
Photoswitchable molecules as key ingredients to drive systems away from the global thermodynamic minimum
Michael
Kathan
 and
Stefan
Hecht
*
and
Stefan
Hecht
*
Department of Chemistry & IRIS Adlershof, Humboldt-Universität zu Berlin, Brook-Taylor-Str.2, 12489 Berlin, Germany. E-mail: sh@chemie.hu-berlin.de
First published on 31st August 2017
Abstract
In order to perform chemical work, molecular systems have to be operated away from thermodynamic equilibrium and therefore require the input of energy. Light is perhaps the most abundant and advantageous energy source that in combination with photoswitches allows for a reversible and hence continuous stimulation of a system. In this review, we illustrate how photoswitchable molecules can be used to escape the global thermodynamic minimum by populating metastable states, from which energy can be transferred and transformed in a controlled fashion. We emphasize the unique feature of photodynamic equilibria, in which population of the states is dictated by the excitation wavelength (and not primarily by temperature), thereby avoiding microscopic reversibility since the photoreaction involves an electronically excited state. Thus, photoswitchable molecular systems can remotely be controlled with high spatial and temporal resolution and in addition their action can be fueled by light.
 Michael Kathan (left) and Stefan Hecht (right) | Michael Kathan (left) studied chemistry at the Freie Universität Berlin and ETH Zürich, with a main research interest in fluorine chemistry and carbon rich materials. After obtaining his master's degree from Freie Universität Berlin under the guidance of Prof. Dieter Lentz, he joined the group of Prof. Stefan Hecht at Humboldt-Universität zu Berlin as a graduate student in 2015. The focus of his doctoral research is on controlling chemical reactivity and adaptive, imine-based materials with light. |
Stefan Hecht (right) studied chemistry at Humboldt-Universität zu Berlin and obtained his PhD from the University of California at Berkeley in 2001, working under the guidance of Prof. Jean M. J. Fréchet. After establishing his own research group at Freie Universität Berlin and a subsequent position as a group leader at the Max-Planck-Institut für Kohlenforschung in Mülheim an der Ruhr, he returned to his alma mater in 2006. His research interests range from synthetic macromolecular and supramolecular chemistry to surface science, with particular focus on utilizing photochromic molecules for remote-controlling materials, devices, and processes. More information at: http://www.hechtlab.de. |
Introduction
In the defining, “golden” era of Physics and Chemistry in the late 19th and early 20th century, the fundamental relationships that (seem to) explain the world around us have been uncovered and formulated. The foundation lies in the four laws of thermodynamics, which describe fundamental properties of matter or more precisely thermodynamic systems. Among them, the Second Law is of particular importance to analyze physical and chemical processes and has been rephrased as the principle of minimum energy, which states that the Gibbs free energy of a closed system will approach a minimum value at equilibrium. As a consequence the most stable state, which can be characterized as the global minimum on the potential energy surface, will predominantly be populated at thermodynamic equilibrium. However, nature, and in particular life, presents itself in many variations manifested in a sheer infinite number of structures with functions that are encoded by their properties and interconnecting processes. Obviously, there are ways to escape thermodynamic equilibrium, at least temporarily.In fact, time and hence kinetics are the key! By stabilizing or trapping alternative, so called metastable states, represented by local minima on the potential energy surface, their conversion to the thermodynamically most stable state can be slowed down or stopped since the height of the barrier dictates the rate of their interconversion. However, processes will only occur if these metastable states can be populated and therefore, in addition to providing kinetic barriers, the system needs to be supplied with energy to drive the processes. It is important to note at this point that in non-classical, non-equilibrium thermodynamics, states at the slope of the potential energy surface, i.e. no local minima, are being considered as dissipative states. In the following, we intentionally exclude this scenario1 and focus on metastable states, i.e. true local minima, as the “energy absorbers” that can dissipate their energy either upon thermal relaxation or by engaging in consecutive chemical processes and thereby fuel the system.2
Even if sufficient energy is supplied to the system, at thermodynamic equilibrium there is an inherent problem related to the population of the states as defined by Boltzmann. While the higher energy state becomes increasingly populated with a decreasing energy gap between the states and with increasing temperature, no population inversion, i.e. predominant population of the higher energy state, is possible – at least at thermodynamic equilibrium. The only option is to leave the thermal ground state potential energy surface upon excitation to a higher energy excited state. This perturbation of thermal population will be “repaired” and the principle of minimum energy will drive the system back to ground state thermal equilibrium. Depending on the type of excitation and hence its energy, this, by the way, constitutes the fundamental concept of spectroscopy and the energy difference between the ground and excited states dictates the type of excitation, i.e. the frequency of the used electromagnetic radiation, and the speed of recovery, i.e. the time-resolution of the measurement. Using this “detour” via an electronically excited state and taking advantage of reversible photochemistry involving photoswitchable molecules, it is possible to attain populations of two interconvertible states that simply cannot be achieved by thermal means! This “flying” over the mountain, i.e. barrier, rather than “climbing” it is the fundamental difference between thermodynamic and photodynamic equilibria and in our opinion a universal principle to generate a system with a population of states that is different from the one at global thermodynamic minimum, i.e. after complete thermal equilibration (Fig. 1).
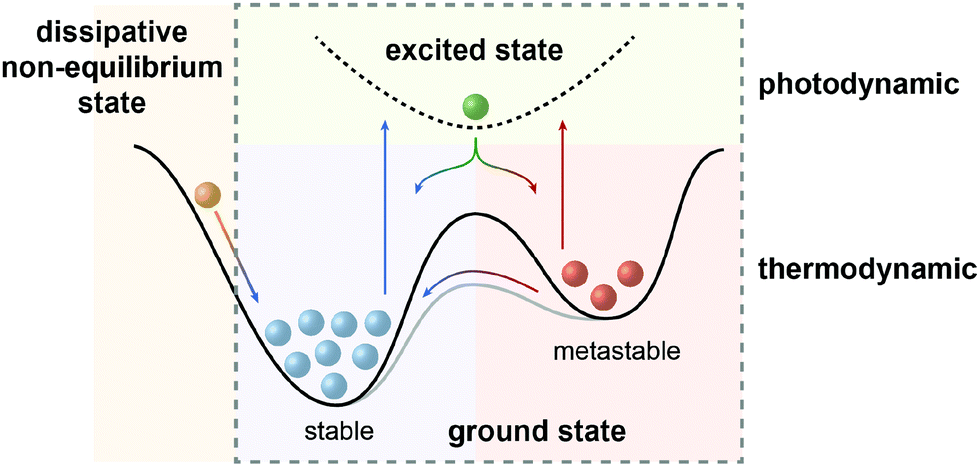 | ||
| Fig. 1 Scope of this review: systems away from thermodynamic equilibrium can be realized either by non-equilibrium thermodynamics involving dissipative states (left) or by equilibrium thermodynamics involving metastable states, which depending on the barrier can either be kinetically trapped or dissipative, i.e. thermally revert to the global minimum (right). The latter case (dashed box) defines photodynamic equilibria, which enable the efficient non-Boltzmann population of the metastable state and are fundamentally different from thermodynamic equilibria (see Fig. 2), as emphasized in this review. | ||
In this review, we outline how photoswitchable molecules can be used to push systems away from their initial/global thermodynamic equilibrium. First, we detail the fundamentals of photodynamic equilibria and introduce photoswitchable molecules. Subsequently, we highlight prominent examples that exploit this principle to drive various molecular processes ranging from chemical reactions over motion and transport to dynamic self-assembly. In all of these cases, we provide an energetic analysis of the system to illustrate the necessity of the photodynamic equilibrium. For this purpose, we consider qualitative potential energy surfaces a very instructive way to describe the energetics (relative stabilities of minima and height of interconnecting barriers defining thermodynamics and kinetics) of the systems. However, we want to emphasize that they should be used with great care and ideally only in the case of simple molecular systems in a constant chemical environment, where enthalpic contributions, which are well represented by the potential energy, dominate and entropy changes can be neglected. Following the discussion of the chosen examples, we finally give our personal outlook on some selected future challenges and prospects.
Thermodynamic vs. photodynamic equilibria
It is rather instructive to illustrate the main difference between thermal and photochemical equilibria by comparing them using potential energy diagrams (Fig. 2). In the classical case of a thermodynamic equilibrium (Fig. 2a), the thermodynamically most stable state, i.e. the global minimum (denoted throughout this review by I), is separated from the metastable state, i.e. the local minimum (II), by a barrier. The energy difference between the two states is given by the Gibbs free energy ΔG⊖, which dictates the equilibrium constant KΔeq, i.e. the population of both states, at a given temperature. The balanced forward and backward reaction rates are the basis of the principle of microscopic reversibility, which has far-reaching consequences, in particular in closed systems where it is manifested in the principle of detailed balance, if only thermal steps are considered.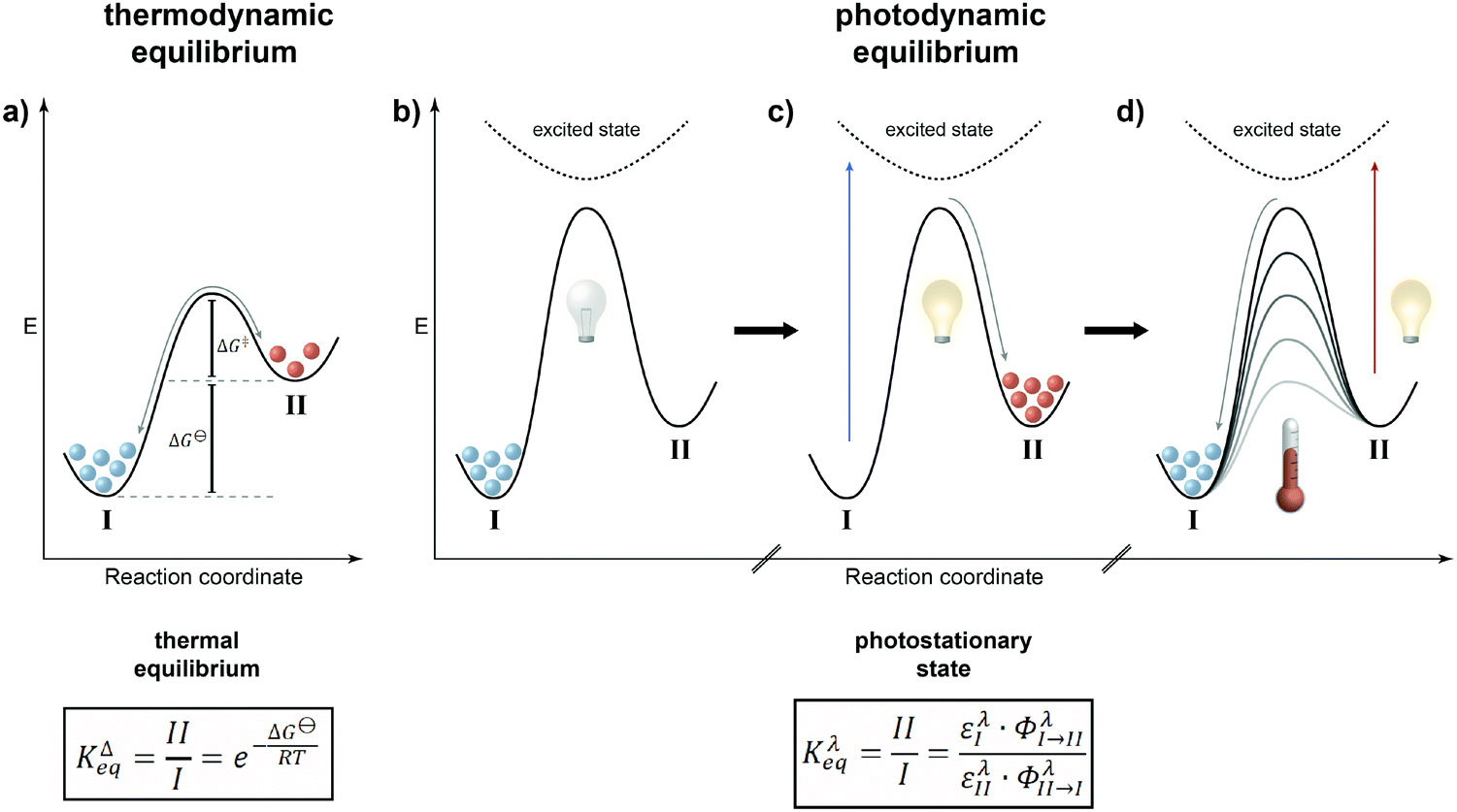 | ||
| Fig. 2 Comparing thermodynamic (a) and photodynamic (b–d) equilibria. (a) Energy levels I and II are populated according to a Boltzmann distribution and given by the equilibrium constant KΔeq, which depends on the levels' energy difference ΔG⊖ and the temperature T. Forward and backward reactions take place continuously with a constant ratio of their rate constants that equals KΔeq and they proceed via the same transition state on the same potential energy surface. The principle of microscopic reversibility applies. (b) Without illumination, only I (ground state) is occupied, as the thermal energy barrier ΔG‡ is too high to be overcome at a given temperature. (c) Light with proper energy (blue arrow) pumps molecules from I in an excited state, from which they relax to II. (d) The back reaction from II to I can either occur photochemically (P-type photochromism), by irradiating the molecules with light of lower energy (red arrow, positive photochromism), or thermally (T-type photochromism), assuming that the barrier ΔG‡ (gray) is low enough to be overcome at a given temperature. In the absence of a thermal back reaction, the equilibrium composition Kλeq is dictated by the ratio of the photochemical forward and backward reactions as given by the molar extinction coefficients ε and reaction quantum yields Φ at the utilized wavelength λ. The equilibrium involves two different pathways and (partially) occurs on the potential energy surface(s) of the excited state(s), thus the principle of microscopic reversibility does not apply. | ||
In the case of a photodynamic equilibrium (Fig. 2b–d), which characterizes the working principle of a photoswitch,3 a high barrier prevents thermal equilibration between both isomers of the photoswitch (at least at reasonable temperatures). However, upon photoexcitation, passing along a higher potential energy surface, and subsequent relaxation, the metastable state (II) can efficiently be populated (Fig. 2c). Return to the initial, thermodynamically more stable state (I) can be achieved upon another photoexcitation and/or thermally, depending on the barrier for the downhill reaction (Fig. 2d). The latter defines whether the photoswitch is of either the P-type, giving rise to a thermally bistable system, or of the T-type, leading to a thermally labile system with a non-negligible thermal half-life. Obviously, this definition implies that there is a continuum spanning from strictly bistable P-type to rapidly thermally reverting T-type photoswitches, which clearly depends on the operating temperature and time duration, i.e. the “(im)patience” of the experimentalist. Importantly, a T-type photoswitch is therefore the prototype of a light-driven “out-of-equilibrium” system that is constantly pumped with photons to populate the metastable state, i.e. the thermally labile isomer, which dissipates its energy by thermal relaxation. Most importantly, the rate of the involved photochemical reaction is not dependent on the intermediate barrier but rather on the likelihood of excitation and the efficiency of the photochemical reaction at the chosen wavelength. In most cases, selective excitation of just one isomer, i.e. I or II, is not entirely possible and hence also in the photochemical case, there are continuous forward and backward reactions occurring even though the ratio of their rate constants, expressed by their extinction coefficients ε multiplied with their reaction quantum yields Φ, remains constant. The composition of the resulting photostationary state (PSS), i.e. the ratio of both isomers of the photoswitchable molecule at a given excitation wavelength λ as characterized by another equilibrium constant Kλeq, therefore does not imply the absence of conversion, i.e. it is not static, but rather dynamic and hence throughout this review we refer to it as photodynamic equilibrium. Note that in T-type photochromic systems, temperature can strongly affect the depopulation of the metastable state, i.e. a thermally labile isomer, depending on the barrier ΔG‡ and associated thermal half-life t1/2.
After highlighting the main difference between thermodynamic and photodynamic equilibria, we want to briefly introduce the main classes of photoswitchable molecules since they are obviously the key component to pump systems away from thermal equilibrium. Molecular photochromism3 is a phenomenon that was first described by Fritzsche in 18674 and is characterized by a light-induced reversible change of the color of a compound. Over the past century, several classes of photochromic molecules have been developed, most importantly the azobenzene,5 spiropyran,6 and diarylethene7 families (Fig. 3).
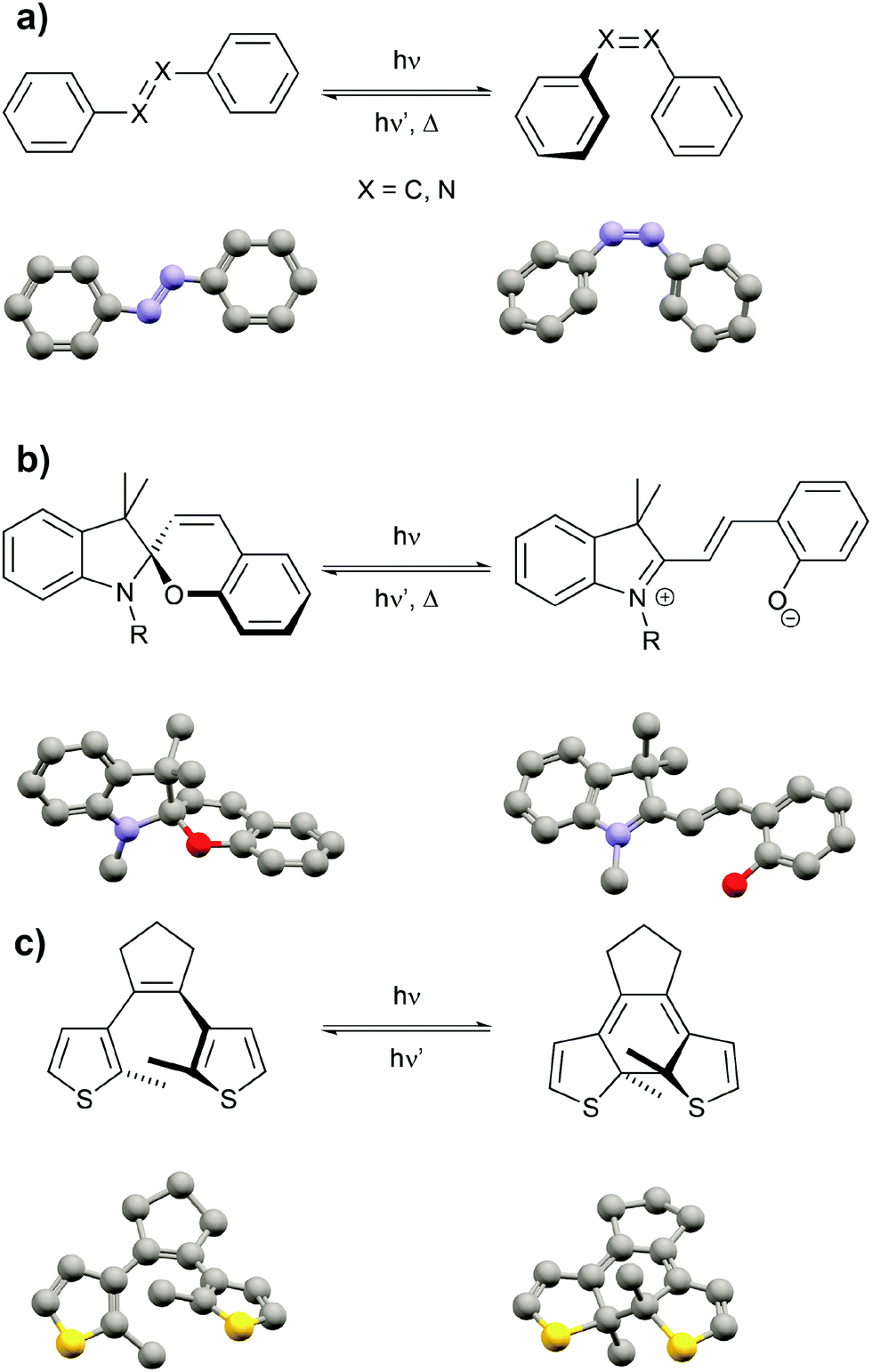 | ||
| Fig. 3 Most prominent families of molecular photoswitches in chronological order of their discovery. (a) Stilbenes and azobenzenes undergoing E/Z photoisomerization associated with large geometry changes, (b) spiropyrans/merocyanines undergoing dynamic C–O bond cleavage/making coupled to E/Z photoisomerization associated with large changes in polarity and basicity, (c) diarylethenes undergoing photoinduced 6π-electrocyclization associated with large electronic changes. | ||
Each of these families has unique features related to their performance, which is dependent on both the intrinsic photoswitching ability and the extent of light-induced property changes, which extend beyond mere changes in color and hence we refer to these molecules as photoswitchable rather than photochromic. The intrinsic photoswitching ability is dictated by not only the operating conditions, in particular the desired switching wavelength(s) and light intensity,8 but also the temperature and surrounding environment, i.e. interactions with solvents or surfaces or neighboring molecules in the bulk. On the one hand, an ideal P-type photoswitch should allow for selective excitation of each isomer at two different switching wavelengths, triggering photochemical forward and backward isomerization reactions with high quantum yields. On the other hand, an ideal T-type photoswitch should allow for selective excitation of the thermodynamically more stable isomer to trigger an efficient photoisomerization process, while reverse isomerization should occur spontaneously with an adjustable rate, i.e. with control over the thermal half-life of the metastable isomer. In addition to switching efficiently, ideally even exploiting catalytic amplification,8,9 the switch should operate reliably over many cycles and hence side reactions giving rise to fatigue have to be minimized.8,10 But even an optimized photoswitch is only useful if switching indeed leads to a large modulation of a desired property and hence a functional system. If large structural changes, for example in the context of photopharmacology11 or photoactuation,12 are desired, azobenzenes, stilbenes or related molecules13 undergoing E/Z photoisomerization and having adjustable thermal barriers are the ideal candidates. If however large electronic changes, for example in optoelectronics,14 are needed, large gap cross-conjugated diarylethenes undergoing 6π-electrocyclization to yield low gap conjugated closed isomers are perfectly suited, in particular since their switching induces very little geometrical changes and therefore also occurs efficiently in the (crystalline) solid. Last but not least, spiropyrans have proven superior as dipole15 and acidity16 switches due to the zwitterionic and basic character of their corresponding merocyanine form. Throughout this review we mostly cover double bond photoisomerization of stilbenes and azobenzenes as a means to drive significant geometry changes as well as photochemical 6π-electrocyclization to induce electronic changes associated with the shifting of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds (tautomerization) and electron density (resonance effects). Although there are many more important features of this fascinating class of molecules, we refer the reader to the full body of literature3,5–7,17 while emphasizing that every switch should be optimized for its intended use.
C double bonds (tautomerization) and electron density (resonance effects). Although there are many more important features of this fascinating class of molecules, we refer the reader to the full body of literature3,5–7,17 while emphasizing that every switch should be optimized for its intended use.
In the following sections we illustrate how photoswitches can be used to drive chemical systems by light. While we primarily focus on an energetic description, we also want to emphasize that photoswitches in addition provide a convenient tool to remotely control a system with high spatial and temporal precision.18
Manipulating chemical equilibria with light
In order to influence reversible thermal reactions with light, the reactivity of one of the involved components should be modulated upon exposure to photons (Fig. 4). Reactivity modulation is reflected in a change of the activation barrier and it can be accomplished either internally by rendering the substrate and the product (an intermediate or a mediator) photoswitchable or externally by employing a photoswitchable catalyst.18,19 While the first approach requires a stoichiometric amount of photons, the second in principle allows for the use of a substoichiometric amount of photons and hence catalytic amplification. In the following we focus entirely on photon-stoichiometric reactivity modulation since we are interested in an energetic analysis of the systems and in particular in the use of light to drive otherwise unfavorable chemical equilibria.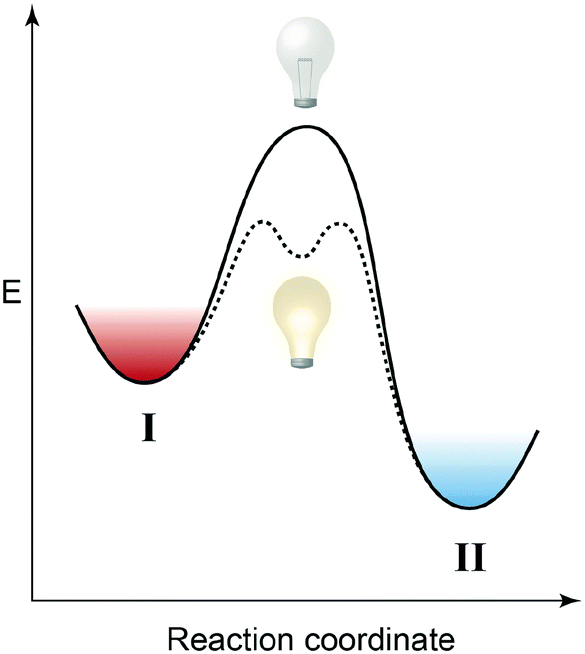 | ||
| Fig. 4 Light-induced reactivity modulation. Photoswitching converts a chemical species into a (more) reactive one, thus lowering the activation barrier ΔG‡ of a thermodynamically favored chemical reaction, i.e. ΔG⊖ < 0. Photoinduced activation/acceleration can involve either molecules, which are consumed or produced during the reaction, or a catalyst and hence occur in a stoichiometric or a substoichiometric (catalytic) fashion. Shown here is a simplified general version of a potential energy diagram, in which the thermodynamic and photodynamic equilibria are not explicitly separated and hence no rigorous energetic analysis is feasible. | ||
In all the examples to be discussed throughout this section, thermodynamic, and more precisely, dynamic covalent equilibria will be coupled to photodynamic equilibria. Depending on your perspective and their designated use the thus created molecular systems display either chemically gated photochromism, i.e. only one of the two thermally interconvertible species is a photoswitch, or light-gated reactivity, i.e. only one of the two photoswitch isomers is thermally reactive. The first is attractive for the generation of multiple switching states, for example in molecular logics,20 while the second allows implementation of a light-triggered remote control in thermal chemistry, for example in (self)healing21 or release22 as illustrated primarily in this section.
In the simplest case, the photoswitching event can enhance or reduce the reactivity in a dynamic covalent reaction. Such a system is illustrated by our recent work on the photomodulation of aldehyde reactivity in imine formation/exchange (Fig. 5).21b,23 Upon light-induced ring-closure both aldehyde termini of a dithienylethene24 become electronically coupled and synergistically enhance each other's electrophilic reactivity. The associated rate increase is translated in an approximately ten times faster formation of bisimine in the closed isomer as compared to the open isomer. This change in kinetics leads to an accelerated crosslinking of amino-functionalized polysiloxanes in UV-irradiated areas and thus allows modulating the self-healing of silicon-rubbers by light.21b Importantly, the photoisomerization only changes the height of the thermal barrier, i.e. ΔG‡, and hence accelerates or decelerates the reaction but does not alter its overall thermodynamics, i.e. ΔG⊖.
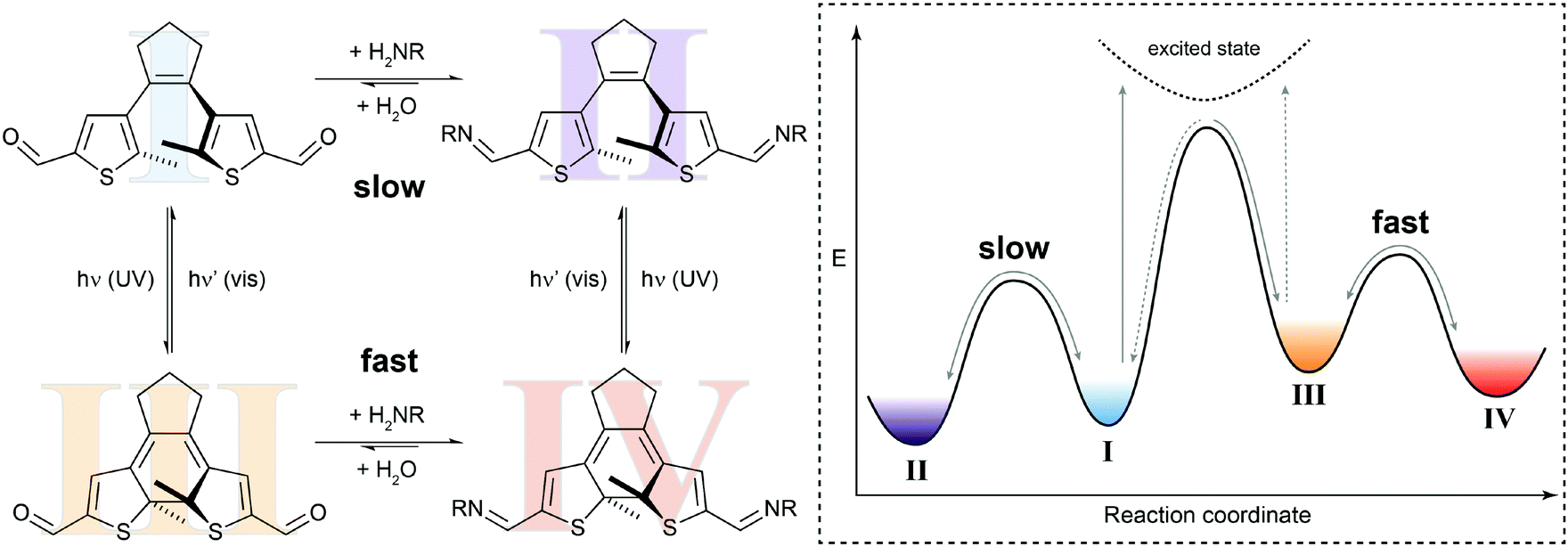 | ||
| Fig. 5 Switching reactivity with light. Diarylethene dialdehyde I reacts slowly with amines to form the respective bisimine II.21b UV-induced ring-closure generates dialdehyde III, in which the reactivity of both carbonyl functionalities is synergistically increased. The thus decreased activation barrier ΔG‡ leads to acceleration of the condensation reaction to yield bisimine IV. The photoisomerization of closed bisimine IV to open bisimine II is not shown in the potential energy diagram. | ||
In a seminal paper, Branda and coworkers combined a thermal Diels–Alder reaction and a photochemical ring-opening/-closure reaction in one molecular system (Fig. 6).22a The initial reaction of an electron-deficient dienophile such as dicyanofumarate with the electron-rich bisthienylfulvene generates the cyclohexene Diels–Alder adduct, which can undergo photoinduced 6π-electrocyclization to the closed isomer. The specific double bond position in the latter prevents the thermal retro Diels–Alder reaction and only after activation with visible light, inducing ring-opening, can the dicyanofumarate be released. Note that the forward cycloaddition process resembles a case of photogated reactivity.
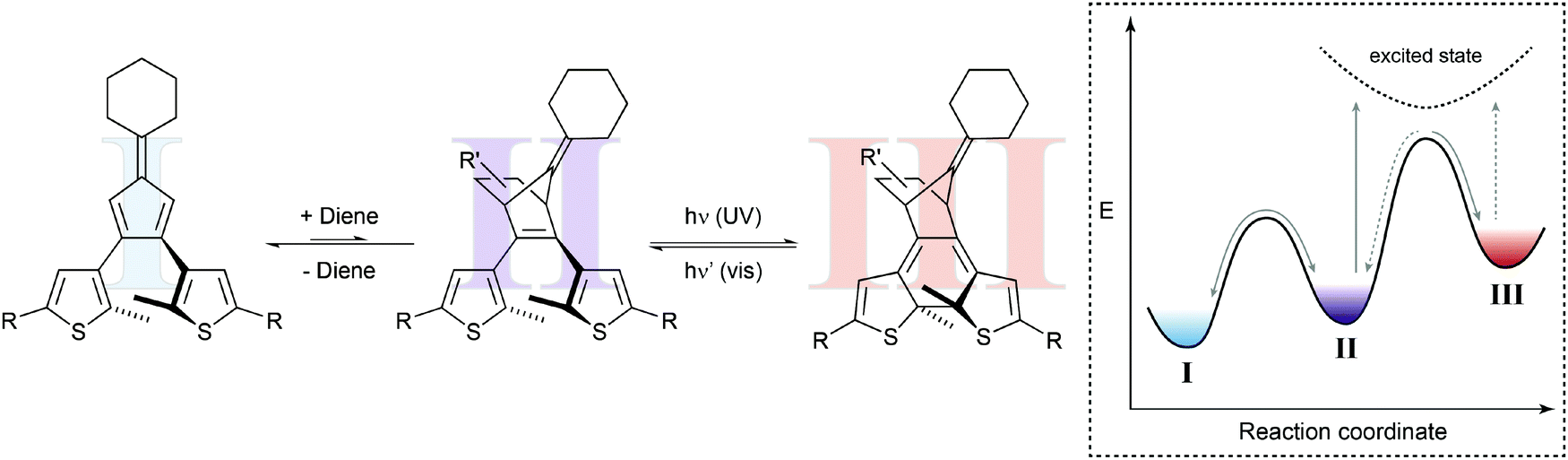 | ||
| Fig. 6 Shifting a chemical equilibrium to one side.22a Dienophile I and a diene are in an endergonic equilibrium with ring-open photoswitch II. Applying UV light leads to electrocyclic ring-closure, forming diarylethene III by simultaneously removing photoswitch II from the thermal up-hill equilibrium with I. According to Le Chatelier's principle, this form of gated photochromism could in principle be used to shift the unfavored Diels–Alder reaction by photochemically trapping cycloadduct II. | ||
Presumably due to the authors’ main interest in light-controlled release, they focused on the latter process while possibly ignoring another important aspect. Their system should in principle allow shifting the thermal Diels–Alder equilibrium by trapping the Diels–Alder adduct in the subsequent photoisomerization. This light-induced removal of the cycloadduct should be compensated for following the equilibrium law, discovered independently by Le Chatelier and Braun and often referred to as Le Chatelier's principle, and thereby shift the Diels–Alder equilibrium to the product side (considering the Diels–Alder adduct of both isomers of the photoswitch). As a result, the composition of the thermal Diels–Alder equilibrium could in principle be changed by the action of light.
In the above example, the thermal Diels–Alder equilibrium could (in principle) be affected by removing or supplying the Diels–Alder product via photochemical ring-closure/opening. However, to truly manipulate the composition of the Diels–Alder equilibrium it would be desirable to furthermore use light to remove or supply one of the substrates, i.e. the dienophile or the diene. Hence, the dynamic covalent Diels–Alder reaction would be controlled by photochemical pre- and post-equilibria. Such a system has been designed by Göstl et al., who replaced a thienyl by a furyl residue in the framework of a diarylethene (Fig. 7).22b Exploiting the different absorption spectra of the two closed Diels–Alder inactive “locked” adducts, the authors could use light of different wavelengths to push the Diels–Alder equilibrium selectively either to the product side or the side of the starting materials, thereby facilitating covalent bond making and bond breaking, respectively. Using this bidirectional control over the thermal Diels–Alder equilibrium, our group could subsequently use this system to photocontrol covalent bond making and breaking, as exemplified by crosslinking and hence healing of dynamic covalent polymer networks21c as well as the release of maleimides in aqueous solution.22c This work furthermore shows that it should indeed be possible to exploit photoswitches in combination with Le Chatelier's principle, to shift thermodynamic equilibria in an otherwise unfavorable direction.
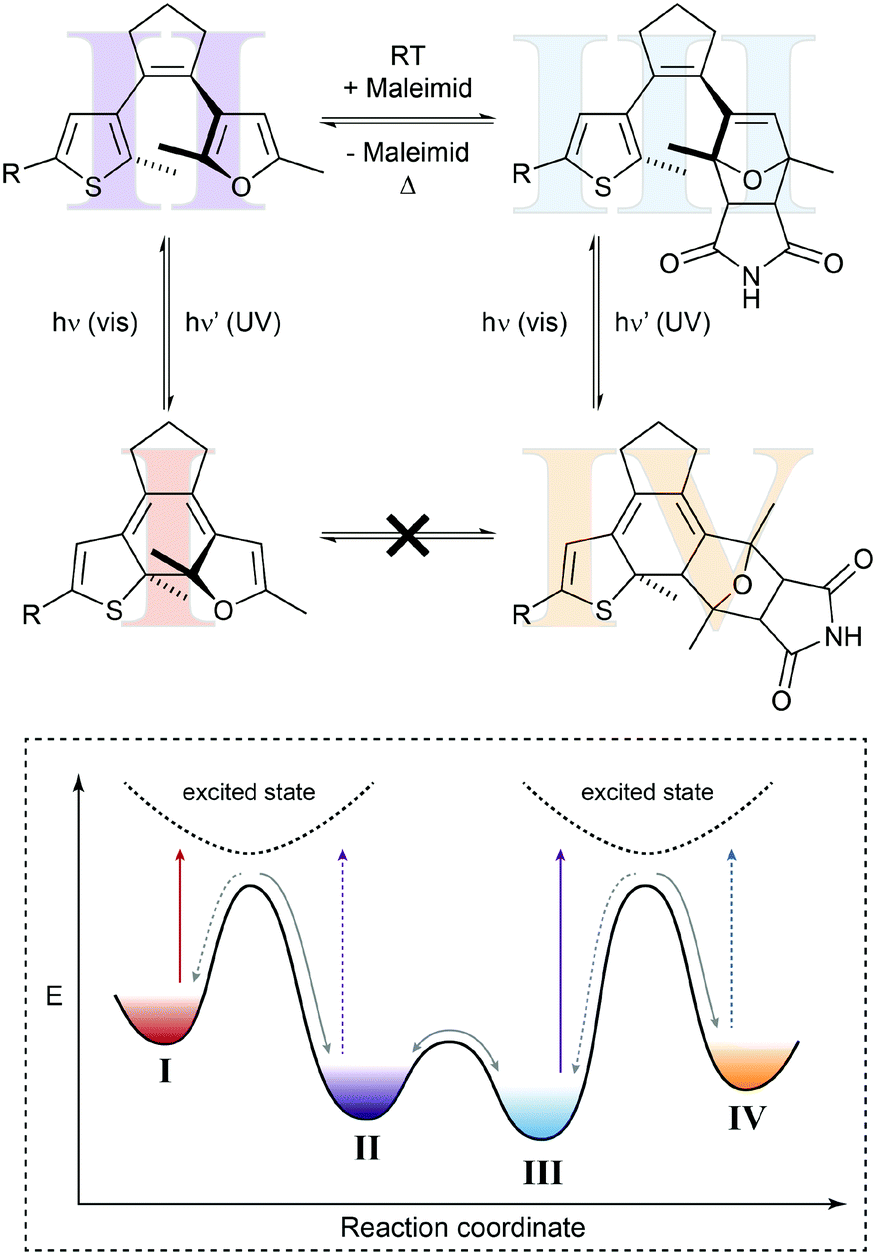 | ||
| Fig. 7 Shifting a chemical equilibrium to both sides.22b Diene II can react with maleimide as the dienophile and is in a dynamic covalent (thermal) equilibrium with Diels–Alder adduct III. Both diarylethene-type photoswitches can undergo electrocyclic ring-closure with UV-light (purple arrows) to form locked furan I or locked Diels–Alder adduct IV, which cannot be converted into each other directly. Due to its more extended and polarized π-system, tetraene I absorbs visible light of lower energy (red arrow), whereas triene IV can be excited with light of higher energy (blue arrow). Taking advantage of Le Chatelier's principle, this allows for directional shifting of the thermal equilibrium by ring-closing II and III with UV-light, while at the same time selectively opening either I or IV with visible light. Cycloaddition is favored by excitation of I, whereas cycloreversion is preferred by excitation of IV. | ||
It is important to note that in the above examples a thermally equilibrated two-state system is coupled to either one or two thermally irreversible photoreactions. The latter affect the absolute (not relative) composition of the initial thermodynamic equilibrium – but not its equilibrium constant. In our analysis we consider both isomers of the photoswitch as one molecular entity, which in its locked form due to the presence of the thermal barrier is able to store the chemical information, i.e. dynamic covalent bond, generated in the thermal equilibrium. This important aspect has been impressively demonstrated in the inspiring work by Leigh and coworkers on their information ratchet (Fig. 8).25 In their cleverly designed rotaxane, the authors introduced a photoswitchable α-methyl stilbene gate between the two different binding sites of the axle. In the E-isomer, the macrocycle can translocate between the two stations, i.e. a thermal equilibrium is established. Photoisomerization, occurring predominantly via an added benzil triplet sensitizer, generates a large population of the thermally stable Z-isomer, which effectively traps the macrocycle on either of the two stations, thereby installing a barrier similarly to Maxwell's Demon. The beauty of the design arises from the fact that the efficiency of the reverse process, i.e. the removal of the gate, depends on the location of the macrocycle and hence the chemical information stored in the two different metastable states. This is due to the efficiency of the Z → E photoisomerization, which is governed by the distance of the Z-configured gate to the macrocycle with an integrated benzophenone-like sensitizer. As a result, the metastable state, in which the macrocycle is stationed more closely to the closed gate, is preferably destroyed, and therefore the macrocycles accumulate on the more distant station. Importantly, the directional translocation of the macrocycles along the axle is not based on changes in thermodynamics (of binding) but is purely kinetic in origin. This distinguishes Leigh's information ratchet from alternative energy ratchets.
From shifting equilibria to continuous work powered by light
The above systems (Fig. 7 and 8) are composed of four states, which define a linear string of coupled thermodynamic and photodynamic equilibria. Such systems will only allow switching between the two ends of the string, i.e. states I and IV, by passing intermediate states II and III. However, in order for a system to perform continuous work,26 the system should be able to return to the initial state via a different path to escape the principle of microscopic reversibility and establish unidirectionality – in other words, it has to recycle and hence a transition from IV back to I is required. In order to re-establish the initial state (or an equivalent), another thermal reaction has to be utilized as nicely illustrated by the light-driven molecular walker of Leigh and coworkers (Fig. 9).27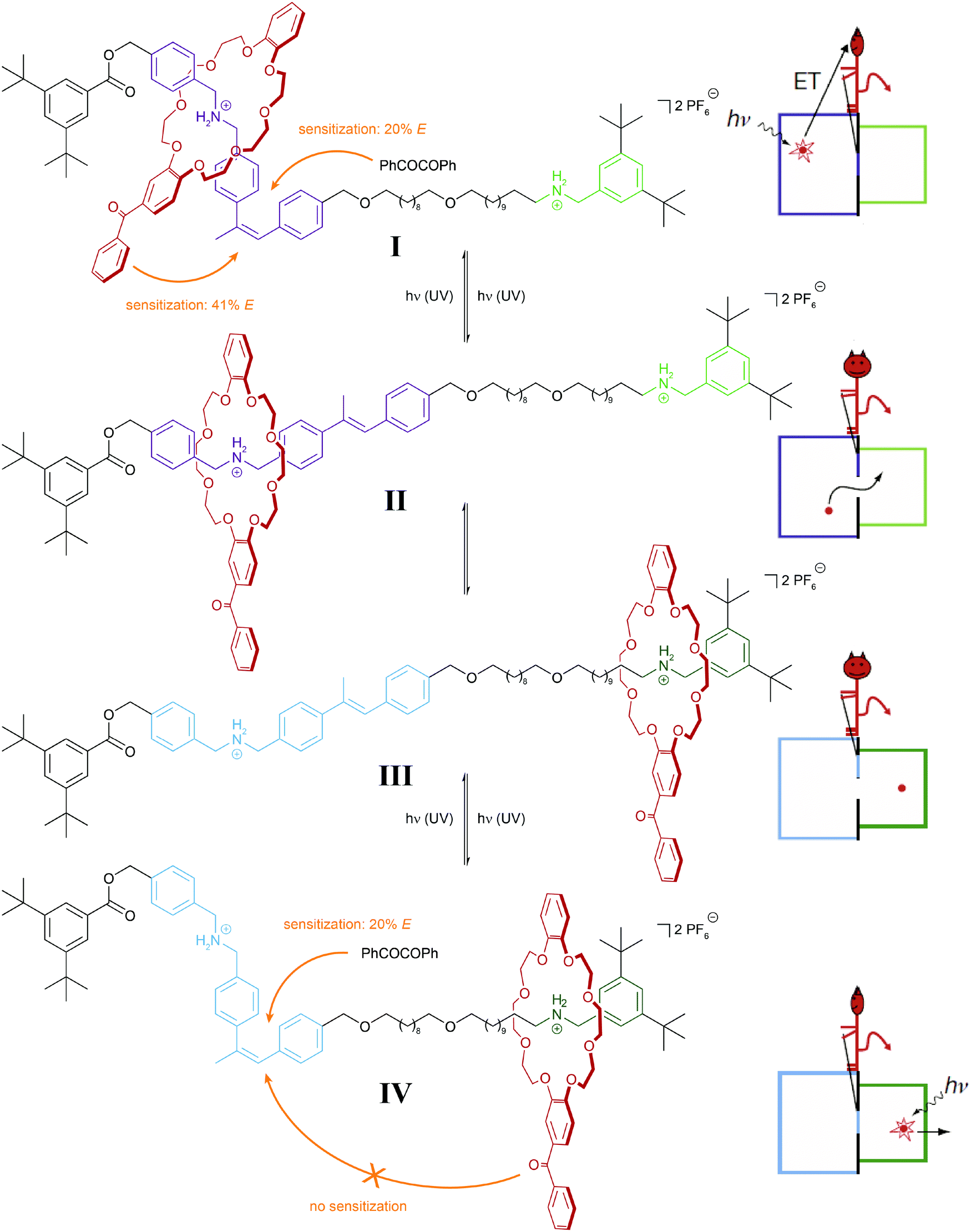 | ||
| Fig. 8 Light-driven molecular information ratchet.25 Photoisomerization of an internal α-methyl stilbene gate traps a crown ether macrocycle on either one or the other ammonium station (states I and IV). Due to the closer proximity of the macrocycle, which carries a photosensitizing benzophenone moiety, to the closed Z-configured gate, state I is more efficiently destroyed and therefore state IV predominates, leading to a net accumulation of macrocycles at the more distant station. In part reproduced from ref. 25 with permission from Nature Publishing Group, copyright 2007. | ||
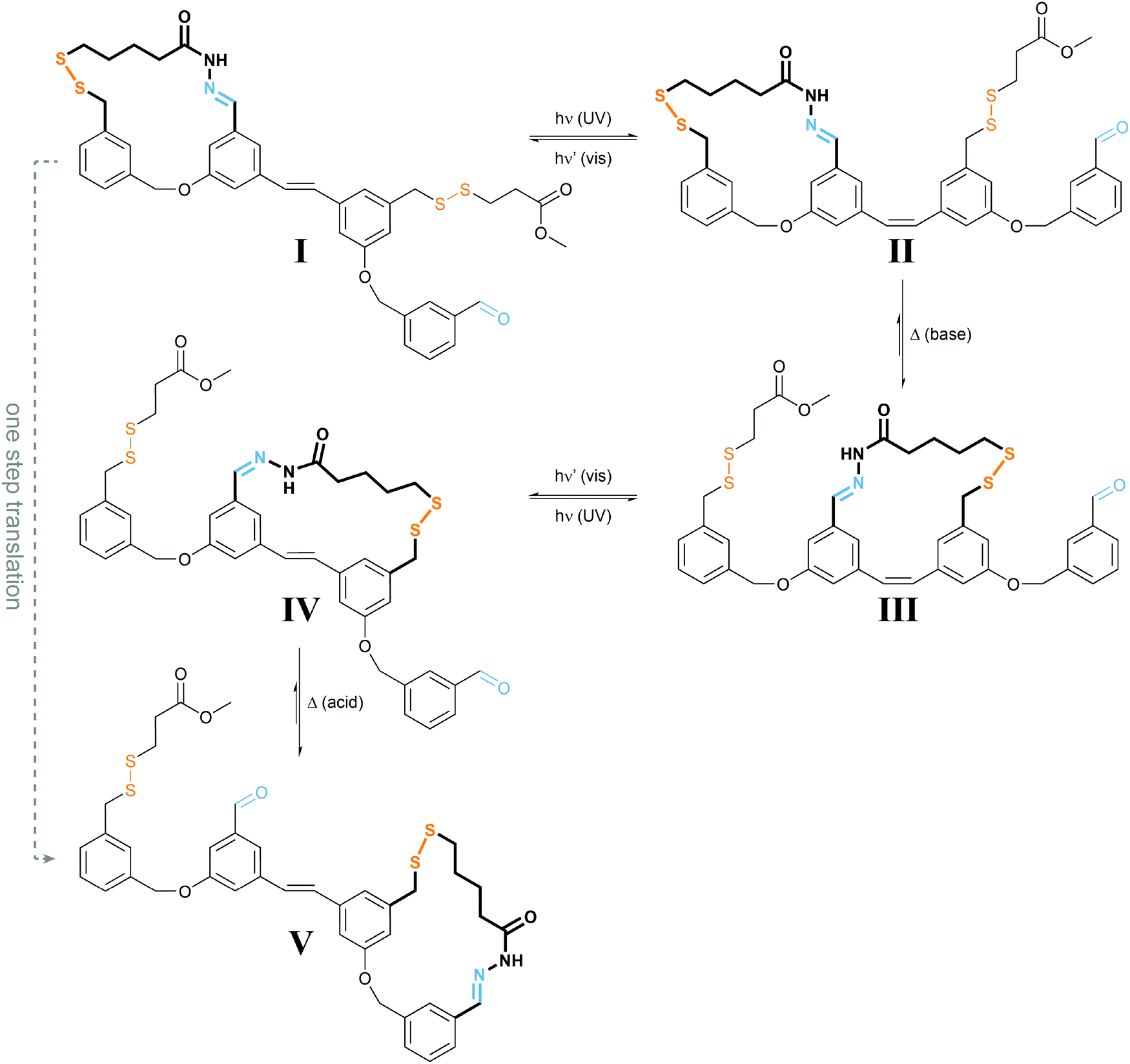 | ||
| Fig. 9 Light powers a molecular walker.27 UV-light induced E → Z photoisomerization of I yields less stable II, which under basic conditions required for disulfide exchange rearranges to the more stable III. Subsequent irradiation with visible light drives E → Z photoisomerization to form strained IV, which under acidic conditions used for acylhydrazone exchange converts into the more stable V, representing the same configuration as I but after net displacement. Overall, the molecular walker, composed of two orthogonal dynamic covalent reactive functionalities, i.e. thiol and hydrazide, has moved over a molecular track composed of a pair of complementary thiol and aldehyde functionalities, which can be addressed separately (acid vs. base), thereby controlling the “stickiness” of the walker’s feet. The energy required for directional movement is supplied by photoisomerization of the molecular stilbene track. | ||
The authors used orthogonal dynamic covalent interactions between the molecular walker and the track, i.e. base-labile disulfide and acid-labile acylhydrazone linkages, to control the site of detachment and of remaining attachment. The light-induced photoisomerization of the stilbene-derived molecular track is used to populate metastable intermediate states II and IV, which convert to the thermodynamically more stable states III and V upon selective disulfide or acylhydrazone exchange, respectively. The overall process enables the successive transfer of chemical information stored in the binding location of the walker and therefore allows for a net directional movement powered by light.
The walker illustrates the above mentioned key design principle, although not in a truly cyclic four state system, in which thermal and photochemical steps are alternating, i.e. pairs of thermodynamic equilibria are coupled via photodynamic ones and the latter enable the overall system to escape the principle of detailed balance. Astumian in his seminal article on the use of microscopic reversibility realized the exception (and hence importance) of photochemistry to escape net-zero thermodynamic cycling, when stating “Unlike photochemical processes, … are all equilibrium processes.”28 In fact, this cyclic design concept has been exploited extensively in the field of molecular machines, which very recently has been recognized with the Nobel Prize in Chemistry.29 In a gedanken experiment one could achieve such a cyclic system by tying the ends of Leigh's track into a loop, on which his walker would continuously move in one direction, given a constant supply of energy (light) and alternating chemical conditions (acid/base catalysis). Two systems that nicely resemble such a hypothetical cyclic walker, at least in a topological way, are two earlier catenane-derived motors by the Leigh group that are driven by light-induced E/Z photoisomerization of fumaramide and maleamide moieties implemented in the track, allowing for control over binding affinities and thus achieving unidirectional movement based on an energy ratchet mechanism.30 To illustrate the power of coupling thermodynamic and photodynamic equilibria in a cyclic and autonomous fashion, we briefly illustrate the working principle of the famous light-driven unidirectional motor created by the Feringa group (Fig. 10),31 resembling a “nano-windmill” fueled by photons (and not wind).
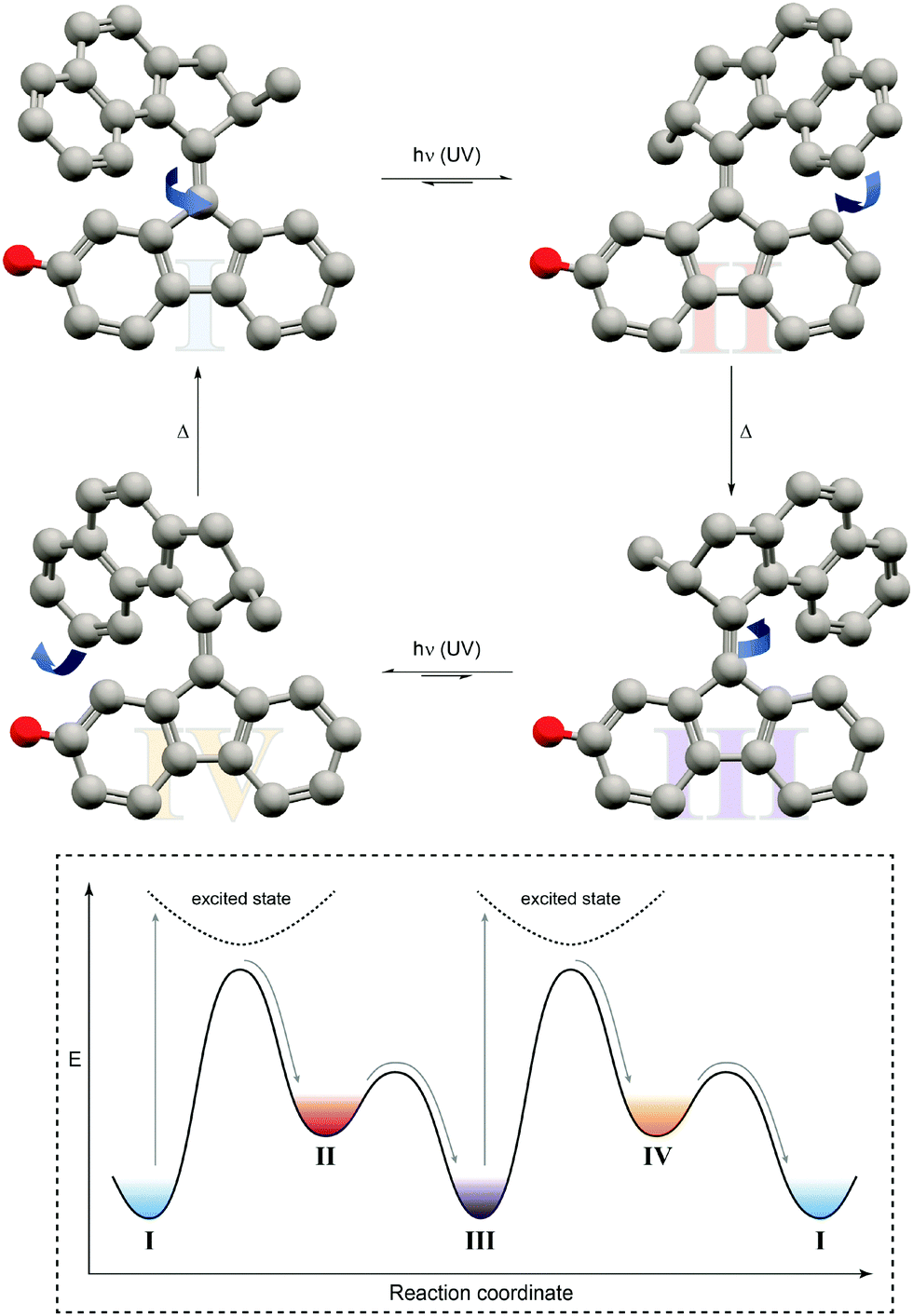 | ||
| Fig. 10 Light-driven unidirectional rotation of a molecular motor.31 Light-induced Z → E photoisomerization of P-Z-I leads to metastable M-E-II, which undergoes a thermal helix inversion to form P-E-III. Subsequent E → Z photoisomerization generates metastable M-Z-IV, which undergoes a second thermal helix inversion back to P-Z-I to close a full rotation cycle. The red dummy atom is used to distinguish between the enantiomers. | ||
In this case, an overcrowded chiral alkene, which adopts two different helicities, i.e. M and P, in both of its geometrical isomers, i.e. E and Z, giving rise to overall four different states has been exploited. Similarly to nature's use of chirality to encode directionality, for example during the processing of DNA, Feringa used it to energetically distinguish between the diastereomeric helicities and hence bias a particular rotation direction. Photoisomerization pumps the system from the more stable state of one configurational isomer into the metastable state of the other, which subsequently relaxes by helix inversion. Subsequent photoinduced back isomerization once again populates the metastable state, which relaxes to the starting point and closes one full turn of the motor rotation. The unidirectional rotation of such a molecular motor has successfully been used to propel a nano-car on a surface (although mediated by excitation through the tip of a scanning tunneling microscope32) or to move microscopic objects in a liquid-crystal by light.33
On the nanoscale, for example on cellular microtubules, directional molecular motion of ATP-fueled motor proteins enables the controlled transport of various chemical entities. While Leigh's walker27 in principle would allow for such molecular transport, the necessary repetitive irradiation-base–irradiation-acid cycles as well as the need for more elaborate track designs would render the approach rather inefficient and complicated. A much simpler example to drive the directional and autonomous transport of a chemical entity, in this case a ring over a linear axle, by light has been devised by Credi and coworkers (Fig. 11).34 In this work, directionality originates from the non-symmetrical axle and the clever design feature that one of the stoppers is photoswitchable to modulate the barrier for dethreading. By a collection of elegant experiments the authors show unidirectional motion of the ring over the axle and hence pumping by applying continuous irradiation.
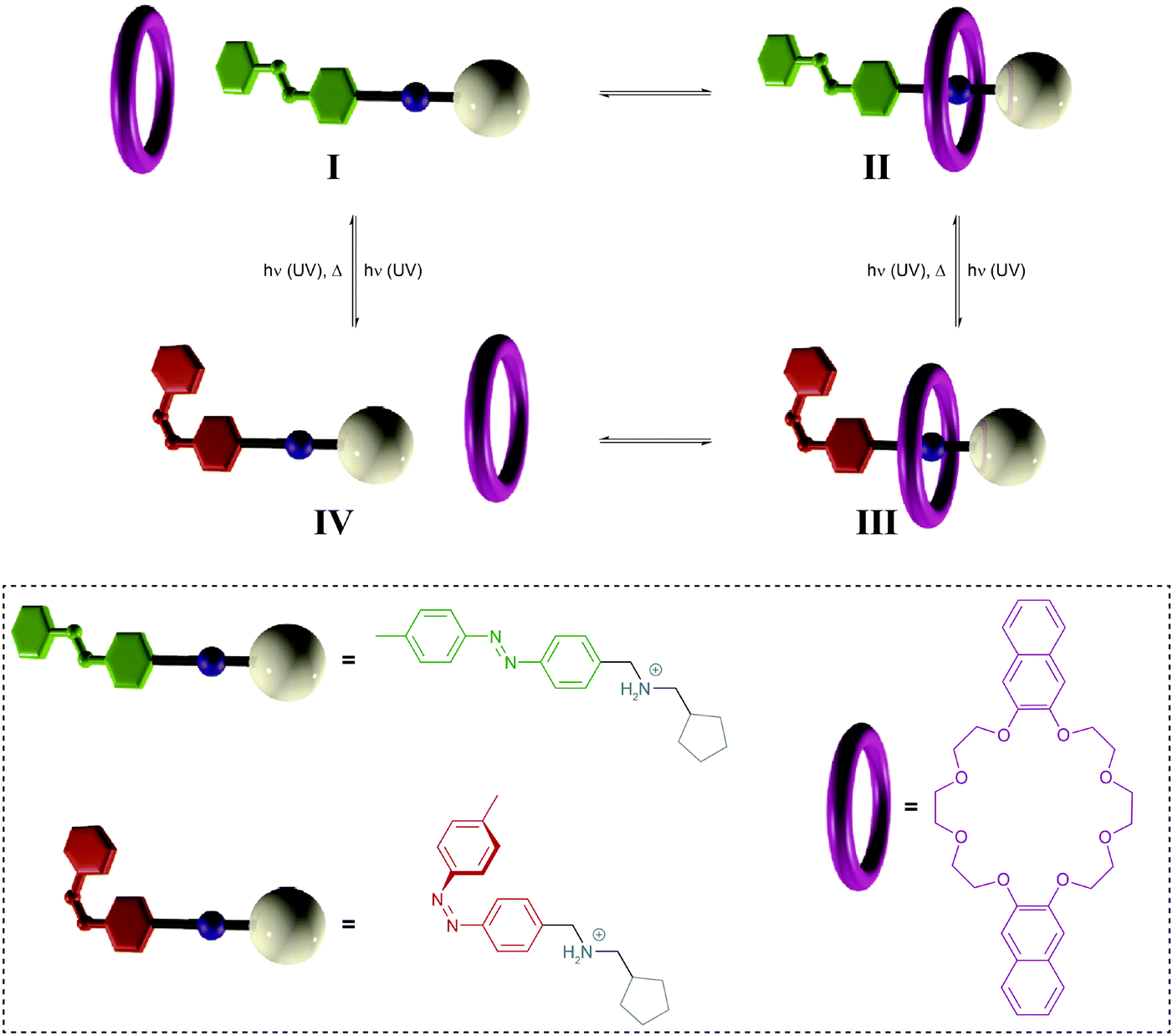 | ||
| Fig. 11 Light-driven pump.34a A crown ether macrocycle threads onto an ammonium axle via the less hindered E-azobenzene terminus (I → II). Subsequent E → Z photoisomerization to generate III blocks the initial entry and destabilizes binding to force dethreading via the cyclopentane stopper (III → IV), thereby leading to a directional movement of the macrocycle over the axle (I → IV). Photochemical Z → E back isomerization re-establishes the initial axle I, ready to undergo another pumping cycle. Adapted from ref. 34a with permission from Nature Publishing Group, copyright 2015. | ||
Chemical transport constitutes an essential part of chemical work in living systems since reactants and products have to actively be pumped in and out of the cell, often against an existing gradient. Typically, natural pumps are being driven by chemical fuels, such as ATP, but light-driven rhodopsin pumps for active proton and ion transport in bacteria are also known.35 Such systems, capable of active, light-driven chemical transport, were first established in an early and seminal work by Shinkai and coworkers (Fig. 12).36
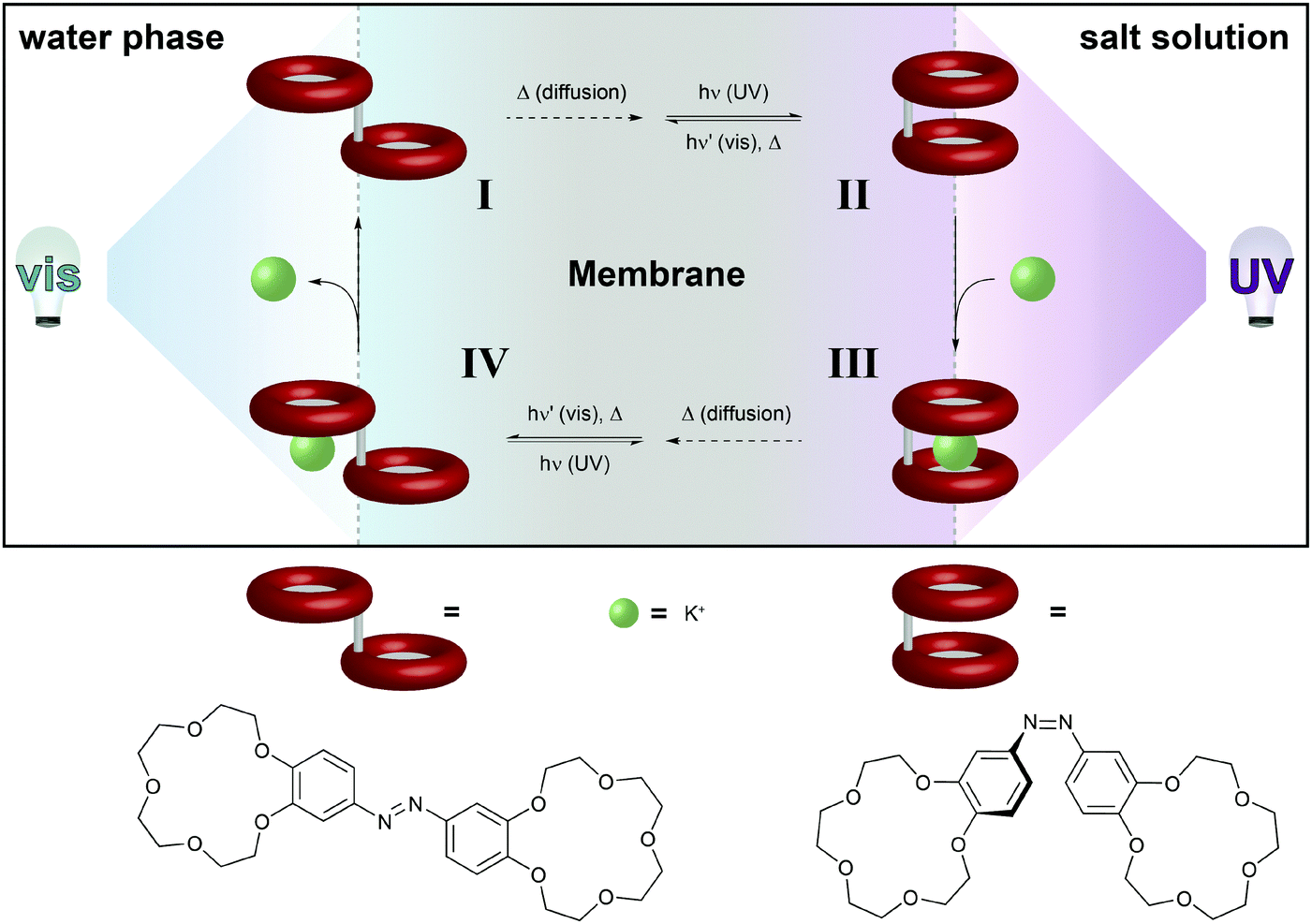 | ||
| Fig. 12 Light-driven chemical transport.36b An intermediate membrane, which contains an azobenzene-bridged crown ether dimer, separates a compartment with distilled water from a compartment with an aqueous potassium salt solution. The interfaces with the water and salt solutions are constantly irradiated with visible and UV-light, respectively. This confines the areas for predominant Z → E and E → Z photoisomerization, respectively, and hence establishes a gradient of weakly K+-binding E- and strongly K+-binding Z-azobenzene across the membrane. Thus, E-I primarily photoisomerizes near the salt solution to generate Z-II, which binds a potassium ion. The formed metal complex Z-III eventually diffuses to the opposite side of the membrane, where photoisomerization converts it back to E-IV, which releases the potassium ion and regenerates E-I. | ||
The basis of this work is an azobenzene-bridged crown ether dimer36a that serves as a photoswitchable supramolecular host. In the Z-isomer, the two crown ether moieties can bind potassium ions in a sandwich fashion, leading to a much higher binding constant as compared to the E-isomer. Pumping however means control over the binding and release and hence the direction of the transport of a chemical species, in this case potassium. Clearly, the symmetry of the system has to be broken and the authors achieve this by using two different irradiation wavelengths at the two opposing interfaces. Therefore, the authors make clever use of the spatial control over photochemistry by confining the two opposing photoisomerization reactions at the opposite interfaces and thereby establishing an E/Z-azobenzene gradient. Exploiting this feature, the authors were able to transport ions following an outer salt concentration gradient yet not against it. On the basis of this concept yet three decades later, Bakker and coworkers used the light-induced basicity changes of a spiropyran/merocyanine photoswitch to pump protons against a gradient, thereby building up a chemical potential and generating a current (Fig. 13).37 Also in other areas, confining the region of forward and backward photoisomerization reactions has proven useful, for example in photoactuation,12 reflected in the bending38 and even the rotation39 of polymer films.
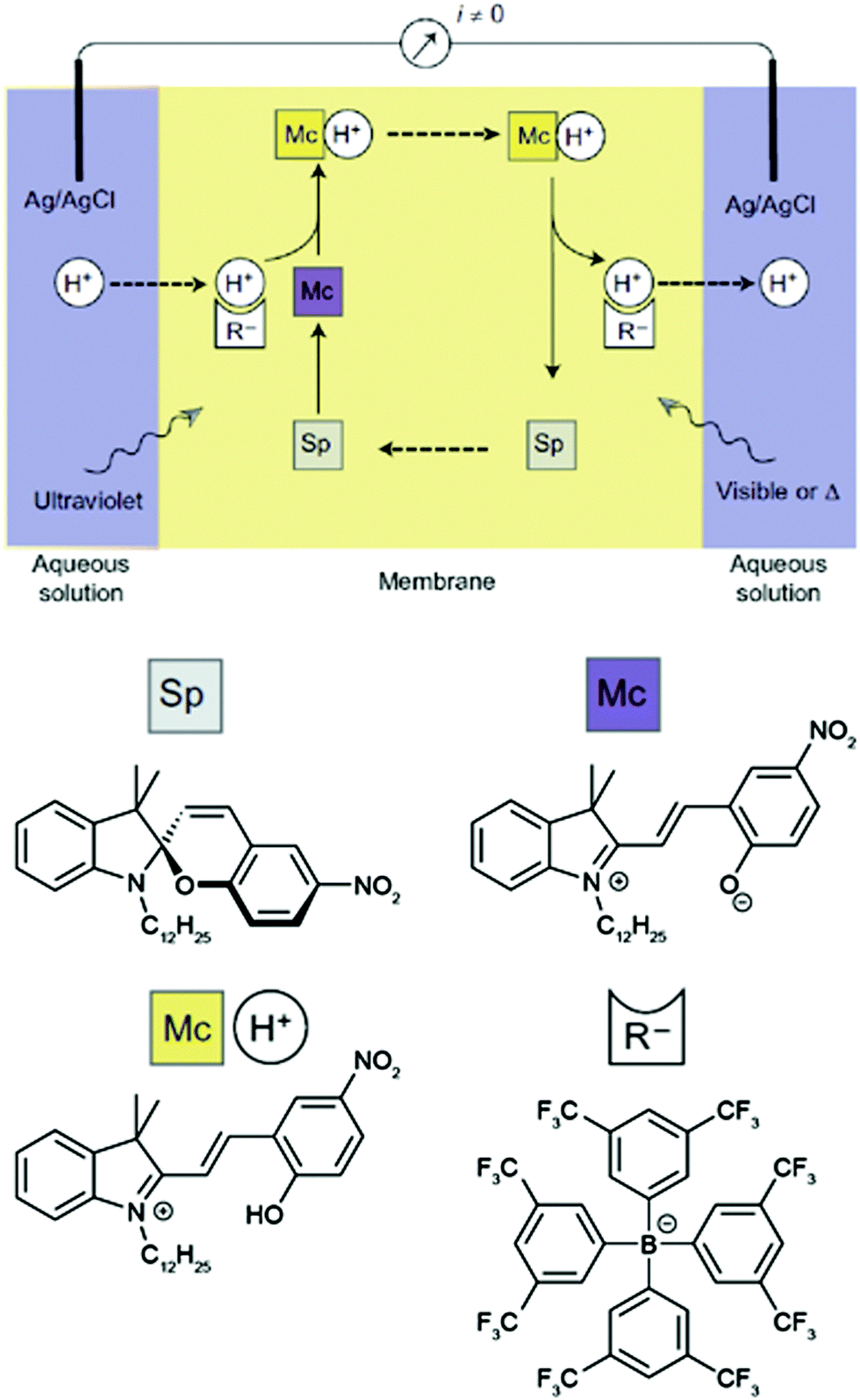 | ||
| Fig. 13 Light-driven proton pump.37 Irradiating an intermediate membrane, which contains a spiropyran/merocyanine photoswitch and separates two aqueous solutions of the same composition, with visible and UV-light, respectively, leads to the formation of a proton gradient that gives rise to a photogenerated current. Adapted from ref. 37 with permission from Nature Publishing Group, copyright 2014. | ||
Light to control assembly and create order
The last examples illustrate clever ways to harness and ideally amplify the molecular photoisomerization events to perform chemical and even mechanical work. Self-assembly constitutes a very powerful mechanism to obtain elaborate functional structures and amplify the properties of the individual components. In order to control and even drive a self-assembly process by light, photoswitchable components have to be integrated into the system. Ideally, the photoswitch mediates the assembly (or disassembly) of a large non-photoswitchable entity. This is conveniently achieved by decorating inorganic nanoparticles with photoswitchable capping ligands, such as azobenzenes, as pioneered by the groups of Grzybowski and Klajn.40 Furthermore, Klajn and coworkers could demonstrate photocontrol over the self-assembly of acid-coated nanoparticles by using an external spiropyran/merocyanine photoswitch to modulate the pH and hence aggregation (Fig. 14).41 In the context of “out-of-equilibrium” systems, the light-induced assembly and disassembly of azobenzene-decorated nanoparticles once again nicely illustrates the coupling of thermodynamic and photodynamic equilibria in a cyclic four state scheme.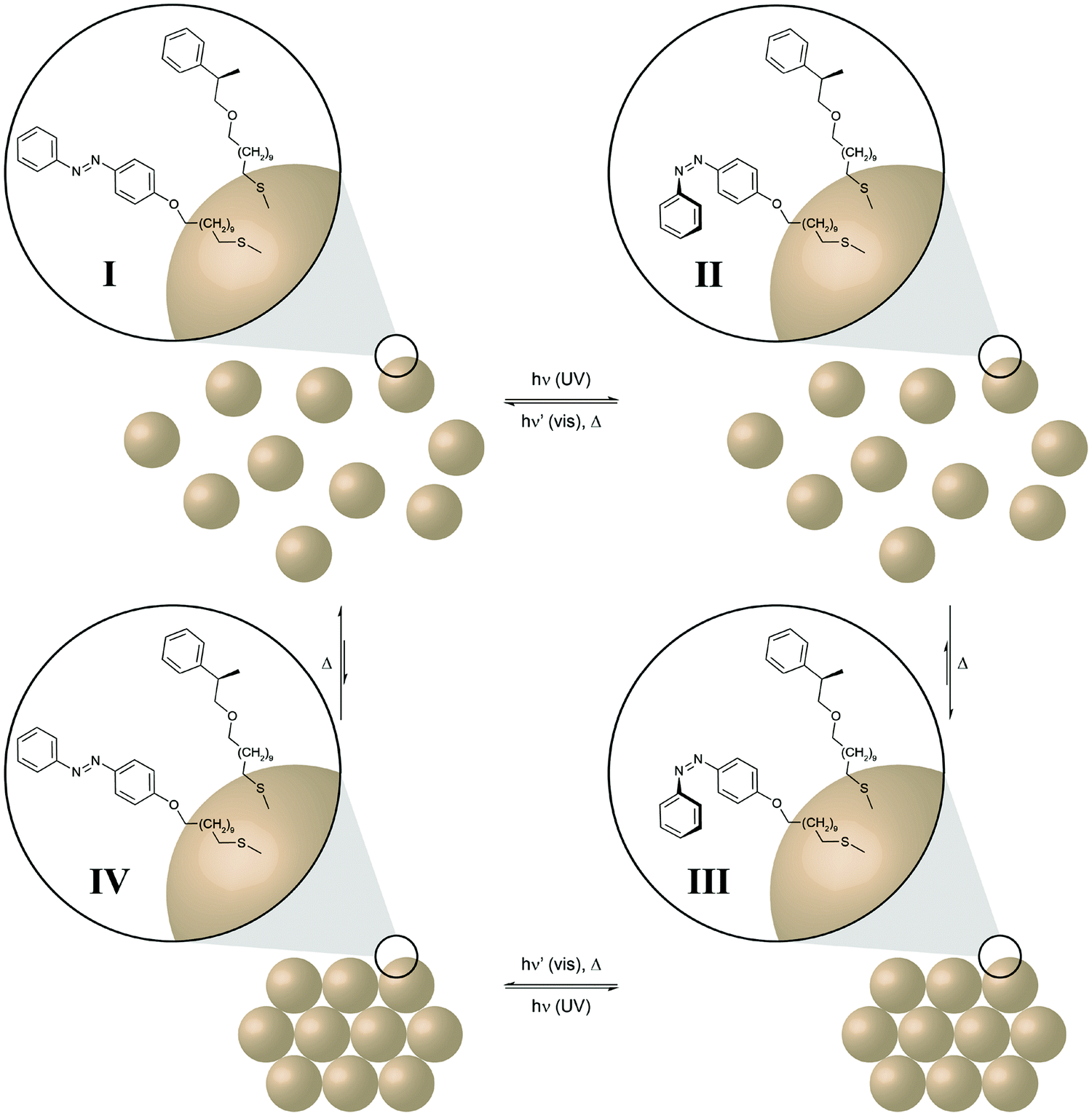 | ||
| Fig. 14 Light drives the self-assembly of nanoparticles.41 Gold nanoparticles are (partially) decorated with photoswitchable azobenzenes and dissolved in a non-polar solvent (toluene). UV-light induced E → Z isomerization of the less polar nanoparticles E-I generates the more polar, metastable nanoparticles Z-II, which subsequently aggregate due to the entropically favored release of the non-polar solvent (toluene) to form the ordered nanoparticle aggregate Z-III. Subsequent Z → E isomerization, triggered either by visible light (P-type, solid barrier) or thermally (T-type, dashed barrier), leads to the metastable nanoparticle aggregate E-IV, which undergoes spontaneous disassembly in the non-polar medium to form the initial individual nanoparticles E-I. | ||
In this case, the initial UV-induced E → Z photoisomerization of the azobenzene in the organic shell induces a dipole change from the less polar E-isomer to the more polar Z-isomer. While the former leads to good solvation by the surrounding non-polar toluene, the latter induces desolvation and the nanoparticles stabilize by the formation of aggregates.42 Hence, E → Z photoisomerization initially pumps the system, i.e. populates the metastable state Z-II, and thus drives formation of the nanoparticle aggregate. Disassembly of the aggregate occurs upon Z → E back isomerization, which can either be driven by visible light or occur thermally depending on the height of the barrier, i.e. the thermal half-life of the Z-isomer. While the former P-type photochromism leads to a kinetically trapped state,43 the latter T-type photochromism depending on the height of the barrier allows for controlled dissipation of the input energy and hence higher order structure formation requires a constant supply of light energy.40b,41,44
Beyond controlling and driving the self-assembly of nanoparticles with light, the Klajn group has successfully been exploiting the confinement within these dynamic aggregates, so called self-assembling “nanoflasks” for uptake and catalysis.43 By decorating the azobenzene-functionalized nanoparticles with chiral molecules, the authors could show chiral discrimination in guest binding. More importantly, they could accelerate chemical reactions within the confined cavities of their nanoparticle aggregates. Because their nanoflasks are dynamic, product inhibition, representing the inherent drawback of most other nanoporous systems, can successfully be overcome through light-driven disassembly and re-assembly cycles.
Outlook: from remote-controlling to driving processes
From the above covered work, we hope that the reader shares our excitement about the opportunities arising from the deliberate incorporation of photoswitchable molecules into functional systems and materials. While we and others have been emphasizing the unique element of spatio-temporal control previously in the context of catalysis,18,19 materials and devices,12,14,17 herein we focus on the use of photoswitches to power molecular processes and analyze them primarily from an energetic perspective. Thereby, we consider photoswitches not only as light-controlled “gates” but as photon-fueled “drivers” of chemical processes!Ciamician's vision of utilizing sunlight to drive the synthesis of fuels and other chemicals45 serves as an inspiration for researchers dedicated to solar-to-chemical energy conversion and storage.46 Chemistry will play a key role in this endeavor and various concepts to harvest the energy of sunlight in chemical compounds should be explored. Nature mostly relies on photosynthesis, involving irreversible photoinduced electron transfer steps from excited states followed by rapid charge separation and subsequent oxidation and reduction events, to drive the uphill processes of water oxidation (directly) and photophosphorylation (indirectly via the generated proton gradient). However, also (thermally) reversible light-induced reactions, most notably the Z → E photoisomerization of the retinal chromophore, can be used to drive the transport of protons and ions to power bacteria or to transduce photon capture into the process of vision, depending on the appropriate protein environment.
The problem in directly harvesting the energy of an excited state relates to its rather short lifetime and the competition with undesired deactivation processes that narrows the mechanistically accessible options to few, very fast chemical processes, in particular (photoinduced) proton and electron transfer. Over the past decade, the latter has successfully put to use in organic synthesis by the advent of photoredox catalysis.47 Using relatively long-lived triplet excited states, their excitation energy can efficiently be transferred to other molecules, most notably to generate singlet oxygen,48 which can undergo unique cycloaddition reactions to give some high-energy products, such as 1,2-dioxetanes that even exhibit chemiluminescence.49
While an alternative use of a metastable ground-state species is certainly much less energy efficient, as most of the light's excitation energy is lost upon return to the ground state, it offers the great advantage that the entire arsenal of conventional, thermal chemical reactions can be utilized.50 Following this approach, the principle challenge boils down to the question on how to utilize the energy stored in the metastable isomer.51 In the simplest scenario, this could involve the release as thermal energy at will as pursued in recent efforts dedicated to molecular solar thermal energy storage.52 However, it would be much more versatile to store this energy in the chemical bonds of other (photochemically inactive) chemical compounds to continuously recycle the photoswitch. To accomplish this, one would need to shift equilibria to the less favorable side, i.e. drive an uphill process. In principle, this should be feasible as shown by some of the examples presented in this review, yet several challenges have to be met before photoswitches will advance to universal solar-to-chemical energy converters.
One of the main challenges lies in the extent of the uphill shifting, similarly to pumps that operate against a chemical potential, i.e. the efficiency of energy conversion. Since shifting an equilibrium uphill will require a large number of photons and stoichiometric amounts of photoswitches, they have to undergo many switching cycles and hence resistance to photo- and thermo-chemically induced degradation is critical. The ideal photoswitch for this task is therefore (i) highly fatigue-resistant, (ii) absorbing in the visible region to utilize sunlight, (iii) negative photochromic, i.e. converting into a less intensely colored metastable isomer, to maximize light-uptake by the system, and (iv) T-type with a tunable thermal half-life. Beyond, it would be extremely attractive to drive chemical reactions uphill by light using photoswitches in a catalytic fashion, i.e. not catalytic in photons – this would violate thermodynamics since the required reaction free energy has to come from somewhere – but catalytic in photoswitch. In principle, this could be achieved by regeneration of the photoswitch using another thermal reaction, thereby amplifying its action and significantly broadening the scope of the conceivable chemical transformations.
In summary, we have highlighted functional molecular systems, in which the incorporation of photoswitchable units enables light to be used as their external power supply. The associated photodynamic equilibria are inherently different from thermodynamic ones as they do not obey microscopic reversibility and hence enable pumping the system away from thermodynamic equilibrium. Broadly speaking, photochemistry allows proper molecules to pass reversibly over thermally insurmountable barriers and hence switch by light. It is our conviction that this key ability to introduce barriers in the right location, control their height, and to jump over them at the right time will guide the future design of systems that operate away from global thermal equilibrium. Hence, photoswitches will (continue to) take center stage in the quest for molecular machinery with ever increasing complexity and more sophisticated function. Let there be light!
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to our dedicated coworkers and collaborators, who have contributed to our research endeavor over the past years. Generous support by the European Research Council (via ERC-2012-STG_308117 “Light4Function”) is gratefully acknowledged. M. K. is indebted to the Studienstiftung des deutschen Volkes for providing a doctoral fellowship.References
- A unique example is described in: (a) J. V. I. Timonen, M. Latikka, L. Leibler, R. H. A. Ras and O. Ikkala, Science, 2013, 341, 253–257 CrossRef CAS PubMed , including the perspective by: ; (b) T. M. Hermans, H. Frauenrath and F. Stellacci, Science, 2013, 341, 243–244 CrossRef CAS PubMed.
- An instructive review is given in: E. Mattia and S. Otto, Nat. Nanotechnol., 2015, 10, 111–119 CrossRef CAS PubMed.
- (a) B. L. Feringa and W. R. Browne, Molecular Switches, John Wiley & Sons, Weinheim, 2011 Search PubMed; (b) H. Dürr and H. Bouas-Laurent, Photochromism: Molecules and Systems, Elsevier, Amsterdam, 2003 Search PubMed; (c) J. C. Crano and R. J. Guglielmetti, Organic Photochromic and Thermochromic Compounds, Kluwer Academic/Plenum Publishers, New York, 1999 Search PubMed; (d) A. V. Eltsov, Organic Photochromes, Consultants Bureau, New York, 1990 Search PubMed.
- J. Fritzsche, Competes Rendus de l'Académie des Sciences, 1867, 69, 1035–1037 Search PubMed.
- First description of E–Z photoisomerization of azobenzene: (a) G. S. Hartley, Nature, 1937, 140, 281 CrossRef CAS . Comprehensive review: ; (b) H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- Pioneering work: (a) E. Fischer and Y. Hirshberg, J. Chem. Soc., 1952, 4522–4524 CAS ; Comprehensive review: ; (b) R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- Pioneering work: (a) M. Irie and M. Mohri, J. Org. Chem., 1988, 53, 803–808 CrossRef CAS . Comprehensive reviews: ; (b) M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed; (c) M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- D. Bléger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef PubMed.
- A. Goulet-Hanssens, M. Utecht, D. Mutruc, E. Titov, J. Schwarz, L. Grubert, D. Bléger, P. Saalfrank and S. Hecht, J. Am. Chem. Soc., 2017, 139, 335–341 CrossRef CAS PubMed.
- M. Herder, B. M. Schmidt, L. Grubert, M. Pätzel, J. Schwarz and S. Hecht, J. Am. Chem. Soc., 2015, 137, 2738–2747 CrossRef CAS PubMed.
- (a) A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC; (b) T. Fehrentz, M. Schönberger and D. Trauner, Angew. Chem., Int. Ed., 2011, 50, 12156–12182 CrossRef CAS PubMed; (c) J. M. Beierle, H. A. V Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114–6178 CrossRef PubMed; (d) W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed; (e) J. Broichhagen, J. A. Frank and D. Trauner, Acc. Chem. Res., 2015, 48, 1947–1960 CrossRef CAS PubMed; (f) M. M. Lerch, M. J. Hansen, G. M. van Dam, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2016, 55, 10978–10999 CrossRef CAS PubMed.
- Y. Zhao and T. Ikeda, Smart Light-Responsive Materials: Azobenzene-Containing Polymers and Liquid-Crystals, John Wiley & Sons, Hoboken, 2009 Search PubMed.
- A review including photoisomerization of non-symmetrical X = Y compounds is given in: (a) D. Fanghänel, G. Timpe and V. Orthman, in Organic Photochromes, ed. A. V. Eltsov, Consultants Bureau, New York, 1990, ch. 2, pp. 105–175 Search PubMed. A recent example is given in: ; (b) D. J. van Dijken, P. Kovaříček, S. P. Ihrig and S. Hecht, J. Am. Chem. Soc., 2015, 137, 14982–14991 CrossRef CAS PubMed.
- X. Zhang, L. Hou and P. Samorì, Nat. Commun., 2016, 7, 11118 CrossRef CAS PubMed.
- (a) Q. Shen, Y. Cao, S. Liu, M. L. Steigerwald and X. Guo, J. Phys. Chem. C, 2009, 113, 10807–10812 CrossRef CAS; (b) M. Levitus, G. Glasser, D. Neher and P. F. Aramendia, Chem. Phys. Lett., 1997, 277, 118–124 CrossRef CAS; (c) M. Bletz, U. Pfeifer-Fukumura, U. Kolb and W. Baumann, J. Phys. Chem. A, 2002, 106, 2232–2236 CrossRef CAS.
- (a) F. M. Raymo and S. Giordani, J. Am. Chem. Soc., 2001, 123, 4651–4652 CrossRef CAS PubMed; (b) Z. Shi, P. Peng, D. Strohecker and Y. Liao, J. Am. Chem. Soc., 2011, 133, 14699–14703 CrossRef CAS PubMed.
- M. M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS PubMed.
- R. Göstl, A. Senf and S. Hecht, Chem. Soc. Rev., 2014, 43, 1982–1996 RSC.
- (a) R. S. Stoll and S. Hecht, Angew. Chem., Int. Ed., 2010, 49, 5054–5075 CrossRef CAS PubMed; (b) B. M. Neilson and C. W. Bielawski, ACS Catal., 2013, 3, 1874–1885 CrossRef CAS; (c) V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341–5370 RSC.
- (a) G. J. Brown, A. P. de Silva and S. Pagliari, Chem. Commun., 2002, 2461–2464 RSC; (b) A. P. De Silva and N. D. McClenaghan, Chem. – Eur. J., 2004, 10, 574–586 CrossRef PubMed.
- (a) A. M. Asadirad, S. Boutault, Z. Erno and N. R. Branda, J. Am. Chem. Soc., 2014, 136, 3024–3027 CrossRef CAS PubMed; (b) M. Kathan, P. Kovaricek, C. Jurissek, A. Senf, A. Dallmann, A. F. Thünemann and S. Hecht, Angew. Chem., Int. Ed., 2016, 55, 13882–13886 CrossRef CAS PubMed; (c) A. Fuhrmann, R. Göstl, R. Wendt, J. Kötteritzsch, M. D. Hager, U. S. Schubert, K. Brademann-Jock, A. F. Thünemann, U. Nöchel, M. Behl and S. Hecht, Nat. Commun., 2016, 7, 13623 CrossRef CAS PubMed.
- (a) V. Lemieux, S. Gauthier and N. R. Branda, Angew. Chem., Int. Ed., 2006, 45, 6820–6824 CrossRef CAS PubMed; (b) R. Göstl and S. Hecht, Angew. Chem., Int. Ed., 2014, 53, 8784–8787 CrossRef PubMed; (c) R. Göstl and S. Hecht, Chem. – Eur. J., 2015, 21, 4422–4427 CrossRef PubMed.
- For a related example, see: D. Wilson and N. R. Branda, Angew. Chem., Int. Ed., 2012, 51, 5431–5434 CrossRef CAS PubMed.
- This dithienylethene has first been described in: L. N. Lucas, J. van Esch, R. M. Kellogg and B. L. Feringa, Chem. Commun., 1998, 2313–2314 RSC.
- V. Serreli, C.-F. Lee, E. R. Kay and D. A. Leigh, Nature, 2007, 445, 523–527 CrossRef CAS PubMed.
- Overviews over molecular machines: (a) V. Balzani, A. Credi and M. Venturini, Molecular Devices and Machines: Concepts and Perspectives for the Nanoworld, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed; (c) A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19–30 RSC; (d) S. Silvi, M. Venturi and A. Credi, Chem. Commun., 2011, 47, 2483–2489 RSC.
- M. J. Barrell, A. G. Campaña, M. von Delius, E. M. Geertsema and D. A. Leigh, Angew. Chem., Int. Ed., 2011, 50, 285–290 CrossRef CAS PubMed.
- R. D. Astumian, Nat. Nanotechnol., 2012, 7, 684–688 CrossRef CAS PubMed.
- For J.-P. Sauvage, F. Stoddart and B. L. Feringa, for the design and synthesis of molecular machines. See: http://www.nobelprize.org/nobel_prizes/chemistry/laureates/2016/.
- (a) D. A. Leigh, J. K. Y. Wong, F. Dehez and F. Zerbetto, Nature, 2003, 424, 174–179 CrossRef CAS PubMed; (b) J. V. Hernández, E. R. Kay and D. A. Leigh, Science, 2004, 306, 1532–1537 CrossRef PubMed.
- (a) N. Koumura, R. W. Zijlstra, R. a van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS PubMed; (b) J. Conyard, A. Cnossen, W. R. Browne, B. L. Feringa and S. R. Meech, J. Am. Chem. Soc., 2014, 136, 9692–9700 CrossRef CAS PubMed.
- T. Kudernac, N. Ruangsupapichat, M. Parschau, B. Maciá, N. Katsonis, S. R. Harutyunyan, K.-H. Ernst and B. L. Feringa, Nature, 2011, 479, 208–211 CrossRef CAS PubMed.
- R. Eelkema, M. M. Pollard, J. Vicario, N. Katsonis, B. S. Ramon, C. W. M. Bastiaansen, D. J. Broer and B. L. Feringa, Nature, 2006, 440, 163 CrossRef CAS PubMed.
- (a) G. Ragazzon, M. Baroncini, S. Silvi, M. Venturi and A. Credi, Nat. Nanotechnol., 2015, 10, 70–75 CrossRef CAS PubMed ; Note that Leigh and coworkers described the first rotaxane that allows to ratchet a particle (macrocycle) energetically uphill and thus drive the system away from equilibrium: ; (b) M. N. Chatterjee, E. R. Kay and D. A. Leigh, J. Am. Chem. Soc., 2006, 128, 4058–4073 CrossRef CAS PubMed.
- H. E. Kato, K. Inoue, R. Abe-Yoshizumi, Y. Kato, H. Ono, M. Konno, S. Hososhima, T. Ishizuka, M. R. Hoque, H. Kunitomo, J. Ito, S. Yoshizawa, K. Yamashita, M. Takemoto, T. Nishizawa, R. Taniguchi, K. Kogure, A. D. Maturana, Y. Iino, H. Yawo, R. Ishitani, H. Kandori and O. Nureki, Nature, 2015, 521, 48–53 CrossRef CAS PubMed . Together with other light-responsive proteins, these rhodopsin pumps and channels constitute the basis of optogenetics, Nature’s Method of the Year 2010: Nat. Methods 2011, 8, 1.
- (a) S. Shinkai, T. Nakaji, T. Ogawa, K. Shigematsu and O. Manabe, J. Am. Chem. Soc., 1981, 103, 111–115 CrossRef CAS; (b) A. Kumano, O. Niwa, T. Kajiyama, M. Takayanagi, K. Kano and S. Shinkai, Chem. Lett., 1983, 1327–1330 CrossRef CAS; (c) S. Shinkai, Pure Appl. Chem., 1987, 59, 425–430 CrossRef CAS.
- X. Xie, G. A. Crespo, G. Mistlberger and E. Bakker, Nat. Chem., 2014, 6, 202–207 CrossRef CAS PubMed.
- (a) Y. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145 CrossRef CAS PubMed; (b) T. J. White, N. V. Tabiryan, S. V. Serak, U. A. Hrozhyk, V. P. Tondiglia, H. Koerner, R. A. Vaia and T. J. Bunning, Soft Matter, 2008, 4, 1796–1798 RSC.
- M. Yamada, M. Kondo, J. Mamiya, Y. Yu, M. Kinoshita, C. J. Barrett and T. Ikeda, Angew. Chem., Int. Ed., 2008, 47, 4986–4988 CrossRef CAS PubMed.
- (a) R. Klajn, K. J. M. Bishop and B. A. Grzybowski, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10305–10309 CrossRef CAS PubMed; (b) R. Klajn, P. J. Wesson, K. J. M. Bishop and B. A. Grzybowski, Angew. Chem., Int. Ed., 2009, 48, 7035–7039 CrossRef CAS PubMed.
- P. K. Kundu, D. Samanta, R. Leizrowice, B. Margulis, H. Zhao, M. Börner, T. Udayabhaskararao, D. Manna and R. Klajn, Nat. Chem., 2015, 7, 646–652 CrossRef CAS PubMed.
- A. Manna, P. L. Chen, H. Akiyama, T. X. Wei, K. Tamada and W. Knoll, Chem. Mater., 2003, 15, 20–28 CrossRef CAS.
- H. Zhao, S. Sen, T. Udayabhaskararao, M. Sawczyk, K. Kučanda, D. Manna, P. K. Kundu, J. Lee, P. Král and R. Klajn, Nat. Nanotechnol., 2016, 11, 82–88 CrossRef CAS PubMed.
- P. K. Kundu, S. Das, J. Ahrens and R. Klajn, Nanoscale, 2016, 19280–19286 RSC.
- G. Ciamician, Science, 1912, 36, 385–394 CAS.
- (a) V. Balzani, A. Credi and M. Venturi, ChemSusChem, 2008, 1, 26–58 CrossRef CAS PubMed; (b) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- Reviews: (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (c) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- S. E. Braslavsky, K. N. Houk and J. W. Verhoeven, Pure Appl. Chem., 1996, 68, 2223–2286 Search PubMed.
- A related argument has been articulated in the context of photoswitchable catalysts: in ref. 19a.
- V. I. Minkin, V. A. Bren and A. E. Lyubarskaya, in Organic Photochromes, ed. A. V. Eltsov, Consultants Bureau, New York, 1990, ch. 5, pp. 229–264 Search PubMed.
- A. Lennartson, A. Roffey and K. Moth-Poulsen, Tetrahedron Lett., 2015, 56, 1457–1465 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |