
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The use of carboxylic acids as traceless directing groups for regioselective C–H bond functionalisation
Marc
Font
 ,
Jacob M.
Quibell
,
Gregory J. P.
Perry
and
Igor
Larrosa
*
,
Jacob M.
Quibell
,
Gregory J. P.
Perry
and
Igor
Larrosa
*
School of Chemistry, University of Manchester, Oxford Road, M13 9PL, Manchester, UK. E-mail: igor.larrosa@manchester.ac.uk
First published on 28th April 2017
Abstract
The ability to selectively functionalise a specific C–H bond is a long-standing challenge due to the ubiquity of such bonds in organic molecules. One of the most common approaches to overcome this obstacle consists of installing directing groups into substrates to direct the functionalisation towards the desired C–H bond, leaving behind the directing group in the molecule. Alternatively, carboxylic acids have been employed as traceless directing groups that are easily removed after carboxylic acid-directed installation of the desired functionality. This review focuses on the development of this concept and its application to organic synthesis during the last decade.
 Marc Font | Marc Font studied Chemistry at the University of Girona, where he was awarded his MChem in 2011. In 2015 he earned his PhD in Chemistry from the University of Girona on the elucidation of the mechanisms of the coinage metal-catalysed cross-coupling reactions under the guidance of Dr Xavi Ribas and Dr Miquel Costas. His PhD studies included a research period with Prof. John F. Hartwig at the University of California at Berkeley. Following this, he joined the group of Prof. Igor Larrosa at the University of Manchester pursuing postdoctoral studies on the development of novel methods for C–H functionalisation. |
 Jacob M. Quibell | Jacob Quibell completed his undergraduate and masters studies at the University of Edinburgh. During this time he spent a year in the University of Alicante working under the supervision of Prof. Francisco Foubelo investigating the use of sulfinylamines in stereoselective nitro-mannich reactions. He then returned to Edinburgh to work with Dr Stephen Thomas on iron-catalysed hydrofunctionalisations. He is now in his second year of his PhD studies with Prof. Igor Larrosa at the University of Manchester focussing on decarboxylative transformations. |
 Gregory J. P. Perry | Gregory Perry received his MChem from the University of Liverpool (UK) where he worked under the supervision of Professor P. Andrew Evans. He then joined the group of Professor Igor Larrosa and was awarded his PhD from the University of Manchester (UK) in 2016. His doctoral studies focused on the development of decarboxylative and C–H transformations for organic synthesis. He is currently a Postdoctoral Fellow at Nagoya University (Japan) working in the lab of Professor Kenichiro Itami. |
 Igor Larrosa | Igor Larrosa graduated in Chemistry from the University of Barcelona (1999) where he also completed an MChem (2000) and a PhD degree (2004) with Profs Felix Urpi and Pere Romea. After a research period in Prof. Erick M. Carreira's laboratories at ETH (Zurich), Igor moved as a Postdoctoral Researcher to Imperial College London to work in Prof. Anthony G. M. Barrett's group. In September 2007 he started his independent career as a Lecturer at Queen Mary University of London. In 2011, Igor was awarded a European Research Council starting grant. Since 2014 Igor holds a Chair in Organic Chemistry at the University of Manchester. |
Introduction
Building molecular complexity from simple, readily available building blocks has long been a fundamental aim of the synthetic chemist. Within organic chemistry C–H bond functionalisation has emerged as one of the most attractive methods for achieving greater intricacy in organic molecules.1–7 The ubiquitous presence of C–H bonds can provide a multitude of natural disconnections for further transformations whilst negating the need for the prefuctionalisation of starting materials, however with these benefits come two inherent limitations: selectively activating just one C–H bond within chemically similar environments and overcoming the inert nature of the bond itself.Finding a solution to these problems has generated a huge amount of interest in the synthetic community over the past few decades and thus far two major strategies have emerged. Firstly, using the innate reactivity of the organic molecule; i.e. using the acidity or nucleophilicity of a C–H bond to guide functionalisation to a specific location. The second strategy has come by way of employing the coordinating ability of other functional groups already installed on the molecule. These directing groups can guide the reactivity of a transition metal to the selected site, allowing the replacement of the C–H bond with C–C, C–O, C–N or other C–heteroatom bonds. These revelations have fuelled the growth of a huge repository of diverse directing groups (Scheme 1(i)), many of which operate at the highest level of reactivity and selectivity.8 However, if these moieties cannot be found in natural or commercially available compounds then the instalment of these groups can have a costly effect on the step and atom economy. This is further exasperated if the removal of the directing group is later on required, creating additional steps in the procedure (Scheme 1(ii)); in many ways reducing the central dogma of C–H activation to redundancy.9 A solution to this has been partially provided by the use of transient, catalytic directing groups which have the ability to tether themselves to a pre-existing part of the molecule and direct the C–H functionalisation before its release (Scheme 1(iii)). Although much progress has been made in the development of these directing groups, mainly through the reversible formation of phosphinite, imine or enamine functionalities, they still require a specific functionality on the molecule on which to tether.10
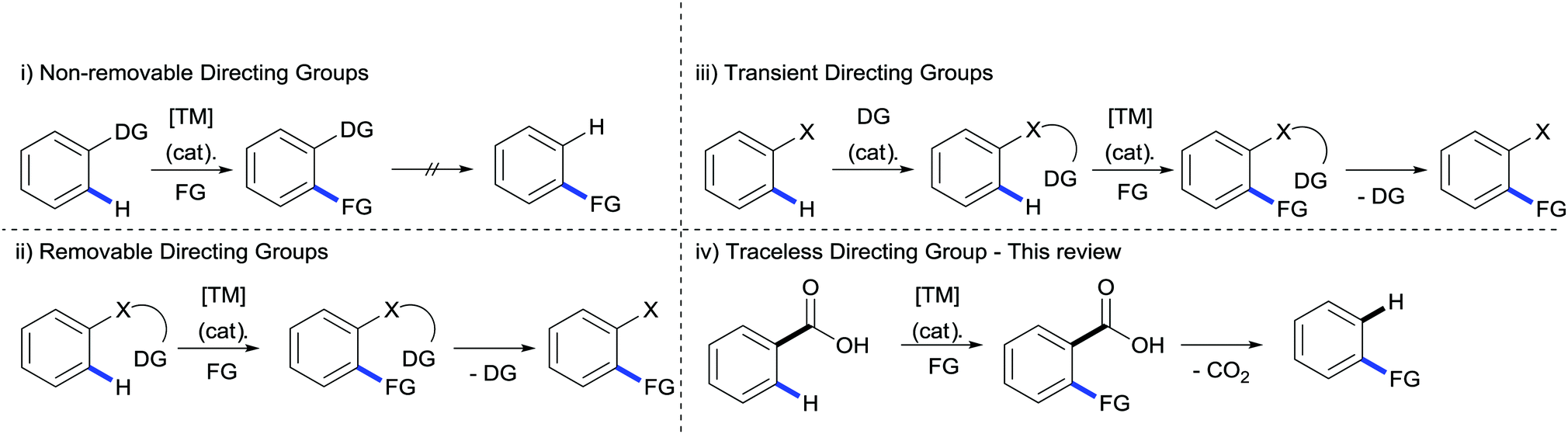 | ||
| Scheme 1 Strategies for directed C–H funtionalisation. FG = functional group reagent. | ||
An ideal situation would be to use a directing group inherent to a number of natural and commercially available compounds which can also be tracelessly removed. Within these specifications an attractive functionality is the carboxylate moiety. The abundancy of carboxylates in organic molecules and the versatility when considering further transformations11 makes it an ideal candidate for directing C–H bond functionalisations; in addition the removal of the carboxylate by extrusion of CO2 allows traceless operation of the directing group (Scheme 1(iv)).12
Carboxylic acids are cheap synthons that can be easily prepared by a number of methods; ranging from the oxidation of a diversity of functional groups such as olefins, aldehydes, alcohols or even alkanes to the hydrolysis of nitriles, acyl chlorides, esters or anhydrides. In addition, their synthesis via formation of organolithium or Grignard reagents followed by quench with CO2 has large precedence in the literature.13 More recently, also modern transition metal catalysis has been applied to carboxylation methods leading to more efficient and greener protocols for the preparation of carboxylic acids.14
This review will mainly focus on the utility of the carboxylate moiety in the ortho-functionalisation of benzoic acids before its subsequent removal via decarboxylation. With this goal in mind it is first pertinent to discuss the trends in transition metal-catalysed protodecarboxylations to better understand the requirements for traceless directing groups in C–H transformations.
Transition metal-promoted protodecarboxylation
Many methods exist for the protodecarboxylation of benzoic acids, however, only examples that bear some relevance to further coupling reactions will be discussed here. For a more comprehensive outlook on protodecarboxylation the reader is directed to some representative reviews.15,16Reports of protodecarboxylation date back to the mid-20th century13 yet the field remained in its infancy until Gooßen et al. disclosed the first copper-catalysed version.17 The Gooßen group showed that the presence of nitrogen donor ligands and an NMP/quinoline solvent mix were required for efficient reactivity. They also observed that electron-deficient benzoic acids were more reactive than electron-rich ones (e.g.1avs.2a, Scheme 2). Interestingly, ortho-substituted benzoic acids generally show higher reactivity than their meta or para-substituted analogues, however, good reactivity was found by employing a modified phenanthroline ligand L1 (e.g.2bvs.2c, Scheme 2). This seminal report paved the way for other similar methods to be developed with changes to the ligand system allowing for lower temperatures18 to be used and microwave heating giving a reduction in reaction times.19 The increased reactivity of ortho-substituted substrates provoked an investigation into the mechanism of these reactions. Through DFT studies the groups of Su20 and Gooßen21 found a plausible transition state 8 in which the Cu inserts into the aryl carboxyl bond (Scheme 3A). Further studies by Su and co-workers showed the effect of the ortho-substituent to be twofold: 1° effect- the extra sterical stress of an ortho-substituent raises the energy of the reactant thus decreasing the overall barrier to decarboxylation (Scheme 3B). 2° effect- ortho-substituents that are capable of coordination to the copper lower the energy of the transition state, decreasing the overall energy barrier to decarboxylation (Scheme 3C).
