under visible-light irradiation. This is the first successful instance of the reduction of CO2 in the absence of electrical or chemical bias using visible light and water alone by a hybrid photocatalytic system consisting of a molecular photocatalyst and a semiconductor photocatalyst. Further investigations for the improvement of the efficiency and durability of the photoelectrochemical cell are currently under way in our laboratory.
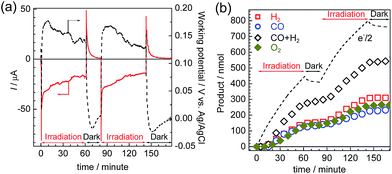 |
| | Fig. 9 Time courses of the photocurrent and working potential of the electrodes (a) and the chemical products (b) during the visible-light irradiation of the photoelectrochemical cell consisting of the RuRe/CuGaO2 photocathode and the CoOx/TaON photoanode under short-circuit conditions. | |
Experimental
Synthesis of CuGaO2 particles
Particles of CuGaO2 powder were prepared using a solid-state reaction method. The precursor materials, i.e., Cu2O (Kanto Chemicals, >92%) and Ga2O3 (Wako Chemicals, 99.99%), were mixed using an agate mortar. The molar ratio of the precursors was set at Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ga = 1:1. The obtained mixture was calcined with a N2 flow of 100 mL min−1 at 1373 K for 5 h to give a polycrystalline powder of CuGaO2.
:Ga = 1:1. The obtained mixture was calcined with a N2 flow of 100 mL min−1 at 1373 K for 5 h to give a polycrystalline powder of CuGaO2.
Fabrication of CuGaO2 electrodes
Fluorine-doped tin oxide (FTO) glass (AGC Fabritech, 15 × 50 mm, 12 Ω sq−1) was sonicated in acetone for 10 min and in methanol for 10 min before use. A suspension of 30 mg of CuGaO2 powder in 300 μL of isopropanol was prepared with sonication for 10 min. Then, 100 μL of the suspension was dropped onto an exposed area of the FTO glass (ca. 5 cm2) and dried in air at room temperature using a drop-casting method and Scotch mending tape as a masking material. The obtained sample was calcined at 773 K for 3 h with a N2 flow of 100 mL min−1. The electrode was cut in half before use. The amount of powder that was deposited was estimated to be approx. 5 mg for each electrode (ca. 2.5 cm2).
Preparation of the hybrid photocathode by the adsorption of the metal complex on the CuGaO2 electrode
RuRe and its model complexes (Ru and Re) (Chart 1) were synthesized in accordance with reported procedures.43 A CuGaO2 electrode was immersed in an acetonitrile solution containing the metal complex (4 mL, 5 μM) overnight. The electrode was washed with acetonitrile after the adsorption procedure. The amount of the metal complex that was adsorbed was estimated from the difference in the absorbance between the solutions at 461 nm before and after the hybridization procedures.
Preparation of the CoOx/TaON photoanode
The CoOx/TaON electrode was prepared in accordance with a reported procedure.46 The outline of the procedure is as follows. TaON powder was prepared by heating Ta2O5 powder with an NH3 flow (20 mL min−1) at 1123 K for 15 h. CoOx nanoparticles (5 wt% as the metal) were loaded onto the TaON particles via impregnation from an aqueous Co(NO3)2 solution, followed by heating at 673 K for 30 min in air. The as-prepared CoOx-loaded TaON particles were deposited on a Ti substrate using electrophoretic deposition. The electrodes were treated with 50 μL of a solution of TaCl5 in methanol (10 mM) and then dried in air at room temperature. After this process had been performed five times, the electrode was heated in an NH3 flow (10 mL min−1) at 723 K for 30 min. The details of the structural characterization of the electrode are shown in our previous paper.
Photoelectrochemical measurements and reduction of CO2 using the RuRe/CuGaO2 photocathode
A three-electrode setup with an HZ-7000 potentiostat (Hokuto Denko) was used throughout the photoelectrochemical measurements and half reactions. A 50 mM aqueous solution of NaHCO3 saturated with CO2 (pH 6.6) was used as an electrolyte. A Pt wire and Ag/AgCl in a saturated aqueous solution of KCl were employed as the counter and reference electrodes, respectively. The counter electrode was separated from the reaction solution using Vycor glass to avoid the influence of the oxidation reaction taking place on the counter electrode. The working electrode (ca. 2.5 cm2) was irradiated using a 300 W Xe lamp (Asahi Spectra MAX-302) with an IR-blocking mirror module. A cutoff filter (HOYA Y48 for irradiation at λex > 460 nm) or a band-pass filter (Asahi Spectra, for the IPCE measurements) was employed to control the irradiation wavelength. The potential against a reversible hydrogen electrode (RHE) (see Fig. 1) was calculated using the Nernst equation (eqn (1)):| | | E (V vs. RHE) = E (V vs. Ag/AgCl) + 0.199 + 0.059 pH. | (1) |
The dependence on the wavelength of the IPCE at a given wavelength was calculated using eqn (2):
| | | IPCE (%) = [(1240/λex)(Ilight − Idark)/Plight] × 100, | (2) |
where
λex and
Plight are the wavelength and power density of the incident light (nm and μW cm
−2), respectively,
Ilight is the current density under irradiation (μA cm
−2), and
Idark is the current density in the dark (μA cm
−2).
The photoelectrochemical reduction of CO2 was conducted using a Pyrex cell sealed with an O-ring and a stirring tip. The total volume of the cell without the stirring tip and electrodes was ca. 130 mL, and 84 mL of the electrolyte solution was utilized; therefore, the estimated volume of the gas phase was ca. 46 mL. The reaction was conducted after purging the system with CO2 for more than 40 min. A cutoff filter (HOYA Y48) was employed for irradiation at λex > 460 nm. Product analyses of CO and H2 in the gas phase and HCOOH in the liquid phase were performed using a gas chromatograph (Inficon MGC3000A) and a Capi-3300I capillary electrophoresis system (Otsuka Electronics), respectively. 13CO2 (CIL, 13C = 99%) and NaH13CO3 (Aldrich, 13C = 99%) were utilized for the labeling experiments. Gas chromatography-mass spectrometry (GC-MS) analysis of the gas phase was conducted using a gas chromatograph equipped with a mass spectrometer (Shimadzu GCMS-QP2010 Ultra).
Photoelectrochemical reduction of CO2 using the hybrid electrochemical cell in the Z-scheme configuration
The reaction was conducted using a Pyrex cell with two chambers (ca. 27 mL for each chamber) that were divided by a Nafion 117 membrane (Aldrich), and the photoelectrodes were installed into each chamber and the cell was sealed with an O-ring. Ag/AgCl in a saturated aqueous solution of KCl was employed as the reference electrode and was placed into the cathode chamber. A 14.5 mL portion of a 50 mM aqueous solution of NaHCO3 was added as an electrolyte to each chamber, which was purged with CO2. A three-electrode setup with an HZ-7000 potentiostat (Hokuto Denko) was used in non-resistance ammeter mode. The RuRe/CuGaO2 photocathode (ca. 3.2 cm2, RuRe 10.5 nmol) was irradiated at λex > 460 nm using a 300 W Xe lamp (Eagle Engineering) with a cutoff filter (HOYA Y48) and a cold mirror (CM-1). The CoOx/TaON photoanode (ca. 3.3 cm2) was irradiated at λex > 400 nm using a 300 W Xe lamp (Asahi Spectra MAX-302) with a cutoff filter (HOYA L42) and an IR-blocking mirror module. The analysis of gas products was performed using gas chromatography (Inficon MGC3000A).
Characterizations
UV-vis diffuse reflectance spectra were recorded using a V-565 spectrophotometer (JASCO) that was equipped with an integral sphere unit. X-ray diffraction (XRD) patterns were recorded using a MiniFlex600 X-ray diffractometer (Rigaku) that was equipped with monochromatic Cu Kα radiation and operated at 15 mA and 40 kV. The scanning electron microscopy (SEM) observations were conducted using an S-4700 microscope (Hitachi High-Tech). An ECSA-3400 X-ray photoelectron spectrometer (Shimadzu) was utilized for the X-ray photoelectron spectroscopy (XPS) measurements. The binding energies were corrected using the C 1s peak (284.6 eV) for each sample.
Conclusions
A novel hybrid photocathode consisting of a CuGaO2 p-type semiconductor and a RuRe supramolecular complex has been developed for the photoelectrochemical reduction of CO2 in aqueous solution. The RuRe/CuGaO2 photocathode displayed photoelectrochemical activity for the conversion of CO2 to CO in an aqueous electrolyte solution with an onset potential of +0.3 V vs. Ag/AgCl. A photoelectrochemical cell comprising the RuRe/CuGaO2 photocathode and a CoOx/TaON photoanode exhibited activity for the visible-light-driven reduction of CO2 using water as a reductant to generate CO, H2, and O2 with no external bias.
