
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic insight into the selective olefin-directed oxidative carbocyclization and borylation by a palladium catalyst: a theoretical study†
Xiao-Wen Zhenga,
Ling Niea,
Ya-Ping Lib,
Shao-Jing Liub,
Feng-Yue Zhaob,
Xin-Yu Zhanga,
Xing-Dong Wanga and
Tao Liu *ab
*ab
aDepartment of Chemistry and Chemical Engineering, Jining University, Qufu 273155, Shandong, China. E-mail: liutao_2005@126.com
bSchool of Chemistry and Chemical Engineering, Qufu Normal University, Qufu 273165, Shandong, China
First published on 17th January 2017
Abstract
A mechanistic study of palladium-catalyzed oxidative carbocyclization and borylation of allenes was carried out by using density functional theory (DFT) calculations. A subtle change in the reaction conditions can cause the reaction to produce either cyclobutene product or alkenylboron compound with high chemoselectivity. Acetic acid solvent gives the alkenylboron compound as the major product, while methanol solvent favors the formation of the cyclobuteneboron product. Our calculations account for the observed solvent-controlled chemoselectivity. Lowering the polarity of the solvent disfavors the olefin insertion step and the formation of cyclobuteneboron product.
1. Introduction
As one of the important synthetic reagents in organic synthesis and medicinal chemistry, the efficient preparation of organoboron compounds has attracted much attention over the years.1 Activation and functionalization of the robust C–H bond in allene offers a promising protocol to synthesize versatile pharmaceutical molecules and functional materials.2,3 Therefore, much attention has been focused on transition-metal-catalyzed functionalization of allenes. Among them, palladium is the most attractive, and many palladium-based synthetic methodologies have been developed in recent decades.4Recently, Bäckvall et al.5 reported the efficient olefin-directed palladium-catalyzed oxidative carbocyclization and borylation of allenes by using 3,4-dienoate (R1) and B2pin2 (R2) as the reactants. A subtle change in the reaction conditions can cause the reaction to produce either cyclobutene product or alkenylboron compound with high chemoselectivity (Scheme 1). Acetic acid solvent gives the alkenylboron compound P1 as the major product, while methanol solvent favors the formation of the cyclobuteneboron product P2.
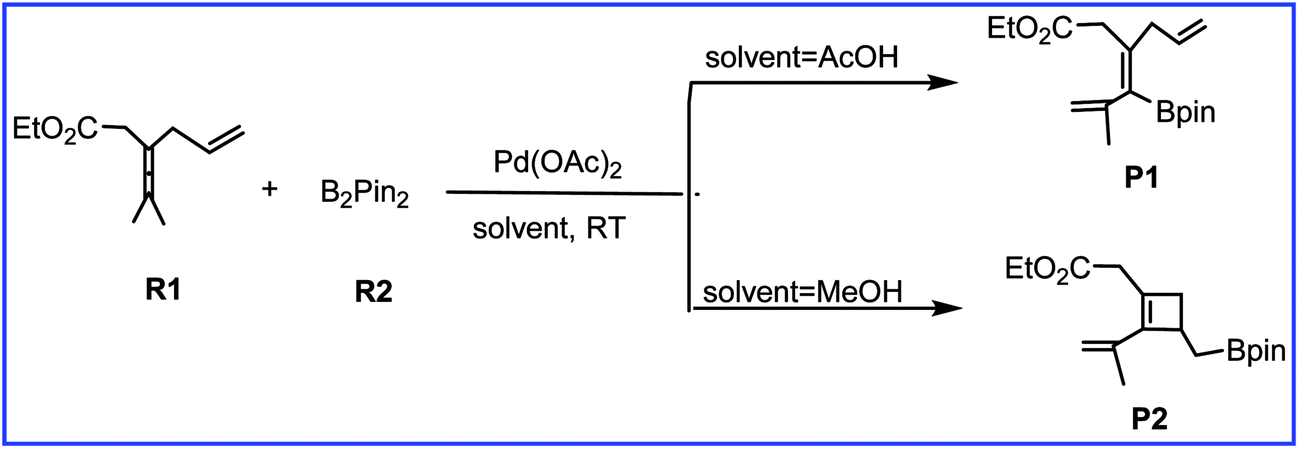 | ||
| Scheme 1 The palladium-catalyzed reaction of R1 with R2 reported by Bäckvall and co-workers. | ||
To account for the observed chemoselectivity, Bäckvall and coworkers postulated the possible reaction mechanisms summarized in Scheme 2. The reaction starts from A formed by the coordination of allene and olefin in R1 to Pd(II) center of catalyst Pd(OAc)2 (cat). Then allene attack involving allenic C–H bond cleavage occurs to afford vinylpalladium B. In the next step, B could undergo an olefin insertion to produce the cyclobutene intermediate C followed by the transmetalation with B2pin2 to form D. Finally, the reductive elimination would give the cyclobutene derivative product P2. In another pathway, the transmetalation of B with B2pin2 and the subsequent reductive elimination could produce product P1.
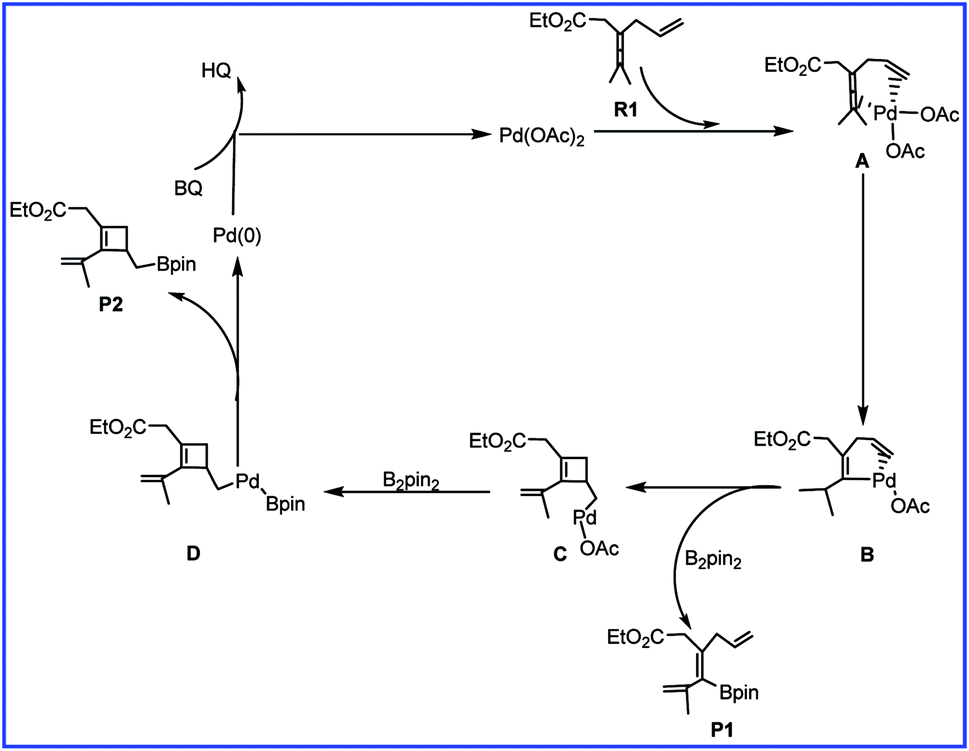 | ||
| Scheme 2 The mechanism of palladium-catalyzed reaction of R1 with R2 reported by Bäckvall and co-workers. | ||
Although a plausible mechanistic pathway has been proposed by Bäckvall et al.,5 several key issues related to the reaction mechanisms remain unanswered. (1) The details of the reaction mechanisms, e.g., the rate-determining step and selectivity-determining step, are still unclear. (2) How do the solvents (MeOH vs. AcOH) alter the reaction pathway to leading to the products P1 and P2?
In this work, we choose density functional theory (DFT) calculations to address all of these questions. We expect a clear understanding for the reaction mechanisms of olefin-directed palladium-catalyzed oxidative carbocyclization and borylation of allenes could benefit in designing new related reactions.
2. Computational details
All of the structures were optimized and characterized as minima or transition states at the B3LYP6/BSI level (BSI designates the basis set combination of LanL2DZ7 for Pd atom, and 6-31G(d,p) for other atoms) in the solvent with solvation effects accounted for by the SMD8 solvent model. According to the experimental conditions, methanol and acetic acid were adopted as the solvents. Harmonic vibrational frequencies were also calculated at the same level of theory to identify all stationary points as minima (zero imaginary frequencies) or transition states (one imaginary frequency). Intrinsic coordinate reaction (IRC)9 calculations were carried out to examine the connection of a transition state with its backward and forward minima when necessary. The energetic results were then further refined by the single-point calculations at the M06 (ref. 10)/BSII level, where BSII denotes the basis set combination of SDD11 for Pd atom and 6-311++G(d,p) for the remaining atoms. In all of the figures that contain energy diagrams, calculated relative Gibbs free energies (kcal mol−1) in the two solvents are presented. Unless otherwise noted, all discussed relative energies in subsequent sections are referred to the Gibbs free energies calculated at the M06/BSII level. All the calculations were performed with the GAUSSIAN 09 (ref. 12) software package.3. Results and discussion
Fig. 1 shows the free energy profiles calculated for C–H activation (R1 + cat → 3), and the geometric structures for selected species are shown in Fig. 2. As shown in Fig. 1, R1 initially coordinates with the Pd metal center of the catalyst cat via the allene and olefin moieties to give intermediate 1, initiating the reaction. This step is exergonic by 17.2 kcal mol−1 in MeOH solvent, suggesting that the step is thermodynamically favorable. Next, one of the acetate ligands in 1 acts as the base to deprotonate the sp3 C–H bond through the transition state TS1 to afford the intermediate 2, with a concerted metalation–deprotonation (CMD) mechanism.13 In TS1, the C–H bond cleavage and Pd–C bond formation occur simultaneously. The distances of C–H bond and Pd–C bond are 1.29 and 2.06 Å, respectively. The free energy barrier of the C–H activation step is calculated to be 11.5 kcal mol−1 in MeOH solvent. Subsequently, dissociation of the coordinated HOAc molecule leads to a more stable 16e complex 3, which is exergonic by of 15.1 kcal mol−1 in MeOH solvent.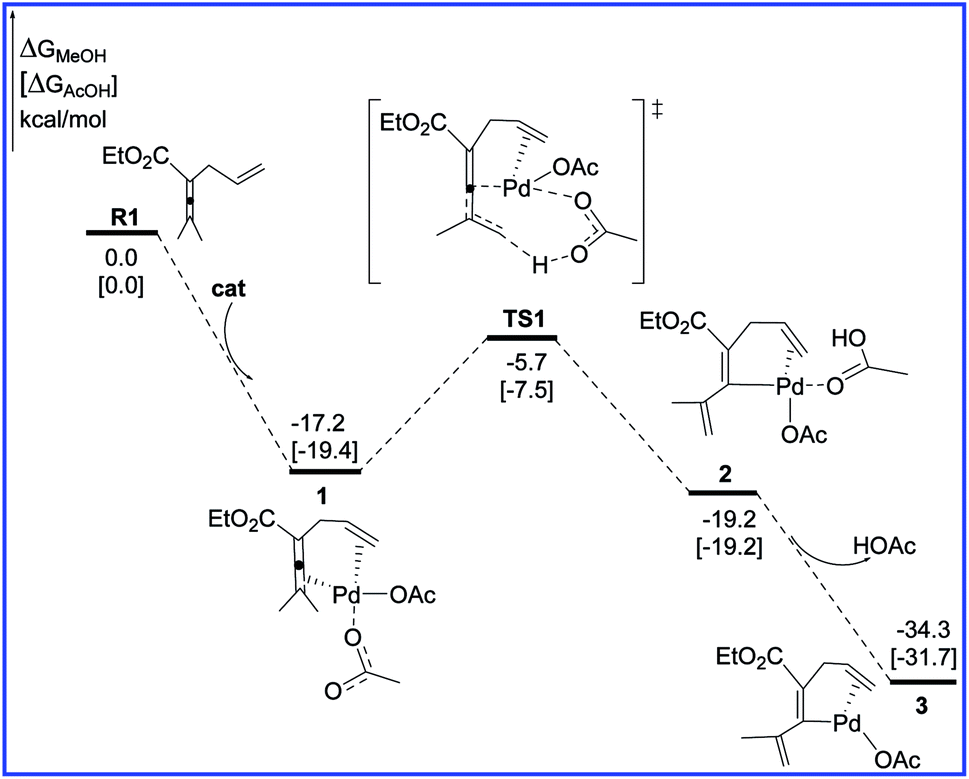 | ||
| Fig. 1 Free energy profiles in methanol [acetic acid] for the C–H activation step in the palladium-catalyzed reaction R1 with R2. | ||
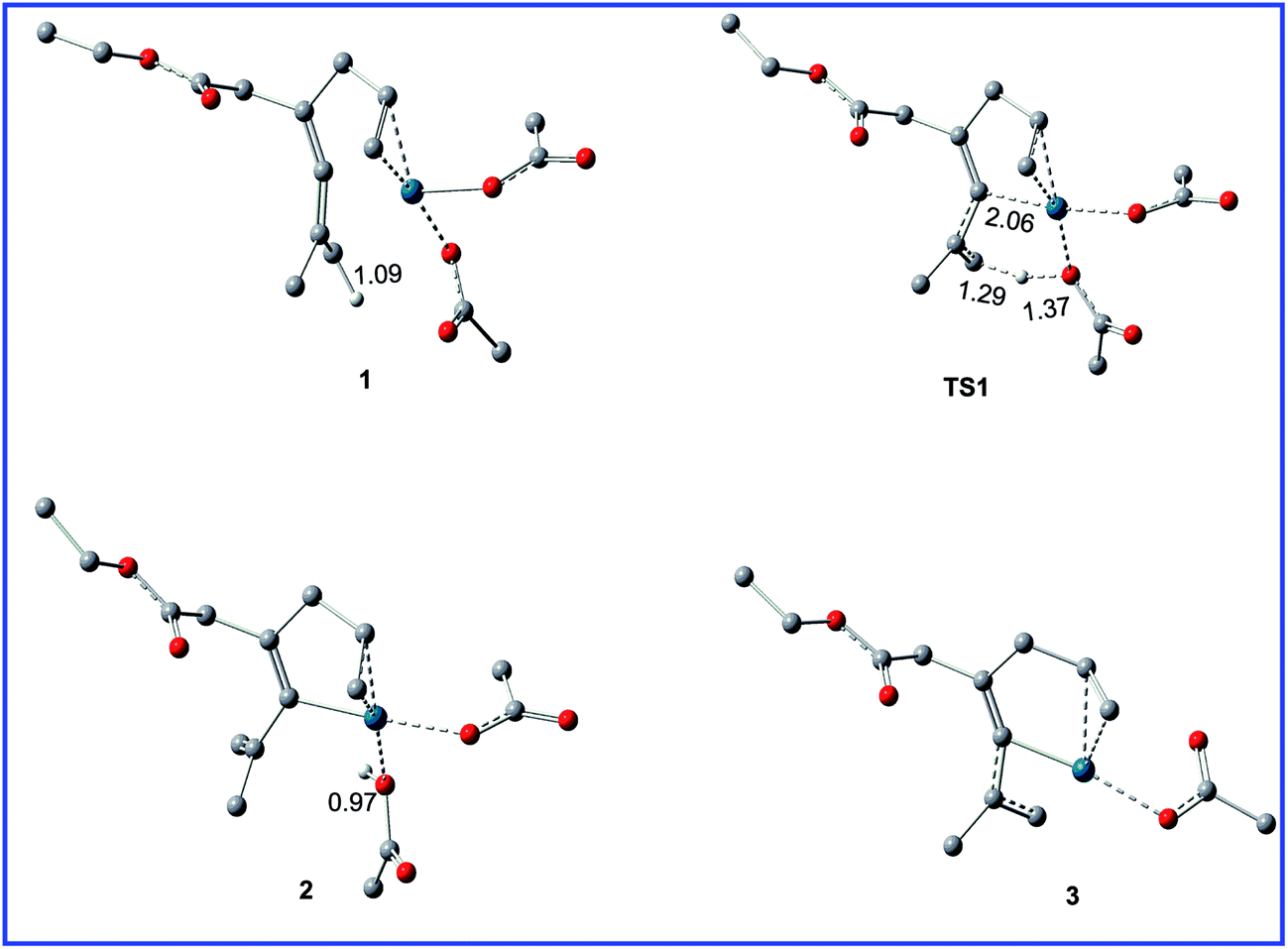 | ||
| Fig. 2 Geometric structures for selected species shown in Fig. 1. The hydrogen atoms not participating in the reaction have been omitted for clarity. Distances are in Å. | ||
The free energy profiles for the transformation from 3 to the intermediates 7 and 11 are calculated and shown in Fig. 3 and the related selected species geometric structures are given in Fig. 4. As shown in Fig. 3, with the addition of R2 to 3, intermediate 4 is produced with the free energy increasing of 6.9 kcal mol−1 (red line). The subsequent transmetalation process is stepwise, and includes two steps. The O center of OAc anion firstly attacks one of the B center in R2 to form intermediate 5, only requiring the activation barrier of 3.9 kcal mol−1. Then transmetalation occurs to lead to intermediate 6 via transition state TS3 by overcoming the energy barrier of 10.1 kcal mol−1. Through dissociating one Bpin-OAc molecule, a more stable intermediate 7 is formed by releasing the free energy of 16.4 kcal mol−1. In another pathway, prior to the addition of R2 to 3, the olefin bond in 3 would inserts into the Pd–C to afford the cyclobutene intermediate 8 through transition state TS4 by overcoming a free energy barrier of 18.3 kcal mol−1 (blue line). Next, with the addition of R2, transmetalation would occur through the transition states TS5 and TS6 to give the intermediate 10 followed by the dissociating Bpin-OAc molecule to form intermediate 11. The calculated free energy barrier for the transmetalation is 6.9 kcal mol−1.
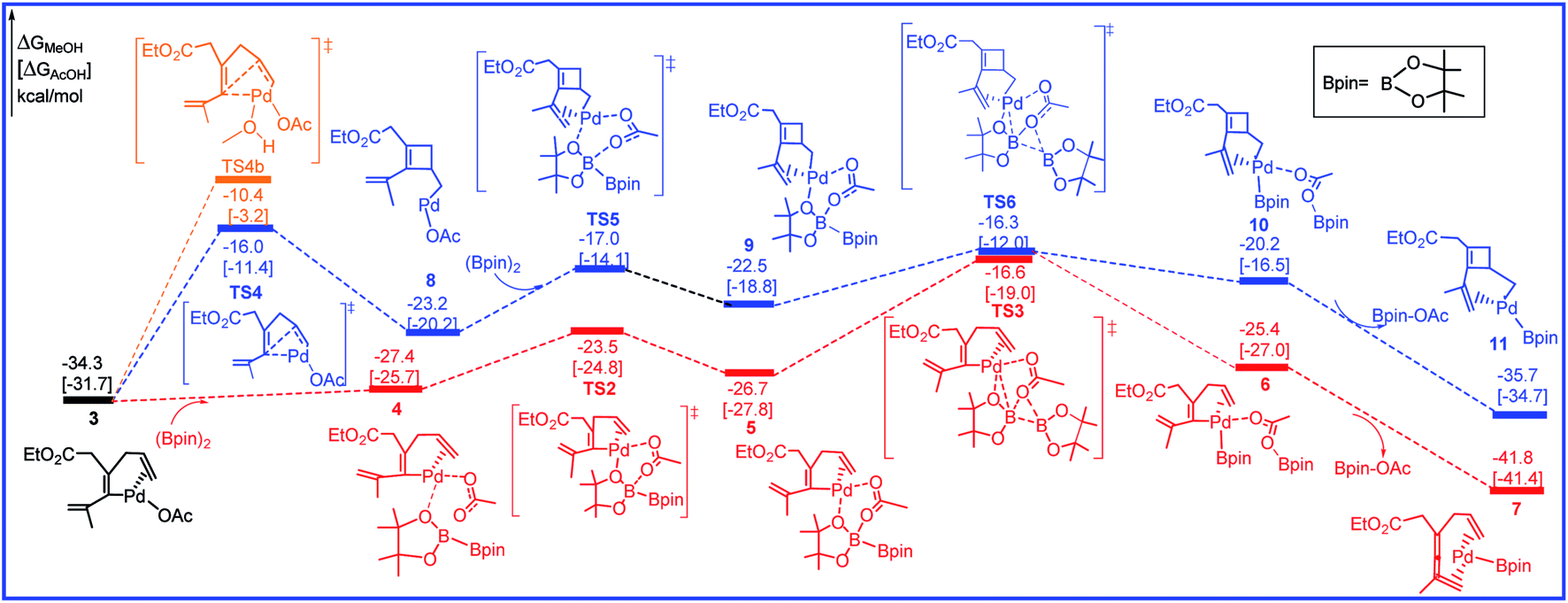 | ||
| Fig. 3 Free energy profiles in methanol [acetic acid] for the transmetalation and olefin insertion steps in the palladium-catalyzed reaction R1 with R2. | ||
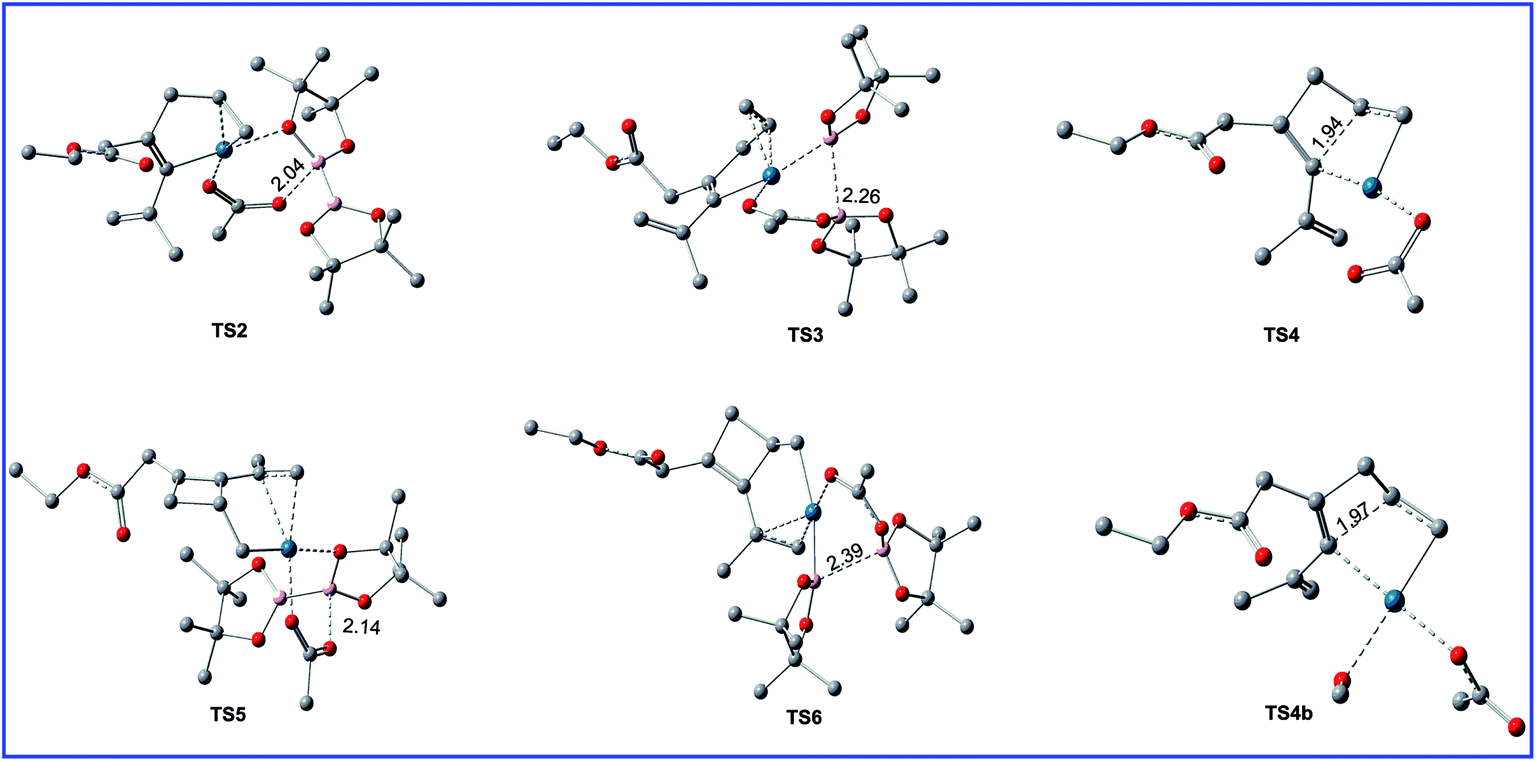 | ||
| Fig. 4 Geometric structures for selected transition states shown in Fig. 3. The hydrogen atoms not participating in the reaction have been omitted for clarity. Distances are in Å. | ||
We have thoroughly studied the possibility of additional methanol coordination in all the intermediates and transition states. For example, in the key olefin insertion step, the transition state with an additional methanol coordination, TS4b, is 5.7 kcal mol−1 higher in free energy as compared to that without the methanol coordination, TS4 (Fig. 3). We have found a similar situation exists for all other species; either no coordination site is available in palladium or the additional methanol coordination is unfavorable in free energy.
The free energy profiles for the reductive elimination step leading to alkenylboron and cyclobuteneboron products are calculated and shown in Fig. 5 and the related selected species geometric structures are given in Fig. 6. As shown in Fig. 5, the last step for the reaction is the C–B reductive elimination from Pd(II), producing the alkenylboron and cyclobuteneboron products and regenerating the Pd(0) catalyst. The two transformations leading to alkenylboron and cyclobuteneboron products are also confirmed both kinetically and thermodynamically favorable. The activation barriers via transition states TS7 and TS8 are 7.2 and 0.9 kcal mol−1 and the reaction energies for the two steps are 23.3 and 29.1 kcal mol−1, respectively.
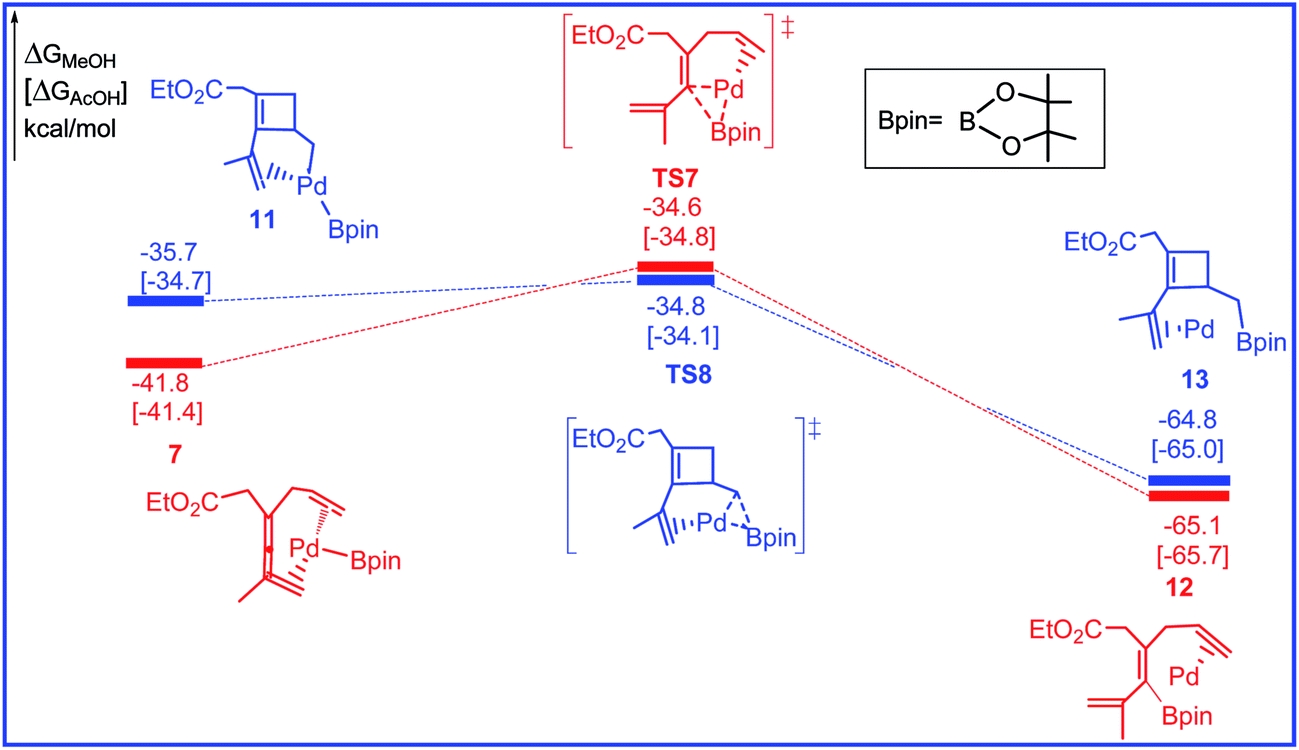 | ||
| Fig. 5 Free energy profiles in methanol [acetic acid] for the reductive elimination step in the palladium-catalyzed reaction R1 with R2. | ||
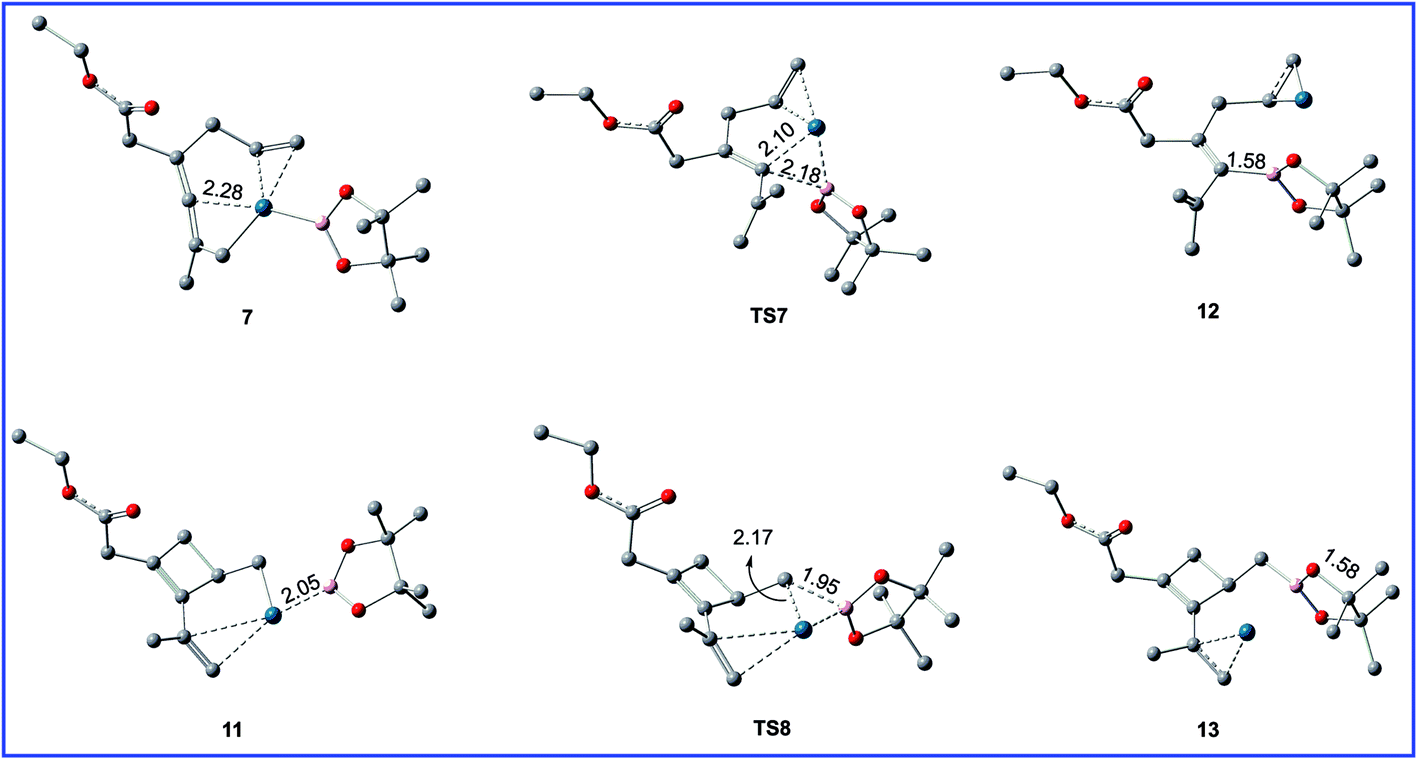 | ||
| Fig. 6 Geometric structures for selected species shown in Fig. 5. The hydrogen atoms not participating in the reaction have been omitted for clarity. Distances are in Å. | ||
As shown in Fig. 1, 3, and 5, the rate- and selectivity-determining step for the reactions leading to alkenylboron and cyclobuteneboron products are transmetalation step (TS3) and olefin insertion step (TS4), respectively. The competition between TS3 and TS4 determines the chemoselectivity between alkenylboron and cyclobuteneboron products. These are very close in energy, and correct prediction of selectivities is very difficult because of inaccuracies in solvation models.14 With methanol solvent (ε = 32.6), the overall energy barriers to produce alkenylboron and cyclobuteneboron products (3 to TS3 and 3 to TS4) are 17.7 and 18.3 kcal mol−1. With acetic acid solvent (ε = 6.3), the overall energy barriers for the transmetalation step to produce alkenylboron product decreases to 12.7 kcal mol−1, while the olefin insertion barrier increases to 20.1 kcal mol−1. These changes suggest that lowering the polarity of solvent disfavors the olefin insertion step and the formation of cyclobuteneboron product. Therefore, the solvent-controlled formation of alkenylboron and cyclobuteneboron product is most likely due to the polarity of methanol. Consistent with this postulate, the thorough experimental examination of solvents by Bäckvall et al. also showed the trend that higher polarity solvents, such as the mixture of MeOH and H2O, favor the formation of cyclobuteneboron product P2.
4. Conclusions
DFT investigations have elucidated the mechanisms of palladium-catalyzed oxidative carbocyclization and borylation of allenes. The chemoselectivity of the reaction is solvent-controlled. Acetic acid solvent gives the P1 as the major product, while methanol solvent favors the formation of the P2 product. The reaction leading to P1 undergoes three steps: C–H activation, transmetalation, and reductive elimination. In contrast, the reaction producing P2 includes C–H activation, olefin insertion, transmetalation, and reductive elimination. Our calculations probed the origins of the observed solvent-controlled chemoselectivity. The lowering the polarity of solvent disfavors the olefin insertion step and the formation of product P2.Acknowledgements
This work was jointly supported by National Natural Science Foundation of China (No. 21303073), the Natural Science Foundation of Shandong Province (No. ZR2014BL013), the Education Department of Shandong Province (No. J14LC58 and J16LC54), National Training Program of Innovation and Entrepreneurship for Undergraduates (No. 201610454011), and Talent Team Culturing Plan for Leading Disciplines of University in Shandong.References
- (a) S. R. Chemler and R. W. Roush, in Modern Carbonyl Chemistry, ed. J. Otera, Wiley-VCH, Weinheim, 2000 Search PubMed; (b) Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, 2nd Revised, ed. D. G. Hall, Wiley-VCH, Weinheim, 2011 Search PubMed.
- S. Ma, in Topics in Organometallic Chemistry, ed. J. Tsuji, Springer, Heidelberg, 2005 Search PubMed.
- (a) R. Zimmer, C. U. Dinesh, E. Nandanan and F. A. Khan, Chem. Rev., 2000, 100, 3067 CrossRef CAS PubMed; (b) J. A. Marshall, Chem. Rev., 2000, 100, 3163 CrossRef CAS PubMed; (c) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2000, 39, 3590 CrossRef CAS; (d) L. K. Sydnes, Chem. Rev., 2003, 103, 1133 CrossRef CAS PubMed; (e) S. Ma, Aldrichimica Acta, 2007, 40, 91 CAS; (f) M. Jeganmohan and C.-H. Cheng, Chem. Commun., 2008, 3101 RSC; (g) S. Ma, Acc. Chem. Res., 2009, 42, 1679 CrossRef CAS PubMed; (h) B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Soc. Rev., 2010, 39, 783 RSC; (i) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994 CrossRef CAS PubMed; (j) C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954 CrossRef CAS PubMed; (k) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074 CrossRef CAS PubMed.
- (a) J. Ye and S. Ma, Acc. Chem. Res., 2014, 47, 989 CrossRef CAS PubMed; (b) J. Piera, K. N. Rhi and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2006, 45, 6914 CrossRef CAS PubMed; (c) J. Piera, A. Persson, X. Caldentey and J.-E. Bäckvall, J. Am. Chem. Soc., 2007, 129, 14120 CrossRef CAS PubMed; (d) Y. Deng, T. Bartholomeyzik and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2013, 52, 6283 CrossRef CAS PubMed.
- Y. Qiu, B. Yang, C. Zhu and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2016, 55, 6520 CrossRef CAS PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee, W. Yang and G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (c) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS; (b) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS; (c) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 284 CrossRef.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- (a) K. J. Fukui, J. Phys. Chem., 1970, 74, 4161 CrossRef CAS; (b) K. Fukui, Acc. Chem. Res., 1981, 14, 363 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- (a) D. Andrae, U. Häussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS; (b) L. E. Roy, P. J. Hay and R. L. Martin, J. Chem. Theory Comput., 2008, 4, 1029 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A. 02, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- D. R. Stuart, P. Alsabeh, M. Kuhn and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 18326 CrossRef CAS PubMed.
- Y. F. Yang, K. N. Houk and Y. D. Wu, J. Am. Chem. Soc., 2016, 138, 6861 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27752g |
| This journal is © The Royal Society of Chemistry 2017 |