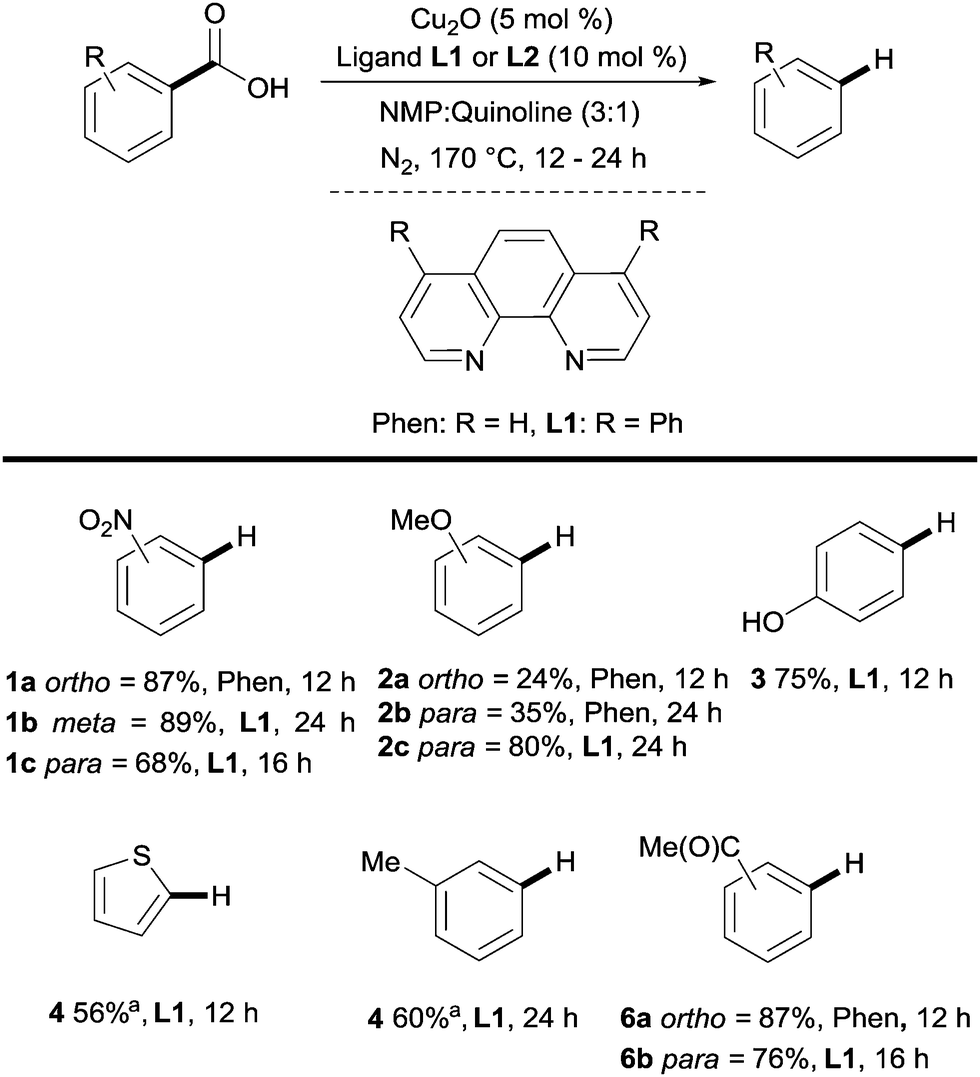 | ||
| Scheme 2 Copper-catalysed protodecarboxylation of aromatic carboxylic acids. a GC yield. | ||
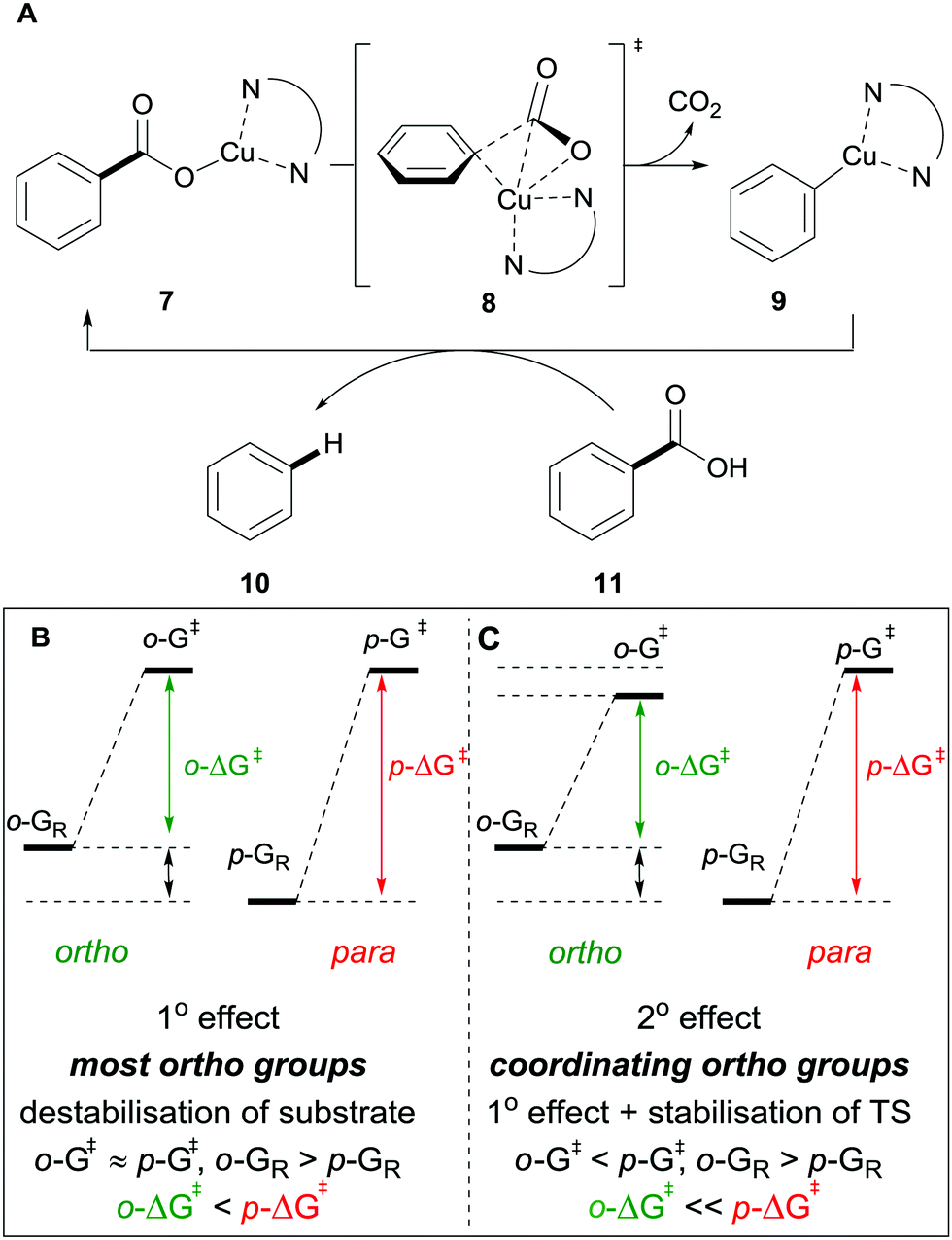 | ||
| Scheme 3 Proposed mechanism for the copper-catalysed protodecarboxylation of aromatic carboxylic acids (A) and delineating the 1° (B) and 2° ortho-effect (C). | ||
Silver was then shown to be a suitable surrogate for the copper-catalysed process with simultaneous publications coming from the groups of Gooßen22 and Larrosa.23,24 These reports showed that both electron withdrawing (e.g.12a, Scheme 4) and electron donating (e.g.16a, Scheme 4) substituents were compatible with the process, however, electron deficient substrates were generally more reactive (Scheme 4). The key difference between Ag- and Cu-catalysed protodecarboxylations is that ortho-substituents are a necessity in Ag-catalysed procedures, but lower temperatures can be used (120 vs. 160 °C).
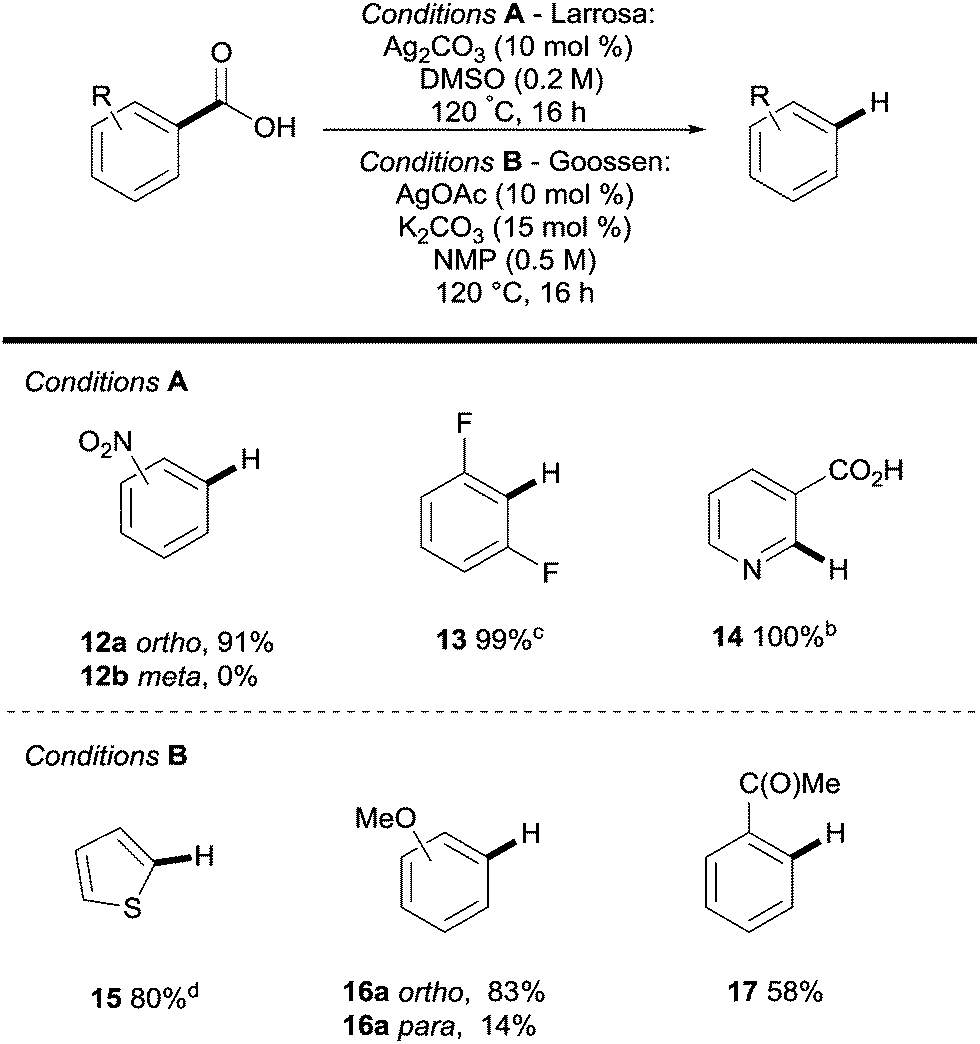 | ||
| Scheme 4 Silver-catalysed protodecarboxylation of aromatic carboxylic acids. a 1H NMR yield. b AcOH (0.05 equiv.). c 160 °C. | ||
DFT studies revealed that the reaction proceeded through a transition state similar to that observed with the copper-catalysed process 19 and the 1° and 2° ortho effects exhibited in the copper systems were also applicable to the silver-catalysed protocols (Scheme 5).
