 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactive oxygen species formed in aqueous mixtures of secondary organic aerosols and mineral dust influencing cloud chemistry and public health in the Anthropocene†
Haijie
Tong
 *a,
Pascale S. J.
Lakey
a,
Andrea M.
Arangio
a,
Joanna
Socorro
a,
Christopher J.
Kampf
ab,
Thomas
Berkemeier
c,
William H.
Brune
d,
Ulrich
Pöschl
a and
Manabu
Shiraiwa
*ae
*a,
Pascale S. J.
Lakey
a,
Andrea M.
Arangio
a,
Joanna
Socorro
a,
Christopher J.
Kampf
ab,
Thomas
Berkemeier
c,
William H.
Brune
d,
Ulrich
Pöschl
a and
Manabu
Shiraiwa
*ae
aMultiphase Chemistry Department, Max Planck Institute for Chemistry, Mainz, Germany. E-mail: h.tong@mpic.de
bInstitute for Organic Chemistry, Johannes Gutenberg University, Mainz, Germany
cSchool of Chemical & Biomolecular Engineering, Georgia Institute of Technology, Atlanta, USA
dDepartment of Meteorology, Pennsylvania State University, University Park, PA, USA
eDepartment of Chemistry, University of California, Irvine, CA, USA. E-mail: m.shiraiwa@uci.edu
First published on 17th February 2017
Abstract
Mineral dust and secondary organic aerosols (SOA) account for a major fraction of atmospheric particulate matter, affecting climate, air quality and public health. How mineral dust interacts with SOA to influence cloud chemistry and public health, however, is not well understood. Here, we investigated the formation of reactive oxygen species (ROS), which are key species of atmospheric and physiological chemistry, in aqueous mixtures of SOA and mineral dust by applying electron paramagnetic resonance (EPR) spectrometry in combination with a spin-trapping technique, liquid chromatography-tandem mass spectrometry (LC-MS/MS), and a kinetic model. We found that substantial amounts of ROS including OH, superoxide as well as carbon- and oxygen-centred organic radicals can be formed in aqueous mixtures of isoprene, α-pinene, naphthalene SOA and various kinds of mineral dust (ripidolite, montmorillonite, kaolinite, palygorskite, and Saharan dust). The molar yields of total radicals were ∼0.02–0.5% at 295 K, which showed higher values at 310 K, upon 254 nm UV exposure, and under low pH (<3) conditions. ROS formation can be explained by the decomposition of organic hydroperoxides, which are a prominent fraction of SOA, through interactions with water and Fenton-like reactions with dissolved transition metal ions. Our findings imply that the chemical reactivity and aging of SOA particles can be enhanced upon interaction with mineral dust in deliquesced particles or cloud/fog droplets. SOA decomposition could be comparably important to the classical Fenton reaction of H2O2 with Fe2+ and that SOA can be the main source of OH radicals in aqueous droplets at low concentrations of H2O2 and Fe2+. In the human respiratory tract, the inhalation and deposition of SOA and mineral dust can also lead to the release of ROS, which may contribute to oxidative stress and play an important role in the adverse health effects of atmospheric aerosols in the Anthropocene.
1. Introduction
Atmospheric particulate matter suspended in the air can strongly affect climate and public health.1,2 Their diameters (Dp) vary from nanometers to tens of micrometers, and they are generally categorized into coarse (Dp > 2.5 μm), fine (Dp < 2.5 μm), and ultrafine (Dp < 0.1 μm) particles. Ambient aerosol particles consisting of inorganic and organic compounds can be emitted both from natural and anthropogenic sources. Coarse particles such as mineral dust and sea salts are injected into the troposphere by direct emission.3 Gaseous volatile organic compounds emitted by vegetation and anthropogenic activities can be oxidised to generate semi-volatile and low volatility products, which can form ultrafine and fine secondary organic aerosols (SOA) by nucleation and gas-to-particle partitioning.4–6Mineral dust is emitted with an annual flux of ∼2000 Tg per year and accounts for a major fraction of atmospheric particulate matter.7 Mineral dust particles can be emitted from arid and semiarid areas and transported over thousands of kilometres.7 They can influence the global radiation budget via scattering and absorption of solar and terrestrial radiation.8,9 In addition, they can also act as cloud condensation or ice nuclei upon interaction with water molecules.10–13 During long-range transport, heterogeneous and multiphase chemistry on mineral dust particles may alter their physical and chemical properties and dust may become internally mixed with SOA by coagulation and condensation.14–18 With respect to health effects, inhalation and deposition of mineral dust into the respiratory tract may cause a broad range of pulmonary, cardio-vascular and occupational lung diseases.19–23
Reactive oxygen species (ROS) is a term describing reactive radicals and molecules including hydroxyl radicals (˙OH), superoxide radicals (O2˙−), hydrogen peroxide (H2O2) and organic radicals.2,24,25 ROS widely exist in the gas and condensed phases. Emerging studies have indicated that particulate matter contain ROS, influencing atmospheric chemistry, air quality and public health.2,26,27 It has also been reported that long-lived reactive oxygen intermediates can be formed during the heterogeneous reactions of ozone with aerosol particles such as pollen proteins, unsaturated hydrocarbons, and polycyclic aromatic hydrocarbons.28–30 Furthermore, combustion-generated particles such as soot contain environmentally persistent free radicals (EPFRs), which are most likely semiquinone radicals.31,32 Ambient particles can release a variety of ROS upon interactions with water,33 amplifying ultrafine particle mediated cellular oxidative stress and cytotoxicity.34,35
Substantial amounts of particle-bound ROS are found on biogenic SOA.36,37 Recent studies have shown that extremely low volatility compounds (ELVOC),38 or highly oxidized molecules (HOM) play an important role in new particle formation and growth of SOA.39,40 They are mostly organic hydroperoxides formed through multigenerational oxidation or autoxidation.41–43 It has also been found that these compounds are labile in the condensed phase.44–46 Recent studies have shown that organic hydroperoxides can decompose to form OH radicals upon UV exposure47 and also under dark conditions, and the formation of OH radicals can be enhanced in the presence of iron ions due to Fenton-like reactions.48
Due to the lack of a quantitative and comprehensive understanding of the interactions of mineral dust and SOA, there is considerable uncertainty over the synergistic effects of SOA and mineral dust on climate and public health in the Anthropocene. In this study, we characterized the formation of ROS by aqueous mixtures of SOA and mineral dust by applying continuous wave electron paramagnetic resonance (CW-EPR) spectroscopy in combination with a spin-trapping technique, liquid chromatography-tandem mass spectrometry (LC-MS/MS), and a kinetic model. Using these approaches, we quantified the ROS yields by SOA formed by different precursors (α-pinene, isoprene, and naphthalene) and five different mineral dusts under different pH and temperature and also with UV exposure.
2. Methods
2.1 Dust and chemicals
The Saharan dust used was obtained from deposits on the Cape Verde islands, its composition (as measured by energy dispersive X-ray spectroscopy) is given by Hanisch et al.49 The individual minerals and clay-minerals ripidolite ((Mg,Fe,Al)6(Si,Al)4O10(OH)8), montmorillonite m{Mg[Si4O10](OH)2} × p(Al,Fe)2[Si4O10], m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :p = 0.8–0.9),49 kaolinite (Al2[Si2O5](OH)4) and palygorskite ((Mg,Al)2[OH|Si4O10]·2H2O + 2H2O) were purchased from the Source Clay Minerals Repository, University of Missouri, Columbia, USA.
:p = 0.8–0.9),49 kaolinite (Al2[Si2O5](OH)4) and palygorskite ((Mg,Al)2[OH|Si4O10]·2H2O + 2H2O) were purchased from the Source Clay Minerals Repository, University of Missouri, Columbia, USA.
The chemicals including isoprene (>99.5%, Sigma-Aldrich), α-pinene (99%, Sigma-Aldrich), naphthalene (99.6%, Alfa Aesar GmbH&Co KG), H2SO4 (≥95%, Fluka), 5-tert-butoxycarbonyl-5-methyl-1-pyrroline-N-oxide (BMPO, high purity spintrap from Enzo Life Sciences GmbH) were used without further purification. The ACS reagent water (14211-1L-F) from Sigma-Aldrich was used as solvent in this study. Micropipettes (50 μL, Brand GmbH&Co KG) were used for the analysis of solution samples. High purity nitrogen gas (99.999%, Westfalen AG) and synthetic air (Westfalen AG) were used for SOA formation.
2.2 SOA formation, characterization, collection, and extraction
SOA particles were generated in a 19 L potential aerosol mass (PAM) chamber50 through ozonolysis of α-pinene, or gas-phase photooxidation of isoprene and naphthalene with OH radicals. SOA generated by the PAM chamber have been shown to be a good surrogate for chamber-generated SOA in terms of their oxidation state, chemical composition, and hygroscopicity.50–52The conditions under which SOA are formed in this study are the same as we used previously and are introduced briefly here.48 Ozonolysis of α-pinene SOA was performed under dry conditions (<5% RH). 1 bar and 50–150 ccm min−1 of N2 were used as a carrier gas to introduce α-pinene into the chamber for a reaction time of about 5 minutes. Isoprene or naphthalene vapours were introduced into the PAM chamber in a similar fashion to the α-pinene with a 1 bar and 250 or 50 ccm min−1 N2 flow as carrier gas. Gas-phase OH radicals were produced via photolysis of ozone and water moisture (30–40% RH). SOA were then formed by reactions of isoprene or naphthalene with OH radicals. Ozone concentrations in the PAM chamber were 10 ± 5 ppm (measured with ozone monitor of model 49i, Thermo Fisher Scientific Inc.), and the gas-phase OH radical exposure in the chamber was estimated to be 0.45–1.3 × 1012 molecules per cm3 per s. The size and mass concentrations of the generated SOA particles were characterized with a scanning mobility particle sizer (SMPS, GRIMM Aerosol Technik GmbH & Co. KG). 47 mm Omnipore Teflon filters (100 nm pore size, Merck Chemicals GmbH) were used to collect the aerosols at a flow rate of 3 L min−1 assisted by a common diaphragm vacuum pump (NMP850KNDC, KNF Neuberger GmbH).
The SOA-loaded filters were placed into a 16 mL vial with mineral dust. SOA particles on the filter were then extracted into ∼1 mL of a 10 mM 5-tert-butoxycarbonyl-5-methyl-1-pyrroline-N-oxide (BMPO, high purity, Enzo Life Sciences GmbH) water solution by stirring with a vortex shaker (Heidolph Reax 1) for 10 minutes at 2500 rpm. BMPO is an efficient spin-trapping agent for ˙OH, O2˙− and organic radicals.33,48,53 The final SOA concentration was ∼1 mM with an assumed average molar mass of 200 g mol−1 for SOA.
For experiments investigating pH effects, aqueous solutions with different pH values were made by diluting concentrated H2SO4 solutions with ultra-pure water. The spintrap, mineral dust, and SOA loaded filters were then added into these solutions for extraction and reactions. The mixtures were shaken for 10 min and centrifuged before the measurement with CW-EPR. A Mettler Toledo SevenCompact™ pH meter (S210) was used for the pH measurement.
2.3 CW-EPR measurement
A CW-EPR spectrometer (EMXplus-10/12, Bruker, Germany) was applied for measurements of BMPO-radical adducts. The parameter set for EPR measurements and the spin counting method used in this study was the same as used in our previous studies:48,54 a modulation frequency of 100 kHz; a modulation amplitude of 0.6 or 1; microwave power of 2.149 mW (20 dB) or 21.17 mW (10 dB); a receiver gain of 40 dB; a time constant of 0.01 ms, and a magnetic field scan of 100 G. Spin concentrations were calculated using the following equation:55where c is a constant determined by a standard sample with a known number of spins, GR = receiver gain, Ct = conversion time, n = number of scans, P = microwave power (W), Bm = modulation amplitude (Gauss), Q = quality factor of resonator, nB = Boltzmann factor for temperature dependence, S = total electron spin, nS = number of spins, and f(B1,Bm) = spatial distribution of the microwave field and the modulation field experienced by the sample.
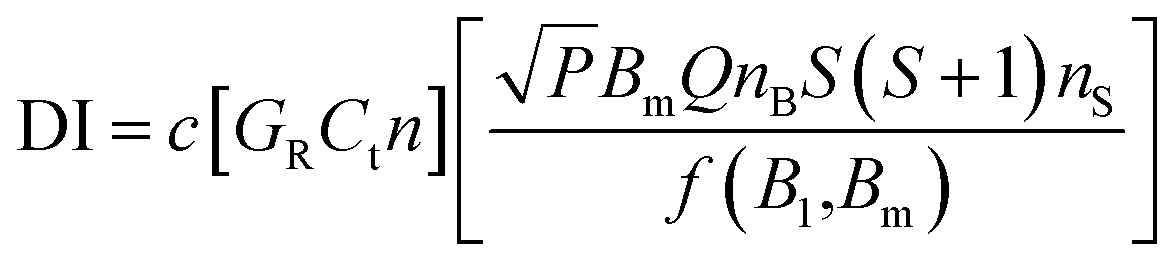
Concentrations of BMPO-radical adduct are reported in the unit of μM. The production rates of radicals can be obtained by dividing concentrations by the extraction time of 300 s. The error bars in Fig. 4–6 are based on repetition of experiments, the estimated deviations from the TEMPOL calibration curve, and the uncertainties in the SOA mass measurements. Potential interferences such as laboratory dust contamination and vial surface catalytic decomposition of H2O2 were found to be negligible based on blank measurements.48 Vials were used only once to avoid contamination from residues.
2.4 LC-MS/MS
The extracts were analysed by liquid chromatography mass spectrometry (LC-MS). The Agilent 1260 Infinity Bio-inert Quaternary LC system consisted of a quaternary pump (G5611A), a HiP sampler (G5667A) and a column thermostat (insert model no. G1316C), and was connected to an electrospray ionization (ESI) source interfaced to a Q-TOF mass spectrometer (6540 UHD Accurate-Mass Q-TOF, Agilent; nominal mass resolution of 30000 at a scan rate of 1 s−1). All modules were controlled by Mass Hunter software (Rev. B.06.00, Agilent).
The analysis of samples was performed using a Zorbax Extend-C18 Rapid resolution HT (2.1 × 50 mm, 1.8 μm) column maintained at 30 °C. The mobile phase consisted of variable mixtures of eluent A (20% (v/v) acetonitrile (HPLC Gradient Grade, Fisher Chemical) in water containing formic acid (0.1% v/v, LC-MS Chromasolv, Sigma-Aldrich)) and eluent B (3% water in acetonitrile), delivered at a flow rate of 0.2 mL min−1 with a total run-time of 50 min. The elution started at 3% B for 3 min followed by a 36 minute linear gradient that raised eluent B to 60%. Eluent B was then increased to 80% in 1 min and initial conditions were restored within 0.1 min, followed by column re-conditioning for 9.9 min before the next run. The injection volume was 95 μL.
The ESI-Q-TOF instrument was employed in the positive ionization mode (ESI+) with 325 °C drying gas temperature, 20 psig nebulizer pressure, 4000 V capillary voltage and 90 V fragmentor voltage. Targeted MS/MS (Table S1†) analysis was employed and the fragmentation of protonated ions was conducted using 10 eV collision energy. Spectra were recorded at a scan rate of 5 s−1 over the mass range of m/z 70–700. The data were processed using the Agilent qualitative data analysis software (B.06.00). Three blank filters were also extracted with 10 mM BMPO water solution and analysed using the same instrumental conditions to ensure the presence of radical adducts in SOA extracts. The radicals that were trapped by BMPO within different types of SOA are listed in Tables S2–S4.† Identification of the different BMPO radical adducts was performed as described previously.48 Briefly, the presence of radical adducts in SOA extracts is determined by their m/z values in the MS spectrum. MS/MS analysis was performed to confirm the radical adducts. In each corresponding MS/MS spectrum, the major fragment ion observed corresponds to the loss of a t-butyl function (–C4H8), which is a characteristic fragment for the BMPO spin trap, indicating the formation of different radical adducts in SOA extracts and as observed in the previous study.48 For several radical adducts, isomers were observed with the same MS and MS/MS spectra but different retention times.
2.5 Kinetic modelling
The concentrations of radicals trapped by BMPO were modelled using the mechanism and reactions shown in Table 1. The reactions in the model included the self-decomposition of organic hydroperoxides (ROOH; R24) and their decomposition due to Fenton-like reactions with iron ions (R20–22). Other important reactions in the model were the trapping reactions of the different radicals by BMPO forming adducts (R16 and R32–35), the destruction of these adducts by decomposition (R16 and R36–38), reactions with iron (R17–19) and reactions with radicals (R26 and R39–43). Literature rate constants were used for the majority of these reactions, but when they were unknown or uncertain, the rate coefficients were determined using the Monte Carlo genetic algorithm (MCGA method) method and sensitivity tests.30,56,57 In the model we assumed that 15% of the total isoprene SOA consisted of ROOH that would decompose to form radicals. This is in agreement with previous work, where it was found that 15% of isoprene SOA, 9% of α-pinene SOA, 10% of β-pinene SOA and 1% of limonene SOA consisted of ROOH that would decompose to form radicals.48 The concentration of transition metal ions due to the presence of dust was parameterized as discussed in the Results and discussion section.| Reaction number | Reaction | Rate constant (cm3 s−1 or s−1) | Reference or comment |
|---|---|---|---|
| a Consistent with the expected trend of OH > OR > R > RO2/O2−/HO2. | |||
| 1 | H2O2 + OH → H2O | k 1 = 5.5 × 10−14 | 80 |
| 2 | OH + OH → H2O2 | k 2 = 8.6 × 10−12 | 81 |
| 3 | OH + HO2 → H2O + O2 | k 3 = 1.2 × 10−11 | 81 |
| 4 | HO2 + HO2 → H2O2 + O2 | k 4 = 1.4 × 10−15 | 82 |
| 5 | H2O2 + HO2 → H2O + O2 + OH | k 5 = 5 × 10−21 | 83 |
| 6 | HO2 + O2− → H2O2 + OH− + O2 | k 6 = 1.6 × 10−13 | 82 |
| 7 | H+ + O2− → HO2 | k 7 = 1 × 10−11 | 84 |
| 8 | HO2 → H+ + O2− | k 8 = 2.3 × 105 | 85 |
| 9 | Fe(II) + H2O2 → Fe(III) + OH− + OH | k 9 = 1.1 × 10−18 | 86 |
| 10 | Fe(II) + OH → Fe(III) + OH− | k 10 = 5.3 × 10−13 | 87 |
| 11 | Fe(III) + H2O2 → Fe(II) + HO2 + H+ | k 11 = 3.3 × 10−24 | 88 |
| 12 | Fe(III) + HO2 → Fe(II) + O2 + H+ | k 12 = 3.3 × 10−18 | 82 |
| 13 | Fe(II) + H2O2 → Fe(IV) + H2O | k 13 = 1.0 × 10−22 | Assumed to be slower than k9 |
| 14 | Fe(IV) + Fe(II) → Fe(III) | k 14 = 6.6 × 10−18 | 81 |
| 15 | BMPO + OH → BMPO–OH | k 15 = 1.6 × 10−12 | Determined from optimizationa |
| 16 | BMPO–OH → product | k 16 = 1.0 × 10−4 | Determined from optimization |
| 17 | All_adducts + Fe(II) → product + Fe(III) | k 17 < 1.0 × 10−19 | Determined from optimization |
| 18 | All_adducts + Fe(III) → product + Fe(II) | k 18 = 9.0 × 10−21 | Determined from optimization |
| 19 | All_adducts + Fe(IV) → product + Fe(III) | k 19 < 1.0 × 10−11 | Insensitive |
| 20 | Fe(II) + ROOH → Fe(III) + RO− + OH | k 20 = 1.0 × 10−21 | Determined from optimization |
| 21 | Fe(II) + ROOH → Fe(III) + RO + OH− | k 21 = 1.0 × 10−21 | Assumed to be the same as k20 |
| 22 | Fe(III) + ROOH → Fe(II) + RO2 + H+ | k 22 = 3.3 × 10−24 | Assumed to be the same as k11 |
| 23 | OH + ROOH → ROH + HO2 | k 23 = 5.5 × 10−14 | Assumed to be the same as k1 |
| 24 | ROOH → RO + OH | k 24 = 2.0 × 10−6 | Determined by optimization |
| 25 | SOA + OH → SOA′ | k 25 = 4.0 × 10−15 | Determined by optimization |
| 26 | All_adducts + OH → product | k 26 = 5.0 × 10−12 | Determined from optimization |
| 27 | Fe(II) + O2 → O2− + Fe(III) | k 27 = 3.3 × 10−19 | Determined from optimization |
| 28 | Fe(II) + O2− + 2H+ → Fe(III) + H2O2 | k 28 = 1.0 × 10−11 | Determined from optimization |
| 29 | O2− + OH → O2 + OH− | k 29 = 1.3 × 10−11 | 89 |
| 30 | RO → R | k 30 = 5 × 105 | 90 |
| 31 | R + O2 → ROO | k 31 = 8.1 × 10−12 | 90 |
| 32 | BMPO + O2−/HO2 → BMPO–O2−/HO2 | k 32 = 2.6 × 10−14 | Determined from optimizationa |
| 33 | BMPO + RO2 → BMPO–OOR | k 33 = 2.6 × 10−14 | Assumed to be the same as k32 |
| 34 | BMPO + OR → BMPO–OR | k 34 = 7.7 × 10−13 | Determined from optimizationa |
| 35 | BMPO + R → BMPO–R | k 35 = 1.5 × 10−13 | Determined from optimizationa |
| 36 | BMPO–O2−/HO2/RO2 + BMPO–O2−/HO2/RO2 → product | k 36 = 4.8 × 10−18 | Determined from optimization |
| 37 | BMPO–R → product | k 37 = 1.0 × 10−4 | Assumed to be the same as k16 |
| 38 | BMPO–OR → product | k 38 = 1.0 × 10−4 | Assumed to be the same as k16 |
| 39 | All_adducts + O2− → products | k 39 = 5.4 × 10−14 | Determined from optimization |
| 40 | All_adducts + HO2 → products | k 40 = 5.4 × 10−14 | Assumed to be the same as k39 |
| 41 | All_adducts + RO2 → products | k 41 = 5.4 × 10−14 | Assumed to be the same as k39 |
| 42 | All_adducts + OR → products | k 42 = 5.0 × 10−12 | Assumed to be the same as k26 |
| 43 | All_adducts + R → products | k 43 < 1.0 × 10−11 | Insensitive |
| 44 | R + R → product | k 44 = 1.0 × 10−12 | 91 |
| 45 | OR + OR → product | k 45 = 3.3 × 10−13 | 92 |
| 46 | RO2 + RO2 → product | k 46 = 1.0 × 10−13 | 93 |
3. Results and discussion
3.1 EPR spectra of aqueous mixtures of SOA and dust
Fig. 1 shows the EPR spectra of the BMPO-radical adducts formed in aqueous solutions of SOA, dust and their mixtures. The black spectra (A, E and I) indicate that isoprene and α-pinene SOA formed mainly OH radicals, whereas superoxide radicals were mainly formed by naphthalene SOA. These three types of SOA also yielded organic radicals. The presence of ˙OH, O2˙− and organic radicals trapped by BMPO was directly detected and confirmed by LC-MS analysis (Tables S1–S3†).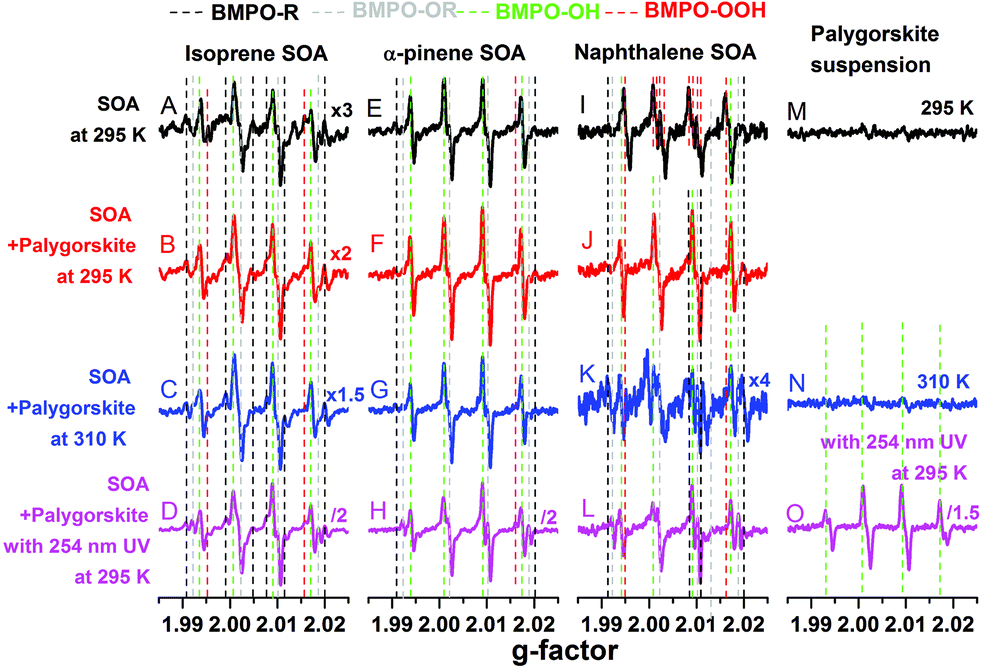 | ||
| Fig. 1 EPR spectra of BMPO-radical adducts of SOA (black lines, except the spectrum M) and aqueous mixtures of isoprene (left column), α-pinene (middle column), and naphthalene (right column) SOA with palygorskite dust in water. The spectra M, N, and O are for the palygorskite suspensions. The red spectra were obtained at 295 K, the blue spectra were obtained at 310 K, and the pink spectra were obtained 15 min after the mixture exposure to 254 nm UV. The different colours of the dashed lines indicate different radical species. | ||
For the palygorskite suspensions, low concentrations of radicals were observed at 295 K (spectrum M) and 310 K (spectrum N), and only OH radicals could be identified upon exposure to 254 nm UV. In addition to the palygorskite, we also measured the effect of four other types of dust on the radical yield of SOA in aqueous mixtures under different conditions, and the EPR spectra are shown in Fig. S1–S3 of the ESI.†
In the aqueous mixtures of SOA and dust at 295 K (red spectra) and 310 K (blue spectra), the dominant radicals formed by isoprene (spectra B and C) and α-pinene (spectra F and G) SOA at 295 (spectra B and F) and 310 (spectra C and G) K were still OH radicals. Significant enhancements in the formation of OH radical were observed after the aqueous mixtures of isoprene (spectrum D) or α-pinene (spectrum H) SOA with palygorskite dust were exposed to 254 nm UV for 15 min. For mixtures of naphthalene SOA and dust, the formation of superoxide seems to be suppressed and the OH radical was the main radical formed at 295.15 K (spectrum J) and at 310 K (spectrum K). Furthermore, no significant enhancement of OH radical formation was observed after the aqueous mixtures were exposed to 254 nm UV for 15 min (spectrum L). It should be noted that 254 nm UV condition may not be relevant to the photochemistry in the troposphere. We plan to investigate the effects of visible light on ROS formation in follow up studies.
The observed formation of OH radicals is most likely due to decomposition of organic hydroperoxides (ROOH), which account for the predominant fraction of isoprene and α-pinene SOA, but have little contribution for naphthalene SOA.58 ROOH are formed via multigenerational gas-phase oxidation and autoxidation, introducing multiple hydroperoxy functional groups forming extremely low volatility organic compounds.38 Due to the low binding energy of the O–O bond, ROOH are known to undergo thermal homolytic cleavage (ROOH → RO˙ + ˙OH). In the presence of transition metal ions such as Fe2+, decomposition of ROOH can be enhanced mainly via Fenton-like reactions leading to heterolytic cleavage of the O–O bond in the following two ways depending on the pH and reaction environments: ROOH + Fe2+ → RO˙ + OH− + Fe3+ or ROOH + Fe2+ → RO− + ˙OH + Fe3+.59 Note that homolytic cleavage can be also catalyzed by iron ions.60,61 The formed alkoxy radicals (RO˙) can be trapped by BMPO (Tables S1–S3†) and secondary reactions between the formed OH and organic compounds may lead to generation of organic radicals.
3.2 Radical concentrations in pure SOA or dust
The total radical concentrations were quantified using the spin-counting technique.33,48 To estimate the relative amount of different types of ROS, the observed EPR spectra were fitted and simulated using the Xenon software.33,54 Four types of radicals have been used to fit the spectra: BMPO–OH (hyperfine coupling constants of aN = 14.3 G, aHβ = 12.7 G, aHγ = 0.61 G), BMPO–OOH (aN = 14.3 G, aH = 8.1 G), BMPO–R (aN = 15.2 G, aH = 21.6 G) and BMPO–OR (aN = 14.5 G, aHβ = 16.6 G).33 The simulated EPR spectrum reproduced the observed spectrum very well with a small residual. The deconvolution of spectra allowed us to estimate the relative contribution of different types of ROS herein.Panels (a) to (c) of Fig. 2 show the determined concentrations of radical types of ROS in SOA extracts (1 mM) and dust suspensions (10 mg mL−1). Isoprene, α-pinene, and naphthalene SOA generated in total ∼0.6 μM, ∼1.6 μM, and ∼1 μM radicals, respectively, at 295 K. At 310 K, ROS concentrations in isoprene SOA increased by about a factor of two, whereas ROS concentrations in α-pinene, and naphthalene SOA stayed almost the same. This may indicate that organic hydroperoxides contained in isoprene SOA are more labile compared to those contained in α-pinene SOA. OH radicals are the dominant radicals formed by isoprene (∼0.5 μM) and α-pinene (∼1.2 μM) SOA at both 295 and 315 K. Smaller amounts of carbon and oxygen-centred organic radicals were also produced. At these two temperatures, naphthalene SOA produced substantial amounts of superoxide radicals (∼0.07–0.1 μM) and small amounts of carbon (<0.02 μM) and oxygen (<0.1 μM) centred organic radicals.
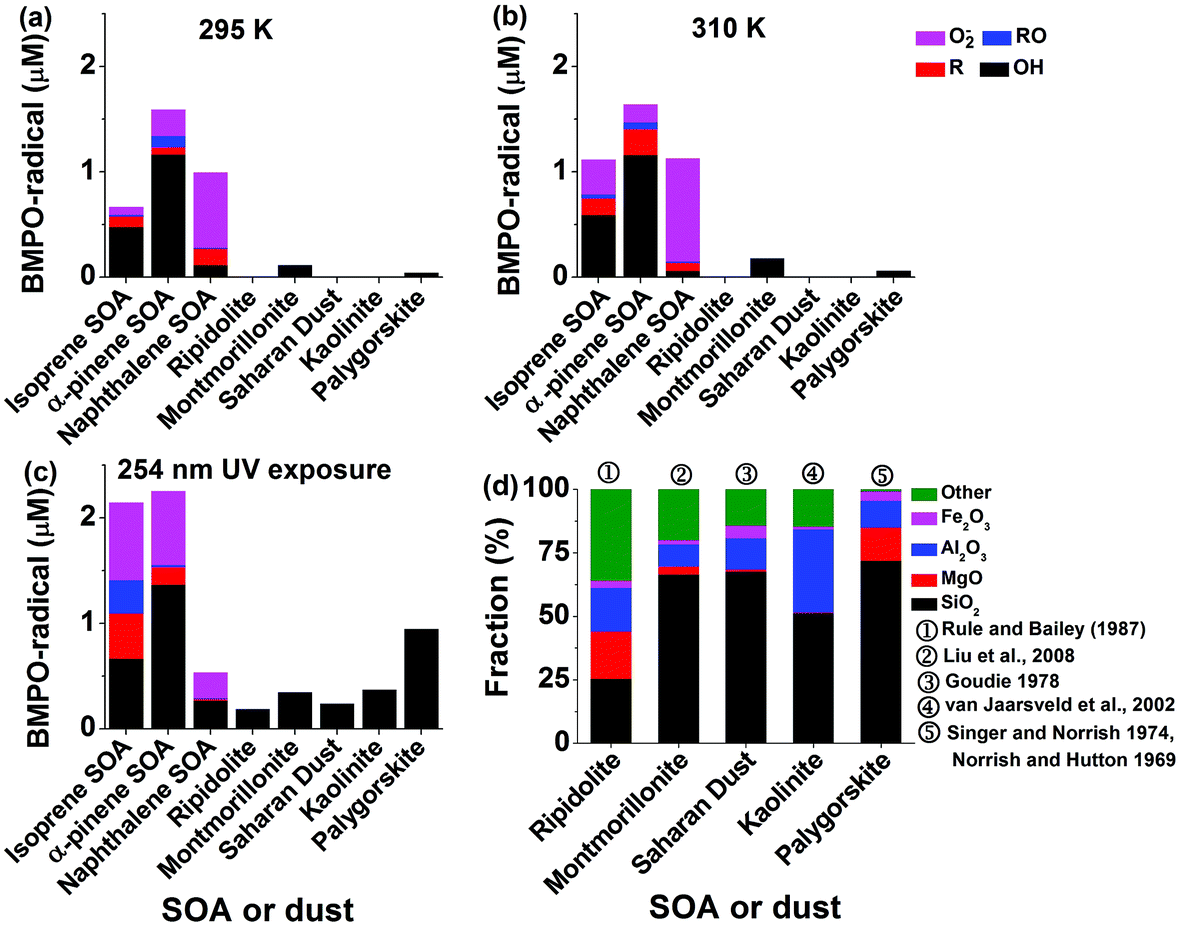 | ||
| Fig. 2 Concentrations of different radicals including ˙OH, O2˙−, carbon- and oxygen centred organic radicals trapped by BMPO in SOA (isoprene, α-pinene, naphthalene) or dust in liquid water at (a) 295 K, (b) 310 K and (c) upon exposure to 254 nm UV for 15 min. (d) Chemical composition of mineral dust (ripidolite, montmorillonite, Saharan dust, kaolinite, palygorskite) as reported in previous studies. The values here are based on dry mineral dust excluding the weight of water. The concentrations of SOA and dust are 1 mM and 10 mg mL−1, respectively. | ||
After the SOA extracts were exposed to 254 nm UV for 15 min, total ROS concentrations produced by isoprene SOA were increased by about a factor of three and those by α-pinene SOA increased by about 50% compared to those at 295 K. A variety of ROS were produced by isoprene and α-pinene SOA with notable increases of superoxide radicals. Such increases of ROS formation may be due to photolysis47 and decomposition of organic hydroperoxides,44 which are highly abundant in isoprene or α-pinene SOA.41,42,47 In contrast, the yield of ROS from naphthalene SOA decreased by a factor of two. Naphthalene SOA are known to contain less organic hydroperoxides58 and these results indicate that the main source of ROS by naphthalene SOA is unlikely to be the decomposition of organic hydroperoxides. Instead, quinones contained in naphthalene SOA are redox active and can trigger formation of superoxide radicals.62 Such redox-active reactions might be hindered under UV irradiation.
For pure dust suspensions, the montmorillonite and palygorskite could produce small amounts of OH radicals at 295 and 310 K (<0.2 μM), whereas other dust did not produce detectable amounts of ROS. We infer that these OH radicals may be produced through Fenton(-like) reactions of dissolved transition metal ions or on the surface of the dust. While exposed to 254 nm UV, the montmorillonite (∼0.3 μM), palygorskite (∼0.9 μM), ripidolite (∼0.1 μM), kaolinite (∼0.4 μM), and Saharan (∼0.3 μM) dust produced significant amounts of OH radicals. We infer that the production of OH radicals was enhanced by photo-Fenton reactions.63Fig. 2d shows the reported chemical structures of mineral dust used in this study. The residue compounds, which are not SiO2, MgO, Al2O3, and Fe2O3, are assigned to be ‘other’. The total abundance of SiO2, MgO, Al2O3, and Fe2O3 increases in ripidolite, montmorillonite, Saharan, kaolinite, and palygorskite, which seems to have a positive correlation with the generated OH radical concentrations.
3.3 Radical concentrations in aqueous mixtures of SOA and dust
Fig. 3 shows yields of radical types of ROS in the aqueous mixtures of SOA with mineral dust with the green dashed lines representing the yield of radicals in pure SOA solutions. In the aqueous mixtures of isoprene and α-pinene SOA with dust, ripidolite caused the strongest enhancement of the radical formation of SOA by a maximum factor of 2.4 for isoprene SOA and a maximum factor of 1.8 for α-pinene SOA at 295 K. At 310 K, an even stronger enhancement has been observed for α-pinene SOA. These enhancements are most likely caused by dissolved transition metal ions, which can cause Fenton(-like) reactions promoting decomposition of SOA components. The effectiveness of dust for enhancing ROS formation by SOA may depend strongly on the chemical composition of the dust and solubility of the transition metal ions. After UV exposure, the ROS concentrations in the aqueous mixtures of isoprene SOA with ripidolite, kaolinite and palygorskite increased significantly, most likely due to ROOH photolysis47 and photo-Fenton reactions.5,63 In the aqueous mixtures of naphthalene SOA with dust, only palygorskite enhanced the ROS formation slightly at 295 and 310 K and ripidolite and palygorskite increased ROS formation under UV exposure. The limited impacts of dust on ROS formation by naphthalene SOA may indicate that transition metal ions do not interfere substantially with ROS formation processes by redox reactions of quinones contained in naphthalene SOA. The extent of the effects of higher temperature and UV exposure is further visualized in Fig. S4,† which shows the ratios of production rates of ROS by SOA at 310 K and upon UV exposure to that at 295 K.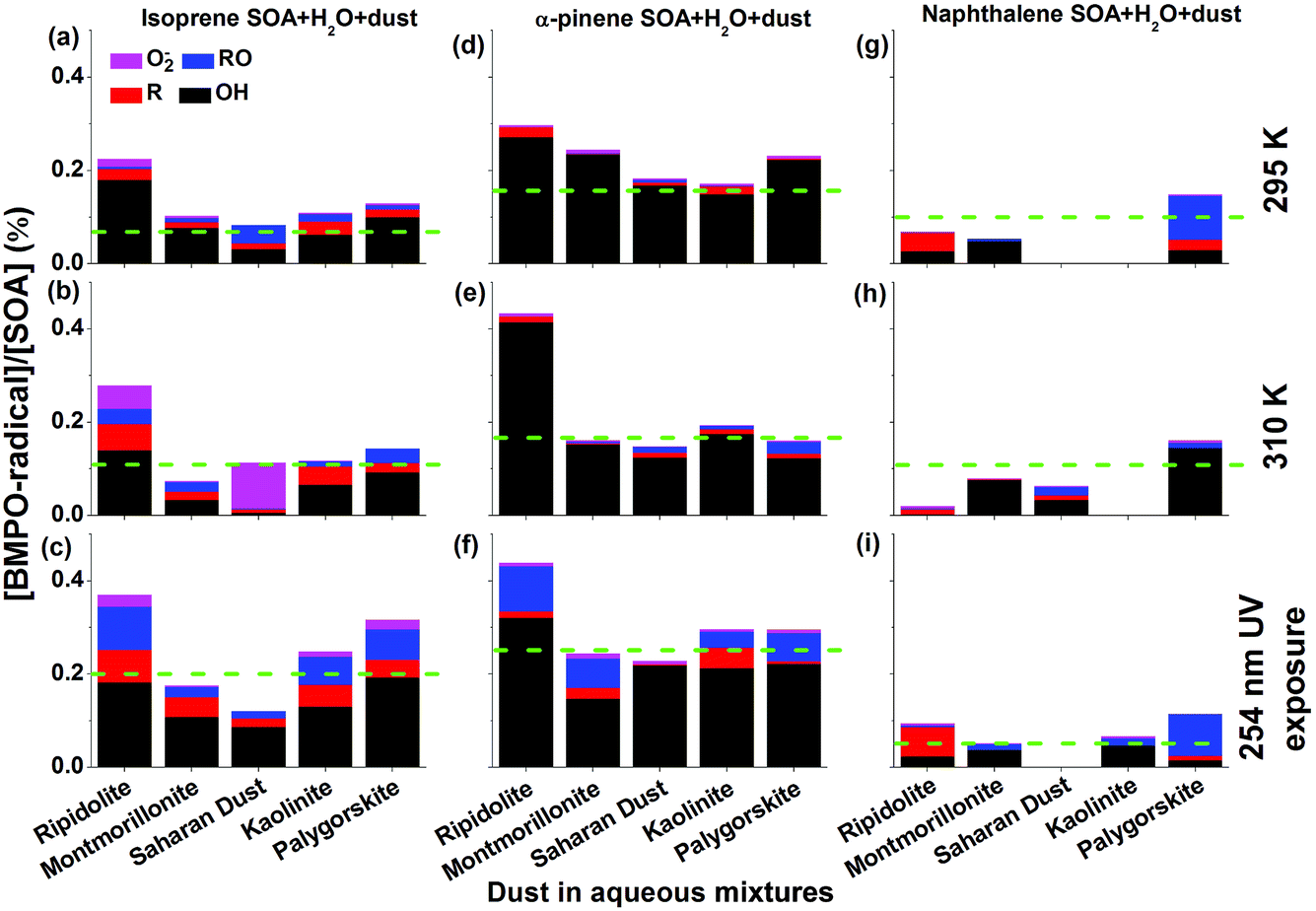 | ||
| Fig. 3 Yield of different radicals including ˙OH, O2˙−, carbon- and oxygen centred organic radicals trapped by BMPO in the aqueous mixtures of dust and (a–c) isoprene, (d–f) α-pinene, and (g–i) naphthalene SOA at (a, d and g) 295 K, (b, e and h) 310 K, and (c, f and i) upon exposure to 254 nm UV for 15 min. The green dashed lines indicate the yield of radicals in pure SOA solutions. | ||
Clearly OH is the dominant radical product for mixtures of dust and isoprene and α-pinene SOA at 295 and 310 K as well as under 254 nm UV exposure. The aqueous mixtures exposed to 254 nm UV showed the highest yield of OH, carbon and oxygen centred radicals. The aqueous mixture of each mineral dust type and α-pinene SOA generated more OH radicals than the mixtures with isoprene SOA. For example, the OH radical yields of the aqueous mixtures of ripidolite dust and isoprene SOA were ∼0.18% at 295 K, ∼0.14% at 310 K, and ∼0.18% after 254 nm UV exposure, whereas the OH radical yields of the aqueous mixture of ripidolite dust and α-pinene SOA were ∼0.27% at 295 K, ∼0.42% at 310 K, and ∼0.32% after 254 nm UV exposure. For the aqueous mixtures of naphthalene SOA with montmorillonite, OH was the dominant radical species at 295 K.
We have investigated the effect of pH on ROS formation by isoprene SOA mixed with three different types of dust (ripidolite, palygorskite, and Saharan dust). Fig. 4 shows the measured radical concentrations with a pH range of 0–6. As the pH of the aqueous mixtures decreases from ∼5.5 to ∼0.3, the total radical concentration increases significantly from ∼0.9 μM to ∼7.3 μM for ripidolite, ∼0.2 μM to ∼5.7 μM for palygorskite, and ∼0.1 μM to ∼17 μM for Saharan dust. Low pH may facilitate an increase in the solubility of transition metal ions64 that can undergo Fenton-like reactions.
 | ||
| Fig. 4 Effects of pH on radical concentrations in aqueous mixtures of isoprene SOA with different dusts (ripidolite, palygorskite, Saharan dust). The concentrations of SOA and all three types of dust are 1 mM and 10 mg mL−1. The error bars are composed of the uncertainties in the SOA mass measurements and the estimated deviations from the TEMPOL calibration curve. The lines connect data points to guide the eye. | ||
3.4 Effect of dust concentrations on radical concentrations in aqueous mixtures of SOA and dust
Fig. 5 shows radical concentrations in water extracts of isoprene SOA mixed with different concentrations of (a) FeSO4 or (b) ripidolite. As the molar ratio of Fe2+ and isoprene SOA increased from ∼0.1 to ∼0.3 (panel a), the concentration of OH and total radical yield of isoprene SOA increased from ∼2 to ∼6 μM and ∼3 to ∼8 μM, respectively. This process can be explained by an enhancement in Fenton-like reactions of iron ions and organic hydroperoxides. As the concentration of Fe2+ increased further, the yield of OH and total radicals decreased, which is induced by the reaction of Fe3+ with the BMPO-radical adducts, leading to decay of BMPO-radical adducts. In the aqueous mixtures of isoprene SOA and ripidolite dust (panel b), the concentration of isoprene SOA was kept at 1 mM, and the concentrations of ripidolite dust were in the range of ∼0.9 to ∼10 mg mL−1. The dissolved transition metal concentrations should have a positive relationship with the dust concentrations. The concentrations of OH and total radicals increase from ∼0.3 to ∼1.8 μM and from ∼0.9 to ∼2.3 μM, respectively, as the mripidolite/mSOA increases from ∼4.5 to ∼50.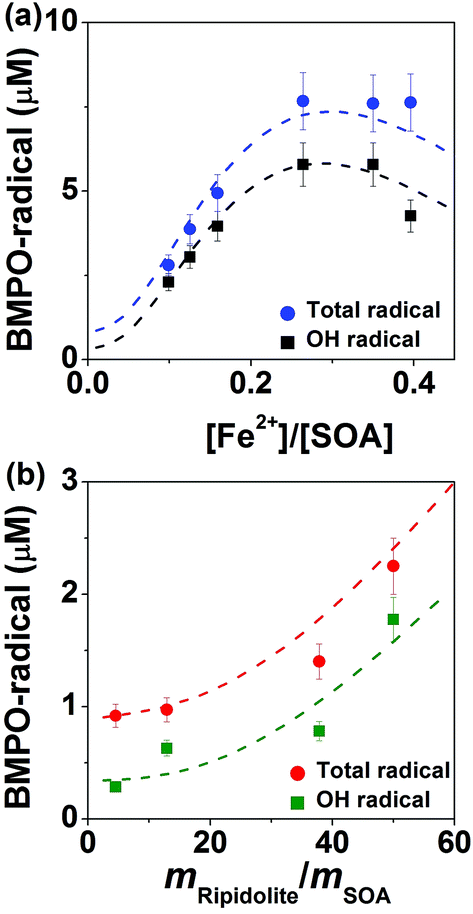 | ||
| Fig. 5 Measured (symbols) and modelled (lines) total radical concentrations in aqueous mixtures of isoprene SOA with Fe2+ or ripidolite dust. (a) Data for aqueous mixtures containing isoprene SOA and FeSO4. The squares and circles represent the experimental data on OH radical and total radicals, respectively. The black and blue dashed lines represent the modelled data on OH radical and total radicals. (b) Data for aqueous mixtures containing isoprene SOA and ripidolite. The squares and circles represent the modelled data on OH radical and total radicals, respectively. The olive and red dashed lines represent the modelled data on OH radical and total radicals. The concentration of SOA is 1 mM or 200 μg mL−1 for all experiments. The error bars are composed of the uncertainties in the SOA mass measurements and the estimated deviations from the TEMPOL calibration curve. ‘mripidolite/mSOA’ means the mass ratio of ripidolite and SOA in the aqueous mixtures under study. | ||
As shown by the dashed lines, this observed behaviour was fully reproduced by the kinetic model, which included in total 46 reactions of ROS and iron chemistry, Fenton-like reactions of organic hydroperoxides, trapping of radicals by BMPO, as well as decay of BMPO-adducts. At very low iron concentrations (<0.02 mM) the self-decomposition of ROOH dominates the radical production. However, as the iron concentration increases, the concentration of adducts increases as the Fenton-like destruction of ROOH becomes important. At very high iron concentrations the destruction of adducts due to reactions with Fe3+ (and to a lesser extent reactions with untrapped radicals) leads to a decrease in the total concentrations of trapped radicals. The modelling results suggested that ripidolite dust decomposed ROOH contained in isoprene SOA in a similar way to Fe2+ and could be parameterised as: [Fe2+] (mM) = 0.0086[Dust] (mg mL−1). These results clearly demonstrate that transition metal ions can significantly enhance ROS formation of isoprene SOA via Fenton-like reactions.
4. Implications and conclusions
The implications of this study are illustrated in Fig. 6 and 7. Mineral dust is emitted from arid and semiarid areas and can be transported over long distances. During long-range transport, dust can be internally mixed with SOA by coagulation and condensation (i.e., gas-particle partitioning of semi-volatile and low volatility compounds). SOA coatings may contain a substantial fraction of highly oxidized multifunctional organic compounds (HOM) and extremely low volatility compounds (ELVOC). These compounds are mostly organic hydroperoxides (ROOH), which are labile and their lifetime is relative humidity dependent.65,66 When such particles undergo aerosol deliquescence or activation to cloud or fog droplets, decomposition of ROOH may be triggered upon interactions with water and transition metal ions that are dissolved from dust, leading to the formation of a variety of radical forms of reactive oxygen species including ˙OH, O2˙−, carbon- and oxygen-centered organic radicals. ROOH decomposition and ROS formation can be especially enhanced by photochemistry. The extent of ROS formation may depend strongly on the chemical composition of SOA and dust; mixing isoprene and α-pinene SOA with ripidolite may lead to high ROS formation, whereas ROS formation by naphthalene SOA would not be affected by the presence of dust. ROS formation will be significantly higher at a lower pH. | ||
| Fig. 6 OH production rate in cloud droplets, pure SOA solutions, and aqueous mixtures of isoprene SOA and ripidolite. The OH production rate in cloud droplets by SOA decomposition compared to the classical Fenton reaction. The data points for β-pinene and limonene SOA, and some data points of the α-pinene isoprene SOA were from Tong et al. (2016).48 The shaded green area represents the possible range in the presence of iron as a function of SOA concentration in the aqueous phase, which is based on the minimum and maximum OH radical production efficiency of SOA. The dashed lines represent OH production rates due to the Fenton reaction from H2O2 with dissolved iron concentrations of 0.1 and 2.5 μM. In the aqueous mixtures, the concentration of isoprene SOA was 1 mM, and the concentration range of ripidolite was from 3 to 10 mg mL−1. The pH = 0 and 3 solutions were made by diluting H2SO4 solutions. The navy color cross shows the OH production rate by the aqueous mixture of 1 mM isoprene SOA and 10 mg mL−1 ripidolite at 310 K. | ||
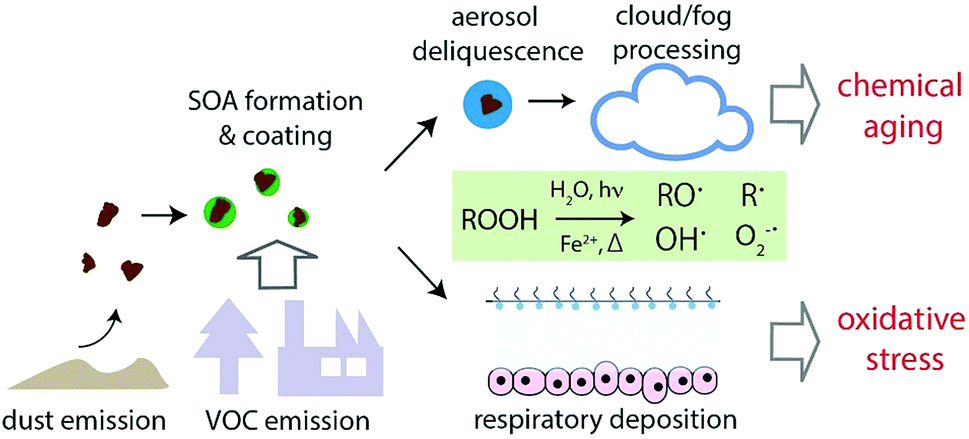 | ||
| Fig. 7 Implications of ROS formation by SOA-coated dust in the aqueous phase. Formation of radicals upon decomposition of organic hydroperoxides (ROOH) induced by heat, UV exposure and Fenton-like reactions of iron ions may lead to rapid chemical aging of SOA particles upon deliquescence and cloud or fog processing in the atmosphere. Inhalation and deposition of such particles in the human respiratory tract may contribute to oxidative stress. | ||
The orange area in Fig. 6 shows the OH production rate by Fenton reactions between Fe2+ and H2O2 forming OH radicals, which has been thought to be the main OH source in cloud droplets,67,68 as a function of H2O2 concentration with typical dissolved iron concentrations in cloud droplets of 0.1–2.5 μM.48,67 The dashed lines represent the OH production rate by Fenton reactions of H2O2 with 0.1 and 2.5 μM iron ions. The green area shows the OH production rate by SOA decomposition in cloud or fog droplets, which ranges between ∼0.01 and 100 nM s−1 depending on SOA precursors and the Fe2+ and SOA concentrations. The pink triangles and navy color crosse show the OH production rate by the aqueous mixtures of 1 mM SOA and ripidolite at 295 and 310 K. It clearly shows that SOA decomposition could be comparably important to the Fenton reaction in most conditions and that SOA can be the main source of OH radicals at low concentrations of H2O2 and Fe2+. In addition, the OH production rate by the aqueous mixtures of SOA and ripidolite can be significantly enhanced at low pH (<3) and elevated temperature (310 K) conditions. The generated OH radicals can play an important role in the chemical aging of organic compounds by inducing reactions of fragmentation, functionalization and oligomerization.69–77
This study also implies that ROS can be formed in lung lining fluid upon inhalation and respiratory deposition of internally mixed SOA–dust particles. Some fractions of ROS may be scavenged by antioxidants contained in the lung lining fluid, which needs to be quantified by further experiments. Nevertheless, excess concentrations of ROS including OH radicals, superoxide, and potentially also carbon- and oxygen-centred organic radicals may cause oxidative stress to lung cells and tissues.2,48,78 Recently, Lakey et al.25 have shown that fine particulate matter containing redox-active transition metals78,79 and SOA can increase ROS concentrations in the lung lining fluid to levels characteristic for respiratory diseases. ROS may induce chemical transformation of biomolecules such as proteins and lipids in lung fluid to form damage associated molecular patterns (DAMPs), which can trigger the toll-like receptor radical cycle causing inflammation.79 Due to the important implications to adverse aerosol health effects, further studies are warranted to characterize the ROS formation efficiency of SOA particles mixed with other types of particles (for example, soot) and to explore multiphase ROS chemistry in lung lining fluid.
Acknowledgements
This work was funded by the Max Planck Society. C. J. Kampf acknowledges financial support by the German Research Foundation (DFG project KA 4008/1-2). We thank John Crowley (Max Planck Institute for Chemistry) for helpful discussion and providing mineral dust samples. Open Access funding provided by the Max Planck Society.Notes and references
- J. H. Seinfeld and S. N. Pandis, Atmospheric chemistry and physics: from air pollution to climate change, John Wiley & Sons, 2016 Search PubMed.
- U. Pöschl and M. Shiraiwa, Chem. Rev., 2015, 115, 4440–4475 CrossRef PubMed.
- U. Pöschl, Angew. Chem., Int. Ed., 2005, 44, 7520–7540 CrossRef PubMed.
- J. Jimenez, M. Canagaratna, N. Donahue, A. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe and N. Ng, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- M. Hallquist, J. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. Donahue, C. George and A. Goldstein, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- M. Kanakidou, J. Seinfeld, S. Pandis, I. Barnes, F. Dentener, M. Facchini, R. V. Dingenen, B. Ervens, A. Nenes and C. Nielsen, Atmos. Chem. Phys., 2005, 5, 1053–1123 CrossRef CAS.
- N. Huneeus, M. Schulz, Y. Balkanski, J. Griesfeller, J. Prospero, S. Kinne, S. Bauer, O. Boucher, M. Chin, F. Dentener, T. Diehl, R. Easter, D. Fillmore, S. Ghan, P. Ginoux, A. Grini, L. Horowitz, D. Koch, M. C. Krol, W. Landing, X. Liu, N. Mahowald, R. Miller, J.-J. Morcrette, G. Myhre, J. Penner, J. Perlwitz, P. Stier, T. Takemura and C. S. Zender, Atmos. Chem. Phys., 2011, 11, 7781–7816 CAS.
- I. Tegen and I. Fung, J. Geophys. Res.: Atmos., 1994, 99, 22897–22914 CrossRef.
- U. Lohmann and J. Feichter, Atmos. Chem. Phys., 2005, 5, 715–737 CrossRef CAS.
- P. J. DeMott, K. Sassen, M. R. Poellot, D. Baumgardner, D. C. Rogers, S. D. Brooks, A. J. Prenni and S. M. Kreidenweis, Geophys. Res. Lett., 2003, 30 DOI:10.1029/2003GL017 410.
- C. Hoose and O. Möhler, Atmos. Chem. Phys., 2012, 12, 9817–9854 CrossRef CAS.
- J. D. Atkinson, B. J. Murray, M. T. Woodhouse, T. F. Whale, K. J. Baustian, K. S. Carslaw, S. Dobbie, D. O’Sullivan and T. L. Malkin, Nature, 2013, 498, 355–358 CrossRef CAS PubMed.
- M. Tang, D. J. Cziczo and V. H. Grassian, Chem. Rev., 2016, 116, 4205–4259 CrossRef CAS PubMed.
- F. J. Dentener, G. R. Carmichael, Y. Zhang, J. Lelieveld and P. J. Crutzen, J. Geophys. Res.: Atmos., 1996, 101, 22869–22889 CrossRef CAS.
- P. R. Buseck and M. POsfai, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3372–3379 CrossRef CAS.
- C. R. Usher, A. E. Michel and V. H. Grassian, Chem. Rev., 2003, 103, 4883–4940 CrossRef CAS PubMed.
- D. M. Cwiertny, M. A. Young and V. H. Grassian, Annu. Rev. Phys. Chem., 2008, 59, 27–51 CrossRef CAS PubMed.
- W. Nie, A. Ding, T. Wang, V.-M. Kerminen, C. George, L. Xue, W. Wang, Q. Zhang, T. Petäjä, X. Qi, X. Gao, X. Wang, X. Yang, C. Fu and M. Kulmala, Sci. Rep., 2014, 4, 6634 CrossRef CAS PubMed.
- C. J. Doelman, R. Leurs, W. C. Oosterom and A. Bast, Exp. Lung Res., 1990, 16, 41–55 CrossRef CAS PubMed.
- E. Hnizdo and V. Vallyathan, J. Occup. Environ. Med., 2003, 60, 237–243 CrossRef CAS PubMed.
- I. Bergdahl, K. Toren, K. Eriksson, U. Hedlund, T. Nilsson, R. Flodin and B. Järvholm, Eur. Respir. J., 2004, 23, 402–406 CrossRef CAS PubMed.
- A. Karanasiou, N. Moreno, T. Moreno, M. Viana, F. De Leeuw and X. Querol, Environ. Int., 2012, 47, 107–114 CrossRef CAS PubMed.
- S. A. Morman and G. S. Plumlee, in Mineral Dust, Springer, 2014, pp. 385–409 Search PubMed.
- K. Apel and H. Hirt, Annu. Rev. Plant Biol., 2004, 55, 373–399 CrossRef CAS PubMed.
- P. S. Lakey, T. Berkemeier, H. Tong, A. M. Arangio, K. Lucas, U. Pöschl and M. Shiraiwa, Sci. Rep., 2016, 6, 32916 CrossRef CAS PubMed.
- J. M. Anglada, M. Martins-Costa, J. S. Francisco and M. F. Ruiz-Lopez, Acc. Chem. Res., 2015, 48, 575–583 CrossRef CAS PubMed.
- J. T. Bates, R. J. Weber, J. Abrams, V. Verma, T. Fang, M. Klein, M. J. Strickland, S. E. Sarnat, H. H. Chang, J. A. Mulholland, P. E. Tolbert and A. G. Russell, Environ. Sci. Technol., 2015, 49, 13605–13612 CrossRef CAS PubMed.
- M. Shiraiwa, Y. Sosedova, A. Rouvière, H. Yang, Y. Zhang, J. P. D. Abbatt, M. Ammann and U. Pöschl, Nat. Chem., 2011, 3, 291–295 CrossRef CAS PubMed.
- C. J. Kampf, F. Liu, K. Reinmuth-Selzle, T. Berkemeier, H. Meusel, M. Shiraiwa and U. Pöschl, Environ. Sci. Technol., 2015, 49, 10859–10866 CrossRef CAS PubMed.
- T. Berkemeier, S. S. Steimer, U. K. Krieger, T. Peter, U. Pöschl, M. Ammann and M. Shiraiwa, Phys. Chem. Chem. Phys., 2016, 18, 12662–12674 RSC.
- W. Gehling and B. Dellinger, Environ. Sci. Technol., 2013, 47, 8172–8178 CrossRef CAS PubMed.
- H. Jia, G. Nulaji, H. Gao, F. Wang, Y. Zhu and C. Wang, Environ. Sci. Technol., 2016, 50, 6310–6319 CrossRef CAS PubMed.
- A. M. Arangio, H. Tong, J. Socorro, U. Pöschl and M. Shiraiwa, Atmos. Chem. Phys., 2016, 16, 13105–13119 CrossRef CAS.
- W. Gehling, L. Khachatryan and B. Dellinger, Environ. Sci. Technol., 2014, 48, 4266–4272 CrossRef CAS PubMed.
- S. Balakrishna, S. M. Lomnicki, K. M. McAvey, R. B. Cole, B. Dellinger and S. A. Cormier, Part. Fibre Toxicol., 2009, 6, 11 CrossRef PubMed.
- X. Chen, P. K. Hopke and W. P. Carter, Environ. Sci. Technol., 2011, 45, 276–282 CrossRef CAS PubMed.
- Y. Wang, H. Kim and S. E. Paulson, Atmos. Environ., 2011, 45, 3149–3156 CrossRef CAS.
- M. Ehn, J. A. Thornton, E. Kleist, M. Sipilä, H. Junninen, I. Pullinen, M. Springer, F. Rubach, R. Tillmann and B. Lee, Nature, 2014, 506, 476–479 CrossRef CAS PubMed.
- M. Riva, Chem, 2016, 1, 526–528 CAS.
- J. Tröstl, W. K. Chuang, H. Gordon, M. Heinritzi, C. Yan, U. Molteni, L. Ahlm, C. Frege, F. Bianchi and R. Wagner, Nature, 2016, 533, 527–531 CrossRef PubMed.
- K. S. Docherty, W. Wu, Y. B. Lim and P. J. Ziemann, Environ. Sci. Technol., 2005, 39, 4049–4059 CrossRef CAS PubMed.
- A. P. Bateman, S. A. Nizkorodov, J. Laskin and A. Laskin, Phys. Chem. Chem. Phys., 2011, 13, 12199–12212 RSC.
- J. D. Crounse, L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard and P. O. Wennberg, J. Phys. Chem. Lett., 2013, 4, 3513–3520 CrossRef CAS.
- S. A. Epstein, S. L. Blair and S. A. Nizkorodov, Environ. Sci. Technol., 2014, 48, 11251–11258 CrossRef CAS PubMed.
- Q. Chen, Y. Liu, N. M. Donahue, J. E. Shilling and S. T. Martin, Environ. Sci. Technol., 2011, 45, 4763–4770 CrossRef CAS PubMed.
- M. Krapf, I. El Haddad, E. A. Bruns, U. Molteni, K. R. Daellenbach, A. S. Prévôt, U. Baltensperger and J. Dommen, Chem, 2016, 1, 603–616 CAS.
- K. Badali, S. Zhou, D. Aljawhary, M. Antiñolo, W. Chen, A. Lok, E. Mungall, J. Wong, R. Zhao and J. Abbatt, Atmos. Chem. Phys., 2015, 15, 7831–7840 CAS.
- H. Tong, A. M. Arangio, P. S. Lakey, T. Berkemeier, F. Liu, C. J. Kampf, W. H. Brune, U. Pöschl and M. Shiraiwa, Atmos. Chem. Phys., 2016, 16, 1761–1771 CAS.
- F. Hanisch and J. N. Crowley, Phys. Chem. Chem. Phys., 2001, 3, 2474–2482 RSC.
- E. Kang, M. Root, D. Toohey and W. Brune, Atmos. Chem. Phys., 2007, 7, 5727–5744 CrossRef CAS.
- A. T. Lambe, P. S. Chhabra, T. B. Onasch, W. H. Brune, J. F. Hunter, J. H. Kroll, M. J. Cummings, J. F. Brogan, Y. Parmar, D. R. Worsnop, C. E. Kolb and P. Davidovits, Atmos. Chem. Phys., 2015, 15, 3063–3075 CrossRef CAS.
- E. A. Bruns, I. El Haddad, A. Keller, F. Klein, N. K. Kumar, S. M. Pieber, J. C. Corbin, J. G. Slowik, W. H. Brune, U. Baltensperger and A. S. H. Prévôt, Atmos. Meas. Tech., 2015, 8, 2315–2332 CAS.
- H. Zhao, J. Joseph, H. Zhang, H. Karoui and B. Kalyanaraman, Free Radical Biol. Med., 2001, 31, 599–606 CrossRef CAS PubMed.
- R. T. Weber, Xenon Data Processing Reference, 2012 Search PubMed.
- G. R. Eaton, S. S. Eaton, D. P. Barr and R. T. Weber, Quantitative EPR, Springer Science & Business Media, 2010 Search PubMed.
- T. Berkemeier, A. J. Huisman, M. Ammann, M. Shiraiwa, T. Koop and U. Pöschl, Atmos. Chem. Phys., 2013, 13, 6663–6686 Search PubMed.
- A. M. Arangio, J. H. Slade, T. Berkemeier, U. Pöschl, D. A. Knopf and M. Shiraiwa, J. Phys. Chem. A, 2015, 119, 4533–4544 CrossRef CAS PubMed.
- K. E. Kautzman, J. D. Surratt, M. N. Chan, A. W. H. Chan, S. P. Hersey, P. S. Chhabra, N. F. Dalleska, P. O. Wennberg, R. C. Flagan and J. H. Seinfeld, J. Phys. Chem. A, 2010, 114, 913–934 CrossRef CAS PubMed.
- S. Goldstein and D. Meyerstein, Acc. Chem. Res., 1999, 32, 547–550 CrossRef CAS.
- T. L. Foster and J. P. Caradonna, J. Am. Chem. Soc., 2003, 125, 3678–3679 CrossRef CAS PubMed.
- Z. Shi, M. D. Krom, S. Bonneville and L. G. Benning, Environ. Sci. Technol., 2015, 49, 1472–1477 CrossRef CAS PubMed.
- R. McWhinney, S. Zhou and J. Abbatt, Atmos. Chem. Phys., 2013, 13, 9731–9744 CrossRef.
- R. G. Zepp, B. C. Faust and J. Hoigne, Environ. Sci. Technol., 1992, 26, 313–319 CrossRef CAS.
- Z. Shi, S. Bonneville, M. Krom, K. Carslaw, T. Jickells, A. Baker and L. Benning, Atmos. Chem. Phys., 2011, 11, 995–1007 CrossRef CAS.
- P. Liu, Y. J. Li, Y. Wang, M. K. Gilles, R. A. Zaveri, A. K. Bertram and S. T. Martin, Proc. Natl. Acad. Sci. U. S. A., 2016, 201603138 Search PubMed.
- Q. Ye, E. S. Robinson, X. Ding, P. Ye, R. C. Sullivan and N. M. Donahue, Proc. Natl. Acad. Sci. U. S. A., 2016, 201604536 Search PubMed.
- L. Deguillaume, M. Leriche, K. Desboeufs, G. Mailhot, C. George and N. Chaumerliac, Chem. Rev., 2005, 105, 3388–3431 CrossRef CAS PubMed.
- H. Herrmann, T. Schaefer, A. Tilgner, S. A. Styler, C. Weller, M. Teich and T. Otto, Chem. Rev., 2015, 115, 4259–4334 CrossRef CAS PubMed.
- B. Ervens, Chem. Rev., 2015, 115, 4157–4198 CrossRef CAS PubMed.
- B. Ervens, B. Turpin and R. Weber, Atmos. Chem. Phys., 2011, 11, 11069–11102 CrossRef CAS.
- Y. Liu, A. Monod, T. Tritscher, A. Praplan, P. DeCarlo, B. Temime-Roussel, E. Quivet, N. Marchand, J. Dommen and U. Baltensperger, Atmos. Chem. Phys., 2012, 12, 5879–5895 CAS.
- V. F. McNeill, Environ. Sci. Technol., 2015, 49, 1237–1244 CrossRef CAS PubMed.
- Y. Lim and B. Turpin, Atmos. Chem. Phys., 2015, 15, 12867–12877 CAS.
- Y. Lim, Y. Tan, M. Perri, S. Seitzinger and B. Turpin, Atmos. Chem. Phys., 2010, 10, 10521–10539 CAS.
- B. Chu, Y. Liu, J. Li, H. Takekawa, J. Liggio, S.-M. Li, J. Jiang, J. Hao and H. He, Environ. Pollut., 2014, 193, 88–93 CrossRef CAS PubMed.
- E. Harris, B. Sinha, D. van Pinxteren, A. Tilgner, K. W. Fomba, J. Schneider, A. Roth, T. Gnauk, B. Fahlbusch and S. Mertes, Science, 2013, 340, 727–730 CrossRef CAS PubMed.
- M. Shiraiwa, T. Berkemeier, K. Schilling-Fahnestock, J. Seinfeld and U. Pöschl, Atmos. Chem. Phys., 2014, 14, 8323–8341 Search PubMed.
- C. C. Winterbourn, Nat. Chem. Biol., 2008, 4, 278–286 CrossRef CAS PubMed.
- K. Lucas and M. Maes, Mol. Neurobiol., 2013, 48, 190–204 CrossRef CAS PubMed.
- H. Christensen, K. Sehested and H. Corfitzen, J. Phys. Chem., 1982, 86, 1588–1590 CrossRef CAS.
- K. Sehested, O. L. Rasmussen and H. Fricke, J. Phys. Chem., 1968, 72, 626–631 CrossRef CAS.
- J. D. Rush and B. H. Bielski, J. Phys. Chem., 1985, 89, 5062–5066 CrossRef CAS.
- W. H. Koppenol, J. Butler and J. W. Vanleeuwen, Photochem. Photobiol., 1978, 28, 655–660 CrossRef CAS.
- J. Divišek and B. Kastening, J. Electroanal. Chem. Interfacial Electrochem., 1975, 65, 603–621 CrossRef.
- J. A. Thornton, L. Jaeglé and V. F. McNeill, J. Geophys. Res.: Atmos., 2008, 113, D05303 CrossRef.
- H. Bataineh, O. Pestovsky and A. Bakac, Chem. Sci., 2012, 3, 1594–1599 RSC.
- Z. Stuglik and Z. P. Zagorski, Radiat. Phys. Chem., 1981, 17, 229–233 CrossRef CAS.
- S. Lewis, A. Lynch, L. Bachas, S. Hampson, L. Ormsbee and D. Bhattacharyya, Environ. Eng. Sci., 2009, 26, 849–859 CrossRef CAS PubMed.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- E. Chevallier, R. D. Jolibois, N. Meunier, P. Carlier and A. Monod, Atmos. Environ., 2004, 38, 921–933 CrossRef CAS.
- M. Simic, P. Neta and E. Hayon, J. Phys. Chem., 1969, 73, 3794–3800 CrossRef CAS.
- D. Carlsson and K. U. Ingold, J. Am. Chem. Soc., 1967, 89, 4891–4894 CrossRef CAS.
- H. Zegota, M. N. Schuchmann, D. Schulz and C. v. Sonntag, Zeitschrift für Naturforschung B, 1986, 41, 1015–1022 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7fd00023e |
| This journal is © The Royal Society of Chemistry 2017 |