
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePyridine sulfinates as general nucleophilic coupling partners in palladium-catalyzed cross-coupling reactions with aryl halides†
Tim
Markovic
a,
Benjamin N.
Rocke
b,
David C.
Blakemore
b,
Vincent
Mascitti
b and
Michael C.
Willis
 *a
*a
aDepartment of Chemistry, University of Oxford, Chemical Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: michael.willis@chem.ox.ac.uk
bMedicine Design, Pfizer Inc., Eastern Point Road, Groton, CT 06340, USA
First published on 28th April 2017
Abstract
Pyridine rings are ubiquitous in drug molecules; however, the pre-eminent reaction used to form carbon–carbon bonds in the pharmaceutical industry, the Suzuki–Miyaura cross-coupling reaction, often fails when applied to these structures. This phenomenon is most pronounced in 2-substituted pyridines, and results from the difficulty in preparing, the poor stability of, and low efficiency in reactions of pyridine-2-boronates. We demonstrate that by replacing these boronates with pyridine-2-sulfinates, a cross-coupling process of unrivalled scope and utility is realized. The corresponding 3- and 4-substituted pyridine variants are also efficient coupling partners. In addition, we apply these sulfinates in a library format to the preparation of medicinally relevant derivatives of the drugs varenicline (Chantix) and mepyramine (Anthisan).
Introduction
The Suzuki–Miyaura cross-coupling reaction,1 in which aryl boronates are combined with aryl halides under the action of a palladium catalyst, is the mainstay of carbon–carbon bond-forming transformations employed in the pharmaceutical industry.2 Factors that have contributed to the success of these reactions include the wide availability and low toxicity of the boron-derived substrates, excellent functional group tolerance and simple experimental protocols, often resulting in highly effective transformations. These reactions allow the logical design of synthetic pathways in which intact ring systems can be joined together in a predictable, efficient manner.3 Unfortunately, this approach often becomes unreliable, or fails completely, when substituted pyridine rings are employed as nucleophilic substrates. This is most pronounced for 2-substituted pyridines, and arises from the difficulty in preparation, poor stability4 and low reaction efficiency of pyridine-2-boronates (Fig. 1b). Pyridine rings are embedded in a large number of drug molecules, which are used to tackle a correspondingly broad range of indications, as diverse as HIV,5 lung cancer6 and arthritis,7 amongst many others (Fig. 1a).8 Substituted pyridine rings are also of importance as ligands in metal-catalyzed reactions9 and as components of functional materials.10 The scale of the challenge in using pyridine derivatives in Suzuki chemistry is evident from a survey of the Pfizer internal electronic laboratory notebook;11 of 358 reactions attempted using pyridine-2-boronates, including boronic acids, boronic esters, and stabilized boronates (e.g. MIDA boronates), only 28 experiments, corresponding to <8% of examples, achieved a yield of at least 20%.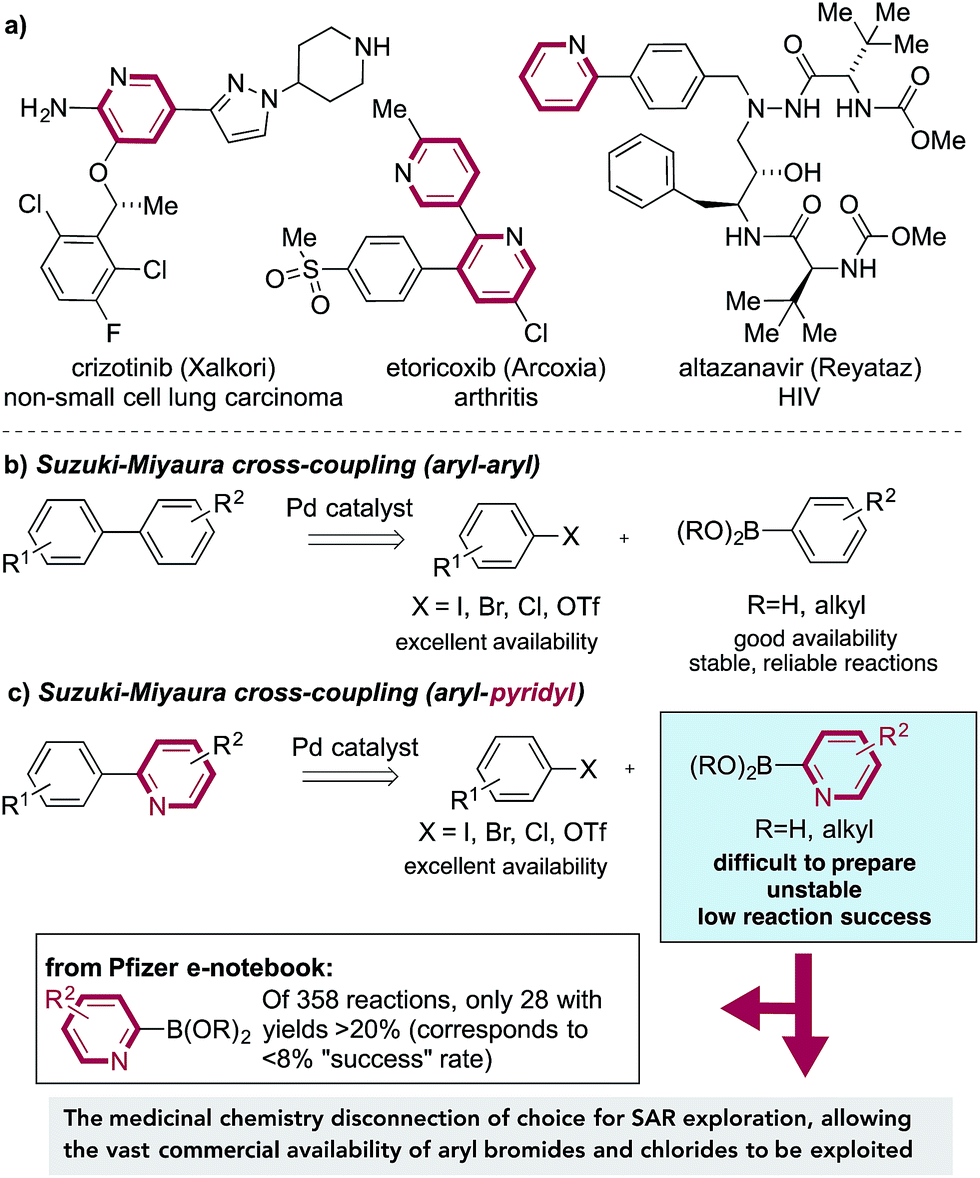 | ||
| Fig. 1 (a) Examples of medicinally relevant pyridine containing molecules; (b) the Suzuki–Miyaura cross-coupling reaction, and (c) application to pyridine derivatives. | ||
Such is the significance of this coupling issue that it is encountered in most pharmaceutical industry projects undertaken. Indeed, an internal survey of Pfizer's medicinal chemists regarding unmet synthetic needs ranked the instability of some heterocyclic boronates (in particular the 2-pyridyl boronate) as the number one synthetic challenge. The failure of Suzuki-based methods to allow access to these target structures therefore has serious consequences, potentially halting the preparation of targeted medicinal agents.
There is of course an alternative disconnection available to access 2-aryl-substituted pyridines, one in which an aryl boronate is combined with a 2-halopyridine. This “reverse coupling” approach is feasible for a smaller number of substrate combinations, but crucially, from a discovery chemistry perspective, this approach fails to exploit the vast numbers of commercially available bromo- and chloro-substituted arenes (relative to the corresponding boronates), and as such allows only a much narrower region of chemical space to be explored. In addition, no such coupling is possible for the synthesis of pyridine–pyridine (or related) linkages, in which by definition one of the coupling partners must be a nucleophilic pyridine. Returning to the use of pyridine 2-boronates, there are a number of reports of alternative strategies to achieve efficient Suzuki reactivity for these substrates, mainly focusing on the use of different boronate derivatives.12 However, due to issues of insufficient scope or the need for substrate-level reaction optimization, no single method has been widely embraced by the chemistry community. Given these considerations, it is evident that there is an urgent need for an efficient, reliable and operationally simple methodology to replace pyridine 2-boronates in cross-coupling chemistry. Desirable features for such a method would include: (i) stable and easy to prepare coupling partners; (ii) efficient reactions with both aryl bromide and chloride substrates; (iii) the ability to prepare the most challenging products, namely linked pyridine-heterocyclic molecules; (iv) tolerance to the use of a single catalyst for all of the various coupling combinations, involving both aryl- and heteroaryl substrates. The collaborative research described in this article delivers chemistry to addresses these issues.