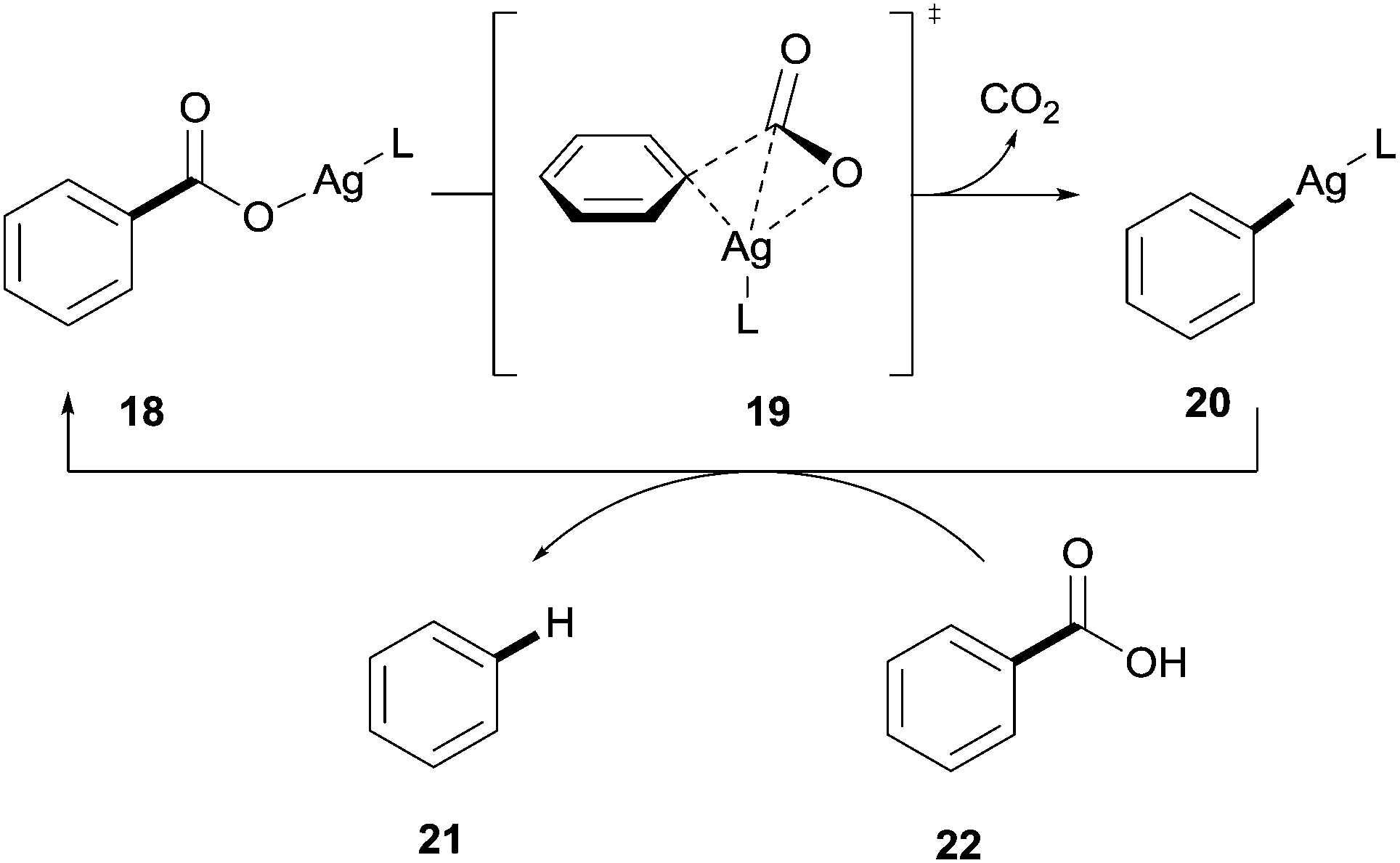 | ||
| Scheme 5 Proposed mechanism for the silver-catalysed protodecarboxylation of aromatic carboxylic acids. | ||
In addition it was revealed that a third effect was in place; it was found that a build-up of negative charge on the carbon ipso to the carboxylate group occurred in the transition state.25 In light of this, it is proposed that benzoic acids bearing electron-withdrawing substituents are more reactive than those bearing electron-donating substituents as they can better stabilise this build-up of negative charge, therefore, facilitating the decarboxylation step.
Palladium has also been shown to be an effective catalyst in the protodecarboxylation of benzoic acids. Kozlowski showed that highly electron rich substrates bearing two ortho-substituents underwent decarboxylation at relatively low temperatures; however this methodology was severely limited. In order for mono-ortho-substituted benzoic acids 28 and less electron rich benzoic acids 29 to undergo decarboxylation a stoichiometric amount of palladium was required (Scheme 6).26 Mechanistic studies by various groups14 have shown a very distinct reaction profile for the palladium system compared to those for copper and silver; firstly the decarboxylation proceeds through a 4-membered transition state with no interaction with the carboxyl carbon (see 31vs.8 and 19, Scheme 7).
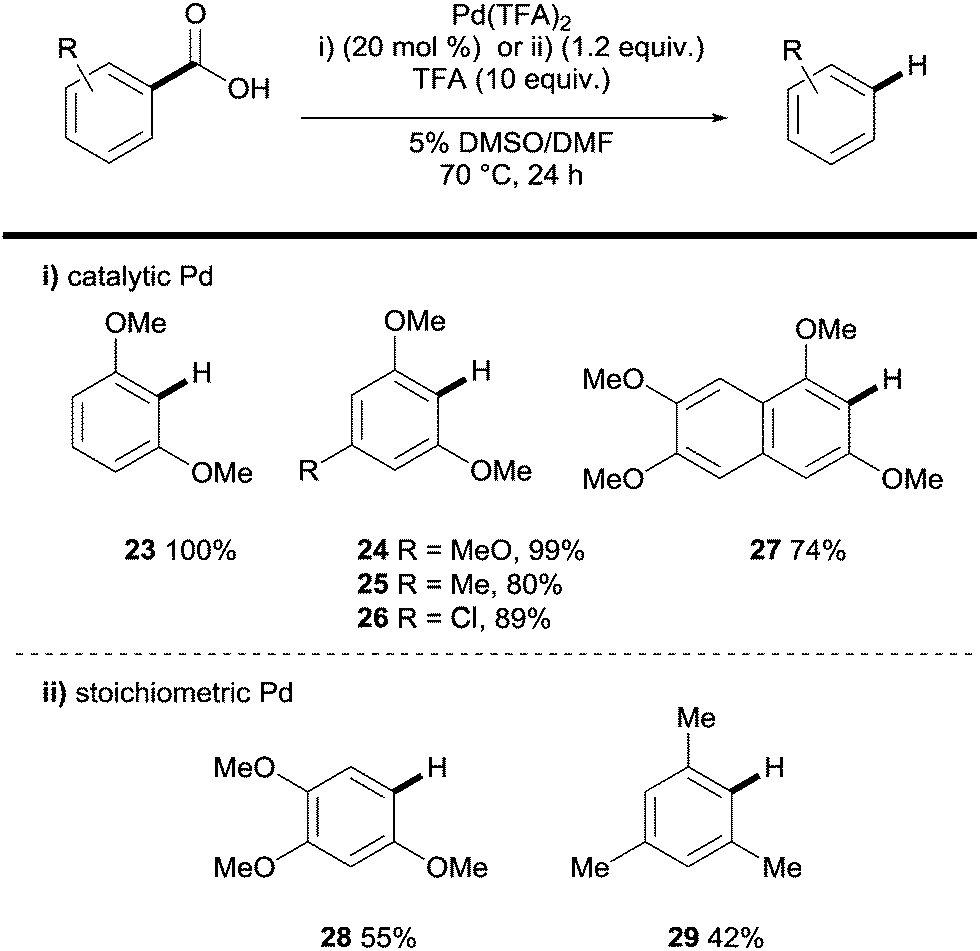 | ||
| Scheme 6 Palladium-promoted protodecarboxylation of aromatic carboxylic acids. | ||
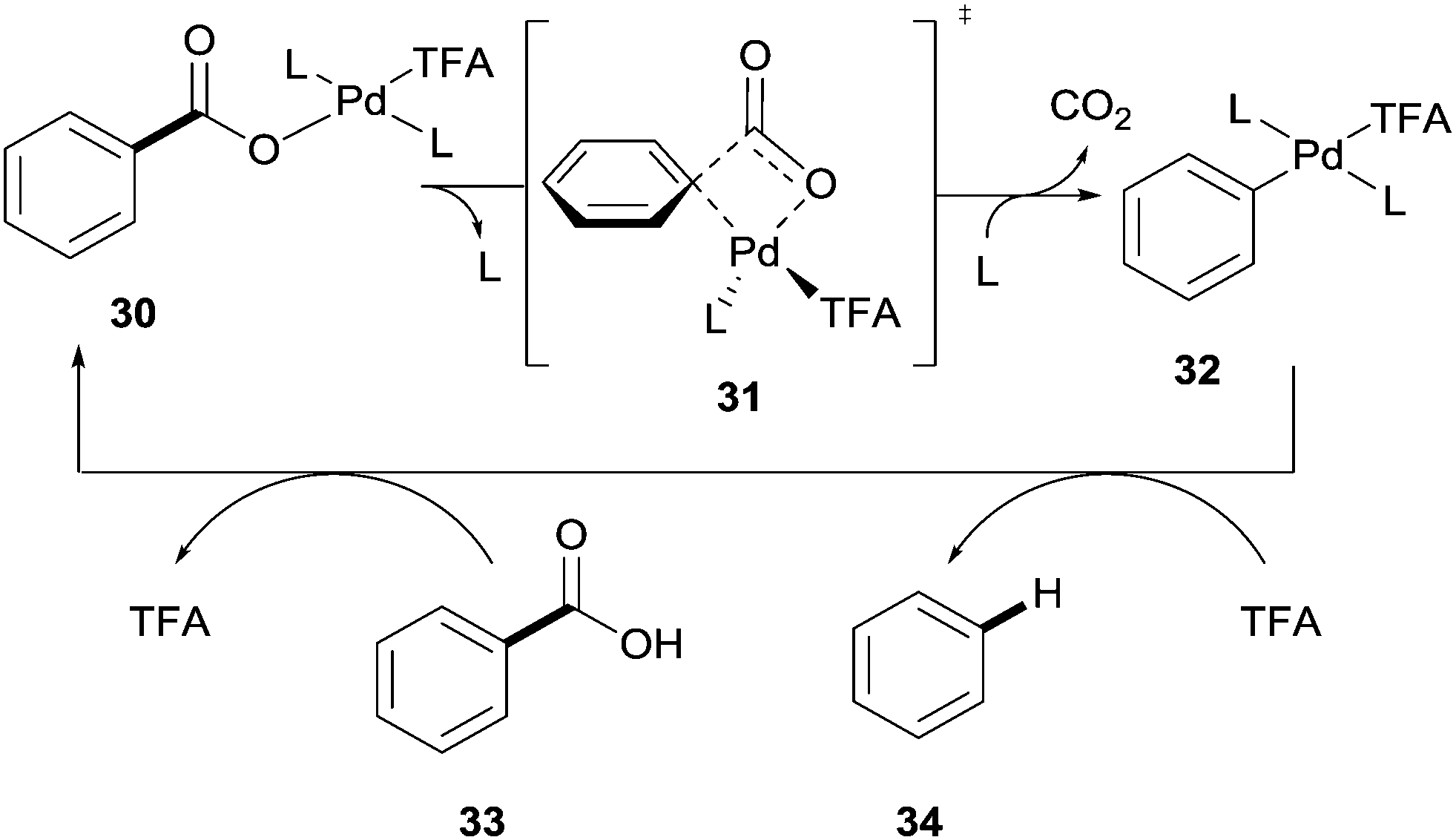 | ||
| Scheme 7 Proposed mechanism for the palladium-catalysed protodecarboxylation of aromatic carboxylic acids. | ||
Secondly the rate determining step was proposed to be the protodemetallation of 32 rather than the decarboxylation. Although it was conceded that this may change between different substrates due to the small difference in barriers (∼2 kcal mol−1) of each step. The authors also noted that the benzoic acid was likely unable to mediate the protodemetallation of the palladium–aryl species and so TFA is necessary in this reaction to facilitate this step.
Another transition metal relevant to coupling reactions that has been shown to promote the protodecarboxylation of benzoic acids is rhodium.27 In this context, Zhao showed that highly activated di-ortho-substituted carboxylic acids could be effectively protodecarboxylated in the presence of Rh(I) species. The protocol is exclusive to fluoro/methoxy substituted benzoic acids and some heteroaromatic carboxylic acids yet is unique in the fact that it uses a phosphine ligand and water as a co-solvent (Scheme 8).
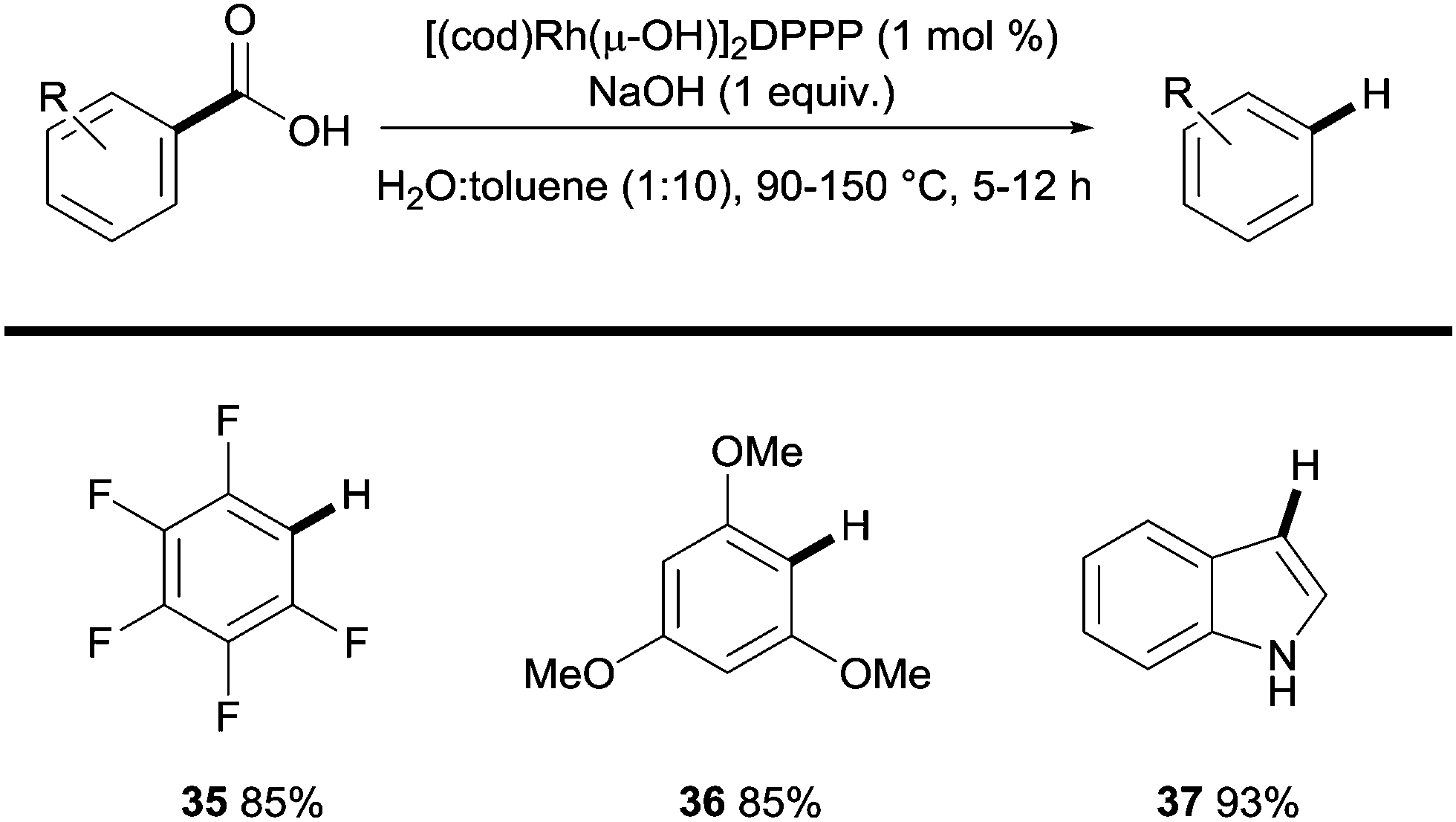 | ||
| Scheme 8 Rhodium-catalysed protodecarboxylation of (hetero)aromatic carboxylic acids. | ||
Carboxylic acids as traceless directing groups
The potential synthetic use of combining decarboxylation of carboxylic acids with their broad use as directing groups12 has incited a boom in methods for the sequential functionalisation/decarboxylation of benzoic acids over the last ten years. These methods can be largely divided into three classes: (1) two-step transformations using multi-metallic systems; (2) single-step transformations using multi-metallic systems and (3) single-step transformations using mono-metallic systems (Scheme 9).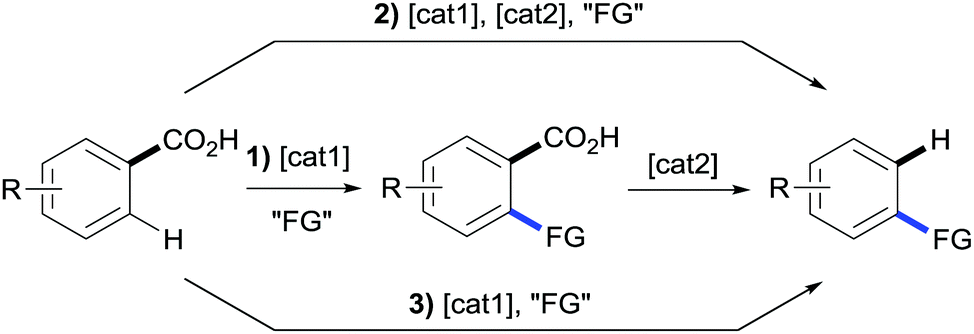 | ||
| Scheme 9 Classification of the methods for directed C–H functionalisation followed by decarboxylation. | ||
Functionalisation/decarboxylation reaction involving two metals and two steps
The first method that revealed the potential of benzoic acids to direct C–H bond functionalisation tracelessly came from Daugulis and co-workers.28 A seminal publication from the group revealed an efficient process for the ortho-arylation of a variety of benzoic acids with electronically diverse aryl chlorides or aryl iodides. Importantly, they then went on to illustrate that the carboxyl group in the biaryl product could be removed via the copper-catalysed protodecarboxylation methodology disclosed by Gooßen et al. (Scheme 10).17 In this example 4-toluic acid 38 was coupled with 1-iodo-3,5-bis(trifluoromethyl)benzene 39via palladium-catalysed ortho C–H activation with a superstoichiometric silver acetate additive. The biaryl carboxylic acid was then isolated prior to being submitted to the conditions for the protodecarboxylation described by Gooßen giving product 40 in a 51% overall yield (Scheme 10). This was the first demonstration of carboxylic acids acting as a traceless directing group in C–H bond functionalisation however the need for purification before removing the carboxylic acid decreased the step economy of the process.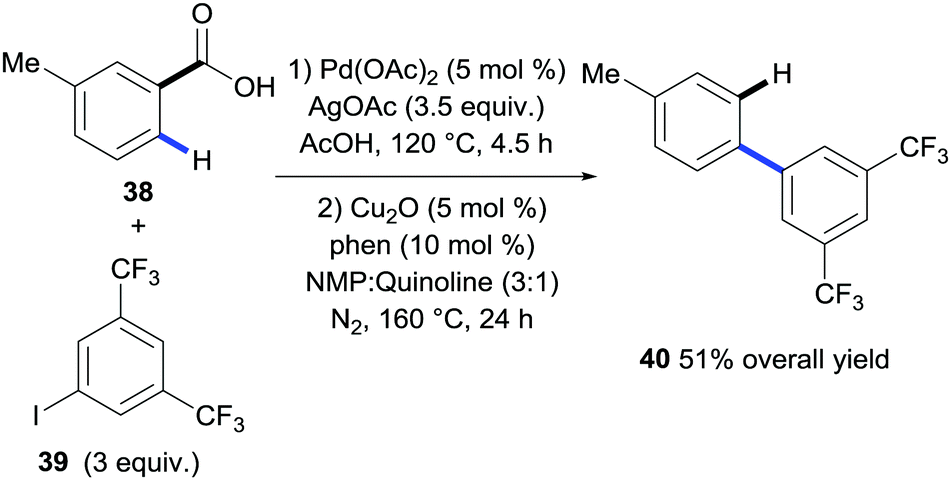 | ||
| Scheme 10 Palladium-catalysed ortho C–H bond arylation of benzoic acids followed by copper-catalysed protodecarboxylation. a 165 °C. b O2 (5 atm). | ||
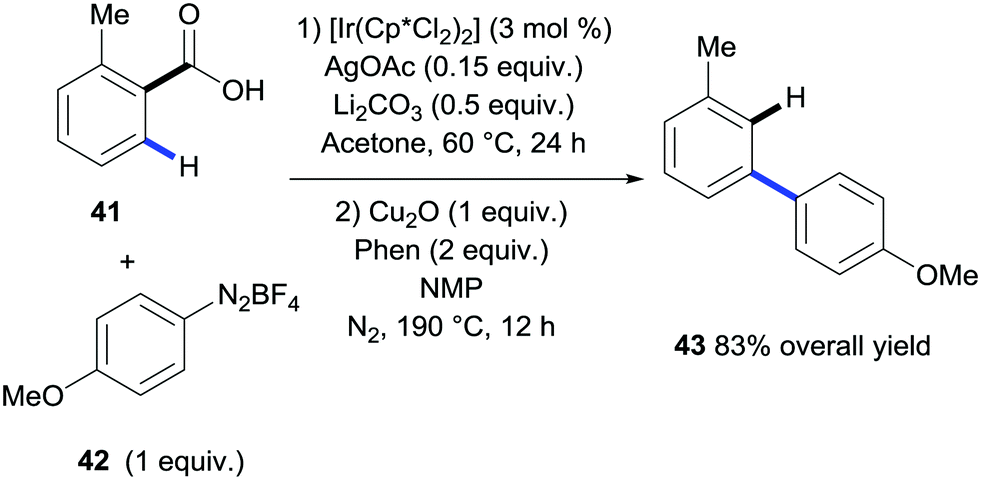
In 2015, Gooßen et al. developed a procedure for the ortho-arylation of benzoic acids catalysed by iridium that was also amenable to subsequent protodecarboxylation in a one-pot fashion (Scheme 11).29 The arylation used arenediazonium salts as the coupling partners with AgCO3 and Li2CO3 as additives. In this case, purification between steps was not required and the resulting meta-functionalised aryl 43 was formed after the reaction mix was subjected directly to the copper-mediated protodecarboxylation conditions developed in their laboratories.
 | ||
| Scheme 11 Iridium-catalysed ortho C–H bond arylation of benzoic acids followed by copper-mediated protodecarboxylation. | ||
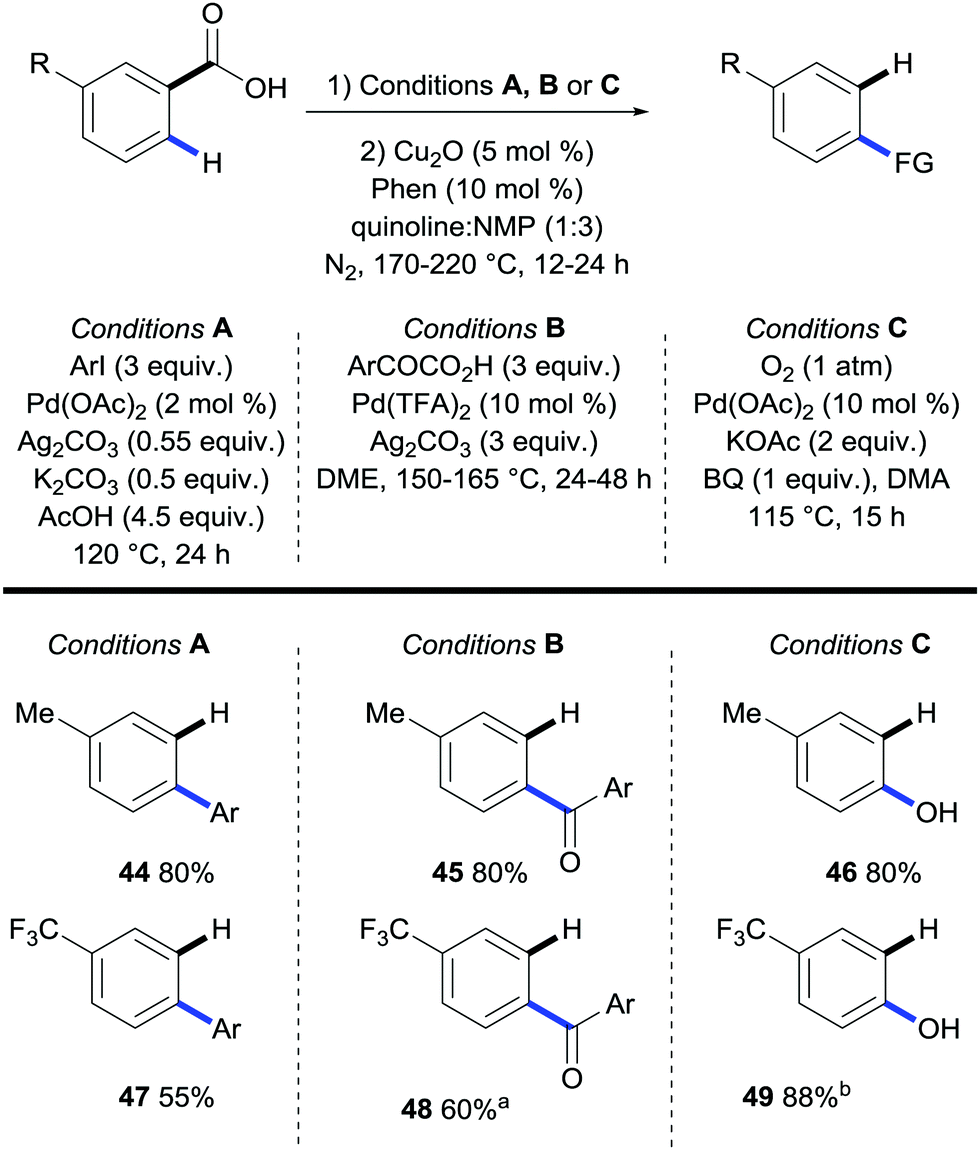
In an analogous publication Zhang extended the scope of the sequential ortho-arylation/protodecarboxylation of meta-substituted benzoic acids (Scheme 12, conditions A).30 Furthermore, they also revealed that, upon changing the reaction conditions, they could effect a carbonylation or hydroxylation sequence, followed by protodecarboxylation. The functionalisation conditions used were originally developed by the groups of Larrosa,31 Ge,32 and Yu33 for the ortho-arylation, carbonylation and hydroxylation of benzoic acids respectively. The novelty of this report by Zhang is the removal of the directing group after the functionalisation to provide a range of para-substituted arenes in good to excellent overall yields. However, a work-up was required between the functionalisation and the decarboxylation steps.
 | ||
| Scheme 12 Sequential palladium-catalysed ortho C–H bond arylation/protodecarboxylation of m-substituted benzoic acids. | ||
Miura and co-workers further developed the system of the traceless directing group into a one-pot process.34,35 Using rhodium to catalyse the coupling followed by silver to facilitate the decarboxylation, the process obviated the requirement of a work-up between steps (Scheme 13). The scope was largely restricted to benzoic acids containing electron-rich substituents; phenyl and fluoro substituents were also shown to react giving moderate yields (Scheme 13, 53, 54) However, both the functionalisation and the protodecarboxylation steps required excess AgOAc.
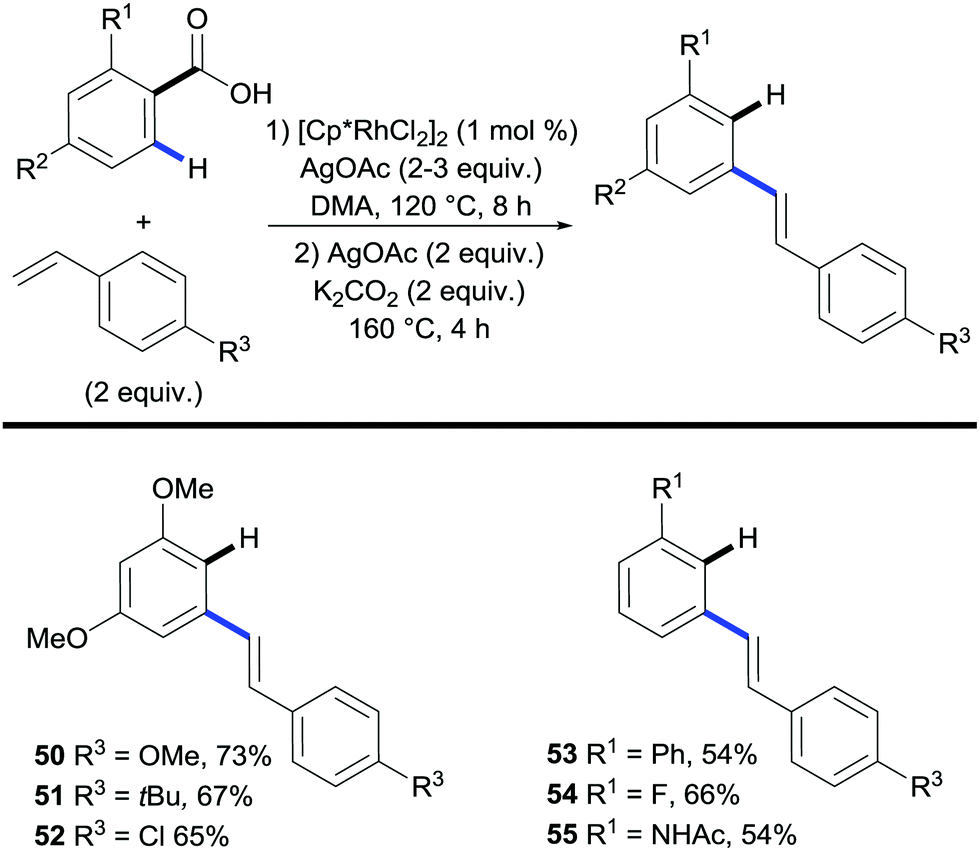 | ||
| Scheme 13 Rhodium-catalysed two-step synthesis of stilbene derivatives. | ||
A protocol for the one-pot C–H amidation/decarboxylation of benzoic acids was reported by Chang and co-workers in which ortho-substituted benzoic acids reacted with sulfonyl azides in the presence of an iridium catalyst followed by a palladium-catalysed protodecarboxylation event (Scheme 14).36 The decision to use a palladium catalyst for the protodecarboxylation step was inspired by the previous report by Kozlowski and co-workers (Scheme 6).26
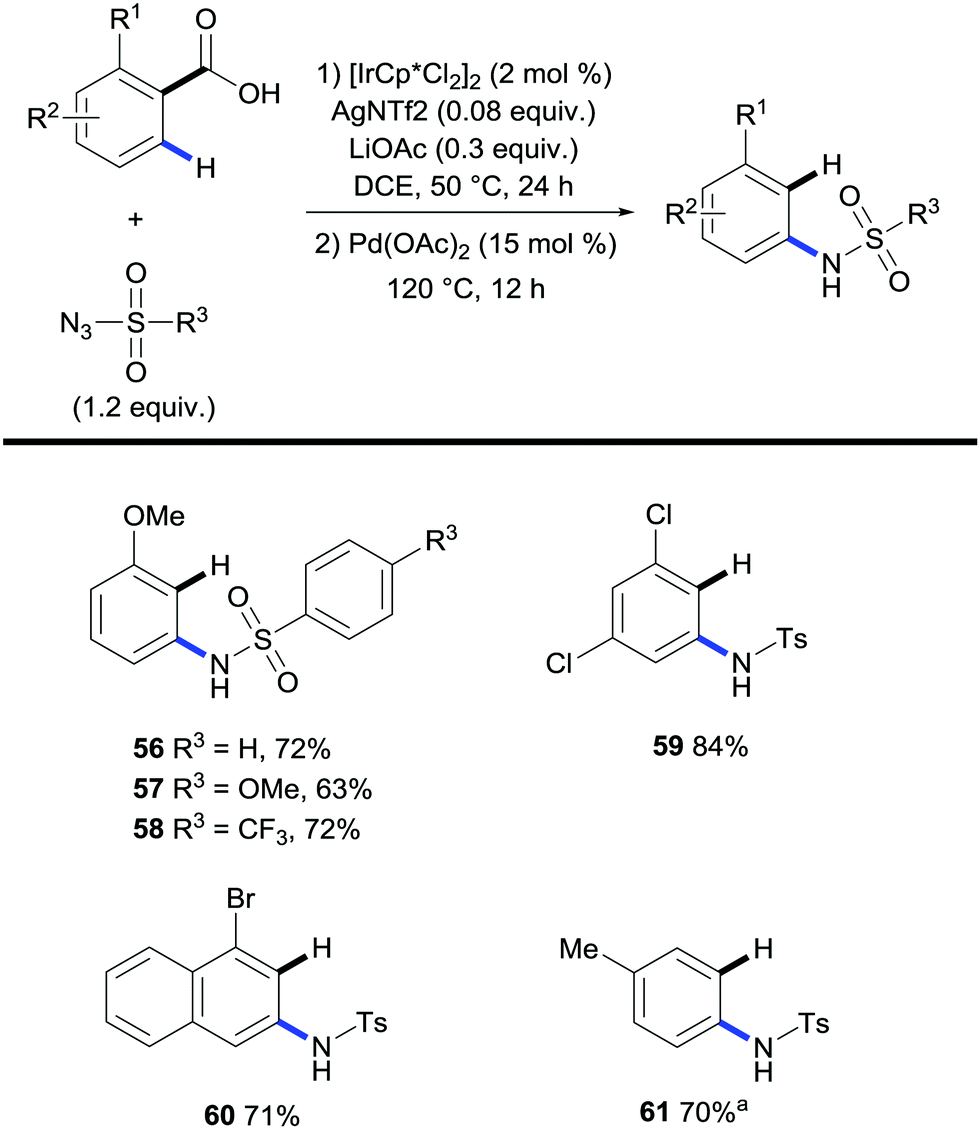 | ||
| Scheme 14 Iridium- and palladium-catalysed 2-step synthesis of N-arylsulfonamides. a Conditions 2nd step: Cu2O (0.5 equiv.), Phen (1 equiv.), DMA, N2, 170 °C, 12 h. | ||
The results yielded meta-substituted arenes in moderate to high yields. Again electron rich benzoic acids were mainly used although some electron deficient benzoic acids such as 2,4-dichlorobenzoic acid (Scheme 14, 59) were also shown to react in high yields. This is quite remarkable given that decarboxylation of electron-deficient substrates by palladium is not favored. They also investigated the feasibility of meta-substituted benzoic acids in the same system; however, the absence of an ortho-substituent with respect to the carboxylate group ensured the lack of reactivity in the palladium-catalysed protodecarboxylation step. The authors went on to show that copper was capable of achieving this transformation although it required stoichiometric amounts of the metal in question (Scheme 14, 61).
Recently Larrosa et al. disclosed a procedure for the ortho-arylation of pyridine carboxylic acids (Scheme 15).37 This methodology allowed for the selective functionalisation at the C3 or C4 positions of the pyridine ring. Pyridines are particularly difficult substrates to undergo functionalisation as the ability for the nitrogen atom to coordinate to the metal often kills any reactivity. It is thought that the inclusion of a carboxyl group in the substrate can out-compete coordination by the nitrogen atom, therefore leading to an efficient and selective functionalisation.
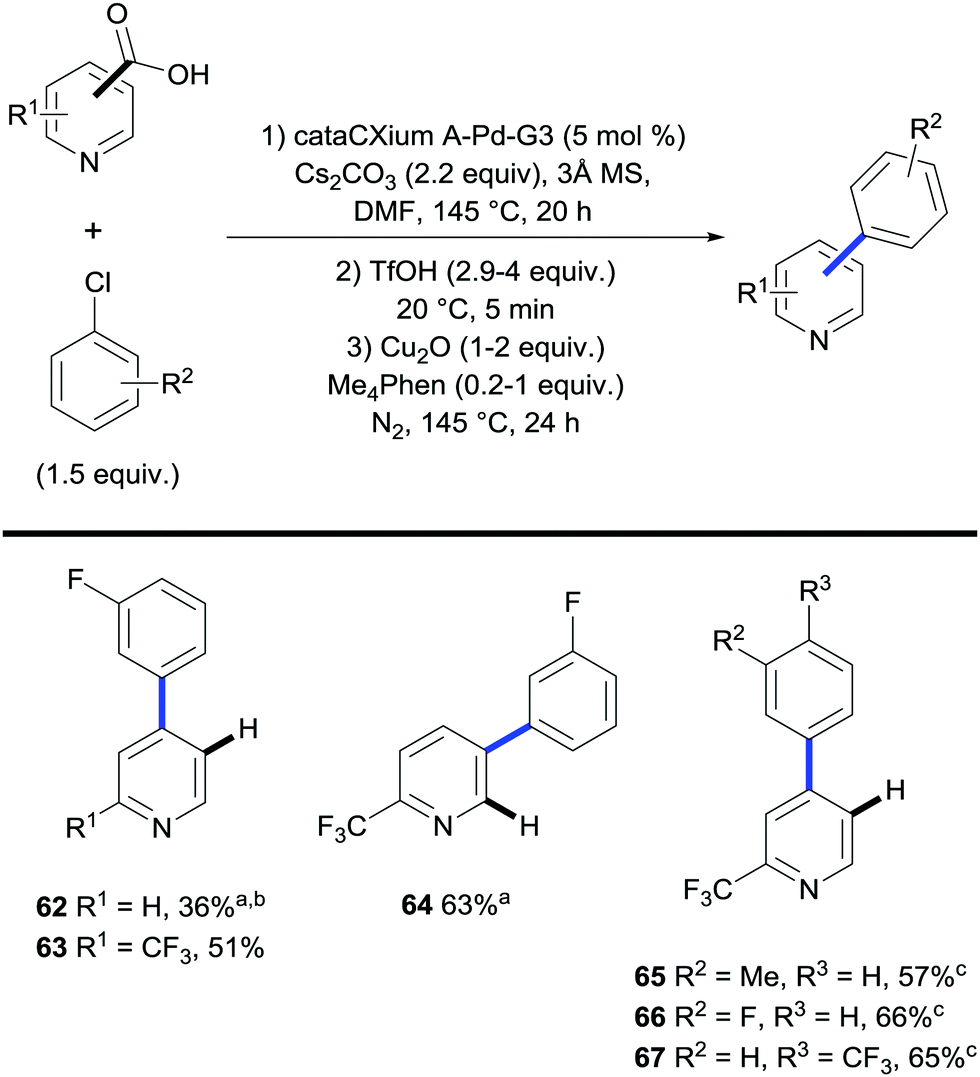 | ||
| Scheme 15 Palladium-catalysed/copper-mediated 2-step synthesis of phenyl pyridines. a Conditions 1st step: reaction performed with Pd(OAc)2 (5 mol%) and PAd2nBu·HI (10 mol%). b 40 h arylation time. c Phen (0.2 equiv.) instead of Me4Phen. | ||
A Buchwald third generation precatalyst (cataCXium A-Pd-G3) was found to be the optimal catalyst for the arylation in a DMF solvent with Cs2CO3 as the base. Both chloroarenes and bromoarenes substituted with electron donating and electron withdrawing groups were found to be suitable electrophiles under these conditions. Generally, electron deficient pyridine carboxylic acids gave better reaction outcomes – likely due to the lower coordinating ability of the N-atom – whereas electron-rich substrates gave diminished yields.
A one-pot arylation/decarboxylation process was then developed giving access to arylated pyridines. Using the previously optimised conditions for arylation the reaction mixture was then subjected to the conditions for copper-mediated decarboxylation using stoichiometric copper oxide and a methyl-substituted phenanthroline ligand Me4Phen. The two conditions were shown to be compatible with each other provided triflic acid was added prior to the decarboxylation step.
Functionalisation/decarboxylation reaction involving two metals and one step
The field of regioselective transition metal-catalysed C–H bond functionalisation via traceless directing groups has evolved towards methods consisting of tandem functionalisation/decarboxylation reactions, thereby accelerating the overall reaction times and simplifying the experimental protocols. Such approaches add extra intricacy to the reaction as the decarboxylation of the starting material must be avoided to obtain synthetically useful reaction outcomes. The viability of this approach was first demonstrated by Miura and co-workers. The authors developed a protocol for the regioselective palladium-catalysed oxidative alkenylation/decarboxylation of (benzo)furan, (benzo)thiophene and N-protected indole and pyrrole carboxylic acids in good to excellent yields with largely good regioselectivity (Scheme 16).38 Alkyl acrylates and styrene proved to be suitable olefin coupling partners for this method. Importantly, this methodology allows for the direct vinylation of 1-methylindole-3-carboxylic acid in the less activated C2 position, whereas, under similar conditions, the alkenylation of N-methylindole proceeds exclusively at the C3 position (Scheme 17A and B). One of the limitations of this method is that selective C3-vinylation of (benzo)thiophene 2-carboxylic acid proved unsuccessful giving rise to mixtures of 2- and 3-olefination products; presumably due to initial protodecarboxylation of the starting heteroaromatic carboxylic acid material followed by subsequent non-directed coupling with the resulting thiophene or benzothiophene although alternatively decarboxylative alkenylation could account for such regioselectivity. However, in a following report by the Miura group, they addressed this issue by developing a rhodium-catalysed protocol that enabled the selective C3-olefination of those substrates as well as benzofuran, N-methylpyrrole and N-methylindole 2-carboxylic acids.35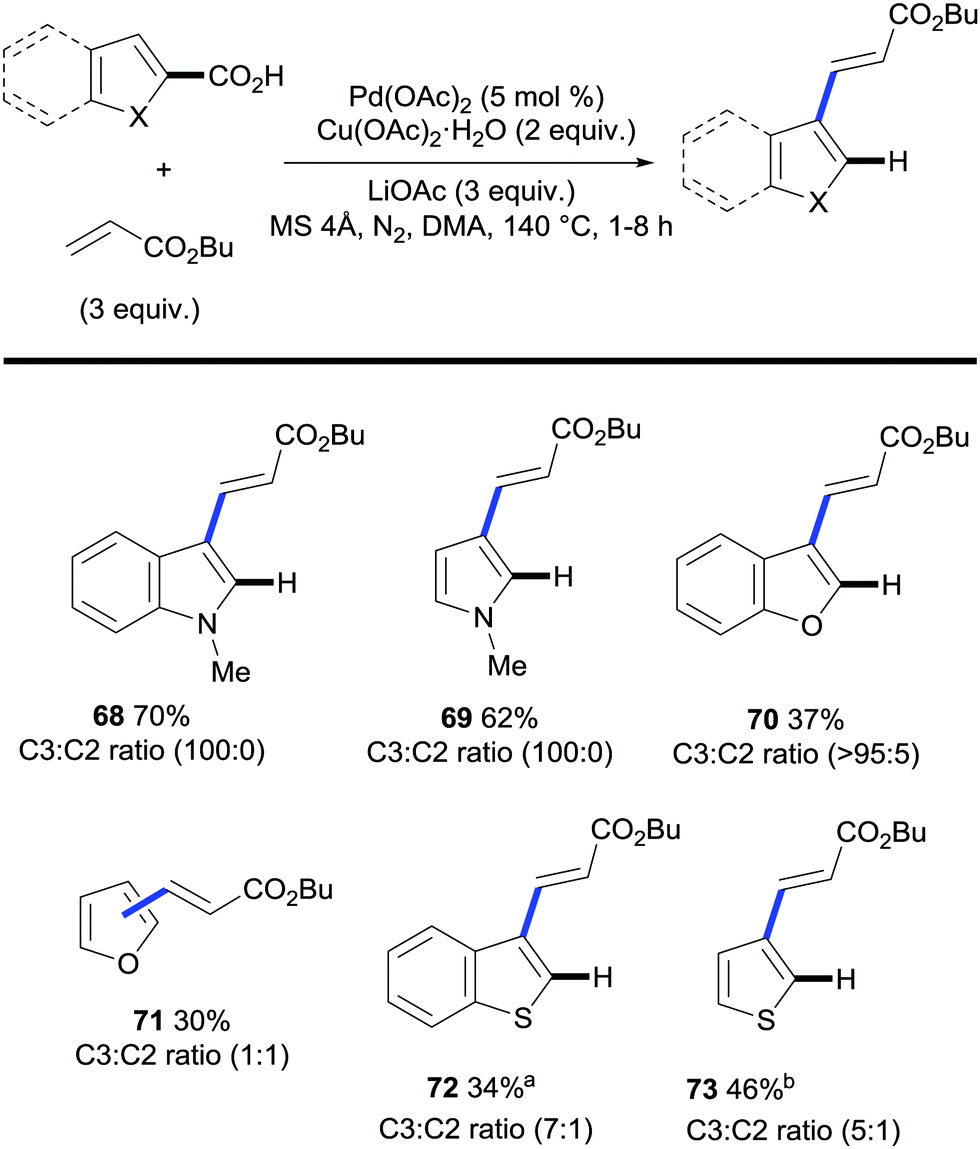 | ||
| Scheme 16 Palladium-catalysed one-step 3-vinylation/decarboxylation of heteroarene carboxylic acids. a 120 °C. b Without LiOAc. | ||
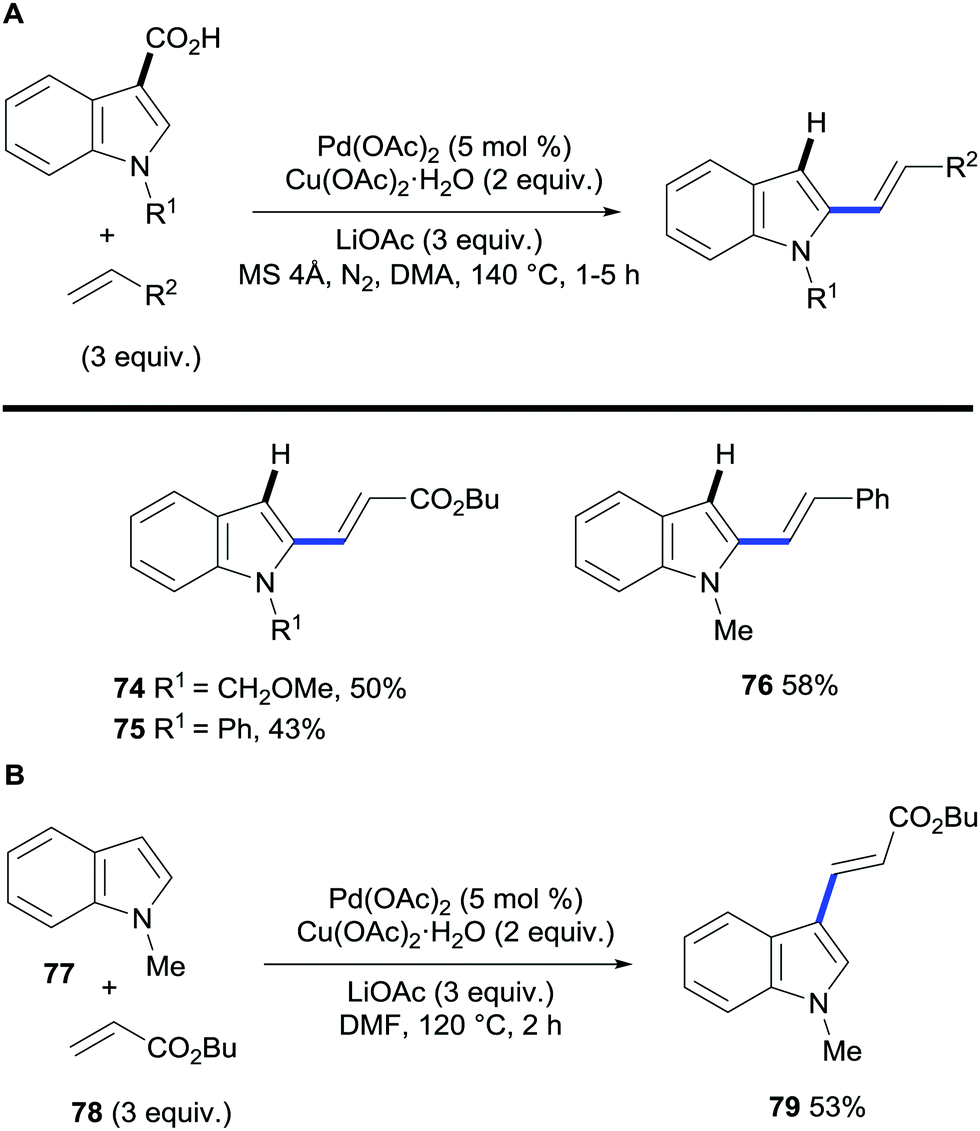 | ||
| Scheme 17 (A) Palladium-catalysed C2-vinylation/decarboxylation of indole-3-carboxylic acids. (B) Palladium-catalysed 3-vinylation of N-methylindole. | ||
Maiti and co-workers took advantage of the carboxylic acid traceless directing group strategy to achieve a regioselective alkenylation of arenes providing a new route towards branched alkenes, achieving the opposite selectivity to the traditional linear alkene-forming Mizoroki–Heck reaction.39 The authors approached the switch in selectivity by using cinnamic acids as the coupling partners. Their protocol involved a palladium/copper system that performed a dehydrogenative arene C–H bond alkenylation/decarboxylation sequence affording 1,1-disubstituted olefins with high selectivity (Scheme 18). The applicability of the method is limited by the use of the arene in vast excess and the poor ortho/meta/para regioselectivity on substituted arenes.