Acknowledgements
This work was supported by JSPS KAKENHI Grant Number JP24107005 in Scientific Research on Innovative Areas “Artificial Photosynthesis (AnApple)”, the Strategic International Collaborative Research Program (SICORP) from JST, and JST CREST Grant Number JPMJCR13L1 in “Molecular Technology”. H. K. thanks JSPS KAKENHI Grant Number JP16K21031 for Young Scientists (B) and K. M. thanks JSPS KAKENHI Grant Number JP16H06441 in Scientific Research on Innovative Areas “Mixed Anion” for their financial support. We thank Prof. Kazunari Domen from the University of Tokyo for the SEM measurements.
Notes and references
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- G. Sahara and O. Ishitani, Inorg. Chem., 2015, 54, 5096–5104 CrossRef CAS PubMed.
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS.
- A. Nakada, K. Koike, T. Nakashima, T. Morimoto and O. Ishitani, Inorg. Chem., 2015, 54, 1800–1807 CrossRef CAS PubMed.
- A. Nakada, K. Koike, K. Maeda and O. Ishitani, Green Chem., 2016, 18, 139–143 RSC.
- B. Gholamkhass, H. Mametsuka, K. Koike, T. Tanabe, M. Furue and O. Ishitani, Inorg. Chem., 2005, 44, 2326–2336 CrossRef CAS PubMed.
- S. Sato, K. Koike, H. Inoue and O. Ishitani, Photochem. Photobiol. Sci., 2007, 6, 454–461 CAS.
- K. Koike, S. Naito, S. Sato, Y. Tamaki and O. Ishitani, J. Photochem. Photobiol., A, 2009, 207, 109–114 CrossRef CAS.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673–15678 CrossRef CAS PubMed.
- Y. Tamaki, K. Watanabe, K. Koike, H. Inoue, T. Morimoto and O. Ishitani, Faraday Discuss., 2012, 155, 115–127 RSC.
- Y. Tamaki, K. Koike, T. Morimoto and O. Ishitani, J. Catal., 2013, 304, 22–28 CrossRef CAS.
- Y. Tamaki, K. Koike, T. Morimoto, Y. Yamazaki and O. Ishitani, Inorg. Chem., 2013, 52, 11902–11909 CrossRef CAS PubMed.
- E. Kato, H. Takeda, K. Koike, K. Ohkubo and O. Ishitani, Chem. Sci., 2015, 6, 3003–3012 RSC.
- Y. Tamaki, K. Koike and O. Ishitani, Chem. Sci., 2015, 6, 7213–7221 RSC.
- T. Arai, S. Sato, K. Uemura, T. Morikawa, T. Kajino and T. Motohiro, Chem. Commun., 2010, 46, 6944 RSC.
- S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240–15243 CrossRef CAS PubMed.
- T. Arai, S. Tajima, S. Sato, K. Uemura, T. Morikawa and T. Kajino, Chem. Commun., 2011, 47, 12664–12666 RSC.
- T. Arai, S. Sato, T. Kajino and T. Morikawa, Energy Environ. Sci., 2013, 6, 1274–1282 CAS.
- K. Maeda, K. Sekizawa and O. Ishitani, Chem. Commun., 2013, 49, 10127–10129 RSC.
- K. Maeda, R. Kuriki, M. Zhang, X. Wang and O. Ishitani, J. Mater. Chem. A, 2014, 2, 15146–15151 CAS.
- T. Arai, S. Sato and T. Morikawa, Energy Environ. Sci., 2015, 8, 1998–2002 CAS.
- R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2015, 54, 2406–2409 CrossRef CAS PubMed.
- K. Maeda, R. Kuriki and O. Ishitani, Chem. Lett., 2016, 45, 182–184 CrossRef CAS.
- R. Kuriki, O. Ishitani and K. Maeda, ACS Appl. Mater. Interfaces, 2016, 8, 6011–6018 CAS.
- K. Sekizawa, K. Maeda, K. Domen, K. Koike and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 4596–4599 CrossRef CAS PubMed.
- F. Yoshitomi, K. Sekizawa, K. Maeda and O. Ishitani, ACS Appl. Mater. Interfaces, 2015, 7, 13092–13097 CAS.
- A. Nakada, T. Nakashima, K. Sekizawa, K. Maeda and O. Ishitani, Chem. Sci., 2016, 7, 4364–4371 RSC.
