
Field responsive materials: photo-, electro-, magnetic- and ultrasound-sensitive polymers
Theodore
Manouras
a and
Maria
Vamvakaki
*ab
aInstitute of Electronic Structure and Laser, Foundation for Research and Technology-Hellas, P.O. Box 1527, 711 10, Heraklion, Crete, Greece. E-mail: vamvakak@iesl.forth.gr
bUniversity of Crete, Department of Materials Science and Technology, P.O. Box 2208, 710 03, Heraklion, Crete, Greece
First published on 26th September 2016
Abstract
Stimuli responsive materials have attracted a great deal of attention during the last few decades and are expected to lead to great advances in the areas of nano- and bio-technology. Chemical and biochemical stimuli that allow one to control the material properties and functions have been extensively investigated; however, recently physical stimuli have been of much interest because they can be applied remotely, they often allow spatiotemporal control and, depending on the required intensity, they can be biocompatible. Progress in this rapidly expanding field may result in advancements in dynamically controlled systems for use, among others, in drug delivery and tissue engineering, artificial muscles and robotics, (bio)sensors and actuators, space and ocean applications and self-healing materials. In this review we highlight recent progress in the emerging field of photo-, electro-, magnetic- and ultrasound-sensitive polymers. Novel synthetic routes to these polymers as well as their responsive properties and functions are presented. Future perspectives and challenges in the field towards the development of more complex systems are also discussed.
 Theodore Manouras | Theodoros Manouras received his B.Sc. in Materials Science from the University of Patras (Greece) in 2005 and his M.Sc. and Ph.D. in Polymer Science from the University of Athens (Greece) in 2007 and 2012, respectively, working under the supervision of Dr P. Argitis (National Center of Scientific Research “Demokritos”, Greece) on the development of novel polymeric materials for lithographic applications and bioapplications. He is currently a research associate at IESL-FORTH (Heraklion-Greece). His current research interests include the synthesis of well-defined polymeric materials for antimicrobial applications, photodegradable biomaterials, smart polymers for drug delivery and lithographic applications of block copolymers and thermo/acid degradable polymers. |
 Maria Vamvakaki | Maria Vamvakaki received her Ph.D. from the University of Sussex (Brighton, UK) in 2008, working under the supervision of Profs S. P. Armes and N. C. Billingham. Shortly after this, she joined the research group of Prof. C. S. Patrickios at the University of Cyprus (Nicosia, Cyprus) as a postdoctoral fellow. In 2001 she joined the Department of Materials Science and Technology of the University of Crete (Heraklion, Greece) in which she has been appointed as an Associate Professor and leads the Materials Chemistry Lab. She has also been an affiliated researcher at FORTH-IESL since 2003. Her current research interests include the synthesis of functional and stimuli-responsive polymeric materials and the self-assembly of macromolecules in solution and at the surface. |
Introduction
“Smart” or responsive polymers are materials that change abruptly their physicochemical properties in response to an applied external stimulus.1 Chemical, physical and biochemical stimuli have been employed to control the material function in an often programmable and complex manner. Chemical stimuli include the solution pH and the addition of salt or metals and so on, whereas biochemical stimuli comprise the presence of biomolecules and bioactive molecules, i.e. proteins, amino acids, polysaccharides, and glycose.2,3 Physical stimuli, including the solution temperature, the application of an external field, such as magnetic, electric and so on, light irradiation, and ultrasonic treatment, have attracted much attention lately, because they possess certain advantages related to their remote and spatiotemporally controlled application.4–6Field-responsive polymers, which undergo reversible or irreversible transitions in response to the applied stimulus, have been proposed for use in a plethora of applications including sensors and actuators,7 biomedicine,8 tissue engineering,9 regenerative medicine,10 microelectronic systems,11 membranes of controlled permeability12 and self-cleaning surfaces.13 The great deal of attention devoted to the area of field-responsive polymers during the last decades can be partially attributed to the development and evolution of new robust synthetic routes such as click chemistries14,15 and orthogonal synthetic strategies,16 as well as controlled polymerization methods including atom transfer radical polymerization (ATRP),17 reversible addition fragmentation chain transfer (RAFT) polymerization18 and nitroxide mediated polymerization (NMP).19 These polymerization techniques have tremendously simplified the synthesis of well-defined and intelligent polymeric materials including block copolymers, star polymers, polymer brushes and gels, whereas click chemistries have enabled the fabrication of more complex macromolecular architectures by coupling the synthetic polymers with nucleic acids, peptides, sugars, proteins or even viruses and cells.
In this review, we highlight recent developments in the area of field-responsive polymeric materials. We will focus on stimuli that allow one to tune the polymer properties in a remote and spatiotemporally controlled fashion. The design parameters, synthetic strategies, properties and functions of stimuli-responsive polymers that are sensitive to an applied electric or magnetic field, to ultrasound or to light irradiation, are reviewed. Although the field is still progressing and new exciting developments are expected in the future, the examples presented herein clearly demonstrate the diversity of the systems and the advances and opportunities that stem from them in numerous practical applications.
Photo-sensitive polymers
Photo-sensitive materials in the form of either photo-responsive or photodegradable polymers have been extensively reported in the literature. Such polymers can change their physicochemical properties or degrade in response to light irradiation of appropriate wavelength and intensity. Light is a readily available trigger and is often inexpensive, i.e. lamps, while being able to be remotely applied and allowing for spatiotemporal control.The most extensively studied photo-responsive polymers in the literature comprise light responsive moieties based on azobenzenes, spiropyrans and spirooxazines, and groups undergoing [2 + 2] cycloaddition upon light irradiation such as cinnamic esters, coumarin and diarylethenes (Fig. 1). The response leads to a light-triggered self-assembly, surface modification or patterning, swelling behavior, shape change or fluorescence emission of the materials, which paves the way for their use in diverse applications in the nanotechnology and biotechnology fields.
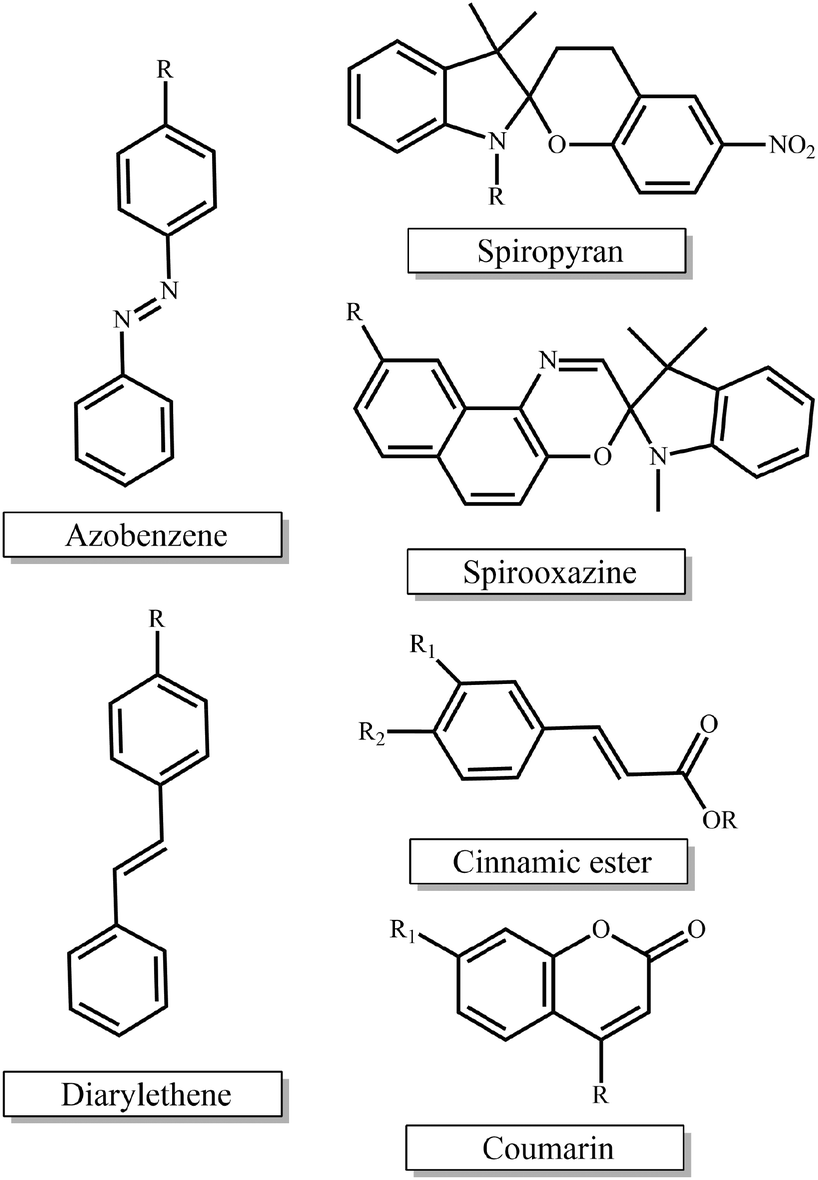 | ||
| Fig. 1 Chemical structures of the light-responsive moieties employed in the synthesis of photo-responsive polymers. | ||
Polymers bearing azobenzene groups
Azobenzene-based copolymers have been extensively employed in the reversible formation and destruction of self-assembled polymer nanostructures in solution using light as a trigger. Two main approaches have been pursued for the organization of such nanostructures; the first involves the self-assembly of a block copolymer in a solvent that is selective for one of the blocks and the second relies on the formation of supramolecular structures based on host–guest interactions of the polymers in solution. In the first approach, poly(ethylene glycol)-b-poly(azobenzene methacrylate) (PEG-b-PAzMA) diblock copolymers were self-assembled in water forming micelles that were shown to exhibit conformational changes from elongated to spherical shape when triggered by either temperature or UV irradiation. These shape changes were used to induce the rupture of giant asymmetric polymersomes by UV illumination. The process follows a nematic-to-coil transition of the hydrophobic block, which leads to an increase of the membrane surface area due to the molecular reorganization of the azobenzene groups and finally the rupture of the asymmetric vesicle membrane by breaking up of the bilayer symmetry. The pore opening process occurs within a few hundreds of milliseconds enabling the release of active substances from the interior of the vesicles which is very attractive for drug delivery applications.20 In a related study, self-assembled vesicles based on photo-responsive azobenzene end-capped poly(ethylene glycol) in water were shown to exhibit a respiring behavior similar to living cells, allowing repeatable transmembrane water transportation upon UV-Vis isomerization of the azobenzene groups. Although the complex mechanism leading to this behavior has not been fully elucidated, the observed changes in membrane permeability are attributed to the polarity switch of the azobenzene groups upon light irradiation, enabling the fabrication of artificial cells with a stimuli controlled function.21 In another example, the self-assembly of more complex azobenzene-based triblock copolymers comprising poly(ethylene oxide) (PEO), polystyrene (PS) and PAzMA blocks in water has been investigated. Triblock copolymers self-assembled into spherical core–shell micelles, which, upon addition of a PS or PAzMA homopolymer, increased in size and also altered their shape and photo-isomerization rate. A photo-induced elongation of the aggregates was found upon their irradiation with linearly polarized laser light, and the deformation increased with the content of the copolymer in AzMA as well as the presence of the PAzMA homopolymer. On the other hand, the photo-isomerization rate increased in the presence of PS and decreased with the addition of a PAzMA homopolymer.22 PEO-b-poly(6-[4-phenylazophenoxy]hexyl methacrylate-co-2-(dimethylamino)ethyl methacrylate) (PEO-b-P(AzMA-co-DMAEMA)) diblock copolymers comprising three different functionalities were also shown to form interesting self-assembled structures in water. These multicomponent systems enable the tuning of the morphology and size of the self-assembled structures in response to different variables and stimuli including the AzMA and DMAEMA content of the copolymers, the addition of beta-cyclodextrin (b-CD) and the light irradiation of the solution, allowing the programmable and cooperative control of the material function.23 Another interesting study was reported by Shen and coworkers who synthesized amphiphilic poly(N-isopropylacrylamide) (PNIPAM)-b-PAzMA block copolymers comprising an azobenzene-based hydrophobic block with 0, 2 and 6 methylene units as spacers between the polymer backbone and the azobenzene moieties. The photo-switching of the vesicles obtained by the self-assembly of the amphiphilic block copolymers in water was investigated. The vesicles lacking the methylene unit spacers did not exhibit a photo-responsive behavior, whereas those with the longer spacer became swollen upon irradiation with UV light. On the other hand, the block copolymer with the 2 methylene unit spacers exhibited an unexpected photo-induced reversible vesicle-to-Janus shape transformation upon irradiation with the light stimulus. This light-controlled bistable shape change was attributed to the stabilization of the Janus metastable shape upon UV irradiation, due to the polarity shift of the azobenzene groups, and represents an attractive approach towards novel more complex nanostructured morphologies.24 An interesting strategy for the synthesis of photo-responsive amphiphilic block copolymers was proposed by Hu et al. who used click chemistry to bind azide-functional trifluoromethoxy-azobenzene onto PEG-b-poly(carbonate) block copolymers bearing alkyne pendant groups on the polycarbonate block. The copolymers self-assembled into spherical micelles in aqueous solution and showed reversible self-assembly and disassembly upon successive UV and visible light irradiation, enabling the light induced encapsulation and release of hydrophobic drugs.25 Similar AB2 shaped amphiphilic block copolymers based on PEG and PAzMA liquid crystalline blocks were prepared by coupling an azide-functional PEG onto an azobenzene polymer bearing anilino functionalities. The novel polymer architectures formed spherical aggregates in water which, upon irradiation with polarized UV light, became elongated in the polarized direction.26,27The photoresponsive properties of the azobenzene units have been also exploited in combination with temperature and pH responsive functionalities, providing facile access to dual- and triple-responsive block copolymers. For example, a thermosensitive PNIPAM block and a photo-responsive poly(4-(6-(4-vinylbenzyloxy)hexyloxy)-2-(4-methoxyl)azobenzene) block were synthesized by RAFT polymerization and were assembled into spherical micelles in water. The abilities to control the size of the micelles using light irradiation and to induce their reversible precipitation in response to changes in the solution temperature provide increased functionality and complexity without compromising their structural precision, which can be advantageous for numerous applications.28 In a related study, photo- and thermo-responsive poly(ethylene glycol) derivatives bearing azobenzene groups were synthesized and the temperature-induced phase transition behavior of the polymer was controlled in a certain temperature range, via photo-irradiation of the solution.29 Another elegant example exploited miktoarm star polymers bearing azobenzene groups and thermo-responsive poly(N,N-diethylacrylamide) arms synthesized by a combination of ATRP and RAFT chemistries. The star polymers formed stable micelles in water at ambient temperature, allowing the UV-triggered release of a model dye (Nile Red) upon the trans-to-cis isomerization of the azobenzene moieties, and precipitated out of the solution above their lower critical solution temperature (27 °C).30 Lately, Pei and coworkers synthesized triple stimuli-responsive poly(DMAEMA-co-6-O-methacryloyl-D-galactopyranose-b-poly(4-(4-methoxy phenylazo)phenoxy methacrylate)) copolymers which respond to temperature, pH and light irradiation using ATRP followed by hydrolysis of the sugar protected moieties. The copolymers formed micelles in water which were stimulated independently by the three different external stimuli. Further effort should focus on the simultaneous and cooperative response of the micelles to the three different stimuli, which could pave the way to biomimetically inspired responsive polymers and further extend their potential applications in many research areas.31
Supramolecular chemistry provides an alternative route enabling the formation of azobenzene-based nanostructures in solution. b-CD–azobenzene host–guest interactions have been widely exploited to obtain complex photo-responsive supramolecular structures. Chen et al. prepared an amphiphilic azobenzene-containing hyperbranched polymer via the carboxylation of the hydroxyl groups of hyperbranched polyphosphate with succinic anhydride, followed by the esterification reaction with 4-hydroxyazobenzene. The polymer formed spherical micelles in water, which dissociated upon the addition of b-CD and enabled the controlled reversible self-assembly and disassembly upon irradiation with UV and visible light, respectively.32 Similar photo-responsive supramolecular linear, hyperbranched and cross-linked polymers have been prepared by the host–guest complexation of azobenzene and b-CD functionalized low molecular weight molecules. The supramolecular nanoassemblies and their optical properties were switched reversibly using UV and visible light irradiation, whereas photo-induced sol–gel transitions were attained for the cross-linked polymer gels.33,34 More complex supramolecular structures can be accessed by exploiting dual b-CD-adamantine and b-CD–azobenzene host–guest interactions. Liu et al. prepared end-functional polystyrene bearing adamantane and azobenzene groups by ATRP and click chemistry. This dual functional homopolymer was combined with poly(ethylene glycol) modified with b-CD end groups, enabling the formation of photo-responsive supramolecular triblock copolymers that self-assembled into micelles in aqueous solution and exhibited reversible assembly–disassembly upon alternate UV-visible light irradiation.35 Dual-responsive linear and cross-linked supramolecular polymers have been prepared using the pillar[5]arene/secondary ammonium salt host–guest interactions. Linear polymers were obtained by the self-assembly of two complementary small molecules, an azobenzene-bridged pillar[5]arene dimer and a bisammonium salt, whereas the assembly of the same pillar[5]arene derivative and a secondary ammonium salt-functionalized poly(methyl acrylate) led to the formation of a polymer network. The dual-responsive behavior of these systems enabled polymer-to-oligomer and polymer-to-monomer transitions upon UV light irradiation and alteration of the solution pH, respectively.36
Other attractive photo-responsive nanostructures, including Janus particles and hollow polymer capsules, have been accessed via microphase separation of two immiscible polymers within dispersed polymer droplets37 and condensation polymerization of bifunctional monomers at the solid–liquid or liquid–liquid interphase, respectively.38,39
Photo-responsive polymer networks are particularly attractive for the development of intelligent systems in drug delivery, tissue engineering, membranes of controlled permeability, sensors and so on. A beautiful example of photo-responsive surface molecular imprinting polymer (SMIP) microspheres was recently reported by Yang et al. The microspheres were synthesized on silica nanoparticles using a water-soluble 4-[(4-methacryloyloxy)-phenylazo]benzenesulfonic acid functional monomer and exhibited good specific affinity towards bisphenol A with high recognition and fast binding and release kinetics upon UV and visible light irradiation.40 In a related study, SMIPs were developed using a photo-responsive functional monomer prepared by covalently connecting azobenzene and 2,4-dichlorophenoxyacetic acid on SiO2 nanoparticles via a sol–gel reaction. The SMIP showed specific affinity for 2,4-dichlorophenoxyacetic acid requiring shorter equilibrium times than conventional molecularly imprinted polymers and a reversible uptake and release upon alternate irradiation with UV and visible light, respectively.41 This photo-controlled molecular recognition behavior has attracted a great deal of research interest and opens new pathways for the use of such systems in the analysis of environmental samples.
Photo-responsive cross-linked polymers can also be used in self-healing materials in actuators and in robotics. Ma et al. reported the synthesis of a photo-healable physical ion gel using a tetra-arm diblock copolymer containing azobenzene groups in a hydrophobic ionic liquid. The gel exhibited a reversible low-temperature gel state and a high temperature sol state with the transition temperature depending on the photo-isomerization state of the azobenzene moiety. The photo-healing of the ion gel was demonstrated by UV light irradiation of the gel damaged areas to induce the gel-to-sol transition followed by visible light irradiation to cause a sol-to-gel transition and the healing of the gel.42 In another example, interpenetrating polymer networks comprising a crosslinked azobenzene polymer and a poly(alkyl methacrylate) polymer were developed and were shown to exhibit reversible bending when irradiated with UV and visible light. The presence of the poly(alkyl methacrylate)s enhanced the photo-responsive and mechanical properties of the networks, while low Tg poly(alkyl methacrylate)s increased the bending speed leading to controlled photo-mobile materials.43
An alternative type of photo-responsive hydrogels which have attracted a great deal of interest is the so-called supramolecular hydrogels. These hydrogels can undergo reversible sol–gel transitions in response to the applied stimulus due their reversible host–guest interactions. Photo-responsive supramolecular hydrogels have been prepared by clicking azobenzene and b-cyclodextrin moieties on two different dextran samples, followed by the directed self-assembly of the functional polymers in solution to form inclusion complexes between trans azobenzene and b-cyclodextrin. The light-responsive hydrogels were used for the in vitro release of model green fluorescent protein after UV irradiation, which induced the azobenzene trans–cis photo-isomerization and the dissociation of the inclusion complexes, leading to the dissolution of the hydrogel (Fig. 2).44 In another approach, Zhou et al. exploited the use of a four arm PEG star with benzaldehyde end-groups for the formation of a supramolecular hydrogel in the presence of α-cyclodextrins (α-CDs). Addition of an azobenzene guest molecule led to a gel-to-sol transition due to the disassembly of the inclusion complexes between the α-CDs and the PEG chains in the presence of the competitive α-CD–azobenzene host–guest interactions and to a photo-responsive reversible gel–sol transition attributed to the photo-isomerization of azobenzene.45 Recently, Wang and coworkers have also prepared similar photo-reversible supramolecular hydrogels from a mixture of α-cyclodextrin and azobenzene substituted poly(acrylic acid)s. The sol–gel transition could be tuned by the polymer concentration, the degree of substitution of the poly(acrylic acid) chain and the length of the spacer of the grafted azobenzene groups, rendering these materials attractive candidates for potential applications in drug delivery, optical sensors and cell culture.46
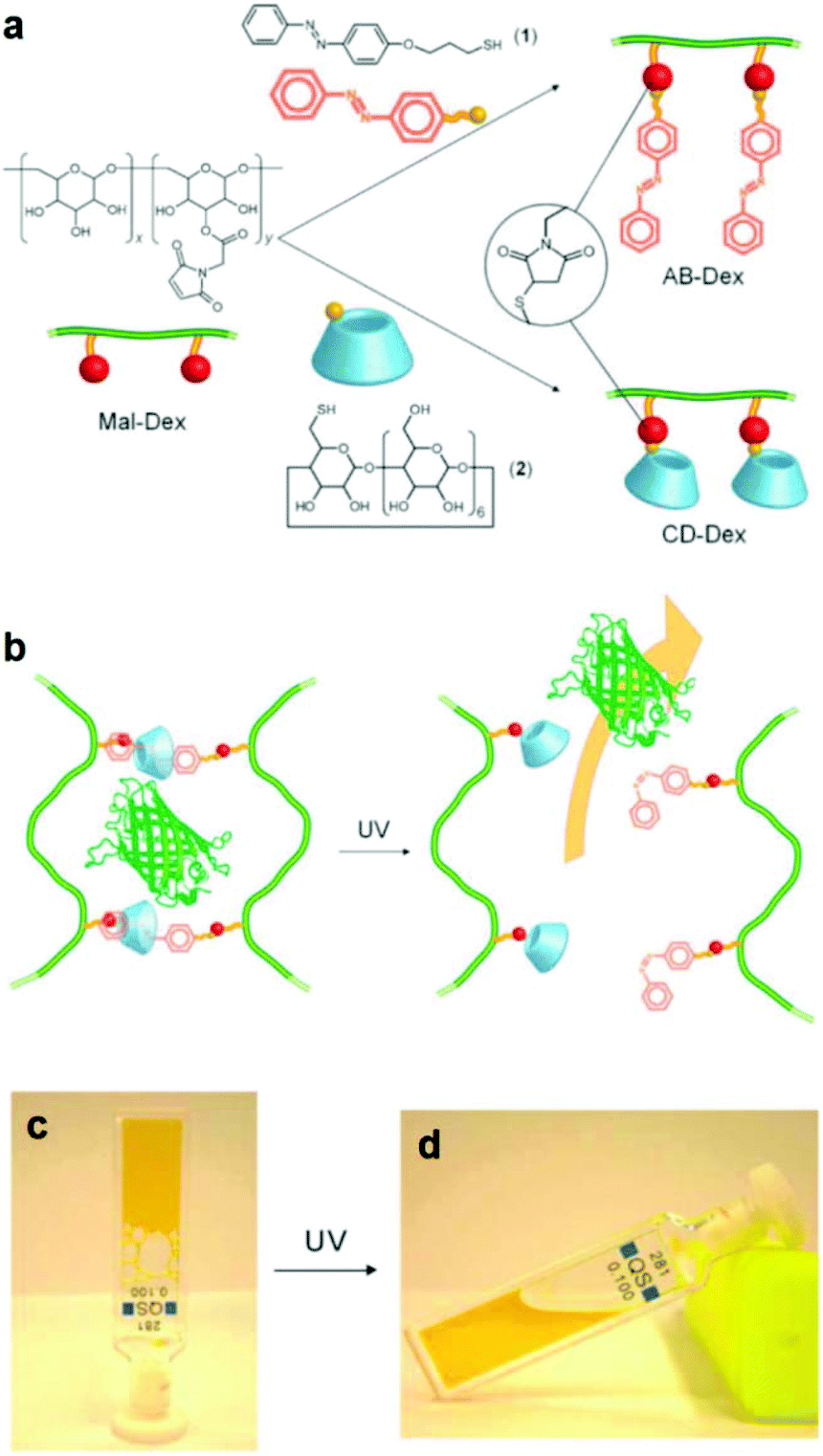 | ||
| Fig. 2 (a) Synthesis of an azobenzene modified dextran (AB-Dex) and α cyclodextrin modified dextran (CD-Dex) via the thiol-maleimide click chemistry on maleimide modified dextran (Mal-Dex). (b) The photo-induced isomerization of the azobenzene moieties upon UV light irradiation causes the dissociation of the supramolecular complexes, resulting in a gel–sol transition and the release of the green fluorescent protein in PBS. Photographs of (c) the supramolecular gel obtained upon mixing the two dextran polymers and (d) the sample after irradiation with UV light. Reproduced from ref. 44 with permission from the Royal Society of Chemistry. | ||
The photo-responsive properties of the azobenzene groups have attracted much attention for the development of photo-active materials. The ability to remotely alter the material morphology, shape and optical properties in a predictable fashion will open new pathways for their use in optical switches, actuators and robotics, holography and optical data storage devices. The light-induced material deformation via a light-to-work transduction has been explored. Modeling of the material photo-active properties is still in its infancy and further effort is still required to master the synthesis–property relationship in order to access predetermined polymer structures.47 Zhu et al. have employed the strong push–pull mechanism on azobenzene chromophores to control the material nanostructure upon light irradiation. Diblock copolymers comprising an azobenzene block and a cholesteryl block were synthesized by RAFT polymerization. A transition from hexagonally-packed nanocylinders to the lamellae morphology was found with increasing the length of the azobenzene block; however the morphology remained unaffected upon the photo-induced reorientation of the azobenzene chromophores within the ordered domains.48 In a related study, PAzMAs bearing strong push–pull azobenzene chromophores were synthesized by RAFT polymerization. The effect of the ligands on the azobenzene moieties was investigated, and the cyanoazobenzene-based polymer exhibited a larger response towards both homogeneous and linearly polarized light than its carboxylazobenzene analogue. This allowed facile patterning of the surface of the material as well as birefringence growth upon light irradiation.49 The change of the optical properties of films comprising poly(meth)acrylates bearing azobenzene side groups was also explored in the development of re-writable paper. Multilayer films consisting of PAzMA and poly(vinyl alcohol) bilayers were spin coated onto glass substrates and were shown to possess tunable reflectivity upon UV irradiation and following thermal relaxation. The concept was based on the very small difference in the refractive index between the trans azobenzene block and poly(vinyl alcohol) which switched off the reflection and the UV-induced transformation from the out-of-plane orientation to an in-plane random orientation of the cis azobenzene groups, and led to the recovery of the reflectance of the film.50 Similar free-standing films were also shown to bend to opposite sides following UV and visible light irradiation due to the reversible trans–cis isomerization of the azobenzene groups.51 The rate, magnitude and reversibility of the material bending process are critical parameters for their use in flexible actuators. Qin and coworkers have reported the formation of a supramolecular network comprising a cationic polymer, poly(diallyldimethylammonium chloride) and electrostatically grafted di-anionic azobenzene groups. The deformation of the polymer on applying the light stimulus to the back of the film was studied. A rapid rolling-up of the film into a tube with a spontaneous shape recovery was seen, as well as a good stability for over 50 cycles, rendering this material attractive for use in artificial muscles and optical micro-tweezers.52 The flexibility of the polymer main chain has been shown to play a significant role in the rapid photo-actuation of the azobenzene side-groups. Highly flexible polysiloxanes bearing azobenzene functionalities have been developed via clicking an azo diazide with a trisiloxane-diyne. The photomechanical bending of the polymers upon exposure to UV light was shown to depend on the polymer molecular weight but was unaffected by the azobenzene substituents, which only influenced the ability of the polymers to exhibit liquid crystalline properties. The presence of the siloxane moieties in these polymers provide additional advantages such as low surface energies and good film forming properties, which, when combined with the photo-induced deformation due to the azobenzene groups, enable their exploitation in many exciting applications.53
The ability to remotely induce surface structuring and surface-relief-gratings on photo-responsive polymer films opens new avenues for their use in liquid mobility in microfluidic devices, light-controlled cell cultures, bioseparation, sensor development and self-cleaning surfaces. The formation of surface patterns on polymer films bearing azobenzene groups using linearly polarized light or interfering laser beams has been reported and was attributed to the development of local stresses on the surface of the film.54,55 Such patterns have been explored in the contact guidance of cells for designing neural networks in vitro. A photo-controlled scaffold comprising an azobenzene based methacrylic copolymer was fabricated on glass substrates. The facile light-induced patterning and erasing of the film surface enabled the topographic guidance of the cells in the culture by the external stimulus.56 In a similar approach, photo-sensitive polysiloxanes have been synthesized and used as a biomimetic analogue to the extracellular matrix (ECM). The writing and erasing on the material surface using optical wavelengths allows controlling the surface topography in a manner that mimics the complexity of natural ECM.57,58
Finally, azobenzene polymer coatings have been developed to control the mobility of water droplets to desired directions and tune the wettability of a surface. Liu et al. have shown that the photo-responsive coatings on rough surfaces can switch the slippery and sticky state of the surface upon visible and UV light irradiation, respectively, leading to the directed motion of the droplet.59 The versatility of the coating method was employed in the fabrication of cotton fabrics with excellent water repellency and light switchable wettability due to the trans–cis photo-isomerization of the azobenzene groups.60 An intriguing approach was introduced by Feng and coworkers to drive the motion of tiny droplets on bio-inspired fibers. An artificial surface patterning that mimics the spindle-knots of wetted spider silk was developed based on a PAzMA-b-poly(methyl methacrylate) (PAzMA-b-PMMA) diblock copolymer. The design of the novel gradient surface enabled the photo-controlled water gathering on the bio-inspired fibers by the cooperative effect of surface roughness and curvature and the photo-responsive wettability of the surface. Visible light drove the water droplets away from the spindle-knots, whereas UV irradiation gathered them towards the spindle-knots, which might open new avenues in the programmable response of artificial surfaces in a manner that mimics the delicate subtlety found in natural systems.61
Hybrid materials, comprising photo-sensitive polymers and inorganic nanoparticles, offer great advantages for theranostic applications. Gold nanoparticles have been widely explored since they combine biosensing and photo-thermal properties. Alvarez et al. reported the synthesis of azobenzene functionalized gold nanoparticles using a photo-responsive elastin-like polymer. The cis–trans photo-isomerization of the azobenzene groups on the gold nanoparticles could only be detected when cyclodextrin functionalized alkanethiol mixed-monolayers were formed on the nanoparticle surface and rendered these multi-functional chromophore–metal nanocomposites attractive for use in the development of multi-stimuli sensitive detectors for biosensing applications.62 In a related study, multifunctional gold nanorod complexes for use in theranostic anticancer treatment were developed. The multifunctional nanorods were functionalized with cyclodextrin for the encapsulation of the drug doxorubicin (DOX), folic acid for cancer cell targeting and a photo-responsive dextran–azobenzene polymer for intracellular controlled drug release. The synergistic effect of UV-controlled drug release and photo-thermal therapy using infrared irradiation led to an enhanced and spatiotemporally controlled anticancer treatment.63
Spiropyran-based polymers
Spiropyrans (SPs) are well-known photochromic and multi-responsive molecules that alter their physicochemical properties in response to several external stimuli such as light irradiation, pH, the presence of metal ions and so on. SP is photo-isomerized to zwitterionic merocyanine (MC) by UV light irradiation and MC turns back into SP upon irradiation with visible light or heating. SPs have been extensively used in the synthesis of multi-responsive polymers exhibiting a reversible response that allows one to control numerous functions such as actuation, encapsulation and release of actives, conductivity, permeability, optical properties and sensing, wettability and colloidal stability and have been proposed for use in biological applications, optical and microfluidic devices, sensors and actuators. Smart engineering of SP-containing polymers is unlimited and the synthetic routes to incorporate these functionalities into polymeric matrices have increased tremendously nowadays. SP groups have been incorporated into various polymeric materials ranging from linear and branched polymers, polymer gels, polymer brushes, colloidal polymer particles and so on.64,65 A careful study was carried out by Ventura et al. who synthesized photo-responsive poly(6-benzospiropyran hexylmethacrylate)s of different architectures by the combination of ATRP of an azo-functionalized methacrylate with click chemistry to attach alkyne SP moieties. The effects of the polymer architecture on the ring-closure kinetics and the reversible photo-switching of the SP groups were investigated. The molecular brush architecture exhibited two times slower kinetics and higher photo-stability compared to the linear and star polymers.66 Dual-responsive polymers are also easily obtainable by combining a SP monomer with other stimuli responsive comonomers. For instance, photo- and temperature dual-responsive block copolymers based on a SP methacrylate (SPMA) block and a di(ethylene glycol)methyl ether methacrylate (DEGMMA) block were synthesized by ATRP and were shown to form micelles with a polySPMA-core and reverse micelles with a polyDEGMMA-core in water. These photo- and temperature-responsive micelles can act as smart nanocarriers for the controlled encapsulation, triggered release and re-encapsulation of model drugs.67 In a related study, SP-based polymers have been employed for cancer therapy and imaging. Multi-responsive micelles based on a pluronic–chitosan copolymer bearing pH-responsive boronate ester groups and redox responsive disulfide bonds have been decorated with SP moieties. Besides the release of the drug in response to the pH and redox stimuli the micelles exhibited a strong fluorescence, attributed to the MC isomer, which verified their successful entry into the intracellular environment.68 Our group has performed extensive work on the synthesis of multifunctional SP-based polymers and hybrid materials. Simple poly(SPMA-co-DMAEMA) random copolymers were synthesized by ATRP and were shown to be responsive to four different stimuli: the solution pH and temperature, the solvent polarity and light irradiation.69 The synergistic effect of light and temperature has been also demonstrated to lead to a complex polymer behavior, which led to the precipitation of the polymer from the aqueous solution at a certain temperature upon irradiation with visible light. In a subsequent study, we used similar random copolymers to decorate the surface of silica particles. The polymer chains were grown from the surface of the inorganic colloids by surface-initiated ATRP leading to the formation of a dense polymer brush. The photo-sensitive polymer shells displayed an inherent fluorescence resonance energy transfer (FRET) process and a biomolecule dependent output FRET signal opening new pathways for their use in remotely-controlled, nanoscale biosensors.70 Moreover, the silica–polymer core–shell nanoparticles were employed in the light-induced supramolecular engineering of hollow polymer capsules in water, based on the reversible formation and disruption of π–π H-type aggregates between the MC isomers within the sterically crowded polymer brush layer, upon irradiation with UV and visible light, respectively (Fig. 3).71 The use of these nanocapsules in pH- and photo-controlled drug delivery is envisaged.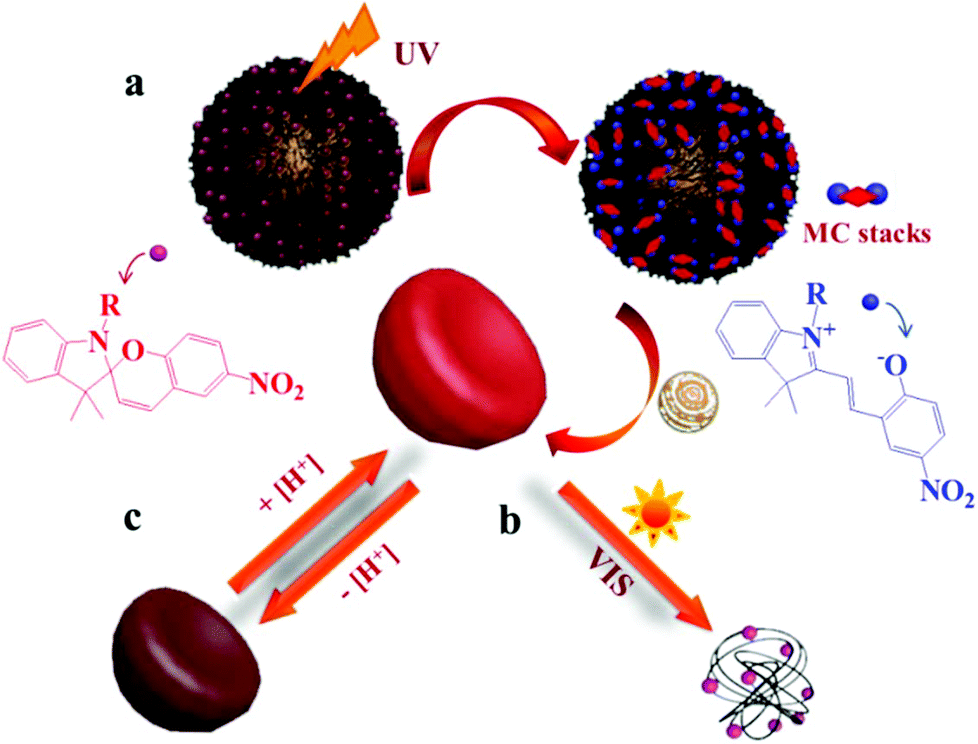 | ||
| Fig. 3 Schematic representation of the (a) UV-induced formation of hollow polymer nanocapsules and (b) visible light-triggered vesicle disruption. (c) Reversible pH-responsive swelling of the hollow nanocapsules.71 | ||
Orthogonal chemistries have been employed for the synthesis of photo-responsive rod–coil diblock copolymers. A regioregular poly(3-hexylthiophene) rod-block was prepared first by quasi-living Grignard metathesis polymerization and was used as a macroinitiator for the ATRP copolymerization of methyl methacrylate and 2-methyl methaspirooxazine to produce the coil-block. The copolymers were photo-active, exhibiting a SP-to-MC isomerization upon UV light irradiation, and could microphase separate in the bulk to provide conducting poly(3-hexylthiophene) domains, which render them particularly attractive for optical and photovoltaic applications.72
SP-based hydrogels that are sensitive to more than one stimulus and display a cooperative function in response to the applied stimuli can be advantageous for many emerging applications. For example, dual light- and temperature-responsive hydrogels comprising PNIPAM functionalized with photo-responsive SP moieties were synthesized and were shown to exhibit both temperature- and light-induced shrinkage, whereas cylindrically shaped hydrogels showed rapid bending due to the MC-to-SP isomerization and the asymmetric light irradiation.73
Photo-patterning of SP polymers on flat and structured surfaces enables one to alter the surface topography, wettability or pore permeability, expanding their applications in the bio- and nano-technology fields. Amphiphilic SP-based copolymers have been used to fabricate honeycomb films. The approach is based on the solubility switch of SP against chloroform vapor, after UV irradiation to produce the MC isomer, resulting in a MC insoluble honeycomb-pattern upon dissolution of the non-irradiated SP polymer.74 Photo-responsive surfaces combining wettability changes with photochromic behavior have been developed by the surface-initiated ATRP of SPMA to grow polymer brushes on silicon and quartz substrates. A large and fast reversible change of the surface wettability and color upon UV and visible light irradiation was demonstrated, enabling their use as smart surfaces.75 In a recent paper, researchers extended the synthetic route to the synthesis of photo-responsive membranes with light controlled permeability and photo-switching properties. In this approach, poly(2-hydroxyethyl methacrylate) brushes were grown first on polyester porous membranes via activators regenerated by electron transfer ATRP followed by the grafting of acid-functionalized SP moieties to obtain the photo-sensitive membranes. The membranes were proposed for applications in filtration, sensors and drug delivery.76
SP-containing organic–inorganic hybrid materials are another class of photo-responsive materials exhibiting unique physicochemical properties derived from the synergistic organic and inorganic components. Hybrid core–shell nanoparticles comprising lanthanide upconversion nanoparticles coated with silica and loaded with an anticancer drug in the core, and a folic acid-functionalized random SP copolymer in the shell, were developed and used in near infrared (NIR)-controlled drug delivery. The tumor targeting ability and bioimaging properties of the particles were studied in vitro, whereas the NIR-triggered drug release was confirmed in vivo in animal experiments, showing the potential of the nanocarriers in anticancer therapy.77 In a recent study, photo-responsive Janus silica–polymer composite nanosheets were developed by growing a SP-based polymer on one side of the nanosheets and hydrophobic octyl groups on the other side. The Janus nanosheets were proposed for use as solid photo-triggered emulsifiers by tuning the hydrophilicity of their SP functionalized side with UV and visible light irradiation.78
Photo-induced [2 + 2] cycloaddition polymers
Light-responsive polymers that undergo reversible photo-crosslinking possess great potential for applications requiring stimulus-controlled swelling, degradation and diffusion behavior such as biomedical applications, i.e. tissue engineering and drug delivery, shape memory and self-healing materials, and so on. Light-induced [2 + 2] cycloaddition reactions have been extensively employed in the photo-controlled cross-linking of coumarin and stilbene containing polymers to alter their physicochemical properties and function.Coumarin groups undergo reversible photo-controlled dimerization upon UV light irradiation at two different wavelengths. UV irradiation at λ > 310 nm results in the coumarin dimerization, observed as a decrease of the coumarin adsorption band at 320 nm, whereas illumination at λ = 254 nm drives the photocleavage of the coumarin dimers and the recovery of the coumarin adsorption at 320 nm. The coumarin photochemistry has been exploited by Sato et al. to prepare dual temperature- and photo-responsive homopolymers using coumarin methacrylates, with ethyleneoxy spacer groups between the methacrylate and coumarin groups, as the functional monomers. The effect of the light-induced [2 + 2] cycloaddition of the coumarin units, upon irradiation with UV light, on the critical solution temperature of the polymer was demonstrated leading to polymer precipitation at a wider temperature range.79 An intriguing approach to multifunctional micelles was reported by Long and coworkers using folic acid bearing PEG-polyurethane copolymers decorated with photo-responsive coumarin moieties and pH-responsive hydrazone groups. The copolymer formed micelles in water which were cross-linked upon UV irradiation by the dimerization of coumarin groups, allowing drug encapsulation efficiencies higher than those for the non-cross-linked analogues. More interestingly, dissociation of the cross-linked micelles at low pH, due to the hydrolysis of the hydrazine groups, released more than 60% of the encapsulated drug compared to 40% for the non-cross-linked micelles. The stimuli-responsive intracellular drug release, combined with the high extracellular stability of the micelles, supports their use as a novel platform in anticancer therapy.80 In another example, photo-responsive microgels have been prepared by the self-assembly of amphiphilic poly(hydroxyethyl acrylate-co-coumaryl acrylate-co-octadecyl acrylate) copolymers in water. The coumaryl acrylate comonomer increased the hydrophobicity of the copolymers leading to smaller microgels, and allowed one to tune the release profile of model dye molecules from the microgels by light irradiation, due to the reversible dimerization of the coumarin units using an appropriate light source.81
A facile route to obtain conducting photo-sensitive polyaniline nanoparticles was proposed by Luo et al. who used photo-responsive poly(vinyl-coumarin-co-2-acrylamido-2-methyl-1-propanesulfonic acid) copolymer micellar templates for the redox polymerization of aniline. The reversible photo-crosslinking of the coumarin groups in the copolymer micelles allowed tuning the size of the conducting nanoparticles upon light irradiation and increased the stability of the nanoparticles, which could find numerous applications in inkjet printing, nanoparticulate emulsifiers and others.82 Shell photo-cross-linked particles bearing coumarin moieties were introduced by Kim's group. They have employed either a proteinoid, composed of lysine and serine, bearing covalently bound coumarin groups or a hydrophobically modified poly(vinyl alcohol) epoxypropoxy coumarin conjugate to form the photo-sensitive shells.83,84 The photo-controlled release of model dyes from the nanoparticles was verified under cyclic irradiation with a 365 nm and a 254 nm light source to induce the cross-linking and de-cross-linking of the polymer shell, respectively, by the photo-reversible dimerization of the coumarin residues. The immobilization of the coumarin conjugated polymer in water channels allowed a similar photo-controlled channel permeation for active molecules.85
Hybrid core–shell mesoporous silica nanoparticles coated with light-responsive polymers bearing coumarin moieties have been also explored as multifunctional nanovehicles in anticancer therapy.86 In a novel approach, two photon disruption of the nanocarriers by a femtosecond NIR light laser (800 nm) was employed to release the drug inside the cells, whereas the strong fluorescence of the polymer enabled the simultaneous imaging of the chemotherapy process, rendering these particles attractive for theranostic applications.87
Stilbenes, cinnamic acid and their derivatives have been also explored in polymeric materials as photo-responsive cross-linking moieties that undergo [2 + 2] cycloaddition reactions upon light irradiation.88–93 The facile photo-chemistry of these polymers has gained a great deal of attention for use in numerous applications ranging from optical and electronic devices and in the biotechnology field.
It is clear from the above that significant progress has been made lately in the development of photo-responsive polymers for different applications in the areas of nano- and bio-technology. The photo-isomerization of azobenzene and the ring-opening reaction of SP moieties have been mostly studied in the synthesis of photo-responsive linear and graft polymers, hydrogels and hybrid materials. However, these photo-induced transformations suffer non-quantitative yields and photo-bleaching of the chromophores upon repeatable irradiation, which have limited their use in practical applications. Other photo-chemistries, including the [2 + 2] cycloaddition of coumarins, stilbenes and cinnamic acids and the [4 + 4] cycloaddition of anthracenes,94–98 have been also explored; however, a great deal of effort is still required to master the properties and functions of these materials in real applications.
Photodegradable polymers
Photo-sensitive polymers have gained much attention over the last 20 years, for applications in which the remote triggering and spatiotemporal control are important. Developments in novel photodegradable polymeric materials has been driven by the rapid evolution of diverse research areas such as photolithography, bio-patterning, tissue engineering and photo-triggered drug delivery. The major challenges in the design of new photodegradable polymers include the sensitivity and the red shifting of the photolysis process to visible or NIR wavelengths. This can be achieved by employing suitable photo-labile segments with low dissociation energies or ultrafast laser sources in multiphoton processes. Below four types of photodegradable polymers are discussed: polymers with photodegradable pendant groups, polymers that undergo main chain scission, polymers containing photodegradable block junctions and polymer networks with photo-labile cross-linkers (Fig. 4).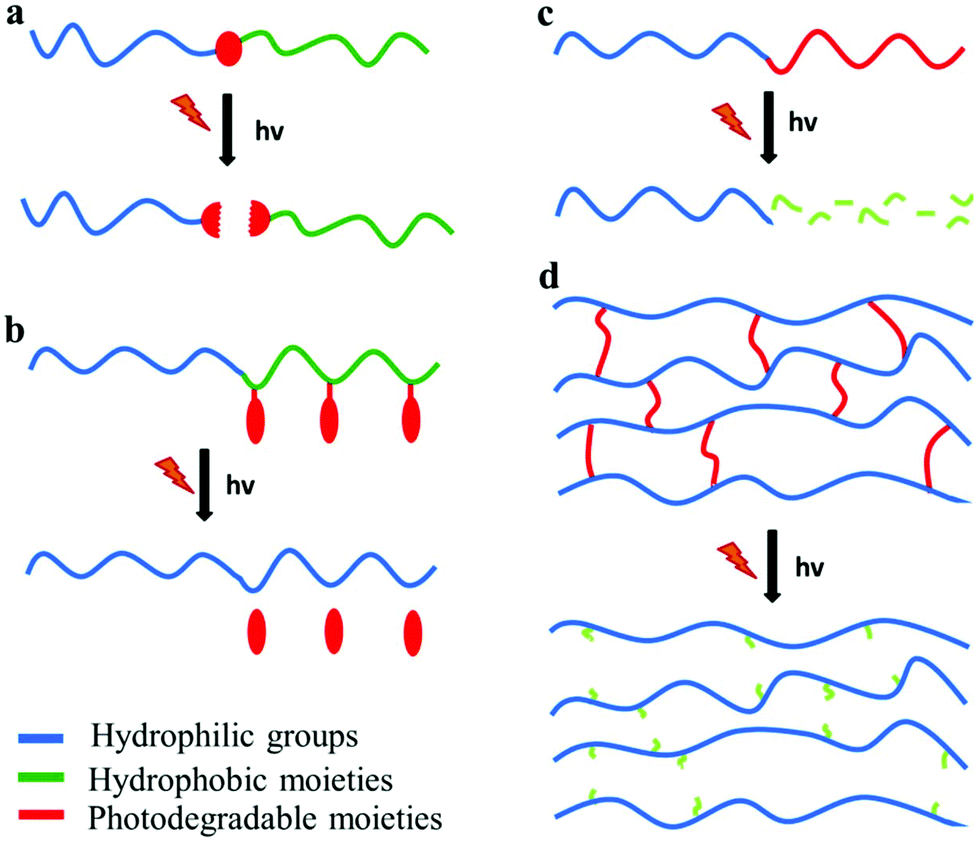 | ||
| Fig. 4 Photodegradable polymeric materials containing (a) a photolabile junction, (b) photo-labile pendant groups, (c) photodegradable backbone and (d) photo-labile cross-linkers. | ||
Pendant side-group cleavage
A common approach to alter the hydrophilicity of a polymer using light is via the cleavage of the pendant side-groups of the polymer. Bonds that can be cleaved using light as a trigger include ester, triazine, disulfide and acetal functionalities. These bonds exhibit low or no absorbance in the deep UV range and usually an absorbing group is introduced nearby in order to enhance the absorption coefficient of the polymer leading to more effective photo-degradation or to the change of the irradiation wavelength. These absorbing groups, also known as chromophores, include aromatic or polyaromatic, coumarin and nitrobenzyl moieties. The most extensively explored functionality is the ortho-nitrobenzyl (ONB) group due to its high sensitivity and the acceptable degradation wavelength (λ = 365 nm). In an intriguing example, Byambaa et al. prepared a photo-reactive surface which allowed the detachment of adhered cells. The surface was covered with a photo-sensitive polymeric layer consisting of a copolymer of 2-methacryloyloxy ethyl phosphorylcholine, butyl methacrylate and a methacrylate bearing a photo-labile ONB group. The photo-induced cleavage of the ONB groups led to the formation of methacrylic acid moieties, which increased the hydrophilicity of the surface, resulting in cell detachment.99 In another approach, ONB esters have been employed as light-triggered solubility and emission modulators of π-conjugated, fluorescent polymers. Photo-cleavage, using either a one- or a two-photon strategy, of the ONB groups from a polyfluorene backbone led to an increase in the emission intensity, while a drastic reduction of the solubility of the polymer enabled surface photo-patterning.100–103An interesting photodegradation strategy was proposed by Almutairi's group using a light-triggered self-immolative polymer. A quinone methide self-immolative moiety was triggered to degrade via multiple light-sensitive ONB ester or coumarin ester groups along the polymeric chain. The photodegradation is induced using either a one- or a two-photon process at relatively low energy dosages.104,105 In a similar method, they explored the acceleration of the hydrolysis of polymer ketal bonds by employing ONB methacrylate moieties along the main chain of the polymer. Photodegradation of the ONB methacrylate moieties produced methacrylic acid groups, which increased the hydrophilicity of the polymer and facilitated ketal hydrolysis.106 Zhu et al. employed the hydrophobic ONB functionalities to prepare physically cross-linked polymeric networks from poly(ONB methacrylate)-b-PEG-b-poly(ONB methacrylate) triblock copolymers. The poly(ONB methacrylate) blocks were converted into water-soluble methacrylic acid moieties upon UV irradiation at 365 nm leading to the destabilization of the physical cross-links and a gel-to-sol transition.107 A more complex architecture was proposed by Yin et al. which comprised temperature- and light-responsive microgels consisting of thermo-responsive NIPAM, photosensitive 5-(2′-(dimethylamino)ethoxy)-2-nitrobenzyl acrylate (DMNA), a FRET donor 4-(2-acryloyl-oxyethylamino)-7-nitro 2,1,3-benzoxadiazole (NBDAE) and a rhodamine B-based FRET acceptor (RhBEA). UV irradiation of the P(NIPAM-DMNA-NBDAE-RhBEA) microgels increased significantly the LCST due to the formation of the hydrophilic acrylic acid moieties, while the phase transition affected the donor–acceptor distance and was monitored by determining the FRET efficiencies of the microgels.108 On the other hand, Fang et al. demonstrated the synthesis of thermo-responsive and photo-sensitive PNIPAM-b-poly(3-methyl-3-nitrobenzyl-trimethylene carbonate) diblock copolymers. The copolymer formed micelles in aqueous solutions which were proposed for the photo-triggered drug release in human cervical HELA cells using indomethacin as a model drug.109
Besides the conventional photo-cleavage reaction of the ONB esters, which leads to hydroxy or aldehyde photo-products, Sinclair et al. and Sobolciak et al. employed the photo-sensitivity of the ONB ester group to form carboxybetaine moieties. In their study, the tertiary amine polymer pendant groups were first quaternized using o-nitrobenzyl 2-chloroacetate followed by ester photo-degradation to produce the zwitterionic and non-toxic polymer.110,111 Moreover, Xi et al. used photo-caged primary amines bearing nitrobenzyl moieties for the photo-induced amine catalyzed thiol-Michael addition. The photo-caged catalyst allowed spatiotemporal control of the reaction, which was employed in surface photo-patterning and two-stage polymer network formation.112,113
Recently, several photodegradable dendrimers have been designed bearing the photodegradable moieties in the core of the dendrimer or as a protecting group in the dendritic side chains or as a linker between the hydrophobic and hydrophilic parts in amphiphilic dendrons. Wang et al. proposed the use of dendronized copolymers of styrene and maleic anhydride pendant with ONB protected poly(amidoamine) dendrons as side groups, for the UV-triggered drug release.114 Kostiainen et al. established a facile method for the preparation of protein–dendron conjugates containing ONB groups and demonstrated the cleavage of the dendron by UV irradiation.115 Similar dendrons were employed in combination with ferritin protein cages which encapsulated iron oxide nanoparticles for magnetic field targeting and UV-induced controlled release.116 In an alternative approach, Nazemi et al. incorporated photodegradable ONB moieties along the dendrimer backbone, which enabled the complete backbone photodegradation to produce small molecule fragments.117
Main chain scission
Polymers exhibiting concerted main chain photodegradation have attracted a great deal of attention recently due to their ability to form low molecular weight fragments under UV or NIR irradiation. These polymers can be employed in a variety of applications including the modification of the surface properties, controlled drug release, photo-patterning and lab-on-a-chip devices. Main chain photodegradable polymers are limited and can be categorized into three main classes, namely polyesters, polytriazines and the recently introduced, by our group, polyacetals.118 Dispinar et al. reported a straightforward method to prepare photodegradable microcapsules by the incorporation of 6-nitroveratroyloxycarbonyl (NVOC) moieties into the shell of polyurea microcapsules. The polyurea microcapsules comprised a photo-cleavable shell and a hydrophobic liquid core. The photo-cleavable NVOC groups in the shell were degraded upon UV irradiation leading to the rupture of the microcapsules and the release of their cargo in solution or in the solid state.119Lv et al. simplified the synthesis of main chain ONB ester-based polymers by the coupling of 2-nitrophenylene glycol and dioyl dichlorides. The polymers were assembled into nanoparticles in a simple emulsion and the direct photo-cleavage of the polymer backbone was demonstrated.120 In a related study, a new family of photodegradable polyurethanes was introduced using 2-nitrophenylene glycol as the photo-labile moiety and diamines as comonomers. The photodegradable polyurethanes were assembled into nanoparticles encapsulating the water insoluble drug Tagalsin G. The photo-triggered release of the drug from the polymeric nanoparticles resulted in an increased cell death from 9% to 67% for RAW 264.7 cells.121 Poly(β-amino esters) (PBAEs) represent an important class of cationic gene delivery materials and have been intensively studied in non-viral gene delivery applications during the last decade. However, these materials suffer uncontrolled DNA release due to the slow degradation of the polyester backbone. Deng et al. proposed a method to overcome this problem by introducing light sensitive ONB ester moieties along the PBAE backbone. The modified PBAEs can efficiently condense and deliver genes intracellularly while upon UV irradiation, at the post-transfection stage, the polymeric chains are rapidly cleaved into small fragments and release the gene.122
Li et al. introduced a novel family of photodegradable polymers based on the ONB ester photochemistry containing allyl, propargyl and epoxy clickable groups and synthesized them by Passerini multicomponent polymerization. Thin films of the random copolymers were first cross-linked by an epoxy–amine reaction and then photo-patterned through a mask using 365 nm light irradiation. The patterns formed were further functionalized by sequential copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC) and thiol–ene click chemistries leading to micro-patterned biomolecules.123
In a different approach, Liu et al. synthesized a photo-labile and biocompatible amphiphilic diblock copolymer 6-arm star based on a cyclotriphosphazene core via ATRP. The star copolymer comprised photo-cleavable ONB ester groups in the inner core, hydrophobic PMMA as the inner arms and hydrophilic poly(poly(ethylene glycol) methyl ether methacrylate) (PPEGMA) as the outer arms. Photo-degradation studies showed that the stability of the star copolymer decreased after UV irradiation, leading to its dissociation and the release of an encapsulated drug under physiological conditions.124
In a similar approach, Han et al. designed an ABA triblock copolymer consisting of hydrophilic PEG as the A end blocks and hydrophobic polyurethane bearing photo-labile ONB ester moieties along the main chain as the B middle block. The block copolymers assembled into micelles in aqueous solutions and underwent fast photo-induced disintegration of the micellar cores leading to the burst release of the loaded hydrophobic drugs.125
Sun et al. designed a new class of alkoxy phenacyl based photodegradable polycarbonates. Homopolymer thin films were employed to create patterns by deep UV irradiation through a photomask whereas polycarbonate-b-PEG diblock copolymer micelles released hydrophobic molecules from their core upon light irradiation.126
Although the photochemistry of small molecules containing acetal or ketal groups has been well known for many years, our group has recently shown for the first time the main chain photo-scission of novel polyketals and polyacetals using fast laser sources. The photodegradation was induced by excitation of the aromatic group which is adjacent to the ketal or acetal bond in the backbone of the polymer. Photochemical degradation was found to take place at very low exposure doses using 248 nm light (two orders of magnitude lower than the ester-based polymers) and led to low molecular weight photo-products. This efficient photodegradation route allowed facile cell detachment and patterning using laser illumination and the application of a photomask. Polyacetal and polyketal homopolymers were employed as substrates to culture mouse fibroblast cells which were harvested from the substrate using a mild laser ablation process and created cell patterns on the cell-culture substrates.127
The facile photodegradation process was also employed to develop a novel drug delivery system based on a block copolymer comprising a hydrophobic polyacetal block and a hydrophilic PEG block. The block copolymer was synthesized by a two-step polycondensation reaction. First, 2-nitroresorcinol was reacted with 1,4-cyclohexanedimethanol divinyl ether catalyzed by pyridinium p-toluenesulfonate and the polyacetal was subsequently end-capped with methoxy terminated PEG (Fig. 5a) at the vinyl ends of the semitelechelic precursor polymer. 2-Nitroresorcinol was chosen as a chromophore in order to red-shift the photodegradation wavelength to the less-harmful-for-the-living-cells range. The degradation of the block copolymer was triggered photo-chemically using UV (193 and 248 nm), UV A (365 nm), visible (532 nm) and NIR (1064 nm) irradiation by single-, double- and multi-photon excitation, respectively. The hydrolysis of the polymer acetal bonds at low pH was also demonstrated (Fig. 5b). Micellar nanoparticles loaded with a potent chemotherapeutic anticancer compound (camptothecin (CPT)) and a phototoxic drug (hematoporphyrin (HP)) were formed in water (Fig. 5c) and tested against HeLa cancer cells using a photo-chemo-induced cell death mechanism. The drug loaded nanoparticles were taken up by the cells via the endocytosis pathway and reached the late endosome, where they released the drug due to their partial degradation in the low pH endosomal environment and their combined laser photolysis. Cell death was concerted by the synergistic excessive reactive oxygen species accumulation (from HP) and the enhanced CPT release from the endosome during laser treatment (Fig. 5d). Cellular uptake increased significantly for the drug loaded nanoparticles while cell death rates were considerably higher for the nanoparticles irradiated with a YAG laser at 532 nm (Fig. 5e).128
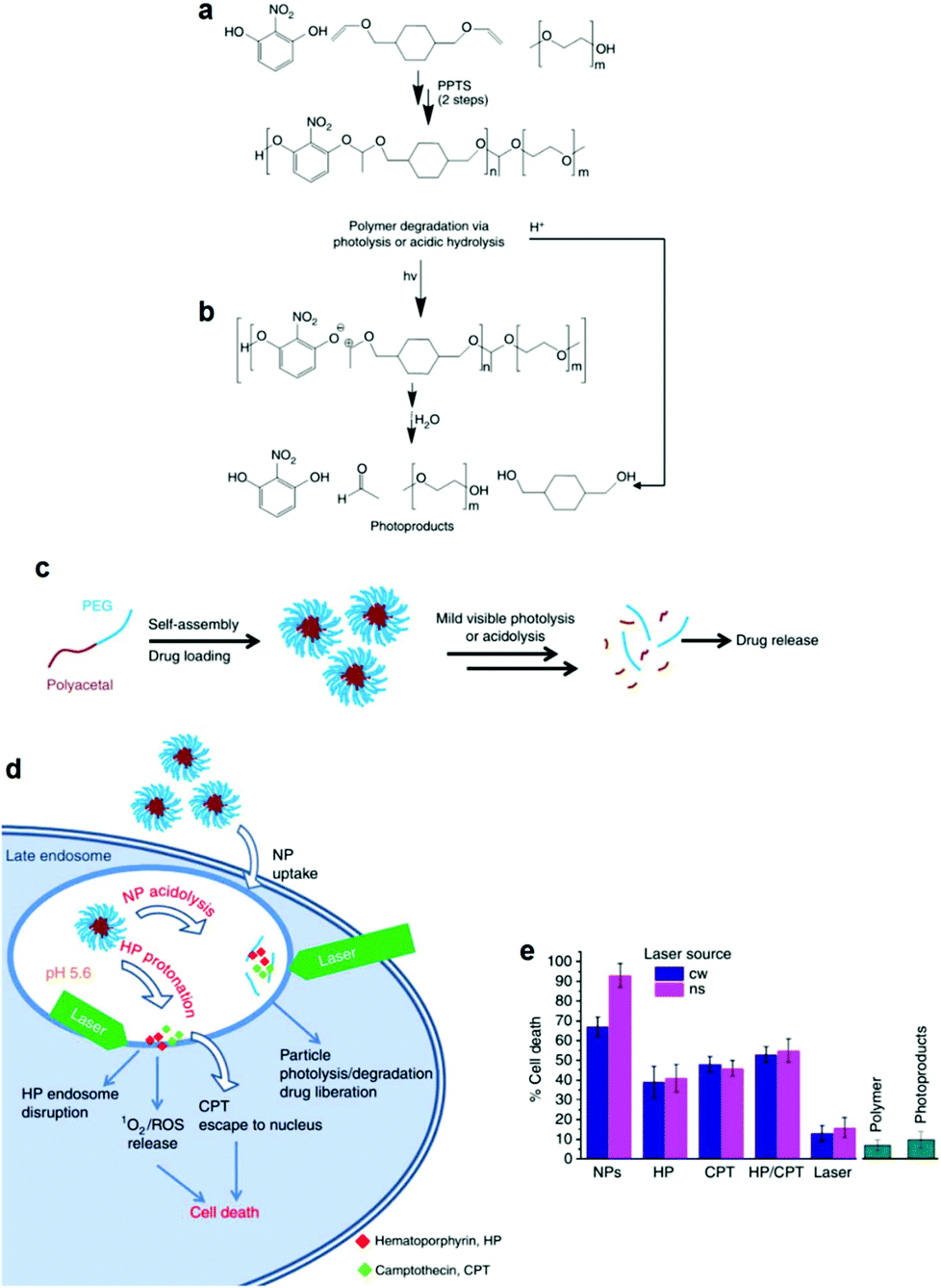 | ||
| Fig. 5 (a) Synthesis of the polyacetal-b-PEG diblock copolymers via a two-step polycondensation reaction. (b) Main chain scission of the polyacetal block via photolysis or acidolysis. (c) Self-assembly of the diblock copolymer in aqueous media and encapsulation of the drug molecules in the core of the micelles, followed by drug release upon light irradiation and/or a decrease of the solution pH. (d) Proposed mechanism for the internalization of the nanoparticles into the HeLa cells followed by particle photo-chemo-lysis. (e) Cell death caused by the photolysis of the nanoparticles using two different laser sources, compared to various control samples.128 | ||
Photodegradable block copolymer junctions
Another type of photodegradable polymers comprises block copolymers connected with a photo-labile junction. ONB esters have been extensively employed as photo-labile junction points connecting two blocks. Kang et al. synthesized photo-cleavable PEO-ONB-PS diblock copolymers by ATRP using a functionalized PEO macroinitiator. The PEO-ONB-PS copolymers formed a microphase separated thin film, which was converted to a porous PS film after irradiation of the block copolymer thin film followed by removal of the cleaved PEO block in a water/methanol mixture.129Substituted polycaprolactones, synthesized by ring opening polymerization using a modified ONB ester group as the initiator, have been employed as biodegradable and biocompatible hydrophobic blocks in the preparation of junction photo-labile amphiphilic copolymers.130–133 Lee et al. synthesized an amphiphilic PNIPAM-ONB-poly(4-substituded-ε-caprolactone) diblock copolymer, which formed spherical micelles in aqueous solution with hydrodynamic sizes <200 nm. The release of Nile Red from the core of the micelles as a function of UV irradiation and the solution temperature was demonstrated, verifying the potential of the system in targeted drug delivery applications.134
Anilkumar et al. synthesized a photo-labile C18-ONB-PEG amphiphile by covalently binding methoxy-carboxy-terminated PEG-2000 with a bromo-nitrobenzyl-functionalized stearyl ester.135 In a similar approach, Cheng et al. used 2-nitrobenzyl 2-pyridinylmethyl borate (NP-B) as the hydrophobic tail in a NP-B-PEG amphiphile. Both amphiphiles formed micelles in aqueous media with encapsulated hydrophobic drugs in their cores, which were effectively released upon UV irradiation.136 In a more sophisticated approach, Ding et al. attached a pesticide on an ONB end-functionalized PEG and employed the self-assembled amphiphiles for the photo-controlled release of the herbicide in aqueous solution.137
Finally, Lu et al. developed a new photosensitive PS-b-PEG block copolymer bearing an acetal photo-cleavable junction between the two blocks. The degradation of the block copolymer under mild irradiation conditions facilitated the formation of nanoporous thin films from microphase separated block copolymers films following light irradiation and removal of the PEG blocks.138
Photo-labile cross-linkers
Photodegradable hydrogels and 3D polymer networks constitute an important class of materials with numerous applications, mainly in biotechnology and medicine, i.e. photodegradable scaffolds for 3D cell culture and tissue/organ reconstruction and photodegradable hydrogels for sensors and actuators. The synthesis of such materials involves the use of bifunctional photodegradable cross-linkers. Anseth's group employed a photodegradable ONB ester based diacrylate cross-linker to form hydrogels via free radical copolymerization with PEG acrylate or Michael-addition polymerization using thiol-functionalized four arm PEG stars. The mechanical integrity of the gels, as quantified by ductility, tensile toughness and shear strain to yield, was demonstrated as well as the light-induced degradation and erosion.139–141 Tsang et al. developed a photodegradable gelatin network using a bifunctional PEG-based cross-linker containing photo-labile ONB ester groups. The gel was accurately patterned, enabling the formation of structures down to the micrometer range. Furthermore, gelatin can be digested by cells and degrades in a suitable time frame for biological applications. The gelatin hydrogel facilitated the attachment, proliferation and elongation of both neonatal rat cardiac fibroblasts and cardiomyocytes. Micro-patterned structures, fabricated by UV lithography through a mask, have led to the alignment of cardiac fibroblasts and cardiomyocytes, mimicking the native myocardium, and rendered the gel an excellent candidate for use in tissue engineering and regenerative medicine.142 In a similar approach, Griffin et al. synthesized a series of PEG-macromers containing photo-labile ONB ester groups. The PEG hydrogels were used to encapsulate and release human mesenchymal stem cells, upon UV irradiation, without compromising the cell viability.143In an alternative approach, photo-labile divinyl functionalized cross-linkers based on ONB ester moieties were used in the synthesis of photodegradable PMMA microgels by miniemulsion polymerization. The photodegradable PMMA microgels were investigated in the light-induced release of active compounds.144 Dual-responsive microgels have gained extensive attention for use in chemical and biomedical applications. ONB ester-based photo-labile cross-linkers have been employed in the synthesis of light and temperature or pH dual stimuli-responsive microgels prepared by redox initiated precipitation polymerization or radical inverse miniemulsion polymerization, respectively. The thermo-responsive behavior was introduced by the copolymerization of the photodegradable cross-linker with N-vinyl-caprolactam and N-hydroxymethyl acrylamide, whereas pH-sensitivity was introduced by the copolymerization of the photodegradable cross-linker with hydroxyethyl methacrylate and methacrylic acid. Changes in the solution temperature or pH caused the reversible swelling of the microgel, while UV irradiation induced the cleavage of the ONB ester cross-links leading to the disintegration of the microgel.145,146
Del Campo's group introduced an interesting route to a positive, a negative and a dual photoresist formulation. Photo-cleavable methoxynitrobiphenyl and p-dialkylaminonitrobiphenyl bearing molecules, mixed with commercially available di-/tri-isocyanates, di/tri alcohols and/or diamines, were spin coated on glass substrates. By tuning the irradiation wavelength on the films, sequential photo-activation of independent cross-linking and depolymerization processes is induced, producing 2D and 3D micro-patterns, by single and two photon excitation, respectively.147,148
Besides the ONB ester-based (meth)acrylate cross-linkers which are suitable for radical, anionic and cationic polymerizations, ONB ester moieties have been also employed in the synthesis of cross-linkers for ring opening metathesis polymerization (ROMP). In this approach, cross-linkers comprising two norbornene units bound onto a photo-labile ONB ester group were designed and used in organogel and hydrogel synthesis. The photo-sensitivity of the cross-linker depended on the stability of the intermediate benzylic radical formed upon cleavage of the ONB ester group.149,150 On the other hand, Suyama et al. proposed novel photodegradable cross-linkers bearing o-acryloxime photo-labile groups. Cross-linked thin films were prepared by the photopolymerization of methyl acrylate with the photo-labile cross-linkers using diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide as the photoinitiator and UV light above 310 nm. The photolysis of the O-cyloxime units in the polymer networks at 254 nm was confirmed by GPC and UV and FTIR spectroscopies.151
It is obvious from the above literature examples that the ONB ester moiety is the most popular photo-labile group in polymer science despite its severe disadvantages including the generation of by-products which absorb light competitively with the cleavage groups and are potentially toxic and the UV light photolysis which limits the penetration depth of the process. To overcome these drawbacks, the development of alternative photo-labile chemistries, i.e. acetals and ketals, which exhibit low photo-degradation thresholds in one-photon or multiphoton processes, is essential for bio-related applications.
Electro-responsive polymers
Electro-active polymers (EAPs) are materials that respond to an applied electric field by changing their size or shape. EAPs have attracted rapidly expanding scientific and technological interest lately due to their potential applications in sensors and actuators, robotics and artificial muscles, optical systems, drug delivery, space, ocean and energy harvesting applications. They are divided into two types: the ionic EAPs, in which the electro-responsiveness is a result of an electric field driven mobility of free ions to create a change in the local concentration of the ions in solution or within the material, and the dielectric EAPs, whose deformation is induced by electrostatic (Coulombic) forces developed between two electrodes. The first type includes the conducting polymers, which are outside the scope of the present review, the ionic polymers and polymer gels, as well as the so-called ionic polymer–metal composites (IPMCs) and the second category comprises the dielectric elastomers and the electrostrictive polymers. Ionic EAPs require low actuation voltages but suffer low deformations and response rates, while they also normally operate under wet conditions, whereas dielectric EAPs exhibit fast response and high deformations and operate under dry conditions, but require high activation fields.152 Main- and side-chain liquid crystalline polymers are another type of electro-responsive materials. However, the alignment of the liquid crystalline domains leads to a weak electro-stimulated effect and the addition of ionic or electronic compounds into these polymers is often employed as a means to maximize their response.153IPMCs are the most extensively studied electro-active materials because they combine low activation voltages with large bending strains, which is very attractive for use in actuators and sensors, and good biocompatibility for biomedical applications. These composites are based on ion exchange polymer membranes bearing covalently bound carboxylate, sulfonate or ammonium ionic groups along the polymer chain. Perfluorinated polymers (i.e. Nafions and Flemions) and styrenic copolymers with good mechanical properties and high ion mobility have been used.154 Sulfonated polyimides have also emerged as attractive candidates due to their self-metallization ability, which leads to the strong adherence of the silver electrodes onto the polymer electrolyte.155 The membrane thickness has been shown to affect the current and displacement of the actuator with larger displacements accessed for thinner membranes, whereas the electrolysis of water enhances further the properties of an IPMC.156
Non-ionic polymers, poly(vinylidene fluoride), polyacrylates and silicone have been also shown to respond to an external electric field in the absence of charge carriers and have been employed as electro-active materials.157 The potential of these systems for robotic and actuator applications was demonstrated in the study by Bhattacharya et al. who developed a poly(dimethyl siloxane)-based four finger micro-gripper resembling the human hand. The device could manipulate micro-objects from less than 1 mm in diameter, up to macro objects with a diameter of 10 mm, with very high accuracy.158 PVDF-based IPMCs have attracted much attention. An electro-actuated muscle was reported by Lu and coworkers based on a poly(styrene-alt-maleimide)/poly(vinylidene fluoride) blend in glycerol which responded at very low frequencies and high potentials in air. The excellent electro-responsive behavior of the blend and the lack of back relaxation under a constant voltage were attributed to the combination of ionic channels within the polymer and the inertia mass of the viscous solvent.159 Another electro-active membrane was developed based on a poly(vinylidene fluoride–trifluoroethylene–chlorofluoroethylene) terpolymer. Maximum displacements were achieved when the field was applied at the periphery of the membrane, causing its buckling.160 Finally, a hybrid electro-active polymer based on poly(vinylidene fluoride) and bacterial cellulose nanowhiskers was used to develop electroactive membranes by electrospinning. The membranes exhibited increased deformation capacity due to the synergistic effect of ion migration and electrochemical doping and have been proposed for use in robotics and sensors.161
The use of bacterial cellulose, which bears hydroxyl polar groups, as an attractive electro-active biopolymer in the development of actuators has been explored. Doping of bacterial cellulose membranes with LiCl or an ionic and a conductive polymer was shown to control the crystallinity and stiffness of the polymer and to lead to much larger bending deformations, rendering the system attractive for use in biomedical applications.162,163
Sulfonated polystyrenes are another type of polymer ionomers used commonly in the development of actuators. Poly(styrene sulfonate-co-ethylene) random copolymers were used for the fabrication of IPMCs. A novel method to increase the bending response of these sulfonated poly(styrene-co-ethylene) random copolymers was proposed via the UV induced grafting of vinyl silane onto the ethylene repeat units followed by the cross-linking of the silane moieties using heat. The cross-linked membranes exhibited even lower water uptake and better microphase separation, which increased the ion channels and facilitated ion migration, thus improving their actuation performance. The good mechanical properties and ion exchange capacities of the ionomer membranes are particularly attractive for use in electromechanical actuation.164,165 The electro-actuation of a sodium-4-vinylbenzenesulfonate-hydroxyethyl methacrylate-acrylonitrile copolymer was tested in phosphate buffer solution and Dulbecco's Modified Eagle's Medium used in cell culture. The response characteristics of the copolymer, exhibiting low voltage and fast response in biologically relevant media, render it a good candidate for the design of biological smart systems.166
In an intriguing approach, Cheedarala et al. designed a bioinspired soft actuator based on a 3D ionic membrane comprising naphthalene-tetracarboxylic dianhydride and sulfonated polyimide block copolymers held together by π–π stacks in an ionic liquid. The interconnected ion transport nanochannels in the membrane increased its ionic conductivity and ion exchange capacity, enabling large strains at low operating voltages compared to the currently available systems.167
Polyelectrolyte hydrogels have been extensively explored as electro-actuating materials for use in soft robotics. Bending, folding and cargo transportation, upon the application of an electric field in aqueous solutions, have been exploited. A re-distribution of the mobile ions in the system in response to the electric field creates osmotic pressure differences which cause the deformation of the gel, whereas the fixed gel charges dictate the direction of deformation and allow one to control the gel motion.168 Electro-active ionic hydrogels in the form of fibers, films and tubules have been extensively investigated for muscle and actuator technologies. For example, electro-responsive hydrogels comprising a semi-interpenetrating network of poly(acrylic acid) and poly(vinyl alcohol) were developed and loaded with a polyethyleneimine and 1-vinylimidazole blend as the electro-active species. The electro-responsive properties of the hydrogels were employed in the effective electro-induced drug release from the polymer and the matrix resilience following the stimulation.169 An alternative approach employed a poly(4-hydroxybutyl acrylate) gel bearing acrylic acid groups as the electro-responsive smart material that could be actuated at very low voltages. This method prevented bubble generation and increased the biocompatibility of the material, allowing its use in an actuating device in cell culture media to sort mouse embryoid bodies. Moreover, it was shown that the pluripotency and differentiation capabilities of the cells are not affected, which opens new pathways for the use of microfluidic actuators in cell biology.170 An interesting study was performed by Jackson et al. using electro-responsive hydrogels for the fabrication of a device to stimulate the occlusion of blood vessels. The hydrogels were based on a pluronic-bismethacrylate gel bearing methacrylic acid moieties as the electro-active groups. The electrically biased hydrogels at the optimum device configuration collapsed uniformly by 230% in salt solution and 320% in Krebs solution compared to the non-biased analogues.171,172 Even hydrolyzed polyacrylamide hydrogels in the form of high aspect ratio fibers have been shown to exhibit electro-active properties and deform under an electric field in salt solution due to the migration of mobile positive ions towards the negative electrode.173,174 Important knowledge of the actuation of electro-active ionic polymer gels has been gained by modelling the effect of the design parameters, such as electric voltage, thickness of the gel, concentration of polyion in the gel, ion concentration in the solution, and degree of cross-linking, on its bending deformation. The optimum conditions to maximize the gel deformation were verified experimentally using a poly(2-acrylamido-2-methylpropane sulfonic acid) hydrogel.175 Advances in gel electro-actuation have led to the design of smart hydrogel walkers. An intriguing approach for the development of walking gel actuators consisted of two oppositely charged gel legs based on an acrylamide polymer network copolymerized with sodium acrylate and quaternized DMAEMA, respectively (Fig. 6). The bending ability of the gels was found to depend on both the mobile charges within the gels and the salt concentration in the surrounding solution. Maximum bending and unidirectional motion was achieved when the osmotic pressure was maximized, i.e. at high effective charge densities of the gels and dilute salt solutions as opposed to high ionisable monomer repeat unit content and concentrated salt solutions.168 Another beautiful example of hydrogel walkers was reported by Yang et al. who used a polyanionic acrylamide gel with 2-methylpropanesulfonic acid ionisable groups, for the transfer of cargo upon the repeatable “on–off” triggering of the gel by an electric field. The arc-loop shaped hydrogel possessed two legs allowing it to walk on the surface and transport its cargo.176
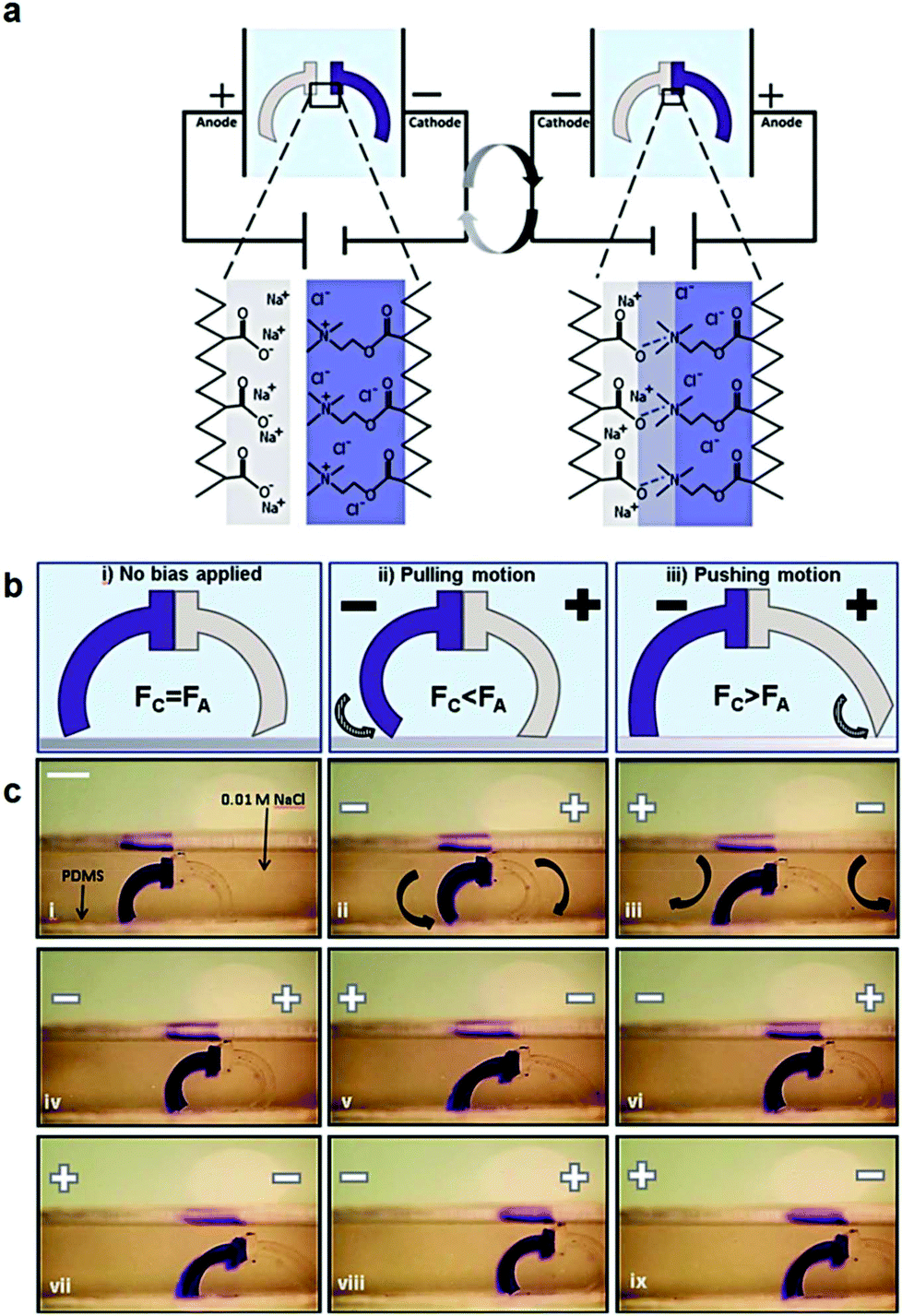 | ||
| Fig. 6 (a) Schematic representation of the charged gel walkers. Under the influence of an electric field, the charged polymer chains across the cationic/anionic gel interface move towards the oppositely charged electrodes, creating strong polyion complexes which promote the gel parts adhesion. On the other hand, reversal of the electric field causes the separation of the gel parts. (b) The intensity of the electric field causes different modes of actuation of the gel walkers. Fc and Fa are the friction forces of the cationic (dark blue) and anionic (light grey) legs, respectively. (c) Photographs of the motion of the gel walkers under a 5 V cm−1 field. Scale bar = 5 mm. Reproduced from ref. 168 with permission from the Royal Society of Chemistry. | ||
An alternative electro-responsive system comprises polyelectrolyte brushes that exhibit tunable swelling behavior upon application of an electric voltage. Anionic poly(acrylic acid) or poly(acrylic acid)-polystyrene gradient copolymer brushes became swollen upon the application of a negative voltage on the substrate, whereas a positive voltage induced a weak brush collapse which was proportional to the applied voltage. Such electro-active polymer brushes are attractive for use as valves or to promote biomolecule adhesion as a function of the electric stimulus.177 The effect of salt concentration and chain stiffness on the electro-responsive behavior of polyelectrolyte brushes has been investigated by Cao et al. In the presence of salt, the application of strong electric fields led to the mobility of a significant amount of anions and cations towards the two opposite electrodes, thus creating an electric field in the direction opposite to the external electric field, which hindered the stretching and shrinkage of the brush. Moreover, the internal electric field became stronger at higher salt concentrations and thus the brush extension or shrinkage decreased further. On the other hand, for the same salt concentration the stiff brushes exhibited a weaker response to the electric field compared to the flexible polymer chains. These findings have a significant impact on the use of brushes for the development of smart valves in nanofluidics for biological and other applications.178
Finally, electro-responsive drug carriers with high responsiveness and drug loading are attractive platforms for use in remote drug delivery. In a pioneering study, Wood et al.179 prepared layer-by-layer films by combining cationic PEI and small anionic Prussian blue nanoparticles, with an average size of around 4 nm, and anionic 14C-dextran sulfate. The electro-active Prussian blue particles were oxidized at mild potentials to their neutral form, which destabilized the film and released the drug. Similar electro-active multilayer films were prepared using the Prussian Blue nanoparticles and a positively antibiotic, gentamicin.180 In all these cases the release of the drug was controlled by the film thickness and the applied field. The disadvantage of this system is the instability of Prussian blue under alkaline conditions, which led to partial non-specific release of the drug and to a shorter lifetime of the film. The increase of the stability of the film was proposed to facilitate its use in long term treatment. In a similar approach, electro-responsive multilayer films employing cationic and anionic derivatives of organometallic poly(ferrocenylsilanes) have been used to release two different dyes, Alexa 488-labeled dextran and tetramethylrhodamine-labeled dextran, from the films upon the electro-triggered reversible redox reaction of iron between ferrocene and ferrocenium, which caused the disassembly of the films.181
Electro-responsive nanoparticulate drug carriers in the form of polymer micelles or microgel particles are particularly interesting for the local accumulation and release of therapeutics. Electro-responsive micellar systems were assembled from poly(ethylene imine) with a ferrocene end-group and encapsulated pyrene, as a model hydrophobic molecule, located at the core of the micelles.182 Application of a small electric field (+0.5 V) signal led to the oxidation of the ferrocene group and to a hydrophobic-to-hydrophilic transition in the core which released the encapsulated compound. In another example, Yuan's group prepared redox-responsive polymer micelles based on host–guest interactions between β-CD functionalized PEG and ferrocene functionalized polylactide or poly(ε-caprolactone). The application of an electric field led to the oxidation of ferrocene to ferrocenium and the de-threatening of the two polymers causing the micelle disassembly. The process is fully reversible upon the application of an opposite reductive voltage which leads to the re-assembly of the micelles enabling the redox-controlled release of a model anticancer drug, paclitaxel.183,184 Ying et al. prepared microgel particles by emulsion copolymerization of DMAEMA, sodium 4-vinylbenzene sulfonate, styrene, acrylate-poly(ethylene glycol)-N-hydroxysuccinimidylester and N,N′-methylene bisacrylamide as the cross-linker. The particles were surface-functionalized with the angiopep-2 ligand, which promoted their brain accumulation and exhibited an electro-responsive behavior upon the application of an electric field, which led to the release of the antiepileptic drug phenytoin sodium both in vitro and in vivo.185
During the last decade, great advances have emerged in the field of electro-active polymers. Polymer actuators and walkers as well as electro-responsive drug delivery systems have been developed and their properties and functions have been studied. Moreover, increased market opportunities for electroactive materials and systems are foreseen. However, further effort is required in the design and synthesis of novel electro-actuated polymers, using orthogonal chemistries, as well as in the construction of novel structures with significantly increased response rates and magnitude to further extend the potential applications of these materials.
Ultrasound-responsive polymers
Ultrasound is the high-frequency waves created by mechanical oscillations of a piezoelectric material when an alternating current is applied. Ultrasound is used in many different fields including ultrasonic devices for object detection and distance measurements, for imaging in medicine and for cleaning, mixing and accelerating chemical processes in industry. The ultrasound frequencies used in the aforementioned applications exceed the threshold of audible human hearing, which is approximately 20 kHz. Ultrasounds are categorized as low (<1 MHz), medium (1–5 MHz) and high (5–10 MHz) frequency ultrasounds. Low frequency ultrasounds have greater penetration depth in tissues and are non-invasive, but they cannot be focused into a small spot and thus, in general, a large focus point is produced. On the other hand, the tissue penetration depth for ultrasounds with frequencies higher than 2 MHz is relatively poor (ten times lower than that of a 200 kHz ultrasound) because of the increased scattering, which is proportional to the increase of the frequency, leading to heating and tissue damage. However, as the frequency increases, the focus point of the ultrasound becomes smaller and the intensity at the focal spot becomes higher, while away from the focal spot the intensity is low and acceptable for the human body. Therefore, high-intensity focused ultrasound (HIFU, frequency range 0.8–3.5 MHz) is emerging as a very interesting external trigger for cancer treatment either by direct application or via the controlled release of anticancer drugs from nanocarriers.The interaction of ultrasounds with tissue fluids can induce thermal and non-thermal effects. The thermal effect can be defined as the transfer of acoustic energy to the tissues, causing a rise of temperature. Heating is intensity and focus dependent. The non-thermal effects of ultrasounds, known as the cavitation effect, are defined as the formation of tiny gas bubbles in the tissues as the result of ultrasound vibration. At low ultrasonic intensities (1–3 W cm2), the bubbles oscillate around some equilibrium size, for many acoustic cycles. At high ultrasonic intensities (>10 W cm2) the bubbles expand after a few acoustic cycles to a radius that is at least twice their initial size, before collapsing violently on compression. This phenomenon is known as transient cavitation. Transient bubble collapse is considered the main source for the chemical and mechanical effects of ultrasonic energy. Each collapsing bubble can be considered as a microreactor in which a shock wave is generated, resulting in a local increase of temperature and pressure. These extremely harsh conditions can temporally increase the cell membrane permeability, resulting in increased drug uptake by the cells and can also disassemble nanocarriers, triggering the release of therapeutic payloads.
Low frequency ultrasounds (typically 20 kHz) have been used in mechano-responsive dendritic186 and polymeric187–189 organogels. Ultrasounds can induce gel-to-gel transitions or can cleave bonds, to transform an organogel into a soluble polymer.190 Schaefer et al. performed an extensive study of the effect of the molecular weight and the contour length of a polymer on its chain-scission during ultra-sonication. PS and poly(norbornene imide alkyne) homopolymers were subjected to pulsed 20 kHz ultrasound waves in order to investigate the chain-scission, and they found a cutoff degree of polymerization below which the polymer chains do not degrade.191 The same ultrasound waves were used to isomerize the SP moieties in a spiropyran–fluorene alternating copolymer. The ultrasound induced SP-to-MC isomerization led to a color change enabling it to modulate the electronic properties of the polymer in a remote manner.192 Chen et al. demonstrated the formation of dual ultrasound- and pH-responsive polymer vesicles from a PEO43-b-poly(2-diethylamino ethyl methacrylate33-co-2-tetrahydrofuranyloxy ethyl methacrylate47) (PEO43-b-P(DEA33-co-TMA47)) diblock copolymer synthesized via ATRP using PEO43-Br as the macroinitiator. The dual-responsive polymer vesicles became smaller in size upon either 40 kHz ultrasound irradiation or by decreasing the solution pH. Moreover, the vesicles were non-cytotoxic at low concentrations (250 mg mL−1) and enabled the controlled release of a loaded anticancer drug by both triggers.193
Lately, HIFU technology has attracted a great deal of attention because it allows external spatiotemporal control for targeted treatment. The HIFU beams are focused to a very small volume, which renders HIFU a promising trigger for targeted drug delivery applications. At the focal point, the ultrasonic intensity is very high whereas at the other areas it remains very weak. The HIFU technology was applied for the first time in humans in 1960, but did not gain clinical acceptance until the 1990s. HIFU treatment has been widely used as a non-contact and remote control method in medical treatment. Compared to other stimuli, HIFU is an ideal trigger because it can be focused and its characteristics can be tuned in a remote manner. The biggest challenge in HIFU technology is the development of novel ultrasound-responsive polymeric materials for the fabrication of optimal drug delivery systems that can target and release their cargo in a controlled fashion. Rapoport's group used a Pluronic P-105 copolymer to form DOX loaded micelles in water. The localized release of the anticancer drug at the tumor site was triggered by focused 1 MHz ultrasound, and the intracellular drug uptake by several types of cancer cells including ovarian carcinoma, breast cancer and promyelocytic leukemia cells as well as by nu/nu male and female mice with internal A2780 tumors was demonstrated. The authors claim that ultrasound irradiation has multiple functions in drug targeting to tumor cells. It enhances drug diffusion through the tumor interstitium, triggers drug release from the micelles in the tumor volume and increases the intracellular uptake of both released and encapsulated drugs at the irradiation site.194–199
In another approach, Zhang et al. prepared a block copolymer comprising a hydrophilic PEO block and a biodegradable, hydrophobic poly(lactic acid) block and employed HIFU to release the entrapped Nile red dye from the core of the spherical PEO-b-PLA micelles in water. The successful tuning of the release profile of the dye by adjusting the duration, intensity and location of the HIFU was verified. The irreversible release of Nile red from the PLA-b-PEG micelles was attributed to the disruption of the micellar structures due to the degradation of the PLA-b-PEG chains by the transient cavitation effect in the HIFU (1.1 MHz) focal spot.200 Zhao and coworkers employed the ultrasound sensitive 2-tetrahydropyranyl methacrylate (THPMA) monomer for the synthesis of the amphiphilic block copolymer. A PEO-b-THPMA block copolymer was synthesized which formed micelles comprising a PTHPMA core and a PEO corona in water. HIFU irradiation induced the cleavage of the tetrahydropyranyl groups to produce a hydrophilic poly(acrylic acid) block, causing the destabilization of the micelles and the release of their cargo (Fig. 7).201 In another approach, a PEO-b-poly(2-(2-methoxyethoxy) ethyl methacrylate-co-THPMA) block copolymer was synthesized which formed micelles above the LCST of the poly(2-(2-methoxyethoxy) ethyl methacrylate) block in water. Ultrasound irradiation of the micellar solution induced the cleavage of the tetrahydropyranyl groups to form acrylic acid moieties, which led to an increase of the LCST of the core forming block and the disassembly of the micelles.202
 | ||
| Fig. 7 (a) Ultrasound triggered cleavage of the hydrophobic tetrahydropyranyl groups to produce hydrophilic methacrylic acid moieties. (b) Schematic diagram of the HIFU setup consisting of: 1. ultrasound generator, 2. acoustic lens transducer, 3. ultrasonic beam, 4. focal spot, 5. water bath, 6. tube reactor, 7. latex membrane, and 8. polymer micelles. Reproduced with permission. Copyright © 2009, American Chemical Society.201 | ||
Li et al. designed an ultrasound and redox dual-responsive polymer comprising a PEO block and a PLA block linked together with a disulfide junction. Disulfide bonds are cleaved in the reducing environment inside the cell by redox reagents such as glutathione (GSH), while they are stable in the oxidizing environment outside the cell. Furthermore, the disulfide bond has a relatively low dissociation energy (ES–S ∼ 268 kJ mol−1) and longer bond length (lS–S ∼ 2.03 Å) compared to the C–C bond (EC–C ∼ 347 kJ mol−1; lC–C ∼ 1.54 Å), which suggests that the –S–S– bonds can be easily cleaved by HIFU (1.1 MHz). As a result of this, the PEO-S-S-PLA micelles were rapidly disrupted by HIFU in a remote manner at room temperature while they slowly disintegrated into unimers in the presence of GSH.203 In a different approach, Liu et al. investigated the thermal effects induced by HIFU (1.1 MHz) on solid polymers. A range of different homopolymers were investigated, including high density polyethylene, low density polyethylene, linear low density polyethylene, polypropylene, polyamide 6, PS and PMMA. The temperature fluctuations of the polymeric samples were recorded by using an infrared camera and suggested that HIFU can cause rapid heating of the sample in a spatiotemporally controlled manner leading to different chemical effects for each polymer.204 The same group studied the HIFU induced thermal effect and shape recovery characteristics of a cross-linked poly(methyl methacrylate-co-butyl acrylate) copolymer. The thermal behavior of the sample was similar to that discussed above for the homopolymers; however, the thickness of the sample had a strong influence on its thermal and shaper recovery properties.205
The above studies suggest that HIFU is a very appealing and promising technology for the spatiotemporally controlled release of therapeutic payloads from nanocarriers. The development of novel polymeric materials exhibiting superior HIFU degradation characteristics is required to produce more reliable ultrasound-responsive polymeric nanoparticles for controlled drug delivery applications.206–208
Magneto-responsive polymers
The development of magnetic nanomaterials has been the source for the discovery of spectacular new phenomena, with potential applications in numerous fields. During the last decade, among the broad spectrum of nanomagnetic materials investigated for various environmental and biomedical applications, organometallic (co)polymers have gained significant attention due to their ability to combine the valuable properties of metals and organic polymers.Rajca et al. prepared for the first time an organic π-conjugated polymer with very large magnetic moment and magnetic order at low temperatures. The polymer possesses alternating connectivity of radical molecules with unequal spin quantum numbers (S) and a large density of crosslinks which allow large net S values for either ferromagnetic or antiferromagnetic exchange coupling between modules. The magnetic characteristics of the polymer include blocking of magnetization and very large magnetic moments at temperatures below 10 K.209 In an intriguing approach, Tew's group synthesized oxanorbornene-based cobalt-containing diblock copolymers via ROMP (Fig. 8). These block copolymers formed microphase separated thin films with cylindrical, lamellar or inverted cylindrical domains. Room temperature ferromagnetic (RTF) materials that retained the block copolymer morphology were generated upon a simple heat treatment of the nanostructured block copolymer thin films. The ordering of the block copolymer film was essential for the ferromagnetic behavior since the parent cobalt containing homopolymer was superparamagnetic even though it contained more cobalt. The RTF properties were attributed to the confinement of the cobalt within the one-dimensional cylinders of the BCP morphology.210,211 In a related study, the same group synthesized metal-containing oxanorbornene diblock copolymers, via ROMP, consisting of an alkyl functionalized block and a block bearing cobalt and ferrocene functionalities. They showed that the RTF properties of the nanostructured block copolymer thin films were affected by the cobalt density within the cylindrical domains and thus the molar ratio of the Co- and Fe-based units in the second block.212
 | ||
| Fig. 8 Magnetic characterization of the oxanorbornene block copolymer (left) and the homopolymer analogue (right). The annealed block copolymer films are ferromagnetic (a, c), whereas the annealed homopolymer thin films are paramagnetic (b, d) measured by using a quantum interference magnetometer. TEM images of the thermally annealed thin films of the block copolymer (f) and the homopolymer (g) show the Co nanoparticles in the cylindrical microdomain morphology of the BCP and distributed randomly in the homopolymer. The scale bar is 100 nm for the low magnification images and 20 nm for the magnified images. Reproduced with permission from ref. 206, Copyright © 2011, Nature Publishing Group. | ||
In another example, Mukherjee et al. synthesized a multifunctional norbornene-based triblock copolymer comprising cobalt, pyrene and PEG-folate motifs as pendant functionalities. PEG served as the hydrophilic block to stabilize the self-assembled polymer nanostructures in aqueous media, whereas the folate functionality enables the effective accumulation of the nanostructures in tissues exposing the folate receptor. Pyrene was used for fluorescence imaging and cobalt facilitated magnetic targeting and imaging using the magnetic resonant imaging (MRI) technique. Cell viability studies verified the biocompatibility of the copolymer.213 In a related study, norbornene-based triblock copolymers comprising DOX, a cobalt carbonyl complex and PEG-biotin as pendant functionalities were synthesized and assembled into capsule-like nanostructures in aqueous media. The nanocarriers entered rapidly into the cancer cells due to the presence of biotin and released DOX in the acidic intracellular environment, leading to an improved drug efficacy. The cobalt complexes enabled the magnetic imaging of the nanocarriers by MRI, as discussed above, rendering them attractive candidates for theranostic applications.214 Finally, Jiang et al. prepared a series of alkyne functionalized poly(4-(phenylethynyl)styrene)-b-PEO-b-poly(4-(phenylethynyl)styrene) (PPES-b-PEO-b-PPES) triblock copolymers by RAFT polymerization and introduced cobalt by the reaction of the alkyne functionalities with Co2(CO)8. The metallated copolymers were cross-linked and formed magnetic cobalt nanoparticles after heating at 120 °C. Immersion of the cross-linked copolymer in water produced a magnetic hydrogel with tunable mechanical properties.215
The above examples verify the significant developments that have emerged in the field of magneto-responsive polymers during the last decade. The combination of the properties of the polymers with those of the metallic materials opens new avenues in multi-responsive and multi-functional systems for advanced applications. However, further effort is still required in the design and synthesis of all-organic magneto-responsive materials that will allow them to enter new unexplored fields.
Conclusions and perspectives
Field responsive materials that are sensitive to the application of a physical stimulus such as light irradiation, an electric or magnetic field and an ultrasound trigger have attracted much attention over the last years. Novel highly-functional polymers with controlled macromolecular characteristics have been attained in this area using precisely controlled polymerization methods and highly-efficient orthogonal chemistries. Polymeric materials ranging from linear and graft polymers to polymer gels and surfaces and hybrid organic–inorganic composites have become available. The properties and functions of these materials upon the application of the appropriate stimulus have been extensively investigated in a broad range of media and for a wide variety of applications. There are numerous reports in the literature on photo-responsive and photodegradable materials for drug delivery and optical/electronic applications. Electroactive materials have emerged as a new and very promising area of research for the development of actuators and robotic systems. Advanced magnetic nanomaterials for use in microelectronics and biomedical applications have attracted much attention. Finally, ultrasound-responsive polymers are perhaps the most appealing systems for targeted and spatiotemporally controlled therapeutic applications.Despite the great advances in the field, there are still tremendous challenges to be overcome in this area and a great deal of effort is required to master the property–function relationship and use these materials in practical applications. The biocompatibility and potential biodegradability of these materials have not been adequately addressed. The synthesis of new biodegradable, field-responsive materials remains a challenge. In addition, the influence of the intensity of the applied fields on the encapsulated drugs and natural biomolecules in the body requires further attention in the biomedical context. On the other hand, the kinetics and magnitude of the stimuli-responsive processes are also of paramount importance for the use of these materials in certain applications i.e. switches, actuators and robotics. Another aspect that has not attracted the required attention so far is related to other physicochemical changes that can be induced in parallel by the applied stimulus, such as pH and temperature changes, which could be unfavorable for certain applications. Future developments in the field should focus on novel designs, syntheses and engineering that will allow one to address the above challenges and that will lead to elaborate materials and intelligent systems of outstanding complexity and functions.
Notes and references
- G. Pasparakis and M. Vamvakaki, Polym. Chem., 2011, 2, 1234–1248 RSC.
- D. Edinger and E. Wagner, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2011, 3, 33–46 CrossRef CAS PubMed.
- I. K. Park, K. Singha, R. B. Arote, Y. J. Choi, W. J. Kim and C. S. Cho, Macromol. Rapid Commun., 2010, 31, 1122–1133 CrossRef CAS PubMed.
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- T. Traitel, R. Goldbart and J. Kost, J. Biomater. Sci., Polym. Ed., 2008, 19, 755–767 CrossRef CAS PubMed.
- W. L. Zhang and H. J. Choi, Polymers, 2014, 6, 2803–2818 CrossRef CAS.
- W. T. S. Huck, Mater. Today, 2008, 11, 24–32 CrossRef CAS.
- P. Bawa, V. Pillay, Y. E. Choonara and L. C. du Toit, Biomed. Mater., 2009, 4, 022001 CrossRef PubMed.
- T. H. Qazi, R. Rai and A. R. Boccaccini, Biomaterials, 2014, 35, 9068–9086 CrossRef CAS PubMed.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2013, 65, 10–16 CrossRef CAS PubMed.
- J. K. Chen and C. J. Chang, Materials, 2014, 7, 805–875 CrossRef CAS.
- J. F. Hester, S. C. Olugebefola and A. M. Mayes, J. Membr. Sci., 2002, 208, 375–388 CrossRef CAS.
- J. A. Howarter and J. P. Youngblood, Adv. Mater., 2007, 19, 3838–3843 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS.
- B. Le Droumaguet and K. Velonia, Macromol. Rapid Commun., 2008, 29, 1073–1089 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- L. Jia and M. H. Li, Liq. Cryst., 2014, 41, 368–384 CrossRef CAS.
- J. H. Hu, H. Yu, L. H. Gan and X. Hu, Soft Matter, 2011, 7, 11345–11350 RSC.
- S. L. Lin, Y. Y. Wang, C. H. Cai, Y. H. Xing, J. P. Lin, T. Chen and X. H. He, Nanotechnology, 2013, 24, 085602 CrossRef PubMed.
- Q. A. Jin, G. Y. Liu, X. S. Liu and J. A. Ji, Soft Matter, 2010, 6, 5589–5595 RSC.
- G. Y. Shen, G. S. Xue, J. Cai, G. Zou, Y. M. Li and Q. J. Zhang, Soft Matter, 2013, 9, 2512–2517 RSC.
- D. Hu, Y. F. Li, Y. L. Niu, L. Li, J. W. He, X. Y. Liu, X. N. Xia, Y. B. Lu, Y. Q. Xiong and W. J. Xu, RSC Adv., 2014, 4, 47929–47936 RSC.
- R. B. Wei, J. Y. Ma, H. X. Zhang and Y. N. He, J. Appl. Polym. Sci., 2016, 133, 43695 Search PubMed.
- R. B. Wei, X. G. Wang and Y. N. He, Chin. Chem. Lett., 2015, 26, 857–861 CrossRef CAS.
- G. X. Huang, J. Zhu, Z. B. Zhang, W. Zhang, N. C. Zhou and X. L. Zhu, J. Macromol. Sci., Part A: Pure Appl. Chem., 2013, 50, 193–199 CrossRef CAS.
- J. Hu, X. Wang and S. H. Zheng, Polym. Adv. Technol., 2012, 23, 1590–1595 CrossRef CAS.
- E. Blasco, B. V. K. J. Schmidt, C. Barner-Kowollik, M. Pinol and L. Oriol, Polym. Chem., 2013, 4, 4506–4514 RSC.
- W. J. Guo, T. S. Wang, X. D. Tang, Q. Zhang, F. Q. Yu and M. S. Pei, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2131–2138 CrossRef CAS.
- C. J. Chen, G. Y. Liu, X. S. Liu, D. D. Li and J. Ji, New J. Chem., 2012, 36, 694–701 RSC.
- R. J. Dong, Y. Liu, Y. F. Zhou, D. Y. Yan and X. Y. Zhu, Polym. Chem., 2011, 2, 2771–2774 RSC.
- S. Y. Dong, L. Y. Gao, J. Y. Li, D. H. Xu and Q. Z. Zhou, Polym. Chem., 2013, 4, 3968–3973 RSC.
- L. C. Liu, L. L. Rui, Y. Gao and W. A. Zhang, Polym. Chem., 2014, 5, 5453–5460 RSC.
- J. Yang, Z. T. Li, Y. J. Zhou and G. C. Yu, Polym. Chem., 2014, 5, 6645–6650 RSC.
- X. R. Zhou, Y. Du and X. G. Wang, ACS Macro Lett., 2016, 5, 234–237 CrossRef CAS.
- V. Marturano, P. Cerruti, C. Carfagna, M. Giamberini, B. Tylkowski and V. Ambrogi, Polymer, 2015, 70, 222–230 CrossRef CAS.
- R. P. Shen, B. Mu, P. C. Du and P. Liu, Soft Matter, 2011, 9, 382–392 CrossRef CAS.
- Y. Z. Yang, Q. Tang, C. B. Gong, X. B. Ma, J. D. Peng and M. H. W. Lam, New J. Chem., 2014, 38, 1780–1788 RSC.
- C. B. Gong, Y. Z. Yang, C. Gao, Q. Tang, C. F. Chow, J. D. Peng and M. H. W. Lam, J. Sol-Gel Sci. Technol., 2013, 67, 442–450 CrossRef CAS.
- X. F. Ma, R. Usui, Y. Kitazawa, H. Kokubo and M. Watanabe, Polymer, 2015, 78, 42–50 CrossRef CAS.
- T. Ube, K. Takado and T. Ikeda, J. Mater. Chem. C, 2015, 3, 8006–8009 RSC.
- K. Peng, I. Tomatsu and A. Kros, Chem. Commun., 2010, 46, 4094–4096 RSC.
- L. P. Zhou, J. X. Li, Q. Luo, J. Y. Zhu, H. X. Zou, Y. Z. Gao, L. Wang, J. Y. Xu, Z. Y. Dong and J. Q. Liu, Soft Matter, 2013, 9, 4635–4641 RSC.
- M. W. Wang, X. J. Zhang, L. Li, J. Y. Wang, J. Wang, J. Ma, Z. Y. Yuan, S. F. Lincoln and X. H. Guo, Macromol. Mater. Eng., 2016, 301, 191–198 CrossRef CAS.
- M. L. Smith, K. M. Lee, T. J. White and R. A. Vaia, Soft Matter, 2014, 10, 1400–1410 RSC.
- Y. Zhu, Y. Q. Zhou, Z. Chen, R. Lin and X. G. Wang, Polymer, 2012, 53, 3566–3576 CrossRef CAS.
- Y. Zhu, Y. Q. Zhou and X. G. Wang, Dyes Pigm., 2013, 99, 209–219 CrossRef CAS.
- M. Moritsugu, T. Ishikawa, T. Kawata, T. Ogata, Y. Kuwahara and S. Kurihara, Macromol. Rapid Commun., 2011, 32, 1546–1550 CrossRef CAS PubMed.
- Y. Kuwahara, M. Kaji, J. Okada, S. Kim, T. Ogata and S. Kurihara, Mater. Lett., 2013, 113, 202–205 CrossRef CAS.
- C. Q. Qin, Y. Y. Feng, W. Luo, C. Cao, W. P. Hu and W. Feng, J. Mater. Chem. A, 2015, 3, 16453–16460 CAS.
- S. Pandey, B. Kolli, S. P. Mishra and A. B. Samui, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1205–1215 CrossRef CAS.
- X. Q. Xue, J. A. Zhu, Z. B. Zhang, N. C. Zhou and X. L. Zhu, React. Funct. Polym., 2010, 70, 456–462 CrossRef CAS.
- R. Y. Zhao, X. P. Zhan, J. Yao, G. N. Sun, Q. D. Chen, Z. Q. Xie and Y. G. Ma, Polym. Chem., 2013, 4, 5382–5386 RSC.
- R. Barille, R. Janik, S. Kucharski, J. Eyer and F. Letournel, Colloids Surf., B, 2011, 88, 63–71 CrossRef CAS PubMed.
- N. Hurduc, A. Macovei, C. Paius, A. Raicu, I. Moleavin, N. Branza-Nichita, M. Hamel and L. Rocha, Mater. Sci. Eng., C, 2013, 33, 2440–2445 CrossRef CAS PubMed.
- L. Rocha, C. M. Paius, A. Luca-Raicu, E. Resmerita, A. Rusu, I. A. Moleavin, M. Hamel, N. Branza-Nichita and N. Hurduc, J. Photochem. Photobiol., A, 2014, 291, 16–25 CrossRef CAS.
- X. J. Liu, M. R. Cai, Y. M. Liang, F. Zhou and W. M. Liu, Soft Matter, 2011, 7, 3331–3336 RSC.
- L. Chen, C. L. He, Y. E. Huang, J. B. Huang, Y. Z. Zhang and Y. Gao, J. Appl. Polym. Sci., 2016, 133, 43540 Search PubMed.
- S. L. Feng, Y. P. Hou, Y. Xue, L. C. Gao, L. Jiang and Y. M. Zheng, Soft Matter, 2013, 9, 9294–9297 RSC.
- R. Alvarez-Rodriguez, F. J. Arias, M. Santos, A. M. Testera and J. C. Rodriguez-Cabello, Macromol. Rapid Commun., 2010, 31, 568–573 CrossRef CAS PubMed.
- D. B. Pacardo, B. Neupane, S. M. Rikard, Y. Lu, R. Mo, S. R. Mishra, J. B. Tracy, G. F. Wang, F. S. Ligler and Z. Gu, Nanoscale, 2015, 7, 12096–12103 RSC.
- L. Florea, D. Diamond and F. Benito-Lopez, Macromol. Mater. Eng., 2012, 297, 1148–1159 CrossRef CAS.
- R. Byrne, C. Ventura, F. B. Lopez, A. Walther, A. Heise and D. Diamond, Biosens. Bioelectron., 2010, 26, 1392–1398 CrossRef CAS PubMed.
- C. Ventura, P. Thornton, S. Giordani and A. Heise, Polym. Chem., 2014, 5, 6318–6324 RSC.
- Q. A. Jin, G. Y. Liu and J. A. Ji, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2855–2861 CrossRef CAS.
- S. Y. Lee, H. Lee, I. In and S. Y. Park, Eur. Polym. J., 2014, 57, 1–10 CrossRef.
- D. S. Achilleos and M. Vamvakaki, Macromolecules, 2010, 43, 7073–7081 CrossRef CAS.
- D. S. Achilleos, T. A. Hatton and M. Vamvakaki, Langmuir, 2016, 32, 5981–5989 CrossRef CAS PubMed.
- D. S. Achilleos, T. A. Hatton and M. Vamvakaki, J. Am. Chem. Soc., 2012, 134, 5726–5729 CrossRef CAS PubMed.
- H. T. Nguyen, L. T. T. Nguyen and T. V. Le, Des. Monomers Polym., 2015, 18, 271–283 CrossRef CAS.
- G. Filipcsei, K. Sumaru, T. Takagi, T. Kanamori and M. Zrinyi, J. Mol. Liq., 2014, 189, 63–67 CrossRef CAS.
- M. Kojima, T. Nakanishi, Y. Hirai, H. Yabu and M. Shimomura, Chem. Commun., 2010, 46, 3970–3972 RSC.
- D. Wang, P. W. Jiao, J. M. Wang, Q. L. Zhang, L. Feng and Z. Z. Yang, J. Appl. Polym. Sci., 2012, 125, 870–875 CrossRef CAS.
- A. C. Pauly, K. Scholler, L. Baumann, R. M. Rossi, K. Dustmann, U. Ziener, D. de Courten, M. Wolf, L. F. Boesel and L. J. Scherer, Sci. Technol. Adv. Mater., 2015, 16, 034604 CrossRef.
- Q. J. Xing, N. J. Li, Y. Jiao, D. Y. Chen, J. Y. Xu, Q. F. Xu and J. M. Lu, RSC Adv., 2015, 5, 5269–5276 RSC.
- Z. Q. Cao, G. J. Wang, Y. Chen, F. X. Lang and Z. Z. Yang, Macromolecules, 2015, 48, 7256–7261 CrossRef CAS.
- E. Sato, Y. Masuda, J. Kadota, T. Nishiyama and H. Horibe, Eur. Polym. J., 2015, 69, 605–615 CrossRef CAS.
- Y. B. Long, W. X. Gu, C. C. Pang, J. B. Ma and H. Gao, J. Mater. Chem. B, 2016, 4, 1480–1488 RSC.
- M. Wang and J. C. Kim, Colloid Polym. Sci., 2013, 291, 2319–2327 CAS.
- J. Luo, Q. Zhou, J. Sun, J. Q. Jiang, X. Zhou, H. W. Zhang and X. Y. Liu, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4037–4045 CrossRef CAS.
- M. K. Kang and J. C. Kim, J. Biomater. Sci., Polym. Ed., 2014, 25, 843–854 CrossRef CAS PubMed.
- M. S. Lee and J. C. Kim, Colloid J., 2013, 75, 668–676 CrossRef CAS.
- M. S. Lee and J. C. Kim, Polym. Eng. Sci., 2014, 54, 227–233 CAS.
- J. Yang, W. D. He, C. He, J. Tao, S. Q. Chen, S. M. Niu and S. L. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3791–3799 CrossRef CAS.
- W. D. Ji, N. J. Li, D. Y. Chen, X. X. Qi, W. W. Sha, Y. Jiao, Q. F. Xu and J. M. Lu, J. Mater. Chem. B, 2013, 1, 5942–5949 RSC.
- S. J. Montgomery, G. Kannan, E. Galperin and S. D. Kim, Macromolecules, 2010, 43, 5238–5244 CrossRef CAS.
- M. Schraub, H. Gray and N. Hampp, Macromolecules, 2011, 44, 8755–8762 CrossRef CAS.
- M. Bender, K. M. Schelkle, N. Jurgensen, S. Schmid, G. Hernandez-Sosa and U. H. F. Bunz, Macromolecules, 2016, 49, 2957–2961 CrossRef CAS.
- K. M. Schelkle, M. Bender, S. Beck, K. F. Jeltsch, S. Stolz, J. Zimmermann, R. T. Weitz, A. Pucci, K. Mullen, M. Hamburger and U. H. F. Bunz, Macromolecules, 2016, 49, 1518–1522 CrossRef CAS.
- S. Li, B. A. Moosa, Y. Chen, W. G. Li and N. M. Khashab, J. Mater. Chem. B, 2015, 3, 1221–1229 Search PubMed.
- D. J. Shi, M. Matsusaki and M. Akashi, J. Controlled Release, 2011, 149, 182–189 CrossRef CAS PubMed.
- L. A. Wells, M. A. Brook and H. Sheardown, Macromol. Biosci., 2011, 11, 988–998 CrossRef CAS PubMed.
- P. Payamyar, M. Servalli, T. Hungerland, A. P. Schutz, Z. K. Zheng, A. Borgschulte and A. D. Schluter, Macromol. Rapid Commun., 2015, 36, 151–158 CrossRef CAS PubMed.
- J. F. Xu, Y. Z. Chen, L. Z. Wu, C. H. Tung and Q. Z. Yang, Org. Lett., 2013, 15, 6148–6151 CrossRef CAS PubMed.
- H. Xie, M. J. He, X. Y. Deng, L. Du, C. J. Fan, K. K. Yang and Y. Z. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 9431–9439 CAS.
- P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, Nat. Chem., 2014, 6, 774–778 CrossRef CAS PubMed.
- B. Byambaa, T. Konno and K. Ishihara, Colloids Surf., B, 2012, 99, 1–6 CrossRef CAS PubMed.
- K. M. Schelkle, S. Becht, S. Faraji, M. Petzoldt, K. Mullen, T. Buckup, A. Dreuw, M. Motzkus and M. Hamburger, Macromol. Rapid Commun., 2015, 36, 31–37 CrossRef CAS PubMed.
- Z. C. Smith, R. H. Pawle and S. W. Thomas, ACS Macro Lett., 2012, 1, 825–829 CrossRef CAS.
- Z. C. Smith, D. M. Meyer, M. G. Simon, C. Staii, D. Shukla and S. W. Thomas, Macromolecules, 2015, 48, 959–966 CrossRef CAS.
- S. W. Thomas, Macromol. Chem. Phys., 2012, 213, 2443–2449 CrossRef CAS.
- N. Fomina, C. McFearin, M. Sermsakdi, O. Edigin and A. Almutairi, J. Am. Chem. Soc., 2010, 132, 9540–9542 CrossRef CAS PubMed.
- N. Fomina, C. L. McFearin, M. Sermsakdi, J. M. Morachis and A. Almutairi, Macromolecules, 2011, 44, 8590–8597 CrossRef CAS PubMed.
- J. Olejniczak, V. A. N. Huu, J. Lux, M. Grossman, S. He and A. Almutairi, Chem. Commun., 2015, 51, 16980–16983 RSC.
- C. C. Zhu and C. J. Bettinger, Macromol. Rapid Commun., 2013, 34, 1446–1451 CrossRef CAS PubMed.
- J. Yin, H. B. Hu, Y. H. Wu and S. Y. Liu, Polym. Chem., 2011, 2, 363–371 RSC.
- J. Y. Fang, Y. K. Lin, S. W. Wang, Y. C. Li and R. S. Lee, React. Funct. Polym., 2015, 95, 46–54 CrossRef CAS.
- P. Sobolciak, M. Spirek, J. Katrlik, P. Gemeiner, I. Lacik and P. Kasak, Macromol. Rapid Commun., 2013, 34, 635–639 CrossRef CAS PubMed.
- A. Sinclair, T. Bai, L. R. Carr, J. R. Ella-Menye, L. Zhang and S. Y. Jiang, Biomacromolecules, 2013, 14, 1587–1593 CrossRef CAS PubMed.
- W. X. Xi, M. Krieger, C. J. Kloxin and C. N. Bowman, Chem. Commun., 2013, 49, 4504–4506 RSC.
- W. X. Xi, H. Y. Peng, A. Aguirre-Soto, C. J. Kloxin, J. W. Stansbury and C. N. Bowman, Macromolecules, 2014, 47, 6159–6165 CrossRef CAS PubMed.
- Z. J. Wang, M. Gao, J. B. Sun, D. H. Liang and X. R. Jia, Macromolecules, 2013, 46, 1723–1731 CrossRef CAS.
- M. A. Kostiainen, J. Kotimaa, M. L. Laukkanen and G. M. Pavan, Chem. – Eur. J., 2010, 16, 6912–6918 CrossRef CAS PubMed.
- M. A. Kostiainen, P. Ceci, M. Fornara, P. Hiekkataipale, O. Kasyutich, R. J. M. Nolte, J. J. L. M. Cornelissen, R. D. Desautels and J. van Lierop, ACS Nano, 2011, 5, 6394–6402 CrossRef CAS PubMed.
- A. Nazemi, T. B. Schon and E. R. Gillies, Org. Lett., 2013, 15, 1830–1833 CrossRef CAS PubMed.
- G. Pasparakis, T. Manouras, P. Argitis and M. Vamvakaki, Macromol. Rapid Commun., 2012, 33, 183–198 CrossRef CAS PubMed.
- T. Dispinar, C. A. L. Colard and F. E. Du Prez, Polym. Chem., 2013, 4, 763–772 RSC.
- C. Lv, Z. Wang, P. Wang and X. J. Tang, Int. J. Mol. Sci., 2012, 13, 16387–16399 CrossRef CAS PubMed.
- C. Lv, Z. Wang, P. Wang and X. J. Tang, Langmuir, 2012, 28, 9387–9394 CrossRef CAS PubMed.
- X. J. Deng, N. Zheng, Z. Y. Song, L. C. Yin and J. J. Cheng, Biomaterials, 2014, 35, 5006–5015 CrossRef CAS PubMed.
- L. Li, X. X. Deng, Z. L. Li, F. S. Du and Z. C. Li, Macromolecules, 2014, 47, 4660–4667 CrossRef CAS.
- X. Liu, Z. C. Tian, C. Chen and H. R. Allcock, Polym. Chem., 2013, 4, 1115–1125 RSC.
- D. H. Han, X. Tong and Y. Zhao, Macromolecules, 2011, 44, 437–439 CrossRef CAS.
- S. Y. Sun, E. A. Chamsaz and A. Joy, ACS Macro Lett., 2012, 1, 1184–1188 CrossRef CAS.
- G. Pasparakis, T. Manouras, A. Selimis, M. Vamvakaki and P. Argitis, Angew. Chem., Int. Ed., 2011, 50, 4142–4145 CrossRef CAS PubMed.
- G. Pasparakis, T. Manouras, M. Vamvakaki and P. Argitis, Nat. Commun., 2014, 5, 3623 Search PubMed.
- M. Kang and B. Moon, Macromolecules, 2009, 42, 455–458 CrossRef CAS.
- J. Y. Fang, S. W. Wang, Y. C. Li and R. S. Lee, J. Polym. Res., 2015, 22, 155 CrossRef.
- E. Cabane, V. Malinova and W. Meier, Macromol. Chem. Phys., 2010, 211, 1847–1856 CrossRef CAS.
- K. Y. Peng, S. W. Wang, M. Y. Hua and R. S. Lee, RSC Adv., 2013, 3, 18453–18463 RSC.
- R. S. Lee, Y. C. Li and S. W. Wang, Carbohydr. Polym., 2015, 117, 201–210 CrossRef CAS PubMed.
- R. S. Lee, S. W. Wang, Y. C. Li and J. Y. Fang, RSC Adv., 2015, 5, 497–512 RSC.
- P. Anilkumar, E. Gravel, I. Theodorou, K. Gombert, B. Theze, F. Duconge and E. Doris, Adv. Funct. Mater., 2014, 24, 5246–5252 CrossRef CAS.
- R. D. Cheng, J. G. Chen, Z. T. Liu, Z. W. Liu and J. Q. Jiang, Macromol. Rapid Commun., 2016, 37, 514–520 CrossRef CAS PubMed.
- K. K. Ding, L. Y. Shi, L. Zhang, T. Zeng, Y. H. Yin and Y. Yi, Polym. Chem., 2016, 7, 899–904 RSC.
- W. Y. Lu, C. Tian, P. Thogaripally, J. Hu and P. F. Wang, Chem. Commun., 2013, 49, 9636–9638 RSC.
- M. W. Tibbitt, B. W. Han, A. M. Kloxin and K. S. Anseth, J. Biomed. Mater. Res., Part A, 2012, 100, 1647–1654 CrossRef PubMed.
- M. W. Tibbitt, A. M. Kloxin and K. S. Anseth, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1899–1911 CrossRef CAS PubMed.
- M. W. Tibbitt, A. M. Kloxin, L. A. Sawicki and K. S. Anseth, Macromolecules, 2013, 46, 2785–2792 CrossRef CAS PubMed.
- K. M. C. Tsang, N. Annabi, F. Ercole, K. Zhou, D. J. Karst, F. Y. Li, J. M. Haynes, R. A. Evans, H. Thissen, A. Khademhosseini and J. S. Forsythe, Adv. Funct. Mater., 2015, 25, 977–986 CrossRef CAS PubMed.
- D. R. Griffin and A. M. Kasko, J. Am. Chem. Soc., 2012, 134, 17833–17833 CrossRef CAS.
- D. Klinger and K. Landfester, Soft Matter, 2011, 7, 1426–1440 RSC.
- S. S. Bian, J. Zheng and W. L. Yang, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1676–1685 CrossRef CAS.
- D. Klinger and K. Landfester, Macromolecules, 2011, 44, 9758–9772 CrossRef CAS.
- L. Garcia-Fernandez, C. Herbivo, V. S. Arranz, D. Warther, L. Donato, A. Specht and A. del Campo, Adv. Mater., 2014, 26, 5012–5017 CrossRef CAS PubMed.
- L. Garcia-Fernandez, A. Specht and A. del Campo, Macromol. Rapid Commun., 2014, 35, 1801–1807 CrossRef CAS PubMed.
- P. Gumbley, X. R. Hu, J. A. Lawrence and S. W. Thomas, Macromol. Rapid Commun., 2013, 34, 1838–1843 CrossRef CAS PubMed.
- X. R. Hu, J. F. Shi and S. W. Thomas, Polym. Chem., 2015, 6, 4966–4971 RSC.
- K. Suyama and H. Tachi, RSC Adv., 2015, 5, 31506–31513 RSC.
- L. J. Romasanta, M. A. Lopez-Manchado and R. Verdejo, Prog. Polym. Sci., 2015, 51, 188–211 CrossRef CAS.
- H. L. Liu, L. Q. Zhang, D. Yang, N. Y. Ning, Y. C. Yu, L. Yao, B. Y. Yan and M. Tian, J. Phys. D: Appl. Phys., 2012, 45, 485303 CrossRef.
- B. Bhandari, G. Y. Lee and S. H. Ahn, Int. J. Precis. Eng. Manuf., 2012, 13, 141–163 CrossRef.
- J. Song, J. H. Jeon, I. K. Oh and K. C. Park, Macromol. Rapid Commun., 2011, 32, 1583–1587 CrossRef CAS PubMed.
- Q. S. He, M. Yu, L. L. Song, H. T. Ding, X. Q. Zhang and Z. D. Dai, J. Bionic. Eng., 2011, 8, 77–85 CrossRef.
- A. Gugliuzza and E. Drioli, J. Membr. Sci., 2013, 446, 350–375 CrossRef CAS.
- S. Bhattacharya, B. Bepari and S. Bhaumik, Mech. Based Des. Struct. Mach., 2014, 42, 312–325 CrossRef.
- J. Lu, S. G. Kim, S. Lee and I. K. Oh, Macromol. Chem. Phys., 2011, 212, 635–642 CrossRef CAS.
- L. Engel, S. Kruk, J. Shklovsky, Y. Shacham-Diamand and S. Krylov, J. Micromech. Microeng., 2014, 24, 125027 CrossRef CAS.
- S. S. Kim and C. D. Kee, Int. J. Precis. Eng. Manuf., 2014, 15, 315–321 CrossRef.
- J. H. Jeon, I. K. Oh, C. D. Kee and S. J. Kim, Sens. Actuators, B, 2010, 146, 307–313 CrossRef CAS.
- W. Kunchornsup and A. Sirivat, Sens. Actuators, A, 2014, 220, 249–261 CrossRef CAS.
- X. L. Wang, I. K. Oh and T. H. Cheng, Polym. Int., 2010, 59, 305–312 CrossRef CAS.
- X. L. Wang, I. K. Oh and L. Xu, Sens. Actuators, B, 2010, 145, 635–642 CrossRef CAS.
- L. Migliorini, T. Santaniello, Y. S. Yan, C. Lenardi and P. Milani, Sens. Actuators, B, 2016, 228, 758–766 CrossRef CAS.
- R. K. Cheedarala, J. H. Jeon, C. D. Kee and I. K. Oh, Adv. Funct. Mater., 2014, 24, 6005–6015 CrossRef CAS.
- D. Morales, E. Palleau, M. D. Dickey and O. D. Velev, Soft Matter, 2014, 10, 1337–1348 RSC.
- S. Indermun, Y. E. Choonara, P. Kumar, L. C. du Toit, G. Modi, R. Luttge and V. Pillay, Int. J. Pharm., 2014, 462, 52–65 CrossRef CAS PubMed.
- G. H. Kwon, Y. Y. Choi, J. Y. Park, D. H. Woo, K. B. Lee, J. H. Kim and S. H. Lee, Lab Chip, 2010, 10, 1604–1610 RSC.
- N. Jackson, N. Cordero and F. Stam, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1523–1528 CAS.
- N. Jackson and F. Stam, J. Appl. Polym. Sci., 2015, 132, 41687 Search PubMed.
- M. Bassil, M. Ibrahim and M. El Tahchi, Soft Matter, 2011, 7, 4833–4838 RSC.
- P. J. Glazer, M. van Erp, A. Embrechts, S. G. Lemay and E. Mendes, Soft Matter, 2012, 8, 4421–4426 RSC.
- C. Jo, H. E. Naguib and R. H. Kwon, Smart Mater. Struct., 2011, 20, 045006 CrossRef.
- C. Yang, W. Wang, C. Yao, R. Xie, X. J. Ju, Z. Liu and L. Y. Chu, Sci. Rep., 2015, 5, 13622 CrossRef PubMed.
- O. V. Borisova, L. Billon, R. P. Richter, E. Reimhult and O. V. Borisov, Langmuir, 2015, 31, 7684–7694 CrossRef CAS PubMed.
- Q. Q. Cao, C. C. Zuo, L. J. Li and G. Yan, Biomicrofluidics, 2011, 5, 044119 CrossRef PubMed.
- K. C. Wood, N. S. Zacharia, D. J. Schmidt, S. N. Wrightman, B. J. Andaya and P. T. Hammond, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2280–2285 CrossRef CAS PubMed.
- D. J. Schmidt, J. S. Moskowitz and P. T. Hammond, Chem. Mater., 2010, 22, 6416–6425 CrossRef CAS PubMed.
- J. Song, D. Janczewski, Y. J. Ma, L. van Ingen, C. E. Sim, Q. L. Goh, J. W. Xu and G. J. Vancso, Eur. Polym. J., 2013, 49, 2477–2484 CrossRef CAS.
- Y. X. Sun, K. F. Ren, Y. X. Zhao, X. S. Liu, G. X. Chang and J. Ji, Langmuir, 2013, 29, 11163–11168 CrossRef CAS PubMed.
- A. C. Feng, Q. Yan, H. J. Zhang, L. Peng and J. Y. Yuan, Chem. Commun., 2014, 50, 4740–4742 RSC.
- L. Peng, A. C. Feng, H. J. Zhang, H. Wang, C. M. Jian, B. W. Liu, W. P. Gao and J. Y. Yuan, Polym. Chem., 2014, 5, 1751–1759 RSC.
- X. Y. Ying, Y. Wang, J. Liang, J. X. Yue, C. L. Xu, L. N. Lu, Z. H. Xu, J. Q. Gao, Y. Z. Du and Z. Chen, Angew. Chem., Int. Ed., 2014, 53, 12436–12440 CAS.
- Z. X. Liu, Y. Feng, Z. C. Yan, Y. M. He, C. Y. Liu and Q. H. Fan, Chem. Mater., 2012, 24, 3751–3757 CrossRef CAS.
- W. G. Miao, L. Qin, D. Yang, X. Jin and M. H. Liu, Chem. – Eur. J., 2015, 21, 1064–1072 CrossRef CAS PubMed.
- X. D. Yu, Q. A. Liu, J. C. Wu, M. M. Zhang, X. H. Cao, S. Zhang, Q. Wang, L. M. Chen and T. Yi, Chem. – Eur. J., 2010, 16, 9099–9106 CrossRef CAS PubMed.
- J. W. Fan, J. Zou, X. He, F. W. Zhang, S. Y. Zhang, J. E. Raymond and K. L. Wooley, Chem. Sci., 2014, 5, 141–150 RSC.
- Y. Q. Min, S. Y. Huang, Y. X. Wang, Z. J. Zhang, B. Y. Du, X. H. Zhang and Z. Q. Fan, Macromolecules, 2015, 48, 316–322 CrossRef CAS.
- M. Schaefer, B. Icli, C. Weder, M. Lattuada, A. F. M. Kilbinger and Y. C. Simon, Macromolecules, 2016, 49, 1630–1636 CrossRef CAS.
- M. Sommer and H. Komber, Macromol. Rapid Commun., 2013, 34, 57–62 CrossRef CAS PubMed.
- W. Q. Chen and J. Z. Du, Sci. Rep., 2013, 3, 2162 Search PubMed.
- Z. G. Gao, H. D. Fain and N. Rapoport, J. Controlled Release, 2005, 102, 203–222 CrossRef CAS PubMed.
- G. A. Husseini, G. D. Myrup, W. G. Pitt, D. A. Christensen and N. A. Y. Rapoport, J. Controlled Release, 2000, 69, 43–52 CrossRef CAS PubMed.
- A. Marin, H. Sun, G. A. Husseini, W. G. Pitt, D. A. Christensen and N. Y. Rapoport, J. Controlled Release, 2002, 84, 39–47 CrossRef CAS PubMed.
- N. Rapoport, Int. J. Pharm., 2004, 277, 155–162 CrossRef CAS PubMed.
- N. Y. Rapoport, D. A. Christensen, H. D. Fain, L. Barrows and Z. Gao, Ultrasonics, 2004, 42, 943–950 CrossRef CAS PubMed.
- N. Y. Rapoport, A. M. Kennedy, J. E. Shea, C. L. Scaife and K. H. Nam, J. Controlled Release, 2009, 138, 268–276 CrossRef CAS PubMed.
- H. J. Zhang, H. S. Xia, J. Wang and Y. W. Li, J. Controlled Release, 2009, 139, 31–39 CrossRef CAS PubMed.
- J. Wang, M. Pelletier, H. J. Zhang, H. S. Xia and Y. Zhao, Langmuir, 2009, 25, 13201–13205 CrossRef CAS PubMed.
- J. Xuan, O. Boissiere, Y. Zhao, B. Yan, L. Tremblay, S. Lacelle, H. S. Xia and Y. Zhao, Langmuir, 2012, 28, 16463–16468 CrossRef CAS PubMed.
- Y. W. Li, R. Tong, H. S. Xia, H. J. Zhang and J. A. Xuan, Chem. Commun., 2010, 46, 7739–7741 RSC.
- B. Liu, H. S. Xia, G. X. Fei, G. Li and W. R. Fan, Macromol. Chem. Phys., 2013, 214, 2519–2527 CrossRef CAS.
- G. Li, G. X. Fei, B. Liu, H. S. Xia and Y. Zhao, RSC Adv., 2014, 4, 32701–32709 RSC.
- Y. S. Kim, Int. J. Hyperthermia, 2015, 31, 225–232 CrossRef PubMed.
- R. Tanbour, A. M. Martins, W. G. Pitt and G. A. Husseini, Curr. Pharm. Des., 2016, 22, 2796–2807 CrossRef CAS PubMed.
- H. S. Xia, Y. Zhao and R. Tong, Adv. Exp. Med. Biol., 2016, 880, 365–384 CrossRef PubMed.
- A. Rajca, J. Wongsriratanakul and S. Rajca, Science, 2001, 294, 1503–1505 CrossRef CAS PubMed.
- Z. M. Al-Badri, R. R. Maddikeri, Y. P. Zha, H. D. Thaker, P. Dobriyal, R. Shunmugam, T. P. Russell and G. N. Tew, Nat. Commun., 2011, 2, 482 CrossRef PubMed.
- Y. P. Zha, R. R. Maddikeri, S. P. Gido and G. N. Tew, J. Inorg. Organomet. Polym. Mater., 2013, 23, 89–94 CrossRef CAS.
- Y. P. Zha, H. D. Thaker, R. R. Maddikeri, S. P. Gido, M. T. Tuominen and G. N. Tew, J. Am. Chem. Soc., 2012, 134, 14534–14541 CrossRef CAS PubMed.
- S. Mukherjee, H. Dinda, L. Shashank, I. Chakraborty, R. Bhattacharyya, J. Das Sarma and R. Shunmugam, Macromolecules, 2015, 48, 6791–6800 CrossRef CAS.
- S. Mukherjee, D. Patra, H. Dinda, I. Chalkraborty, L. Shashank, R. Bhattacharyya, J. Das Sarma and R. Shunmugam, Macromolecules, 2016, 49, 2411–2418 CrossRef CAS.
- B. Y. Jiang, W. L. Hom, X. Y. Chen, P. Q. Yu, L. C. Payelka, K. Kisslinger, J. B. Parise, S. R. Bhatia and R. B. Grubbs, J. Am. Chem. Soc., 2016, 138, 4616–4625 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |