
Photo-responsive polymers: synthesis and applications
Olivier
Bertrand
and
Jean-François
Gohy
*
Institute of Condensed Matter and Nanosciences (IMCN), Bio- and Soft Matter (BSMA), Université catholique de Louvain, Place L. Pasteur, 1, 1348 Louvain-la-Neuve, Belgium. E-mail: jean-francois.gohy@uclouvain.be
First published on 15th August 2016
Abstract
In this review, we highlight the progress realized in recent years on light-responsive polymers. More precisely, we provide some guidelines towards the rational design of photo-responsive block copolymers and we present the different photo-responsive moieties that have been used so far. We also discuss the different types of irreversible and reversible responses encountered by photo-responsive polymers. Finally, we discuss the application of light responsive polymers in material science.
 Olivier Bertrand (on the left) and Jean-François Gohy (on the right) | Olivier Bertrand got his PhD from Prof. Jean-François Gohy and Prof. Charles-André Fustin group (Université catholique de Louvain) in 2013, working on photo-responsive polymers. After his PhD thesis he obtained a Post-doctoral position in the group of Prof. David M. Haddleton (University of Warwick), followed by a position of Senior Researcher in the group Prof. Jean-François Gohy, working on redox sensitive polymers. Jean-François Gohy got his PhD from Prof. Robert Jérôme's group (University of Liège, Belgium) in 1999. He then obtained a “Chargé de Recherches” position from the Belgian National Foundation for Scientific Research and started to work on stimuli-responsive block copolymer micelles in the same group. In 2001–2002, he was post-doc in the group of Prof. Ulrich S. Schubert at the Eindhoven University of Technology (The Netherlands) working on metallo-supramolecular polymers. Since 2003, he is Professor at the catholic University of Louvain (Belgium) in the Institute of Condensed Matter and Nanosciences. In 2008–2010, he was part-time professor at the Eindhoven University of Technology and invited professor at the University of Bordeaux (France) in 2009. He is author or co-author of 200 scientific papers, most of them in the field of functional block copolymers. |
Introduction
Stimuli-responsive polymers are a topic of intense research since the behaviour of these polymers can be altered by a variation in their surrounding media.1–4 By definition, stimuli-responsive polymers are able to display a sharp modification of their physical and chemical properties when submitted to a chemical of physical stimuli, such as temperature, pH, redox, ionic strength, light etc.Among all the available stimuli light has recently attracted much attention since the stimulus can be localized in time and space and it can also be triggered from outside the system. Moreover, most of the photochemical processes involved do not require additional reagents, and by-products are limited in most of the cases. The irradiation parameters, such as light intensity and wavelength and irradiation time, can easily be modulated to adequately comply with the system.5–10
In material science, the targeted applications drastically influence the polymer synthesis. Reversibly, the nature, composition and architecture of the polymer discriminate which type of application can be targeted. It is thus very important to know precisely which type of material properties are desired to correctly design stimuli-responsive polymers. In the case of photo-responsive polymers (PRPs), it is very important to know what type of photo-responsive moieties is introduced in the polymer and what is the response of these moieties to a light irradiation. The nature of the photochemical reaction is thus the key parameter for a rational design of photo-responsive materials. Several photosensitive moieties, such as o-nitrobenzyl esters, may undergo an irreversible transformation during the irradiation whereas others can react reversibly (e.g. azobenzenes). A second parameter that influences the targeted application is the location of the photo-responsive moiety in the copolymer. Fig. 1 depicts the effect of the photo-responsive group location on the behaviour of block copolymer micelles in solution. The effect of light irradiation on micellar nanoobjects can vary from the stabilisation of the nanostructures to the degradation of the polymer chains and thus the destruction of the objects.
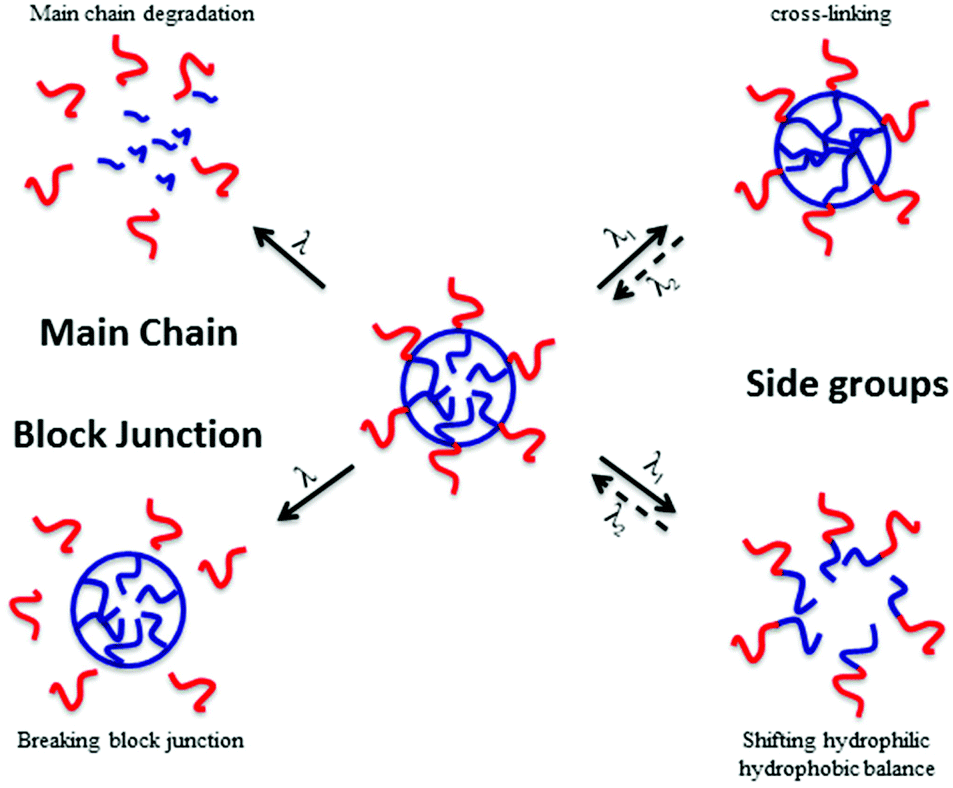 | ||
| Fig. 1 Effect of the photo-responsive moieties onto the behaviour of micelles in solution under irradiation (reproduced from ref. 8 with permission from the ACS). | ||
In this review, some guidelines towards the rational design of photo-responsive polymers (PRPs) depending on the targeted application are presented. In the first section, different applications of photo-responsive polymers in solution will be presented. We will also discuss the different types of photosensitive moieties introduced in polymers and the physical or chemical changes induced by light irradiation. Different strategies to induce the disruption of micelles with the combination of light and photo-responsive groups will be detailed as well as strategies to stabilize micellar structures. Moreover, recent advances in light control release of drugs will be presented at the end of the section. The third section of this review presents photo-responsive polymer brushes. The use of photo-responsive block copolymers in thin films is detailed in the next section. The creation of photoresists based on PRPs and techniques to pattern surfaces will be presented. Moreover several examples of nanoporous thin films based on photo-responsive block copolymers will be illustrated. Different applications of photo-responsive polymer networks will be then detailed. Finally, the formation of nanostructured materials and nanopatterned surfaces from photo-initiated polymerization processes will be briefly discussed.
Applications of PRPs in solution
The main envisioned application of PRPs in solution is light controlled delivery with micelles since light can penetrate to a certain extent into the skin. This characteristics of light can be employed to deliver active molecules (drugs) in a precise area of the body.1,2,7,8,76,77 With this idea in mind, the simplest design consists of a block copolymer micelle containing the drug encapsulated into a photo-responsive hydrophobic micellar core (Fig. 2).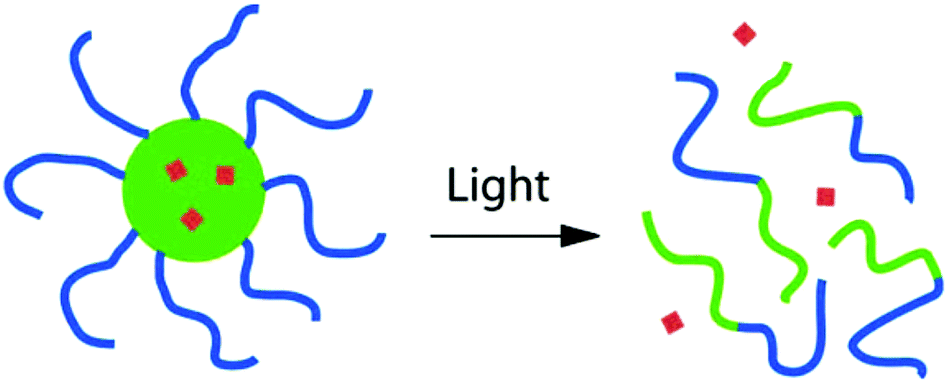 | ||
| Fig. 2 Light induced release of drugs from a photo-responsive block copolymer micelle (reproduced from ref. 13 with permission from the ACS). | ||
The light irradiation on such a system could result in the reversible of irreversible transformation of the hydrophobic core into a hydrophilic one. This shift in the hydrophobic/hydrophilic balance can induce the disruption of the micelles and thus the release of the drug in solution. The most common design of photo-responsive block copolymer for this application consists of a hydrophilic block, which forms the micellar corona, and a hydrophobic block bearing the photo-responsive side groups.
Irreversible light induced disruption or formation of micelles
Fig. 3 presents the different photo-responsive groups that have been incorporated into poly(methacrylate)s for irreversible light induced disruption of micelles. The changes in chemical structure of the photosensitive moieties in response to light irradiation are also depicted. Upon light irradiation, pyrenylmethyl esters undergo a photolysis reaction requiring the presence of water or a protonic solvent while the photocleavage of o-nitrobenzyl esters proceeds via a Norrish II type intra-molecular rearrangement with no need of additional reactant or solvent.9 Furthermore, o-nitrobenzyl esters undergo photocleavage upon the absorption of two near infrared (NIR) photons. Although the NIR photo-dissociation of o-nitrobenzyl containing micelles has been demonstrated, the process efficiency is much lower than with UV irradiation due to a much smaller absorption cross-section for the two NIR photons.13 In order to address this problem, the chromophores have been changed to esters of (diethylamino)methylcoumarinyl which have the advantage to feature a larger two-photon absorption cross-section.39
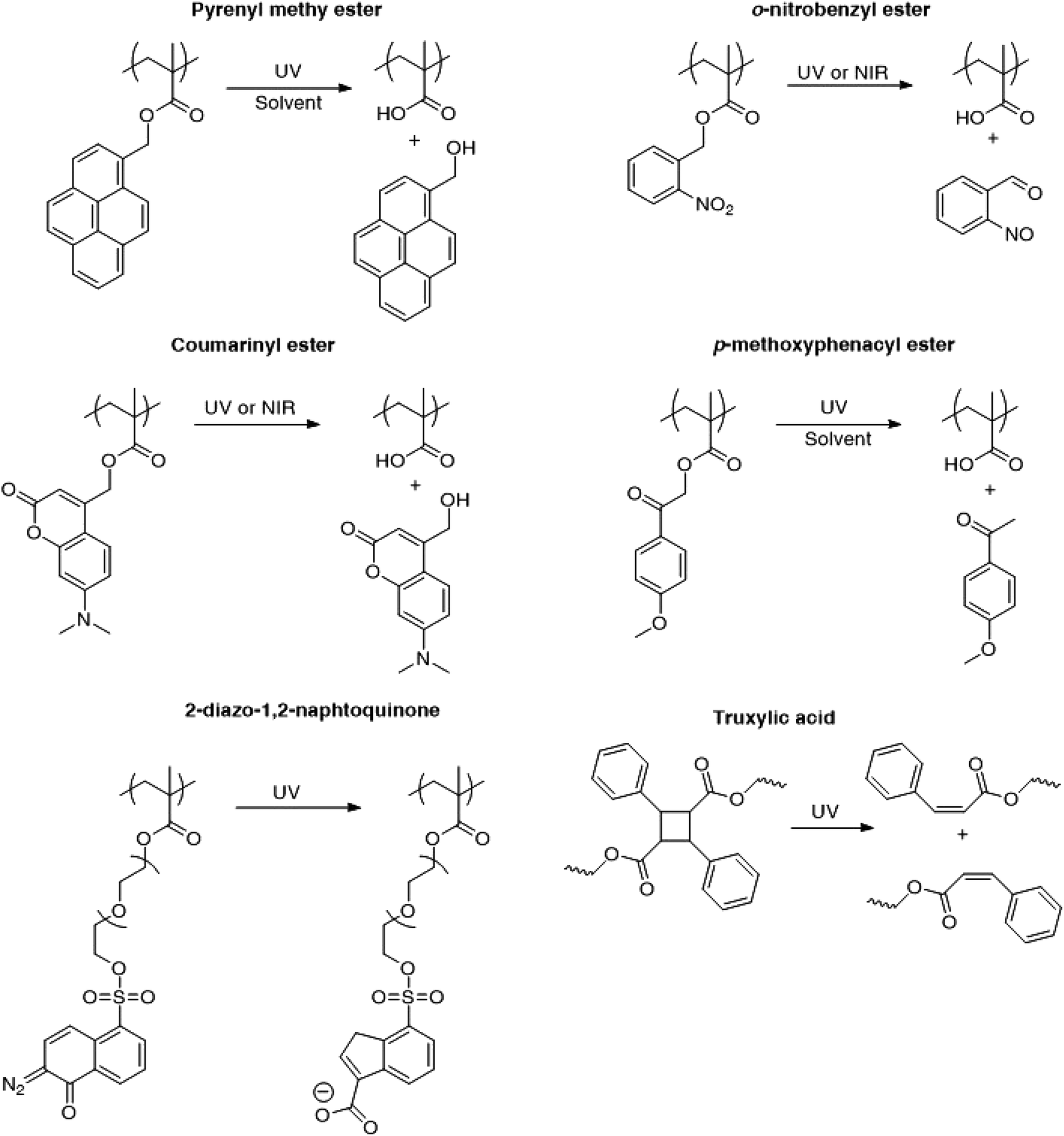 | ||
| Fig. 3 Different types of photo-responsive moieties that have been employed for the irreversible light-induced disruption of aqueous micelles. | ||
A similar approach for the light disruption of micelles was realised with polymer containing diazonaphtoquinones.45,46 Ji and coworkers synthesized poly(ethylene oxide)-block-poly[(oligoethylene glycol) methacrylate-random-(2-diazo-1,2-naphthoquinone oligoethylene glycol) methacrylate] (PEO-b-P(OEGMA-r-DNQMA)) via a post modification approach, where the 2-diazo-1,2-naphthoquinone group was grafted on the terminal hydroxyl function of the POEGMA block. This polymer was self-assembled into micelles with a PEO corona and a P(OEGMA-r-DNQMA) core.45 The irradiation of the micelles triggered the transformation of the 2-diazo-1,2-naphthoquinone moiety into 3-indenecarboxylic acid via a Wolff rearrangement.78 The shift in the hydrophobic/hydrophilic balance induces the micelle disruption and the ability of the system to release active molecules was demonstrated with coumarin 102 loaded micelles.