Results and discussion
We recently initiated a program exploring the catalytic introduction of sulfur dioxide into organic molecules,13 and during the development of a palladium-catalyzed sulfinylative route to diaryl sulfones,14 we observed the formation of desulfinylated side-products, resulting in biaryl formation. Significantly, these side-products were more common when heterocycle-derived sulfinates were implicated as intermediates, relative to the corresponding carbocyclic variants. Intrigued by the apparent accelerated loss of sulfur dioxide from the heterocycle derivatives, we instigated the present investigation to determine if we could exploit desulfinylation in the development of a synthetically useful cross-coupling process for pyridine derivatives. The use of benzene-derived sulfinates as nucleophiles in palladium-catalyzed desulfinylative cross-coupling reactions is documented.15 However, examples of heterocycle-derived substrates are limited, and focus on the use of electron-rich five-membered variants.16The preparation of pyridine sulfinates is straightforward with a number of synthetically useful approaches available. For example, metallation of the parent pyridine followed by trapping with sulfur dioxide or a sulfur dioxide surrogate17 and the use of Cu-mediated or Pd-catalyzed sulfinylative processes are both viable routes.13b,18 However, for this study, our preferred method was the oxidation of the corresponding thiol using sodium hydroxide/hydrogen peroxide mixtures.19 This method exploits commercially available (or readily prepared) thiols and is a high yielding, operationally simple process. We began our investigation into the cross-coupling reactivity of pyridine sulfinates by studying the reaction between pyridine-3-sulfinate 1a and 4-bromotoluene, employing a variety of catalysts and reaction conditions. The optimized reaction conditions involved the use of a catalyst generated from the combination of palladium acetate and tricyclohexyl phosphine (Table 1). Although the sulfinate substrate itself is anionic, the use of an additional inorganic base was crucial to achieve an efficient reaction; potassium carbonate was optimal, delivering the 3-arylated pyridine product (2a) in 99% yield. Although some flexibility in the metal cation partner of the sulfinate was possible, with lithium, sodium and potassium sulfinates all being competent substrates, the identity of the base was more constrained with only potassium and cesium carbonate being effective. A reaction temperature of 150 °C was needed to achieve complete conversion.
|
|
||
|---|---|---|
| Entry | Variation from (eqn (1)) | Yield |
| a Reaction conditions: 1a (2.0 equiv.), 4-bromotoluene (1.0 equiv.), K2CO3 (1.5 equiv.), Pd(OAc)2 (5 mol%), PCy3 (10 mol%). Isolated yields. | ||
| 1 | Cs2CO3 in place of K2CO3 | 90% |
| 2 | Li2CO3 in place of K2CO3 | 0% |
| 3 | No base added | 0% |
| 4 | K sulfinate used in place of 1a | 77% |
| 5 | 100 °C | 29% |
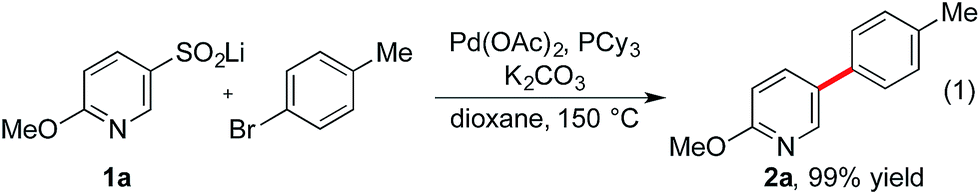
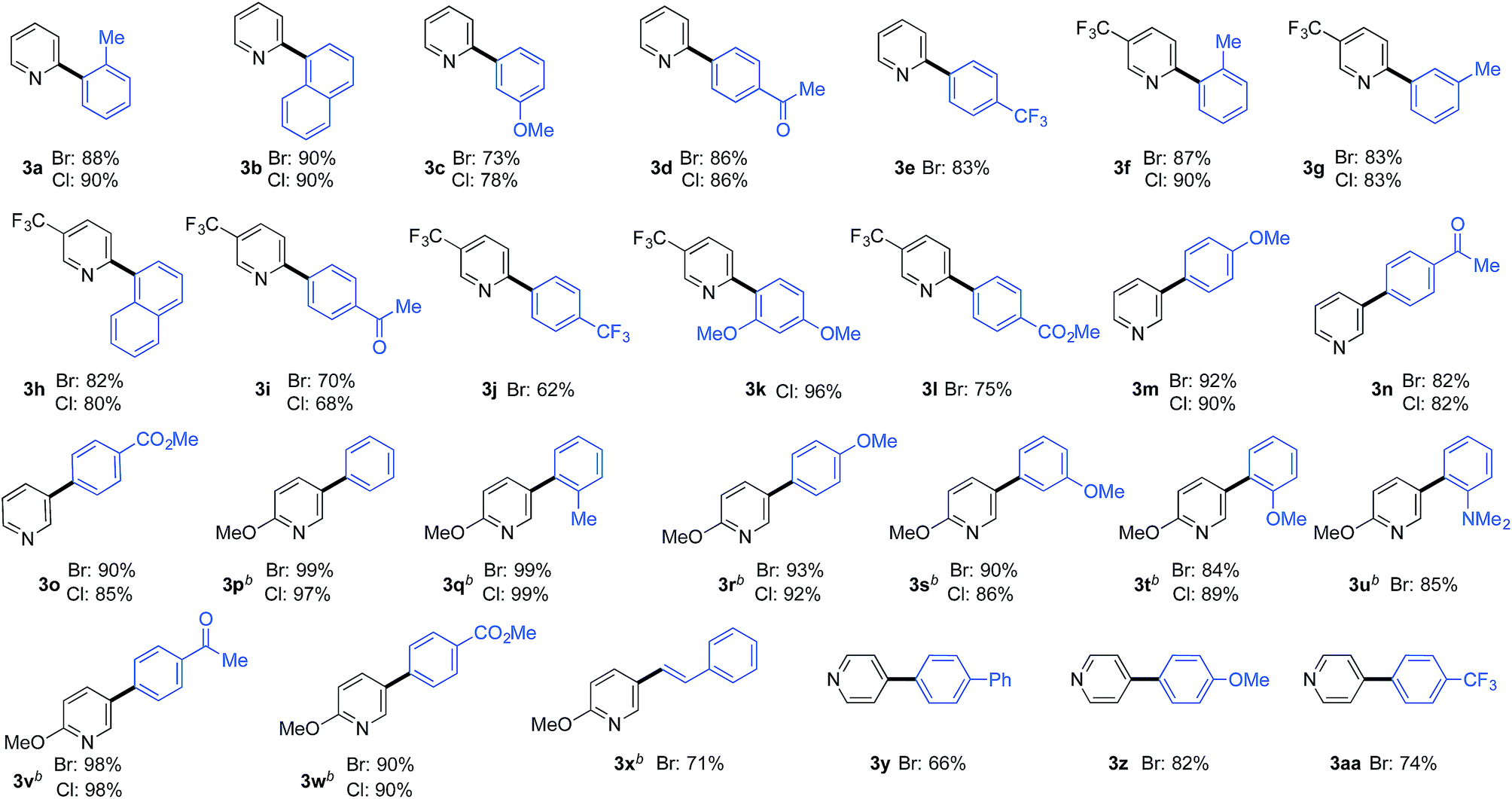
Pyridine-2-sulfinates bearing a variety of substituents were readily prepared and evaluated in coupling reactions with 4-bromo and 4-chlorotoluene (Table 2). Pleasingly, the simplest unsubstituted pyridine 2-sulfinate delivered the expected coupled product (2b) in excellent yields from both the bromo- and chloro-coupling partners. The ability to routinely employ chloro-derivatives has benefits from both cost of goods and availability perspectives.20 Although the simple unadorned pyridine-sulfinate derivative superficially appears to be the most straightforward example to employ in coupling chemistry, the lack of substituents to moderate the basicity and coordinating ability of the nitrogen atom in fact adds to the challenge of successfully using this substrate.11 The remainder of the examples feature substituted 2-pyridyl sulfinates (2c–2l) and show that both electron-donating and electron-withdrawing substituents can be tolerated in a variety of positions around the pyridine core, in all cases providing the coupled products in high yields. The ability to prepare Cl-derivative 2l is particularly notable, as the Cl-substituent provides a useful functional group for further manipulation. In addition, pyridine-3-sulfinate itself (2m), as well as derivatives featuring both electron-donating and -withdrawing substituents performed well with both the Br- and Cl-coupling partners (2a, 2n), as did the unsubstituted 4-pyridine derivative (2o).

A large selection of substituted benzene-derivatives were readily employed as coupling partners with the parent pyridine-2-sulfinate (3a–e), 5-trifluoromethylpyridine-2-sulfinate (3f–l), pyridine-3-sulfinate (3m–o), 6-methoxypyridine-3-sulfinate (3p–x), and pyridine-4-sulfinate (3y–aa) (Table 3a), demonstrating excellent tolerance to the nature and position of substituents. Notable amongst these examples are substrates bearing sterically demanding ortho-substituents (3a,f,k,q,t,u), as well as electronically challenging substrates such as the 2,4-dimethoxy-chlorobenzene substrate used to deliver coupled product (3k). An alkenyl halide coupling partner was also successfully used (3x).
