
A general polymer-assisted strategy enables unexpected efficient metal-free oxygen-evolution catalysis on pure carbon nanotubes†
You
Zhang‡
a,
Xuliang
Fan‡
a,
Junhua
Jian‡
a,
Dingshan
Yu
 *a,
Zishou
Zhang
*a and
Liming
Dai
*b
*a,
Zishou
Zhang
*a and
Liming
Dai
*b
aKey Laboratory for Polymeric Composite and Functional Materials of Ministry of Education, Key Laboratory of High Performance Polymer-based Composites of Guangdong Province, School of Chemistry Sun Yat-Sen University, Guangzhou, 510275, China. E-mail: yudings@mail.sysu.edu.cn; zhzish@mail.sysu.edu.cn
bDepartment of Macromolecule Science and Engineering, Case Western Reserve University, Cleveland, OH 44106, USA. E-mail: liming.dai@case.edu
First published on 22nd August 2017
Abstract
A conceptually new and general strategy was, for the first time, proposed to significantly boost the electrocatalytic activity of metal-free pure carbon nanotubes (CNTs) towards the oxygen evolution reaction (OER) by simple polymer wrapping without introducing any heteroatom dopants, functional groups, or edge defects into the graphitic structure. Our strategy is straightforward, efficient, green, and easy to be scaled up. After wrapping pure CNTs with a certain class of electrochemically inert polymers (i.e. poly(ethylene-alt-maleic acid), poly(vinyl alcohol), poly(vinyl acetate), poly(ethylene glycol)) with polar oxygen-containing groups (i.e. –COOH, –OH, –COOCH3, –O–) through noncovalent interactions, a series of advanced metal-free composite membrane catalysts were easily achieved, which yielded unexpected, surprisingly high OER activity – on par with the commercial noble RuO2 catalyst, though pure CNTs have rather poor OER activity. Combined experimental and computational studies revealed that the observed superb OER activity could be attributed to a synergistic effect of intrinsic topological defects in the CNTs as active centers and the coated polymer layer as a co-catalyst to optimize the adsorption energies of intermediates for improving the OER energetics.
Broader contextDeveloping low-cost efficient catalysts for the oxygen evolution reaction (OER) is a key prerequisite for the large-scale deployment of many renewable energy technologies, including rechargeable metal–air batteries and water splitting. Herein, we demonstrate a conceptually new and general polymer-assisted strategy to boost the OER performance of pure multi-walled CNTs (MWCNTs) without introducing heteroatom dopants, functional groups, or edge defects into the graphitic structures. After simply wrapping pure MWCNTs with a certain class of electrochemically inert polymers (i.e. poly(ethylene-alt-maleic acid)) with polar oxygen-containing groups (i.e. –COOH) through noncovalent interactions, the resultant composite membrane catalysts exhibited surprisingly high OER performance – on par with the benchmark RuO2 catalyst, though pure MWCNTs have poor OER activity. Combined experimental and computational studies revealed that the observed superb OER activity was attributed to a synergistic effect of intrinsic topological defects in the CNTs as the main active centers and the coated polymer layer as a co-catalyst for improved OER energetics. Our processing is straightforward and easy to be scaled up. Our finding provides new insights on efficient metal-free oxygen catalysis, which can be used as guidance for other catalytic reactions. The proposed methodology can serve as a general approach to the development of low-cost metal-free catalysts for energy-related and many other applications. |
The electrochemical oxygen evolution reaction (OER) is a pivotal process for a number of key renewable energy systems, including rechargeable metal–air batteries and water splitting.1–6 Nevertheless, OER often suffers from sluggish kinetics on the anode, and highly active catalysts are often required to reduce the overpotential for the anodic oxidation reaction.1–3 The current state-of-the-art OER electrocatalysts are noble metal oxides, such as IrO2 and RuO2. However, their scarcity and prohibitive cost severely hinder the widespread applications of the renewable energy technology. Thus, enormous efforts have been directed towards the development of low-cost and efficient OER alternatives, such as transition metal/metal compounds,7–13 perovskites,14,15 layered double hydroxides,16,17 and carbon-based materials.1–3,18 Despite some progress, most non-precious metal-based OER catalysts usually suffer from low conductivities and complicated fabrication procedures.19 Furthermore, for sustainable oxygen evolution with reduced environmental impact, it is urgent to reduce the metal usage and exploit new catalytic systems with little or no metal. In this context, metal-free carbon catalysts, such as carbon nanotubes (CNTs) and graphene, are particularly attractive for electrocatalytic water oxidation due to their low cost, high conductivity, good activity, operation stability, facile preparation, and eco-friendliness.1,20–22 Reported carbon-based OER catalysts mainly include heteroatom-doped carbon,23–27 surface oxidized carbon,28,29 and defective carbon.30–33 It was found that the introduction of dopants (i.e. N, P, S), functional groups (i.e. C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), or edge defects into the graphitic framework can improve the OER activity of nanocarbons by inducing charge redistribution, molecular orbital distortion, and/or electron transfer.23,34 Compared to the benchmark noble metal OER catalysts (e.g., IrO2, RuO2), however, the OER performance of the carbon-based counterparts still needs to be further improved. Besides, the specific role of topological defects, intrinsically associated with certain nanocarbons (i.e. CNTs), in the OER cannot be underrated, but still remains elusive. On the other hand, the preparation of these previous carbon catalysts often involves high temperature with high energy consumption and harmful chemical treatments, which are tedious and difficult to be scaled up. Thus, it is highly desirable, though challenging, to develop new facile and effective strategies to further boost the OER performance of metal-free carbon catalysts and understand the underlying catalytic mechanisms.
O), or edge defects into the graphitic framework can improve the OER activity of nanocarbons by inducing charge redistribution, molecular orbital distortion, and/or electron transfer.23,34 Compared to the benchmark noble metal OER catalysts (e.g., IrO2, RuO2), however, the OER performance of the carbon-based counterparts still needs to be further improved. Besides, the specific role of topological defects, intrinsically associated with certain nanocarbons (i.e. CNTs), in the OER cannot be underrated, but still remains elusive. On the other hand, the preparation of these previous carbon catalysts often involves high temperature with high energy consumption and harmful chemical treatments, which are tedious and difficult to be scaled up. Thus, it is highly desirable, though challenging, to develop new facile and effective strategies to further boost the OER performance of metal-free carbon catalysts and understand the underlying catalytic mechanisms.
Herein, we demonstrate a conceptually new and general polymer-assisted strategy to dramatically boost the OER performance of metal-free pure multi-walled CNTs (MWCNTs). Unlike all the previously reported carbon-based OER catalysts, this newly developed strategy does not destroy the structural integrity of the CNT graphitic network. By simply wrapping pure CNTs with a certain class of electrochemically inert polymers (i.e. poly(ethylene-alt-maleic acid) (PEMAc), poly(acrylic acid) (PAA), poly(vinyl alcohol) (PVA), poly(vinyl acetate) (PVAc), poly(ethylene glycol) (PEG)) containing polar oxygen-carrying groups (i.e. –COOH, –OH, –COOCH3, –O–) without any charge transfer, we can create a favorable environment at the polymer@CNT interface for stabilizing the OER intermediates via hydrogen bonding to modulate the OER kinetics and eventually impart surprisingly high OER activities to the CNTs. Despite the fact that pure CNTs have very poor OER activity and the used polymers are electrochemically inert, the free-standing composite membrane catalysts derived from the polymer-wrapped CNTs exhibited unexpected, dramatically improved OER activities in alkaline media – superior to almost all existing metal-free carbon catalysts and even comparable to the benchmark RuO2 catalyst with similar mass loading. Combined experimental and computational studies reveal the critical role of the intrinsic topological defects in CNTs as active centers and polar oxygen groups in the wrapped polymer chains as a “kinetics accelerator” for generating the observed remarkable OER activity. The ease with which pure CNTs can be transformed into efficient metal-free OER catalysts simply by polymer wrapping opens new avenues for low-cost, large-scale preparation of various effective metal-free catalysts. Although CNTs and graphene wrapped with specific charge-transferrable polymers (e.g., polyelectrolytes, PDDA) have been reported to show catalytic activities for the oxygen reduction reaction (ORR),22,34 the possible effects of electrochemically inert polymers on the OER kinetics of CNTs, in particular, and the possibility of such polymer-wrapped carbon material as efficient OER catalysts have not been explored yet.
Fig. 1a shows a schematic illustration of the general procedure to prepare a series of different free-standing polymer/MWCNT composite membranes (denoted as polymer@CNTs). A certain class of polymers with various polar oxygen-containing groups, including PEMAc, PVA, PAA, PVAc, and PEG, was used for wrapping CNTs via noncovalent interactions. Prior to use, the as-received MWCNTs (Nanocyl NC 7000) were thoroughly purified to remove the residual metal impurities, if any, by refluxing in hydrochloric acid, which, unlike the oxidative purification with concentrated nitric or sulfuric acid, can avoid the introduction of surface oxygen groups on the CNTs, as confirmed by X-ray photoelectron spectroscopy (XPS) results (Fig. 1e and Fig. S1, ESI†). Here, a high frequency, low power sonic bath (Elmasonics, 40 kHz) was used for dispersing the CNTs and the samples were sonicated in an ice bath with a sonication time of only 20 min in the dispersion solvent of acetone or water. The effect of the sonication on the surface structure and activity of the CNTs was examined by Raman and XPS spectroscopy as well as electrochemical testing. It was found that the sonication effect was negligible under our mild sonication conditions with a short sonication time (see details in Fig. S2 and S3, ESI†). In a typical experiment, a given amount of purified MWCNTs was firstly mixed with the polymer in a desirable solvent (i.e. acetone, water) under sonication to form a homogenous dispersion. Thereafter, the mixture dispersion was simply poured onto a clean vessel, followed by the complete evaporation of the solvent and mechanically peeling to produce a free-standing composite membrane. Different CNT amounts (95 wt%, 90 wt%, and 80 wt%) were used for producing the composite membranes with each of the aforementioned specific polymers, being denoted as polymer@CNTs95, polymer@CNTs90, and polymer@CNTs80, respectively.
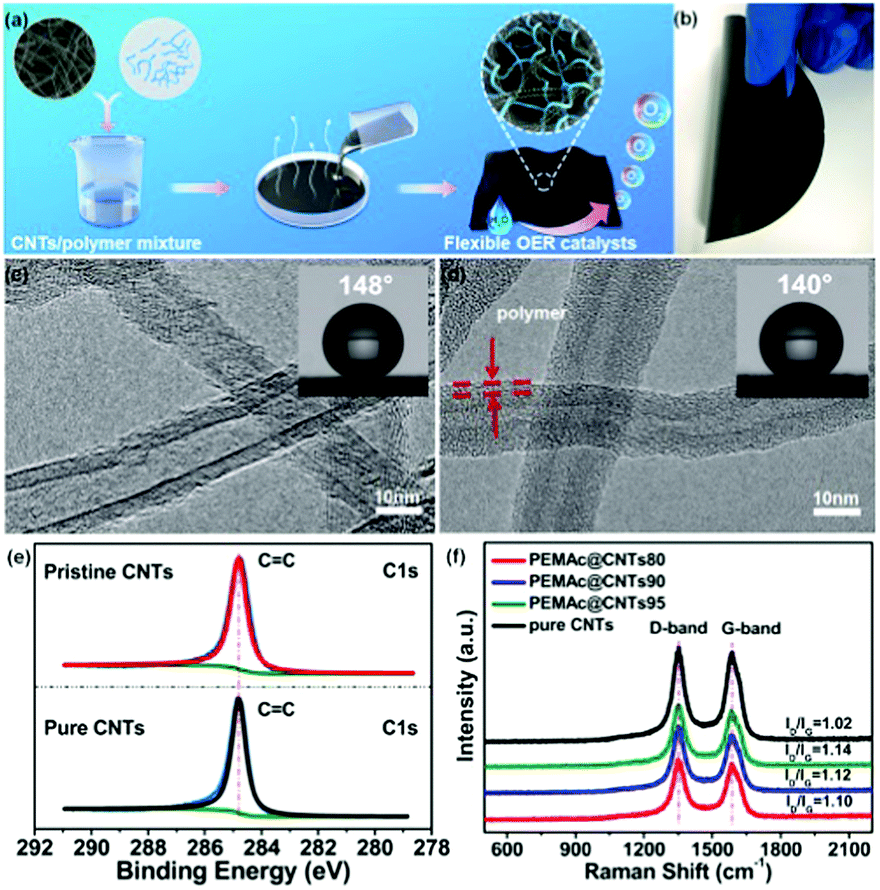 | ||
| Fig. 1 (a) Schematic illustration of the preparation procedure for flexible OER electrocatalysts based on polymer@CNT composite membranes. (b) A photograph of the flexible free-standing PEMAc@CNTs90 membrane that is 12.56 cm2 in size. (c and d) Representative HRTEM images of pure CNTs and PEMAc@CNTs90; the inset shows the water contact angle of the sample. (e) High resolution XPS C1s spectra for pristine and purified CNTs. (f) Raman spectra of purified CNTs, PEMAc@CNTs80, PEMAc@CNTs90, and PEMAc@CNTs95. | ||
The as-produced membrane (e.g., PEMAc@CNTs90) showed a good mechanical flexibility (Fig. 1b). It is noted that our produced membranes are already 12.56 cm2 in size (Fig. 1b). This method is straightforward and readily adapted to much larger size membranes. Fig. 1c and d present high-resolution transmission microscopic (HRTEM) images of pure CNTs and PEMAc@CNTs90, which show a ∼2 nm thick polymer coating-layer along the nanotube length in PEMAc@CNTs90 through the hydrophobic interaction between the polymer backbone and the nanotube surface.35 The thickness of the polymer coating layer can be easily regulated by varying the nanotube amount (Fig. S4 and S5, ESI†). The coating thickness increased with decreasing CNT content, leading to a thick polymer coating (thickness of ∼3 nm) at 80 wt% CNT content. The insets of Fig. 1c and d show the water contact angles for the pure CNT and PEMAc@CNTs90 membranes, respectively. Clearly, the contact angle of the composite membrane was smaller than that of the pure CNT membrane, due to the presence of hydrophilic carboxyl groups in the PEMAc side chains around the polymer-coated CNTs. This should reduce the intrinsic van der Waals force of the nanotubes to form a good CNT dispersion.35 Nevertheless, the polymer coating layer is too thin to manifest precise hydrophilicity relative to the pure polymers and the ultrathin coating has little effect on the electron transfer of the composite electrodes, as confirmed by electrochemical impedance spectra (Fig. S6, ESI†).
The surface chemistry of the as-produced samples was explored by XPS. As shown in Fig. S1a–c (ESI†), for the purified CNTs, a pronounced C1s peak was observed and no peaks from possible metal impurities and heteroatom dopants were detected in the XPS spectrum. It is noted that the presence of a trace amount of O (rather low atomic ratio of O/C: 0.014%) for the purified CNTs was possibly credited to physically absorbed oxygen,36,37 since CNTs are well known to be susceptible to oxygen adsorption even at a pressure as low as 10−8 to 10−10 Torr typical for the XPS measurements.36,37 Furthermore, the high-resolution XPS C1s spectra for the purified CNTs are very similar to those of the pristine CNTs and highly ordered pyrolytic graphite (HOPG) and can be fitted into only one CC peak without any other oxygen-carrying component (Fig. 1e). These results verify that our purified CNTs are indeed free from surface oxygen groups (i.e. C–O, CO).36–38 Upon PEMAc modification, the intensity of the O1s peak for the PEMAc@CNTs significantly increased, which arises from the polymer chain with oxygen-containing groups anchored on the nanotube surface. The Raman spectra for the purified CNTs and PEMAc@CNTs shown in Fig. 1f and Fig. S7 (ESI†) revealed almost the same pattern with the D and G bands centered at ∼1350 and ∼1580 cm−1 respectively. Notably, the peak positions for both the D and G bands almost remained unchanged after polymer wrapping, implying that there is no charge-transfer interaction between the two components, since the presence of charge-transfer interaction between the CNT and the polymer would result in a shift in the G band position.39,40
The electrocatalytic OER properties of the as-obtained samples were evaluated by linear sweep voltammetric (LSV) measurements in 1 M KOH solution. Similar to previous reports regarding carbon-based OER catalysts,23,28,30,33 the applied potential range in our case is ∼1.0–1.75 V vs. RHE, which avoids possible electrochemical etching of the CNTs at an overly high potential in KOH during the OER test (see Fig. S20, ESI†).28 For comparison, similar measurements were also performed on the pristine CNTs, purified CNTs, and RuO2. All the potentials cited in this study were calibrated with respect to a reversible hydrogen electrode (RHE). We tested the PEMAc@CNT catalysts with different CNT contents and found that the PEMAc@CNTs90 catalyst exhibited an optimized OER activity with the lowest overpotential (η) of 298 mV at 10 mA cm−2 (Fig. 2a). This value of η is comparable to that of the commercial noble RuO2 catalyst (294 mV) and significantly lower than those of the pristine CNTs (466 mV) and purified CNTs (511 mV) with the identical mass loading, indicating a giant enhancement in the OER activity after the polymer wrapping. Remarkably, PEMAc@CNTs90 outperformed most previously reported metal-free carbon-based OER catalysts and even many transition-metal-based OER catalysts with similar mass loading (Fig. 2c, Tables S2 and S3, ESI†).
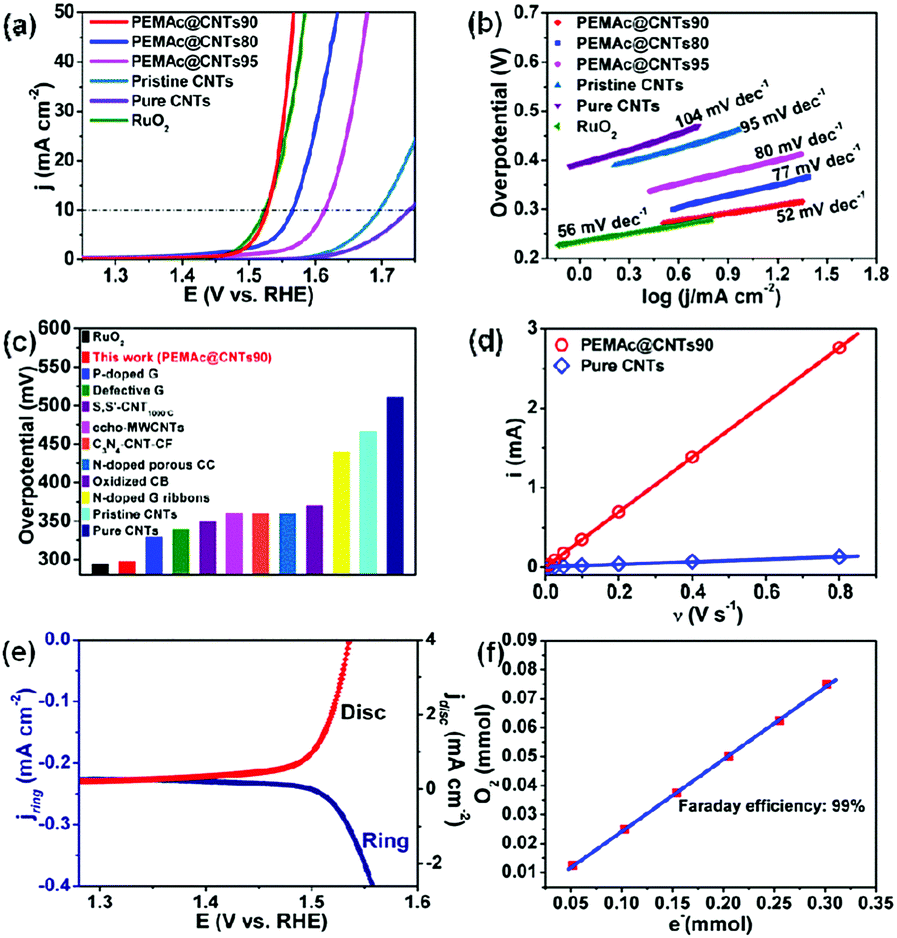 | ||
| Fig. 2 (a) LSV curves normalized to the geometric area of the electrodes for the OER of PEMAc@CNTs90, PEMAc@CNTs80, PEMAc@CNTs95, pristine CNTs, purified CNTs, and RuO2 in 1 M KOH solution with the same mass loading of 0.3 mg cm−2. (b) The corresponding Tafel plots. (c) OER overpotentials of various metal-free electrocatalysts for comparison at 10 mA cm−2 in 1 M KOH. (d) The anodic capacitance currents plotted as a function of scan rate, the data points were fitted to a linear regression enabling the estimation of double-layer capacitance. (e) Detection of O2 evolution from PEMAc@CNTs90 using RRDE measurements, the O2 generated during the anodic polarization scan was reduced at the Pt ring at 0.4 V. (f) Comparison of evolved O2vs. the amount of consumed e− during the course of electrolysis. | ||
Fig. 2b further shows an ultrasmall Tafel slope of 52 mV dec−1 for PEMAc@CNTs90 relative to other samples, including purified CNTs (104 mV dec−1), pristine CNTs (95 mV dec−1) and RuO2 (56 mV dec−1). Taken together, the above results suggest that the polymer wrapping has not only significantly reduced the catalytic overpotential but also boosted the reaction kinetics for the oxygen evolution reaction. To further understand the effect of the polymer wrapping on the CNT catalyst towards OER activity, the electrochemically active surface areas (ECSA) of pure CNTs and PEMAc@CNTs90 were estimated to be 4 and 86 cm2, respectively (Fig. 2d and Fig. S8, ESI†). Despite the marked increase in ECSA, the correcting LSV data for the ECSA shows that the OER performance of the composite catalyst is still much superior to that of pure CNTs (Fig. S9, ESI†). At 0.5 mA cmECSA−2, there is a 105 mV overpotential decrease for PEMAc@CNTs90. At η = 350 mV, PEMAc@CNTs90 delivers a ∼9 times higher current density (jECSA) than that of pure CNTs. This indicates that the increase in ECSA is not the sole cause for the drastically improved OER activity of the polymer@CNT catalyst and the wrapping polymer indeed intrinsically facilitates OER at the CNTs. Thus, the coated polymer layer acts as a co-catalyst to promote the OER at the CNTs. To determine the origin of the superb OER activity, SCN− ions were introduced into the electrolyte to poison the metal-based (i.e. Fe) catalytic sites,33 if any, which led to no activity loss (Fig. S10, ESI†), thus, the observed superior OER activity for PEMAc@CNTs90 arose from metal-free active centers, excluding the role of possible metal-impurities towards the OER over our composite catalysts (see Table S1, ESI†).
To further validate the OER efficiency of PEMAc@CNTs90, we performed rotating ring disk electrode (RRDE) measurements in N2-saturated KOH electrolyte (Fig. 2e). The potential for the Pt-ring electrode is held at 0.4 V to monitor the evolved oxygen at the PEMAc@CNTs90 modified disk electrode. When the potential was scanned above the onset potential of the OER (1.46 V), the ORR current was simultaneously detected at the ring electrode and increased concomitantly with the OER current at the disk electrode, implying that oxygen was generated at the disk electrode (Fig. S11, ESI†). The Faradaic efficiency was determined by the volumetric method at 10 mA cm−2, which reached up to a 99% Faradaic efficiency, indicating that oxygen was the only gaseous product of the electrode reaction (Fig. 2f).28 The chronopotentiometric response of PEMAc@CNTs90 showed that the current largely remained stable over 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s during the OER reaction at 5 mA cm−2 (Fig. S12, ESI†). The appearance of a sustained current indicates that the polymer layer is stable on the CNT surface, as verified by HRTEM observation (Fig. S13, ESI†). Gas chromatographic tests on the gas product from the water electrolysis using PEMAc@CNTs90 showed no other possible oxidation products. These results, coupled with the high Faraday efficiency of 99%, unambiguously verify that the newly developed PEMAc@CNT composites are highly efficient, selective, and durable OER catalysts in alkaline media.
000 s during the OER reaction at 5 mA cm−2 (Fig. S12, ESI†). The appearance of a sustained current indicates that the polymer layer is stable on the CNT surface, as verified by HRTEM observation (Fig. S13, ESI†). Gas chromatographic tests on the gas product from the water electrolysis using PEMAc@CNTs90 showed no other possible oxidation products. These results, coupled with the high Faraday efficiency of 99%, unambiguously verify that the newly developed PEMAc@CNT composites are highly efficient, selective, and durable OER catalysts in alkaline media.
Interestingly, similar enhancement in the OER performance for the PEMAc@CNTs was also observed on pure CNTs wrapped by other polymers, including PAA, PVAc, PVA, and PEG (Fig. 3). Prior to the OER test, the morphology, microstructure, and stability of these newly developed polymer@CNT membranes were investigated (Fig. S1, S4, S5, S12 and S14, ESI†). Compared to purified CNTs, these polymer@CNT composite catalysts exhibited dramatically enhanced OER activities with a significantly reduced overpotential at 10 mA cm−2 from 511 mV for pure CNTs to 344 mV for PAA@CNTs90, 356 mV for PVA@CNTs90, 373 mV for PVAc@CNTs90, and 390 mV for PEG@CNTs90 (Fig. 3a and Fig. S14, ESI†). Also, the Tafel slopes for all of these polymer@CNT composite catalysts are smaller than that of the purified CNTs (Fig. 3b). These results reveal the versatility for the polymer-wrapping strategy developed in this work.
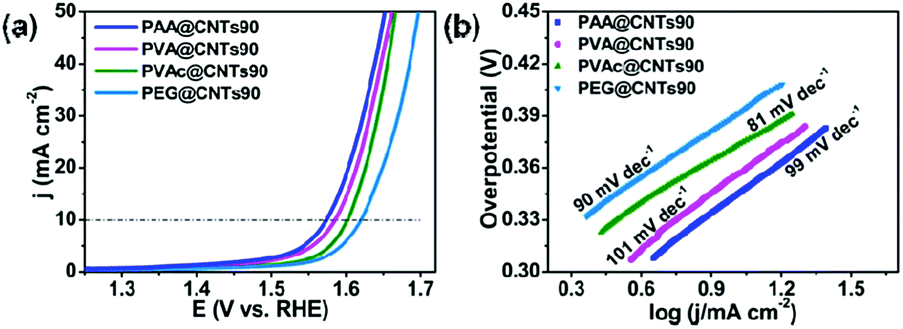 | ||
| Fig. 3 (a) LSV curves for the OER of PAA@CNTs90, PVA@CNTs90, PVAc@CNTs90 and PEG@CNTs90 in 1 M KOH. (b) The Tafel plots. | ||
For our composite catalysts, the employed polymers are electrochemically inert without OER activity (Fig. S15, ESI†) and have no charge-transfer capability, as verified by the Raman spectra (Fig. 1f and Fig. S7, ESI†).22,34 Thus, the OER active sites for our composite should come from the CNTs. Previous studies have shown that possible OER active sites for metal-free nanocarbon catalysts can be created by heteroatom-dopants, surface oxygen groups (i.e. CO), and defective sites induced by chemical functionalization.1,30–33 Unlike all previous carbon-based OER catalysts, however, our polymer@CNT catalysts do not destroy the CNT structure and retain the graphitic structure integrity, which excludes the presence of heteroatom dopants and surface oxygen groups as evidenced by detailed XPS analysis (Fig. 1e and Fig. S1, ESI†). Also, more recent work has experimentally confirmed that individual MWCNTs were not electrochemically etched and/or oxidized within a certain potential range of ∼1.0–1.8 V vs. RHE in 1 M KOH28 and even after a long-term OER durability test, the MWCNTs remained almost unaffected in terms of their chemical