- R. Kuriki, H. Matsunaga, T. Nakashima, K. Wada, A. Yamakata, O. Ishitani and K. Maeda, J. Am. Chem. Soc., 2016, 138, 5159–5170 CrossRef CAS PubMed.
- K. Muraoka, H. Kumagai, M. Eguchi, O. Ishitani and K. Maeda, Chem. Commun., 2016, 52, 7886–7889 RSC.
- K. Wada, M. Eguchi, O. Ishitani and K. Maeda, ChemSusChem, 2017, 10, 287–295 CrossRef CAS PubMed.
- G. Sahara, R. Abe, M. Higashi, T. Morikawa, K. Maeda, Y. Ueda and O. Ishitani, Chem. Commun., 2015, 51, 10722–10725 RSC.
- G. Sahara, H. Kumagai, K. Maeda, N. Kaeffer, V. Artero, M. Higashi, R. Abe and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 14152–14158 CrossRef CAS PubMed.
- J. He, H. Lindström, A. Hagfeldt and S.-E. Lindquist, J. Phys. Chem. B, 1999, 103, 8940–8943 CrossRef CAS.
- N. Kaeffer, J. Massin, C. Lebrun, O. Renault, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2016, 138, 12308–12311 CrossRef CAS PubMed.
- S. Nandy, A. Banerjee, E. Fortunato and R. Martins, Rev. Adv. Sci. Eng., 2013, 2, 273–304 CrossRef.
- H. Kawazoe, M. Yasukawa, H. Hyodo, M. Kurita, H. Yanagi and H. Hosono, Nature, 1997, 389, 939–942 CrossRef CAS.
- H. Yanagi, H. Kawazoe, A. Kudo, M. Yasukawa and H. Hosono, J. Electroceram., 2000, 4, 407–414 CrossRef CAS.
- K. Ueda, T. Hase, H. Yanagi, H. Kawazoe, H. Hosono, H. Ohta, M. Orita and M. Hirano, J. Appl. Phys., 2001, 89, 1790–1793 CrossRef CAS.
- A. Renaud, B. Chavillon, L. Le Pleux, Y. Pellegrin, E. Blart, M. Boujtita, T. Pauporté, L. Cario, S. Jobic and F. Odobel, J. Mater. Chem., 2012, 22, 14353–14356 RSC.
- M. Yu, G. Natu, Z. Ji and Y. Wu, J. Phys. Chem. Lett., 2012, 3, 1074–1078 CrossRef CAS PubMed.
- J. W. Lekse, M. K. Underwood, J. P. Lewis and C. Matranga, J. Phys. Chem. C, 2012, 116, 1865–1872 CAS.
- M. Lee, D. Kim, Y. T. Yoon and Y. Il Kim, Bull. Korean Chem. Soc., 2014, 35, 3261–3266 CrossRef CAS.
- Y. Ueda, H. Takeda, T. Yui, K. Koike, Y. Goto, S. Inagaki and O. Ishitani, ChemSusChem, 2015, 8, 439–442 CrossRef CAS PubMed.
- The value of
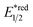 of RuRe was calculated using the reported values of
of RuRe was calculated using the reported values of 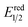 and E00 from ref. 31 and was converted to potential vs. Ag/AgCl.
and E00 from ref. 31 and was converted to potential vs. Ag/AgCl.
- Y. Kuramochi and O. Ishitani, Inorg. Chem., 2016, 55, 5702–5709 CrossRef CAS PubMed.
- M. Higashi, K. Domen and R. Abe, J. Am. Chem. Soc., 2012, 134, 6968–6971 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc00940b |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Go
Sahara
a,
Kazuhiko
Maeda
a,
Go
Sahara
a,
Kazuhiko
Maeda

 .44 These results obviously indicate that the photocurrent was induced by the injection of the electrons from the CuGaO2 electrode into the excited Ru photosensitizer unit of RuRe, as shown in Fig. 3. To conclude this section, we successfully synthesized the new hybrid photocathode (RuRe/CuGaO2) for the reduction of CO2 with an onset potential that was approximately 0.4 V more positive in comparison to that reported for the RuRe/NiO photocathode.
.44 These results obviously indicate that the photocurrent was induced by the injection of the electrons from the CuGaO2 electrode into the excited Ru photosensitizer unit of RuRe, as shown in Fig. 3. To conclude this section, we successfully synthesized the new hybrid photocathode (RuRe/CuGaO2) for the reduction of CO2 with an onset potential that was approximately 0.4 V more positive in comparison to that reported for the RuRe/NiO photocathode.