The combination of light responsive polymers with other stimuli-responsive polymers can significantly broaden the scope of applications of such micellar systems. For example, this allows the formation of materials displaying multiple micellisation processes (MMP). In this respect, Zhao and coworkers synthesised a thermoresponsive polymers exhibiting a lower critical solution temperature (LCST) behavior bearing o-nitrobenzyl groups.15,16
The irradiation of such a system is expected to increase the overall LCST of the copolymer compared to the corresponding homopolymer with no photoresponsive groups. More precisely a poly(ethylene oxyde)-block-poly(ethoxytri(ethyleneglycol)acrylate-random-o-nitrobenzylacrylate) (PEO-b-P(ETEGA-r-NBA)) block copolymer has been prepared by ATRP. The irradiation of this system induces a shift in the LCST from 25 to 36 °C due to the formation of hydrophilic carboxylic acid functions in the PETEGA backbone (Fig. 4a).15 This strategy was also successfully applied to the multiple sol–gel–sol transition of micellar gels (Fig. 4b).16
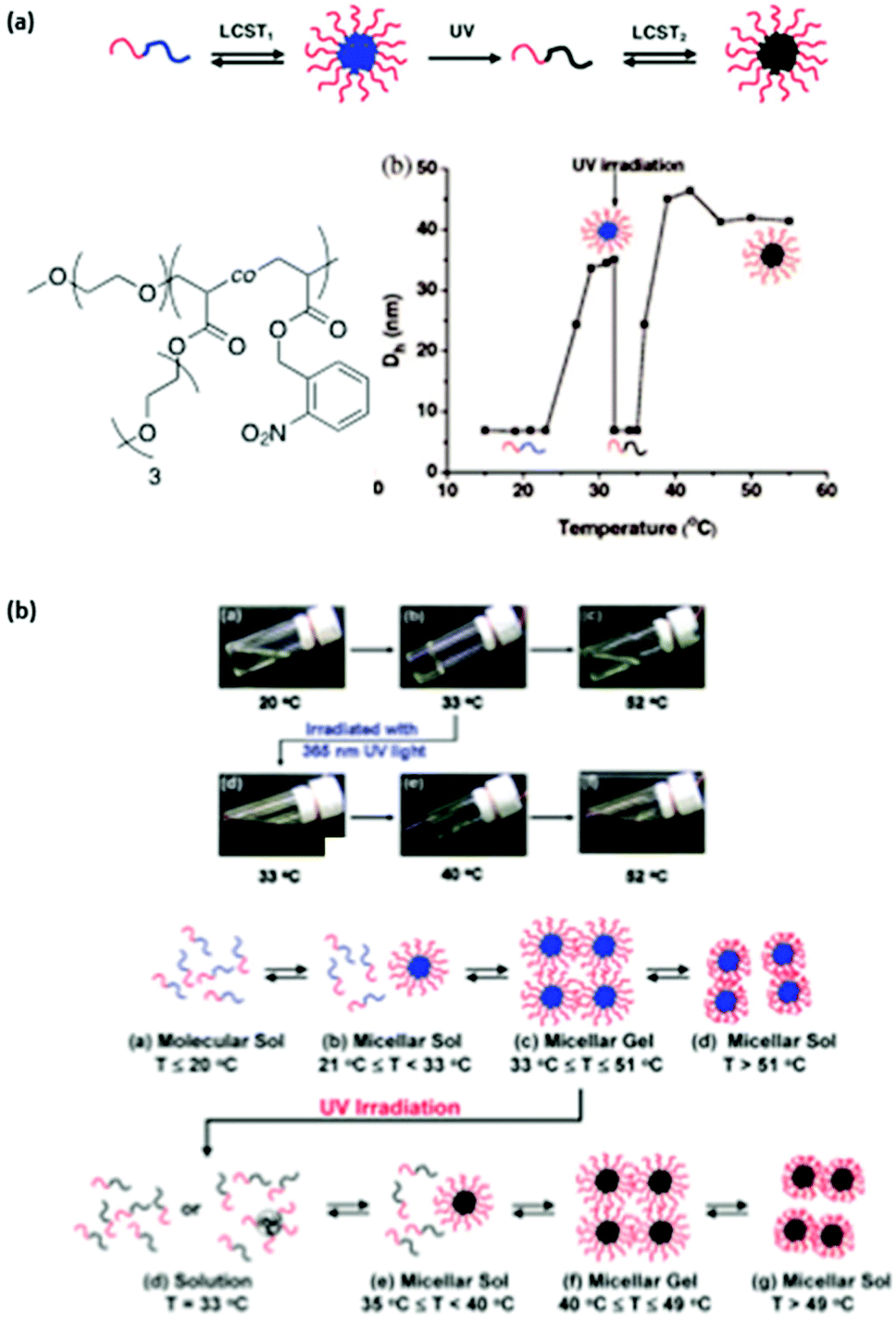 | ||
| Fig. 4 Multiple micellisation process: (a) schematic representation of the MMP, chemical structure of PEO-b-P(ETEGA-r-NBA) and changes in the micellar hydrodynamic diameter with temperature and light irradiation; (b) digital optical picture of an aqueous solution of PEO-b-P(ETEGA-r-NBA) at different temperatures and schematic representation of the multiple sol–gel–sol transitions (reproduced from ref. 15 and 16 with permission from the ACS). | ||
Light-responsiveness can also be coupled to other stimuli as recently demonstrated by Gohy and coworkers with a poly(p-methoxyphenacyl methacrylate)-block-poly((oligo ethylene glycol)methacrylate) diblock copolymer (PMPMA-b-POEGMA). Phenacyl esters also undergo a cleavage of their ester bound under irradiation. The advantage of phenacyl esters compared to o-nitrobenzyl esters is their chemical structure that does not contain nitro-aromatic group. Therefore, their polymerisation by controlled radical polymerisation techniques is possible.29,42
Upon dissolution in water, PMPMA-b-POEGMA forms micelles with a PMPMA core and a POEGMA corona. Light irradiation of the micelles induces the cleavage of the phenacyl esters, i.e. the transformation of PMPMA into PMAA, and thus the disruption of the initial micelles. The accordingly obtained PMAA-b-POEGMA copolymers show multi-responsive behaviour to pH and calcium ions while addressing the PMAA block and to temperature and phosphate (PO43−) ions due to the salting out the of the PEG side chains of the POEGMA block (Fig. 5).43
 | ||
| Fig. 5 A multi-responsive micellar system based on PMPMA-b-POEGMA diblock copolymers (EDTA: ethylenediaminetetraacetic acid sodium salt, reprinted with permission from ref. 43 with permission from the ACS). | ||
In all the above mentioned examples a quantitative photocleavage is considered to happen in order to trigger the disruption of the micelles and thus the release of the active molecules in solution. However, Lui and Dong demonstrated that this assumption is not correct. They proved that even if the photocleavage of micelles formed with poly(S-(o-nitrobenzyl)-L-cysteine)-block-poly(ethylene oxide) diblock copolymer led only to the shrinkage of the micellar core and not to micelle disruption, doxorubicin was gradually released in the aqueous medium.25 This experiment clearly demonstrates that the swelling or modifications in the characteristic size of micellar compartments as well as morphological transitions may induce the release of drugs from micellar cores.
Other strategies for releasing molecules were developed based on an irreversible shift in the hydrophobic/hydrophilic balance. In those cases the release is induced by the photocleavage of a hydrophobic collapsed corona.14,79 Zhao and coworkers polymerised o-nitrobenzyl methacrylate by ATRP on the surface of cross-linked poly(ethylene oxide)-block-poly(coumarin methacrylate-random-methylmethacrylate) inverse micelles (PEO-b-P(CMA-r-MMA)). The accordingly obtained PEO-b-P(CMA-r-MMA)-b-PNBMA micelles were loaded with gadolinium tetraphenylporphyrin (GdTPP), a magnetic resonance imaging probe. The irradiation of the micellar solution led to the photocleavage of the nitrobenzyl side groups and to the expansion of the corona. This coronal expansion allowed the diffusion of the GdTPP in solution.14
Light may also be used to irreversibly induce the formation of micelles instead of disrupting them. In this case, the photocleavable block copolymer is dissolved in a non-selective solvent. The irradiation of this polymer sample switches the hydrophobic/hydrophilic balance of the photo-responsive block and renders it insoluble in the considered solvent and thus induces the formation of micelles. This strategy could be of high interest to encapsulate the desired molecule into a micellar core and thus separate it from the surrounding solution. This strategy has been documented by Gohy and coworkers for copolymers constituted of a polystyrene (PS) block and either a poly(o-nitrobenzylacrylate) (PNBA)21 or a poly(dimethoxy-o-nitrobenzylacrylate) (PDMNBA)20 photocleavable block. In chloroform, those block copolymers are dissolved as unimers. However, irradiation at 300 nm (for PNBA) or at 350 nm (for PDMNBA) results in the photocleavage of the o-nitrobenzyl esters in PNBA or PDMNBA, respectively, to form a poly(acrylic acid) block (PAA). Since PAA is not soluble in chloroform the copolymer forms micelles with a PAA core and a PS corona. This photo-driven micellisation process can be advantageously used to encapsulate molecules of interest. The concept has been demonstrated by using a fluorescent coumarin 343 during the micellisation process (Fig. 6). This dye bears a tertiary amine group and is able to interact with the carboxylic acid moieties of PAA. Moreover, the photo-driven encapsulation process has been demonstrated to be more efficient than the encapsulation of the coumarin realised by direct dissolution of a PAA-b-PS copolymer in chloroform. The sore point of the irreversible light induced micellisation is its non-suitability to aqueous media. Indeed, all the photocleavable moieties used up to now in this approach are characterised by the transformation of a hydrophobic group into a hydrophilic carboxylic acid.
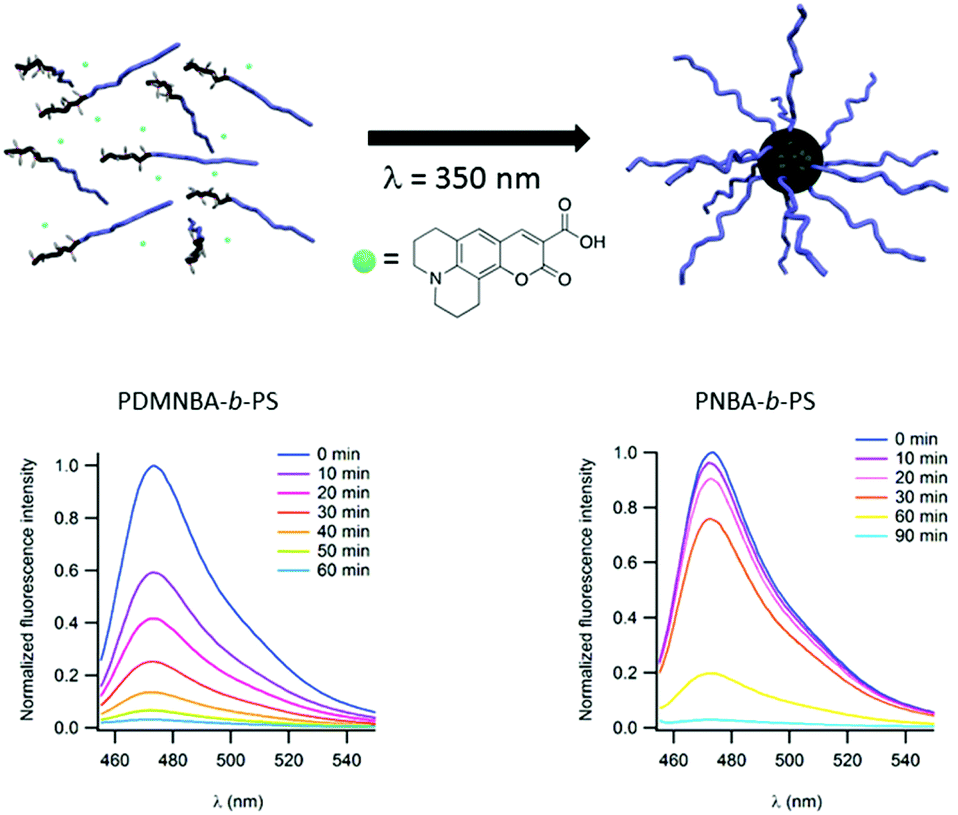 | ||
| Fig. 6 Schematic representation of the irreversible light induced micellisation and evolution of the fluorescence of coumarin 343 during the light driven encapsulation into micellar cores for PNBA-b-PS (left) and PDMNBA-b-PS (right) (reproduced from ref. 7 with permission from the RSC). | ||
The disruption of micelles via the degradation of the micellar core may also be obtained with the so-called light triggerable self-immolative polymers. In this case, the polymer chain degrades itself via an end-to-end depolymerisation mechanism in response to the cleavage of a stabilising end-cap from the polymer terminus.30,80 In this regard, Lui et al. designed poly(benzyl carbamate)-block-poly(N,N-dimethylacrylamide) diblock copolymers (PBC-b-PDMA) end capped with light (o-nitrobenzyl carbamate and perylen-3-yl carbamate) or redox (disulfide bond) sensitive groups.80 Upon dissolution in water, these polymers self-assemble in vesicles with a PDMA inner and outer corona and a PBC wall. The irradiation of these vesicles triggers the cleavage of the light sensitive end-cap and initiate the PBC depolymerisation and thus the vesicle disruption (Fig. 7). A similar strategy was applied to poly(glyoxylate) polymers by Gillies and coworkers. An ABA triblock copolymer was synthesized with o-nitrobenzyl ester junctions between the PEO outer A blocks and the poly(ethyl glyoxylate) central B block (PEtG).30
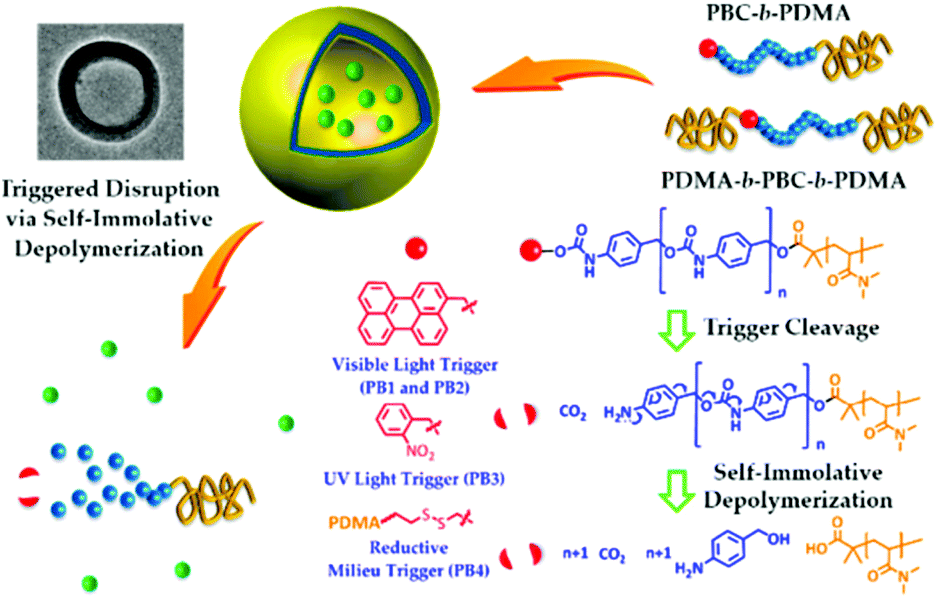 | ||
| Fig. 7 Light induced disruption of micelles with self-immolating PBC-b-PDMA copolymers (reproduced from ref. 80 with permission from the ACS). | ||
Another strategy towards irreversible light induced disruption of micelles consists in the introduction of a single photocleavable group between the core and the corona-forming blocks of a photo-responsive block copolymer. Those systems differ from the aforementioned PRPs in their modus operandi. Indeed, the photocleavage of the polymer junction does not result in the direct disruption of the micellar core. Light liberates the coronal chains from the micellar cores that tend to aggregate and precipitate out from the solution due to the lack of steric stabilisation. This strategy was successfully applied for block copolymers containing o-nitrobenzyl ester35,37,38 and truxillic acid73 derivatives. Even if this strategy is not the best suited for light induced drug release from spherical micelles, the self-assembly of such PRPs in vesicles is a promising approach. Indeed, the removal of coronal chains in vesicles results in the destabilisation of the vesicular wall, the subsequent breaking of the vesicles into small hydrophobic parts and finally in the release of the molecules initially encapsulated in the vesicles. This approach was successfully carried out by Katz and coworkers that demonstrated the release of guest molecules from poly(ε-caprolacone)-block-poly(ethylene oxide) where the junction is a 2-nitrophenylalanine.81 A similar approach was used by Meier and coworkers to prepare light-responsive polymersomes able to deliver a variety of payloads including drugs, proteins, enzymes and DNA.82
The key point of this approach is to design synthetic strategies allowing for precisely introducing in high yield the photocleavable group at the block junction. An interesting synthetic route to introduce the photocleavable group at the junction between the two blocks was developed by our group. We designed a photocleavable ATRP initiator, which can react on an end-functionalized azide block via a CuAAC while the second block is simultaneously polymerized by ATRP.36 This synthetic route was employed for the synthesis of various block copolymers, including poly(tert-butyl acrylate)-block-poly(dimethyaminoethyl methacrylate) copolymers (PtBA-b-PDMAEMA).32 Those block copolymers were designed for the production of hollow nanocapsules. Spherical micelles with a photocleavable o-nitrobenzyl junction at the interface between the PtBA core and the PDMAEMA corona were obtained upon dissolution of the copolymer in water. The nanocapsule membrane was formed by the crosslinking of the PDMAEMA corona with a diiodo compound (bis(2-iodoethoxy)ethane, BIEE). The shell cross-linked micelles were irradiated to break the junction and the core forming PtBA chains were extracted from the PDMAEMA nanocapsules. Moreover, the stimuli responsive behavior of the nanocages was demonstrated by variation in the solution pH and temperature. Finally, we demonstrated that it is possible to functionalize the internal cavity of the nanocapsules (Fig. 8).
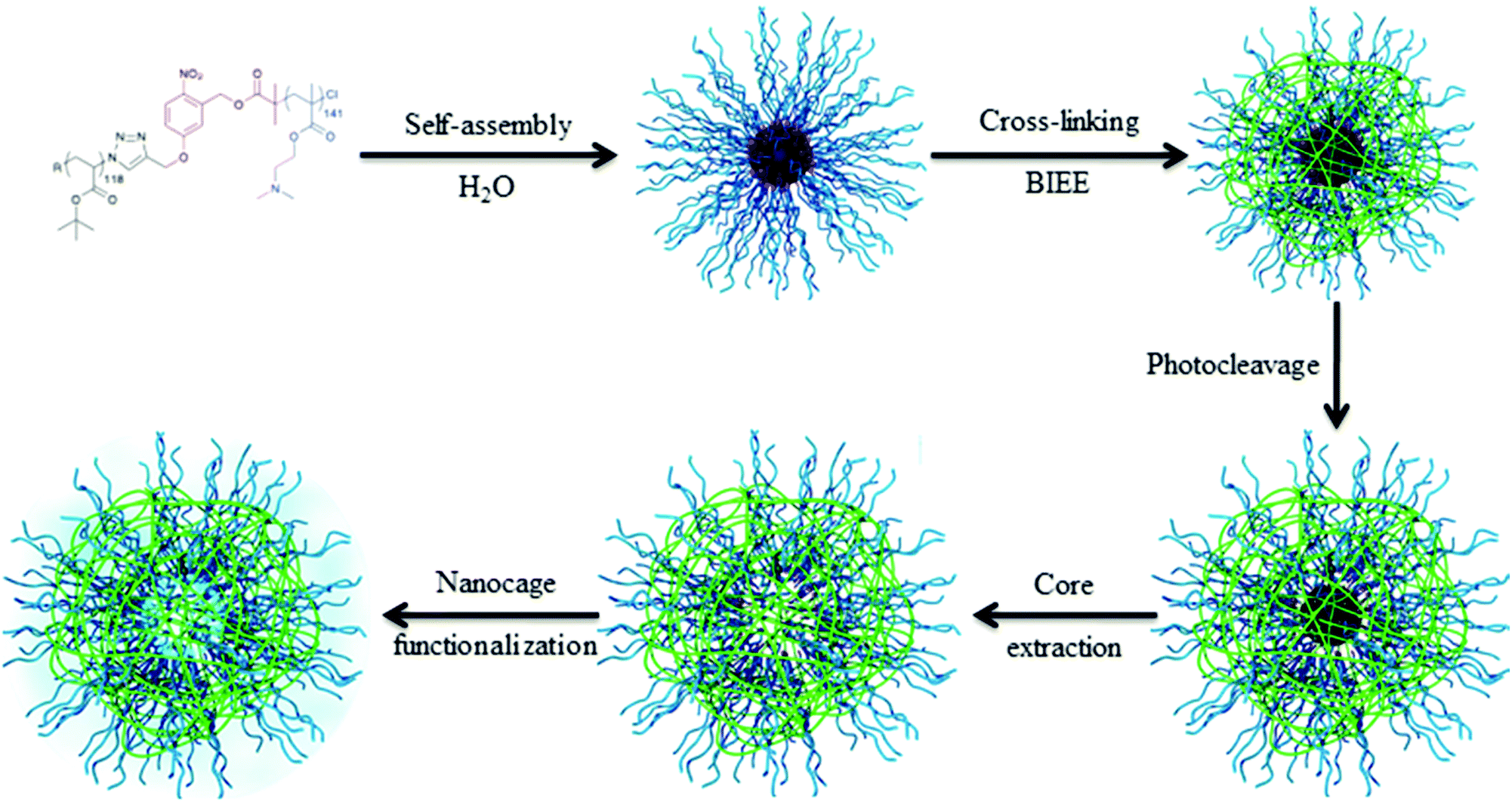 | ||
| Fig. 8 Schematic representation of the strategy toward nanocage formation (reproduced from ref. 32 with permission from the ACS). | ||
Reversible light induced disruption or formation of micelles
Light controlled drug delivery from micelles containing reversible light-responsive moieties was also considered in the recent years. The use of reversible moieties directly allows the light driven formation of micelles in addition to the disruption of micelles. Although this strategy appears to be more powerful than the irreversible micelle disruption, reversible light sensitive groups generally display a weaker shift in the hydrophobic/hydrophilic balance when submitted to light irradiation than irreversible photo-responsive groups. Moreover, the slow kinetics at which the photoactive moieties respond to light or relax to their initial state in the absence of light could be a severe limitation to their use in practical applications. Finally, the reversible characteristics of these groups are still subject to debate, since the initial characteristic features of the system are usually not fully recovered during irradiation cycles. Fig. 9 presents several photochromic molecules that were introduced into polymeric structures for the light induced disruption and formation of micelles, including azobenzene, spiropyrane and dithienylethene. Those groups are characterised by a reversible photoisomerisation reaction upon irradiation to UV or visible light.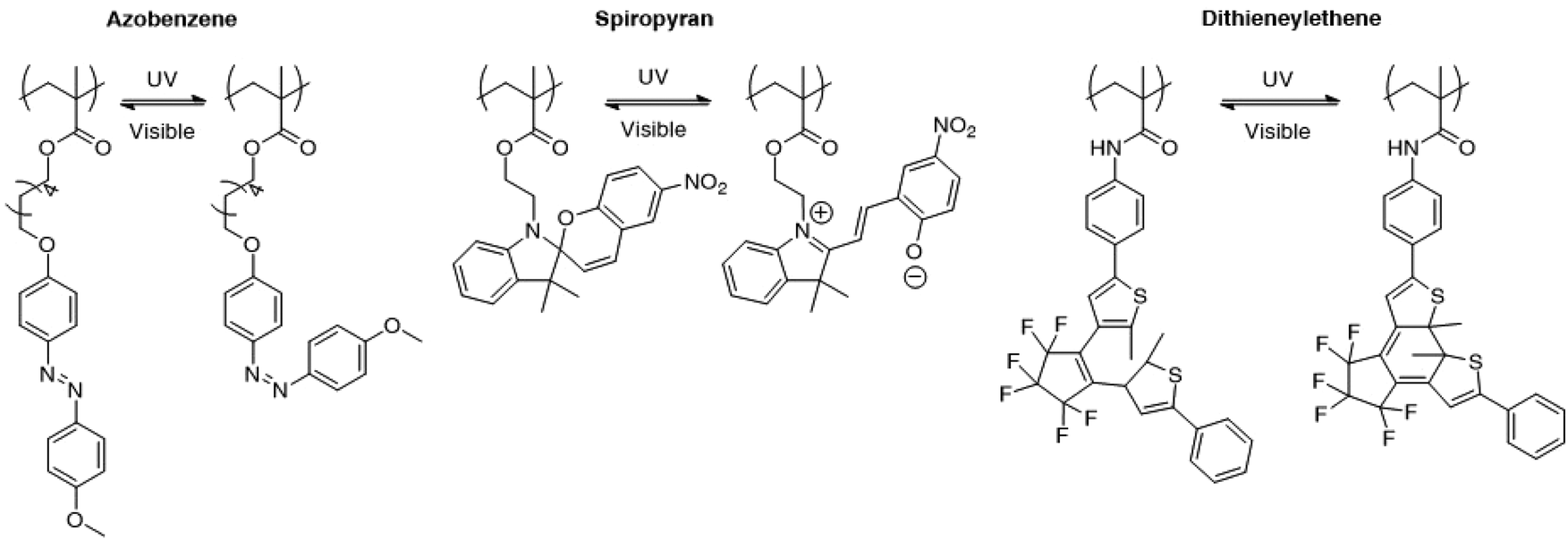 | ||
| Fig. 9 Photo-isomerisable groups for the reversible light-induced disruption or formation of micelles. For each group, both photoisomers are shown. | ||
Among the photoisomerisable groups, azobenzenes and their derivatives constitute by far the most studied.83–85 Upon light irradiation, azobenzenes undergo a reversible trans–cis isomerisation of their nitrogen double bound (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–). In this process, the apolar trans isomer is converted into the polar cis one upon UV irradiation while the reversible process is triggered by visible light. This strategy has been used by various authors to promote the formation or the disruption of micelles47,48,51,52 and to modify the LSCT/UCST behaviour of thermo-responsive polymers.53,86–88 For example, Zhao and coworkers designed a diblock copolymer containing a random poly(tert-butyl acrylate-random-acrylic acid) (P(tBA-r-AA)) block and on the other side a poly(methacrylate) bearing azobenzenes as pendent groups (PMAz).48 The dissolution of this polymer in water/dioxane mixtures promoted the formation of micelles and vesicles, depending on the dioxane content. Upon UV irradiation, the azobenzene side groups in the apolar trans isomers (dipole moment = 0D) were converted into their cis form (dipole moment = 4.4D). This increase in the block polarity resulted in the disruption of the micelles to form unimers in solution. The subsequent visible irradiation promoted the isomerisation into the trans form and the micellar aggregates were reformed.
N–). In this process, the apolar trans isomer is converted into the polar cis one upon UV irradiation while the reversible process is triggered by visible light. This strategy has been used by various authors to promote the formation or the disruption of micelles47,48,51,52 and to modify the LSCT/UCST behaviour of thermo-responsive polymers.53,86–88 For example, Zhao and coworkers designed a diblock copolymer containing a random poly(tert-butyl acrylate-random-acrylic acid) (P(tBA-r-AA)) block and on the other side a poly(methacrylate) bearing azobenzenes as pendent groups (PMAz).48 The dissolution of this polymer in water/dioxane mixtures promoted the formation of micelles and vesicles, depending on the dioxane content. Upon UV irradiation, the azobenzene side groups in the apolar trans isomers (dipole moment = 0D) were converted into their cis form (dipole moment = 4.4D). This increase in the block polarity resulted in the disruption of the micelles to form unimers in solution. The subsequent visible irradiation promoted the isomerisation into the trans form and the micellar aggregates were reformed.
As stated above, the hydrophobic/hydrophilic shift triggered by the isomerisation process is not always sufficient to induce the complete disruption of the micelles. It can however induce a deformation or morphological changes for the irradiated micellar aggregates.50,52,89,90 In this respect, Li et al. designed a poly(N-isopropylacrylamide)-block-poly(acrylate) copolymer bearing azobenzene as side groups (PNIPAM-b-PAz) that formed polymersomes in THF/water mixture (50/50 vol%). They observed a photo-triggered fusion process upon irradiation.50 Wang and coworkers observed the deformation of micellar aggregates alongside the laser polarization direction when they irradiated a solution of PMAz-b-PMAA copolymer in THF/water mixtures (Fig. 10).52
 | ||
| Fig. 10 TEM images of PMAz-b-PMAA micellar aggregates before and after the irradiation with Ar+ laser beam for 30 min (scale bar = 1 μm), the arrow represents the polarization direction of the laser beam (reproduced from ref. 52 with permission from Elsevier). | ||
Other promising photosensitive groups for the reversible formation or disruption of micelles are hydrophobic spiropyrans. They undergo a photoisomerisation to form a hydrophilic zwitterionic merocyanine when irradiated with UV light. The reverse isomerisation is realized with visible light (620 nm). In comparison to azobenzenes, the hydrophobic/hydrophilic shift is more pronounced due to the zwitterionic form. This characteristic features makes them valuable for micellisation and disruption applications. This approach has been used by Matyjaszewski and coworkers with a poly(ethylene oxide)-block-poly(methacrylate) whose methacrylate block is bearing spiropyran (SP) side-chains (PEO-b-PMSP).57 The disruption of the micelles was observed upon UV irradiation whereas the reformation of the micelles was carried out by visible light irradiation. Moreover they demonstrated that this approach could be used for the release/encapsulation of guest molecules. They used fluorescence spectroscopy to observe the signal of coumarin 102 during the UV induced release and the re-encapsulation by visible light (up to almost 50% of re-encapsulation of released dye molecules). A similar approach was used by Das and coworkers for a block copolymer composed of PMSP and a glycopolymer58 and by Mezzenga et al. for a copolymer containing a PEO block and a poly(L-glutamic acid) block doped with spiropyran groups.62
All the examples of reversible PRPs discussed in this section involve many photo-responsive moieties attached as pendent groups on a polymer backbone. Similarly to the irreversible micelle disruption, the use of block copolymers containing a single reversible photo-responsive junction was considered. This was achieved by the formation of an inclusion complex between azobenzene and cyclodextrin (CD) each positioned at one end of a polymer chain.55,56 Yuan and coworkers designed a copolymer composed of a CD-capped poly(ε-caprolactone) and an azobenzene-capped poly(acrylic acid).56 When the two homopolymers were added in aqueous solution, supramolecular PCL-CD/Az-PAA micelles were formed via homopolymer orthogonal self-assembly based on host–guest interaction (Fig. 11). The disruption of those micelles was achieved under UV irradiation, where the azobenzene isomerises in its trans form and is expulsed from the cavity. This strategy was also applied by Barner-Kowollik et al. for the production of a poly(N-(2-hydroxypropyl)methacrylamide)-CD/Az-poly(N,N-dimethylacrylamide)-CD/Az-poly(N-(2-hydroxypropyl)methacrylamide) triblock copolymer (PHPMA-CD/Az-PDMAAm-CD/Az-PHPMA). This system allows the reversible formation and disruption of micelles via light and temperature triggers.55
 | ||
| Fig. 11 Supramolecular photosensitive PAA-Azo/PCL-a-CD block copolymer based on homopolymer orthogonal self-assembly via a host–guest junction (reproduced from ref. 7 with permission from the RSC). | ||
Another system of photo-induced controlled release of molecules was achieved by the introduction of azobenzenes in the polymer backbone. Tenhu and coworkers synthesized a poly(azocalix[4]arene) which comprises exclusively calix[4]arenes locked in the cone conformation and joined by azo-bridges. Irradiation of this polymer induces slight changes in the cone conformation of the calix[4]arene units. This in turn affects the degree of host–guest interaction between the calixarene and a pyridinium-based guest, which is decreased when the polymers are in the cis-rich state.91 They also demonstrated that this photo-isomerism drastically influences the behavior of the polymer in solution. Indeed, a poly(azocalix[4]arene) substituted with tetraethylene glycol monomethyl ether chains displays a LSCT behaviour in water and an UCST behaviour in alcohols. The authors demonstrated that the cloud point of this polymer can be modulated depending on the trans/cis ratio of the azobenzene groups.92
Irreversible photo-induced crosslinking of micelles
The stabilisation of micelles via the crosslinking of either their core or corona is a topic of high interest.93 Indeed, micelles are dynamic structures that disintegrate themselves below their critical micelle concentration. The crosslinking of the core/corona is the easiest way to stabilise those structures. In this regard, the light-induced crosslinking is a valuable tool since chemical reagents and undesired by-products are avoided. Light-induced crosslinking of micelles and vesicles was first reported by Liu and coworkers who used the [2 + 2] photocycloaddition of cinnamic esters for photo-crosslinking.69,70 They self-assembled polyisoprene-block-poly(2-cinnamoylethyl methacrylate) (PI-b-PCEMA) copolymers in hexane/THF mixtures to form vesicles with a PCEMA wall and a PI corona. They subsequently used the photo-crosslinking of 2-cinnamoylethyl methacrylate to induce the stabilisation of the vesicle walls.70 Since the PI chains formed the outer and the inner layers of the vesicles, ozonolysis could either partially of totally “shave” those hairy nano-objects (Fig. 12). More recently the photo-crosslinking of cinnamic esters was combined with the irreversible photocleavage of a polymer junction. To reach this goal, a poly(ethylene oxide)-block-poly(2-(perfluorooctyl)ethyl methacrylate)-block-poly(2-cinnamoylethyl methacrylate) (PEO-b-PFOEMA-b-PCEMA) triblock copolymer was synthesized by ATRP. The photocleavable o-nitrobenzyl group was introduced by the modification of the PEO block to obtain the ATRP macroinitiatior. The dissolution of this triblock copolymer in a water/THF mixture (80/20 vol%) promoted the formation of micelles with a PCEMA core, a PFOEMA shell and a PEO corona. The photo-crosslinking and photocleavage of the junction allowed the production of PFOEMA-b-PCEMA nanoparticles exhibiting at the same time oil and water repellent behaviour.71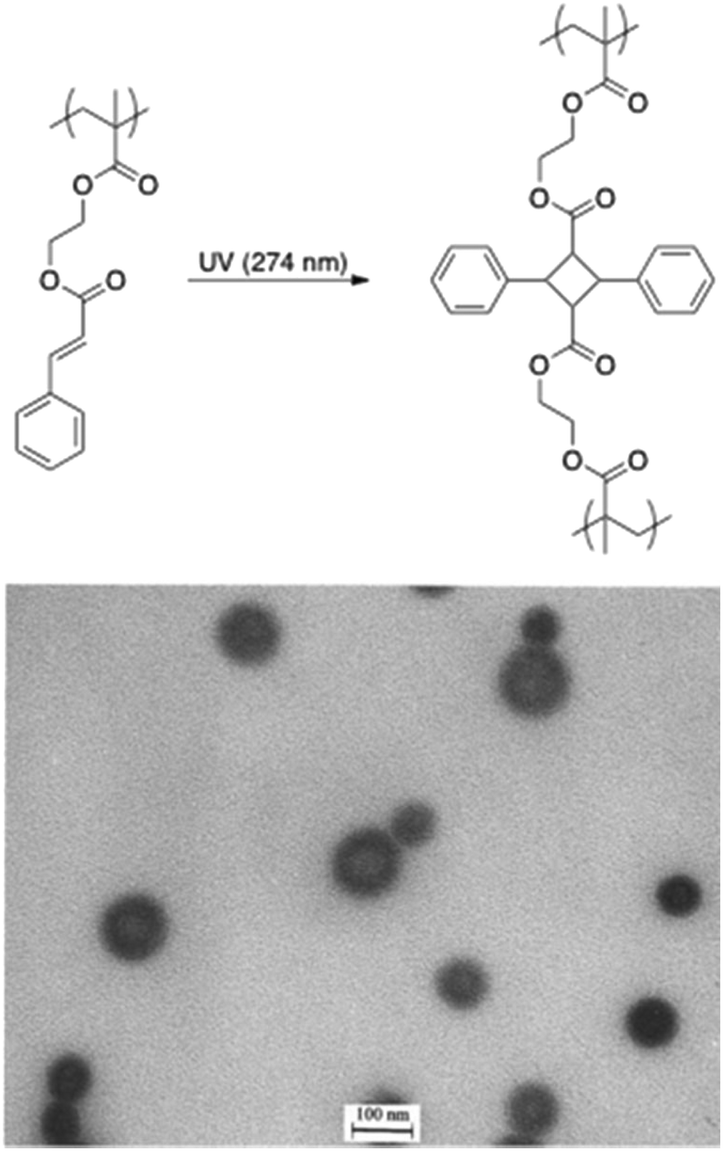 | ||
| Fig. 12 Photodimerisation of cinnamic esters and TEM picture of fully shaved nanospheres made from PI-b-PCEMA (reproduced with permission from ref. 70 with permission from the ACS). | ||
The irreversible crosslinking of cinnamic esters was also used for the production of hollow nanocapsules. Yusa and coworkers adopted a strategy relying on the use of a poly(ethylene oxide)-block-poly((3-acryl-amidopropyl)trimethylammonium chloride-random-(2-cinnamoylethyl methacrylate)) (PEO-b-P(APTAC-r-CEMA)) and a micellar template to produce the nanocapsules.94 In a first step they prepared a micellar template of poly(2-acrylamido-2-methylpropanesulfonic acid)-block-poly(sodium 6-acrylamidohexanoate) (PAMPS-b-PAaH) in acidic water. The addition of the PEO-b-P(APTAC-r-CEMA) copolymer in this solution led to the deposition of the latter on the PAMPS-b-PAaH micellar structure due to the electrostatic interactions between the PAaH and the P(APTC-r-CEMA) blocks. The UV irradiation triggered the crosslinking between the cinnamic ester functions. Finally the cavitation of the nanocapsules was realized in basic medium by the dissolution and the extraction of the micellar template.
It has to be noted that the denomination “cinnamic ester” is used to characterize the uncoupled moieties whereas “truxillic acid” is used to characterize the [2 + 2] cycloadduct.73 Although, the coupling of cinammic esters is often referred as irreversible, it is not the case. Indeed, the coupling of cinnamic esters into their truxillic derivatives, is promoted by an UV irradiation with a wavelength above 260 nm whereas the reversed reaction is triggered by deep UV irradiation (λ < 260 nm, see section “Application of PRPs networks: gels and hydrogels”).72,73 The examples of photo-crosslinking presented above are here referred as irreversible since the reversibility of the cycloaddition has not been demonstrated by the authors.
Another example of irreversible photocrosslinking involving the o-nitrobenzyl group was developed by Liu and coworkers.18 The authors used a poly(ethylene oxide)-block-poly(2-nitrobenzyloxycarbonylaminoethyl methacrylate) (PEO-b-PNBOC). The self-assembly of this copolymer in water led to the formation of vesicles with a PEO corona and a photocleavable PNBOC core. Similarly to the o-nitrobenzyl ester, the photocleavage of the nitrobenzyl carbamate led to the release of the corresponding primary amine. Surprisingly, the irradiation of the vesicles at pH = 7.4 did not promoted their disruption. Indeed, an increase of the vesicle hydrodynamic radius was monitored by dynamic light scattering (DLS) and the vesicles were still observable by transmission electron microscopy (TEM) after the irradiation. The authors postulate that the primary amines generated after the photocleavage were able to react with esters functions and induced a crosslinking via an amidation reaction (Fig. 13).
 | ||
| Fig. 13 Design of vesicles exhibiting photo-triggered “traceless” crosslinking (reproduced from ref. 18 with permission from Wiley). | ||
Reversible photo-induced crosslinking of micelles
The reversible photo-crosslinking of micellar structures is advantageous for some applications, such as the release of active molecules. Indeed, the release of guest molecules from crosslinked cores may be slow and inefficient. Reversible crosslinking allows a process where the micelles have a good stability and display a limited undesired release of the encapsulated molecules, while the non-crosslinked state could be specifically triggered to activate the delivery of the encapsulated species.This strategy was explored by Zhao and coworkers who introduced coumarin derivatives in amphiphilic block copolymers in order to stabilize either the core or the shell of the accordingly formed micelles.64,65 Coumarin moieties are indeed able to undergo a reversible [2 + 2] photocycloaddition upon UV irradiation. A copolymer composed of a water-soluble PEO block and of a poly(methacrylate) block incorporating coumarin side-chains (PCMA) was first synthesized by ATRP. The self-assembly of this copolymer in micelles with a photo-crosslinkable PCMA core and a PEO corona was achieved in water.64 Photo-crosslinking was triggered by irradiating the micelles at wavelengths above 310 nm while photo-de-crosslinking occurred upon irradiation at wavelengths below 260 nm. Although the photo-crosslinking and de-crosslinking appear to be incomplete, general reversibility could be observed (Fig. 14). They also demonstrated that the crosslinking limits the diffusion of guest molecules from the micellar core in comparison to the non-crosslinked micelles.
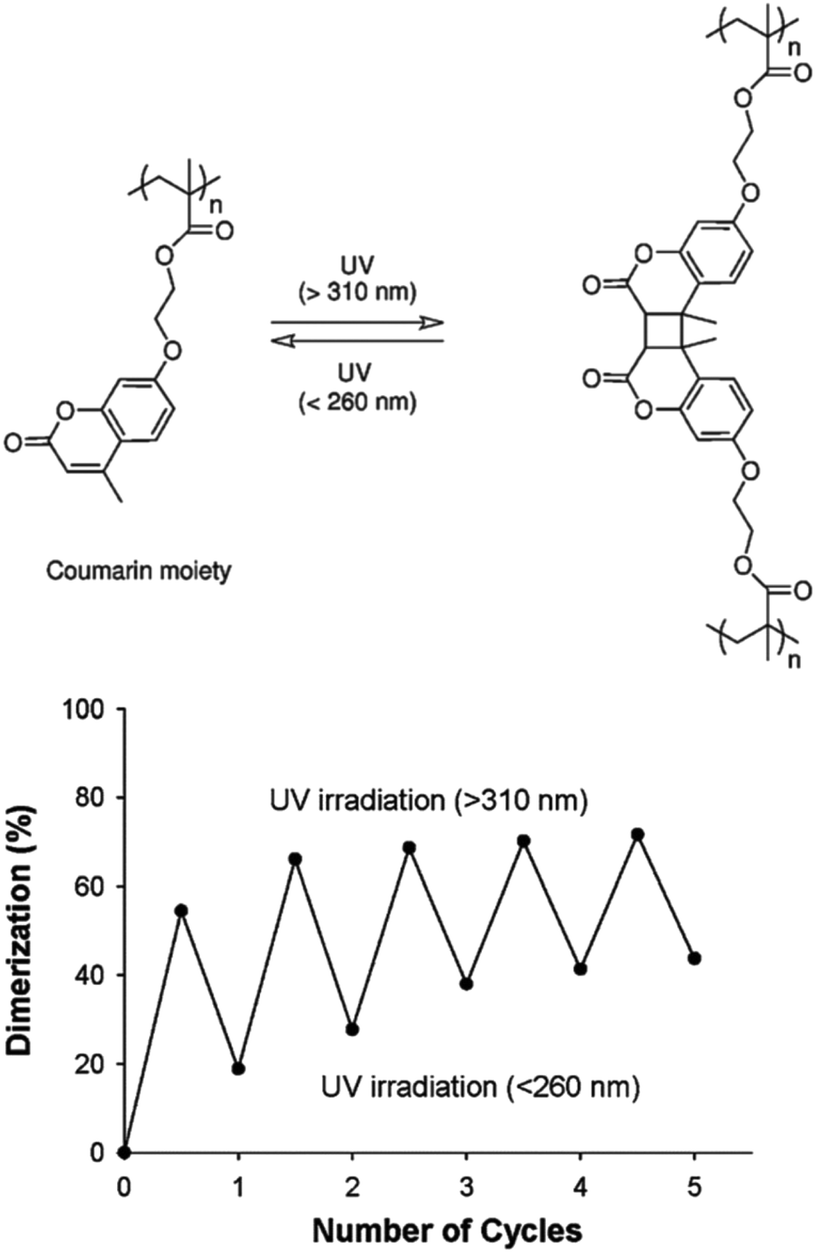 | ||
| Fig. 14 Photo-induced dimerisation of coumarin moieties in PCMA (left). Reversible photodimerisation of the coumarin moieties in PEO-b-PCMA micelles exposed to alternated illuminations (right) (reproduced from ref. 64 with permission from the ACS). | ||
Corona-photo-crosslinked micelles were also reported by Zhao and coworkers from a poly(dimethylaminoethyl methacrylate)-block-poly(methyl methacrylate-random-coumarin methacrylate) (PDMAEMA-b-P(MMA-r-CMA)).65 Micelles with a PDMAEMA core and a P(MMA-r-CEMA) corona were produced by the quaternisation of the PDMAEMA block with HCl in THF. The outer P(MMA-r-CMA) corona was then photo-crosslinked and the periphery of those micelles was further decorated by extra PDMAEMA and polystyrene chains grafted by a subsequent ATRP of DMAEMA or styrene initiated by chloride groups remaining at the surface of the crosslinked shell.
Additional stimuli-responsive character can be added to reversibly photo-crosslinked micelles. The basic design to achieve this goal is to introduce one or two blocks displaying LCST behaviour for instance and bearing a small amount of coumarin derivatives as side groups for photo-crosslinking. In this respect, Zhao and coworkers described a diblock copolymer composed of a PEO block and a coumarin-doped poly(2-(2-methoxyethoxy)ethyl methacrylate) (PMEO2MA) known for its LCST behaviour.66 The micellisation was induced by the elevation of the temperature above the LCST of PMEO2MA in water. The subsequent irradiation of the micelles led to the crosslinking of the coumarin derivatives in the micellar core. The crosslinking of the coumarin moieties in the core prevents dissociation of the micellar structure below the LCST of PMEOMA, giving rise to the formation of microgel nanoparticles. In addition, with the reversible photoreaction, the size of the nano-objects can by tuned the degree of crosslinking that determines the swelling of the micelles.
Similarly, these authors described large reversible volume changes in vesicles prepared from the same approach.67 A copolymer formed of a coumarin-containing PDMAEMA block and a thermo-responsive poly(N-isopropylacrylamide) (PNIPAM) block was used in this study. Large vesicles with a PNIPAM membrane were formed in water from this copolymer above the LCST of the PNIPAM block. After photodimerisation of the coumarin groups, the vesicles were cooled down below the LCST of PNIPAM and underwent expansion up to 700% due to the hydration of the PNIPAM membrane while retaining their structural integrity due to crosslinking.
PRPs for drug delivery applications: limitations and future trends
In the previous sections, the use of photo-responsive copolymers in solution was highlighted. It is clear that one of the key applications of micelles from PRPs is their use as nanocarriers for drug delivery. However a careful choice of the materials should be done. In this respect, the biocompatible and biodegradable characteristic features are mostly required since the nanocarriers will obviously interact with the metabolism of the patient. Considering the examples described so far in this chapter, PEO has been mostly considered as the corona-forming block. Indeed, PEO is a major player for bio-applications since it is biocompatible polymer (although this is still controversial) and his highly hydrophilic behaviour ensures a good stabilisation of the nanocarriers. Moreover, PEO inhibits protein adsorption on its surface thanks to the so-called “stealth” effect. However, less attention has been paid until now on the choice of the hydrophobic blocks incorporating the photo-responsive moieties. Most often photochromic groups were introduced on poly(methacrylate)s which do not complies with the biocompatible and biodegradable desired characteristics. As presented above, recent studies are focusing on the use of poly(amino acids) sequences modified by photocleavable moieties as hydrophobic blocks for PRPs.62The toxicity of the photochromic group itself and of the photoreaction products has to be also carefully considered. For example, the toxicity of o-nitrobenzyl esters is poorly documented. Moreover, the products of their photodegradation contain a nitrosobenzaldehyde which not only might be toxic on its own, but also absorbs UV light and degrades further into other ill-defined products. In this regards, Doris and coworkers studied the cytotoxicity of nitrosobenzaldehyde derivatives.95 They self-assembled a stearyl-orthonitrobenzyl-PEO copolymer to form micelles and studied the copolymer toxicity on cancerous cells. They afterwards used light irradiation to cleave the junction and form a carboxylic terminated PEO and a stearyl-nitrosobenzaldehyde. They demonstrated that the photocleavage drastically decreases the micellar concentration at which the cells are viable (Fig. 15).
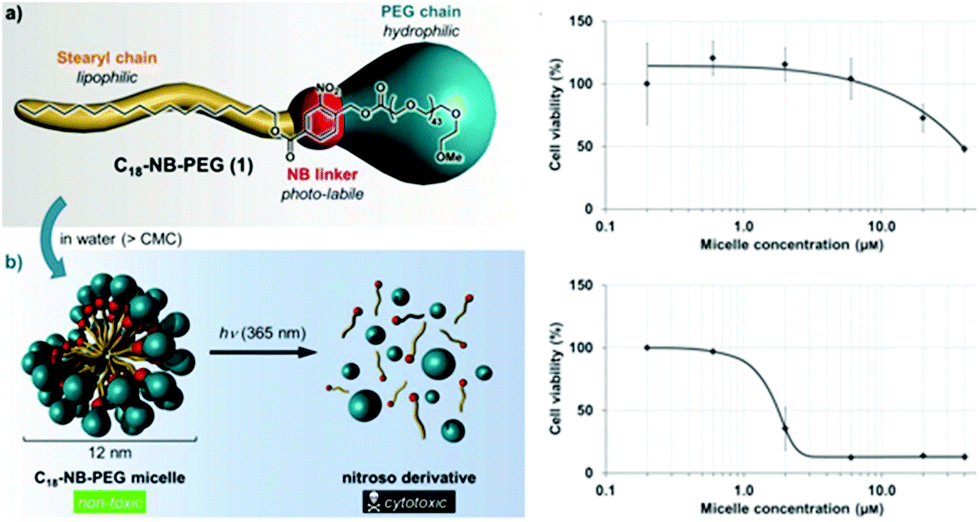 | ||
| Fig. 15 Cytotoxicity of o-nitrobenzyl esters and nitrosobenzaldehyde derivatives: schematic representation of the experiments and effect of the micelle concentration on the cell viability before (top) and after irradiation (bottom) (reproduced from ref. 95 with permission from Wiley). | ||
An obvious way to get rid off the toxicity of the products of the photo-induced reaction would be to consider light-cleavable drug–PRP conjugates. The irradiation of such a conjugate would release the active drugs and not undesired toxic photocleavage products. A recent example of this strategy has been reported for the anticancer drug 5-fluorouracil which has been linked covalently to coumarin side groups via UV-promoted cycloaddition in the hydrophobic micellar core. Further irradiation at a wavelength of 254 nm allowed the transformation of the hydrophobic drug-bearing blocks in the core into hydrophilic ones and thus in the disruption of the micelles accompanied by the release of the drug.96
Besides the aforementioned issues, the choice of the wavelength to trigger photoreactions is also critical for biomedical applications. In most of the examples presented earlier the UV irradiations are mostly used to trigger either irreversible or reversible photo-induced reactions. Such radiations are problematic for biomedical applications since they might cause damages to healthy cells during their application and because their penetration into living tissues is rather limited. This issue was raised in the early stage of the research on photo-responsive materials for drug delivery application.39 One of the solutions to this problem is the utilization of near infra-red (NIR) light that is a lower energy radiation with a reduced absorption and scattering by biological media and hence deeper penetration. However the use of NIR light requires that the photocleavable moieties are able to undergo a two-photon absorption process for the same energy as the corresponding one-photon absorption in the UV or visible spectral region, which is possible only for a limited number of photo-responsive groups. Moreover the photoreactions induced by two-photon absorption of NIR are generally slower and inefficient due to the typically low two-photon absorption cross-sections of the chromophores. Therefore, the use of a femtosecond pulse laser is generally needed to observe significant two-photon absorption, which might be a technical limitation for applications.
To address this problem, alternatives have recently emerged in the literature. A possibility to enhance the efficiency of NIR light excitation is to use a sensitizer approach. In this respect, lanthanide-doped up-converting nanoparticles that absorb NIR light and convert it into UV or visible light are an interesting alternative. These particles also display a more efficient two-photon absorption since their excitation occurs via sequential, multiple absorptions with real energy levels. This approach was adopted by Zhao and coworkers, which encapsulated NaYF4:TmYb up-converting nanoparticles into the o-nitrobenzyl-containing core of aqueous block copolymer micelles loaded with model molecules (Fig. 16).97 The accordingly loaded micelles were exposed to NIR light at 980 nm and the photons emitted at λ = 350 nm by the up-converting nanoparticles were used for the irreversible photocleavage of the o-nitrobenzylesters. This process thus led to the disruption of the micelles and the release of the initially encapsulated payloads. Although once would believe that the UV light emitted by up-converting nanoparticles might also cause damaging effect on surrounding tissues or biomolecules, such an effect can be considered negligible. Indeed, the UV light is emitted from the interior of micelles during the irradiation and most of these photons should be absorbed by surrounding photosensitive moieties in the polymer instead of interacting with biomolecules. The potential of this strategy for drug delivery applications and cancer therapy was successfully demonstrated for polymeric micelles based on spiropyran58 and coumarin.98
 | ||
| Fig. 16 (a) Schematic illustration of using NIR light excitation of up-converting nanoparticles to trigger the disruption of micelles. (b) NIR light-triggered photoreaction (reproduced with permission from ref. 97 with permission from the ACS). | ||
Application of PRPs at surfaces
Light-responsive polymers were extensively used for the production of smart surfaces.99,100 The ability of such polymers to switch their properties on demand is interesting for surface patterning, control of the surface wettability (self-cleaning surfaces)61 and non-sticking surfaces.101 In this regard photo-responsive moieties such as o-nitrobenzyl,102,103p-methoxyphenacyl,41,44 spiropyran61,104,105 and azobenzene101,106,107 were introduced in polymer brushes and their response to light irradiation was studied.One example of surface patterning using photo-responsive polymers was presented by del Campo and coworkers. They synthesized a poly(dimethoxynitrobenzyl methacrylate) brush via surface initiated atom transfer radical polymerisation (SiATRP).102 The irradiation of the surface through a photolithographic mask followed by the washing of the surface to remove the photocleaved chromophores allowed the production of the patterned surface. Moreover, since the photocleavage of the o-nitrobenzyl side groups results in a pH sensitive poly(methacrylic acid) brush (PMAA), the thickness of the pattern could be modulated depending on the pH (Fig. 17). The same authors used a similar strategy to produce an amine functionalized patterned surface based on protected poly(2-aminoethyl methacrylate).103 Voit and coworkers developed a similar strategy based on p-methoxyphenacyl ester. They synthesized poly(p-methoxyphenacyl methacrylate-random-methyl methacrylate-random-glycidyl methacrylate) P(MPMA-r-MMA-r-GLY) and poly(p-methoxyphenacyl methacrylate-random-methyl methacrylate-random-3-(trimethoxysilyl)propyl methacrylate) P(MPMA-r-MMA-r-SIL) terpolymers by free radical polymerisation.41,44 The copolymers were chemically anchored into boron doped silica surfaces. The irradiation of the surfaces with UV lamps allowed the formation of patterned surfaces with P(MAA-MMA) chains. The authors verified the film patterning by atomic force microscopy (AFM) and fluorescence spectroscopy.
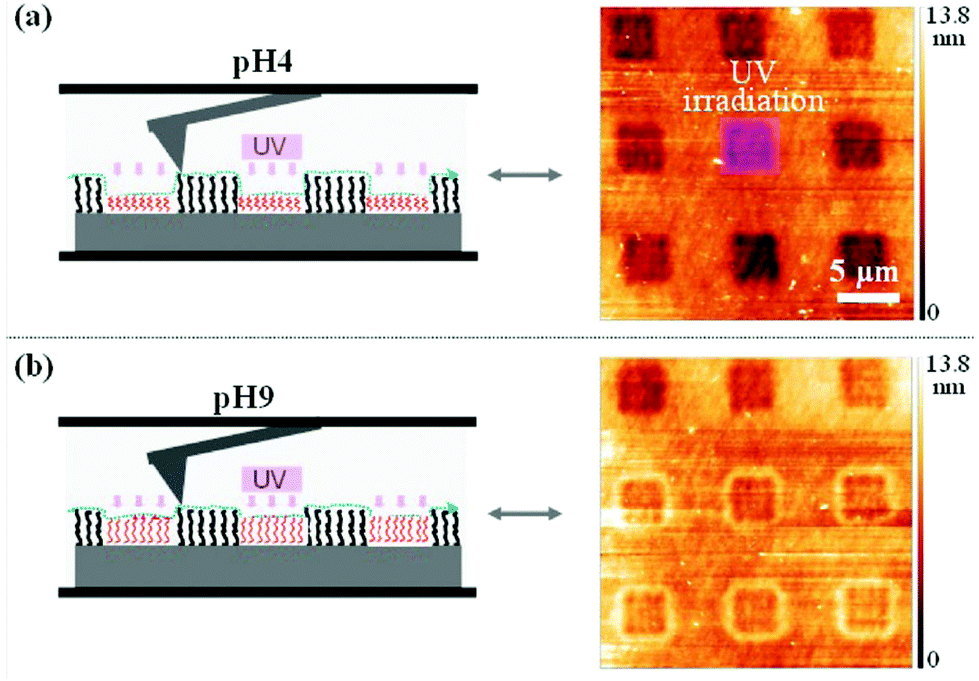 | ||
| Fig. 17 Scanning force microscopy image of the PDMNBA patterned surface at pH = 4 (a) and pH = 9 (b) (reproduced form ref. 102 with permission from the ACS). | ||
Surface wettability can be controlled with photo-responsive polymers. This was demonstrated by Padeste and coworkers.61 Their strategy relies on the post modification of a poly(glycidyl methacrylate) (PGLY) surface with spiropyran moieties. The polymer brushes were synthesized via extreme ultraviolet lithography (EUV). Fluoropolymer foils were irradiated with EUV lithography (λ = 13.5 nm, 92.5 eV) to crack chemical bonds and generate radicals on the surface of the foils. Using these radicals as initiators, PGLY brushes were covalently grafted from the surface. The spyropyran groups were finally grafted on the brush via the nucleophilic attack of the spiropyran amine on the epoxy ring of the PGLY. This technique allows the production of polymer brushes whose dry thickness exceeds that of brushes produced by controlled radical polymerisation. The spiropyran-modified surface was used to modulate surface wettability. Indeed when the surface is irradiated with visible light the photo-responsive moieties adopt their hydrophobic spiropyran form whereas the irradiation by UV light induces the transformation of the spiropyran into its hydrophilic merocyanine form. The reversibility of the hydrophobic/hydrophilic character of the surface was demonstrated by static contact angle of a water droplet (Fig. 18).
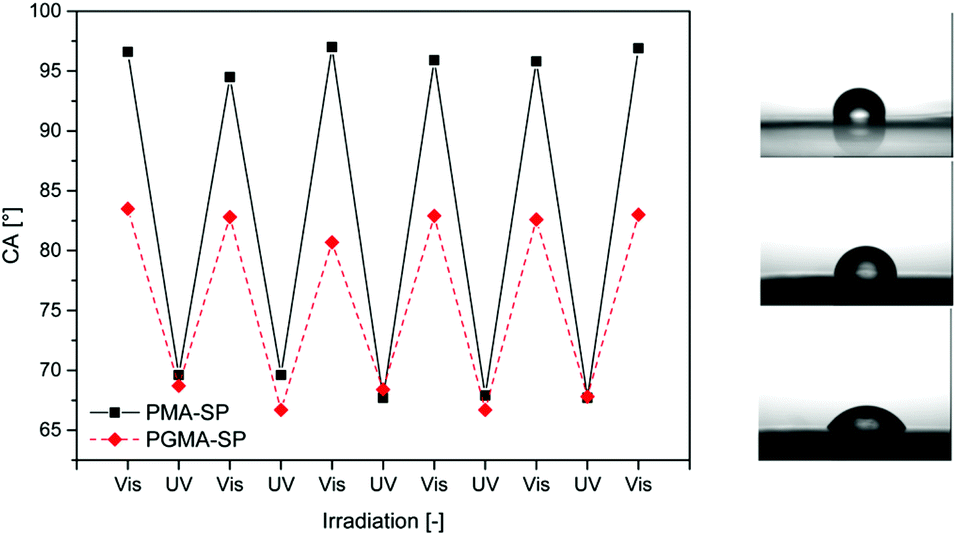 | ||
| Fig. 18 Light induced modification of surface wettability with a spiropyran containing polymer brush (reproduced from ref. 61 with permission from the ACS). | ||
One interesting feature of the spiropyran group can be further used for the photo-detection of the solvent polarity. Indeed, the merocyanine isomers of nitro-substituted spiropyran derivatives present negative solvatochromism, i.e. their absorption bands undergo a hypsochromic (blue) shift in solvents of increasing polarity (Fig. 19a). This feature is interesting for the development of novel miniaturized analytical platforms. The group of Benito-Lopez functionalized fused silica microcapillaries with spiropyran-polymer brushes via ROMP.105 The irradiation by UV lamps of these microcapillaries under continuous flow of different solvents demonstrated the possibility to monitor the solvent polarity. Indeed a change in the UV-Visible absorption spectrum of the PSP capillary was observed when MeOH, EtOH, ACN, acetone, THF, and toluene were passed through the microcapillary after UV irradiation (Fig. 19b). Moreover, variations in the local polarity of the medium may macroscopically be observed (Fig. 19c).
 | ||
| Fig. 19 (a) Negative solvatochromism of the merocyanine isomers of spiropyrans, (b) evolution of the absorption spectra of the spiropyran-functionalized capillary and (c) pictures of the microcapillary when irradiated for 20 s with UV light while different solvents are passed through the microcapillary in continuous flow (reproduced from ref. 105 with permission from the ACS). | ||
Application of PRPs in thin films
In addition to the formation of micelles in solution and of polymer brushes, photo-responsive polymers were used for the formation of thin films.108–111 Two major applications for PRPs in thin films will be presented here: thin film patterning and creation of porous membranes.One example of surface patterning using photo-responsive polymers was presented by Irvine and coworkers. They used a poly(o-nitrobenzyl methacrylate-random-methyl methacrylate-random-ethylene glycol methacrylate) triblock copolymer (P(NBMA-r-MMA-r-EGMA)) to prepare thin film photoresists.112 The photoresist was formed by the successive deposition onto a substrate via spin coating of a film of poly(allylamine hydrochloride) (3 nm) followed by a film of the P(NBMA-r-MMA-r-EGMA) photoresist (130 nm). The irradiation of the thin film led to the cleavage of the o-nitrobenzyl side groups and thus the formation of a P(MMA-r-MMA-r-EGMA) copolymer. The thin film was further washed with a phosphate buffer solution to remove the P(MMA-r-MMA-r-EGMA) copolymer and to create the patterned surface. The conjugation of biotin to the hydroxyl end groups of PEGMA units allowed selective immobilization of streptavidin (SAv-TR and SAv-FITC) on the surface and led to multicomponent protein patterning (Fig. 20). The same authors studied the effect of the photoresist composition on the UV-exposed photoresist solubility. They demonstrated that depending on its composition, the P(MMA-r-MMA-r-EGMA) copolymer can be washed with aqueous solutions of pH = 7.5 to 5.5.113
 | ||
| Fig. 20 Schematic procedure of the dual streptavidin patterning with the P(NBMA-r-MMA-r-EGMA) photoresist and fluorescence spectroscopy of the dual streptavidin patterned surface (a) SAv-TR fluorescence, (b) SAv-FITC fluorescence and (c) overlay (reproduced from ref. 112 with permission from the ACS). | ||
Another interesting photoresist was developed by Ionov and Diez who utilized the combination of thermoresponsive and photo-responsive copolymers for the patterning of thin polymer films. They synthesized a series of copolymers composed of N-isopropylacrylamide and o-nitrobenzyl acrylate.114 Depending on the o-nitrobenzyl acrylate content, the LCST of these polymers can vary up to 50 °C before and after UV irradiation. This temperature-dependent solubility behaviour was used to pattern thin films by UV irradiation. Sequential removal of irradiated and non-irradiated areas could be achieved by washing steps at different temperatures, allowing the patterning of proteins.
Recently del Campo and coworkers developed a photoresist based on a polyurethane containing an o-nitrobenzyl ester.29 This strategy allows the fabrication of a cheap and simple positive photoresist since the photolabile group (1-(2-nitrophenyl)-1,2-ethanediol) is commercially available and can be easily introduced for the synthesis of polyurethane based materials.
Two major strategies can be applied to form nanoporous thin films from photo-responsive block copolymers. The nanoporous channels can be formed either by the irradiation of a block copolymer with a sequence bearing photocleavable side groups or by the illumination of a block copolymer with a photo-responsive junction between the two blocks (Fig. 21). The underlying strategy in both cases is the self-assembly of the block copolymer into the desired structure followed by the cleavage of the photo-responsive groups to release the side groups or the minor block and their removal to create the porosity.
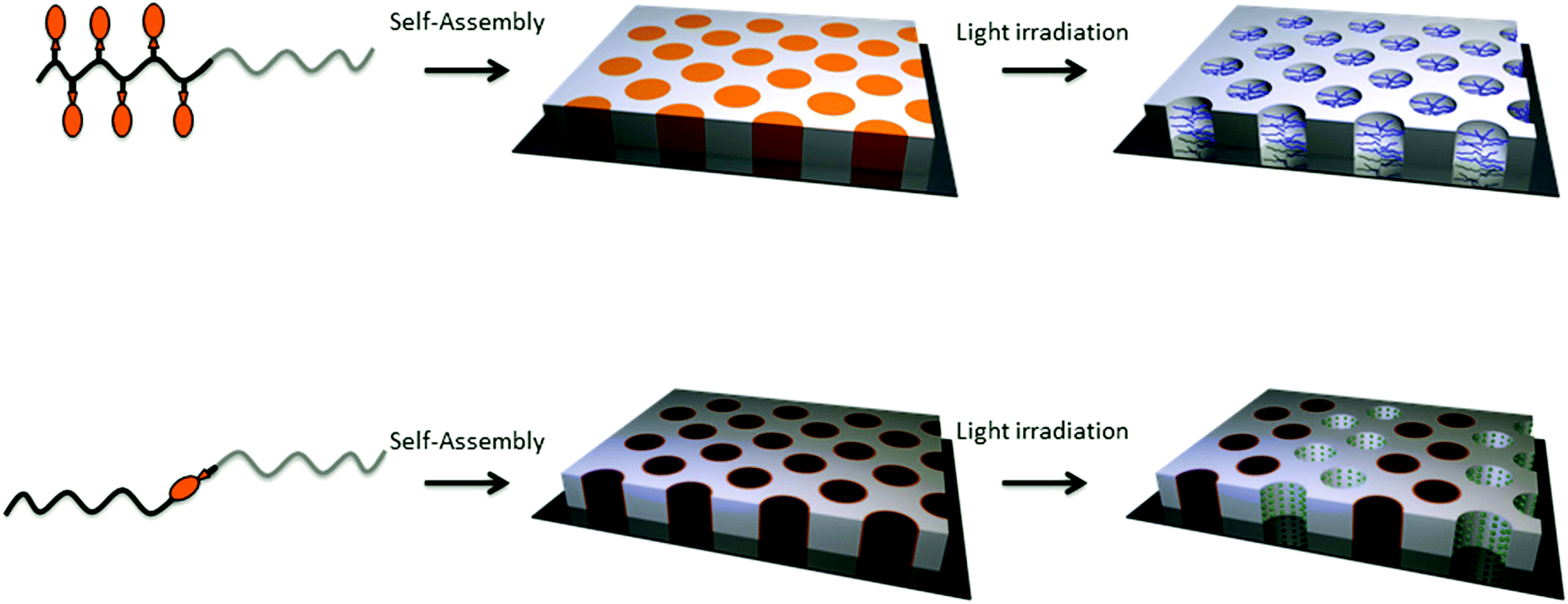 | ||
| Fig. 21 Strategies for the formation of nanoporous thin films based on photo-responsive block copolymers. | ||
The first example of nanoporous thin films based on photo-responsive block copolymers was developed by Russell and Penelle with PS-b-PMMA diblock copolymerd where the two blocks were linked by an anthracene dimer (AA).75 Anthracene is able to undergo a [4 + 4] photodimerisation when irradiated at 366 nm whereas the photo-cycloadduct can be cleaved by irradiation at λ = 280 nm or a temperature higher than 180 °C.74 The authors developed a convergent synthesis for the production of their PMMA-AA-PS copolymer as depicted in Fig. 22. The PS-A was synthesised by anionic polymerisation and the functional chain-end was introduced by quenching the polymerisation with an anthracene derivative. The A-PMMA was synthesized by anionic polymerisation where the anthracene moiety acted as an initiator. The copolymer was obtained after the UV photocoupling of the two homopolymers. The sore point of this approach is the formation of undesired PS-AA-PS and PMMA-AA-PMAA polymers, which have to be extracted from the PMMA-AA-PS block copolymer.75 The authors demonstrated that the self-assembly of the PMMA-AA-PS copolymer in thin films followed by the thermal cleavage of the anthracene dimer and the extraction of the PMMA block led to the formation of nanoporous PS film.74 Although the cleavage by UV irradiation in solution was demonstrated, no characterisation of the thin films under UV irradiation was mentioned.
 | ||
| Fig. 22 UV photocoupling of anthracene end-functionalised PS and PMMA (reproduced from ref. 75 with permission from the ACS). | ||
The first successful use of light to produce nanoporous thin films was realized by Kang and Moon with a PS-b-PEO block copolymer, where the PS block is linked to the PEO block by an o-nitrobenzyl ester junction.115 The copolymer was synthesised by the ATRP of styrene initiated with a PEO macroinitator bearing the photocleavable group. The self-assembly of this block copolymer led to a thin film with a PS matrix and PEO cylinders oriented perpendicular to the substrate. The irradiation of the film at 350 nm induced the cleavage of the o-nitrobenzyl junction and the PEO was selectively extracted from the PS matrix with a methanol/water mixture. The film porosity was demonstrated by TEM and SEM measurements.
A versatile synthetic strategy for the introduction of o-nitrobenzyl ester at the junction of block copolymer was developed by our group.36 The strategy consists in “clicking”, by CuAAC, a photocleavable ATRP initiator on an end-functionalized azide block while, in a simultaneous one-pot process, the second block is polymerised by ATRP. An advantage of this strategy is that the synthesis avoids the preparation of macroinitiators, which are often more difficult to synthesise than small molecule initiators. The versatility of this approach was demonstrated by the synthesis of PEO-b-PtBA, and PS-b-PMMA photocleavable block copolymers. Moreover, this strategy was used for the synthesis of PS-b-PEO copolymers linked by o-nitrobenzyl esters and carbamates.33,34 In both cases the PS-b-PEO block copolymers were self-assembled to obtain thin films with a cylindrical morphology, where the matrix is composed of PS and the cylinders of PEO. After irradiation of the films at 300 nm and the extraction of the PEO with a methanol/water mixture, nanoporous PS thin films were obtained. The film porosity was demonstrated by TEM and SEM measurements. In addition to the formation of nanoporosity, the thin film formation strategy based on o-nitrobenzyl junction allows the formation of functional pores. Indeed, thin films made of o-nitrobenzyl esters or carbamates respectively allow the formation of nanopores functionalized with carboxylic acids34 or primary amines.33 The thin films functionality was demonstrated by the selective grafting of pyrenyl (COOH pores) and coumarin (NH2 pores) chromophores into patterned nanoporous films (Fig. 23).
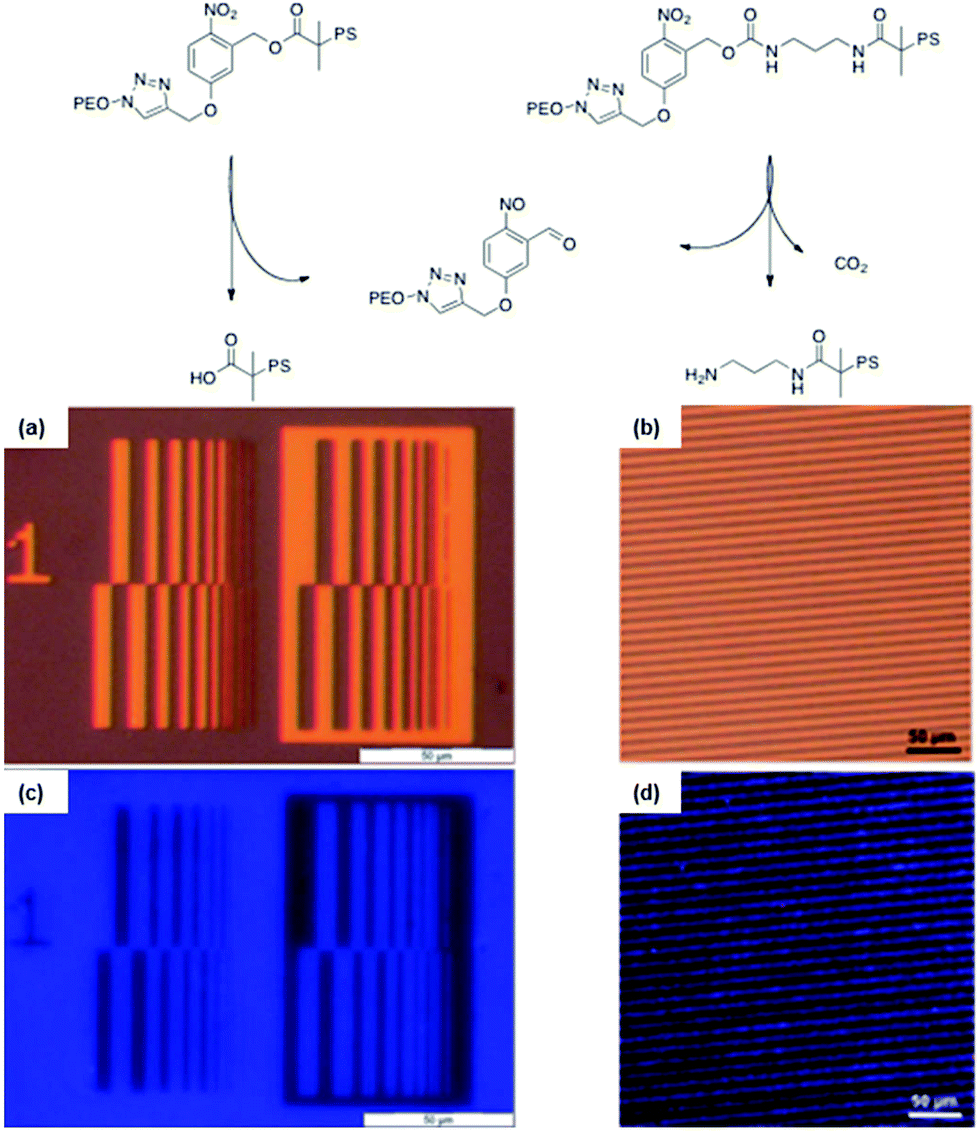 | ||
| Fig. 23 Strategy used to create nanoporous membranes with either carboxylic acids or primary amines functional groups. (a and c) Optical microscopy images of the used photolithographic masks and (b and d) fluorescence microscopy images of the corresponding pattern transferred to a thin film from the block copolymer (reproduced from ref. 33 with permission from the RSC and ref. 34 with permission from Wiley). | ||
Theato, Coughlin, and coworkers adopted a similar approach for the synthesis of linear photocleavable block copolymers by combining RAFT polymerisation and a intermacromolecular CuAAC reaction.116–118 This strategy was used for the synthesis of PS-b-PEO copolymers and the formation of nanoporous thin films. The nanoporous thin films were obtained in three steps: the self-assembly of the block copolymer, the photocleavage of the o-nitrobenzyl junction and the extraction of the PEO block from the thin films. The film porosity was demonstrated by TEM and AFM measurements. Moreover, the accordingly obtained nanoporous thin films were used as templates for preparation of silica nanodots.116
The same authors designed a triblock copolymer based on ethylene oxide, maleimide pentafluorophenyl ester (MAIPFP) and styrene. The photocleavable o-nitrobenzyl junction was introduced via a CuAAC reaction between an azide terminated PEO and the photocleavable RAFT chain transfer agent (CTA). This macroCTA was then used for the polymerisation of MAIPFP and styrene. Due to the similarity of the reactivity ratios of maleimide monomers and styrene, the MAIPFP monomer was precisely incorporated in an alternating fashion with styrene. The excess of styrene was then polymerised to give a triblock copolymer with the following composition PEO-b-P(MAIPFP-alt-S)-b-PS.118 The copolymer was self-assembled into thin films forming a cylindrical morphology with highly ordered hexagonally packed cylinders oriented perpendicular to the substrate. Moreover, the interface between the matrix and the cylinder is composed of the P(MAIPFP-alt-S) reactive blocks. After irradiation of the thin film and extraction of PEO, a PS nanoporous film with reactive functional groups on the pore walls was obtained. To demonstrate the presence of reactive functional groups on the pore walls, the film was modified with 2-aminoethyl-ferrocene via activated ester-amine conjugation chemistry followed by an acidic washing to remove physically absorbed 2-aminoethyl-ferrocene. The modified nanoporous films were then treated with oxygen plasma to decompose all the organic materials while converting ferrocene to iron oxide at the same time and producing iron oxide nanotori (also called nanorings or nanodonuts).
One final example of nanoporous thin films based of PRPs was developed by our group with a copolymer bearing o-nitrobenzyl side groups.21 We synthesized a PNBA-b-PS diblock copolymer by the combination of ATRP and a post modification reaction. Thin films from these block copolymers with PNBA cylinders in a PS matrix were obtained after spin coating and solvent annealing of the films. The annealing with benzene/nitromethane mixed solvents allowed the formation of PNBA cylinders perpendicular to the surface. The o-nitrobenzyl side groups were then selectively removed by exposing the films to UV irradiation and the 2-nitrosobenzaldehyde photoproducts were extracted by soaking the films in pure methanol. The accordingly obtained nanoporous PS films display PAA hairy pores The accessibility of the remaining carboxylic acids was then proven by reacting them with a fluorescent dye, demonstrating thereby the possibility to perform controlled chemistry inside the cylindrical pores.
Application of PRPs networks: gels and hydrogels
Photo-responsive (co)polymers are vastly used for drug release, nanofabrication and sensing applications, but their application is not limited to these topics. Indeed, photo-responsive groups were also introduced in polymeric material for the formation of gels and hydrogels.119–123 Photo-responsive gels can be classified into two categories: the gels based on physical crosslinking and gels based on chemical crosslinking. In the first category the gel is composed of polymer chains crosslinked between each other by covalent bonds whereas the second category is characterised by physical crosslinking, which is the consequence of the formation of clusters of collapsed chains in the considered solvent.Turro and coworkers were the first to design a crosslinked network based on o-nitrobenzyl.124 The PRPs were synthesised in two steps: first a four-arm star polymer containing an o-nitrobenzyl linker was produced by the ATRP of tert-butyl acrylate followed by the post modification of the terminal bromide by an azide. In the second step, the accordingly obtained PtBA was crosslinked with difunctional alkynes under CuAAC conditions to form 1,2,3-triazole linkages (Fig. 24a). The UV irradiation of this network allows the degradation of the material. Yamamoto and coworkers developed a polymeric system based on PEO and coumarin.125 Their synthetic strategy relies in the esterification of dihydroxy PEO with coumarins bearing phenyl-3,5-diacyl chloride functions. The formation of the hydrogel was promoted by the irradiation of the polymer at wavelengths >280 nm, which induced the dimerisation of the coumarin units. They also demonstrated that the degree of swelling of the hydrogel could be influenced by the irradiation time, i.e. the dimerization degree (Fig. 24b).
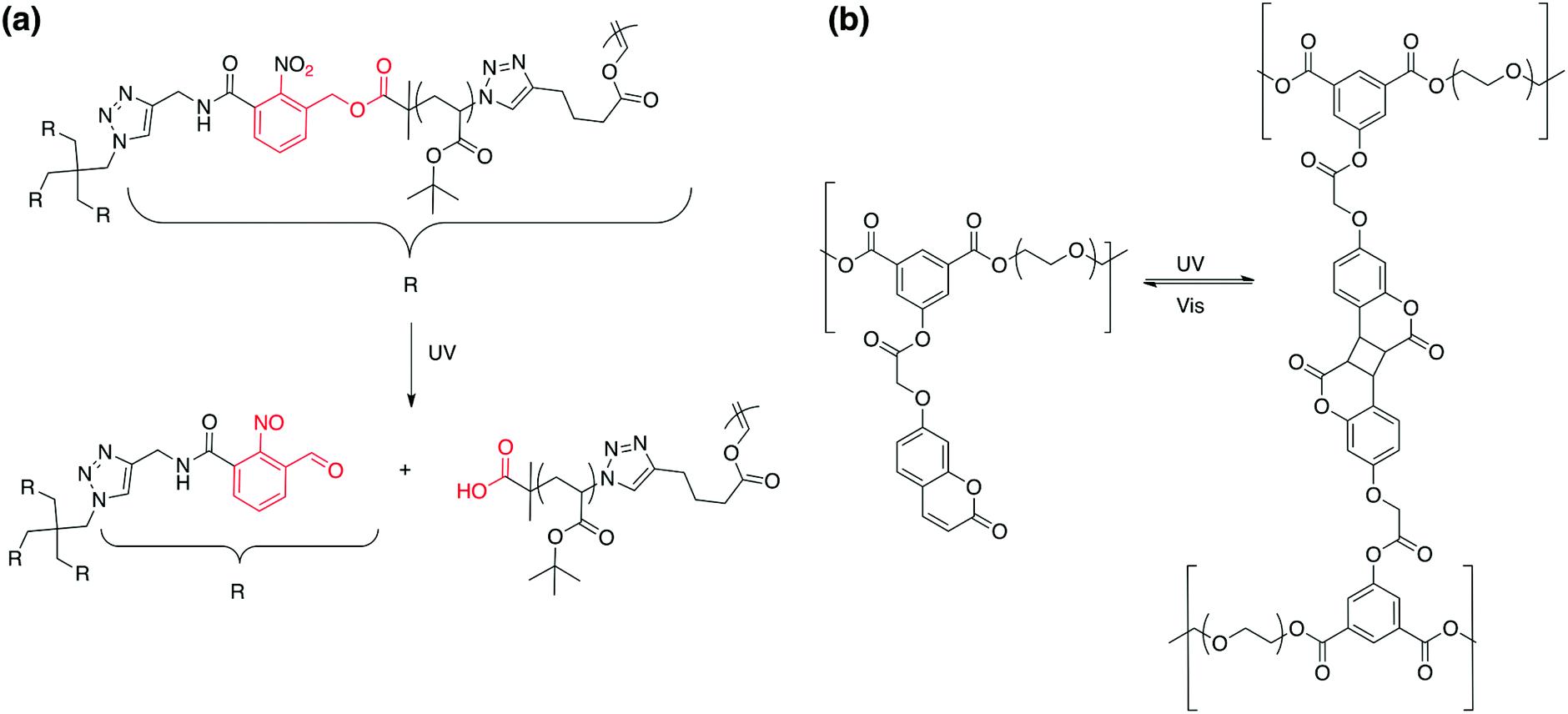 | ||
| Fig. 24 Photo-responsive chemical hydrogels based on (a) o-nitrobenzyl esters and on (b) coumarins. | ||
An interesting characteristic feature of hydrogels compared to micelles and nanocapsules is that the cavities in the hydrogels allow the encapsulation of large guest molecules, such as proteins and enzymes. Zhao and coworkers used this characteristics for the release on demand of guest molecules from photocleavable hydrogels by NIR light using upconverting nanoparticles.126 They designed a polymer based on PEO, polyacrylamide and o-nitrobenzyl esters. The hydrogel based on this polymer was loaded with NaYF4:TmYb up-converting nanoparticles and fluorescein isothiocyanate bovine serum albumin (FITC-BSA). The UV irradiation of the hydrogel by NIR light at 980 nm led to the emission of the photons at λ = 350 nm by the up-converting nanoparticles and to the irreversible photocleavage of the o-nitrobenzyl esters. The release of FITC-BSA in solution was monitored by fluorescence spectroscopy.
Langer and coworkers designed a polymeric network based on cinnamic esters for shape memory applications (Fig. 25 left). This polymer network is obtained by the copolymerisation of n-butylacrylate (BA) and hydroxyethyl methacrylate (HEMA), ethyleneglycol-1-acrylate-2-cinnamic ester (HEA-CA) with poly(propylene glycol) dimethacrylate as crosslinker.72 When the photo-responsive copolymer is stretched, the coil segments of the polymer chains are elongated between the crosslinking points (Fig. 25a). The irradiation of the elongated polymer with UV light (λ > 260 nm) induces the coupling between the cinnamic moieties and thus the formation of a new photo-responsive network, which partially fixes the stretching of the chains (Fig. 25b). The photo de-crosslinking of the network is triggered by the irradiation of the gel at wavelengths below 260 nm. As a result, the fixed elongated film can recover to the original permanent shape (Fig. 25c).
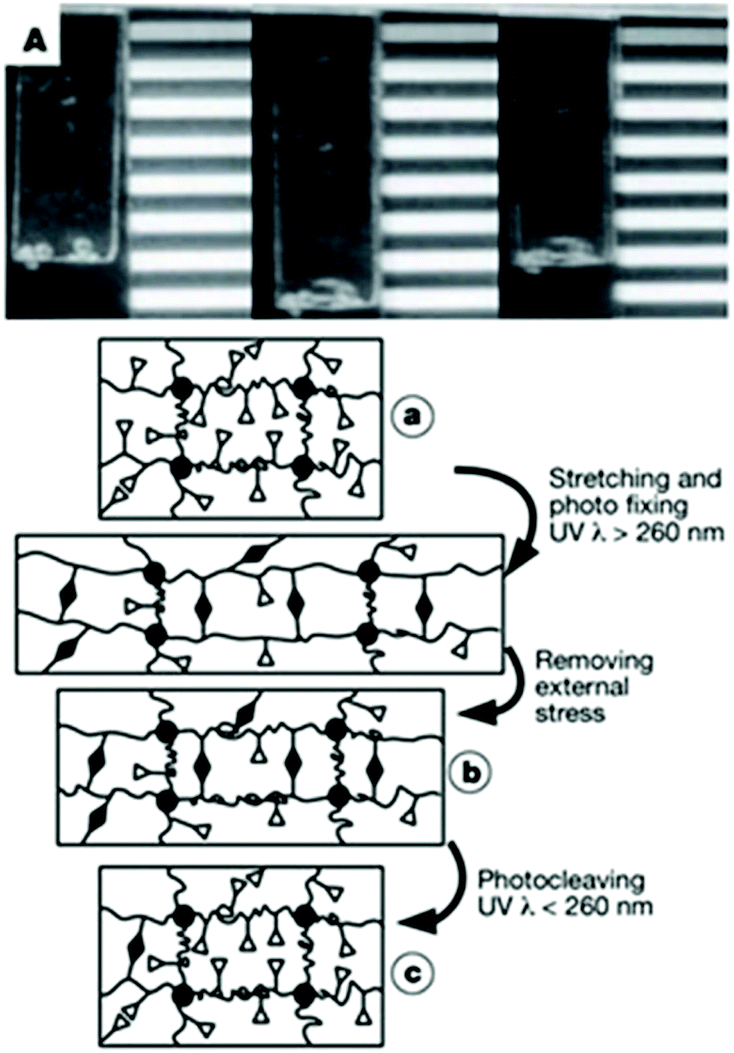 | ||
| Fig. 25 Shape memory polymers based on cinnamic esters: (top) photograph of the shape memory polymer material (permanent, temporary and recovered shapes) and (bottom) schematic representation of the molecular mechanism of the shape memory effect (reproduced from ref. 72 with permission from NPG). | ||
Bettinger and coworkers were interested in the light remodelling of physically cross-linked hydrogels. In this respect, they studied the formation of hydrogels based on PNBA-b-PEO-b-PNBA127 and P(MMA-r-CMA)-b-PEO-b-P(MMA-r-CMA)128 triblock copolymers. The copolymers were synthesised by a post modification approach where o-nitrobenzyl bromide and 6-bromo-4-chloromethyl-7-hydroxycoumarin were grafted onto a PMAA-b-PEO-b-PMAA triblock copolymer. The hydrogels were prepared by precipitation of the copolymer in water. This precipitation led to the self-assembly of the PBNA/P(MMA-r-CMA) blocks to form hydrophobic clusters linked by PEO chains (Fig. 18). The irradiation of the polymer led to the photocleavage of the nitrobenzyl/coumarin side groups and the formation of PMAA-b-PEO-b-PMAA (Fig. 26). Since the PMAA-b-PEO-b-PMAA is soluble in water, the gel disintegrates. The hydrogel disintegration was monitored with the evolution of the storage modulus of the hydrogel during the photocleavage of the side groups in the case of the PNBA-b-PEO-b-PNBA copolymer.127 The authors also demonstrated that the formation of microstructures on the surface of the hydrogels is possible by photolithography techniques.128
 | ||
| Fig. 26 Schematic representation of the light induced disintegration of physically crosslinked hydrogels (light blue segments, red squares and blue squares respectively represent the PEO chains, the NBA functions and the MAA functions, reproduced from ref. 127 with permission from the ACS). | ||
Building nanostructures with light: thinking outside the box
In this review, we presented the different photo-responsive groups that can be incorporated into block copolymers and the type of nanostructures that can be obtained via the irradiation of such polymers. In addition, some other strategies relying on the use of light to synthesize block copolymers that further self-assemble into nanostructures are worth being mentioned. One of these strategies relies on the synthesis of polymeric nanostructures via photo-induced control radical polymerisations.129–132 Those polymerisation techniques allow the control of the polymerisation via a light irradiation. The potential of these techniques was recently demonstrated by Boyer and coworkers.133 By using a selective photo-activation of thiocarbonylthio compounds with different types of chromophores, they were able to produce block copolymers with perfectly controlled block composition. They also demonstrated that it was possible via this approach to generate complex macromolecular architectures. By mixing two different types of chromophores able to selectively activate two different RAFT chain transfer agents, they were able to synthesize a polymer brush by the successive irradiation of the polymerisation mixture at different wavelengths.Building nanostructures with photo-polymerisation is not limited to the control of the composition and architecture of the polymer but also to the formation of micelles in solution or for the functionalisation of surfaces. An example of photo-initiated polymerisation-induced self-assembly (photo-PISA) was described recently by the group of Zhang and co-workers. They use a PEG modified CTA to photo-initiate the polymerisation of 2-hydroxypropyl methacrylate (HPMA) in water. Since HPMA is soluble in water, the polymerisation can occur and a PEG-b-PHPMA copolymer was produced. On the other hand PHPMA is insoluble in water and thus the polymerisation induces self-assembly into micelles.134 Moreover, the morphology of the self-assembled nanostructures can be modulated via this photo-PISA technique by varying the degree of polymerisation of the hydrophobic block. Indeed, Zhang et al. were able to produce spherical micelles, worms, lamellae, unilamellar vesicles, jellyfishes, branched worms, and multilamellar vesicles with their PEG-b-PHPMA copolymers (Fig. 27). A similar example was recently published by Boyer and co-workers with the photo-PISA of poly((oligo ethylene glycol) methacrylate)-block-poly (benzyl methacrylate) (POEGMA-b-PBzMA) in ethanol/acetonitrile mixtures.135 They also were able to generate different morphologies depending on the degree of polymerisation of the PBzMA block.
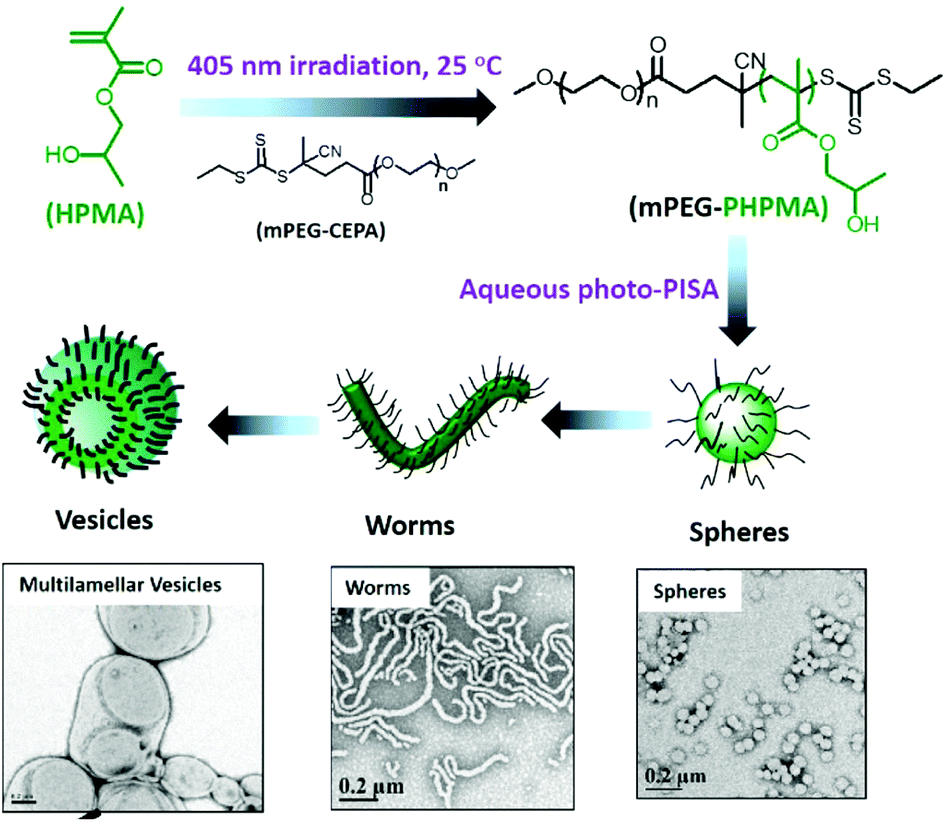 | ||
| Fig. 27 Preparation of various nano-objects via aqueous photo-PISA and TEM image of the nanostructures (adapted from ref. 134 with permission from the ACS). | ||
As stated in the introduction, light is a stimulus that can be localized in time and space and this characteristic is a real added value to produce well-defined patterned surfaces. In the previous section we presented light-induced patterning techniques relying on the modification of the polymer nature. Hawker and co-workers adopted a strategy relying on the use of photo-polymerisation and irradiation mask in order to produce patterned surfaces.136,137 These authors used a silicon surface modified by a polymerisation initiator. The irradiation of the substrate through a photo-mask in the presence of an Iridium photo-catalyst allowed the growth of PMMA polymer brushes in the exposed areas (Fig. 28). Moreover, depending on the type of used photo-mask, patterned structures displaying an on–off behaviour or brushes with a gradient height profile could be obtained.
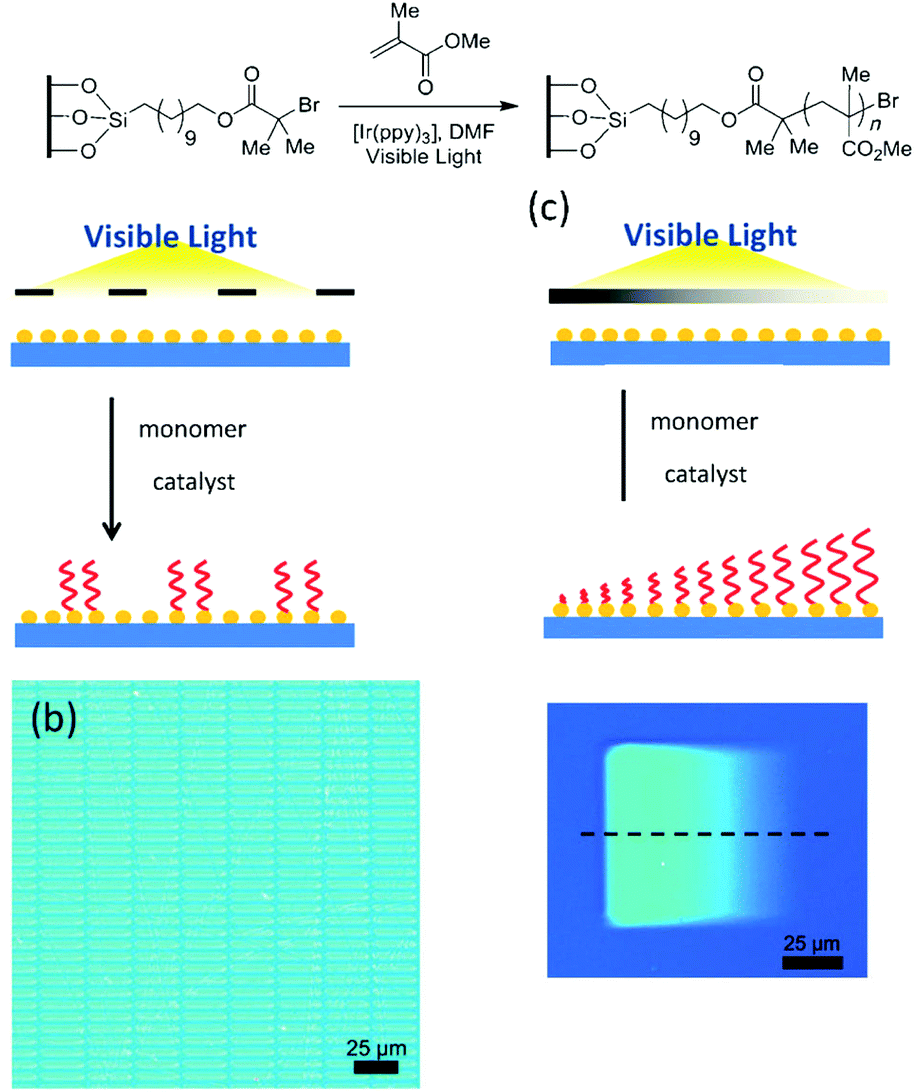 | ||
| Fig. 28 Patterning surfaces with photo-polymerisation: schematic of the photo-patterning process and optical microscopy image of the patterned surfaces (reproduced from ref. 136 with permission from Wiley). | ||
This strategy was also successfully applied for the production of patterned surfaces with coumarin and fluorescein molecules.137 In this case, a N-hydroxysuccinimide (NHS) ester derivative was first polymerised under irradiation through the photo-mask, then the patterned polymer brushes were functionalised with the coumarin. In a second step, a monomer bearing biotin groups was polymerised to cover the rest of the surface. Then the surface was functionalised with avidine-fluorescein. They also demonstrated that this strategy could be employed to stabilise iron nanoparticles on a patterned surface.137
Conclusions
In this review, we have discussed the different photoresponsive groups that can be incorporated into block copolymers. Firstly, we have focused on self-assembly and dis-assembly of the light-responsive block copolymers in dilute solutions to form or to disintegrate micellar structures. We have classified the different systems based on the reversible or irreversible character of their photo-induced structural or morphological changes. We have also discussed systems where light is used to stabilise the micellar structure by crosslinking.In order to provide the reader a general overview of the different PRPs that have been detailed in this chapter, Table 1 summarizes the different photo-responsive moieties introduced in polymer materials. The effect of light irradiation is presented for each photo-responsive group and the reversible character of the photochemical reaction is stated. The design of each PRP is described emphasizing the location of the photo-responsive group: as side-groups, incorporated in the main chain or included as a single group at the block junction. Finally, the polymerisation techniques used to synthesize PRPs are listed.
| Photosensitive group | Effect of light | Reversibility | Location | Polymerisation technique |
|---|---|---|---|---|
| AP: anionic polymerisation; ATRP: atom transfer radical polymerisation; FRP: free radical polymerisation; RAFT: reversible addition–fragmentation chain transfer polymerisation; ROP: ring opening polymerisation; SGP: step-growth polymerisation. The structure of the photosensitive moieties can be found in Fig. 3 and 9. | ||||
| Pyrenylmethyl ester | Photocleavage | No | Side chain11,12 | ATRP11,12 |
| o-Nitrobenzyl ester | Photocleavage | No | Side chain,13–25 main chain26–30 and at block junction31–38 | ATRP,13–17,31–36 RAFT,18,19,37,38 SGP,26–29 SET-LRP,24 ROP,25 and post modification20–23,30 |
| Coumarinyl ester | Photocleavage | No | Side chain39 and main chain40 | ATRP39 and SGP40 |
| p-Methoxyphenacyl ester | Photocleavage | No | Side chain41–43 and main chain40 | SGP,40 FRP41,44 and ATRP42,43 |
| Diazonaphtoquinone | Wolff rearrangement | No | Side chain45,46 | Post modification45,46 |
| Azobenzene | Photoisomerisation | Yes | Side chain,47–52 main chain53,54 and at block junction55,56 | ATRP,47–49 RAFT,50,51,55,56 SGP54 and post modification52,53 |
| Spiropyran | Photoisomerisation | Yes | Side chain57–61 | ATRP,57,58 FRP,59 SET-LRP60 and post modification60,61 |
| Dithienylethene | Photoisomerisation | Yes | Side chain63 | FRP63 |
| Coumarin | Photocycloaddition | Yes | Side chain64–68 | ATRP64–67 and RAFT68 |
| Truxillic acid or cinnamic ester | Photocycloaddition | No | Side chain69–72 and at block junction73 | ATRP73 and post modification69–72 |
| Anthracene | Photocycloaddition | Yes | At block junction74,75 | AP74,75 |
It is clear that a wide variety of photochromic moieties as well as synthetic strategies to introduce them in defined locations of macromolecular architectures are now made available to the chemist. However many challenges still need to be addressed, especially as far as applications of those systems are concerned. In this respect, we have shortly discussed the obstacles to overcome for the application of such systems for drug delivery applications. We have also discussed the formation of photo-responsive surfaces and how nanoporous thins films with interesting functionalities can be obtained from light-responsive block copolymers. Finally, we have discussed the use of photo-responsive polymers to make interesting stimuli-responsive gels. A whole library of different light-responsive copolymers with adjustable properties is now available. It is clear that those polymers will be extensively used by material scientists in the upcoming years to create new materials with unique properties.
Acknowledgements
The authors thank the SPW DG06 for financing the BATWAL and HYB2HYB projects.Notes and references
- A. P. Esser-Kahn, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Macromolecules, 2011, 44, 5539–5553 CrossRef CAS.
- B. P. Timko, T. Dvir and D. S. Kohane, Adv. Mater., 2010, 22, 4925–4943 CrossRef CAS PubMed.
- F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3–23 CrossRef CAS.
- H. G. Börner, H. Kühnle and J. Hentschel, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1–14 CrossRef.
- S. Chatani, C. J. Kloxin and C. N. Bowman, Polym. Chem., 2014, 5, 2187–2201 RSC.
- Q. Yan, D. Han and Y. Zhao, Polym. Chem., 2013, 4, 5026–5037 RSC.
- J.-F. Gohy and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7117–7130 RSC.
- Y. Zhao, Macromolecules, 2012, 45, 3647–3657 CrossRef CAS.
- H. Zhao, E. S. Sterner, E. B. Coughlin and P. Theato, Macromolecules, 2012, 45, 1723–1736 CrossRef CAS.
- J.-M. Schumers, C.-A. Fustin and J.-F. Gohy, Macromol. Rapid Commun., 2010, 31, 1588–1607 CrossRef CAS PubMed.
- J. Jiang, X. Tong and Y. Zhao, J. Am. Chem. Soc., 2005, 127, 8290–8291 CrossRef CAS PubMed.
- S. Menon and S. Das, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4448–4457 CrossRef CAS.
- J. Jiang, X. Tong, D. Morris and Y. Zhao, Macromolecules, 2006, 39, 4633–4640 CrossRef CAS.
- M. Lepage, J. Jiang, J. Babin, B. Qi, L. Tremblay and Y. Zhao, Phys. Med. Biol., 2007, 52, N249–N255 CrossRef CAS PubMed.
- X. Jiang, C. A. Lavender, J. W. Woodcock and B. Zhao, Macromolecules, 2008, 41, 2632–2643 CrossRef CAS.
- X. Jiang, S. Jin, Q. Zhong, M. D. Dadmun and B. Zhao, Macromolecules, 2009, 42, 8468–8476 CrossRef CAS.
- J.-M. Schumers, C.-A. Fustin, A. Can, R. Hoogenboom, U. S. Schubert and J.-F. Gohy, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6504–6513 CrossRef CAS.
- X. Wang, G. Liu, J. Hu, G. Zhang and S. Liu, Angew. Chem., Int. Ed., 2014, 53, 3138–3142 CrossRef CAS PubMed.
- Y. Li, Y. Qian, T. Liu, G. Zhang and S. Liu, Biomacromolecules, 2012, 13, 3877–3886 CrossRef CAS PubMed.
- O. Bertrand, J.-M. Schumers, C. Kuppan, J. Marchand-Brynaert, C.-A. Fustin and J.-F. Gohy, Soft Matter, 2011, 7, 6891–6896 RSC.
- J.-M. Schumers, O. Bertrand, C.-A. Fustin and J.-F. Gohy, J. Polym. Sci., Part A: Polym. Chem., 2011, 50, 599–608 CrossRef.
- X. Liu, J. He, Y. Niu, Y. Li, D. Hu, X. Xia, Y. Lu and W. Xu, Polym. Adv. Technol., 2015, 26, 449–456 CrossRef CAS.
- Q. Jin, T. Cai, Y. Wang, H. Wang and J. Ji, ACS Macro Lett., 2014, 3, 679–683 CrossRef CAS.
- S. M. Abdellatif Soliman, C. Nouvel, J. Babin and J.-L. Six, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2192–2201 CrossRef CAS.
- G. Liu and C.-M. Dong, Biomacromolecules, 2012, 13, 1573–1583 CrossRef CAS PubMed.
- D. Han, X. Tong and Y. Zhao, Macromolecules, 2011, 44, 437–439 CrossRef CAS.
- J. T. McIntosh, A. Nazemi, C. V. Bonduelle, S. Lecommandoux and E. R. Gillies, Can. J. Chem., 2015, 93, 126–133 CrossRef CAS.
- D. Han, X. Tong and Y. Zhao, Langmuir, 2012, 28, 2327–2331 CrossRef CAS PubMed.
- L. García-Fernández, A. Specht and A. del Campo, Macromol. Rapid Commun., 2014, 35, 1801–1807 CrossRef PubMed.
- B. Fan, J. F. Trant, A. D. Wong and E. R. Gillies, J. Am. Chem. Soc., 2014, 136, 10116–10123 CrossRef CAS PubMed.
- J. Xuan, D. Han, H. Xia and Y. Zhao, Langmuir, 2014, 30, 410–417 CrossRef CAS PubMed.
- O. Bertrand, E. Poggi, J.-F. Gohy and C.-A. Fustin, Macromolecules, 2014, 47, 183–190 CrossRef CAS.
- C. G. Gamys, J.-M. Schumers, A. Vlad, C.-A. Fustin and J.-F. Gohy, Soft Matter, 2012, 8, 4486–4493 RSC.
- J.-M. Schumers, A. Vlad, I. Huynen, J.-F. Gohy and C.-A. Fustin, Macromol. Rapid Commun., 2011, 33, 199–205 CrossRef PubMed.
- E. Cabane, V. Malinova and W. Meier, Macromol. Chem. Phys., 2010, 211, 1847–1856 CrossRef CAS.
- J.-M. Schumers, J.-F. Gohy and C.-A. Fustin, Polym. Chem., 2010, 1, 161–163 RSC.
- H. Zhou, Y. Lu, H. Qiu, G. Guerin, I. Manners and M. A. Winnik, Macromolecules, 2015, 48, 2254–2262 CrossRef CAS.
- Y. Gao, H. Qiu, H. Zhou, X. Li, R. Harniman, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2015, 137, 2203–2206 CrossRef CAS PubMed.
- J. Babin, M. Pelletier, M. Lepage, J.-F. Allard, D. Morris and Y. Zhao, Angew. Chem., Int. Ed., 2009, 48, 3329–3332 CrossRef CAS PubMed.
- E. A. Chamsaz, S. Sun, M. V. S. N. Maddipatla and A. Joy, Photochem. Photobiol. Sci., 2014, 13, 412–421 CAS.
- M. Millaruelo, K. J. Eichhorn, B. Sieczkowska and B. Voit, Langmuir, 2006, 22, 9436–9445 CrossRef CAS PubMed.
- O. Bertrand, J.-F. Gohy and C.-A. Fustin, Polym. Chem., 2011, 2, 2284–2292 RSC.
- O. Bertrand, C.-A. Fustin and J.-F. Gohy, ACS Macro Lett., 2012, 1, 949–953 CrossRef CAS.
- M. Millaruelo, L. M. Eng, M. Mertig, B. Pilch, U. Oertel, J. Opitz, B. Sieczkowska, F. Simon and B. Voit, Langmuir, 2006, 22, 9446–9452 CrossRef CAS PubMed.
- C.-J. Chen, G.-Y. Liu, Y.-T. Shi, C.-S. Zhu, S.-P. Pang, X.-S. Liu and J. Ji, Macromol. Rapid Commun., 2011, 32, 1077–1081 CrossRef CAS PubMed.
- Y. Yuan, Z. Wang, P. Cai, J. Liu, L.-D. Liao, M. Hong, X. Chen, N. Thakor and B. Liu, Nanoscale, 2015, 7, 3067–3076 RSC.
- G. Wang, X. Tong and Y. Zhao, Macromolecules, 2004, 37, 8911–8917 CrossRef CAS.
- X. Tong, G. Wang, A. Soldera and Y. Zhao, J. Phys. Chem. B, 2005, 109, 20281–20287 CrossRef CAS PubMed.
- E. Blasco, B. V. K. J. Schmidt, C. Barner-Kowollik, M. Piñol and L. Oriol, Polym. Chem., 2013, 4, 4506–4514 RSC.
- W. Su, Y. Luo, Q. Yan, S. Wu, K. Han, Q. Zhang, Y. Gu and Y. Li, Macromol. Rapid Commun., 2007, 28, 1251–1256 CrossRef CAS.
- T. Ueki, Y. Nakamura, T. P. Lodge and M. Watanabe, Macromolecules, 2012, 45, 7566–7573 CrossRef CAS.
- D. Wang, J. Liu, G. Ye and X. Wang, Polymer, 2009, 50, 418–427 CrossRef CAS.
- P. J. Roth, F. D. Jochum, F. R. Forst, R. Zentel and P. Theato, Macromolecules, 2010, 43, 4638–4645 CrossRef CAS.
- S. Wiktorowicz, V. Aseyev and H. Tenhu, Polym. Chem., 2012, 3, 1126–1129 RSC.
- B. V. K. J. Schmidt, M. Hetzer, H. Ritter and C. Barner-Kowollik, Macromolecules, 2013, 46, 1054–1065 CrossRef CAS.
- Q. Yan, Y. Xin, R. Zhou, Y. Yin and J. Yuan, Chem. Commun., 2011, 47, 9594–9596 RSC.
- H.-I. Lee, W. Wu, J. K. Oh, L. Mueller, G. Sherwood, L. Peteanu, T. Kowalewski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2007, 46, 2453–2457 CrossRef CAS PubMed.
- S. Menon, R. M. Ongungal and S. Das, Polym. Chem., 2013, 4, 623–628 RSC.
- Q. Xing, N. Li, Y. Jiao, D. Chen, J. Xu, Q. Xuand and J. Lu, RSC Adv., 2015, 5, 5269–5276 RSC.
- Y.-N. Zhou, Q. Zhang and Z.-H. Luo, Langmuir, 2014, 30, 1489–1499 CrossRef CAS PubMed.
- M. Dübner, N. D. Spencer and C. Padeste, Langmuir, 2014, 30, 14971–14981 CrossRef PubMed.
- V. K. Kotharangannagari, A. Sánchez-Ferrer, J. Ruokolainen and R. Mezzenga, Macromolecules, 2011, 44, 4569–4573 CrossRef CAS.
- S.-J. Lim, C.-J. Carling, C. C. Warford, D. Hsiao, B. D. Gates and N. R. Branda, Dyes Pigm., 2011, 89, 230–235 CrossRef CAS.
- J. Jiang, B. Qi, M. Lepage and Y. Zhao, Macromolecules, 2007, 40, 790–792 CrossRef CAS.
- J. Babin, M. Lepageand and Y. Zhao, Macromolecules, 2008, 41, 1246–1253 CrossRef CAS.
- J. He, X. Tong and Y. Zhao, Macromolecules, 2009, 42, 4845–4852 CrossRef CAS.
- J. He, X. Tong, L. Tremblay and Y. Zhao, Macromolecules, 2009, 42, 7267–7270 CrossRef CAS.
- J. Luo, Q. Zhou, J. Sun, J. Jiang, X. Zhou, H. Zhang and X. Liu, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4037–4045 CrossRef CAS.
- A. Guo, G. Liu and J. Tao, Macromolecules, 1996, 29, 2487–2493 CrossRef CAS.
- J. Ding and G. Liu, Chem. Mater., 1998, 10, 537–542 CrossRef CAS.
- M. Rabnawaz and G. Liu, Macromolecules, 2012, 45, 5586–5595 CrossRef CAS.
- A. Lendlein, H. Jiang, O. Jünger and R. Langer, Nature, 2005, 434, 879–882 CrossRef CAS PubMed.
- H. Yang, L. Jia, Z. Wang, A. Di-Cicco, D. Lévy and P. Keller, Macromolecules, 2011, 44, 159–165 CrossRef CAS.
- J. T. Goldbach, K. A. Lavery, J. Penelle and T. P. Russell, Macromolecules, 2004, 37, 9639–9645 CrossRef CAS.
- J. T. Goldbach, T. P. Russell and J. Penelle, Macromolecules, 2002, 35, 4271–4276 CrossRef CAS.
- N. Fomina, J. Sankaranarayanan and A. Almutairi, Adv. Drug Delivery Rev., 2012, 64, 1005–1020 CrossRef CAS PubMed.
- A. Rösler, G. W. M. Vandermeulen and H.-A. Klok, Adv. Drug Delivery Rev., 2012, 64, 270–279 CrossRef.
- H. Meier and K.-P. Zeller, Angew. Chem., Int. Ed. Engl., 1975, 14, 32–43 CrossRef.
- S. Li, S. Ji, Z. Zhou, G. Chen and Q. Li, Macromol. Chem. Phys., 2015, 216, 1192–1200 CrossRef CAS.
- G. Liu, X. Wang, J. Hu, G. Zhang and S. Liu, J. Am. Chem. Soc., 2014, 136, 7492–7497 CrossRef CAS PubMed.
- J. S. Katz, S. Zhong, B. G. Ricart, D. J. Pochan, D. A. Hammer and J. A. Burdick, J. Am. Chem. Soc., 2010, 132, 3654–3655 CrossRef CAS PubMed.
- E. Cabane, V. Malinova, S. Menon, C. G. Palivan and W. Meier, Soft Matter, 2011, 7, 9167–9176 RSC.
- D. Wang and X. Wang, Prog. Polym. Sci., 2013, 38, 271–301 CrossRef CAS.
- H. Yu and T. Kobayashi, Molecules, 2010, 15, 570–603 CrossRef CAS PubMed.
- K. G. Yager and C. J. Barrett, J. Photochem. Photobiol., A, 2006, 182, 250–261 CrossRef CAS.
- F. D. Jochum and P. Theato, Macromolecules, 2009, 42, 5941–5945 CrossRef CAS.
- Y. Zhao, L. Tremblay and Y. Zhao, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4055–4066 CrossRef CAS.
- J. He, L. Tremblay, S. Lacelle and Y. Zhao, Polym. Chem., 2014, 5, 5403–5411 RSC.
- D. Wang, G. Ye and X. Wang, Macromol. Rapid Commun., 2007, 28, 2237–2243 CrossRef CAS.
- K. Han, W. Su, M. Zhong, Q. Yan, Y. Luo, Q. Zhang and Y. Li, Macromol. Rapid Commun., 2008, 29, 1866–1870 CrossRef CAS.
- S. Wiktorowicz, H. Tenhu and V. Aseyev, Polym. Chem., 2013, 4, 2898–2906 RSC.
- S. Wiktorowicz, H. Tenhu and V. Aseyev, Macromolecules, 2013, 46, 6209–6216 CrossRef CAS.
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- M. Inoue, K. Noda and S.-I. Yusa, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2596–2603 CrossRef CAS.
- P. Anilkumar, E. Gravel, I. Theodorou, K. Gombert, B. Thézé, F. Ducongé and E. Doris, Adv. Funct. Mater., 2014, 24, 5246–5252 CrossRef CAS.
- Q. Jin, F. Mitschang and S. Agarwal, Biomacromolecules, 2011, 12, 3684–3691 CrossRef CAS PubMed.
- B. Yan, J.-C. Boyer, N. R. Branda and Y. Zhao, J. Am. Chem. Soc., 2011, 133, 19714–19717 CrossRef CAS PubMed.
- S. Kumar, J.-F. Allard, D. Morris, Y. L. Dory, M. Lepage and Y. Zhao, J. Mater. Chem., 2012, 22, 7252–7257 RSC.
- T. Chen, R. Ferris, J. Zhang, R. Ducker and S. Zauscher, Prog. Polym. Sci., 2010, 35, 94–112 CrossRef CAS.
- H.-I. Lee, J. Pietrasik, S. S. Sheiko and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 24–44 CrossRef CAS.
- J. Deng, X. Liu, W. Shi, C. Cheng, C. He and C. Zhao, ACS Macro Lett., 2014, 3, 1130–1133 CrossRef CAS.
- J. Cui, T.-H. Nguyen, M. Ceolín, R. Berger, O. Azzaroni and A. del Campo, Macromolecules, 2012, 45, 3213–3220 CrossRef CAS.
- J. Cui, O. Azzaroni and A. del Campo, Macromol. Rapid Commun., 2011, 32, 1699–1703 CrossRef CAS PubMed.
- Y. S. Park, Y. Ito and Y. Imanishi, Macromolecules, 1998, 31, 2606–2610 CrossRef CAS.
- L. Florea, A; McKeon, D. Diamond and F. Benito-Lopez, Langmuir, 2013, 29, 2790–2797 CrossRef CAS PubMed.
- H. A. Haque, S. Nagano and T. Seki, Macromolecules, 2012, 45, 6095–6103 CrossRef CAS.
- A. Dirani, X. Laloyaux, A. E. Fernandes, B. Mathy, O. Schicke, O. Riant, B. Nysten and A. M. Jonas, Macromolecules, 2012, 45, 9400–9408 CrossRef CAS.
- E. A. Jackson and M. A. Hillmyer, ACS Nano, 2010, 4, 3548–3553 CrossRef CAS PubMed.
- A. del Campo and E. Arzt, Chem. Rev., 2008, 108, 911–945 CrossRef CAS PubMed.
- S. Krishnamoorthy, C. Hinderling and H. Heinzelmann, Mater. Today, 2006, 9, 40–47 CrossRef CAS.
- C. Park, J. Yoon and E. L. Thomas, Polymer, 2003, 44, 6725–6760 CrossRef CAS.
- J. Doh and D. J. Irvine, J. Am. Chem. Soc., 2004, 126, 9170–9171 CrossRef CAS PubMed.
- J. S. Katz, J. Doh and D. J. Irvine, Langmuir, 2006, 22, 353–359 CrossRef CAS PubMed.
- L. Ionov and S. Diez, J. Am. Chem. Soc., 2009, 131, 13315–13319 CrossRef CAS PubMed.
- M. Kang and B. Moon, Macromolecules, 2009, 42, 455–458 CrossRef CAS.
- H. Zhao, W. Gu, E. Sterner, T. P. Russell, E. B. Coughlin and P. Theato, Macromolecules, 2011, 44, 6433–6440 CrossRef CAS.
- H. Zhao, W. Gu, M. W. Thielke, E. Sterner, T. Tsai, T. P. Russell, E. B. Coughlin and P. Theato, Macromolecules, 2013, 46, 5195–5201 CrossRef CAS.
- H. Zhao, W. Gu, R. Kakuchi, Z. Sun, E. Sterner, T. P. Russell, E. B. Coughlin and P. Theato, ACS Macro Lett., 2013, 2, 966–969 CrossRef CAS.
- J. A. Johnson, N. J. Turro, J. T. Koberstein and J. E. Mark, Prog. Polym. Sci., 2010, 35, 332–337 CrossRef CAS.
- M. Behl, M. Y. Razzaq and A. Lendlein, Adv. Mater., 2010, 22, 3388–3410 CrossRef CAS PubMed.
- I. Tomatsu, K. Peng and A. Kros, Adv. Drug Delivery Rev., 2011, 63, 1257–1266 CrossRef CAS PubMed.
- E. S. Dragan, Chem. Eng. J., 2014, 243, 572–590 CrossRef CAS.
- C. A. DeForest and K. S. Anseth, Angew. Chem., Int. Ed., 2012, 51, 1816–1819 CrossRef CAS PubMed.
- J. A. Johnson, M. G. Finn, J. T. Koberstein and N. J. Turro, Macromolecules, 2007, 40, 3589–3598 CrossRef CAS.
- M. Nagata and Y. Yamamoto, React. Funct. Polym., 2008, 68, 915–921 CrossRef CAS.
- B. Yan, J.-C. Boyer, D. Habault, N. R. Branda and Y. Zhao, J. Am. Chem. Soc., 2012, 134, 16558–16561 CrossRef CAS PubMed.
- C. Zhu and C. J. Bettinger, Macromolecules, 2015, 48, 1563–1572 CrossRef CAS.
- C. Zhu and C. J. Bettinger, J. Mater. Chem. B, 2014, 2, 1613–1618 RSC.
- J. T. Trotta and B. P. Fors, Synlett, 2016, 702–713 CAS.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou, A. Simula, J. Godfrey, M. Li, G. Nurumbetov, P. Wilson and D. M. Haddleton, Macromolecules, 2014, 47, 3852–3859 CrossRef CAS.
- N. Corrigan, J. Xu and C. Boyer, Macromolecules, 2016, 49, 3274–3285 CrossRef CAS.
- J. Xu, S. Shanmugam, C. Fu, K.-F. Aguey-Zinsou and C. Boyer, J. Am. Chem. Soc., 2016, 138, 3094–3106 CrossRef CAS PubMed.
- J. Tan, H. Sun, M. Yu, B. S. Sumerlin and L. Zhang, ACS Macro Lett., 2015, 4, 1249–1253 CrossRef CAS.
- J. Yeow, O. R. Sugita and C. Boyer, ACS Macro Lett., 2016, 5, 558–564 CrossRef CAS.
- J. E. Poelma, B. P. Fors, G. F. Meyers, J. W. Kramer and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 52, 6844–6848 CrossRef CAS PubMed.
- B. P. Fors, J. E. Poelma, M. S. Menyo, M. J. Robb, D. M. Spokoyny, J. W. Kramer, J. H. Waite and Craig J. Hawker, J. Am. Chem. Soc., 2013, 135, 14106–14109 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |