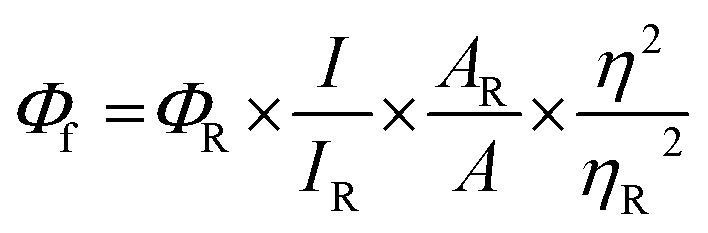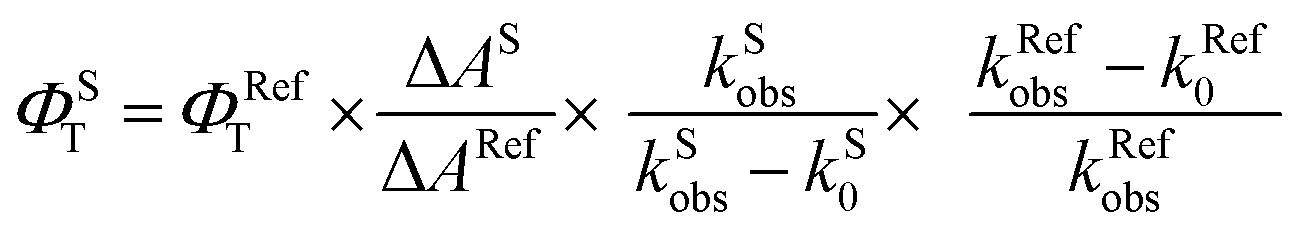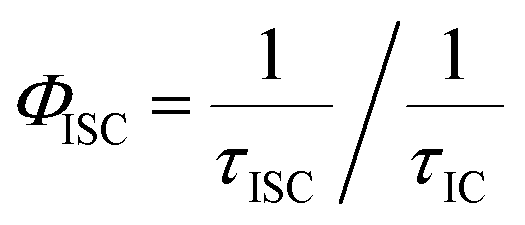DOI:
10.1039/C6SC05126J
(Edge Article)
Chem. Sci., 2017,
8, 1776-1782
Enhanced intersystem crossing in core-twisted aromatics†
Received
21st November 2016
, Accepted 19th December 2016
First published on 20th December 2016
Abstract
We describe the design, bottom-up synthesis and X-ray single crystal structure of systematically twisted aromatics 1c and 2d for efficient intersystem crossing. Steric congestion at the cove region creates a nonplanar geometry that induces a significant yield of triplet excited states in the electron-poor core-twisted aromatics 1c and 2d. A systematic increase in the number of twisted regions in 1c and 2d results in a concomitant enhancement in the rate and yield of intersystem crossing, monitored using femtosecond and nanosecond transient absorption spectroscopy. Time-resolved absorption spectroscopic measurements display enhanced triplet quantum yields (ΦT = 10 ± 1% for 1c and ΦT = 30 ± 2% for 2d) in the twisted aromatics when compared to a negligible ΦT (<1%) in the planar analog 3c. Twist-induced spin–orbit coupling via activated out-of-plane C–H/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibrations can facilitate the formation of triplet excited states in twisted aromatics 1c and 2d, in contrast to the negligible intersystem crossing in the planar analog 3c. The ease of synthesis, high solubility, access to triplet excited states and strong electron affinity make such imide functionalized core-twisted aromatics desirable materials for organic electronics such as solar cells.
C vibrations can facilitate the formation of triplet excited states in twisted aromatics 1c and 2d, in contrast to the negligible intersystem crossing in the planar analog 3c. The ease of synthesis, high solubility, access to triplet excited states and strong electron affinity make such imide functionalized core-twisted aromatics desirable materials for organic electronics such as solar cells.
Introduction
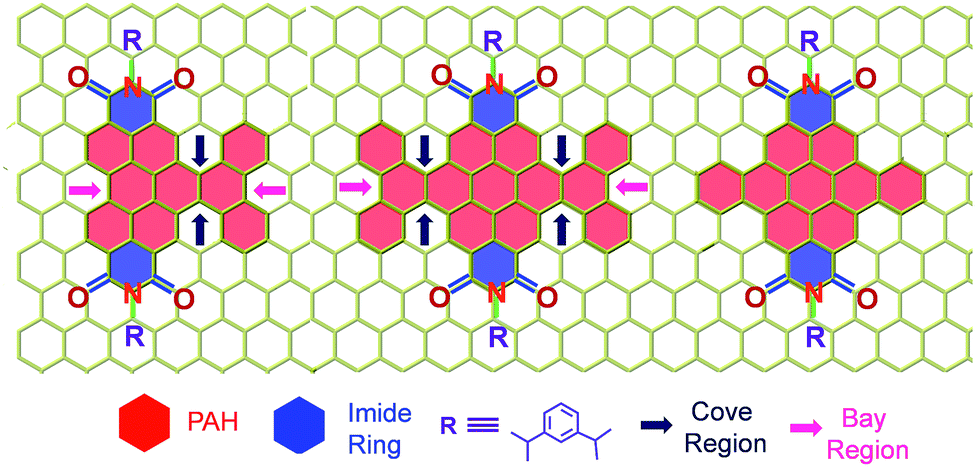
Carbon based contorted nanostructures1 are emerging as vital components for optoelectronic devices,2 drug-delivery,3 catalysis4 and sensors.5 Planar nanostructures of carbon continue to attract immense interest for diverse applications6 despite a low singlet–triplet energy gap and weak spin–orbit coupling (SOC) diminishing intersystem crossing (ISC) in graphene.7 Enhancement of SOC in graphene has been achieved by dilute hydrogenation,8 fluorination,9 or proximity to WS2.10 Graphene grown on Cu, gold intercalated graphene grown on Ni,11 and Pb intercalated graphene grown on Ir12 show strong SOC (ca. 20–100 meV). Heavy adatoms (with partially filled p orbitals) deposited on a graphene lattice, also induce large intrinsic SOC.13 Hydrogenation of graphene generates non-planar sp3 sites that are responsible for the induced SOC, whereas other adatoms exhibit a heavy atom effect in promoting the ISC in graphenoid structures. Interestingly, curvature dependent excited state properties such as ISC were observed in fullerene derivatives.14 State-of-the-art theoretical and experimental investigations15 validate the importance of twists/nonplanarity in enhancing the SOC in graphenoid structures (Table S1 (ESI†)).16 Systematic incorporation of twists to activate ISC in heavy atom free17 sp2 hybridized graphenoid structures18 (Scheme 1) has received less attention.7 Though the effect of non-planarity on SOC is established in organic molecules,19 the phenomenon could not be generalized due to the observation of quantitative fluorescence in highly twisted chromophores.20
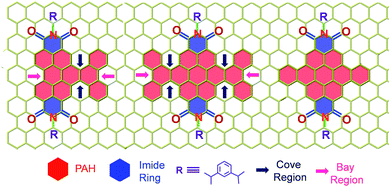 |
| | Scheme 1 Molecular structure of the twisted derivatives 1c and 2d along with the planar derivative 3c (arrows indicate the cove and bay regions in the derivatives). | |
Our on-going interest in core-twisted21 organic chromophores22 prompted us to study the role of twisting in triplet formation. Recent efforts from our group on bay substitution with multiple bromine atoms revealed the core-twisted geometry of perylenediimide which results in an enhancement in triplet generation.21 Though core-twisting of perylenediimide through bay substitution is well established, the studies were not focused on the intersystem crossing properties of the materials.23 To isolate the influence of twisting from the heavy atom effect, it is imperative to impart heavy atom-free twisting in the chromophoric structure. Bottom-up approaches to synthesize non-planar aromatics,24 that include hexabenzocoronene,25 hexabenzoovalene,26 dibenzotetrathienocoronene,27 octabenzocircumbiphenyl,28 dimeric29 and core-twisted perylenediimides,30 are still of emerging interest for diverse opto-electronic applications. Hydrogen–hydrogen repulsion induced steric congestion at the cove region of the extended perylenediimide chromophore resulted in core-twisted aromatics 1c and 2d (Scheme 1) having π-extension lengths of 1.1–1.6 nm. The presence of imide in the nonplanar derivatives 1c and 2d improves (i) electron affinity;31 (ii) access to precisely functionalized edges32 and (iii) chemical/thermal/photochemical stability.33 We herein report the first systematic investigation on “twist-only” induced intersystem crossing (kISC = 1 × 109 s−1 for 1c and kISC = 4 × 1010 s−1 for 2d) in imide functionalized core-twisted aromatics. Time-resolved absorption spectroscopic measurements display enhanced triplet quantum yields (ΦT = 10 ± 1% for 1c and ΦT = 30 ± 2% for 2d) in twisted aromatics when compared to a negligible ΦT (<1%) in the planar analog 3c.
Results and discussion
Synthesis and characterization
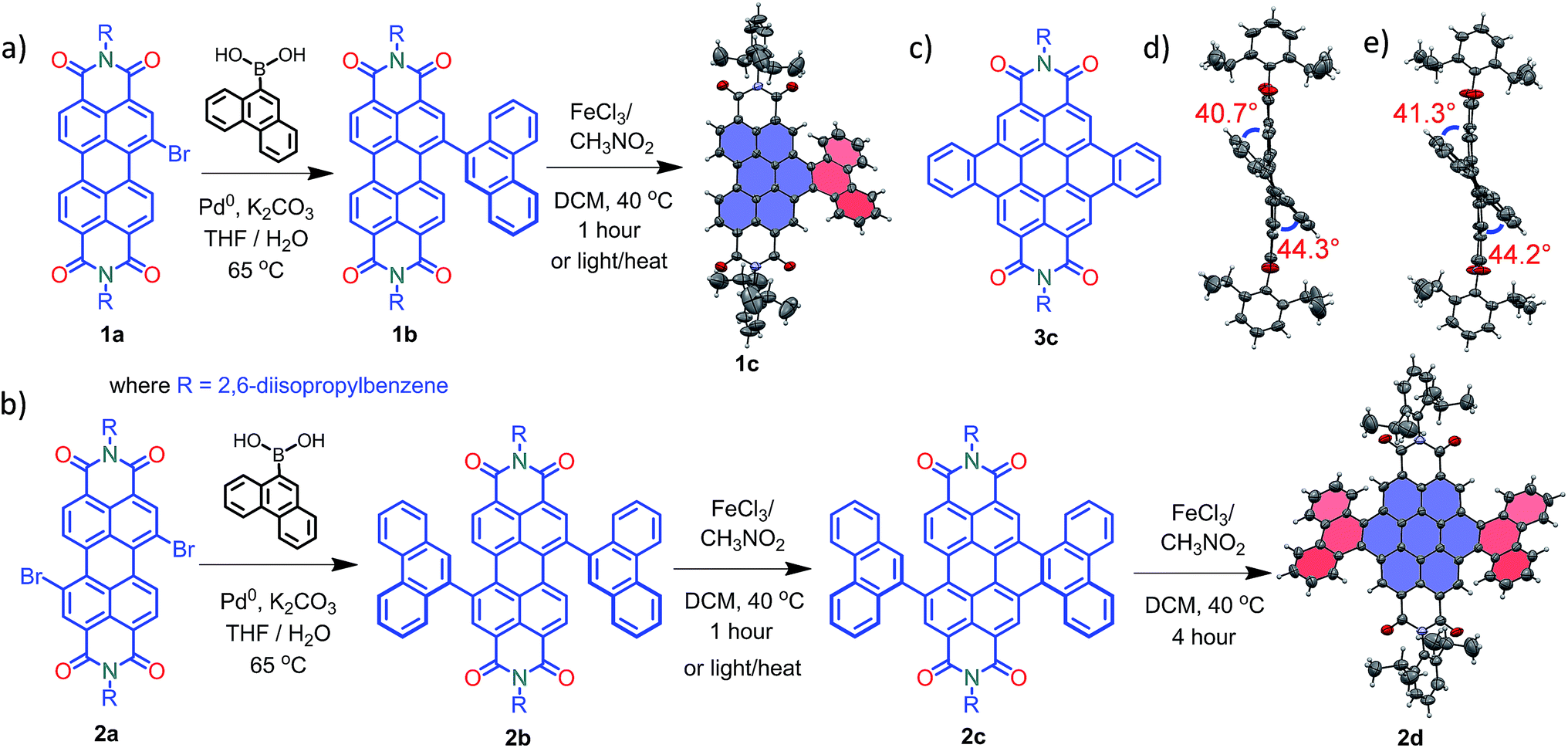
Compounds 1c and 2d were synthesized via Suzuki coupling of one and two phenanthrene units, respectively, with perylenediimide (PDI) followed by the metal catalyzed Scholl dehydrogenation reaction (Fig. 1a and b). Bromination and imidisation of 1 were performed by following the procedure reported elsewhere.211a and 2a were treated with one and two equivalents of 9-phenanthreneboronic acid to yield 1b and 2b respectively (ESI†). Further, 1b and 2b underwent the Scholl reaction with FeCl3 in dry DCM/CH3NO2 solution under nitrogen. Between 0–30 °C, cyclized products from 1b and 2b were formed in low yield (<1%). At a higher temperature (40 °C), the desired products 1c and 2d were obtained in 50% and 40% yields, respectively, under continuous nitrogen flow (Fig. S1–S4, ESI†). Intermediate 2c (5% yield) was also isolated during the reaction and characterized using spectroscopic methods. A model planar derivative 3c (Fig. 1c) was synthesized via Suzuki coupling of two benzene units with 1,7-dibromo PDI (2a) followed by the Scholl dehydrogenation reaction. Red fluorescent single crystals of 1c and 2d were obtained from chloroform and toluene solutions, respectively.
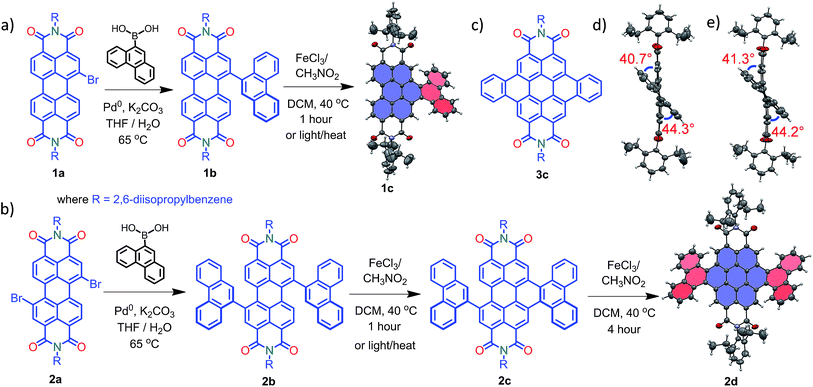 |
| | Fig. 1 Synthetic route to the core-twisted aromatics (a) 1c and (b) 2d; (c) molecular structure of the planar analog 3c; (d) and (e) twist angles at the cove regions in 1c and 2d. | |
Crystal structure analysis
Compound 1c crystallized in the tetragonal space group P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3, containing 4 molecules per unit cell (Table S2, ESI†). Repulsion between the H1′ and H8′ atoms of the phenanthrene unit and the H2 and H11 atoms of the PDI unit at the cove region (Fig. S5a, ESI†) resulted in twisting of the chromophore 1c. The twist angle was calculated from the angle between the perylenediimide and phenanthrene planes in the crystal structure (Fig. S5b and d, ESI†). The twist angles at the two cove regions of compound 1c were found to be 44.3° and 40.7° (Fig. 1d). Compound 2d possessing a waggling1 conformation crystallized in the triclinic space group P
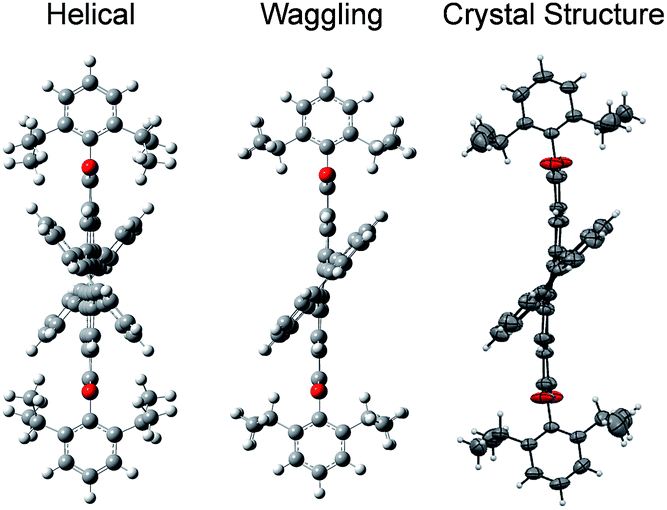
3, containing 4 molecules per unit cell (Table S2, ESI†). Repulsion between the H1′ and H8′ atoms of the phenanthrene unit and the H2 and H11 atoms of the PDI unit at the cove region (Fig. S5a, ESI†) resulted in twisting of the chromophore 1c. The twist angle was calculated from the angle between the perylenediimide and phenanthrene planes in the crystal structure (Fig. S5b and d, ESI†). The twist angles at the two cove regions of compound 1c were found to be 44.3° and 40.7° (Fig. 1d). Compound 2d possessing a waggling1 conformation crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with one molecule per unit cell (Table S2, ESI†). Repulsion between the H1′ and H8′ atoms of the phenanthrene units and the hydrogen atoms in the ortho region (H2, H5, H8 and H11) of PDI (Fig. S5c, ESI†) twist the chromophore 2d at the 4 cove regions with a twist angle of 44.2° and 41.3° (Fig. 1e). To evaluate the thermodynamic stability between the helical and waggling1 (Fig. 2) conformations of the derivative 2d, we conducted density functional theory (DFT) calculations at the B3LYP/6-311G++(d,p) level.34 From the DFT calculations, it is estimated that the waggling conformer of 2d is thermodynamically more stable than the helical conformer by 17.5 kcal mol−1, which is in agreement with the waggling conformation obtained from the single crystal X-ray structure (Fig. 2). Single crystal X-ray structure analysis of 1c and 2d revealed a core-twisted polycyclic skeleton having 9 and 13 aromatic rings, respectively.
, with one molecule per unit cell (Table S2, ESI†). Repulsion between the H1′ and H8′ atoms of the phenanthrene units and the hydrogen atoms in the ortho region (H2, H5, H8 and H11) of PDI (Fig. S5c, ESI†) twist the chromophore 2d at the 4 cove regions with a twist angle of 44.2° and 41.3° (Fig. 1e). To evaluate the thermodynamic stability between the helical and waggling1 (Fig. 2) conformations of the derivative 2d, we conducted density functional theory (DFT) calculations at the B3LYP/6-311G++(d,p) level.34 From the DFT calculations, it is estimated that the waggling conformer of 2d is thermodynamically more stable than the helical conformer by 17.5 kcal mol−1, which is in agreement with the waggling conformation obtained from the single crystal X-ray structure (Fig. 2). Single crystal X-ray structure analysis of 1c and 2d revealed a core-twisted polycyclic skeleton having 9 and 13 aromatic rings, respectively.
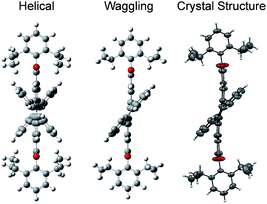 |
| | Fig. 2 Possible helical and waggling conformations of the compound 2d. | |
Electrochemical properties
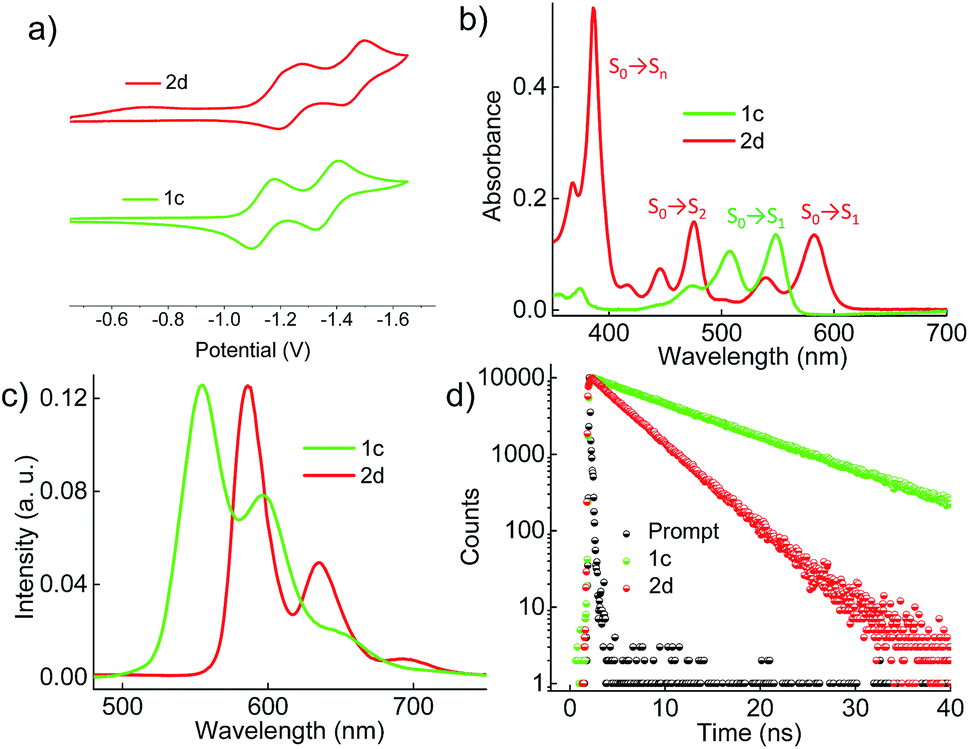
Cyclic voltammograms (0.1 M, nBu4NPF6 in DCM) exhibited reversible reduction peaks (Fig. 3a) at −1.14 and −1.37 V for 1c, and −1.22 and −1.46 V for 2d (Table S3, ESI†) with reference to the Fc/Fc+ electrode. The reduction potentials of the derivatives 1c and 2d are more negative than those of the model derivative 3c (−1.00 and −1.24 V), indicating that the 1c and 2d derivatives are significantly weaker electron acceptors. The highest occupied molecular orbitals (HOMOs) are distributed over the whole π system of the derivatives 1c, 2d and the model derivative 3c (Fig. S6, ESI†). In contrast, the lowest unoccupied molecular orbitals (LUMOs) spread only at the coronenediimide core, due to the presence of electron withdrawing imide groups.
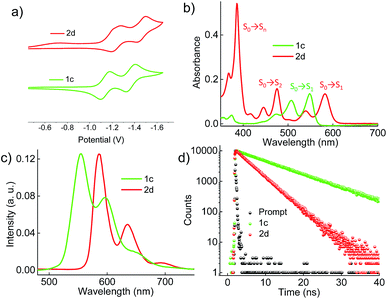 |
| | Fig. 3 (a) Electrochemical measurement of the derivatives in dry DCM with reference to the Fc/Fc+ electrode, (b) UV-Vis absorption spectra, (c) emission (λex = 480 nm), and (d) time-dependent fluorescence spectra (λex = 480 nm and monitored at the respective emission maxima) of the derivatives 1c and 2d in toluene. | |
Photophysical characterization
By virtue of their twisted nature, derivatives 1c and 2d with large π-surface dissolve well in common organic solvents like chloroform, dichloromethane and toluene. The UV-Vis absorption spectrum (Fig. 3b) of 1c in toluene shows peaks centered at 475, 505 and 545 nm corresponding to π → π* (HOMO → LUMO) transitions (Table S4, ESI†). Derivative 2d in toluene exhibits a π → π* transition at (i) 582 and 539 nm corresponding to S0 → S1 (HOMO → LUMO); (ii) 475, 445 and 416 nm corresponding to S0 → S2 (HOMO−1 → LUMO); and (iii) 385 nm corresponding to S0 → Sn (HOMO → LUMO+1), in agreement with DFT calculations (Fig. 3b). The model derivative 3c exhibits peaks centered at 460 and 490 nm corresponding to π → π* (HOMO → LUMO) transitions as reported earlier.35 Upon excitation at 480 nm, 1c shows vibronically resolved emission (Fig. 3c) centered at 555, 597 and 652 nm with a fluorescence quantum yield (Φf) of 70%. Temperature dependent emission (λex = 480 nm) and excitation (λem = 600 nm) spectra of 1b in toluene indicated the evolution of a new species having fluorescence emission features identical to that of 1c (Fig. S7a and b, ESI†). Spectroscopic analysis confirms the photocyclization of 1b in toluene (ca. 1 μM) at higher temperature to yield 1c. When compared to 1c, derivative 2d exhibits red-shifted emission centered at 586, 635 and 696 nm with a Φf value of 40%. The model derivative 3c shows vibronically resolved emission centered at 510, 550 and 580 nm with a Φf value of 85% (Table S4 and Fig. S8a, ESI†). Partial reduction in the Φf values of 1c and 2d when compared with 3c could be attributed to the non-radiative decay pathways arising from the nonplanar nature of the chromophores 1c and 2d.16a Upon excitation at 480 nm, derivatives 1c and 2d in toluene exhibit fluorescence lifetimes (Fig. 3d) of 10 and 5.4 ns, respectively. The model derivative 3c in toluene shows a monoexponential fluorescence lifetime of 5.5 ns upon excitation at 480 nm (Fig. S8b, ESI†).
Nanosecond transient absorption measurements
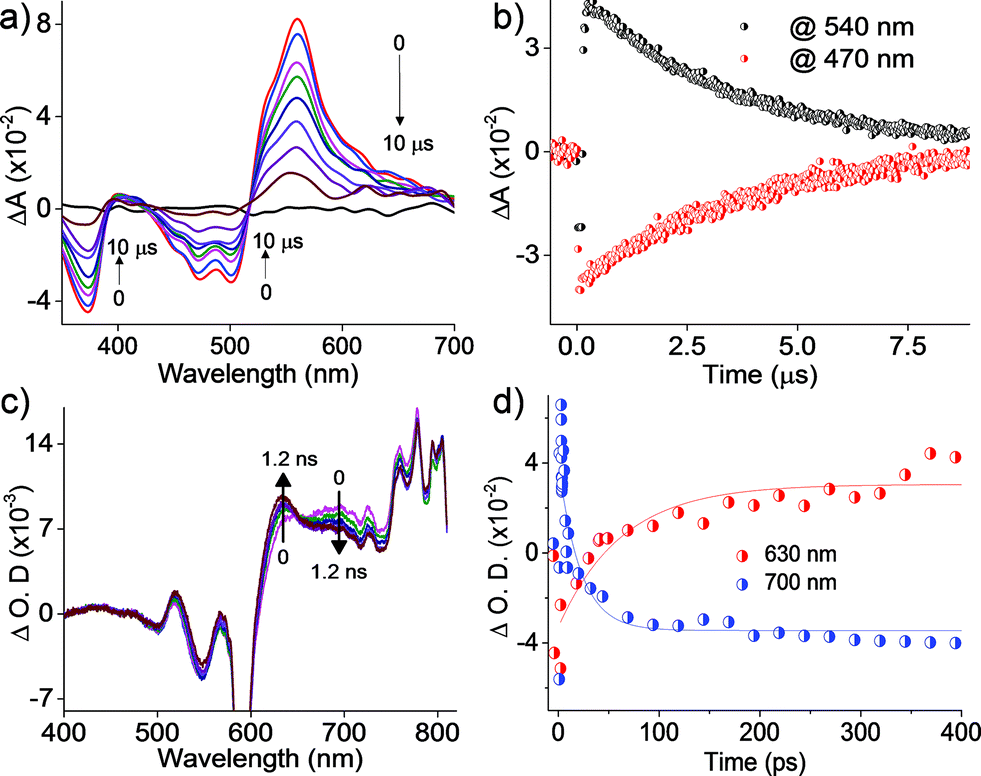
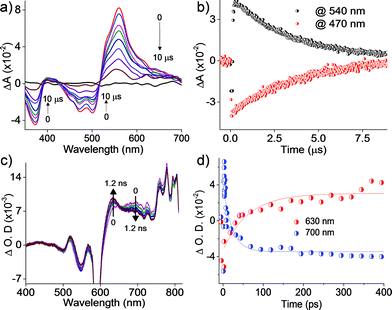
Further insights into the excited state deactivation in core-twisted derivatives came from nanosecond and femtosecond transient absorption measurements. Upon excitation with a 10 ns laser pulse at 355 nm, 1c in toluene (Fig. 4a) exhibited negative absorption peaks centered at 380, 470 and 510 nm corresponding to ground state depletion (S0 → Sn). Due to the stronger positive signal at 520–620 nm, ground state bleaching from 520 to 570 nm is not seen in the nTA spectra compared to the UV-Vis absorption spectra. Observed twin absorption centered at 400 and 580 nm with a single exponential decay lifetime of 3.7 μs (Fig. 4b) is attributed to a triplet excited state in 1c. Compound 2d in toluene (Fig. S9a, ESI†) showed ground state depletion at 390, 480 and 590 nm, consistent with the UV-Vis absorption spectrum. Transient absorption corresponding to a triplet excited state is observed at 340, 420, 560 and 610 nm with a lifetime of 19.6 μs (Table S4 and Fig. S9b, ESI†). The existence of a triplet excited state in 1c and 2d was further confirmed by quenching of the transient spectra by oxygen purging. In contrast, the planar derivative 3c in toluene exhibited negligible transient absorption upon excitation at 355 nm. Triplet quantum yields (ΦT) of 1c and 2d were calculated to be 10 ± 1% and 30 ± 2% (Table S4, ESI†), respectively, employing a triplet–triplet energy transfer method.21 Significant enhancement in the ΦT values of 1c and 2d when compared to the model derivative 3c is attributed to the twist induced SOC, as reported earlier.19 However, ISC was reported by Flamigni and coworkers in unsymmetrically substituted planar perylene derivatives which could be attributed to the nπ* to ππ* transition arising from bay imidisation.36 Attempts to record phosphorescence in the core-twisted derivatives 1c and 2d was not successful in DCM–EtOH glass at low temperature (77 K). However the derivatives 1c and 2d exhibited phosphorescence in DCM–EtOH–CH3I (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.1) glass at low temperature (77 K, Fig. S10, ESI†).
:1:0.1) glass at low temperature (77 K, Fig. S10, ESI†).
 |
| | Fig. 4 (a) nTA spectra and (b) corresponding decay profile of 1c upon excitation at 355 nm in toluene; (c) fTA spectra and (d) corresponding decay profile of 1c upon excitation at 300 nm in toluene. | |
Femtosecond transient absorption measurements
To unravel the kinetics of intersystem crossing, the core-twisted derivatives 1c and 2d in toluene were excited with a 110 fs laser pulse, at 300 nm. Femtosecond transient absorption (fTA) spectra of 1c and 2d showed a sharp negative absorption at 600 nm, corresponding to the second harmonic of the pump laser (2λex = 600 nm). Photoexcitation of 1c at 300 nm (Fig. 4c) displayed negative absorption at 505 and 545 nm along with positive absorption peaks centered at 510, 550, 630, 680 and 720 nm. Negative absorption observed at 505 and 545 nm could be attributed to ground state depletion consistent with the UV-Vis absorption spectrum. Singular value decomposition (SVD) of ΔA versus time and the wavelength based three-dimensional map of 1c followed by global analysis yielded three principle components (Fig. S11, ESI†). Negative absorption centered at 560 nm with a lifetime of 9.5 ns is ascribed to stimulated emission. Positive absorption centered at 720 nm corresponds to S1 → Sn transitions that decay with a lifetime of 70 ps (kIC = 0.14 × 1011 s−1; Fig. 4d). The rise time of an emerging positive absorption peak at 630 nm is estimated to be 1 ns (kISC = 1 × 109 s−1) and is ascribed to a T1 → Tn transition. Upon excitation at 300 nm, 2d in toluene (Fig. S9c, ESI†) showed ground state depletion at 475, 532 and 585 nm, consistent with the ground state absorption spectrum. SVD followed by global analyses of the positive absorption bands centered at 455, 516, 552 and 610–800 nm consist of three principal components (Fig. S12, ESI†). Negative absorption centered at 600 nm, with a lifetime of 5.1 ns is attributed to stimulated emission. The right singular vector at 720 nm decays with a lifetime of 6.5 ps (kIC = 1.54 × 1011 s−1) and corresponds to a S1 → Sn transition (Fig. S9d, ESI†). During the decay centered at 720 nm, concomitant appearance of a new band at 630 nm is observed (Fig. S9c, ESI†). The emerging band at 630 nm with a rise time (τISC) of 25 ps (kISC = 4 × 1010 s−1) is attributed to a T1 → Tn transition in the derivative 2d. According to the rates of internal conversion (kIC) and intersystem crossing (kISC), the efficiency of ISC (ΦISC = kISC/kIC) is calculated to be 7.1% and 26% for 1c and 2d, respectively, which is in agreement with the ΦT calculated from the triplet–triplet energy transfer method. Quantum chemical calculations (Fig. S13, ESI†) indicate that out of plane CC and C–H vibrations (νop) can allow efficient ISC from a ππ* type singlet to a ππ* type triplet driven by Herzberg–Teller vibronic coupling in the core-twisted derivatives 1c and 2d.37
Conclusions
In conclusion, we report the design and synthesis of solution processable electron deficient core-twisted aromatics, 1c and 2d. Femtosecond and nanosecond transient absorption measurements revealed “twist-only” induced ultrafast ISC in the non-planar derivatives 1c and 2d. Enhanced out of plane CC and C–H vibrations facilitate efficient ISC with ΦT values of 10 ± 1% and 30 ± 2% in the derivatives 1c and 2d, respectively, driven by Herzberg–Teller vibronic coupling. A higher kISC of 4 × 1010 s−1 for doubly twisted 2d, when compared to a kISC of 1 × 109 s−1 for singly twisted 1c, clearly establishes the role of non-planarity in facilitating ISC in the reported derivatives 1c and 2d. Ease of solution processability and activated triplet excited states in the twisted aromatics 1c and 2d are beneficial for solar energy conversion by virtue of their long-lived triplet excited states. Current efforts in our laboratory are directed towards developing twisted chromophores for high performance opto-electronic devices.
Experimental section
Spectral measurements
Absorption spectra were recorded using a Shimadzu UV-3600 UV-VIS-NIR while emission (fluorescence/phosphorescence) and excitation spectra were performed using a Horiba Jobin Yvon Fluorolog spectrometer. All spectroscopic experiments were performed using standard quartz cuvettes with path length of 1 cm for solutions in dried and distilled solvents. The solution state fluorescence quantum yields were determined using optically matched solutions. Fluorescein dissolved in ethanol (Φf = 0.79)38 was used as a standard. Φf values for the samples were calculated as follows,| | 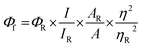 | (1) |
Nanosecond transient absorption measurements
Laser flash photolysis experiments with nitrogen purged solutions were carried out using Applied Photophysics Model LKS-60 laser kinetic spectrometer with the second and third harmonic (355 nm and 532 nm, pulse duration ≈10 ns) of a Quanta Ray INDI-40-10 series pulsed Nd:YAG laser. Triplet states of the contorted aromatics 1c and 2d in toluene were confirmed using measurements of oxygen purged solutions through nanosecond flash photolysis studies. Triplet quantum yields39 upon direct photoexcitation (355 nm) were determined using [Ru(bpy)]Cl2 in methanol as standard (ΦT = 1), with nonsaturating laser intensities. Equal volumes of a 0.2 mM solution of β-carotene were added to optically matched solutions of the reference and the sample. The equation for the triplet quantum yield is given by,| | 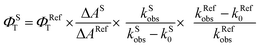 | (2) |
where, ΦST and ΦRefT denote the triplet quantum yields of the sample and reference, respectively; ΔAS and ΔARef are transient absorption intensities of β-carotene in the sample and reference, respectively; kSobs and kS0 are decay rates of the sample transient species before and after the addition of β-carotene. kRefobs and kRef0 are decay rates of the reference transient species before and after the addition of β-carotene.
Femtosecond transient absorption measurements
A Spectra-physics Tsunami Oscillator (80 MHz, 800 nm) was used as the seed for a Spectra-Physics Spitfire Regenerative amplifier (1 kHz, 4 mJ). A fraction of the amplified output was used to generate a 300 nm pump pulse. A residual 800 nm pulse was sent through a delay line inside an ExciPro pump-probe spectrometer from CDP Systems. A rotating CaF2 plate (2 mm thickness) was used to generate a continuum of white light from the delayed 800 nm pulse. The continuum of white light was split into two and the streams were used as probe and reference pulses. Transient absorption spectra were recorded using a dual diode array detector with a 200 nm detection window. Sample solutions were prepared in a rotating sample cell with a path length of 400 μm. IRF was determined by solvent (10% benzene in methanol) two photon absorption and was found to be approximately 130 fs at about 530 nm. Energy per pulse incident on the sample is attenuated, employing 80% neutral density filter when required. Toluene solution of the derivatives 1c and 2d were pumped with 300 nm, 200 nJ, ∼110 fs laser pulses and probed with the white light. Singular value decomposition (SVD) of ΔA versus time and a wavelength based three-dimensional map of the derivatives 1c and 2d was obtained from the fTA measurements. For SVD, the fTA spectra of 1c and 2d were constructed into a matrix using the Origin graphics software program (Version 8.5; MicroCal, Inc., Northampton, MA). Global analyses of the fTA spectra of the derivatives 1c and 2d were carried out using Glotaran (version 1.2).40
Efficiency of intersystem crossing (ΦISC) could be estimated from the rates of internal conversion (kIC) and intersystem crossing (kISC) as follows,41
| | 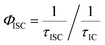 | (3) |
where,
τISC is the rate of intersystem crossing and
τIC is the rate of internal conversion; extracted from the fTA spectra analysis.
X-ray crystallography
High-quality specimens of appropriate dimensions were selected for the X-ray diffraction experiments. Crystallographic data collected are presented in the ESI.† Single crystals were mounted using oil (Infineum V8512) on a glass fibre. All measurements were made on a CCD area detector with graphite monochromated MoKα radiation. The data was collected using a Bruker APEXII detector and processed using an APEX2 from Bruker. All structures were solved by direct methods and expanded using Fourier techniques. The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were included in idealized positions, but not refined. Their positions were constrained relative to their parent atom using the appropriate HFIX command in SHELXL-97. The full validation of CIFs and structure factors of 1c and 2d were performed using the CheckCIF utility and found to be free of major alert levels. 3D structure visualization and the exploration of the crystal packing of the derivatives were carried out using Mercury 3.1.
Computational methods
Ground-state optimised structures and harmonic oscillator frequencies were computed using density functional theory (DFT) at the Becke's three parameter functional in combination with the Lee–Yang–Parr correlation functional (B3LYP) and 6-311++G(d,p) basis set. Vertical excitation energies and oscillator strengths were calculated employing time-dependent DFT (TD-DFT) at the B3LYP/6-311++G(d,p) level of theory. All computations were performed with the Gaussian 09 program suite.34
Acknowledgements
The authors thank the Nanobiotechnology Task Force, DBT, Govt. of India for the support, BT/PR5761/NNT/106/599/2012; A. P. Andrews for X-ray analysis; and K. Nagaraj for femtosecond transient absorption measurements. K. N. acknowledges the University Grants Commission (UGC), India; A. R. M. thanks the Council of Scientific & Industrial Research (CSIR) for the fellowship; and K. M. thanks INSPIRE for the fellowship.
Notes and references
- M. Ball, Y. Zhong, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. Xiao and C. Nuckolls, Acc. Chem. Res., 2015, 48, 267–276 CrossRef CAS PubMed.
- J. R. Sanchez-Valencia, T. Dienel, O. Groning, I. Shorubalko, A. Mueller, M. Jansen, K. Amsharov, P. Ruffieux and R. Fasel, Nature, 2014, 512, 61–64 CrossRef CAS PubMed.
- Z. Liu, A. C. Fan, K. Rakhra, S. Sherlock, A. Goodwin, X. Chen, Q. Yang, D. W. Felsher and H. Dai, Angew. Chem., Int. Ed., 2009, 48, 7668–7672 CrossRef CAS PubMed.
- N. Suzuki, Y. Wang, P. Elvati, Z.-B. Qu, K. Kim, S. Jiang, E. Baumeister, J. Lee, B. Yeom, J. H. Bahng, J. Lee, A. Violi and N. A. Kotov, ACS Nano, 2016, 10, 1744–1755 CrossRef CAS PubMed.
- A. Modi, N. Koratkar, E. Lass, B. Wei and P. M. Ajayan, Nature, 2003, 424, 171–174 CrossRef CAS PubMed.
-
(a) V. Urbanova, F. Karlicky, A. Matej, F. Sembera, Z. Janousek, J. A. Perman, V. Ranc, K. Cepe, J. Michl, M. Otyepka and R. Zboril, Nanoscale, 2016, 8, 12134–12142 RSC;
(b) A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- M. L. Mueller, X. Yan, J. A. McGuire and L.-S. Li, Nano Lett., 2010, 10, 2679–2682 CrossRef CAS PubMed.
- J. Balakrishnan, G. Kok Wai Koon, M. Jaiswal, A. H. Castro Neto and B. Ozyilmaz, Nat. Phys., 2013, 9, 284–287 CrossRef CAS.
- S. Irmer, T. Frank, S. Putz, M. Gmitra, D. Kochan and J. Fabian, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 115141 CrossRef.
- A. Avsar, J. Y. Tan, T. Taychatanapat, J. Balakrishnan, G. K. Koon, Y. Yeo, J. Lahiri, A. Carvalho, A. S. Rodin, E. C. O'Farrell, G. Eda, A. H. Castro Neto and B. Ozyilmaz, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- D. Marchenko, A. Varykhalov, M. R. Scholz, G. Bihlmayer, E. I. Rashba, A. Rybkin, A. M. Shikin and O. Rader, Nat. Commun., 2012, 3, 1232 CrossRef CAS PubMed.
- M. Kralj, Nat. Phys., 2015, 11, 11–12 CrossRef CAS.
- C. Weeks, J. Hu, J. Alicea, M. Franz and R. Wu, Phys. Rev. X, 2011, 1, 021001–021015 Search PubMed.
- J. W. Arbogast and C. S. Foote, J. Am. Chem. Soc., 1991, 113, 8886–8889 CrossRef CAS.
- T. Vosch, J. Hofkens, M. Cotlet, F. Köhn, H. Fujiwara, R. Gronheid, K. Van Der Biest, T. Weil, A. Herrmann, K. Müllen, S. Mukamel, M. Van der Auweraer and F. C. De Schryver, Angew. Chem., Int. Ed., 2001, 40, 4643–4648 CrossRef CAS PubMed.
-
(a) F. D. Lewis and X. Zuo, J. Am. Chem. Soc., 2003, 125, 8806–8813 CrossRef CAS PubMed;
(b) D. Huertas-Hernando, F. Guinea and A. Brataas, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 155426 CrossRef.
- O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed.
- Y. Li, Z. Jia, S. Xiao, H. Liu and Y. Li, Nat. Commun., 2016, 7, 11637 CrossRef PubMed.
- K. Schmidt, S. Brovelli, V. Coropceanu, D. Beljonne, J. Cornil, C. Bazzini, T. Caronna, R. Tubino, F. Meinardi, Z. Shuai and J.-L. Brédas, J. Phys. Chem. A, 2007, 111, 10490–10499 CrossRef CAS PubMed.
-
(a) Y. Zagranyarski, L. Chen, D. Jänsch, T. Gessner, C. Li and K. Müllen, Org. Lett., 2014, 16, 2814–2817 CrossRef CAS PubMed;
(b) F. Würthner, Pure Appl. Chem., 2006, 78, 2341 CrossRef;
(c) J. Hofkens, T. Vosch, M. Maus, F. Köhn, M. Cotlet, T. Weil, A. Herrmann, K. Müllen and F. C. De Schryver, Chem. Phys. Lett., 2001, 333, 255–263 CrossRef CAS;
(d) Z. Chen, U. Baumeister, C. Tschierske and F. Würthner, Chem.–Eur. J., 2007, 13, 450–465 CrossRef CAS PubMed.
- K. Nagarajan, A. R. Mallia, V. S. Reddy and M. Hariharan, J. Phys. Chem. C, 2016, 120, 8443–8450 CAS.
-
(a) A. R. Mallia, P. S. Salini and M. Hariharan, J. Am. Chem. Soc., 2015, 137, 15604–15607 CrossRef CAS PubMed;
(b) R. T. Cheriya, A. R. Mallia and M. Hariharan, Energy Environ. Sci., 2014, 7, 1661–1669 RSC.
-
(a) A. J. Jimenez, M.-J. Lin, C. Burschka, J. Becker, V. Settels, B. Engels and F. Wurthner, Chem. Sci., 2014, 5, 608–619 RSC;
(b) Y. Cai, L. Huo, X. Sun, D. Wei, M. Tang and Y. Sun, Adv. Energy Mater., 2015, 5, 1500032–1500036 CrossRef;
(c) P. Osswald and F. Würthner, J. Am. Chem. Soc., 2007, 129, 14319–14326 CrossRef CAS PubMed.
-
(a) A. K. Dutta, A. Linden, L. Zoppi, K. K. Baldridge and J. S. Siegel, Angew. Chem., Int. Ed., 2015, 54, 10792–10796 CrossRef CAS PubMed;
(b) T. Fujikawa, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 7763–7768 CrossRef CAS PubMed;
(c) X. Qiao, D. M. Ho and R. A. Pascal, Angew. Chem., Int. Ed., 1997, 36, 1531–1532 CrossRef CAS;
(d) Y.-Z. Tan, B. Yang, K. Parvez, A. Narita, S. Osella, D. Beljonne, X. Feng and K. Müllen, Nat. Commun., 2013, 4, 1–7 Search PubMed;
(e) K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739–744 CrossRef CAS PubMed.
- S. Xiao, M. Myers, Q. Miao, S. Sanaur, K. Pang, M. L. Steigerwald and C. Nuckolls, Angew. Chem., Int. Ed., 2005, 44, 7390–7394 CrossRef CAS PubMed.
- K. Baumgartner, A. L. Meza Chincha, A. Dreuw, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2016, 55, 15594–15598 CrossRef PubMed.
- C.-Y. Chiu, B. Kim, A. A. Gorodetsky, W. Sattler, S. Wei, A. Sattler, M. Steigerwald and C. Nuckolls, Chem. Sci., 2011, 2, 1480–1486 RSC.
- S. Xiao, S. J. Kang, Y. Wu, S. Ahn, J. B. Kim, Y.-L. Loo, T. Siegrist, M. L. Steigerwald, H. Li and C. Nuckolls, Chem. Sci., 2013, 4, 2018–2023 RSC.
- Y. Wu, Y. Zhen, Y. Ma, R. Zheng, Z. Wang and H. Fu, J. Phys. Chem. Lett., 2010, 1, 2499–2502 CrossRef CAS.
-
(a) Y. Zhong, M. T. Trinh, R. Chen, W. Wang, P. P. Khlyabich, B. Kumar, Q. Xu, C.-Y. Nam, M. Y. Sfeir, C. Black, M. L. Steigerwald, Y.-L. Loo, S. Xiao, F. Ng, X. Y. Zhu and C. Nuckolls, J. Am. Chem. Soc., 2014, 136, 15215–15221 CrossRef CAS PubMed;
(b) D. Sun, D. Meng, Y. Cai, B. Fan, Y. Li, W. Jiang, L. Huo, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2015, 137, 11156–11162 CrossRef CAS PubMed;
(c) M. Gsänger, J. H. Oh, M. Könemann, H. W. Höffken, A.-M. Krause, Z. Bao and F. Würthner, Angew. Chem., Int. Ed., 2010, 49, 740–743 CrossRef PubMed;
(d) Z. Chen, M. G. Debije, T. Debaerdemaeker, P. Osswald and F. Würthner, ChemPhysChem, 2004, 5, 137–140 CrossRef CAS PubMed;
(e) R. Schmidt, J. H. Oh, Y.-S. Sun, M. Deppisch, A.-M. Krause, K. Radacki, H. Braunschweig, M. Könemann, P. Erk, Z. Bao and F. Würthner, J. Am. Chem. Soc., 2009, 131, 6215–6228 CrossRef CAS PubMed;
(f) Y. Zhong, B. Kumar, S. Oh, M. T. Trinh, Y. Wu, K. Elbert, P. Li, X. Zhu, S. Xiao, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2014, 136, 8122–8130 CrossRef CAS PubMed.
- S. Sanyal, A. K. Manna and S. K. Pati, J. Phys. Chem. C, 2013, 117, 825–836 CAS.
- L. Dai, Acc. Chem. Res., 2013, 46, 31–42 CrossRef CAS PubMed.
- M. Yoonessi, Y. Shi, D. A. Scheiman, M. Lebron-Colon, D. M. Tigelaar, R. A. Weiss and M. A. Meador, ACS Nano, 2012, 6, 7644–7655 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Wallingford, CT, USA, 2009 Search PubMed.
- C. Lütke Eversloh, C. Li and K. Müllen, Org. Lett., 2011, 13, 4148–4150 CrossRef PubMed.
-
(a) L. Flamigni, A. Zanelli, H. Langhals and B. Böck, J. Phys. Chem. A, 2012, 116, 1503–1509 CrossRef CAS PubMed;
(b) B. Ventura, H. Langhals, B. Bock and L. Flamigni, Chem. Commun., 2012, 48, 4226–4228 RSC.
- B. R. Henry and W. Siebrand, J. Chem. Phys., 1971, 54, 1072–1085 CrossRef CAS.
- D. F. Eaton, Pure Appl. Chem., 1988, 60, 1107–1114 CrossRef CAS.
- W. E. Ford and P. V. Kamat, J. Phys. Chem., 1987, 91, 6373–6380 CrossRef CAS.
- J. J. Snellenburg, S. P. Laptenok, R. Seger, K. M. Mullen and I. H. van Stokkum, J. Stat. Software, 2012, 49, 1–22 Search PubMed.
- Y. Wu, Y. Zhen, Y. Ma, R. Zheng, Z. Wang and H. Fu, J. Phys. Chem. Lett., 2010, 1, 2499–2502 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Includes details of the synthesis, structural information for all of the compounds (NMR, elemental analysis and mass spectra) and experimental details for the photophysical studies. CCDC 1402604 and 1402605. For ESI and crystallographic data in CIF or other electronic formats see DOI: 10.1039/c6sc05126j |
|
| This journal is © The Royal Society of Chemistry 2017 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*