compositions and structures,28 which is similar to our experimental observation by TEM and XPS for our composite catalysts after the durability test (1.0–1.70 V vs. RHE) (Fig. S13 and S20, ESI†). Meanwhile, the role of metal impurities was also ruled out, as elucidated above (Fig. S10 and Table S1, ESI†). Thus, for our CNTs free from dopants and oxygen groups, there are two possible sites: non-defective sites (perfect surface) and intrinsic topological defects (TDs, i.e. pentagon and heptagon carbon rings), while recent studies have identified intrinsic TDs as effective active sites for OER over dopant (i.e. O, N)-free carbons.30,33 Based on the above fact, intrinsic TDs (i.e. topological Stone–Wales defects) in the CNTs are thus considered as the main OER active center for our polymer@CNT catalysts, as validated by our theoretical calculation (vide infra, Fig. 4 and Fig. S16–S18, ESI†) and supported by recent literature.30,33 Despite the absence of oxygen groups in our purified CNTs (Fig. S1, ESI†), it is noted that all the polymers used in this study possess oxygen-containing polar groups, including –COOH, –OH, –COOCH3, and –O–, that are prone to interact with the H-carrying OER intermediates (e.g., OH* and OOH*) through H-bonding interactions,41 to facilitate the adsorption of the OER intermediates at the polymer@CNT interface. Previous theoretical studies demonstrated that the OER activities of carbon materials are mainly driven by the energetics of the OER intermediates (OH*, O*, and OOH*) on the catalyst surface and the transformation from O* to OOH* is often the rate determining step.30,33Fig. 4a shows that the PEMAc layer could stabilize both OH* and OOH* by hydrogen bonding, thus regulating the chemisorption of intermediates at the PEMAc@CNT interface by creating a stabilized local environment to accelerate the OER kinetics,41 as verified by our theoretical modeling (vide infra, Fig. 4c–e). This is consistent with our experimental observation that the OER overpotential was significantly reduced with a concomitant sharp decline in the Tafel slope after introducing the polymer layer onto the CNTs (Fig. 2a and b and Fig. S8, ESI†). Also, the 3D porous membrane structures (Fig. S19, ESI†) facilitate the mass transport.
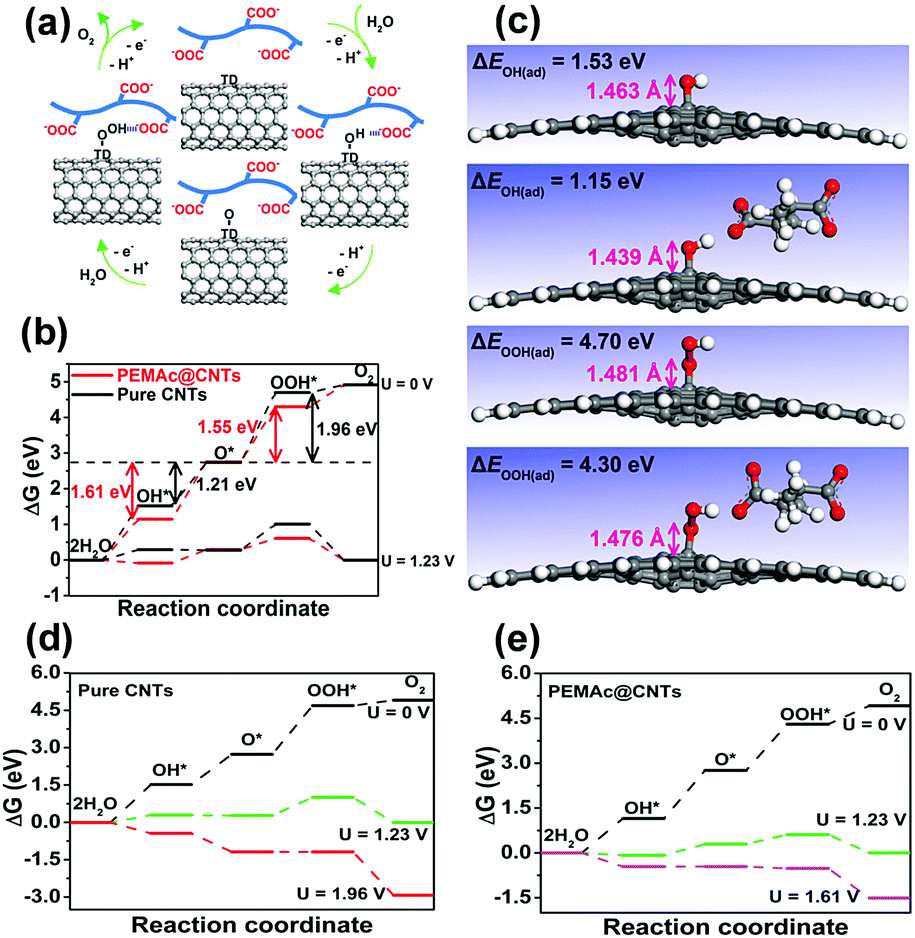 | ||
| Fig. 4 (a) Proposed reaction mechanism at the PEMAc–CNT interface in alkaline media. (b) Standard free energy diagrams of the OER at U = 0 V and U = 1.23 V for PEMAc@CNTs and pure CNTs. (c) The optimized geometries of pure CNTs and PEMAc@CNTs with intermediate adsorbate OH* or OOH*. (d) Standard free energy diagram for elementary reactions of the OER over pure CNTs, and (e) PEMAc@CNTs with topological defects on the carbon plane as active centers. | ||
To gain further insight into the exceptionally enhanced OER performance for the polymer@CNT catalysts, we performed density functional theory (DFT) calculations and identified that topological Stone–Wales defects, the most common and predominant TDs in CNTs,42 were the most effective OER active sites and other sites were ineffective (see details in ESI,† Fig. S16–S18). The free energy diagrams of OER substeps on the pure CNTs and PEMAc@CNTs are predicted in Fig. 4b. For the pure CNTs, the adsorption of OOH* species on the CNTs is too weak and thus the transformation from O* to OOH* is the rate determining step (ΔGO* → OOH* = 1.96 eV), similar to previous investigations.33 In contrast, for PEMAc@CNTs, the adsorption energies for both OH* and OOH* species exhibit a marked decline from 1.53 and 4.70 eV to 1.15 and 4.30 eV, respectively. This is accompanied by reductions in the bond lengths of both C–OH and C–OOH from 1.463 and 1.481 Å to 1.439 and 1.476 Å, respectively (Fig. 4c), and the reduced bond lengths for C–OH and C–OOH imply that the PEMAc layer indeed renders a favourable local environment to stabilize OH* and OOH* through hydrogen bonding.41 The above binding energy changes eventually lead to a notable decrease in the free energy of OOH* formation (ΔGO* → OOH* = 1.55 eV) and an obvious increase in the free energy of O* formation (ΔGOH* → O* = 1.61 eV), which is regarded as the rate determining step for oxygen evolution on the polymer@CNT composite catalysts. Clearly, compared to the pure CNTs (Fig. 4b, 1.96 eV), the polymer@CNT composite catalysts displayed a substantially lower activation barrier (1.61 eV), which coincides with the much lower calculated overpotential (0.38 V) for the composite system than that of the pure CNTs (0.73 V) (Fig. 4d and e and Fig. S18, ESI†). The above results indicate that simple wrapping of certain polymers with polar oxygen groups onto CNTs can regulate the adsorption energies of the OER intermediates towards the optimal values to enhance the energetics for the OER, facilitating O2 evolution.
Conclusions
In summary, we have, for the first time, demonstrated that a certain class of polymers with polar oxygen groups can be utilized to functionalize CNTs simply through polymer wrapping to afford a series of advanced metal-free OER catalysts with high performance. Although pure CNTs have very little OER activity and the polymers used are electrochemically inert without charge-transfer capability, the derived polymer-wrapped CNT composite membranes demonstrated drastically reduced overpotential and improved kinetics for the OER. Unlike previous carbon-based catalysts, the polymer-wrapped CNT composite catalysts developed in this study represent the first success to boost the OER performance for metal-free CNTs without any damage or charge-transfer to the inherent nanotube structure. Remarkably, the optimal PEMAc@CNT catalyst exhibited a low overpotential of 298 mV at 10 mA cm−2 with an ultrasmall Tafel slope of 52 mV dec−1 in alkaline media, in sharp contrast to the pure CNTs (511 mV, 104 mV dec−1). Our catalyst surpasses almost all existing metal-free carbon-based OER catalysts and even many transition-metal-based catalysts reported to date and is on par with the benchmark RuO2 (294 mV, 56 mV dec−1) with similar mass loading. Combined experimental and computational studies reveal an important role in the observed superb OER performance co-played by both the intrinsic topological defects in CNTs and oxygen-containing groups along the adsorbed polymer chains. Specifically, the topological Stone–Wales defects of CNTs are identified to act as the main active sites while the wrapped polymer layer functions as a co-catalyst to stabilize the OER intermediates by hydrogen bonding, optimizing the adsorption energies of the intermediates and eventually improving the OER energetics. Our preparation method is simple, efficient, and easy to be scaled up, possessing overwhelming advantages over the current complicated fabrication process of OER catalysts. Our results provide new insights on efficient metal-free oxygen catalysis, which can be used as guidance for other catalytic reactions. Thus, the methodology developed herein serves as a general approach to the design and development of various low-cost, high-performance metal-free catalysts for energy-related and many other applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the Natural Science Foundation of China (No. 51573214, 51573213 and 51303215), the Youth 1000 Talent Program of China, and the Pearl River Nova Program of Guangzhou (201610010163).References
- X. E. Liu and L. M. Dai, Nat. Rev. Mater., 2016, 1, 16064 CrossRef CAS.
- L. M. Dai, Y. H. Xue, L. T. Qu, H. J. Choi and J. B. Baek, Chem. Rev., 2015, 115, 4823 CrossRef CAS PubMed.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060 RSC.
- Z. L. Wang, X. F. Hao, Z. Jiang, X. P. Sun, D. Xu, J. Wang, H. X. Zhong, F. L. Meng and X. B. Zhang, J. Am. Chem. Soc., 2015, 137, 15070 CrossRef CAS PubMed.
- J. Wang, H. X. Zhong, Z. L. Wang, F. L. Meng and X. B. Zhang, ACS Nano, 2016, 10, 2342 CrossRef CAS PubMed.
- Z. X. Tao, T. Wang, X. J. Wang, J. Zheng and X. G. Li, ACS Appl. Mater. Interfaces, 2016, 8, 35390 CAS.
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780 CrossRef CAS PubMed.
- K. Xu, P. Z. Chen, X. L. Li, Y. Tong, H. Ding, X. J. Wu, W. S. Chu, Z. M. Peng, C. Z. Wu and Y. Xie, J. Am. Chem. Soc., 2015, 137, 4119 CrossRef CAS PubMed.
- H. B. Tao, L. W. Fang, J. Z. Chen, H. B. Yang, J. J. Gao, J. W. Miao, S. L. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 9978 CrossRef CAS PubMed.
- X. F. Xiao, C. T. He, S. L. Zhao, J. Li, W. S. Lin, Z. K. Yuan, Q. Zhang, S. Y. Wang, L. M. Dai and D. S. Yu, Energy Environ. Sci., 2017, 10, 893 CAS.
- J. Wang, H. X. Zhong, Y. L. Qin and X. B. Zhang, Angew. Chem., Int. Ed., 2013, 52, 5248 CrossRef CAS PubMed.
- J. Wang, K. Li, H. X. Zhong, D. Xu, Z. L. Wang, Z. Jiang, Z. J. Wu and X. B. Zhang, Angew. Chem., Int. Ed., 2015, 54, 10530 CrossRef CAS PubMed.
- H. X. Zhong, J. Wang, F. L. Meng and X. B. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9937 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383 CrossRef CAS PubMed.
- J. I. Jung, H. Y. Jeong, J. S. Lee, M. G Kim and J. Cho, Angew. Chem., Int. Ed., 2014, 53, 4582 CrossRef CAS PubMed.
- M. Gong, Y. G. Li, H. L. Wang, Y. Y. Liang, J. Z. Wu, J. G. Zhou, J. Wang, T. Regier, F. Wei and H. J. Dai, J. Am. Chem. Soc., 2013, 135, 8452 CrossRef CAS PubMed.
- M. Görlin, P. Chernev, J. Ferreira de Araújo, T. Reier, S. Dresp, B. Paul, R. Krähnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603 CrossRef PubMed.
- J. J. Zhao, Y. M. Liu, X. Quan, S. Chen, H. M. Zhao and H. T. Yu, Electrochim. Acta, 2016, 204, 169 CrossRef CAS.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347 CrossRef CAS PubMed.
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760 CrossRef CAS PubMed.
- D. S. Yu, Q. Zhang and L. M. Dai, J. Am. Chem. Soc., 2010, 132, 15127 CrossRef CAS PubMed.
- S. Y. Wang, D. S. Yu and L. M. Dai, J. Am. Chem. Soc., 2011, 133, 5182 CrossRef CAS PubMed.
- J. T. Zhang, Z. H. Zhao, Z. H. Xia and L. M. Dai, Nat. Nanotechnol., 2015, 10, 444 CrossRef CAS PubMed.
- J. P. Lai, S. P. Li, F. X. Wu, M. Saqib, R. Luque and G. B. Xu, Energy Environ. Sci., 2016, 9, 1210 CAS.
- C. G. Hu and L. M. Dai, Adv. Mater., 2017, 29, 1604942 CrossRef PubMed.
- M. S. Balogun, W. T. Qiu, H. Yang, W. J. Fan, Y. C. Huang, P. P. Fang, G. R. Li, H. B. Ji and Y. X. Tong, Energy Environ. Sci., 2016, 9, 3411 CAS.
- A. M. EI-Sawy, I. M. Mosa, D. Su, C. J. Guild, S. Khalid, R. Joesten, J. F. Rusling and S. L. Suib, Adv. Energy Mater., 2016, 6, 1501966 CrossRef.
- X. Y. Lu, W. L. Yim, B. H. R. Suryanto and C. Zhao, J. Am. Chem. Soc., 2015, 137, 2901 CrossRef CAS PubMed.
- B. H. R. Suryanto and C. Zhao, Chem. Commun., 2016, 52, 6439 RSC.
- Y. Jia, L. Z. Zhang, A. J. Du, G. P Gao, J. Chen, X. C. Yan, C. L. Brown and X. D. Yao, Adv. Mater., 2016, 28, 9532 CrossRef CAS PubMed.
- Y. F. Jiang, L. J. Yang, T. Sun, J. Zhao, Z. Y. Lyu, O. Zhuo, X. Z. Wang, Q. Wu, J. Ma and Z. Hu, ACS Catal., 2015, 5, 6707 CrossRef CAS.
- H. L. Jin, H. H. Huang, Y. H. He, X. Feng, S. Wang, L. M. Dai and J. C. Wang, J. Am. Chem. Soc., 2015, 137, 7588 CrossRef CAS PubMed.
- C. Tang, H. F. Wang, X. Chen, B. Q. Li, T. Z. Hou, B. S. Zhang, Q. Zhang, M. M. Titirici and F. Wei, Adv. Mater., 2016, 28, 6845 CrossRef CAS PubMed.
- S. Y. Wang, D. S. Yu, L. M. Dai, D. W. Chang and J.-B. Baek, ACS Nano, 2011, 5, 6202 CrossRef CAS PubMed.
- N. G. Sahoo, S. Rana, J. W. Cho, L. Li and S. H. Chan, Prog. Polym. Sci., 2010, 35, 837 CrossRef CAS.
- Q. Chen, L. Dai, M. Gao, S. Huang and A. Mau, J. Phys. Chem. B, 2001, 105, 618 CrossRef CAS.
- K. M. Lee, L. Li and L. Dai, J. Am. Chem. Soc., 2005, 127, 4122 CrossRef CAS PubMed.
- H. Li, T. Xu, C. Wang, J. Chen, H. Zhou and H. Liu, Diamond Relat. Mater., 2006, 15, 1228 CrossRef CAS.
- S. Y. Wang, D. S. Yu and L. M. Dai, J. Am. Chem. Soc., 2011, 133, 5182 CrossRef CAS PubMed.
- H. J. Shin, S. M. Kim, S. M. Yoon, A. Benayad, K. K. Kim, S. J. Kim, H. K. Park, J. Y. Choi and Y. H. Lee, J. Am. Chem. Soc., 2008, 130, 2062 CrossRef CAS PubMed.
- S. Zhang, P. Kang, S. Ubnoske, M. K. Brennaman, N. Song, R. L. House, J. T. Glass and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 7845 CrossRef CAS PubMed.
- L. G. Zhou and S. Q. Shi, Appl. Phys. Lett., 2003, 83, 1222 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ee01702b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |