
A post-cyclotetramerization strategy towards novel binuclear phthalocyanine dimers†
Kang
Wang
a,
Chunhua
Huang
a,
Houhe
Pan
a,
Nagao
Kobayashi
b and
Jianzhuang
Jiang
*a
aBeijing Key Laboratory for Science and Application of Functional Molecular and Crystalline Materials, Department of Chemistry, University of Science and Technology Beijing, Beijing 100083, China. E-mail: jianzhuang@ustb.edu.cn
bFaculty of Textile Science and Technology, Shinshu University, Ueda 386-8567, Japan
First published on 24th October 2016
Abstract
A new and efficient post-cyclotetramerization strategy was developed for the synthesis of binuclear phthalocyanine dimers sharing one common pyrazine moiety, for the first time, paving a new way towards the design and synthesis of novel conjugated oligomeric phthalocyanine derivatives with various application potentials.
Inspired by the successful isolation of the initial binuclear porphyrin dimers connected by alkenyl, alkynyl, furandiyl, and pyrazino[2,3-g]quinoxaline conjugated linkages,1a,2a a large number of porphyrin oligomers with a conjugated electronic structure showing superior electronic and photonic properties over their mononuclear counterparts and special applications in molecular wires and photosynthetic antenna models have been fabricated from different monomeric porphyrin units.1–3 Phthalocyanines are the most important synthetic analogues of porphyrins. However, different from the porphyrin chromophore, the bridging atoms of meso-positions are four nitrogen atoms rather than four carbon atoms and four benzene moieties are fused onto the pyrrole periphery in phthalocyanine, which lead to the very harder functionalization of phthalocyanine ligands in comparison with the porphyrin chromophore, rendering the construction of π-conjugated phthalocyanine oligomers only through the cyclotetramerization pathway with 1,2,4,5-tetracyanobenzene as the necessary precursor, Scheme S1 (ESI†).4 Nevertheless, random cyclotetramerisation of 1,2,4,5-tetracyanobenzene with another phthalonitrile induces the formation of a statistical mixture including mononuclear phthalocyanine as the major product and various oligomers as the minor product. This in turn leads to the problem of difficult isolation and purification of the aimed π-conjugated phthalocyanine oligomers. Actually, only homoleptic binuclear phthalocyanine dimers and few examples of homoleptic trinuclear phthalocyanine trimers have been isolated thus far.4 The synthesis of heteroleptic phthalocyanine dimers is even more challenging since the monomeric unsymmetric ABBB-type phthalocyanine analogue with two cyano groups has to be prepared in the first stage, which then reacts with the second phthalonitrile precursor, giving the target heterobinuclear compound in quite low yield via twice statistically-driven cyclocondensation.5
Obviously, developing a new synthesis strategy besides cyclotetramerization towards π-conjugated phthalocyanine oligomers becomes highly desired in this field. In the present paper, a new and efficient post-cyclotetramerization strategy was developed for the first time towards the synthesis and characterization of unprecedented binuclear phthalocyanine dimers sharing one common pyrazine moiety M1Pc(OR1)6–M2Pc(OR1)6 (M1–M2 = 2H–2H, Zn–2H, Zn–Zn, OR1 = 2,6-dimethylphenoxy) (1–3), H2Pc(SR2)6–H2Pc(SR2)6 (4), and H2Pc(OR1)6–H2Pc(SR2)6 (5) (SR2 = hexylthio), Scheme 1, in an unexpected good reaction yield as high as 72%, paving a new way towards novel conjugated oligomeric phthalocyanine derivatives with various application potentials.
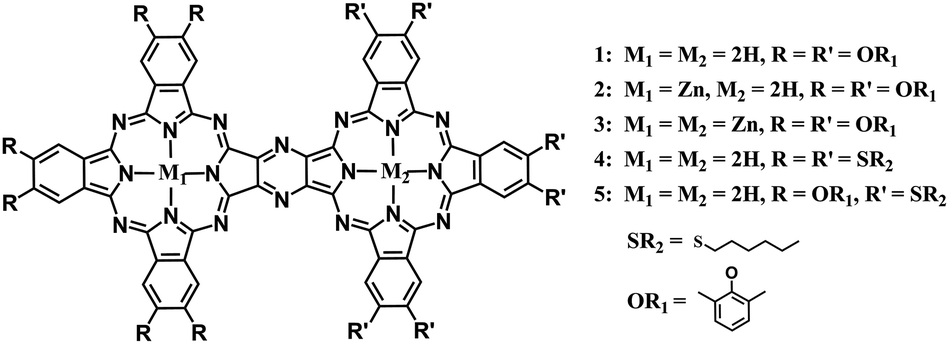 | ||
| Scheme 1 Molecular structure of binuclear phthalocyanine dimers 1–5. | ||
For the initial purpose of synthesizing a mixed phthalocyanine–porphyrin-fused dimer,6 unsymmetrical ABBB-type diamino-Pc 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 and dione porphyrin6b monomers were prepared according to the published procedures. Surprisingly, a reaction between 6 and the dione porphyrin monomer in CH2Cl2 at room temperature in the presence of 0.1% trifluoroacetic acid (TFA) as a catalyst afforded the metal free homobinuclear phthalocyanine H2Pc(OR1)6–H2Pc(OR1)6 (1) in the yield of ca. 10% as the main product and only a trace amount of the targeted phthalocyanine-porphyrin-fused dimer, suggesting the effective self-condensation reaction of diamino-Pc 6 under the present reaction conditions. With diamino-Pc 6 as the sole mononuclear precursor, metal free binuclear phthalocyanine 1 was isolated in the yield of 12% under exactly the same reaction conditions, Scheme 2. More interestingly, the increase in the proportion of TFA from 0.1% led to a significant increase in the yield of 1 with the highest value of 72% being achieved in the CH2Cl2/TFA ratio of 80:20, Fig. 1. In order to further understand the role of TFA in the formation of 1, other acid species including acetic acid, propanoic acid, H2SO4, and HCl instead of TFA were employed to carry out the above-mentioned self-condensation reaction, however, all of which failed to afford the dimer 1. Nevertheless, only a trace amount of 1 could be detected by MALDI-TOF spectrometry for the self-condensation reaction of 6 in the absence of TFA. These results clearly reveal the key role of the TFA catalyst in the formation of phthalocyanine dimer 1.
7 and dione porphyrin6b monomers were prepared according to the published procedures. Surprisingly, a reaction between 6 and the dione porphyrin monomer in CH2Cl2 at room temperature in the presence of 0.1% trifluoroacetic acid (TFA) as a catalyst afforded the metal free homobinuclear phthalocyanine H2Pc(OR1)6–H2Pc(OR1)6 (1) in the yield of ca. 10% as the main product and only a trace amount of the targeted phthalocyanine-porphyrin-fused dimer, suggesting the effective self-condensation reaction of diamino-Pc 6 under the present reaction conditions. With diamino-Pc 6 as the sole mononuclear precursor, metal free binuclear phthalocyanine 1 was isolated in the yield of 12% under exactly the same reaction conditions, Scheme 2. More interestingly, the increase in the proportion of TFA from 0.1% led to a significant increase in the yield of 1 with the highest value of 72% being achieved in the CH2Cl2/TFA ratio of 80:20, Fig. 1. In order to further understand the role of TFA in the formation of 1, other acid species including acetic acid, propanoic acid, H2SO4, and HCl instead of TFA were employed to carry out the above-mentioned self-condensation reaction, however, all of which failed to afford the dimer 1. Nevertheless, only a trace amount of 1 could be detected by MALDI-TOF spectrometry for the self-condensation reaction of 6 in the absence of TFA. These results clearly reveal the key role of the TFA catalyst in the formation of phthalocyanine dimer 1.
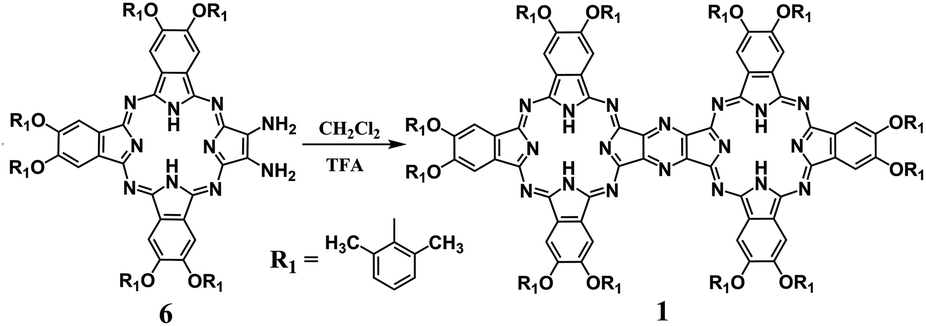 | ||
| Scheme 2 Synthesis of the metal-free homobinuclear phthalocyanine 1 in CH2Cl2 in the presence of TFA at room temperature. | ||
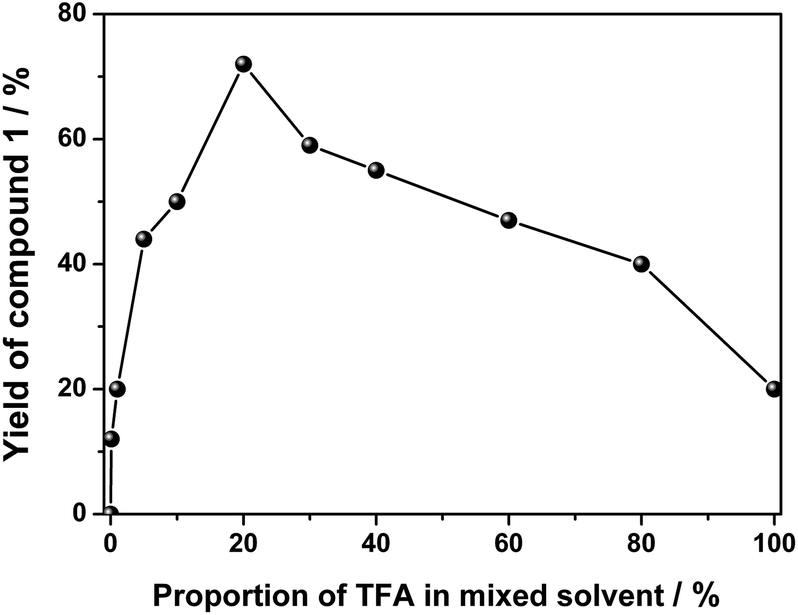 | ||
| Fig. 1 Change of the yield of 1 as a result of the proportion of TFA in the mixed solvent of CH2Cl2 and TFA at room temperature. | ||
To reveal the generality of this newly developed synthetic strategy, the zinc complex of diamino-Pc 7 instead of the metal free species 6 was utilized as the precursor to carry out the self-condensation reaction, resulting in the isolation of the homobinuclear phthalocyanine zinc dimer ZnPc(OR1)6–ZnPc(OR1)6 (3) also in good yield, 56%. In addition, condensation between the metal free diamino-Pc 6 and its zinc complex 7 led to the isolation of the heterobinuclear phthalocyanine dimer ZnPc(OR1)6–H2Pc(OR1)6 (2) in the yield of 41% in addition to the homobinuclear phthalocyanine dimers 1 and 3 in the yield of 10 and 8.0%, respectively, Scheme S2 (ESI†). Nevertheless, mixed condensation of metal free diamino-Pc precursor 6 with another metal free diamino-Pc precursor 8 bearing different peripheral substituents induced the isolation of the heterobinuclear phthalocyanine dimer H2Pc(OR1)6–H2Pc(SR2)6 (5), 7.4%, in addition to the homobinuclear phthalocyanine dimers 1 and H2Pc(SR2)6–H2Pc(SR2)6 (4) in the yield of 30 and 21%, Scheme 3. Obviously, it seems strange at the first glance for the significantly lower yield of the heterobinuclear phthalocyanine dimer 5 in comparison with that for the analogue 2. This, however, could be rationalized on the basis of the very much different reaction activity between the two different monomeric diamino-Pc precursors 6 and 8 bearing different peripheral substituents. In contrast, despite having the different central metal ions, the same peripheral substituents in the two monomeric diamino-Pc precursors 6 and 7 endow them with quite similar reaction activity. As a result, random condensation reaction between these two precursors led to the isolation of heterobinuclear phthalocyanine dimer 2 in obviously higher yield than those for the homoleptic counterparts 1 and 3. Conversely, both the diamino-Pc precursors 6 and 8 were inclined to carry out self-condensation rather than cross-condensation reaction, resulting in the isolation of the heterobinuclear dimer 5 in lower yield than their homobinuclear phthalocyanine dimers 1 and 4. Actually, to the best of our knowledge, only phthalocyanine dimers/trimers sharing a common benzene/naphthalene/anthracene moiety have been reported thus far.4,5 As a consequence, the phthalocyanine dimeric compounds 1–5 sharing a common pyrazine moiety described in the present work represent the unprecedented type of binuclear phthalocyanine derivatives, affording new members in the family for further functional investigations.
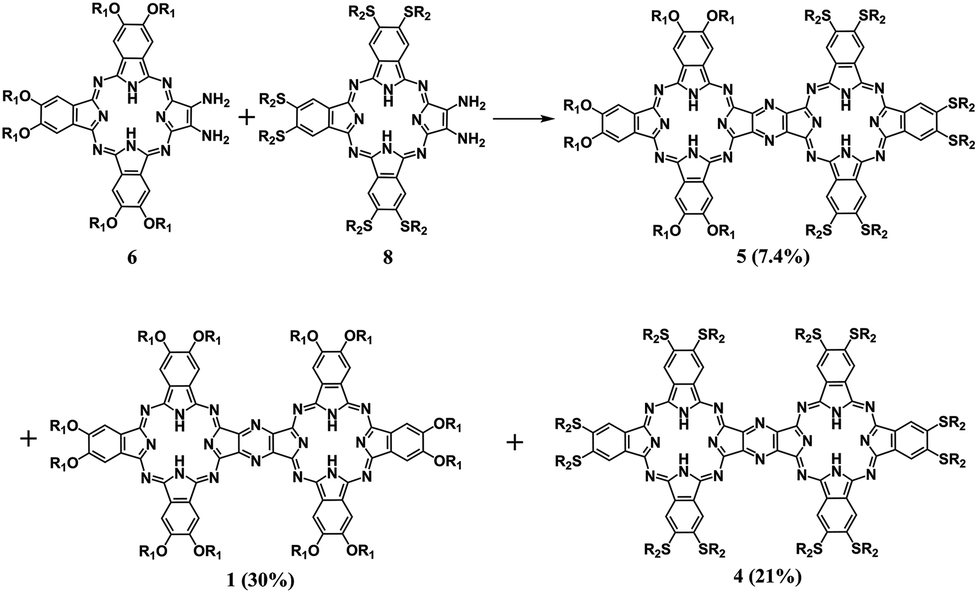 | ||
| Scheme 3 Reaction between diamino-Pcs 6 and 8 in CH2Cl2/TFA (80:20) at room temperature (OR1 = 2,6-dimethylphenoxy, SR2 = hexylthio). | ||
The newly prepared binuclear phthalocyanine dimers 1–5 gave satisfactory elemental–analytical results. Their MALDI-TOF mass spectra clearly show intense signals for the corresponding molecular ion [M]+. The isotopic pattern closely resembled the simulated one as exemplified by the spectrum of 1 given in Fig. S1 (ESI†). These compounds (except for 4 due to its intensive aggregation nature in common organic solvents including CH2Cl2, CHCl3, THF, DMF, toluene, and chlorobenzene) were further characterized with a range of spectroscopic methods including NMR, electronic absorption, and magnetic circular dichroism (MCD) spectroscopy. Fig. S2–S5 (ESI†) show the 1H NMR spectra of 1–3 and 5, and all the signals could be unambiguously assigned with the result as tabulated in Table S1 (ESI†).
Fig. 2 and S6 (ESI†) show the electronic absorption and MCD spectra of 1–3 and 5 in either CHCl3 or CHCl3–pyridine (100:1) with the corresponding data summarized in Table S2 (ESI†). As can be seen, the dimeric compounds 1–3 with the same peripheral substituents display an intense near IR Q band at ca. 830 nm and an intense Soret band at ca. 360 nm. In comparison with the monomeric Pcs (which usually exhibit the Q band at ca. 680 nm),4c the Q band of 1 is significantly red-shifted by ca. 150 nm due to the extended π-electron system of the conjugated binuclear phthalocyanine dimer, which can be explained by the exciton coupling type interaction of the two constituent Pc units.4b,i,j However, compared to the homobinuclear phthalocyanine analogue linked by a benzene moiety,4c,i the Q band of 1 is blue-shifted by ca. 20 nm owing to the stabilized highest occupied molecular orbital and a slight decrease of the practical π-system due to the two nitrogen atoms in the pyrazine moiety.8 This is also true for 2 and 3, Fig. S6 and Table S2 (ESI†). From the Faraday B MCD terms of opposite sign, the long- and short-axis polarized Q transitions of 1 lie at around 830 and 720 nm, while those of 5 lie at ca. 840 and 685 nm, Fig. 2, revealing the characteristic electronic transitions to the nondegenerate excited states.4f This is also true for 2 and 3, Fig. S6 (ESI†).
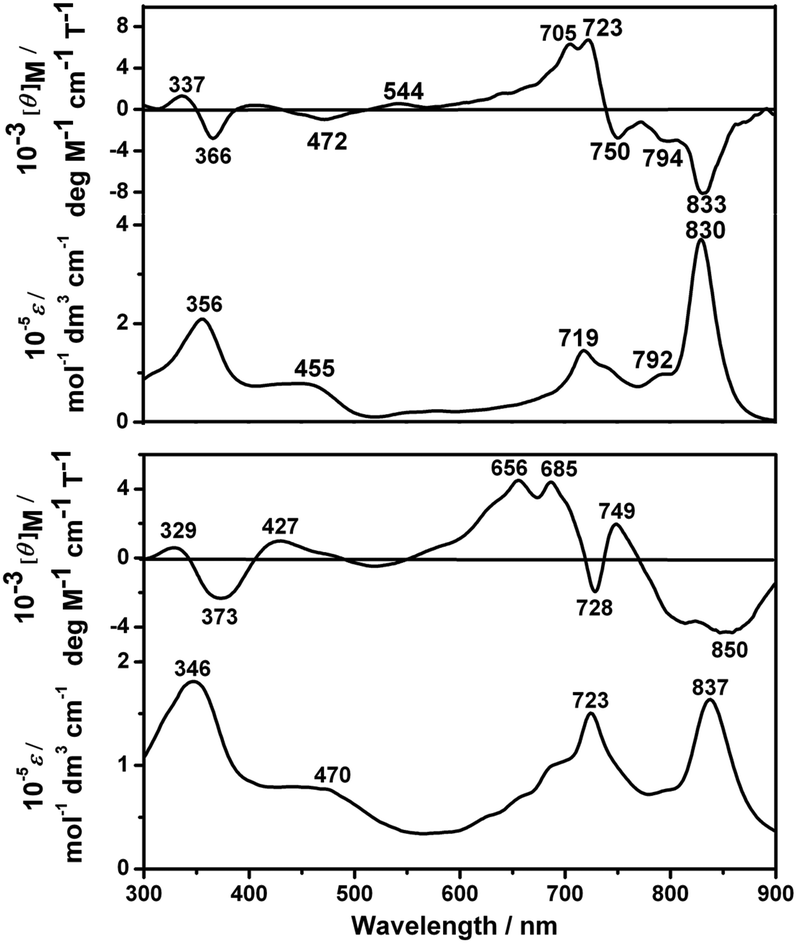 | ||
| Fig. 2 Electronic absorption and MCD spectra of 1 (top) and 5 (bottom) in CHCl3. | ||
Single crystals of ZnPc(OR1)6–ZnPc(OR1)6 (3) suitable for X-ray diffraction analysis were obtained by slow diffusion of methanol into the solution of this compound in CHCl3 with a drop of pyridine. 3 crystallizes in the orthorhombic system with a Pbca space group containing four homobinuclear phthalocyaninato zinc molecules coordinated with two pyridine molecules per unit cell. Detailed crystal and structural data are listed in Table S3 (ESI†). Fig. 3 shows the molecular structure of 3, disclosing its binuclear phthalocyanine nature in an unambiguous manner. As can be seen, in the dimeric molecule each zinc ion locates in the center of the tetrapyrrole ring coordinated by four isoindole N atoms and one pyridine N atom, forming a five-coordinate pentagonal pyramid geometry around the zinc ion. This leads to two N4 planes in the binuclear phthalocyaninato zinc molecule slightly domed with the average dihedral angle of the individual isoindole ring with respect to the corresponding N4 mean plane being 4.33°. In addition, owing to the extended π-electron system, the binuclear phthalocyaninato zinc molecule adopts a slightly ruffled conformation with a mean deviation of 0.16 Å from planarity for the 74 atoms of the binuclear phthalocyanine core.
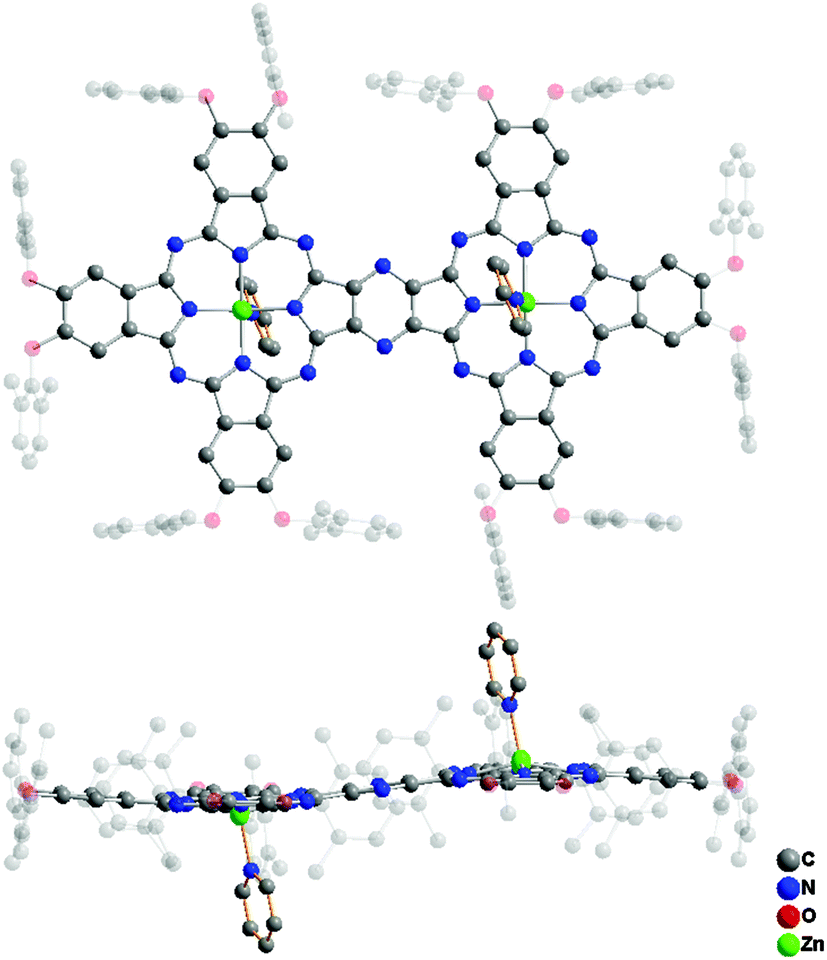 | ||
| Fig. 3 Molecular structure of ZnPc(OR1)6–ZnPc(OR1)6 (3) in top-view and side-view with all the hydrogen atoms omitted for clarity. | ||
In conclusion, a new post-cyclotetramerization strategy was developed for the first time towards the synthesis of an unprecedented type of π-conjugated binuclear phthalocyanine dimeric compound sharing one common pyrazine moiety, paving a new pathway towards the design and synthesis of novel π-conjugated oligomeric phthalocyanine derivatives with various application potentials.
Financial support from the National Key Basic Research Program of China (Grant No. 2013CB933402), the Natural Science Foundation of China (Grant No. 21290174 and 21401009), JSPS (Grant No. JP15H00910), the Beijing Municipal Commission of Education, and the University of Science and Technology Beijing is gratefully acknowledged.
Notes and references
- (a) D. P. Arnold, Synlett, 1999, 296–305 Search PubMed; (b) D. P. Arnold and L. J. Nitschinsk, Tetrahedron, 1992, 48, 8781–8792 CrossRef CAS; (c) D. P. Arnold and G. A. Heath, J. Am. Chem. Soc., 1993, 115, 12197–12198 CrossRef CAS; (d) D. P. Arnold, D. A. James, C. H. L. Kennard and G. Smith, J. Chem. Soc., Chem. Commun., 1994, 2131–2132 RSC; (e) D. P. Arnold and D. A. James, J. Org. Chem., 1997, 62, 3460–3469 CrossRef CAS; (f) D. P. Arnold and D. A. James, J. Org. Chem., 1998, 63, 4856–4856 CrossRef CAS.
- (a) M. J. Crossley and P. L. Burn, J. Chem. Soc., Chem. Commun., 1987, 39–40 RSC; (b) M. J. Crossley and P. Thordarson, Angew. Chem., Int. Ed., 2002, 41, 1709–1712 CrossRef CAS; (c) M. J. Crossley and L. A. Johnston, Chem. Commun., 2002, 1122–1123 RSC; (d) R. Paolesse, L. Jaquinod, F. D. Sala, D. J. Nurco, L. Prodi, M. Montalti, C. D. Natale, A. D'Amico, A. D. Carlo, P. Lugli and K. M. Smith, J. Am. Chem. Soc., 2000, 122, 11295–11302 CrossRef CAS; (e) H. Uoyama, K. S. Kim, K. Kuroki, J.-Y. Shin, T. Nagata, T. Okujima, H. Yamada, N. Ono, D. Kim and H. Uno, Chem. – Eur. J., 2010, 16, 4063–4074 CrossRef CAS PubMed.
- (a) A. Tsuda and A. Osuka, Science, 2001, 293, 79–82 CrossRef CAS PubMed; (b) T. Sakurai, K. Shi, H. Sato, K. Tashiro, A. Osuka, A. Saeki, S. Seki, S. Tagawa, S. Sasaki, H. Masunaga, K. Osaka, M. Takata and T. Aida, J. Am. Chem. Soc., 2008, 130, 13812–13813 CrossRef CAS PubMed; (c) S. Ito, S. Hiroto, S. Lee, M. Son, I. Hisaki, T. Yoshida, D. Kim, N. Kobayashi and H. Shinokubo, J. Am. Chem. Soc., 2015, 137, 142–145 CrossRef CAS PubMed; (d) T. Kamada, N. Aratani, T. Ikeda, N. Shibata, Y. Higuchi, A. Wakamiya, S. Yamaguchi, K. S. Kim, Z. S. Yoon, D. Kim and A. Osuka, J. Am. Chem. Soc., 2006, 128, 7670–7678 CrossRef CAS PubMed; (e) Y. Rao, T. Kim, K. H. Park, F. Peng, L. Liu, Y. Liu, B. Wen, S. Liu, S. R. Kirk, L. Wu, B. Chen, M. Ma, M. Zhou, B. Yin, Y. Zhang, D. Kim and J. Song, Angew. Chem., Int. Ed., 2016, 55, 6438–6442 CrossRef CAS PubMed; (f) J. Song, S. Y. Jang, S. Yamaguchi, J. Sankar, S. Hiroto, N. Aratani, J.-Y. Shin, S. Easwaramoorthi, K. S. Kim, D. Kim, H. Shinokubo and A. Osuka, Angew. Chem., Int. Ed., 2008, 47, 6004–6007 CrossRef CAS PubMed.
- (a) C. C. Leznoff, H. Lam, S. M. Marcuccio, W. A. Nevin, P. Janda, N. Kobayashi and A. B. P. Lever, J. Chem. Soc., Chem. Commun., 1987, 699–701 RSC; (b) N. Kobayashi, H. Lam, W. A. Nevin, P. Janda, C. C. Leznoff, T. Koyama, A. Monden and H. Shirai, J. Am. Chem. Soc., 1994, 116, 879–890 CrossRef CAS; (c) S. Makarov, C. Litwinski, E. A. Ermilov, O. Suvorova, B. Rçder and D. Wöhrle, Chem. – Eur. J., 2006, 12, 1468–1474 CrossRef CAS PubMed; (d) K. Wang, D. Qi, H. Wang, W. Cao, W. Li, T. Liu, C. Duan and J. Jiang, Chem. – Eur. J., 2013, 19, 11162–11166 CrossRef CAS PubMed; (e) Y. Asano, J. Sato, T. Furuyama and N. Kobayashi, Chem. Commun., 2012, 48, 4365–4367 RSC; (f) C. Huang, K. Wang, J. Sun and J. Jiang, Dyes Pigm., 2014, 109, 163–168 CrossRef CAS; (g) J. Yang and M. Mark, Tetrahedron Lett., 1993, 34, 5223–5226 CrossRef CAS; (h) M. Handa, N. Kataoka, Y. Ito, T. Tonomura, I. Hiromitsu, T. Sugimori, K. Sogabe and K. Kasuga, Bull. Chem. Soc. Jpn., 2004, 77, 1647–1648 CrossRef CAS; (i) N. Kobayashi, T. Fukuda and D. Lelievre, Inorg. Chem., 2000, 39, 3632–3637 CrossRef CAS PubMed; (j) N. Kobayashi and H. Ogata, Eur. J. Inorg. Chem., 2004, 906–914 CrossRef CAS; (k) T. Ikeue, N. Sawada, N. Matsumoto, A. Miyazaki, T. Sugimori, M. Koikawa, I. Hiromitsu, K. Yoshino, M. Mikuriya, Y. Kataoka and M. Handa, J. Porphyrins Phthalocyanines, 2014, 18, 708–714 CrossRef CAS; (l) T. V. Dubinina, N. E. Borisova, M. V. Sedova, L. G. Tomilova, T. Furuyama and N. Kobayashi, Dyes Pigm., 2015, 117, 1–6 CrossRef CAS.
- (a) K. Ishii, N. Kobayashi, Y. Higashi, T. Osa, D. Lelièvre, J. Simon and S. Yamauchi, Chem. Commun., 1999, 969–970 RSC; (b) M. Calvete and M. Hanack, Eur. J. Org. Chem., 2003, 2080–2083 CrossRef CAS; (c) N. Kobayashi, Y. Higashi and T. Osa, J. Chem. Soc., Chem. Commun., 1994, 1785–1786 RSC; (d) N. Kobayashi and H. Ogata, Eur. J. Inorg. Chem., 2004, 906–914 CrossRef CAS; (e) N. Kobayashi, Y. Higashi and T. Osa, Chem. Lett., 1994, 1813–1816 CrossRef CAS; (f) M. J. F. Calvete, D. Dini, S. R. Flom, M. Hanack, R. G. S. Pong and J. S. Shirk, Eur. J. Org. Chem., 2005, 3499–3509 CrossRef CAS.
- (a) Y. Zhang, J. Oh, K. Wang, C. Chen, W. Cao, K. H. Park, D. Kim and J. Jiang, Chem. – Eur. J., 2016, 22, 4492–4499 CrossRef CAS PubMed; (b) K. Wang, D. Qi, J. Mack, H. Wang, W. Li, Y. Bian, N. Kobayashi and J. Jiang, Chin. J. Inorg. Chem., 2012, 28, 1779–1789 CAS.
- (a) D. Wöhrle, M. Eskes, K. Shigehara and A. Yamada, Synthesis, 1993, 194–196 CrossRef; (b) H. Pan, C. Chen, K. Wang, W. Li and J. Jiang, Chem. – Eur. J., 2015, 21, 3168–3173 CrossRef CAS PubMed; (c) T. Ikeue, T. Fukahori, T. Mitsumune, K. Ueda, K. Fujii, S. Kurahashi, M. Koikawa, T. Sugimori, I. Hiromitsu, K. Yoshino, M. Mikuriya and M. Handa, Inorg. Chim. Acta, 2014, 409, 433–440 CrossRef CAS.
- P. Zimcik, V. Novakova, K. Kopecky, M. Miletin, R. Z. U. Kobak, E. Svandrlikova, L. Váchová and K. Lang, Inorg. Chem., 2012, 51, 4215–4223 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, NMR and IR results, electrochemical properties, spectroscopic and electrochemical data, and crystallographic data for 3 (CIF). CCDC 1476231. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qi00408c |
| This journal is © the Partner Organisations 2017 |