
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
G-quadruplex DNA targeted metal complexes acting as potential anticancer drugs
Qian
Cao
a,
Yi
Li
a,
Eva
Freisinger
b,
Peter Z.
Qin
*c,
Roland K. O.
Sigel
*b and
Zong-Wan
Mao
*a
aMOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry and Chemical Engineering, Sun Yat-Sen University, Guangzhou 510275, China. E-mail: cesmzw@mail.sysu.edu.cn
bUniversity of Zurich, Department of Chemistry, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland. E-mail: roland.sigel@chem.uzh.ch
cDepartment of Chemistry, University of Southern California, Los Angeles, California 90089, USA. E-mail: pzq@usc.edu
First published on 30th September 2016
Abstract
Although cisplatin and its analogues have been widely utilized as anticancer metallodrugs in clinics, their serious side effects and damage to normal tissues cannot be avoided because cisplatin kills cancer cells by attacking genomic DNA. Thus the design of metallodrugs possessing different actions of anti-cancer mechanism is promising. G-quadruplex nucleic acid, which is formed by self-assembly of guanine-rich nucleic acid sequences, has recently been considered as an attractive target for anticancer drug design. The basic unit of a G-quadruplex is a G-quartet, a planar motif generated from four guanine residues pairing together through Hoogsteen like hydrogen bonds. DNA G-quadruplex (G4) structures exist in the chromosomal telomeric sequences and the promoter regions of numerous genes, including oncogenetic promoters. Formation of G4 structures within the 3′-overhang of telomeric DNA can inhibit the telomerase activity, which is silent in normal cells but up-regulated in most cancer cells, thus significantly shortening telomeres and preventing cancer cell proliferation and immortalization. Intramolecular G4 structures formed within the oncogene promoter regions can effectively inhibit oncogenen transcription and expression. Thus rational design of small molecular ligands to selectively interact, stabilize or cleave G4 structures is a promising strategy for developing potent anti-cancer drugs with selective toxicity towards cancer cells over normal ones. This review will highlight the recent development of G4-interacting metal complexes, termed G4-ligands, discussing their binding modes with G-quadruplex DNA and their potential to serve as anticancer drugs in the medical field.
Introduction to the international collaborationThe collaboration between Prof. Zong-Wan Mao from Sun Yat-Sen University, P. R. China and Prof. Roland K. O. Sigel from the University of Zurich, Switzerland officially began in January, 2014. The international collaborative research project titled “Chemical Biology Research of New Metallodrugs for Cancer Therapy” is supported by the Science and Technology Program of Guangdong Provincial Government [20130501c]. With the rapid development of tumor molecular pharmacology, molecular targeted anti-tumor drugs have become a hot spot in the research of cancer therapy. This international collaborative research project combines the computer simulation and in vitro drug screening platform to design a series of metallodrugs that are systematic and have structural diversity, which can target specific nucleic acid structures (e.g. G-quadruplexes), key proteins (DNA topoisomerase, telomerase, CDK kinase) associated with the occurrence and development of tumor. With the advantages of both laboratories, the structural–functional relationship, interaction modes, co-crystallization, and mechanisms of action of these newly designed metallodrugs are intensively studied, and their in vitro and in vivo anti-tumor activities are comprehensively evaluated. |
1. Introduction
In 1978 cisplatin was approved by the FDA as an anticancer drug for clinical use. Nowadays cisplatin and its analogues are very effective chemotherapeutic agents in treating testicular and ovarian cancers.1 However, they have several serious side effects including nephrotoxicity and ototoxicity, and their clinical efficacy is limited by cisplatin-resistant tumor cells. The main reason for this is assigned to their action mechanism: cisplatin-based drugs attack genetic DNAs, resulting in the dysfunction of transcription, translation and other processes, ultimately causing tumor cell death. However, such attacks do not distinguish between tumor and normal cells, and hence serious side effects occur due to damage of normal tissues.2,3 Since then, the development of metallodrugs that possess different actions of mechanism has attracted great interest.2,4 One promising strategy for the design of metallodrugs is to explore new potential biological targets rather than genomic DNA.G-quadruplex nucleic acids, which have structural features very different from the regular double helix, have recently gained increasing interest as targets for anticancer drugs. A G-quadruplex is formed by self-assembly of guanine-rich nucleic acid sequences, the basic unit of which is the G-quartet, a planar motif generated from four guanine residues pairing together through Hoogsteen like hydrogen bonds.5 The structures and topologies of G-quadruplex nucleic acids have been well investigated and widely reviewed.6,7 As shown in Fig. 1, a series of planar G-quartets stack with each other and are connected by the intervening sequences (termed loops), forming the G-quadruplex (G4) structure. Both DNA and RNA G4 structures are inherently stabilized by the presence of alkali–metal cations (most often Na+ or K+ ion) coordinated by the guanine carbonyl oxygen atoms pointing towards the inner channel formed at the centre of G-quartets.8 G-quadruplex structures can be formed not only intra-molecularly within single stranded nucleic acid sequences but also inter-molecularly from two or more individual strands. Depending on the distinct ways the exterior loops connecting the G-quartets and the relative orientation of the tetra-stranded helices, G-quadruplexes display a wide range of topologies (ca. parallel, anti-parallel, hybrid of parallel and anti-parallel, etc.). Indeed, NMR and crystallography have been widely used to explore the structures of G-quadruplexes, some of which have been successfully reported either as native nucleic acids or as complexes of G4 nucleic acids binding with small molecules.6,7 The distinctive structures of G-quadruplexes offer a great opportunity for specific molecular recognition. Recently, the existence of G-quadruplex DNA has been quantitatively visualized on chromosomes in human cells,9 further motivating efforts to understand the biological implications and the structural–functional relationship of G4 structures.
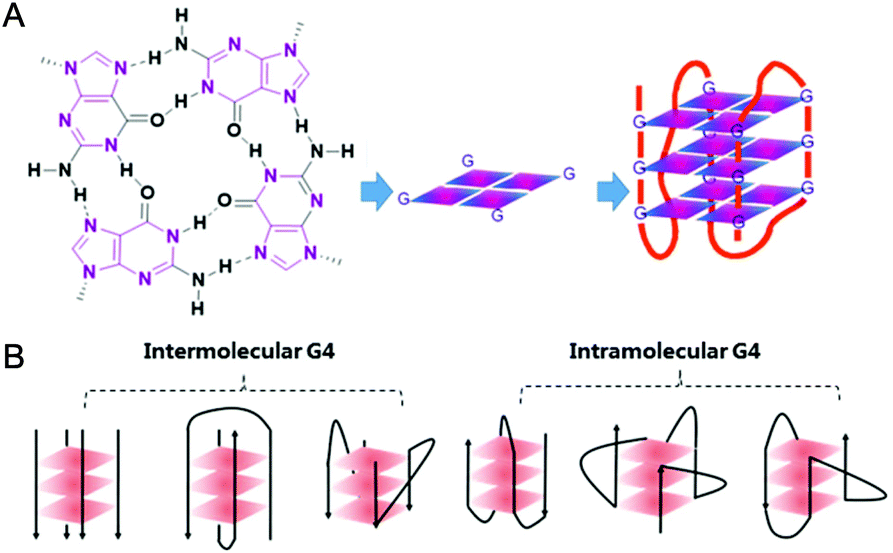 | ||
| Fig. 1 (A) Schematic representation of a G-quartet and its stacking to form an intramolecular G-quadruplex structure. (B) Different topologies of G-quadruplex arranged intermolecularly and intramolecularly. | ||
2. What are the biological implications of G4 and G4-ligands?
G-quadruplex DNA predominantly exists in the chromosomal telomeric sequences and the promoter regions of numerous genes, such as the oncogene bcl-2, VEGF, c-myc, c-kit.10–15 Human telomeric (hTel) DNA consists of the tandem d(TTAGGG) repeats and a single stranded 3′-overhang of 100–200 bases.16–18 In normal somatic cells human telomeric DNA is shortened at a rate of 50–200 bp per cell division cycle, and the accumulated telomere shortening ultimately results in cell senescence and apoptosis. However, telomere maintenance and elongation rather than telomere shortening dominates in cancer cells which is the basis for cancer cell proliferation and immortalization.16 This is attributed to telomerase activity which is silent in normal cells but up-regulated with high activity in 85–90% of cancer cells.19 Active telomerase hybridizes with the 3′-overhang of the telomeric DNA to elongate the telomere thus maintaining the telomeric DNA integrity in cancer cells. Formation of G4 structures within the 3′-overhang of telomeric DNA blocks telomerase hybridization, resulting in the inhibition of telomerase activity thereby interfering with telomere maintenance in cancer cells.20 G-quadruplex DNA formed in promoter regions also displays significant biological roles especially in modulating gene transcription.10–14 Prior to transcription the duplex DNA transiently unwinds to release the G-rich single strand from the complementary C-rich strand. Once the single-stranded G-rich sequence folds into a stable G-quadruplex, its access to the promoter will be blocked thereby down-regulating transcription. Thus rational design of small molecule ligands to selectively interact and stabilize either or both hTel G4 and promoter G4 of oncogenes has been identified as a promising strategy for the development of anti-cancer drugs with selective toxicity towards cancer cells over normal ones.21Although much research has focused on G4 DNA, G-quadruplex RNA exists within the non-coding telomeric r(UUAGGG) repeats (TERRA) and untranslated regions (5′-UTRs) of mammalian mRNA sequences.22–25 G-quadruplex formation within the nascent RNA blocks the progress of the ribosome complex formation thereby down-regulating translation.24 Compared to equivalent G4 DNA sequences, G4 RNA is much more stable, invariably folding into parallel topology, and widely distributed in the entire cell including cytoplasm which makes it easier to access than DNA. Thus G-quadruplex RNA is also considered a potential target for anticancer drugs, and lately considerable research effort has focused on RNA-directed drug design that can selectively target G-quadruplex RNA.26,27
Indeed, the development of small molecules that can recognize and bind to the G-quadruplex with high affinity and specificity over duplex nucleic acids, termed G4-ligands, has become a progressively large field of research with rapidly increasing numbers of reported molecules as well as excellent reviews.28,29 The representative member is a first-in-class in vivo G4-ligand, named as CX3543 (also known as Quarfloxin), which has reached phase II clinical trials for treating neuroendocrine tumors and carcinoid tumors.30 Besides purely organic G4-ligands, metal complexes acting as small molecule G4-ligands have recently attracted a lot of interest, as they interact strongly and selectively with quadruplex nucleic acids.28,31 Compared with organic molecules, metal complexes possess characteristic structural features, various charges, and additional electromagnetic properties providing advantages for the construction of G4-ligands. For example, the synthesis is very regular and much easier; the geometry is variable and controllable (e.g. planar, octahedral, tetragonal pyramidal, etc.) which is predominately determined by the coordination geometry surrounding the metal centre. Such a variety of geometries provides a greater number of action modes: planar molecules favour p-stacking with G-quartets, including end-stacking and intercalation, but also alternative modes such as groove/loop binding, electrostatic interactions, and direct coordination to bases or the phosphate backbone are possible. In addition, the central metal ions and suitable substituted ligands offer cationic properties to the entire molecule which is preferable for stronger electrostatic interactions with electronegative nucleic acids and easier cell permeability. Certain metal complexes possess interesting optical, magnetic, or catalytic properties and thus show additional functions and anti-cancer properties. For example, optical properties including large stokes shifts, high quantum yields, long-lived luminescence, and good photostability allow one to trace the behaviour of these metal complexes in real-time in living cells, thereby at the same time acting as theranostic agents and G-quadruplex probes. These unique properties of metal complexes make them ideal candidates for constructing novel G4-ligands.
This review highlights the recent developments of G4-targeting metal complexes, termed G4-ligands, discussing their binding modes with the G-quadruplex, their inhibition effect on telomerase activity, their interference with gene transcriptional and translational regulation, and their potential to act as anticancer drugs in the clinics. Although G4 RNA also possesses significant biological roles in cancer biology, to the best of our knowledge the only report of metal complexes acting as RNA G4-ligands is a bimetallic platinum(II)-modified perylene derivative reported by Bierbach et al.32 Thus this review will only cover the reported metal complexes acting as DNA G4-ligands. Generally speaking, these compounds achieve their anticancer potential by inducing or stabilizing G-quadruplex formation through various binding modes, thus affecting gene transcription and expression (promoter G4) or inhibiting telomerase activity (telomere G4). In a few cases, metal-containing G4-ligands can even induce cross-linking or cleavage of G-quadruplex DNA, ultimately achieving anticancer properties.
3. Methods for studying ligand/G-quadruplex interactions
A wide variety of biophysical and biochemical techniques have been employed to investigate the interactions of G4-ligands and their effect on telomerase activity and gene expression regulation.33 Usually the combination of more than one technique is required for a detailed understanding of G4-ligand interactions. Optical spectroscopy, including UV-visible (UV-Vis), fluorescence resonance energy transfer (FRET), and circular dichroism (CD), is one of the routinely used techniques capable of determining, e.g., the melting temperature of the G-quadruplex in the presence of G4-ligands via the spectral changes, thus providing crucial information about G4-ligand interactions, such as stoichiometry, stabilization potency and especially the selectivity for quadruplex in comparison to duplex DNA. These optical methods are rapid, non-destructive and only require small amounts of material. Moreover, the G-quadruplex has various topologies and CD spectroscopy can monitor conformational transitions. For example, the characteristic spectrum of an antiparallel G4 structure shows a positive band at 295 nm and a negative band at 265 nm, while both a negative band at 240 nm and a positive band at 275 nm are signatures of a parallel G4 structure. Taking a propeller-shaped trinuclear Pt(II) complex {[Pt(dien)]3(ptp)}(NO3)6,34 which has recently been reported by our group as an example (complex 165 in Fig. 15), FRET melting curves and stabilization temperature (ΔTm) values obtained in the presence of 0.5 μM Pt(II) complex showed that this complex has little effect on the duplex DNA (ΔTm = 0.4 °C), but induces an appreciable increase in thermal stability (ΔTm = 30.2 °C) in hTel G4 sequences, indicating excellent selectivity towards the G-quadruplex. Titration of this complex to the hTel d[AG3(T2AG3)3] sequence, in the presence of either K+ or Na+, induces characteristic CD signatures of an antiparallel G4 structure even at a low complex/DNA ratio (r = 0.2–1.5), indicating that this complex likely induces and stabilizes the antiparallel G4 topology.To quantitatively analyse G4-ligand interactions, two complementary methods, isothermal titration calorimetry (ITC) and surface plasmon resonance (SPR), have been extensively used to determine the kinetic and thermodynamic parameters of the binding events, including binding affinity, dissociation constant, enthalpic and entropic contributions of the binding process, etc.33 These parameters provide crucial information for understanding the driving force for ligand/quadruplex complex formation, evaluating the selectivity of the G4-ligands, and to some degree revealing the binding modes. However, all the biophysical techniques mentioned above cannot provide precise structural information. For the development of excellent G4-ligands with high affinity and specificity, it is important to determine the structural parameters of ligand/quadruplex interactions including precise binding sites and binding modes at the molecular level. Such structural information can be obtained by X-ray diffraction and NMR spectroscopy, and until now a variety of resolved structures of ligand/G-quadruplex complexes have been reported and reviewed.8,29,35–37 Among them, the first crystal structure of a hTel G4 DNA bound to a metal–salphen complex (copper or nickel) was reported by Campbell et al. in 2012.38
Besides the described biophysical methods, several in vitro biochemical methods have also been developed to assess the capability of G4-ligands to induce and stabilize the G-quadruplex, to inhibit telomerase activity, to accelerate telomeric shortening, and to inhibit oncogene expression. Here we focus on two usually employed PCR-based methods, the PCR-stop assay and in vitro TRAP (Telomere Repeat Amplification Protocol) assay.33 In a PCR-stop assay the G4-ligand induced formation and stabilization of a G-quadruplex structure within the DNA sequence will reduce or even inhibit DNA synthesis, resulting in decreased formation or complete absence of the PCR product. The IC50 value obtained in a PCR-stop assay indicates the concentration of G4-ligands required to achieve 50% inhibition of the amplification reaction. Telomerase activity can be evaluated by the in vitro TRAP assay, which involves three steps: initial primer elongation by telomerase in the absence or presence of G4-ligands, removal of the G4-ligands and then PCR amplification of the telomerase elongation products. The obtained IC50-TRAP value is defined as the concentration of G4-ligands required for inhibiting telomerase activity by 50%. Taking again the propeller-shaped {[Pt(dien)]3(ptp)}(NO3)6 complex as an example (vide infra, complex 165 in Fig. 15),34 increasing concentrations of the Pt(II) complex indeed decrease the amount of PCR products (dsDNA) with a complete PCR stop observed in the presence of 3.0 μM of Pt(II) complex, confirming that this complex can induce and stabilize hTel G4 structure. Simultaneously this complex exhibits effective inhibition towards telomerase activity in a concentration-dependent manner with an IC50-TRAP value of 16.0 ± 0.40 μM.
In addition to the techniques described above for investigating metal complex/G-quadruplex interactions and the anticancer properties of G4-targeted metal complexes, other efficient and robust biophysical techniques and in vitro assays are also available to monitor the ligands/G-quadruplex interactions, such as mass spectrometry, surface-enhanced Raman spectroscopy, single-molecule fluorescence imaging, equilibrium dialysis, gel electrophoresis, translational assay as well as molecular modelling. These are not described in detail here as their applications to metal complexes have been limited.
4. G-quadruplex targeting metal complexes
4.1. Cisplatin derivatives – platination of G-quadruplex
In spite of the disadvantages of its unselective binding to genetic DNA, cisplatin is still the most successful anticancer drug in clinical use. Thus the development of rationally designed cisplatin derivatives acting as specific DNA G4-ligands also attracts a lot of attention. Similar to cisplatin, these derivatives also contain labile groups (Cl, H2O, etc.) making direct coordination of the platinum centre to G4 DNA nucleobases highly probable. This type of coordination is traditionally denoted as platination and can occur either at a single-site or lead to cross-linking of two nucleobases, in most cases, guanines. For example, cross-linking of two guanine bases or of an adenine-and a guanine base was observed after platination of a preorganized G-quadruplex with [Pt(NH3)2(H2O)2](NO3)2 (cis or trans), the action pattern of which is similar to cisplatin.39 A dimetallic cisplatin derivative (1 in Fig. 2) can also cross-link two guanine bases and the cross-linking position was located at the terminal G-quartets.40 In addition, an interesting Pt–ACRAMTU complex 2 (Fig. 2) showed an abnormal kinetic preference for platination at adenine (N7 site) over guanine, and more interestingly, HPLC analysis of the reaction between DNA and Pt–ACRAMTU showed that the amount of the Pt–G4 adducts exceeds the corresponding Pt–duplex DNA adducts, indicating some binding selectivity of this complex towards G4 DNA.41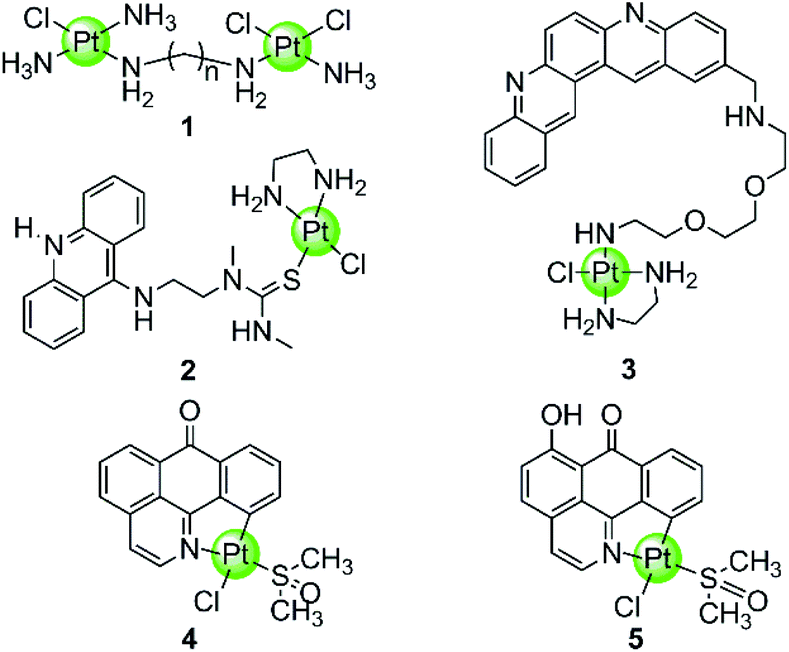 | ||
| Fig. 2 Cisplatin derivatives reported as G4-ligands, the binding mode of which is mainly direct platination (single-site coordination or cross-linking of two nucleobases).40–43 Complex 3 utilized a double noncovalent/covalent binding mode.42 | ||
Cisplatin derivatives containing a planar aromatic ligand suitable for π-stacking with the G-quartets allows interactions with the G-quadruplex through a double noncovalent/covalent binding mode. For example, a Pt(II)–MPQ complex 3 (Fig. 2) was constructed by linking cisplatin with a planar quinacridine aromatic moiety through a long hydrophilic linker, the length of which was suitable for spanning the length of the G-quartet stacks.1,42 Such structural features allow the quinacridine plane to end-stack on one face of the G-quartets and the Pt(II) metal centre to platinate a tetrad guanine base on the opposite face, ultimately stabilizing the antiparallel topology of a 22-mer G4 DNA. Very recently, two organoplatinum complexes 4–5 (Fig. 2) have also been reported containing both the labile chloride atom and a π-conjugated planar aromatic ligand (1-azabenzanthrone or 6-hydroxyloxoisoaporphine alkaloid), which allowed platination at guanine nucleobases of the G-quadruplex as well as the non-covalent π-stacking with G-quartets.43 The in vitro and in vivo anticancer efficacies of these two Pt(II) complexes were also investigated in cisplatin-resistant tumor cells and xenograft models, respectively.
4.2. Metalloporphyrins and derivatives
In the examples presented above, the Pt(II) metal centre coordinates directly to G4 DNA bases. However, most of the reported metal-containing G4-ligands interact with G4 DNA through non-covalent binding. The earliest reported metal complexes acting as G4-ligands are metalloporphyrins, the binding mode of which was proposed to be π-stacking on top of the terminal G-quartets (termed end-stacking), similar to the free porphyrins.44–46 TMPyP4, a cationic meso-methylpyridinium-substituted porphyrin, was reported in 1998 to be capable of stabilizing the human telomeric G4 DNA by end-stacking on the terminal G-quartets and inhibiting telomerase activity (IC50-TRAP = 6.5 ± 1.4 μM).47 The cationic properties of TMPyP4 enable electrostatic interactions with the negative backbone of G4 DNA, and hence a higher binding affinity is observed compared to porphyrin. Subsequently, a series of TMPyP4 complexes with various metal centres, including main-group metals and transition metals (Ni(II), Mn(III), Mn(V)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, Mg(II), Cu(II), Zn(II), Pd(II), Pt(II), Fe(III), Co(II), In(III), etc.) have been synthesized and investigated. All these metal–TMPyP4 complexes (6–16 in Fig. 3) efficiently stabilize the hTel quadruplex and inhibit telomerase activity in vitro, due to the cationic charge and the π-stacking abilities (either end-stacking or intercalation) of the complexes described above. The stoichiometric ratio of ligand/G-quadruplex was found to be 2
O, Mg(II), Cu(II), Zn(II), Pd(II), Pt(II), Fe(III), Co(II), In(III), etc.) have been synthesized and investigated. All these metal–TMPyP4 complexes (6–16 in Fig. 3) efficiently stabilize the hTel quadruplex and inhibit telomerase activity in vitro, due to the cationic charge and the π-stacking abilities (either end-stacking or intercalation) of the complexes described above. The stoichiometric ratio of ligand/G-quadruplex was found to be 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in most cases.45,46
:1 in most cases.45,46
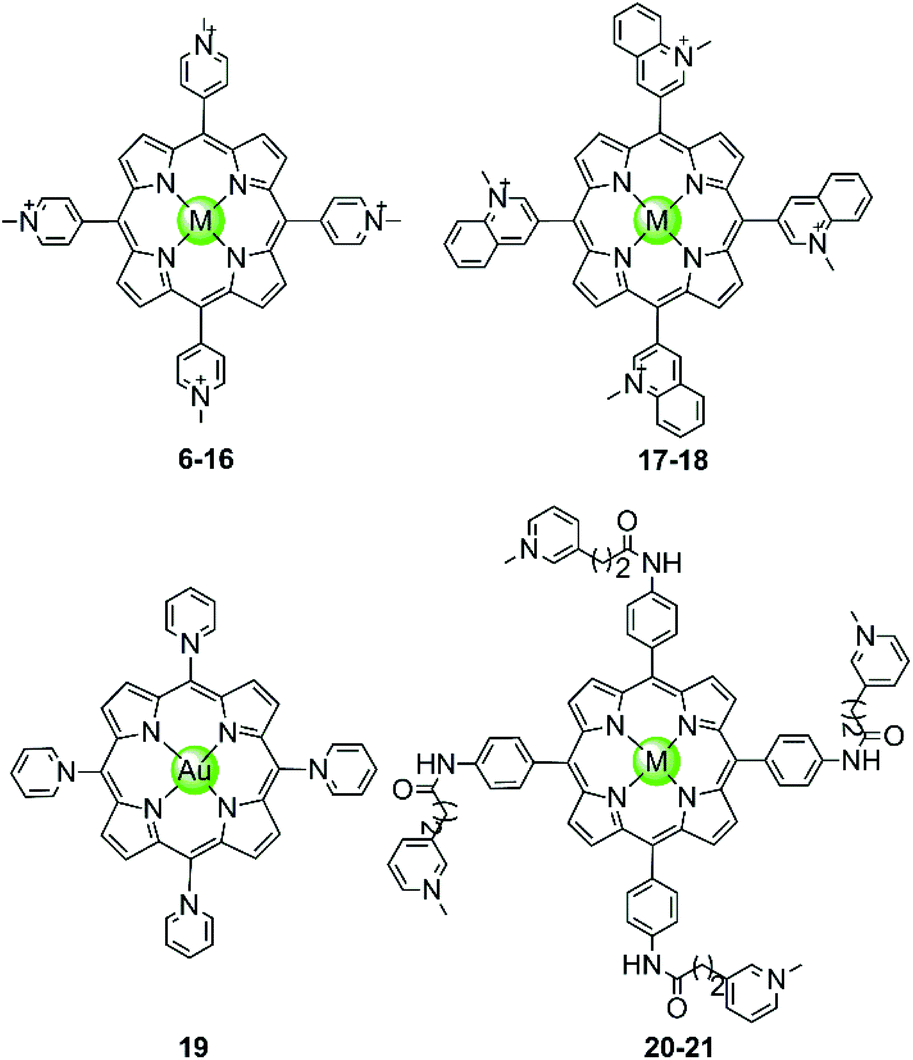 | ||
| Fig. 3 Metal–TMPyP4 complexes and derivatives reported as G4-ligands. The metal centres in complexes 6–16 are Ni(II), Mn(III), Mn(V)O, Mg(II), Cu(II), Zn(II), Pd(II), Pt(II), Fe(III), Co(II), In(III), in 17–18 Cu(II) and Zn(II), and in 20–21 Mn(III) and Ni(II), respectively.44–54 | ||
The metal centre plays a critical role in the ligand/G-quadruplex interactions including binding affinity, specificity, as well as telomerase inhibiting activity. For example, although the nickel(II)–TMPyP4 complex 6 was reported as a potent inhibitor of telomerase with an IC50-TRAP value of 5 μM, its binding affinity for G4 DNA is unfortunately one order of magnitude lower than that for duplex DNAs (106 M−1vs. 107 M−1) in a SPR assay.48,49 In comparison, the manganese(III)–TMPyP4 complex 7 displays similar telomerase inhibition potency to the free TMPyP4 ligand itself, but shows at least 10-fold better selectivity for the quadruplex over duplex DNA.48,49 Changing the metal centre from Mn(III) to Zn(II) maintains the selectivity for quadruplex as well as the telomerase inhibition potency.50 Interestingly, the Zn–TMPyP4 complex 11 was found to be capable of inducing the formation of parallel G4 topology for some specific sections of single-stranded DNA.50 Cu(II)–TMpyP4 complex 10 and its analogue 17 (Fig. 3) also attracted attention because copper is an essential element for human living.51,5210 and 17 both stabilize the hTel quadruplex and inhibit telomerase activity in vitro. Upon titration of the parallel G-quadruplex, a large hypochromic effect in the UV-Vis spectra accompanied by an induced negative band at 240 nm in the CD spectra was observed, indicating a binding mode via the intercalation of the square-pyramidal geometry between the G-quartets of the G-quadruplex. Gold(III)–TMPyP4 complex 19 was also reported as an hTel G4 stabilizer and a potent telomerase inhibitor, which was proven by the effective inhibition of the PCR amplification of a G4 sequence in the PCR-stop assay and the observed 57% inhibition rate of telomerase, Moreover, this Au(III) complex showed low cytotoxicity (IC50 > 50 μM) against normal nasopharyngeal cells.
Besides the metal centre, the choice of the meso substituents on the porphyrin is another critical parameter influencing the interaction with G4. For example, when one or two meso-methylpyridinium groups of the TMPyP4 are replaced by one or two long 4-aminoquinoline moieties, new metalloporphyrins were constructed, probably enhancing the G4 affinity and cell wall penetration (Fig. 4). Such modified manganese(III) complexes 22 and 28 show greater telomerase inhibition than the unmodified Mn(III)–TMPyP4 complex 7, with lower IC50-TRAP values of 11.5 and 8.6 μM, respectively.49 The binding rate was found to be fast in kinetic experiments suggesting the stacking of the quinoline substituents with a G-quartet. When one meso-methylpyridinium group of the TMPyP4 is replaced by a simple polyamine side chain, the Mn(II) complexes 24 show a relatively high affinity (nearly 108 M−1) for quadruplex DNA, although this was not accompanied by improved selectivity.48 Upon replacement of all four meso-methylpyridinium groups of the TMPyP4 ligand by four flexible bulky cationic moieties, the most impressive pentacationic manganese(III)–porphyrin complex 20 was constructed, which has a high affinity (nearly 108 M−1) for quadruplex DNA as well as 10000-fold selectivity towards G4 over duplex DNA.54 Such a high selectivity can also be explained by the steric effect of the bulky side arms preventing the intercalation of the complex into the duplex DNA as similarly described for porphyrin-bridged tetranuclear platinum complexes. In contrast, applying the same modifications mentioned above to the Ni–TMPyP4 analogues 21, 23, 25 does not increase the G4 affinity, the selectivity or the telomerase inhibition activity. These modified Ni–TMPyP4 analogues cause inhibition of telomerase-mediated elongation of the telomere primer at 7–39 μM in the TRAP assay, and hence the inhibition effect is even lower than that of the unmodified Ni–TMPyP4 complex 6.49
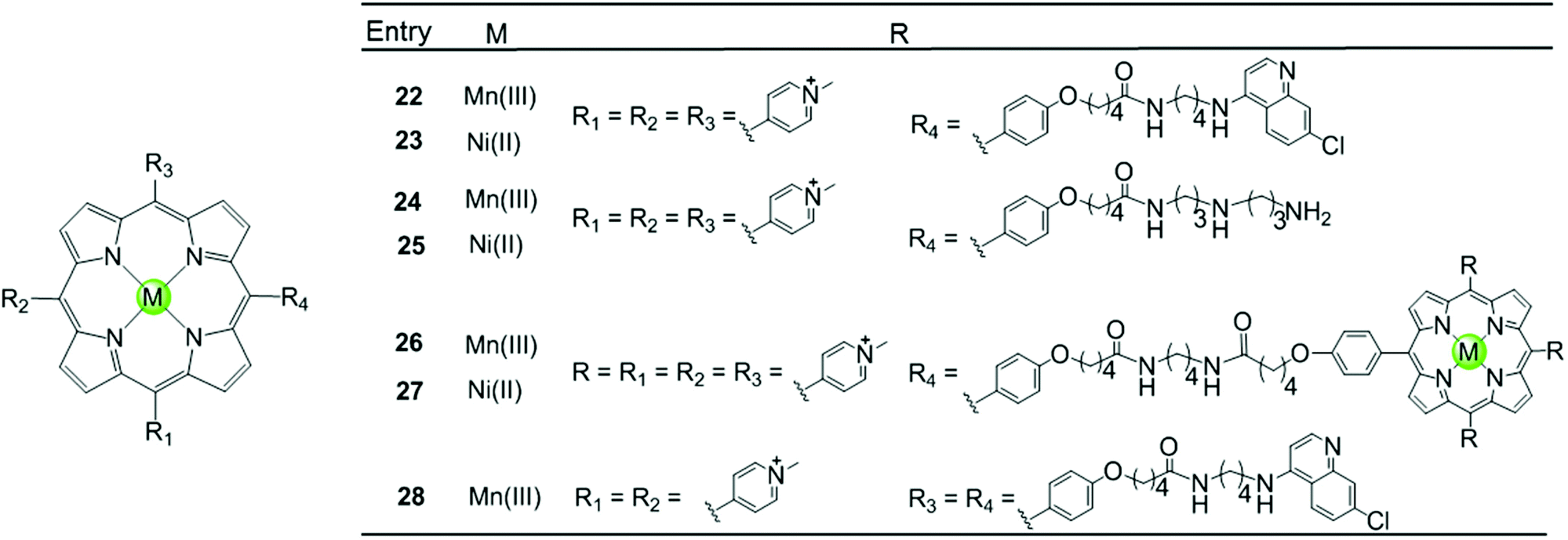 | ||
| Fig. 4 Metalloporphyrins with various meso-substituents reported as G4-ligands.48,49 | ||
In addition, binuclear manganese/nickel–porphyrin derivatives 26–27 were synthesized with a linker of appropriate length linking the symmetric porphyrinic dimer. This design proposes a sandwich-type binding mode with the quadruplex DNA structure in theory, but these complexes were shown to be inefficient for stabilizing or discriminating G4 over the duplex.48
Our group also reported two clover-like shaped, porphyrin-bridged tetranuclear platinum complexes 29–30 (Fig. 5).55 Different from the complexes mentioned above, the Pt(II) ions are coordinated at the side arms providing high positive charges in the compounds and probably increasing the overall steric hindrance, which helps end-on stacking and prevents intercalation into duplex DNA. Indeed, both of them were found to effectively stabilize various kinds of G-quadruplexes (hTel, c-myc, c-kit and bcl2) in the parallel topology with high affinity but displayed negligible effects on the duplex. The π–π end-stacking binding mode was proven by the hypochromic effect in UV/Vis spectra and unquenched fluorescence upon titration of the G4 sequence. Furthermore, the maximum binding ratio of 4:1 ([complex]:[G4]) indicates the presence of other binding modes such as groove interactions. The clover-like shaped platinum complexes showed excellent anticancer activity that was attributed to a dual effect, the inhibition of telomerase activity (IC50-TRAP = 1.46 μM and 0.25 μM) and the repression of oncogene expression, ultimately inducing apoptosis and G2/M phase arrest in HeLa cells. Another special case is the ruthenium(II) polypyridyl complex 31 having a pyridine ligand attached to a porphyrin.56 Although the Ru(II) metal core determines the octahedral geometry of this compound, the planar porphyrin ligand results in a high affinity to the hTel G4 accompanied by hypochromism effects in UV/Vis spectra.
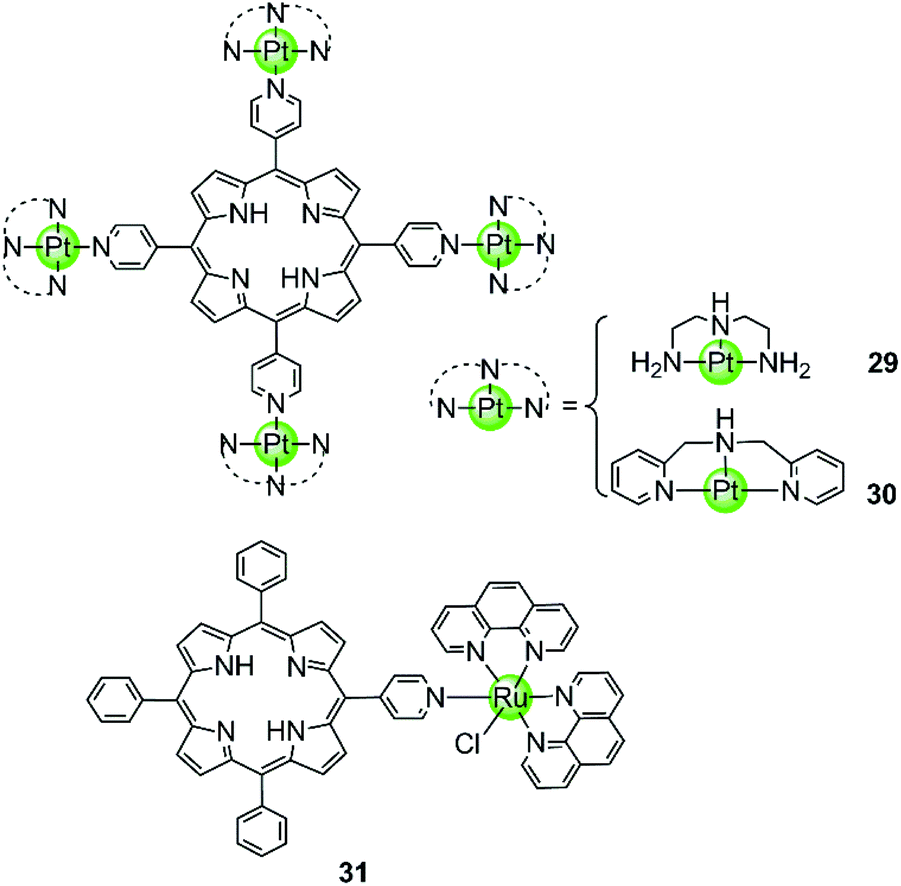 | ||
| Fig. 5 Porphyrin-bridged tetranuclear Pt(II) complexes and a mononuclear Ru(II) complex containing a porphyrine-like ligand.55,56 | ||
These investigations indicate that the central aromatic core (TMPyP4 moiety) might be predominantly responsible for the interactions between metalloporphyrines and G4. The side arms also play significant roles, helping to improve the binding affinity and selectivity over the duplex depending on the rational design.
4.3. Metallophthalocyanines and derivatives
Phthalocyanine, a porphyrin derivative featuring aromatic rings fused to each pyrrole moiety of the basic porphyrin skeleton with nitrogen atoms in the meso-positions, provides a more extended aromatic surface preferred for end-stacking on a terminal G-quartet. A series of metallophthalocyanines, especially with zinc(II) and nickel(II) as metal centres, have been reported as novel and potent telomerase inhibitors (Fig. 6).57–61 The phthalocyanine skeleton of these complexes was modified by introducing four or eight cationic oxygen/sulfur armed quaternary ammonium on the fused aromatic rings (complex 32–39), increasing the number of cationic charges and enhancing the steric hindrance, both of which made the complexes more favourable G4 binders. Compared to the corresponding metalloporphyrins, these metallophthalocyanines display enhanced binding affinities and selectivity for the G-quadruplex over duplex DNA. For example, the Zn(II) complex 38 with eight cationic quaternary ammonium exhibits strong electrostatic interactions with grooves or loops, contributing to the approximately 6-fold G-quadruplex selectivity over duplex DNA and very effective telomerase inhibition (IC50-TRAP = 0.23 μM). Moreover, this complex prefers to induce conformational transitions from antiparallel to parallel G-quadruplex even in alkali–metal deficient buffer.60 By comparison, the Zn(II) analogue 47 with fewer positive charges (4+) and less steric hindrance prefers an antiparallel G4 topology in alkali–metal deficient buffer.61 A series of Zn(II) complexes with amido-armed phthalocyanines (41–46) were also reported with excellent affinities for hTel G4 as well as a metallophthalocyanine was reported in 2009: the guanidinium–parallel G4 with high affinity. When interacting with c-myc G-quadruplex DNA the dissociation constant is only 2 nM, which is the strongest binding interaction among all reported small molecule-based G4-ligands.57,58 These investigations indicate that both the planar phthalocyanine moiety and the highly positively charged side arms contribute to the favourable binding to G-quadruplex as well as the telomerase inhibition potential.57,58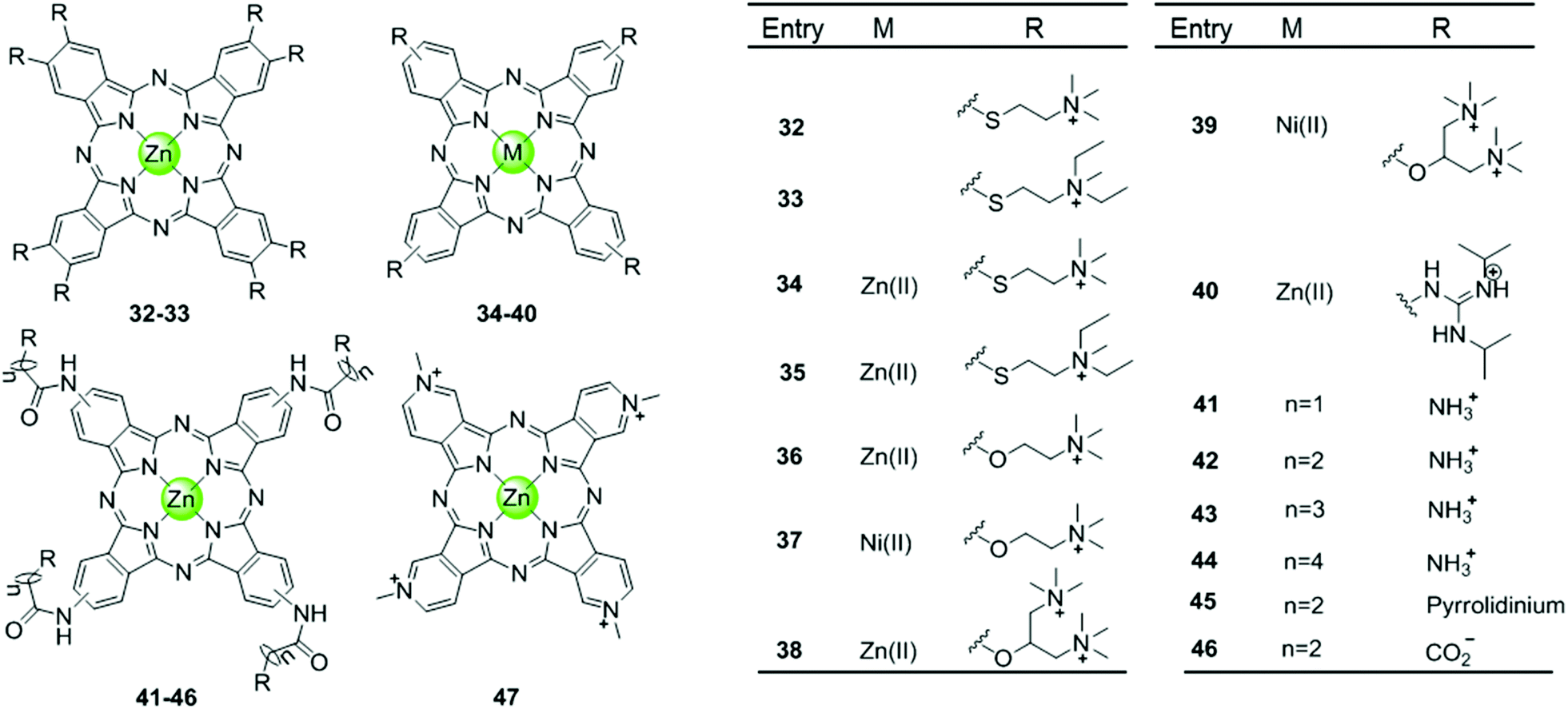 | ||
| Fig. 6 Metallophthalocyanines and derivatives with four or eight cationic side arms reported as G4-ligands.57–61 | ||
4.4. Metallocorroles and derivatives
Another porphyrin derivative is the corrole ligand, which provides additional geometries and electronic properties for the effective stabilization of transition metal ions in high oxidation states. A series of metallocorroles, mainly with Cu(II) or Mn(III) as metal centres (Fig. 7), were reported as effective G4-ligands and potent telomerase inhibitors.62–64 Interestingly, these complexes have typically a saddle-type geometry opposed to the planar metalloporphyrins. One example is the water-soluble, saddle-shaped, meso-methylpyridinium-substituted Mn(III)–corrole complex 48.62,63,65 Due to the favourable shape and high electron deficiency, this Mn(III)–corrole complex exhibits 64-fold selectivity towards G4 over duplex DNA according to the binding constant values and prefers to induce hybrid G4 topology according to the CD characteristic signatures. In a PCR-stop assay, this complex effectively induces and stabilizes hTel and c-myc G-quadruplex DNA with the IC50 values of 2.37 and 1.52 μM, respectively. The Cu(II)–corrole complex with the same meso-methylpyridinium substitution as the Mn(III)–corrole complex 48 was reported to be as effective and selective in G4 stabilization, the activity of which was slightly lower than that of the corresponding Mn(III)–corrole complex.62 Its binding constant to G-quadruplex DNA was found to be 50-fold greater than that to duplex DNA, and the IC50 values obtained in the PCR stop assay are 3.51 and 2.74 μM, respectively, for hTel and c-myc. CD spectra showed that this complex also prefers to induce the parallel-to-hybrid conformational transitions of the hTel G4 sequence. Further Cu(II)–corrole complexes (49–54), modified by introducing three meso-substituted benzene ring-armed pyridinium or quaternary ammonium moieties through different linkers into the corrole skeleton, were also investigated as G4-ligands.62 Such modifications increase the number of cationic charges which promotes electrostatic interactions with the negatively charged DNA backbone as mentioned above. Both CD spectra and PCR-stop assay indicated that these complexes are good at stabilizing G4 in the presence of micromolar Na+ concentration, and that some of them induce antiparallel G4 topology.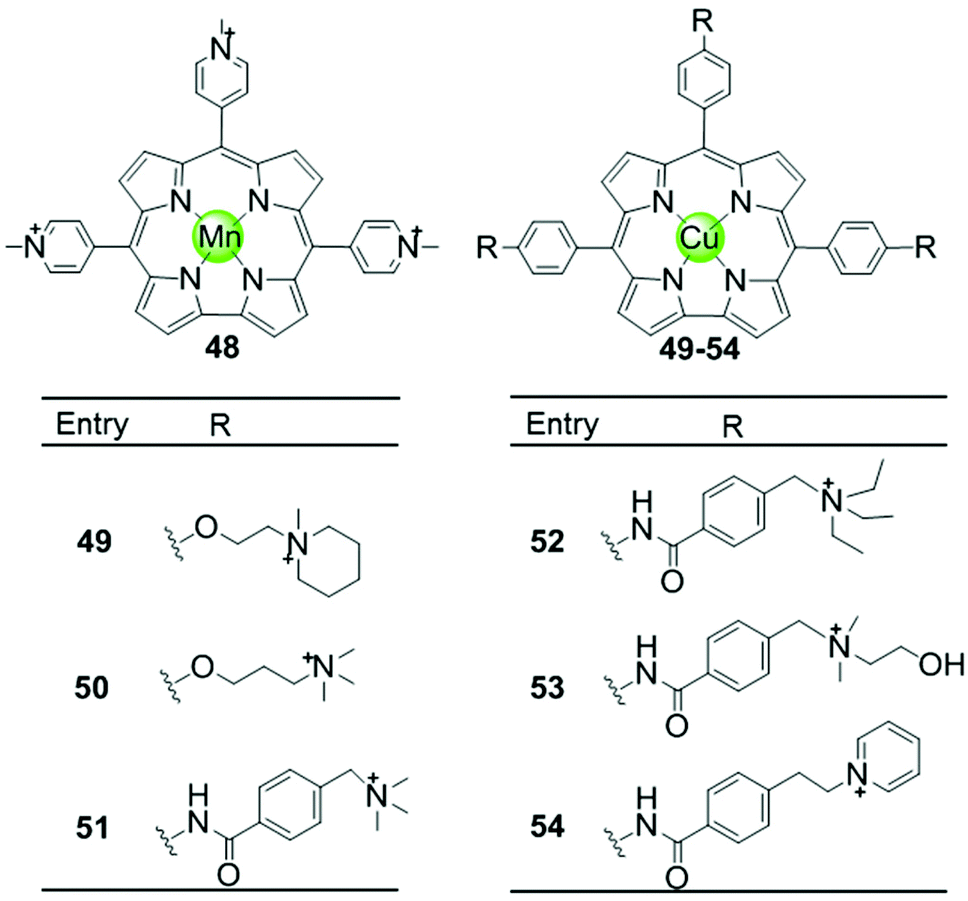 | ||
| Fig. 7 Metallocorroles with cationic side arms reported as G4-ligands.62–65 | ||
4.5. Metal–salphen and metal–salen complexes
Besides macrocyclic metal complexes, planar metal complexes with nonmacrocyclic polydentate ligands acting as effective G4-ligands have also attracted ample attention. A representative group of these complexes are metal–salen and metal–salphen complexes, which have previously been proven to be capable of intercalating into duplex DNA through π–π stacking.66,67 A series of comparative metal–salphen complexes with similar polydentate ligands but different metal ion centres were constructed (Fig. 8). Crystal structures revealed that Ni(II) complexes 55–64 and 73–82, Cu(II) complex 65 and Pt(II) complexes 68–72 have a planar geometry,68–71 whereas the penta-coordinated V(IV)O complex 67 has a quasi-planar geometry69,72 and the Zn(II)(H2O) complex 66 is non-planar.69 According to FRET, FID and SPR assays, all these planar metal–salphen complexes are effective G4 stabilizers whereas non-planar metal–salphen complexes only show low G4 affinity or specificity. For example, the melting temperature of telomeric G4 was increased by 33, 21.5 and 10.5 °C in the presence of the planar Ni complex 55, the Cu complex 65 and the quasi-planar VO complex 67, respectively, whereas non-planar Zn(II)(H2O) complex 66 only increased the melting temperature of G4 by 1.4 °C. These planar compounds were found to be highly selective for the G-quadruplex over duplex DNA, e.g. 50-fold selectivity of Ni–salphen complexes as reflected by the equilibrium dissociation constants of: 0.1–1.0 μM for the G-quadruplex vs. 2.0 μM for duplex DNA. The interaction mode of the planar metal–salphen complexes with the G-quadruplex was proposed based on molecular modelling: the planar aromatic surface of the complex stacks on the terminal G-quartet with the exterior cationic side chains inserted into the opposite grooves. It is worth mentioning that a co-crystallization of a typical square-planar Ni–salphen compound 57 complexed with the telomeric G4 DNA in parallel topology was successfully obtained.68 The TRAP assay also proved the potent telomerase inhibition activity of these metal–salphen complexes resulting in IC50 values in the low micromolar or even nanomolar range.
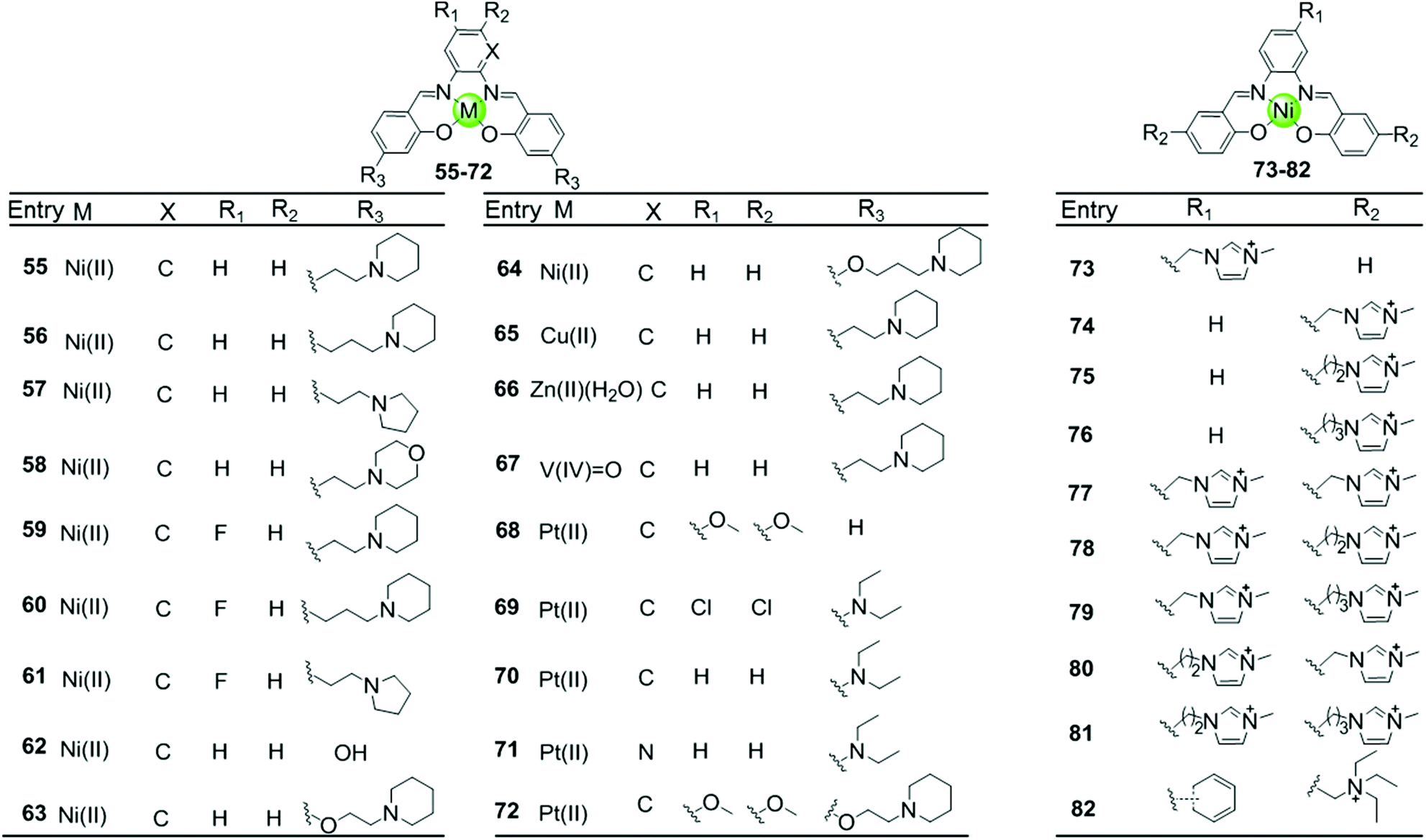 | ||
| Fig. 8 Metallosalphens with various side arms reported as G4-ligands.66–73 | ||
Despite the inability of the salphen ligand itself to π-stack with and stabilize G-quartets due to its flexible geometry, the coordination with suitable metal ions often induces a planar geometry and arranges the aromatic rings around the central metal ion in an optimal structure to enhance π–π stacking between the metal–salphen complexes and G4 structures. Moreover, the modification of the salphen ligand in metal complexes by, for example positively charged quaternary ammoniums, alkyl–imidazolium side chains, or cyclic amine-based side chains which can be protonated under physiological conditions, enhances the binding affinity with G4 by electrostatic interactions of the protonated side arms with the loops and grooves of the DNA backbones. All these factors (overall geometry, charge, modification on the salphen ligand) also dictate the selectivity of the metal–salphen complexes towards G-quadruplexes. For example, the Pt(II)–salphen complex 72 with cyclic amine-based side chains was reported to effectively stabilize the c-myc promoter G4 structure by end-stacking with the terminal G-quartets, resulting in the inhibition of oncogene expression both in a cell-free system and in cultured cells.73 It is worth mentioning that this complex modified with cyclic amine-based side arms exhibited a 10-fold higher inhibition activity than the non-modified complex. Moreover, several Pt(II)–salphen complexes such as 72 possess additional fluorescence emissive properties capable of showing their cellular uptake and localization in living cells by using confocal microscopy.71
A series of Ni(II), Cu(II) and Pt(II) based metal–salen complexes 83–91 with suitably modified salen ligands were also reported to act as efficient G4-ligands, illustrating again the significance of the planar geometry and modifications on the side arms for improving G4 binding (Fig. 9).73–75 For example, a planar Ni(II)–salen complex 87 with quaternary ammonium side chains is capable of selectively stabilizing an oncogene promoter G4 over duplex DNA, as shown by UV/Vis, CD and FRET assays.74 Another two interesting Ni(II)–salen complexes 90–91 with cyclic amine-based side chains were also investigated, the salen ligand of which contained the meso-1,2-diphenylethylenediamine moiety.75 Compared with the corresponding Ni(II)–salphen complex 63–64, the presence of the meso-1,2-diphenylethylenediamine moiety provides steric hindrance eliminating the possibility of intercalation with duplex DNA, thus offering a slightly better selectivity for either a unimolecular or intermolecular G-quadruplex over duplex DNA.
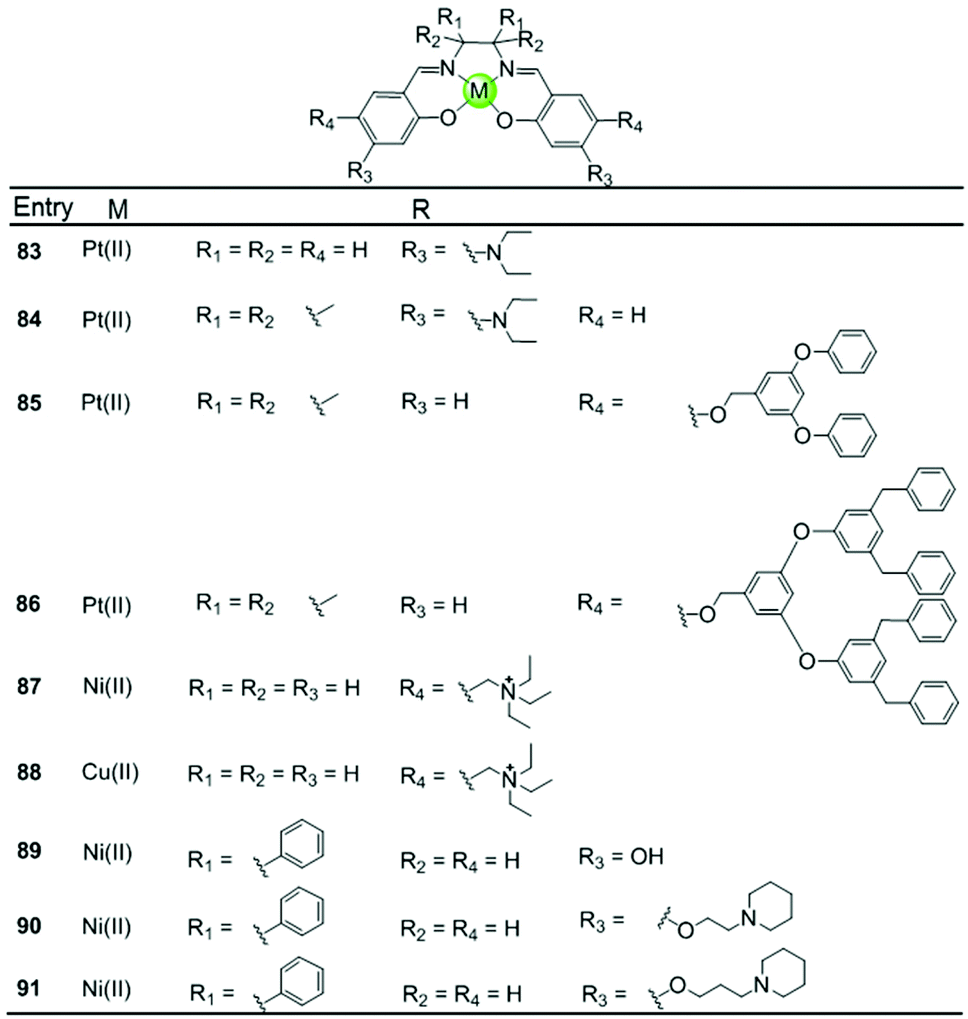 | ||
| Fig. 9 Metallosalens with various side arms reported as G4-ligands.73–75 | ||
4.6. Metal–phenanthroline complexes and derivatives
Another representative group of planar non-macrocyclic polydentate metal complexes are square-planar Pt(II)–phenanthroline complexes, in which the phenanthroline moiety is replaced by analogues such as bipyridine, phenylpyridine, dipyridophenazine, or phenanthroimidazole (Fig. 10). The Pt(II) centre with its positive charges is essential for DNA binding, the square-planar geometry of these complexes again promote π–π stacking with G-quartets. Comparative studies illustrated that ligands possessing an extended π surface favour G4 interactions. For example, both Pt(II) complexes 94 with bis-phenathroline76 or phenanthroline–ethylenediamine77,78 stabilize G-quadruplex structures, the interaction being stronger than with the bis-bipyridine and bipyridine–ethylenediamine analogues 92–93.76–78 Upon replacement of the phenanthroline by a phenanthroimidazole moiety, the latter possessing an extended π-delocalized surface and an aromatic pendant, the selectivity of the platinum complexes 102–103 towards G4 improved and the affinity constants for G4 binding were two orders of magnitude larger than binding to duplex DNA.77,79,80 Organoplatinum complexes usually exhibit excellent photophysical properties, e.g. organoplatinum(II)–dipyridophenazine complexes 96–99 with C-deprotonated 2-phenylpyridine ligands. Upon interaction with G4 DNA, the emission intensity of the complexes is greatly enhanced.81 These photophysical experiments also provide additional evidence supporting the end-stacking binding mode for G4 DNA. The binding affinity in the order of 106–107 M−1 for G4 DNA is stronger than for the phenanthroline complexes and at the same time potent inhibition of telomerase activity in in vitro TRAP assays is observed.81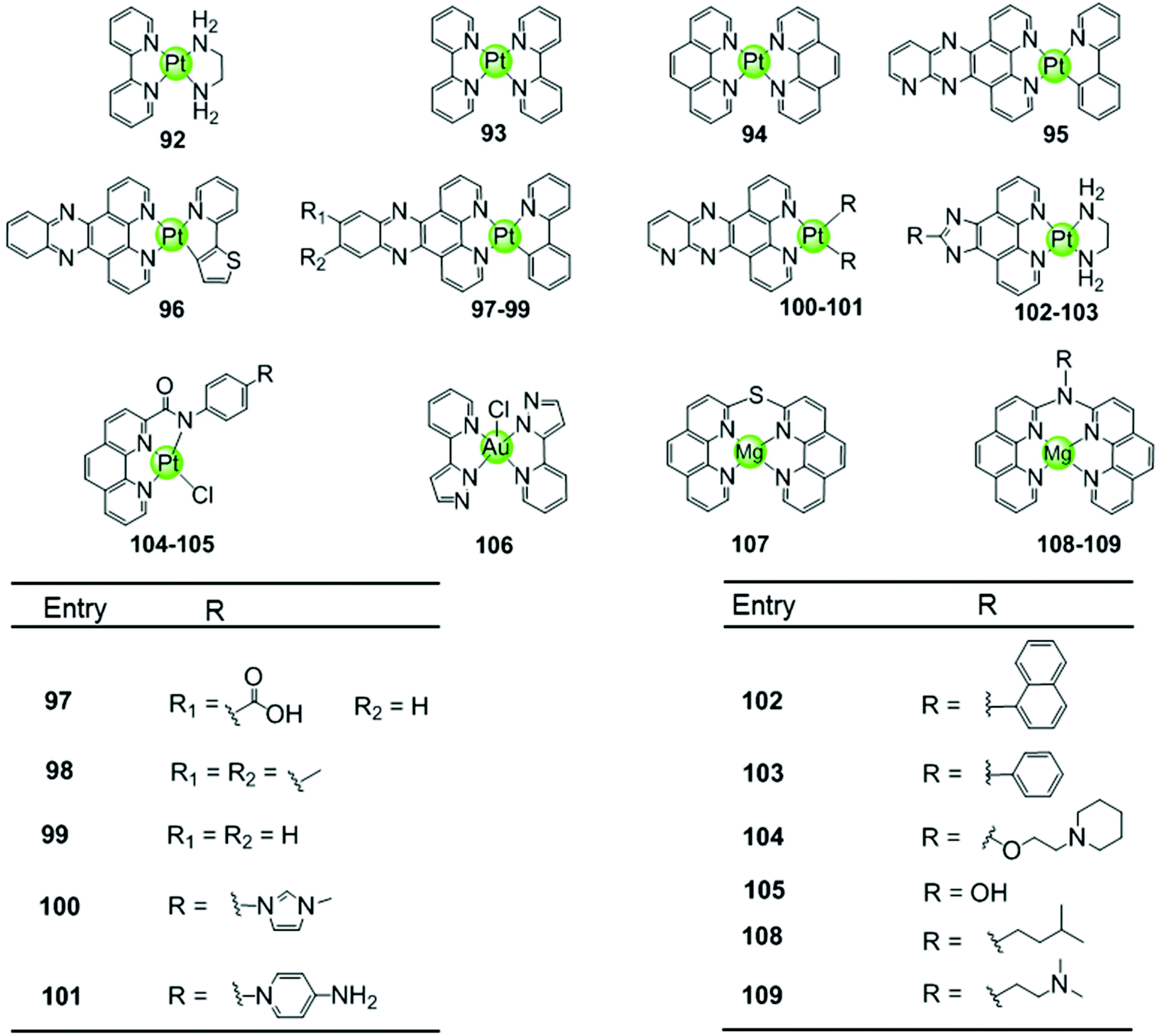 | ||
| Fig. 10 Metal–phenanthroline complexes and derivatives reported as G4-ligands. The bidentate ligands are, e.g. ethylenediamine (dien), bipyridine (bpy), phenanthroline (phen), dipyridophenazine (dppz), phenanthroimidazol, phenylpyridine (ppy). Several Pt(II) complexes are also luminescent G4-ligands.76–84 | ||
Similar phenanthroline complexes with other metal centres 106–109 are also effective G4 DNA binders capable of distinguishing G4 and duplex DNA (Fig. 10).82,83 For example, the Mg(II) complex with bis-phenathroline selectively increases the melting temperature of G4 DNA but displays only a negligible effect on the melting temperature of duplex DNA, illustrating selective G4 stabilization.82 In the case of the Ni(II) complex with the phenanthroline–ethylenediamine ligand, the in situ formation of the complex/hTel G4 DNA adduct was directly observed by electrospray ionization mass spectrometry (ESI-MS).83 A penta-coordinated Au(III) pyrazolylpyridine complex 106 interacts strongly and selectively with quadruplex DNA, in particular with c-myc, through π–π stacking. This is the first example of an Au(III) complex based G4-ligand.84
4.7. Metal–terpyridine complexes and derivatives
Metal–terpyridine complexes with Cu(II), Zn(II), Pt(II) and Pd(II) ion centres can act as effective G4-ligands (Fig. 11 and 12).85–88 The central metal ion, the number of aromatic rings, as well as the number and position of the substituents on the terpyridine scaffold play critical roles for G4 recognition and stabilization. A series of comparative studies of metal–terpyridine complexes demonstrated that the binding affinity and selectivity for G4 DNA depend mainly dependent on the geometry of the complex. Here the effective G4 stabilizers must exhibit at least one planar aromatic surface accessible for π–π stacking interactions with G-quartets. Thus the capability of the zinc(II) terpyridine complexes 110–111 for G4 binding was poor due to their nonplanarity as reported by Teulade-Fichou et al.85 whereas similar Cu(II) 113–114 and Pt(II) complexes 131–132 as reported by Bertrand et al.85 and Wang et al.86, respectively, are good G4 stabilizers due to their planar geometry.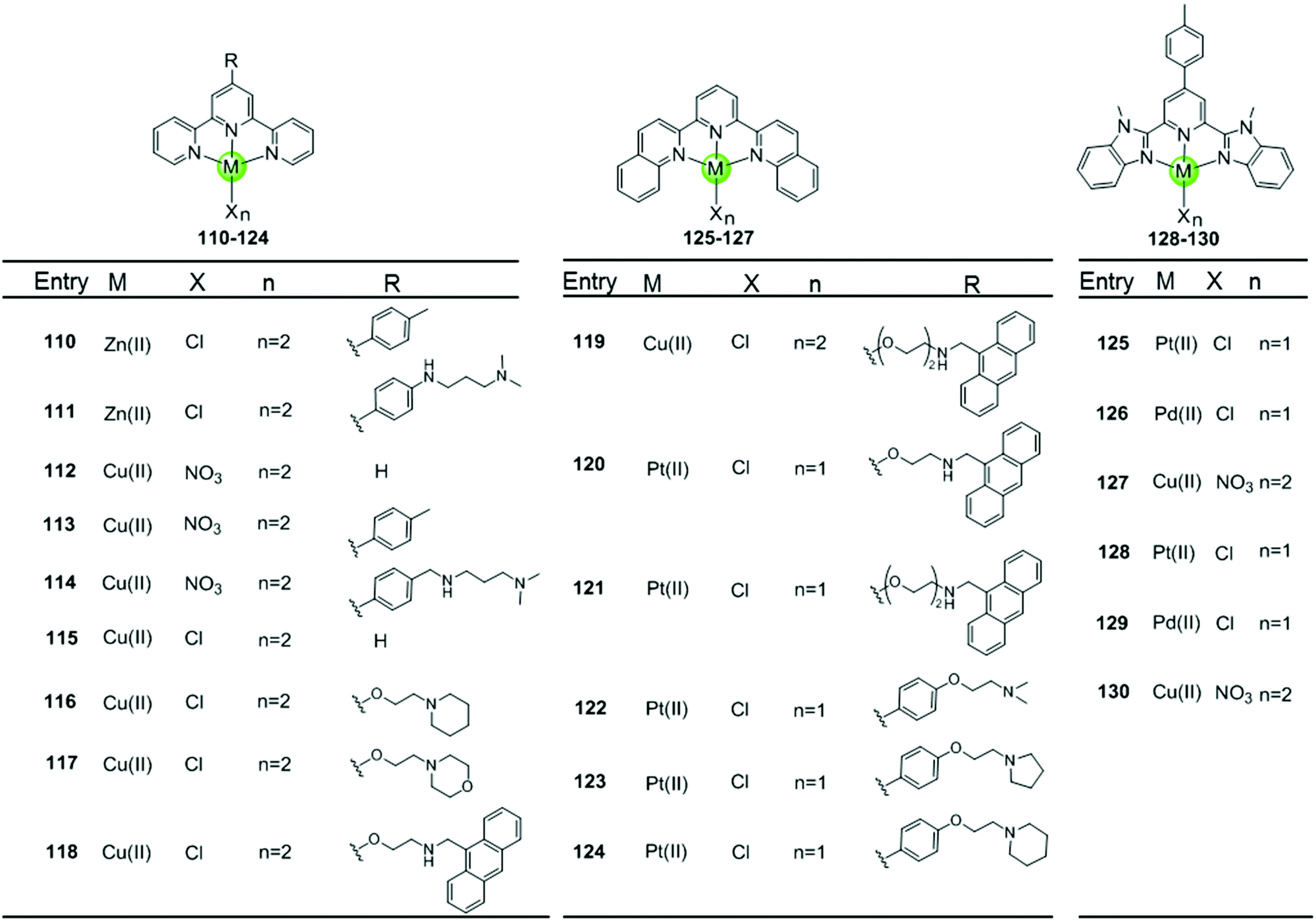 | ||
| Fig. 11 Metal–terpyridine complexes and derivatives reported as G4-ligands.85–89 | ||
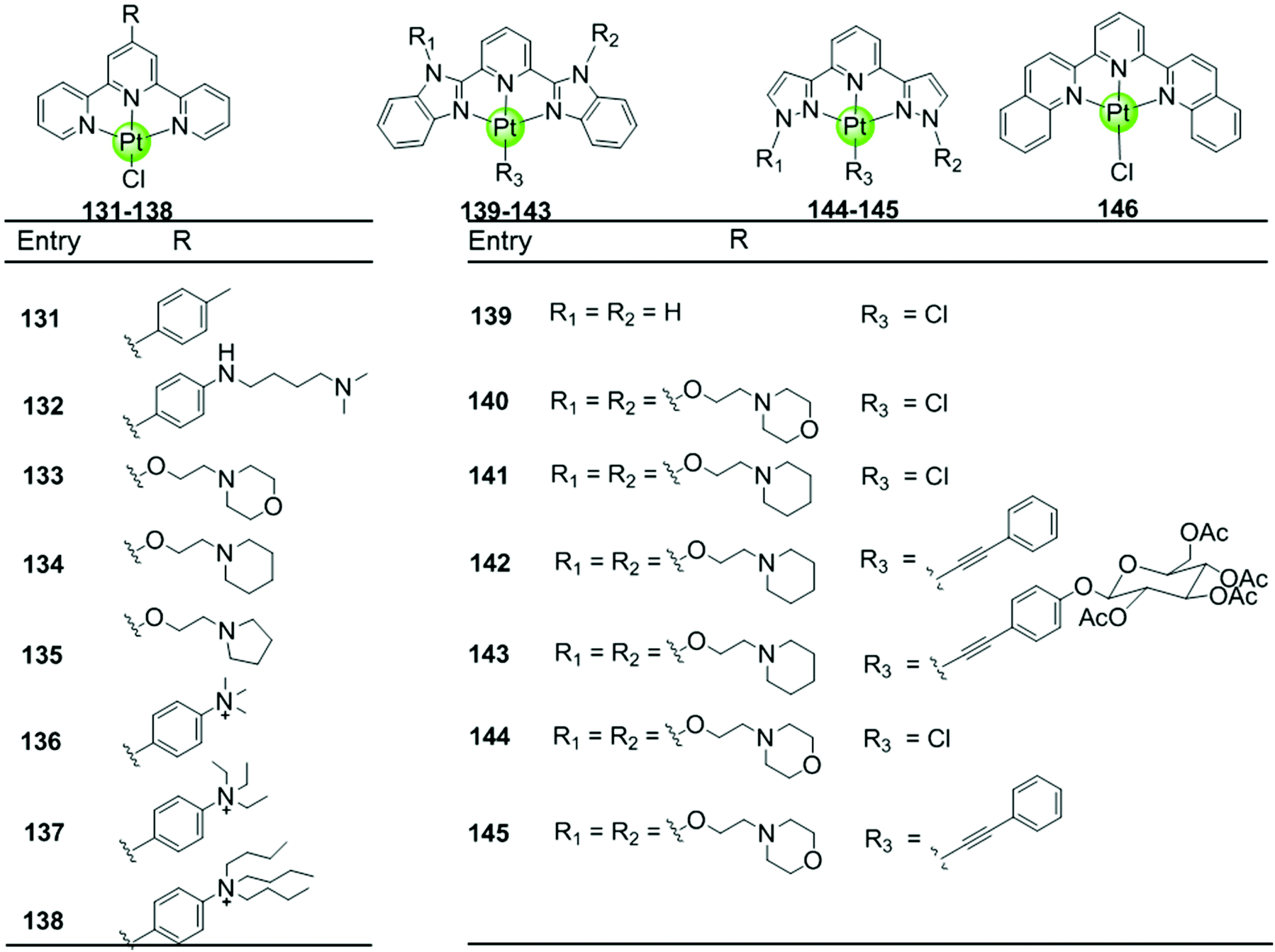 | ||
| Fig. 12 Platinum(II)–terpyridine complexes and derivatives reported as G4-ligands, the monodentate ligand is either a labile chloride or an alkynyl moiety.85–88,90–92 | ||
The modification of terpyridine by attaching additional aromatic rings, charged peralkylated ammonium, cyclic amine-based side chains, or another suitable pendant improves the binding affinity and selectivity of the metal–terpyridine complexes for G4-quadruplex.46–88 For example, Bertrand et al. designed a range of copper–terpyridine complexes, among them complexes with ttpy 113 and ctpy 114, that display a better affinity and selectivity for G4 DNA (ΔTm = 15.3 and 10.0 °C, respectively) than complex 112 which carries a simple tpy ligand (ΔTm = 1.0 °C).85 Another example is a group of Cu(II)– and Pt(II)–terpyridines 118–121 constructed by Gama et al., in which the terpyridine is tethered with a planar anthracene moiety via different linkers.87 All these complexes are potent G4 stabilizers and telomerase inhibitors, exhibiting good affinity and selectivity for the G-quadruplex over duplex DNA. It is interesting to note that the strength of the ligand/G4 interactions increases with the linker size between the anthracenyl moiety and the terpyridine chelating unit.
The extension of the planar aromatic surface of the terpyridine scaffolds also enhances the binding affinity and specificity of metal complexes for G4 DNA. Several N-donor tridentate (terpyridine-like) ligands with a more extended planar aromatic surface than the simple terpyridines were designed. The corresponding Pd(II), Cu(II) and Pt(II) complexes 125–129 exhibit an enhanced binding affinity and specificity for G4 DNA (Fig. 11).89 It is interesting to note that the Pd(II) complex 129 exhibits a better stabilization effect on G4 DNA than the corresponding Pt(II) and Cu(II) complexes 128 and 130 according to the FRET assay. The ESI-MS and UV/Vis measurements suggest that the Pd(II) complex possesses the greater ability to effectively coordinate DNA bases at room temperature whereas the Pt(II) complex predominately binds non-covalently to G4 DNA.
Here we concentrate on platinum(II)–terpyridine complexes as these are most widely studied (131–146 in Fig. 12).85,88,90,91 Besides very few examples with a sulfur atom-armed cyclic amine as the monodentate ligand, most reported platinum(II)–terpyridine complexes can be divided into two main groups: the monodentate ligand is either a labile chloride or an alkynyl moiety, both ensuring a planar geometry of the complex optimal for G4 interactions through π stacking. The presence of the labile chloride allows the possibility of direct coordination of the platinum(II)–terpyridine complex to G4 DNA.85,88,90,91 For example, the complexes 131–132 can interact with a telomeric G4 sequence with good affinity–selectivity via platination of the adenine residues in the loops.42 However, a labile chloride does not ensure platination of G4, for e.g. the complex 146,89 carrying a bis-quinolino modification on the terpyridine, not only exhibits a larger π-delocalized surface but also extensive steric hindrance, resulting in no platination of G4 DNA.
Members of the second group 142–145 with an alkynyl moiety as the monodentate ligand, not only act as good G4 stabilizers with high affinity and selectivity but also exhibit excellent optical properties. Hence the Pt(II) terpyridyl alkynyl complexes can be additionally used to monitor the G-quadruplex.86,92 Furthermore, the terpyridine ligands of all these Pt(II) complexes were modified with additional aromatic rings, or cyclic amine- or peralkylated ammonium-based side chains, resulting in an enhanced binding affinity and superior selectivity for G4 structures over duplex DNA.
4.8. Octahedral metal complexes with planar ligands
With few exceptions, all metal complexes described above possess planar geometry due to the planar macrocyclic or nonmacrocyclic polydentate ligands coordinated to various metal centres. Several octahedral metal complexes such as Ru(II), Ir(III), Fe(III) ion centres with planar ligands have also been investigated as potent G4-ligands (Fig. 13), e.g. ruthenium(II) polypyridyl complexes. Ruthenium complexes were initially developed as alternatives to platinum anticancer drugs because of their prominent DNA binding properties and outstanding anticancer activity.2,93 One of the most recently examined complexes 147 [Ru(bpy)2(dppz)]2+ is known as a molecular “light switch” for DNA that can intercalate between the duplex DNA base pairs by π–π stacking.94,95 This observation prompted scientists to direct efforts towards the construction of Ru(II) complexes as G4-ligands. Recent research shows that the complexes [Ru(bpy)2(dppz)]2+147 and [Ru(phen)2(dppz)]2+148 serve as a prominent molecular “light switch” not only for duplex DNA but also for G4 DNA in Na+ or K+ containing buffer.96,97 However, the affinity and selectivity of these complexes towards G4 DNA was rather weak and their G4 stabilization effect was not prominent (ΔTm < 1 °C with complex/G4 = 0.75:1). An enhancement of the stabilization effect and selectivity for the G-quadruplex could be achieved by suitably modifying the polypyridyl ligand of the Ru complexes. For example, the ruthenium complex 149, [Ru(bpy)2dppz-idzo]2+ with an imidazolone substituent on the dppz ligand, was constructed,98 exhibiting not only a remarkable “light switch” effect for G4 DNA in K+ containing solution (300-fold enhancement in emission) but also a powerful ability to induce and stabilize the formation of the antiparallel G4 structure in buffer solution without alkali–metal ions.98 Its powerful G4 stabilization ability was evident from the significant increase in the melting temperature of G4 DNA being stronger than that of the classic [Ru(bpy)2(dppz)]2+ complex, implying its great potential to act as a telomerase inhibitor and an anti-cancer agent. Two other Ru(II) polypyridyl derivatives 150–151 bearing flexible cyclic amine-based side chains on the large planar aromatic ligand were also constructed by Chao et al.99 Measurements by using CD spectroscopy, FRET and PCR-stop assays proved that these complexes could effectively bind with and stabilize G-quadruplex structures (ΔTm = 5–15 °C), thus exhibiting a concentration-dependent inhibitory effect on telomerase activity (IC50-TRAP = 100–500 nM), ultimately resulting in long-term anti-proliferation of cancer cells. Similarly, [Ru(dppz)2(bpy)]2+ derivatives 153–154 bearing two dppz ligands and one modified bpy moiety with two quaternary ammonium pendants, again exhibited stronger interactions with G4 DNA than the classic [Ru(bpy)2(dppz)]2+ and increased the melting temperature of G4 DNA by 7.0–9.4 °C.100
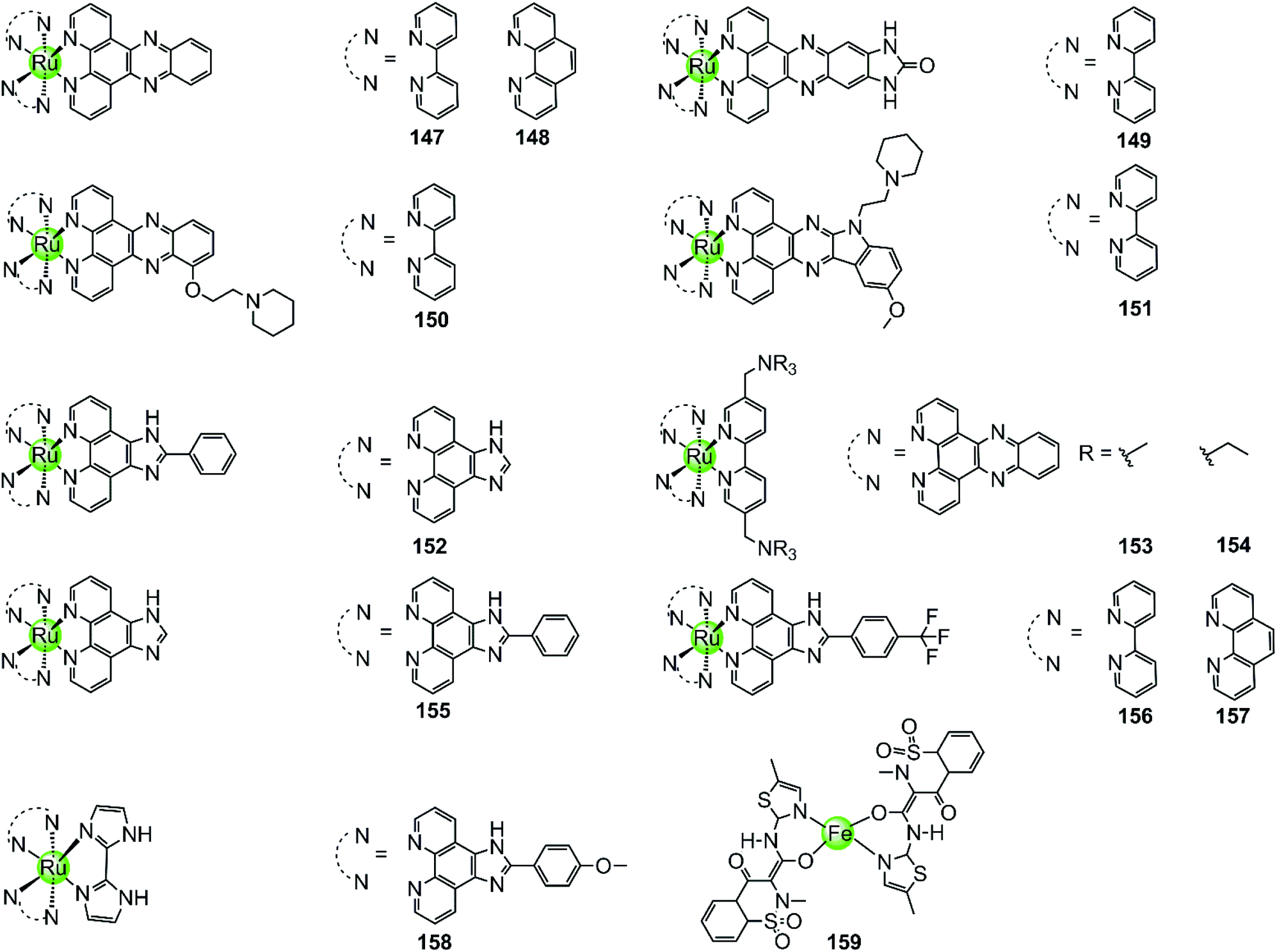 | ||
| Fig. 13 Octahedral ruthenium(II) polypyridyl complexes and a Fe(III) complex containing two meloxicam ligands reported as G4-ligands.94–104,106 | ||
Lately, a series of Ru(II) polypyridyl complexes 155–158 containing the phenyl-imidazo-[4,5f][1,10] phenanthroline ligand were also shown to stabilize the formation of G-quadruplex structures, such as hTel G4 and c-myc G4 (ΔTm = 9–18 °C), via optical spectroscopy, FRET and PCR-stop assays, resulting in effective inhibition of telomerase in the TRAP assay.101–104 Moreover, cellular studies such as cytotoxicity and flow cytometric analysis of mitochondrial membrane potential found that these complexes inhibited the growth of cancer cells through effectively promoting cell apoptosis, resulting in antiproliferative activities at low micromolar ranges comparable with cisplatin.
A series of octahedral cyclometallic Ir(III) complexes have been constructed as molecular “light switch” for G4 DNA.105 Upon binding with a G-quadruplex, these complexes exhibit a luminescence enhancement, the magnitude of which is higher than that upon binding with duplex DNA. The difference in the emission intensity of these Ir(III) complexes suggest a moderate selectivity for the G-quadruplex over duplex DNA, but their G4-mediated anticancer activity was not discussed.
The octahedral Fe(III) complex 159 contains two meloxicam ligands and also induces and stabilizes the hTel G4 structure with good affinity and selectivity as shown by UV/Vis, fluorescence and CD spectroscopy as well as PCR-stop assays.106 Although the binding constant between 159 and hTel G4 (4.53 × 105 M−1) is smaller than that of most Pt(II) complexes, it is an order of magnitude higher than that for the interaction with duplex DNA. According to molecular modelling, the binding mode between the Fe(III) complex and the G4 structure is end-stacking of the meloxicam ligand on the terminal G-quartet.
Because of the octahedral geometry of these metal complexes, the metal centre and the entire molecule is unlikely to end-stack on or intercalate into the G-quartets. It is actually the aromatic ligand with its large π-delocalized planar surface, such as the meloxicam and the dppz ligand, that adopts the end-stacking or intercalation binding mode with G-quartets. At the same time the cationic properties of the entire molecule caused by the metal centre further enhanced the affinity to the negatively charged DNA backbone at the grooves and loops.
4.9. Exceptions: introduction of metal centres reducing the binding capability of organic G4-ligands
In all cases discussed above, regardless of whether the initial ligand was planar or not, the introduction of the metal ion centres resulted in a stronger interaction with G4 DNA by offering a more optimal molecular geometry and cationic properties. The rational modification of the ligand can further improve the affinity and selectivity towards G4 DNA. However, there are also adverse cases in which the introduction of the metal ion breaks the initially planar π-delocalized surface of the ligand, resulting in a dramatic weakening of the binding with G4 DNA.107,108 A representative member is the bisquinolinium ligand 160, which has a large π-delocalized surface and is one of the most selective G4 binders with highest affinity. However, addition of Cu(II) to a solution of a G4-bisquinolinium mixture leads to unfolding of the G4 structure into a single strand meaning that the stabilizing effect of the bisquinolinium ligand is dramatically weakened by the coordination of Cu(II) (Fig. 14).107 Presumably this effect is due to the change of the planar geometry of the free bisquinolinium ligand to a tilted arrangement upon coordination of Cu(II).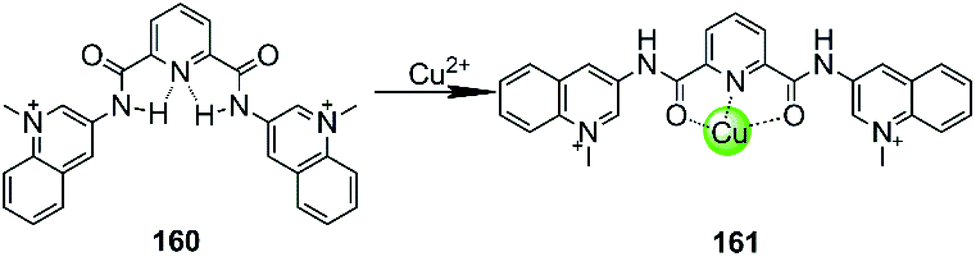 | ||
| Fig. 14 Bisquinolinium is a selective G4-ligand and its coordination to Cu(II) causes a planar-to-linear conformational transition.107,108 | ||
4.10. Multinuclear metal assemblies
Besides the monometallic complexes, a series of rationally designed multinuclear metal assemblies are also described as effective G4-ligands and potent polymerase inhibitors. Their binding modes can also be end-stacking, groove binding, loop binding and others depending on the geometry of the supramolecules.Multinuclear platinum complexes are the most widely investigated supramolecular G4-ligands. In previous studies dimetallic Pt(II) and Pd(II) complexes with bis-carboxamido pyridines were synthesized as potent G4-ligands because of their planar surface offering favourable geometry for stacking with G4 DNA. However, the dimetallic Pt(II) structure 172 only exists in the solid state but dissociates in solution to the monometallic complex. It is actually this monometallic complex that subsequently interacts with the G-quadruplex.109 However, the related dimetallic palladium(II) complex 173 with bis-carboxamido pyridines is not a good G4-ligand, probably because of its poor solubility.109 A number of monometallic variants of this dimetallic Pd(II) complex with various side arms were also described, but only the positively charged complex containing the tertiary amine-modified side arms can moderately stabilize hTel G4 DNA.109
The first stable multimetallic G4-ligand and telomerase inhibitor described is the tetranuclear platinum(II) molecular square 162 reported by Kieltyka et al., which is a representative of the class of multinuclear metal assemblies with flat surfaces.110 This square arrangement has four [Pt(en)]2+ at the corners and four 4,4′-bipyridyl as bridging ligands. This complex has a flat surface for effective end-stacking to the terminal G-quartet and is highly positive charged for strong electrostatic interactions with the DNA backbone. The ethylenediamine ligands of the Pt(II) ions at the corners also allow hydrogen bonding with the DNA backbone, further improving the binding affinity with G4. As a result, this complex strongly stabilizes hTel G4 (ΔTm = 34.5 °C, FRET assay) with high selectivity, the telomerase inhibition activity also being very high (IC50-TRAP = 0.2 μM, in vitro TRAP assay). Based on this representative compound, our group also synthesized a series of four-nuclear Pt(II) assemblies with different bridging ligands trying to understand the structure–activity relationship for G4 interactions.111,112 For example, the platinum supramolecular square 164 with pyridyl as the bridging ligand also stabilizes G4 DNA (hTel and c-kit promoter) by end-stacking with the terminal G-quartets (binding ratio of complex/DNA = 2:1), but the selectivity is not prominent. FRET assays showed a complex-induced increase in melting temperature by ca. 27.4 °C for hTel and 8.5 °C for duplex DNA, respectively.111 This effect was assigned to the relatively smaller size of the flat surface compared to the previously reported 4,4′-bipyridyl bridging analogues 162, the geometry size of the latter was perfectly matching the size of G-quartets (10.8 Å). Further studies of the four-nuclear Pt(II) quasi-cubes 163 exhibit enhanced selectivity towards G4 over duplex DNA.112 The complex-induced increase in melting temperature was ca. 33.5 °C for hTel and less than 1 °C for duplex DNA, with a binding constant for G4 being two orders of magnitude higher than that for duplex DNA. The binding ratio between complex 163 and G4 was unexpectedly high (complex/DNA = 6:1) and molecular docking studies suggested both end-stacking and groove-binding modes. All these Pt(II) squares and Pt(II) quasi-cubes acted as effective telomerase inhibitors, with IC50-TRAP values ranging from 0.12 to 0.35 μM.
The first stable tri-nuclear and di-nuclear Pt(II) complexes 165–166 acting as effective G4-ligands and telomerase inhibitors were synthesized by our group, the Pt(II) corners of which are bridged by a (quaternized-)trigeminal chelating ligand.34,113,114 Both FRET and ITC assays indicated high affinity and excellent selectivity of these complexes for hTel G4 (ΔTm > 30 °C) over promoter c-myc and bcl-2 (ΔTm < 6.0 °C) as well as duplex DNA (ΔTm < 1.5 °C). The PCR-stop and in vitro TRAP assays indicate that these complexes effectively induced hTel G4 formation thereby effectively inhibiting telomerase activity with IC50-TRAP values in the micromolar range. Interestingly, the V-shaped bi-nuclear Pt(II) complexes 166 favour to induce the hybrid parallel/antiparallel hTel G4 topology while the propeller-shaped trinuclear platinum(II) complexes allow the recognition of different G4 topologies with different Pt(II) corners, possibly due to their flexibility.114 For example, the complex with diethylenetriamine Pt(II) corners stabilizes antiparallel G4, while the complex with bis-(2-pyridylmethyl)amine Pt(II) corners stabilizes parallel G4. Based on the V-shaped bi-nuclear Pt(II) complex, we reported the first nitroxide tagged G4-ligand 167 as a new strategy for investigating detailed interactions between small molecules and the G-quadruplex.113 Using the nitroxide moiety as a spin label, the inter-spin distance between the two G4-bound 167 could be measured by using electron paramagnetic resonance (EPR) spectroscopy. Combining the EPR-measured data with molecular docking revealed that this complex predominately binds to the neighbouring-grooves as hTel adopts the antiparallel conformation. Very recently, four dinuclear Pt(II)–terpyridine complexes 168–171 were also reported to act as efficient G4-ligands, with high selectivity (ΔTm up to 17 °C) over duplex DNA (ΔTm = 1 °C).115
Multimetallic supramolecules based on non-platinum metal centres, such as Ru(II), Ni(II), Cu(II), Zn(II), Fe(II), Tb(III), or Ce(IV), were also investigated as G4-ligands (Fig. 16–19). Among them, the multimetallic Ru(II) complexes possess additional excellent fluorescence emissive properties and their interaction with G4 DNA is in some cases accompanied by great changes in their optical properties. For example, the binuclear Ru(II) complexes 175–176 (Fig. 16) are virtually non-luminescent in aqueous solution, however upon binding with the G4 DNA (d[AG3(T2AG3)3]) in the presence of K+ ions the emission is significantly enhanced (150-fold) accompanied by a blue shift of 30 nm.97,116 It is worth mentioning that the emission enhancement of the Ru(II) complexes is only observed upon binding to G4 structures containing lateral loops that are at least 3 base pairs long. Thus it is proposed that the possible G4 binding mode is “end-pasting” or “threading” through the G4 lateral loops, in addition to partial intercalation. More interestingly, these dinuclear Ru(II) complexes can be obtained in enantiomerically pure forms and thus offer novel enantio-selectivity for interacting with hTel G4 DNA of antiparallel basket-like topology.117
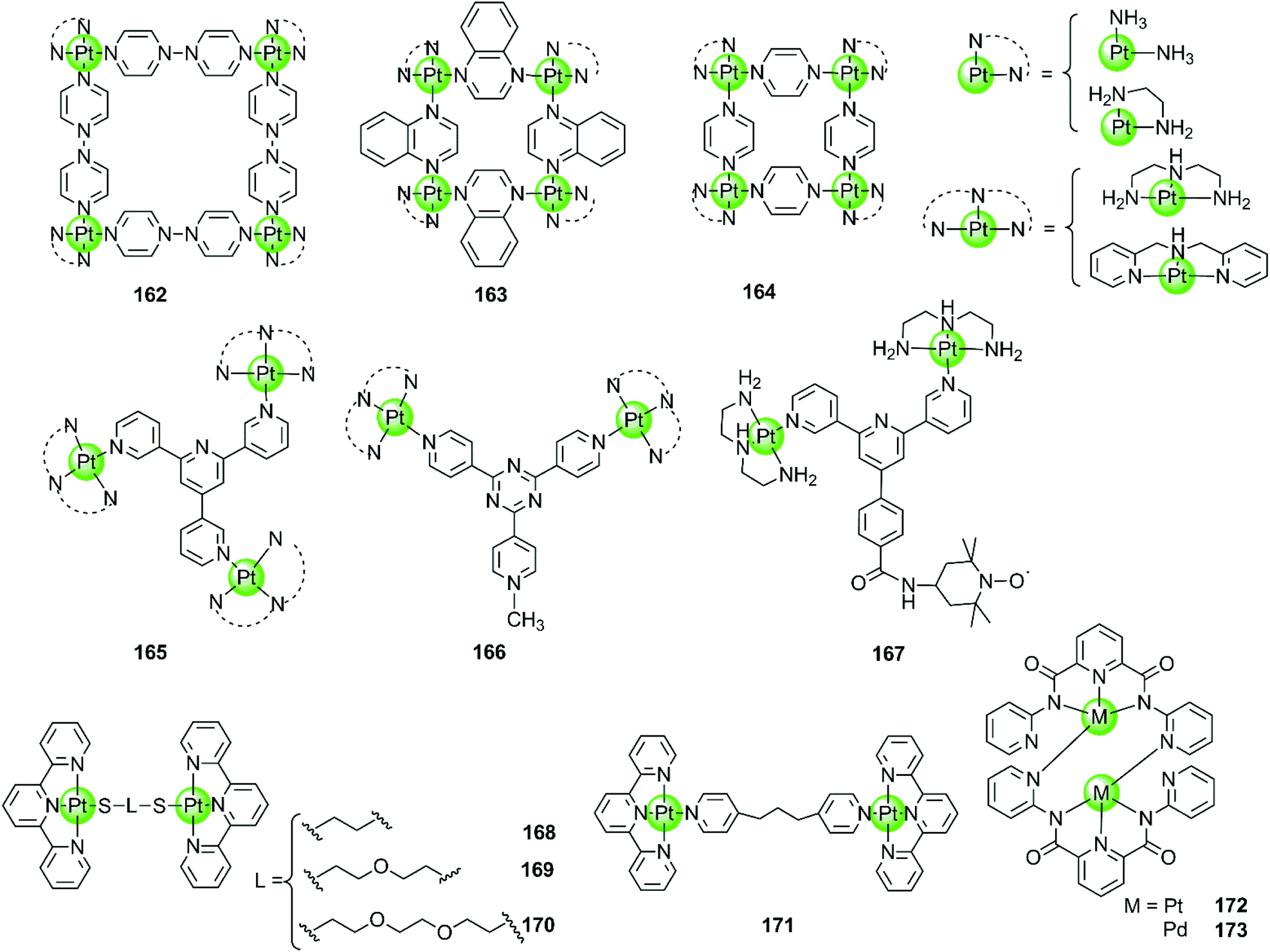 | ||
| Fig. 15 Multimetallic platinum(II) complexes reported as selective G4-ligands. Complex 167 was utilized for EPR measurement thus precisely determining the binding mode and binding sites of the Pt(II) complex.109–115 | ||
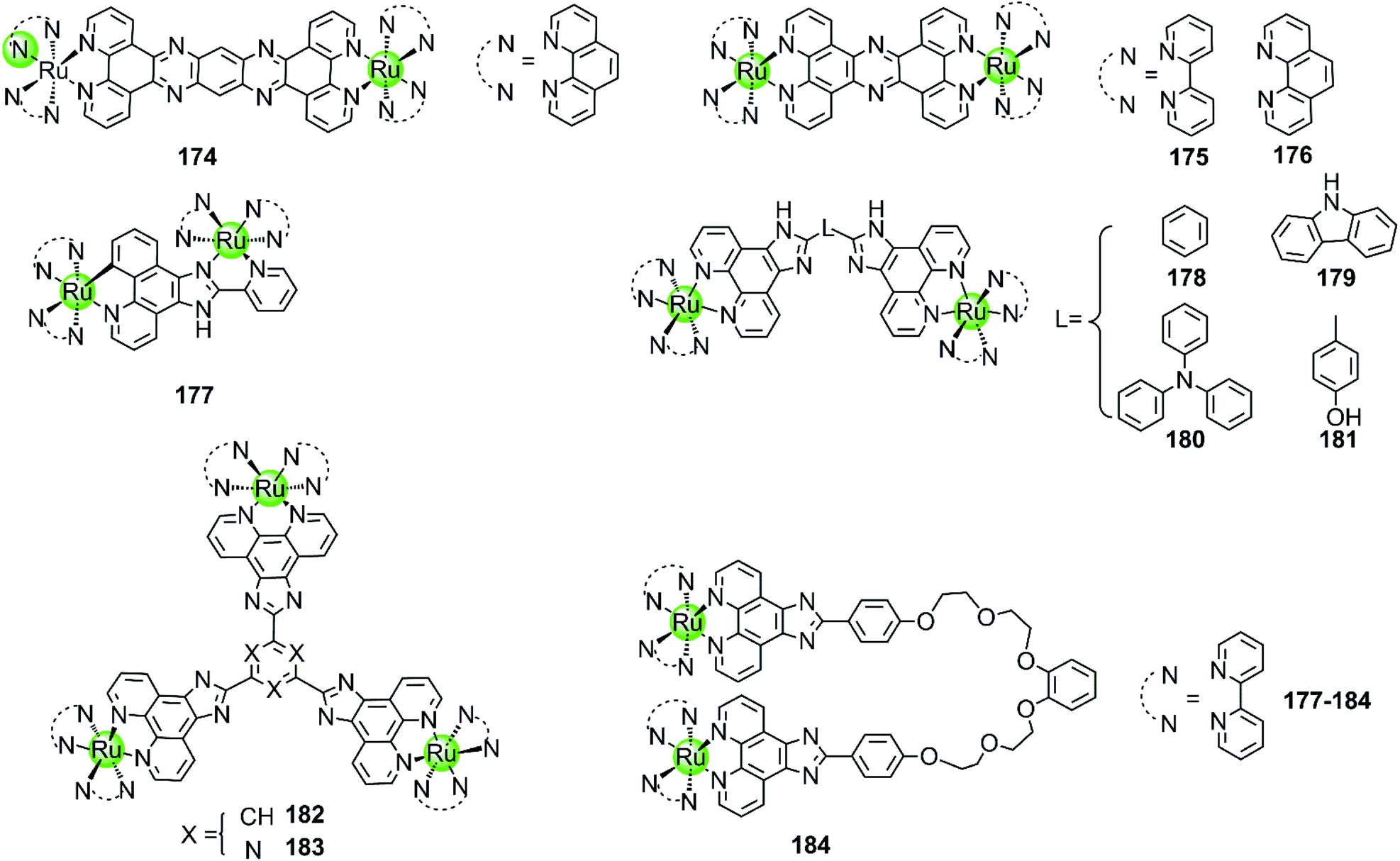 | ||
| Fig. 16 Multimetallic ruthenium(II) polypyridyl complexes reported as G4-ligands. The bridging ligands are either rigid or flexible.116–121 | ||
 | ||
| Fig. 17 Other examples of bimetallic complexes with homogeneous or heterogeneous metal centres reported as G4-ligands;122,123 Monometallic Zn(II) macrocyclic complexes utilize the thymine residues as the primary mode of recognition.124–126 | ||
 | ||
| Fig. 18 The cylinder-like bimetallic Ni(II) complex exhibits excellent chirality-based selectivity,129 and terbium(III)–amino acid complexes capable of binding with i-motif DNA.130 | ||
 | ||
| Fig. 19 Metal complexes acting as G4 DNA-cleavage agents.131–136 | ||
Although the complexes discussed above moderately stabilize G4 DNA (ΔTm = 3.8–5.4 °C in thermal melting experiments), their selectivity towards G4 over duplex DNA is not optimal: they can also bind to duplex DNA with high affinity, resulting in a moderate emission enhancement (50-fold) as well. Changing the bridging ligand tppz to obip, a second-generation luminescent bimetallic complex 177 is obtained, displaying relatively better selectivity towards G4 DNA. The emission enhancement of 178 with G4 is one order of magnitude higher than that for duplex DNA, and the complex/G4 interaction increases the melting temperature by 5.8–9.4 °C in the presence of alkali metal cations.118 Chao et al. constructed a series of comparative dinuclear Ru(II) complexes 178–181 with larger sized bridging ligands, exhibiting high selectivity for binding with G4 over duplex DNA. These complexes can induce and stabilize antiparallel hTel G4 (ΔTm = 12–24 °C) in the presence or absence of K+ with 1:1 stoichiometry, leading to promising inhibition effects on telomerase activity and cancer cell proliferation.119 In addition, two trigeminal ligand bridged trinuclear ruthenium(II) complexes 182–183 were also reported to moderately stabilize G4 DNA (ΔTm = 14–19 °C with complex/DNA = 5:1) but the information about the selectivity between G4 and duplex DNA is not available.120 These investigations indicate that bridging ligands with appropriately sized planar surfaces are essential for the strong and selective interaction of the multimetallic Ru(II) complexes with the G-quadruplex.
All the multimetallic Ru(II) complexes described above are bridged by a rigid, planar bridging ligand with extensive aromaticity. In addition, a novel bimetallic Ru(II) complex 184 is reported in which two octahedral fluorescent Ru(II) monomers are connected with a partially flexible chain (noncyclic crown ethers).121 Compared to other bimetallic Ru(II) complexes, this complex shows a highly distinct fluorescence upon binding to G4 compared to duplex DNA, the intensity difference of which can even be distinguished by the naked eye. This indicates a relatively high selectivity toward G4, the binding constant of which is about one order of magnitude higher than that for duplex DNA. This complex also moderately stabilizes the G4 structure (ΔTm = 12.7 °C) and was also considered as a potent telomerase inhibitor.
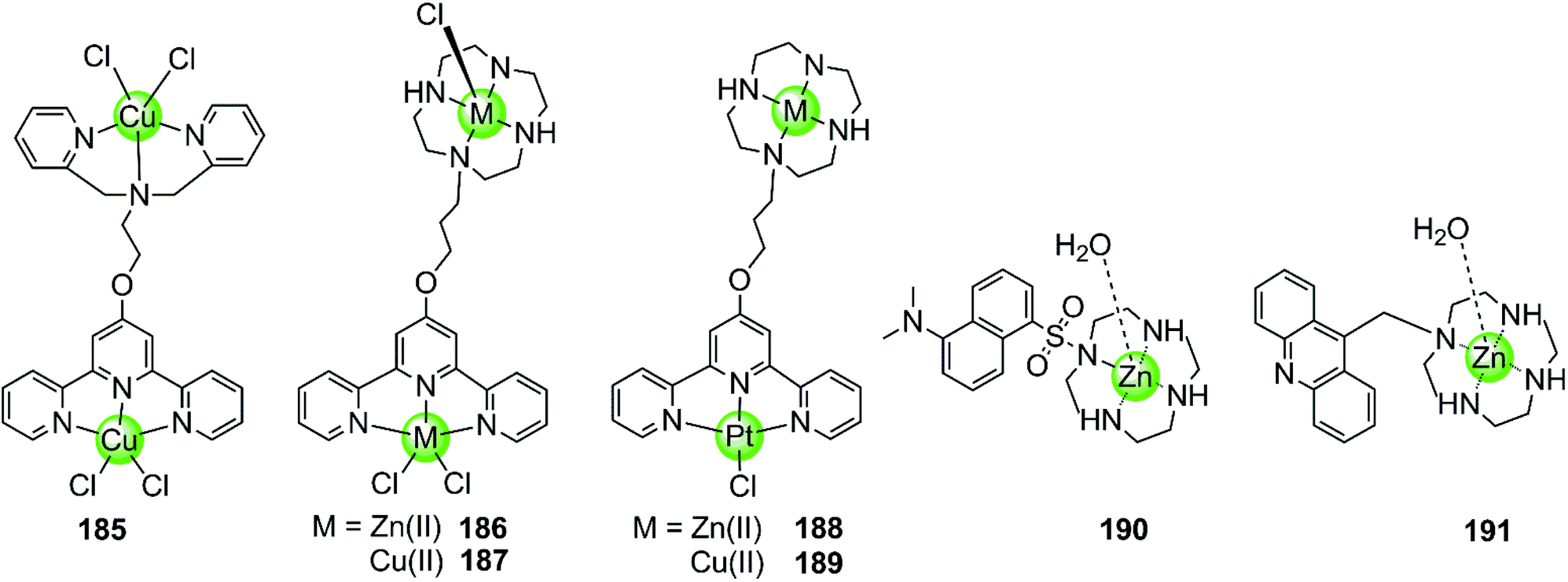
Besides multimetallic Pt(II) and Ru(II) complexes, several bimetallic terpyridyl-containing complexes with either homogeneous or heterogeneous metal centres were also reported as effective G4 binders (Fig. 17). For example, a dinuclear Cu(II)–terpyridine complex 185 effectively binds G4 DNA with very high affinity and stabilizes the antiparallel topology, and its selectivity for G4 is 100-fold higher than that for duplex DNA.122 Bimetallic complexes 186–189 with either homogeneous or heterogeneous metal centres, e.g. Zn(II)/Zn(II),Cu(II)/Cu(II), Pt(II)/Zn(II), Pt(II)/Cu(II), were also constructed based on a novel terpyridine–cyclen ligand, both the terpyridne and cyclen moieties of which possessed efficient metal chelating properties. These dinuclear complexes all exhibited higher binding affinities towards G4 compared to their monometallic counterparts.123 In addition, Morrow et al. reported two Zn(II) macrocyclic complexes 190–191, the structures of which were similar to the monometallic counterparts of complex 186 with the nonplanar dansyl group and acridine group pendents, respectively.124–126 Complex 190 shows 110-fold selectivity in binding to hTel G4 over duplex DNA, evidenced by an increase in the fluorescence and a simultaneous shift in emission upon G4 interaction. ITC, fluorescence and NMR spectroscopy give a complex/G4 stoichiometry of 2:1 and indicate one complex binding to two spaced thymines within two separate loops in the hTel G4. Notably, this is the first reported metal complex-based hTel G4-ligand utilizing the thymine residues as the primary mode of recognition. Although loop bindings have also been found in several previously reported G4-ligands, they were mainly the secondary mode of interaction and driven through aromatic stacking.127,128 As a comparison, complex 191 containing an acridine pendent shows stronger binding to the G-quadruplex but indiscriminately binds to duplex DNA as well.
A novel cylinder-like bimetallic Ni(II) complex 192 (Fig. 18) with triple diimine ditopic ligands was constructed by Qu and co-workers, exhibiting a novel chirality-based selectivity for hTel G4 structure over the duplex as well as other different types of G4 DNA.129 Only the P-enantiomer of this supramolecular cylinder selectively stabilizes hTel G4 DNA and simultaneously induces an antiparallel-to-hybrid conformational transition in the presence of Na+. The stoichiometry ratio complex/G4 was determined to be 1:1 and an S1 nuclease cleavage assay was performed to determine whether the complex binds with an end-stacking binding mode with the cylinder stacking on top of the terminal G-quartet via its extensive hydrophobic exterior. In addition, the positively charged metal ion centres allow strong electrostatic interactions with loops and grooves, so that the termini of the hTel G4 are protected from the S1 nuclease cleavage. This example paved the way towards the development of a new class of G4-targeted chiral binders and anticancer drug candidates. In addition, two bimetallic terbium(III)–amino acid complexes 193–194 are capable of binding hTel G4 DNA as well as i-motif DNA, although the binding constant and stabilization effect were as good as found for previously described metal complexes.130
4.11. Metal assemblies as G4 DNA-cleavage agents
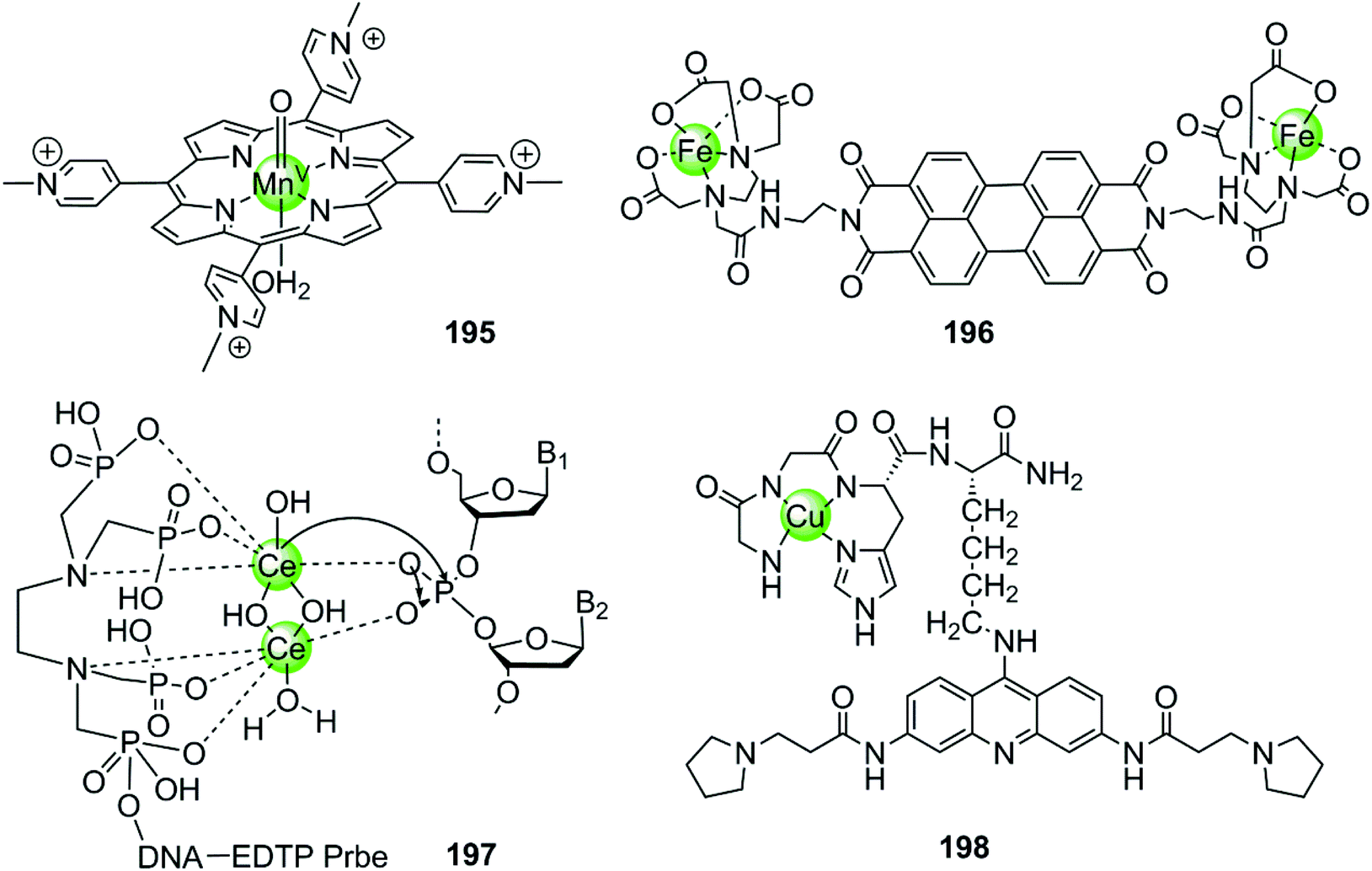
In the examples presented above, the metal complexes effectively stabilize certain G-quadruplex structures thus inhibiting telomerase activity or oncogene expression, ultimately interfering with telomere maintenance and preventing the proliferation of cancer cells effectively. However, this strategy usually requires long-term treatment to significantly shorten the telomere length of cancer cells, because each round of cell division only reduces 50–200 nt of the telomere length. Thus several metal-containing G4-ligands have recently been reported to induce direct cleavage of G4 DNA as another anticancer strategy. The first reported metal complex acting as a G4 DNA cleavage agent was a highly reactive high-valent oxo-manganese porphyrin species 195, which was formed in situ from the Mn(III)–TMPyP4 complex 7 in the presence of the oxygen atom donor, KHSO5 (Fig. 19).131 During the redox reaction only one face of the porphyrin is solvent accessible which is consistent with the partial stacking of the metalloporphyrin on the external side of the terminal G-quartet. The in situ formed Mn(V)O species from the Mn(III)–TMPyP4/KHSO5 system can act as nuclease mediating oxidative cleavage of the bound hTel G4 sequence. Such oxidative damage consisted of both guanine oxidation within the interacting G-quartet and the 1′-carbon hydroxylation of deoxyriboses carrying thymidine residues located on the neighboring single-stranded loop. However, this highly reactive Mn(V)O species cleaves both hTel G4 and duplex DNA indiscriminately, indicating that improved targeting and selective cleavage towards hTel G4 structures are still required.131
Afterwards a water-soluble dimetallic Fe(II)–EDTA complex 196 bridged by the planar polyaromatic ligand perylene was constructed as a selective G4 DNA cleavage agent (Fig. 19).132,133 The bridging perylene ligand is known as an effective G4 binder, allowing π-stacking of the dimetallic complex on the terminal G-quartets, and two Fe(II)-EDTA cores bind with the opposite grooves. Upon interaction, this complex selectively cleaves the G-quadruplex over duplex DNA in the presence of the reducing agent, dithiothreitol, probably through a hydroxyl radical mechanism.132,133
The dimetallic Ce(IV) complex 197 with ethylenediamine–tetramethylene phosphonic acid (EDTP) has recently been reported as a selective DNA-cleavage agent for intermolecular G-quadruplex DNA (Fig. 19).134 This complex can induce the assembly of a highly stable (3 + 1) intermolecular G4 structure by covalently binding to the G-rich sequence, and such a Ce(IV)-EDTP–DNA conjugate can further induce sequence-specific hydrolytic cleavage at a specific phosphodiester site of the hTel G4 DNA backbone. However, this complex possesses some inherent disadvantages e.g. low cellular uptake, instability to natural nucleases, self-cleavage, which limit its further applications and investigations in vivo.
Very recently, a novel DNA-cleaving agent 198 has been reported to be capable of selectively cleaving intramolecular hTel G4 DNA.135,136 This complex is constructed by coupling an amino-terminal copper binding motif peptide GGHK to an acridine-based G4 ligand (Fig. 19). The acridine moiety promotes the selective binding of the complex towards hTel G4 over duplex DNA, and helps in positioning the catalytic metallodrug moiety CuGGH in close proximity to G4 DNA. Based on this G4 targeting property, complex 198 could selectively and efficiently induce irreversible cleavage on the G4 structure rather than other structural states of telomeric DNA in the presence of redox co-reagents. The 3′overhang cleavage product was generated from both hydrolysis and oxidative cleavage mechanisms. Data from molecular modelling and MALDI-MS suggested the major selective cleavage sites at A1-G2 and T6-A7 nucleotides. As a result, complex 198 caused the significant shortening of telomeric DNA, cellular senescence and apoptosis in MCF7 cell lines.
5. Conclusions
G-quadruplex nucleic acids are formed by self-assembly of guanine-rich sequences and can be found in telomeres, oncogene promoters, and non-translated RNA regions. They possess unique structures distinct from the well-known double helical structures adopted by most genomic DNA. Such distinct structures offer a great opportunity for selective molecular recognition toward the G-quadruplex over genomic duplex DNA. Moreover, formation of G4 nucleic acids interferes with numerous biological pathways, including maintenance of telomeres, regulation of oncogene transcription and translation and so on. Once intramolecular G4 is formed within the human telomeric DNA sequence, the activity of telomerase is indirectly inhibited due to the loss of the substrate, resulting in telomere shortening in cancer cells. Because telomerase is silent in normal cells but up-regulated in most cancer cells contributing to their immortality, molecules capable of inhibiting or down-regulating telomerase activity thereby has the potential for selective toxicity toward cancer cells over normal cells. Intramolecular G4 formed within the oncogene promoter regions could lead to the inhibition of oncogene expression, thus exhibiting selective toxicity toward cancer cells as well. Thus G4 nucleic acids have attracted a lot of attention as potential clinical targets for the development of new types of anticancer agents.Small molecules that can recognize and interact with the G-quadruplex with high affinity and specificity, termed G4-ligands, have been systematically investigated over the past few decades. Their capabilities to act as G-quadruplex stabilizers, telomerase activity inhibitors, oncogene transcription and translation modulators, as well as DNA-cleavage agents have been assessed, thus opening new avenues for cancer chemotherapy. This review summarizes the various families of metal complexes reported in the literature that have G4 DNA targeting properties so as to exhibit potent anticancer effects through selectively stabilizing or cleaving the G-quadruplex over duplex DNA. The majority of understanding has focused on the G4 DNA at telomeres and promoter regions although G-quadruplex motifs are definitely present in other regions of the genome. These metal complexes possess peculiar tridimensional geometries, such as square-planar in most cases, square-based pyramidal, cube-like shape or even an octahedral structure, and the metal ions must arrange the scaffold into the optimal geometry for G4 binding. In fact, different metal centres coordinating with the same organic ligand can generate metal complexes with overall distinct G4 binding properties.
The interactions between the metal complexes and G-quadruplex have been characterized by numerous biophysical methods revealing the strength and specificity of these G4-ligand interactions. A summary of current knowledge of metal-containing G4-ligands concludes three main binding sites: the G-quartets, the grooves, and the loops of the G-quadruplex. The large majority of known G4-ligands exhibit a preference for the first binding site, usually end-stacking of the metal complex scaffold onto one of the terminal G-quartets. To optimize this end-stacking binding mode, metal complexes reported in the literature generally present a suitably sized planar surface, which is larger than that involved in DNA intercalators so as to eliminate the possibility of intercalation binding mode. In some cases the scaffold of metal complexes is flexible but attains planarity upon end-stacking onto the external G-quartet. On the other hand, the presence of bulky flexible side chains at the periphery of the central planar core increases the steric hindrance of the metal complexes, which could prevent the intercalation mode of G4-ligands between DNA base pairs. Moreover, positively charged or protonable side chains can not only enhance electrostatic interaction strength with the negatively charged DNA phosphate backbone but also improve the fitting of the molecule into the grooves, loop residues or cavities, resulting in an increase in the affinity of the G4-ligands. All these structural design considerations are beneficial for increasing the selectivity of metal complexes for G4 over duplex DNA.
Although the majority of G4-ligands reported so far recognize G-quartets and present end-stacking binding mode, this interaction can only allow a selective discrimination between G4 and duplex DNA, it can hardly distinguish one G4 structure over another. This is not therapeutically effective enough. As a consequence more promising binding modes, such as groove binding and loop binding, are favourable for the enhancement of selectivity. Because different G-quadruplex topologies endow grooves and loops with various dimensions and accessibilities, G4-ligands presenting specific groove or loop binding modes have great potential to discriminate among different G4 DNA derived from various G-rich sequences. It is a big challenge to combine targeting of the grooves/loops with targeting of the G-quartets and to date this has only provided limited success. In these examples multiple binding modes can occur simultaneously, but groove/loop binding are mainly the secondary mode of interaction and usually driven through the primary mode of interaction end-stacking. Only a few bimetallic Pt(II), bimetallic Ru(II) and macrocyclic Zn(II) complexes utilize the grooves and loops as the primary mode of recognition. This provides great inspiration for the design of the next generation of G4-ligands that depends less on general G-quartet but more on specific groove/loop recognition.
Besides π stacking and groove/loop binding, metal complexes containing labile groups (Cl, H2O, etc.) can also make direct coordination of the metal centre to G4 DNA nucleobases, similar to the platination of duplex DNA by cisplatin. This type of coordination either occurs at a single nucleotide, in most cases guanines, or causes cross-linking of two nucleobases, probably enhancing the affinity and specificity of the metal complexes to G4 DNA, similar to the compounds 131–132. Regardless of the kind of binding mode, these metal-containing G4-ligands effectively stabilize certain G-quadruplex structures, thus inhibiting telomerase activity or oncogene expression, ultimately interfering with telomere maintenance and inhibiting the proliferation of cancer cells. On the other hand, several rationally designed metal complexes have very recently been reported possessing the property to selectively cleave hTel G4 over duplex DNA through either oxidative damage or hydrolysis mechanism. These cleavage processes require the presence of redox co-reagents – hydrolysis cleavage and oxidative damage. This is considered as another anticancer strategy, which can significantly shorten the telomere length of cancer cells in a relatively shorter term rather than other G4 stabilizers.
From all these studies, the rational design of metal complexes to selectively interact with, stabilize or cleave G-quadruplex structures has evolved as a promising strategy for the development of anticancer drugs with selective toxicity towards cancer cells over normal ones. However, the G4-ligand development has been dominated by in vitro biophysical and biochemical investigations, the understanding of G4-ligand activity and selectivity in the in vivo environment still remains a major challenge. After so many years only one promising in vivo G4-ligand, Quarfloxin, has reached phase II clinical trials for treating neuroendocrine tumors and carcinoid tumors.30 Upon its binding with the G-quadruplex structure in the ribosomal DNA (rDNA) template, the interaction between nucleolin protein and rDNA is disrupted, resulting in the inhibition of rRNA biogenesis and ribosome synthesis in cancer cells. Similar ribosomal DNA G4-targeted metal complexes have not been reported. This may shed light on the development of rDNA G4-targeted metal complexes for clinical use.
The structures of G4 sequences in vivo are highly dynamic, which may be in its linear form or interacting with proteins. In this sense there are two critical questions that need to be addressed for the development of G4-targeted drugs: what are the in vivo targets of these G4-ligands and how to control their accessibility? Thus it is important for the G4-ligands not only to recognize the G4 arrangement already organized but also to reach the target and induce the formation of G4 conformation. Several metal complexes have been reported capable of inducing the formation of certain G4 conformations and those possessing distinct photophysical properties can be used as “light switches” for G-quadruplexes to monitor the occurrence of G4 structures even in living cells. Thus it is probably possible to design G4-ligands capable of simultaneously inducing and real-time tracing the occurrence of G4 structures during cancer therapy. The chirality-based selectivity of metal complexes is also very attractive for designing G4-ligands: different enantiomers can recognize different G4 topologies and even show differential uptake mechanisms and cellular localization. The utilization of these metal complexes as in vivo probes for G4 DNA or as unique photosensitizers is also likely to be an active area.
Additionally, an important first step in designing selective G4 DNA-cleavage agents has been taken by application of a catalytic metallodrugs’ strategy, in which metal complexes act as an artificial nuclease mediating the hydrolysis or oxidative cleavage of telomeric DNA. The combination of metallodrugs and G4 DNA binding may also catalyze oxidation directing toward small substrates present in the bulk, thus becoming a DNAzyme. For example, a heme cofactor of natural enzymes known as iron(III) protoporphyrin IX (or Hemin) is endowed with G-quadruplex binding capability with high affinity, and the hemin-binding G4 DNA aptamer has been found to exhibit peroxidase-like activity which can be used as a sensitive probe for the identification of single nucleotide polymorphisms by giving a color signal.137,138 This may open new avenues for the application of metal-containing G4-ligands as attractive catalytic labels in biosensing.
In summary, development of metal-containing G4-ligands as novel anti-cancer drugs is a rapidly growing field. Despite making some progress, to date the majority of metal-containing G4-ligands do not have realistic drug-like structures and their in vivo applications are still very rare. Thus it is expected that further advancement in this field will focus on progress in medicinal studies and in the next few years we would like to see new generations of metal-containing G4-ligands enter clinical trials.
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China [21231007, 21401217, 21328101, 21572282], 973 Program [2014CB845604], Guangdong Provincial Government [2013B051000047, 20130501c, 207999], the Science and Technology Program of Guangzhou [201504281600481] and the Fundamental Research Funds for the Central Universities. We also thank the Swiss National Science Foundation [to EF and RKOS] and the University of Zurich [EF and RKOS]. We thank Dr Cui-Xia Xu for her help in constructing endnote library and drawing some figures.References
- B. Lippert, in Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug, John Wiley & Sons, Inc., 1999 Search PubMed.
- I. Kostova, Curr. Med. Chem., 2006, 13, 1085–1107 CrossRef CAS PubMed.
- Y.-P. Ho, S. C. F. Au-Yeung and K. K. W. To, Med. Res. Rev., 2003, 23, 633–655 Search PubMed.
- P. C. A. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 Search PubMed.
- M. Gellert, M. N. Lipsett and D. R. Davies, Proc. Natl. Acad. Sci. U. S. A., 1962, 48, 2013–2018 Search PubMed.
- D. J. Patel, A. T. Phan and V. Kuryavyi, Nucleic Acids Res., 2007, 35, 7429–7455 Search PubMed.
- S. Burge, G. N. Parkinson, P. Hazel, A. K. Todd and S. Neidle, Nucleic Acids Res., 2006, 34, 5402–5415 Search PubMed.
- N. H. Campbell and S. Neidle, Met. Ions Life Sci., 2012, 10, 119–134 Search PubMed; H. G. Miserachs, D. Donghi, R. Börner, S. Johannsen and R. K. O. Sigel, Distinct differences in metal ion specificity of RNA and DNA G-quadruplexes, 2016, in revision Search PubMed; E. Largy, J.-L. Mergny and V. Gabelica, Met. Ions Life Sci., 2017, 17, 203–258 Search PubMed.
- G. Biffi, D. Tannahill, J. McCafferty and S. Balasubramanian, Nat. Chem., 2013, 5, 182–186 CrossRef CAS PubMed.
- Y. Qin and L. H. Hurley, Biochimie, 2008, 90, 1149–1171 Search PubMed.
- H. Fernando, A. P. Reszka, J. Huppert, S. Ladame, S. Rankin, A. R. Venkitaraman, S. Neidle and S. Balasubramanian, Biochemistry, 2006, 45, 7854–7860 Search PubMed.
- S. Rankin, A. P. Reszka, J. Huppert, M. Zloh, G. N. Parkinson, A. K. Todd, S. Ladame, S. Balasubramanian and S. Neidle, J. Am. Chem. Soc., 2005, 127, 10584–10589 Search PubMed.
- A. Siddiqui-Jain, C. L. Grand, D. J. Bearss and L. H. Hurley, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11593–11598 CrossRef CAS PubMed.
- A. Rangan, O. Y. Fedoroff and L. H. Hurley, J. Biol. Chem., 2001, 276, 4640–4646 Search PubMed.
- A. K. Todd, S. M. Haider, G. N. Parkinson and S. Neidle, Nucleic Acids Res., 2007, 35, 5799–5808 Search PubMed.
- E. H. Blackburn, C. W. Greider, E. Henderson, M. S. Lee, J. Shampay and D. Shippen-Lentz, Genome, 1989, 31, 553–560 Search PubMed.
- C. W. Greider and E. H. Blackburn, Cell, 1987, 51, 887–898 Search PubMed.
- J. Shampay, J. W. Szostak and E. H. Blackburn, Nature, 1984, 310, 154–157 Search PubMed.
- M. A. Blasco, Eur. J. Cell Biol., 2003, 82, 441–446 Search PubMed.
- D. Sun, B. Thompson, B. E. Cathers, M. Salazar, S. M. Kerwin, J. O. Trent, T. C. Jenkins, S. Neidle and L. H. Hurley, J. Med. Chem., 1997, 40, 2113–2116 Search PubMed.
- S. Balasubramanian and S. Neidle, Curr. Opin. Chem. Biol., 2009, 13, 345–353 Search PubMed.
- Y. Xu, T. Ishizuka, T. Kimura and M. Komiyama, J. Am. Chem. Soc., 2010, 132, 7231–7233 Search PubMed.
- Y. Xu, K. Kaminaga and M. Komiyama, J. Am. Chem. Soc., 2008, 130, 11179–11184 Search PubMed.
- S. Kumari, A. Bugaut, J. L. Huppert and S. Balasubramanian, Nat. Chem. Biol., 2007, 3, 218–221 Search PubMed.
- K. Halder and J. S. Hartig, Met. Ions Life Sci., 2011, 9, 125–139 Search PubMed.
- D. Gomez, A. Guédin, J.-L. Mergny, B. Salles, J.-F. Riou, M.-P. Teulade-Fichou and P. Calsou, Nucleic Acids Res., 2010, 38, 7187–7198 Search PubMed.
- A. Bugaut, R. Rodriguez, S. Kumari, S.-T. D. Hsu and S. Balasubramanian, Org. Biomol. Chem., 2010, 8, 2771–2776 Search PubMed.
- S. N. Georgiades, N. H. Abd Karim, K. Suntharalingam and R. Vilar, Angew. Chem., Int. Ed., 2010, 49, 4020–4034 Search PubMed.
- N. Campbell, G. W. Collie and S. Neidle, in Current Protocols in Nucleic Acid Chemistry, John Wiley & Sons, Inc., 2012 Search PubMed.
- K. Papadopoulos, A. Mita, A. Ricart, D. Hufnagel, D. Northfelt, D. Von Hoff, L. Darjania, J. Lim, C. Padgett and R. Marschke, Am. Assoc. Cancer Res., 2007, 6, B93–B93 Search PubMed.
- J. Zhang, F. Zhang, H. Li, C. Liu, J. Xia, L. Ma, W. Chu, Z. Zhang, C. Chen, S. Li and S. Wang, Curr. Med. Chem., 2012, 19, 2957–2975 Search PubMed.
- L. Rao, J. D. Dworkin, W. E. Nell and U. Bierbach, J. Phys. Chem. B, 2011, 115, 13701–13712 Search PubMed.
- P. Murat, Y. Singh and E. Defrancq, Chem. Soc. Rev., 2011, 40, 5293–5307 Search PubMed.
- C.-X. Xu, Y. Shen, Q. Hu, Y.-X. Zheng, Q. Cao, P. Z. Qin, Y. Zhao, L.-N. Ji and Z.-W. Mao, Chem. – Asian J., 2014, 9, 2519–2526 Search PubMed.
- N. H. Campbell and G. N. Parkinson, Methods, 2007, 43, 252–263 Search PubMed.
- S. Neidle and G. N. Parkinson, Biochimie, 2008, 90, 1184–1196 Search PubMed.
- G. N. Parkinson, F. Cuenca and S. Neidle, J. Mol. Biol., 2008, 381, 1145–1156 Search PubMed.
- N. H. Campbell, N. H. A. Karim, G. N. Parkinson, M. Gunaratnam, V. Petrucci, A. K. Todd, R. Vilar and S. Neidle, J. Med. Chem., 2012, 55, 209–222 Search PubMed.
- S. Redon, S. Bombard, M. A. Elizondo-Riojas and J. C. Chottard, Nucleic Acids Res., 2003, 31, 1605–1613 Search PubMed.
- I. Ourliac-Garnier, M.-A. Elizondo-Riojas, S. Redon, N. P. Farrell and S. Bombard, Biochemistry, 2005, 44, 10620–10634 Search PubMed.
- L. Rao and U. Bierbach, J. Am. Chem. Soc., 2007, 129, 15764–15765 Search PubMed.
- H. Bertrand, S. Bombard, D. Monchaud and M.-P. Teulade-Fichou, Nucleic Acids Symp. Ser., 2008, 52, 163–164 Search PubMed; H. Bertrand, S. Bombard, D. Monchaud and M.-P. Teulade-Fichou, J. Biol. Inorg. Chem., 2007, 12, 1003–1014 Search PubMed.
- Z.-F. Chen, Q.-P. Qin, J.-L. Qin, Y.-C. Liu, K.-B. Huang, Y.-L. Li, T. Meng, G.-H. Zhang, Y. Peng, X.-J. Luo and H. Liang, J. Med. Chem., 2015, 58, 2159–2179 Search PubMed.
- G. N. Parkinson, R. Ghosh and S. Neidle, Biochemistry, 2007, 46, 2390–2397 Search PubMed.
- D.-F. Shi, R. T. Wheelhouse, D. Sun and L. H. Hurley, J. Med. Chem., 2001, 44, 4509–4523 Search PubMed.
- E. Izbicka, R. T. Wheelhouse, E. Raymond, K. K. Davidson, R. A. Lawrence, D. Sun, B. E. Windle, L. H. Hurley and D. D. Von Hoff, Cancer Res., 1999, 59, 639–644 Search PubMed.
- R. T. Wheelhouse, D. Sun, H. Han, F. X. Han and L. H. Hurley, J. Am. Chem. Soc., 1998, 120, 3261–3262 Search PubMed.
- I. M. Dixon, F. Lopez, J.-P. Estève, A. M. Tejera, M. A. Blasco, G. Pratviel and B. Meunier, ChemBioChem, 2005, 6, 123–132 Search PubMed.
- A. Maraval, S. Franco, C. Vialas, G. Pratviel, M. A. Blasco and B. Meunier, Org. Biomol. Chem., 2003, 1, 921–927 CAS.
- A. J. Bhattacharjee, K. Ahluwalia, S. Taylor, O. Jin, J. M. Nicoludis, R. Buscaglia, J. Brad Chaires, D. J. P. Kornfilt, D. G. S. Marquardt and L. A. Yatsunyk, Biochimie, 2011, 93, 1297–1309 Search PubMed.
- S. E. Evans, M. A. Mendez, K. B. Turner, L. R. Keating, R. T. Grimes, S. Melchoir and V. A. Szalai, J. Biol. Inorg. Chem., 2007, 12, 1235–1249 Search PubMed.
- L. R. Keating and V. A. Szalai, Biochemistry, 2004, 43, 15891–15900 CrossRef CAS PubMed.
- R. W.-Y. Sun, C. K.-L. Li, D.-L. Ma, J. J. Yan, C.-N. Lok, C.-H. Leung, N. Zhu and C.-M. Che, Chem. – Eur. J., 2010, 16, 3097–3113 Search PubMed.
- I. M. Dixon, F. Lopez, A. M. Tejera, J.-P. Estève, M. A. Blasco, G. Pratviel and B. Meunier, J. Am. Chem. Soc., 2007, 129, 1502–1503 Search PubMed.
- X.-H. Zheng, Q. Cao, Y.-L. Ding, Y.-F. Zhong, G. Mu, P. Z. Qin, L.-N. Ji and Z.-W. Mao, Dalton Trans., 2015, 44, 50–53 Search PubMed.
- W.-J. Mei, X.-Y. Wei, Y.-J. Liu and B. Wang, Transition Met. Chem., 2008, 33, 907–910 Search PubMed.
- A. Membrino, M. Paramasivam, S. Cogoi, J. Alzeer, N. W. Luedtke and L. E. Xodo, Chem. Commun., 2010, 46, 625–627 Search PubMed.
- J. Alzeer, B. R. Vummidi, P. J. C. Roth and N. W. Luedtke, Angew. Chem., Int. Ed., 2009, 48, 9362–9365 Search PubMed.
- L. Zhang, J. Huang, L. Ren, M. Bai, L. Wu, B. Zhai and X. Zhou, Bioorg. Med. Chem., 2008, 16, 303–312 Search PubMed.
- L. Ren, A. Zhang, J. Huang, P. Wang, X. Weng, L. Zhang, F. Liang, Z. Tan and X. Zhou, ChemBioChem, 2007, 8, 775–780 Search PubMed.
- D. P. N. Goncalves, R. Rodriguez, S. Balasubramanian and J. K. M. Sanders, Chem. Commun., 2006, 4685–4687 Search PubMed.
- B. Fu, D. Zhang, X. Weng, M. Zhang, H. Ma, Y. Ma and X. Zhou, Chem. – Eur. J., 2008, 14, 9431–9441 Search PubMed.
- Z. Gershman, I. Goldberg and Z. Gross, Angew. Chem., Int. Ed., 2007, 46, 4320–4324 Search PubMed.
- I. H. Wasbotten, T. Wondimagegn and A. Ghosh, J. Am. Chem. Soc., 2002, 124, 8104–8116 Search PubMed.
- J. Bendix, H. B. Gray, G. Golubkov and Z. Gross, Chem. Commun., 2000, 1957–1958 Search PubMed.
- J. G. Muller, L. A. Kayser, S. J. Paikoff, V. Duarte, N. Tang, R. J. Perez, S. E. Rokita and C. J. Burrows, Coord. Chem. Rev., 1999, 185, 761–774 Search PubMed.
- S. S. Mandal, U. Varshney and S. Bhattacharya, Bioconjugate Chem., 1997, 8, 798–812 Search PubMed.
- J. E. Reed, A. A. Arnal, S. Neidle and R. Vilar, J. Am. Chem. Soc., 2006, 128, 5992–5993 Search PubMed.
- A. Arola-Arnal, J. Benet-Buchholz, S. Neidle and R. Vilar, Inorg. Chem., 2008, 47, 11910–11919 Search PubMed.
- L. Lecarme, E. Prado, A. De Rache, M.-L. Nicolau-Travers, R. Bonnet, A. V. D. Heyden, C. Philouze, D. Gomez, J.-L. Mergny, H. Jamet, E. Defrancq, O. Jarjayes and F. Thomas, Inorg. Chem., 2014, 53, 12519–12531 Search PubMed.
- N. H. Abd Karim, O. Mendoza, A. Shivalingam, A. J. Thompson, S. Ghosh, M. K. Kuimova and R. Vilar, RSC Adv., 2014, 4, 3355–3363 Search PubMed.
- Y. L. Jiang and Z. P. Liu, Mini-Rev. Med. Chem., 2010, 10, 726–736 Search PubMed.
- P. Wu, D.-L. Ma, C.-H. Leung, S.-C. Yan, N. Zhu, R. Abagyan and C.-M. Che, Chem. – Eur. J., 2009, 15, 13008–13021 CrossRef CAS PubMed.
- A. Terenzi, D. Lotsch, S. van Schoonhoven, A. Roller, C. R. Kowol, W. Berger, B. K. Keppler and G. Barone, Dalton Trans., 2016, 45, 7758–7767 Search PubMed.
- K. J. Davis, C. Richardson, J. L. Beck, B. M. Knowles, A. Guedin, J.-L. Mergny, A. C. Willis and S. F. Ralph, Dalton Trans., 2015, 44, 3136–3150 Search PubMed.
- J.-T. Wang, X.-H. Zheng, Q. Xia, Z.-W. Mao, L.-N. Ji and K. Wang, Dalton Trans., 2010, 39, 7214–7216 Search PubMed.
- R. Kieltyka, J. Fakhoury, N. Moitessier and H. F. Sleiman, Chem. – Eur. J., 2008, 14, 1145–1154 Search PubMed.
- J. Talib, C. Green, K. J. Davis, T. Urathamakul, J. L. Beck, J. R. Aldrich-Wright and S. F. Ralph, Dalton Trans., 2008, 1018–1026 Search PubMed.
- S. E. Pierce, R. Kieltyka, H. F. Sleiman and J. S. Brodbelt, Biopolymers, 2009, 91, 233–243 Search PubMed.
- K. J. Castor, J. Mancini, J. Fakhoury, N. Weill, R. Kieltyka, P. Englebienne, N. Avakyan, A. Mittermaier, C. Autexier, N. Moitessier and H. F. Sleiman, ChemMedChem, 2012, 7, 85–94 CrossRef CAS PubMed.
- D.-L. Ma, C.-M. Che and S.-C. Yan, J. Am. Chem. Soc., 2009, 131, 1835–1846 Search PubMed.
- S. Bianco, C. Musetti, A. Waldeck, S. Sparapani, J. D. Seitz, A. P. Krapcho, M. Palumbo and C. Sissi, Dalton Trans., 2010, 39, 5833–5841 Search PubMed.
- C. Musetti, L. Lucatello, S. Bianco, A. P. Krapcho, S. A. Cadamuro, M. Palumbo and C. Sissi, Dalton Trans., 2009, 3657–3660 RSC.
- K. Suntharalingam, D. Gupta, P. J. Sanz Miguel, B. Lippert and R. Vilar, Chem. – Eur. J., 2010, 16, 3613–3616 Search PubMed.
- H. Bertrand, D. Monchaud, A. De Cian, R. Guillot, J.-L. Mergny and M.-P. Teulade-Fichou, Org. Biomol. Chem., 2007, 5, 2555–2559 Search PubMed.
- P. Wang, C.-H. Leung, D.-L. Ma, S.-C. Yan and C.-M. Che, Chem. – Eur. J., 2010, 16, 6900–6911 Search PubMed.
- S. Gama, I. Rodrigues, F. Mendes, I. C. Santos, E. Gabano, B. Klejevskaja, J. Gonzalez-Garcia, M. Ravera, R. Vilar and A. Paulo, J. Inorg. Chem., 2016, 160, 275–286 Search PubMed.
- C. Wei, L. Ren and N. Gao, Int. J. Biol. Macromol., 2013, 57, 1–8 Search PubMed.
- E. Largy, F. Hamon, F. Rosu, V. Gabelica, E. De Pauw, A. Guédin, J.-L. Mergny and M.-P. Teulade-Fichou, Chem. – Eur. J., 2011, 17, 13274–13283 CrossRef CAS PubMed.
- K. Suntharalingam, A. J. P. White and R. Vilar, Inorg. Chem., 2009, 48, 9427–9435 Search PubMed.
- J.-T. Wang, Y. Li, J.-H. Tan, L.-N. Ji and Z.-W. Mao, Dalton Trans., 2011, 40, 564–566 Search PubMed.
- C. Yu, K. H.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Chem. Commun., 2009, 3756–3758 Search PubMed.
- N. P. E. Barry, N. H. Abd Karim, R. Vilar and B. Therrien, Dalton Trans., 2009, 10717–10719 Search PubMed.
- R. M. Hartshorn and J. K. Barton, J. Am. Chem. Soc., 1992, 114, 5919–5925 CrossRef CAS.
- A. E. Friedman, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1990, 112, 4960–4962 Search PubMed.
- S. Shi, X. Geng, J. Zhao, T. Yao, C. Wang, D. Yang, L. Zheng and L. Ji, Biochimie, 2010, 92, 370–377 Search PubMed.
- C. Rajput, R. Rutkaite, L. Swanson, I. Haq and J. A. Thomas, Chem. – Eur. J., 2006, 12, 4611–4619 Search PubMed.
- J.-L. Yao, X. Gao, W. Sun, S. Shi and T.-M. Yao, Dalton Trans., 2013, 42, 5661–5672 Search PubMed.
- G. Liao, X. Chen, J. Wu, C. Qian, H. Wang, L. Ji and H. Chao, Dalton Trans., 2014, 43, 7811–7819 Search PubMed.
- J. Sun, Y. An, L. Zhang, H.-Y. Chen, Y. Han, Y.-J. Wang, Z.-W. Mao and L.-N. Ji, J. Inorg. Chem., 2011, 105, 149–154 Search PubMed.
- Z. Zhang, Q. Wu, X.-H. Wu, F.-Y. Sun, L.-M. Chen, J.-C. Chen, S.-L. Yang and W.-J. Mei, Eur. J. Med. Chem., 2014, 80, 316–324 Search PubMed.
- Q. Li, J. Zhang, L. Yang, Q. Yu, Q. Chen, X. Qin, F. Le, Q. Zhang and J. Liu, J. Inorg. Chem., 2014, 130, 122–129 Search PubMed.
- Y. Xia, Q. Chen, X. Qin, D. Sun, J. Zhang and J. Liu, New J. Chem., 2013, 37, 3706–3715 Search PubMed.
- X. Chen, J.-H. Wu, Y.-W. Lai, R. Zhao, H. Chao and L.-N. Ji, Dalton Trans., 2013, 42, 4386–4397 Search PubMed.
- D.-L. Ma, H.-Z. He, K.-H. Leung, D. S.-H. Chan and C.-H. Leung, Angew. Chem., Int. Ed., 2013, 52, 7666–7682 CrossRef CAS PubMed.
- M. Ebrahimi, T. Khayamian, H. Hadadzadeh, B. E. Sayed Tabatabaei, Z. Jannesari and G. Khaksar, J. Biomol. Struct. Dyn., 2015, 33, 2316–2329 CAS.
- D. Monchaud, P. Yang, L. Lacroix, M.-P. Teulade-Fichou and J.-L. Mergny, Angew. Chem., Int. Ed., 2008, 120, 4936–4939 Search PubMed.
- D. Monchaud and M. P. Teulade-Fichou, Org. Biomol. Chem., 2008, 6, 627–636 Search PubMed.
- J. E. Reed, A. J. P. White, S. Neidle and R. Vilar, Dalton Trans., 2009, 2558–2568 RSC.
- R. Kieltyka, P. Englebienne, J. Fakhoury, C. Autexier, N. Moitessier and H. F. Sleiman, J. Am. Chem. Soc., 2008, 130, 10040–10041 Search PubMed.
- X.-H. Zheng, Y.-F. Zhong, C.-P. Tan, L.-N. Ji and Z.-W. Mao, Dalton Trans., 2012, 41, 11807–11812 Search PubMed.
- X.-H. Zheng, H.-Y. Chen, M.-L. Tong, L.-N. Ji and Z.-W. Mao, Chem. Commun., 2012, 48, 7607–7609 Search PubMed.
- C.-X. Xu, X. Zhang, Y.-W. Zhou, H. Wang, Q. Cao, Y. Shen, L.-N. Ji, Z.-W. Mao and P. Z. Qin, Chem. – Eur. J., 2016, 22, 3405–3413 Search PubMed.
- C.-X. Xu, Y.-X. Zheng, X.-H. Zheng, Q. Hu, Y. Zhao, L.-N. Ji and Z.-W. Mao, Sci. Rep., 2013, 3, 2060 Search PubMed.
- D. L. Ang, B. W. J. Harper, L. Cubo, O. Mendoza, R. Vilar and J. Aldrich-Wright, Chem. – Eur. J., 2016, 22, 2317–2325 Search PubMed.
- T. Wilson, M. P. Williamson and J. A. Thomas, Org. Biomol. Chem., 2010, 8, 2617–2621 Search PubMed.
- T. Wilson, P. J. Costa, V. Félix, M. P. Williamson and J. A. Thomas, J. Med. Chem., 2013, 56, 8674–8683 Search PubMed.
- S. Shi, J. Liu, T. Yao, X. Geng, L. Jiang, Q. Yang, L. Cheng and L. Ji, Inorg. Chem., 2008, 47, 2910–2912 Search PubMed.
- L. Xu, X. Chen, J. Wu, J. Wang, L. Ji and H. Chao, Chem. – Eur. J., 2015, 21, 4008–4020 Search PubMed.
- L. Xu, G.-L. Liao, X. Chen, C.-Y. Zhao, H. Chao and L.-N. Ji, Inorg. Chem. Commun., 2010, 13, 1050–1053 CrossRef CAS.
- L. Xu, D. Zhang, J. Huang, M. Deng, M. Zhang and X. Zhou, Chem. Commun., 2010, 46, 743–745 Search PubMed.
- K. Suntharalingam, A. J. P. White and R. Vilar, Inorg. Chem., 2010, 49, 8371–8380 Search PubMed.
- V. S. Stafford, K. Suntharalingam, A. Shivalingam, A. J. P. White, D. J. Mann and R. Vilar, Dalton Trans., 2015, 44, 3686–3700 Search PubMed.
- I. M. A. del Mundo, K. E. Siters, M. A. Fountain and J. R. Morrow, Inorg. Chem., 2012, 51, 5444–5457 Search PubMed.
- K. E. Siters, M. A. Fountain and J. R. Morrow, Inorg. Chem., 2014, 53, 11540–11551 Search PubMed.
- K. E. Siters, S. A. Sander and J. R. Morrow, Dalton Trans., 2015, 44, 3708–3716 RSC.
- F. Doria, M. Nadai, M. Folini, M. Scalabrin, L. Germani, G. Sattin, M. Mella, M. Palumbo, N. Zaffaroni, D. Fabris, M. Freccero and S. N. Richter, Chem. – Eur. J., 2013, 19, 78–88 Search PubMed.
- N. H. Campbell, M. Patel, A. B. Tofa, R. Ghosh, G. N. Parkinson and S. Neidle, Biochemistry, 2009, 48, 1675–1680 Search PubMed.
- H. Yu, X. Wang, M. Fu, J. Ren and X. Qu, Nucleic Acids Res., 2008, 36, 5695–5703 Search PubMed.
- H. Xu, H. Zhang and X. Qu, J. Inorg. Chem., 2006, 100, 1646–1652 Search PubMed.
- C. Vialas, G. Pratviel and B. Meunier, Biochemistry, 2000, 39, 9514–9522 Search PubMed.
- W. Tuntiwechapikul and M. Salazar, Biochemistry, 2001, 40, 13652–13658 CrossRef CAS PubMed.
- W. Tuntiwechapikul, J. T. Lee and M. Salazar, J. Am. Chem. Soc., 2001, 123, 5606–5607 Search PubMed.
- Y. Xu, Y. Suzuki, T. Lönnberg and M. Komiyama, J. Am. Chem. Soc., 2009, 131, 2871–2874 Search PubMed.
- Y. Jin, M. A. Lewis, N. H. Gokhale, E. C. Long and J. A. Cowan, J. Am. Chem. Soc., 2007, 129, 8353–8361 Search PubMed.
- Z. Yu, M. Han and J. A. Cowan, Angew. Chem., Int. Ed., 2015, 54, 1901–1905 Search PubMed.
- M. Deng, D. Zhang, Y. Zhou and X. Zhou, J. Am. Chem. Soc., 2008, 130, 13095–13102 Search PubMed.
- G. Pratviel, Coord. Chem. Rev., 2016, 308, 460–477 Search PubMed.
| This journal is © the Partner Organisations 2017 |