| (b) Variation of heteroaryl coupling partners with pyridine sulfinates |
|---|
|
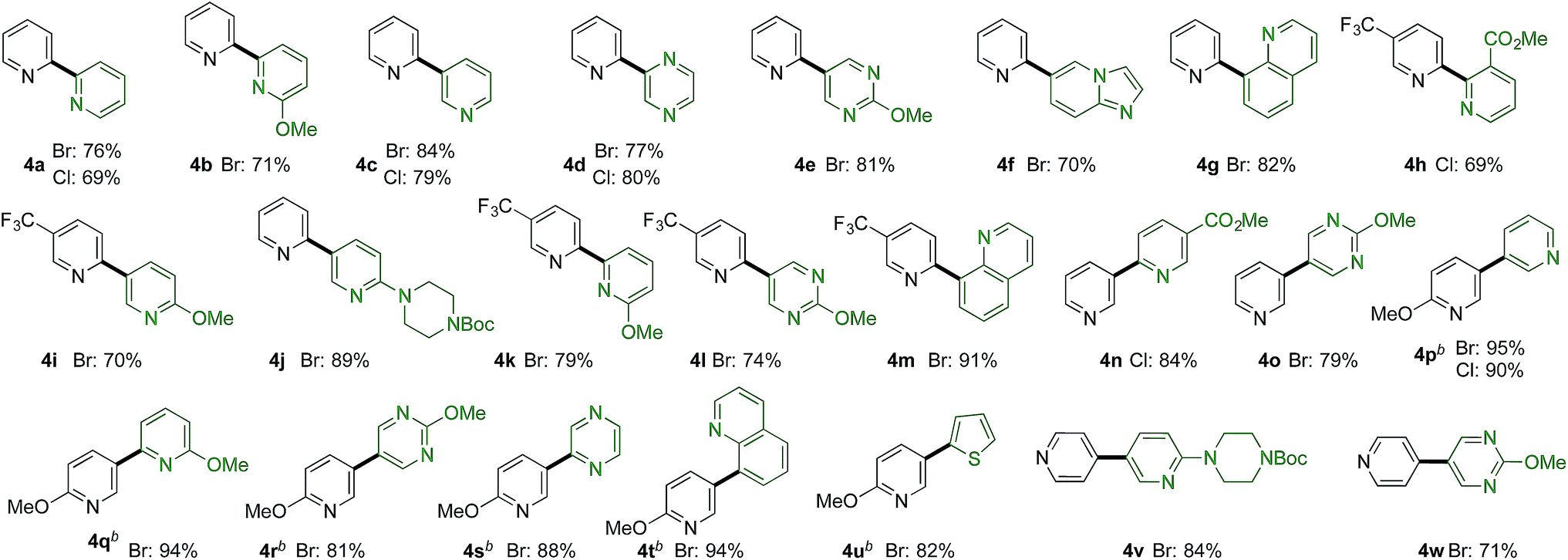
More challenging than combining pyridine-sulfinates with benzene-derivatives is to combine them with heteroaromatic coupling partners to deliver linked bis-heterocyclic products (Table 3b). Again, product 4a appears deceptively simple, but the challenge in achieving the union of two unsubstituted pyridine units in a 2,2′-linkage is significant using conventional coupling strategies. 2,2′-Linked bipyridines featuring a variety of substituents were also prepared (4b,h,k), and are of interest as this motif is the most common connection found in bipyridine ligands for metal-mediated catalysis. In addition to a variety of substituted pyridine coupling partners, heterocycles such as pyrazines (4d), pyrimidines (4e,l), quinolines (4g,m) and imidopyridine (4f) could be combined with pyridine-2-sulfinates without incident. Pyridine-3-sulfinates (4n–u) and pyridine-4-sulfinate (4v,w) were similarly tolerant, allowing access to a broad range of linked heterocycles.
The reactions reported in Tables 2 and 3 were routinely performed on a 0.2 mmol scale in sealed reaction tubes. Although we have never experienced a pressure event during these reactions, for larger scale experiments we deemed it prudent to move to an atmospheric pressure reaction vessel. Accordingly, 7 grams (20 mmol) of bipyridine 4j were prepared using a standard round-bottom flask equipped with a reflux condenser. For this larger scale transformation we employed modified reaction conditions, and required only 1 mol% of palladium and 2 mol% of phosphine ligand, and only 1.5 equivalents of the sulfinate to deliver the coupled product in 92% yield. Dibutyl ether was used as solvent, and the reaction was heated to 140 °C (Scheme 1).
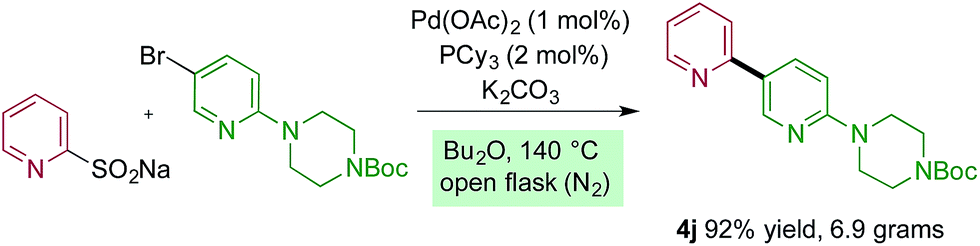 | ||
| Scheme 1 Preparative scale synthesis of bipyridine 4j. Reaction conditions pyridine sodium sulfinate (1.5 equiv.), aryl halide (1.0 equiv.), Pd(OAc)2 (1 mol%), PCy3 (2 mol%), K2CO3 (1.5 equiv.), Bu2O, 140 °C, 16 hours. | ||
Tables 2 and 3 illustrate the utility of pyridine sulfinate nucleophiles to produce a wide range of structurally varied coupled products. Further, with a focus on possible applications in medicinal chemistry we explored this methodology in the context of library chemistry, a key enabler of rapid Structure Activity Relationship (SAR) exploration in early drug discovery programs. The primary goal of library chemistry is to rapidly synthesize and purify a defined set of analogues using a broad set of readily available reactants. The analogues prepared are subsequently tested in vitro to refine the medicinal chemist's understanding of SAR. In this approach, reaction scope and synthetic efficiency (speed) are of prime importance over isolated yield, and therefore automated mass-triggered HPLC is used for rapid isolation of the minimum amount of product necessary for testing. In this context, we prepared a collection of pyridyl-coupled derivatives of the smoking cessation treatment varenicline.21 Specifically, a chlorinated derivative (5) of varenicline was combined with eight pyridyl sulfinates (Scheme 2). The sulfinates were used after two months of storage under ambient conditions, thus establishing their credentials as stable, storable building blocks. We also performed an array experiment in the reverse format. In this case we prepared a complex lithium 2-pyridyl sulfinate (6), which was combined with seven pyridine or pyridine-like heteroaryl bromides to produce derivatives of mepyramine, an antihistamine compound that targets the H1-receptor.22 We used a variation of the optimized reaction conditions for these reactions, employing only 1.2 equivalents of the sulfinate, compared to the 2.0 equivalents used in the standard conditions. This change reflects the complexity of the sulfinate, and the need to use valuable, late-stage sulfinates such as 6 in the most economical way possible. For the reasons mentioned above, yields obtained in array-type automated syntheses are often lower than isolated reactions, and not representative of the robustness of a specific transformation. In both the mepyramine and varenicline arrays all of the reactants returned the desired coupling products, thereby validating the potential of this novel methodology for library chemistry applications.
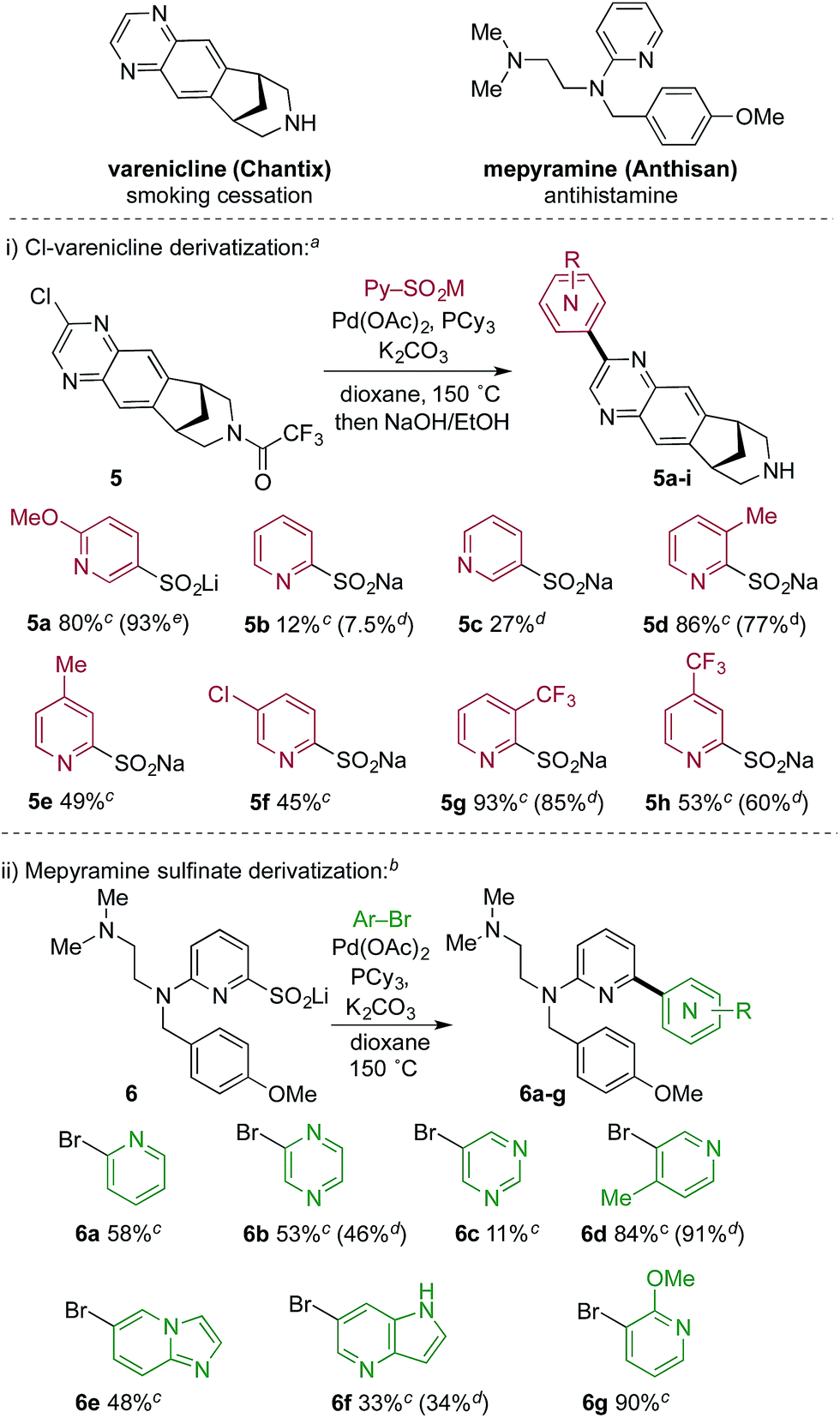 | ||
| Scheme 2 Application of pyridine sulfinate coupling reaction to medicinal chemistry targets. aReaction conditions for varenicline library: 0.10 mmol scale, varenicline-Cl (1.0 equiv.), sulfinate (2.0 equiv.), Pd(OAc)2 (5 mol%), PCy3 (7.5 mol%), K2CO3 (2.0 equiv.), 12 h in a sealed 0.5–2 mL microwave vial in a 150 °C aluminum block; then, in the same pot, 1.0 M aq. NaOH and EtOH and heated at 50 °C for 1 h. bReaction conditions for mepyramine library: 0.083 mmol scale, Het-Br (1.0 equiv.), sulfinate core (1.2 equiv.), Pd(OAc)2 (5 mol%), PCy3 (7.5 mol%), K2CO3 (2 equiv.), 12 h in a sealed 0.5–2 mL microwave vial in a 150 °C aluminum block. cYield after automated HPLC purification. dNMR yield based on analysis of the crude product prior to purification. eYield from an isolated single experiment. | ||
Conclusions
We have shown that pyridine sulfinates are stable and straightforward to prepare nucleophilic coupling partners for palladium catalyzed cross-coupling reactions with aryl and heteroaryl halides. The scope with respect to the halides coupling partner is considerable, and allows the preparation of a broad range of linked pyridines. We have shown that application of the method to medicinally relevant molecules is possible, including in the context of library synthesis. As the ability to employ pyridine derivatives in efficient cross-coupling processes is essential for medicinal chemistry, we anticipate that these attributes will lead to brisk exploitation of this methodology. It is likely that these reagents could also find application in related transition metal catalyzed processes for which aryl and heteroaryl boronates are employed.Acknowledgements
Financial support for this work was provided by the EPSRC and Pfizer, Inc. In addition, we acknowledge helpful discussions with Yuan Zhang, and automated purification by Jacob Belval, Joseph Cantone, and Bhagyashree Khunte, all of Pfizer Medicinal Sciences. BNR, DCB, and VM are employees of Pfizer Inc. and may own stocks in the company.Notes and references
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef.
- (a) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC; (b) D. G. Brown and J. Bostrom, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- C. C. C. Johansson Seechurn, A. DeAngelis and T. J. Colacot, in New Trends in Cross-Coupling: Theory and Applications, The Royal Society of Chemistry, 2015, pp. 1–19, 10.1039/9781782620259-00001.
- P. A. Cox, A. G. Leach, A. D. Campbell and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2016, 138, 9145–9157 CrossRef CAS PubMed.
- G. Bold, A. Fässler, H.-G. Capraro, R. Cozens, T. Klimkait, J. Lazdins, J. Mestan, B. Poncioni, J. Rösel, D. Stover, M. Tintelnot-Blomley, F. Acemoglu, W. Beck, E. Boss, M. Eschbach, T. Hürlimann, E. Masso, S. Roussel, K. Ucci-Stoll, D. Wyss and M. Lang, J. Med. Chem., 1998, 41, 3387–3401 CrossRef CAS PubMed.
- J. J. Cui, M. Tran-Dubé, H. Shen, M. Nambu, P.-P. Kung, M. Pairish, L. Jia, J. Meng, L. Funk, I. Botrous, M. McTigue, N. Grodsky, K. Ryan, E. Padrique, G. Alton, S. Timofeevski, S. Yamazaki, Q. Li, H. Zou, J. Christensen, B. Mroczkowski, S. Bender, R. S. Kania and M. P. Edwards, J. Med. Chem., 2011, 54, 6342–6363 CrossRef CAS PubMed.
- T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, P. W. Collins, S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, J. N. Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert, A. W. Veenhuizen, Y. Y. Zhang and P. C. Isakson, J. Med. Chem., 1997, 40, 1347–1365 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801–805 CrossRef CAS PubMed.
- D. Saccone, C. Magistris, N. Barbero, P. Quagliotto, C. Barolo and G. Viscardi, Materials, 2016, 9, 137 CrossRef.
- D. Blakemore, in Synthetic Methods in Drug Discovery, The Royal Society of Chemistry, 2016, vol. 1, pp. 1–69 Search PubMed.
- (a) K. L. Billingsley and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 4695–4698 CrossRef CAS PubMed; (b) Y. Yamamoto, M. Takizawa, X. Q. Yu and N. Miyaura, Angew. Chem., Int. Ed., 2008, 47, 928–931 CrossRef CAS PubMed; (c) J. Z. Deng, D. V. Paone, A. T. Ginnetti, H. Kurihara, S. D. Dreher, S. A. Weissman, S. R. Stauffer and C. S. Burgey, Org. Lett., 2009, 11, 345–347 CrossRef CAS PubMed; (d) G. R. Dick, E. M. Woerly and M. D. Burke, Angew. Chem., Int. Ed., 2012, 51, 2667–2672 CrossRef CAS PubMed; (e) P. B. Hodgson and F. H. Salingue, Tetrahedron Lett., 2004, 45, 685–687 CrossRef CAS; (f) K. Chen, R. Peterson, S. K. Math, J. B. LaMunyon, C. A. Testa and D. R. Cefalo, Tetrahedron Lett., 2012, 53, 4873–4876 CrossRef CAS; (g) W. Ren, J. Li, D. Zou, Y. Wu and Y. Wu, Tetrahedron, 2012, 68, 1351–1358 CrossRef CAS.
- (a) B. Nguyen, E. J. Emmett and M. C. Willis, J. Am. Chem. Soc., 2010, 132, 16372–16373 CrossRef CAS PubMed; (b) E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2014, 53, 10204–10208 CrossRef CAS PubMed; (c) A. S. Deeming, C. J. Russell and M. C. Willis, Angew. Chem., Int. Ed., 2016, 55, 747–750 CrossRef CAS PubMed; (d) A. Shavnya, S. B. Coffey, A. C. Smith and V. Mascitti, Org. Lett., 2013, 15, 6226–6229 CrossRef CAS PubMed; (e) A. Shavnya, K. D. Hesp, V. Mascitti and A. C. Smith, Angew. Chem., Int. Ed., 2015, 54, 13571–13575 CrossRef CAS PubMed.
- E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2013, 52, 12679–12683 CrossRef CAS PubMed.
- (a) C. Zhou, Q. Liu, Y. Li, R. Zhang, X. Fu and C. Duan, J. Org. Chem., 2012, 77, 10468–10472 CrossRef CAS PubMed; (b) D. H. Ortgies, A. Barthelme, S. Aly, B. Desharnais, S. Rioux and P. Forgione, Synthesis, 2013, 45, 694–702 CrossRef CAS; (c) D. H. Ortgies, A. Hassanpour, F. Chen, S. Woo and P. Forgione, Eur. J. Org. Chem., 2016, 2016, 408–425 CrossRef CAS.
- (a) S. Sévigny and P. Forgione, Chem.–Eur. J., 2013, 19, 2256–2260 CrossRef PubMed; (b) S. Sévigny and P. Forgione, New J. Chem., 2013, 37, 589 RSC; (c) D. Mangel, C. Buonomano, S. Sevigny, G. Di Censo, G. Thevendran and P. Forgione, Heterocycles, 2015, 90, 1228–1239 CrossRef CAS.
- B. Skillinghaug, J. Rydfjord and L. R. Odell, Tetrahedron Lett., 2016, 57, 533–536 CrossRef CAS.
- J. M. Baskin and Z. Wang, Tetrahedron Lett., 2002, 43, 8479–8483 CrossRef CAS.
- T. Kamiyama, S. Enomoto and M. Inoue, Chem. Pharm. Bull., 1988, 36, 2652–2653 CrossRef CAS.
- Data taken from the eMolecules database using the MNMiner interface (September, 2016). Commercially available (hetero)aryl iodides 59
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 463; (hetero)aryl bromides 528562; (hetero)aryl chlorides 1404653.
463; (hetero)aryl bromides 528562; (hetero)aryl chlorides 1404653. - J. W. Coe, P. R. Brooks, M. G. Vetelino, M. C. Wirtz, E. P. Arnold, J. Huang, S. B. Sands, T. I. Davis, L. A. Lebel, C. B. Fox, A. Shrikhande, J. H. Heym, E. Schaeffer, H. Rollema, Y. Lu, R. S. Mansbach, L. K. Chambers, C. C. Rovetti, D. W. Schulz, F. D. Tingley and B. T. O'Neill, J. Med. Chem., 2005, 48, 3474–3477 CrossRef CAS PubMed.
- C. P. Huttrer, C. Djerassi, W. L. Beears, R. L. Mayer and C. R. Scholz, J. Am. Chem. Soc., 1946, 68, 1999–2002 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc00675f |
| This journal is © The Royal Society of Chemistry 2017 |