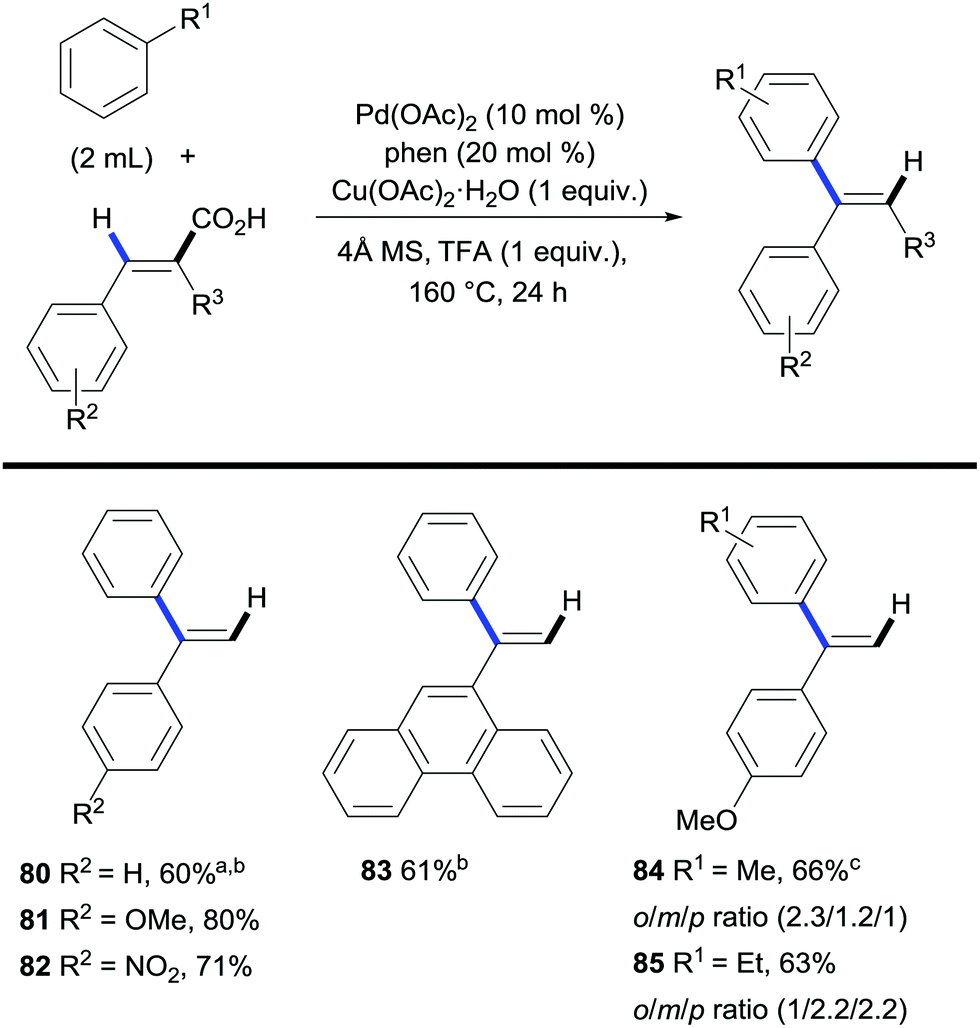 | ||
| Scheme 18 Palladium-catalysed branched C–H bond alkenylation of arenes with cinnamic acids. a Reaction performed with Pd(OPiv)2, bpy. b 140 °C. Reaction performed with bpy, Ac2O (1 equiv.), Cu(OAc)2·H2O. | ||
Hong and co-workers approached the synthesis of (hetero)acenes from di(hetero)aryl acetic acids and ethyl acrylate by developing a palladium-catalysed cascade reaction involving a C–H bond functionalisation/decarboxylation sequence (Scheme 19).40 This reaction is believed to be initiated with the carboxylate-directed palladium-catalysed C–H vinylation reaction, followed by C–H bond cyclisation. Upon decarboxylation and isomerisation of the double bond, the final (hetero)acene scaffold was obtained. This protocol gives rise to a new step- and atom-economical route towards complex linearly fused polycyclic aromatic hydrocarbons with very specific substitution patterns.
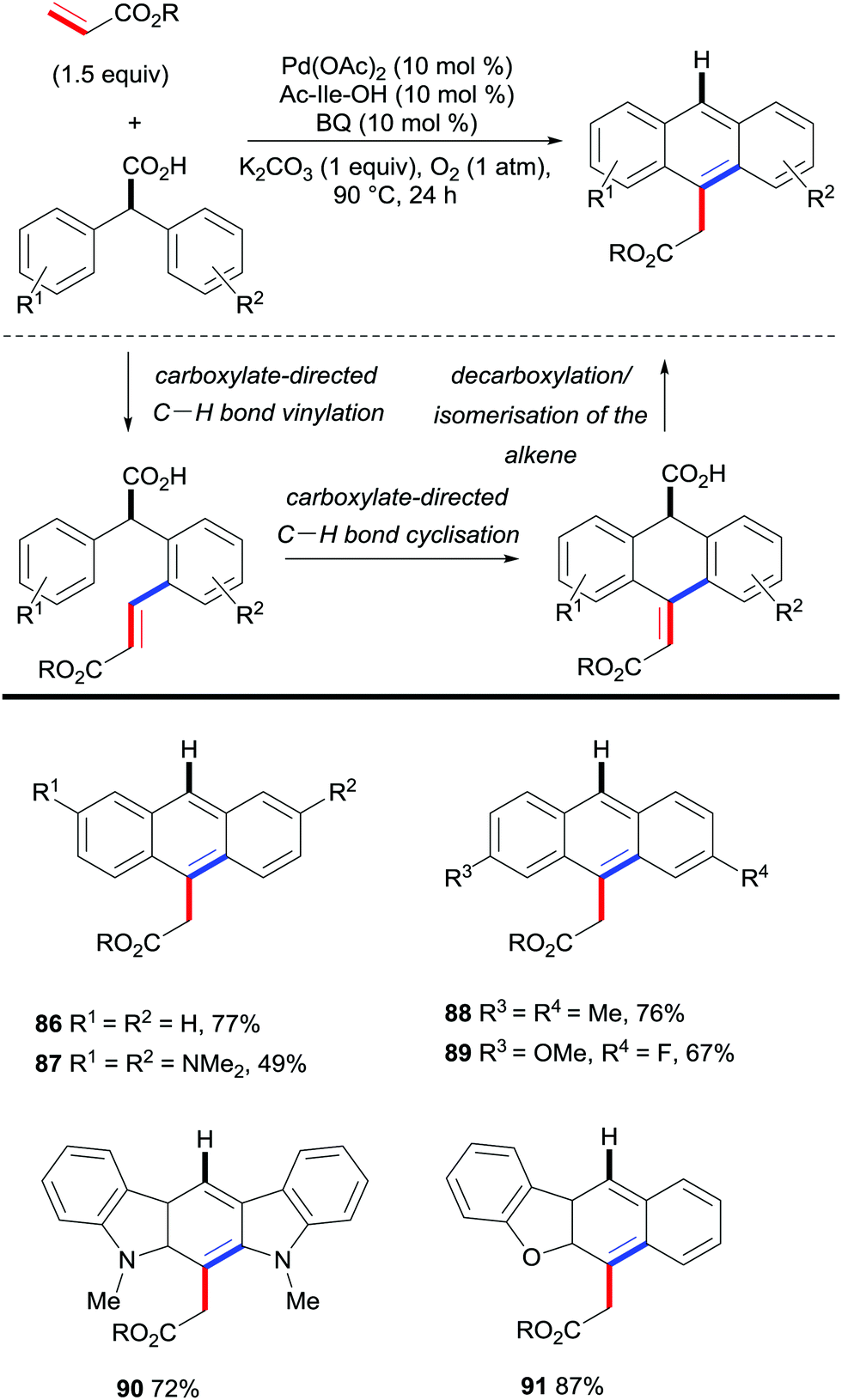 | ||
| Scheme 19 Palladium-catalysed synthesis of (hetero)acenes by reaction of di(hetero)aryl acetic acids with ethyl acrylate. | ||
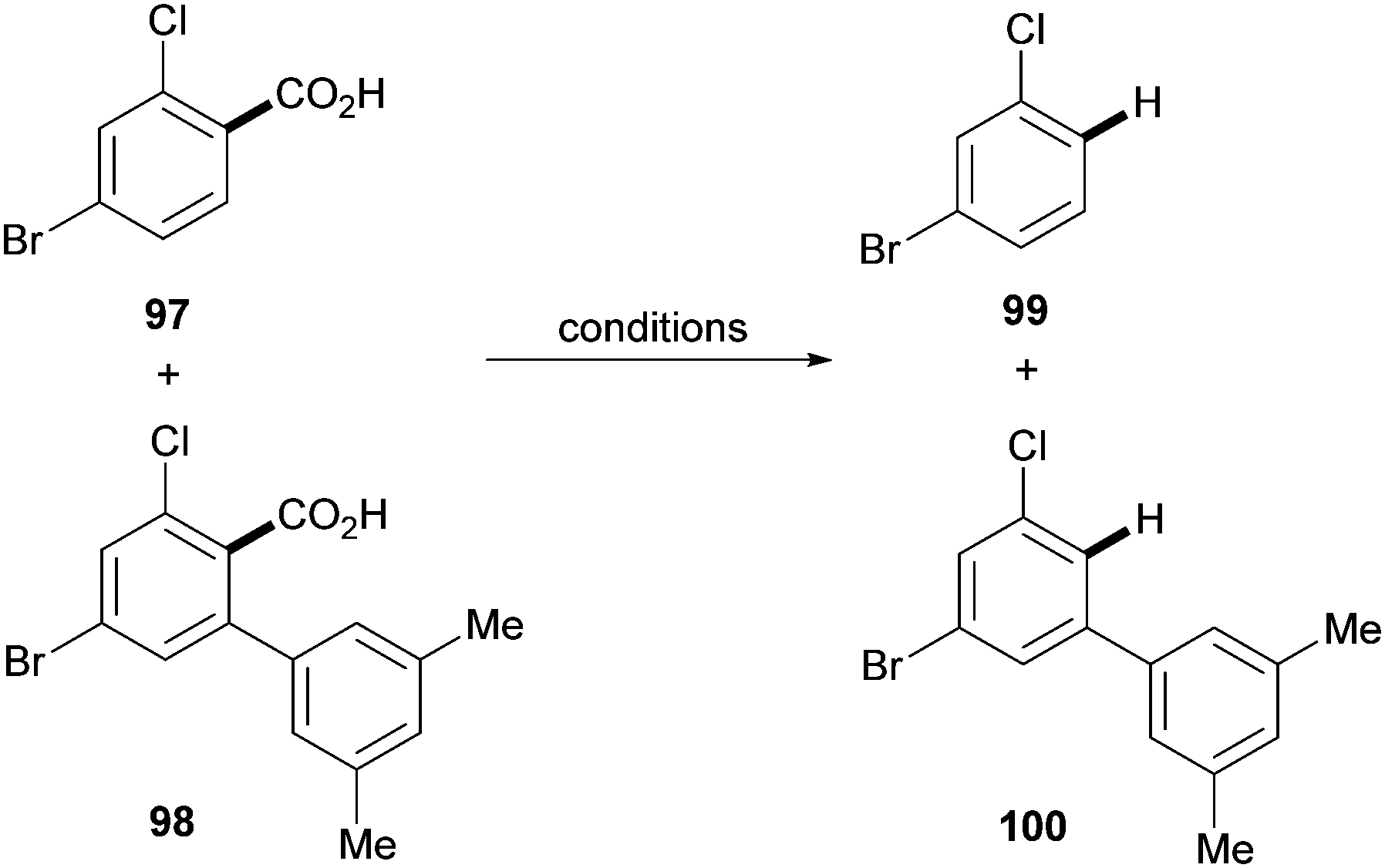
As highlighted by Miura's and Maiti's examples, one of the main features of the aforementioned approach is the ability to achieve regioselectivities that are often unattainable by using traditional methods. This was further demonstrated in 2011 in a paper by Larrosa and co-workers.41 The authors applied the traceless directing group strategy to ortho-substituted benzoic acids to achieve meta-substituted biaryl scaffolds using aryl iodides as the coupling partners and Pd(OAc)2 as the catalyst (Scheme 20). This protocol exploited the preference of palladium to decarboxylate electron rich bis-ortho-substituted benzoic acids (Scheme 6) to tackle the selective protodecarboxylation of the arylated benzoic acid product over the benzoic acid starting material. This provided the desired reactivity obtaining the meta-arylated product. Remarkably, the scope of the reaction included electron-deficient benzoic acids, which were previously considered highly deactivated substrates for the palladium-catalysed decarboxylation step (e.g.92 and 94–96, Scheme 20). The authors demonstrated that palladium selectively decarboxylates the arylated products by submitting both the starting benzoic acid 97 and arylation product 98 to different silver- and palladium-promoted decarboxylations. While the silver-catalysed conditions developed within the Larrosa group23 afforded 3 times more decarboxylation of the starting benzoic acid 97 than that of the arylated product 98 (Table 1, entry 1), employing silver in AcOH at 130 °C did not afford any decarboxylation product (Table 1, entry 2). Palladium instead was shown to be able to selectively decarboxylate the arylation product 98 in the arylation/decarboxylation reaction conditions (Table 1, entry 3) with no decarboxylation of starting benzoic acid 97. In addition, when both benzoic acids were submitted to the reaction conditions only the arylation product 98 underwent decarboxylation (Table 1, entry 4). The authors also found that the protodecarboxylation of the arylated product can be avoided by adding sub-stoichiometric amounts of K2CO3 to the reaction providing a reliable route towards 2-aryl-6-substituted benzoic acids.31
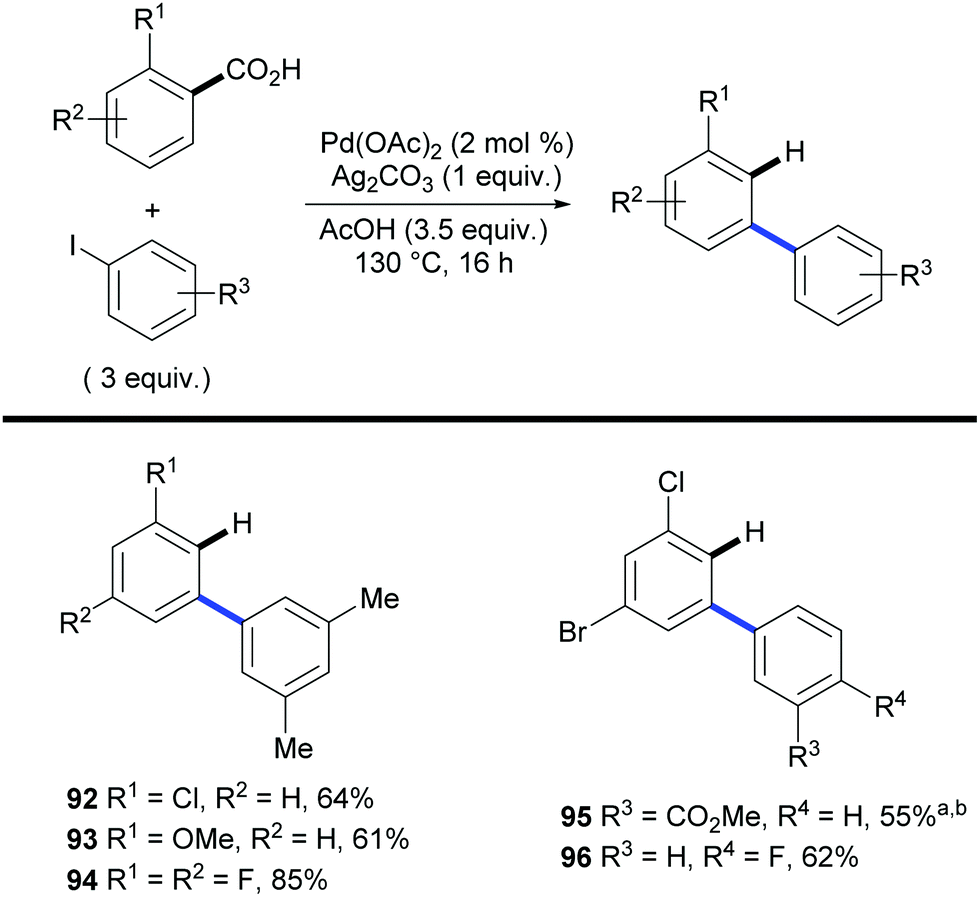 | ||
| Scheme 20 Synthesis of m-substituted biaryls via palladium-catalysed C–H bond arylation/decarboxylation of 2-substituted benzoic acids. a After 16 h, 0.25 equiv. of Ag2CO3 in 2.5 ml DMSO added and stirred for 3 h at 130 °C. b 7 equiv. AcOH. | ||
|
|
|||
|---|---|---|---|
| Entry | Conditions | Yield 99 (%) | Yield 100 (%) |
| 1 | 10 mol % Ag2CO3, DMSO, 120 °C, 4 h | 18 | 5 |
| 2 | 1 equiv. Ag2CO3, AcOH, 130 °C, 2 h | 0 | 0 |
| 3 | 2 mol % Pd(OAc)2, AcOH, 130 °C, 2 h | 0 | 22 |
| 4 | 2 mol % Pd(OAc)2, 1 equiv. Ag2CO3, AcOH, 130 °C, 2 h | 0 | 5 |
Next, Larrosa and co-workers investigated the tandem arylation/decarboxylation of widely available salicylic acids. By finely tuning the reaction conditions they found PEPPSI™–IPr to be a superior catalyst for this transformation yielding a wide array of meta-arylated phenol compounds, which have traditionally been a challenging class of compounds to access (Scheme 21).42
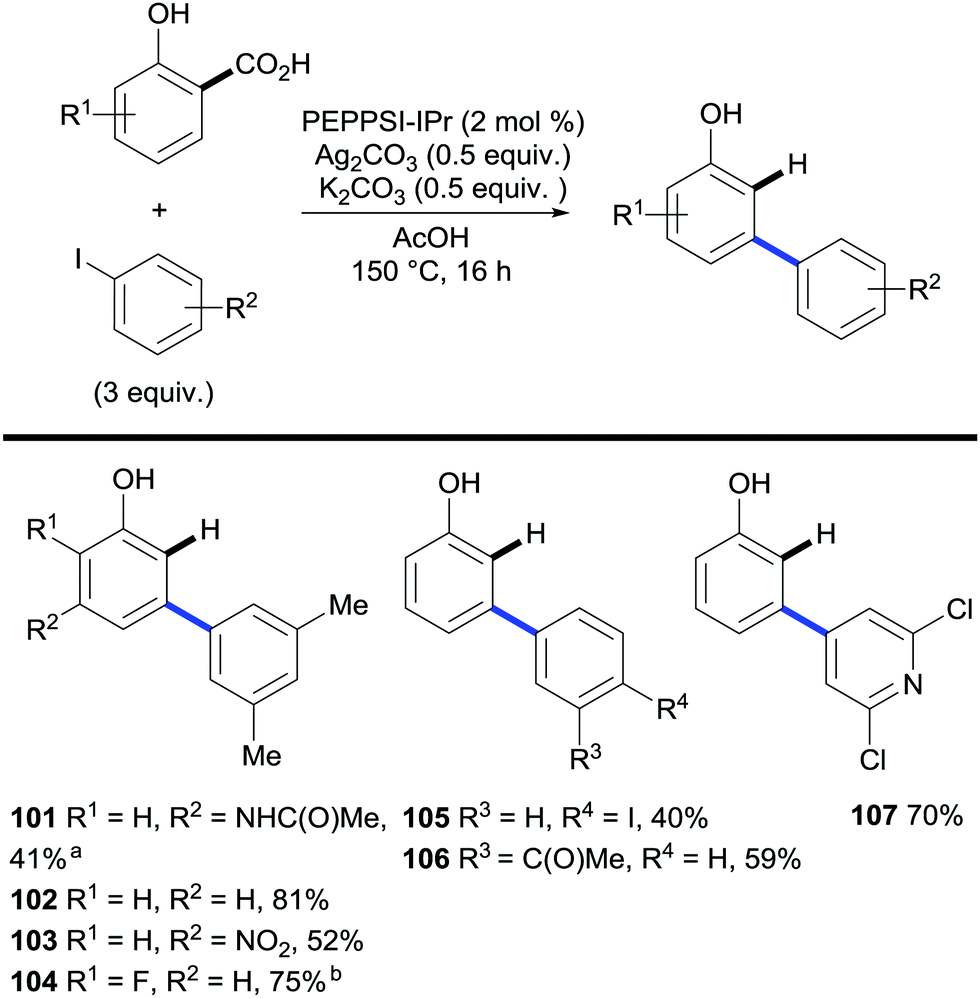 | ||
| Scheme 21 Synthesis of m-arylated phenols via palladium-catalysed C–H bond arylation/decarboxylation of salicylic acids. a Without K2CO3. b 1 ml AcOH. | ||
Importantly, the authors also demonstrated that this methodology tolerates the replacement of the silver-based halide scavenger by an ammonium organic salt (NMe4Cl) leading to a more environmentally friendly and cheaper protocol.43 Taking advantage of this protocol the authors designed a novel approach for the meta-arylation of phenols consisting of a two-step one-pot synthetic sequence involving an initial Kolbe–Schmitt carboxylation followed by the aforementioned tandem arylation/decarboxylation reaction without exacerbating the reaction scope and yields (Scheme 22).44 This example shows that by using the traceless directing group relay strategy a new mode of reactivity for phenols is enabled and their inherent ortho/para selectivity for electrophilic aromatic substitutions can be overridden. Alternatively, salicylaldehydes have been demonstrated to be suitable building blocks of meta-arylated phenols. In a related strategy, Larrosa and co-workers reported the domino oxidation/C–H arylation/protodecarboxylation reaction of salicylaldehydes. Investigation on the nature of this transformation revealed the implication of palladium in the oxidation step indicating a plausible Wacker-type process.45
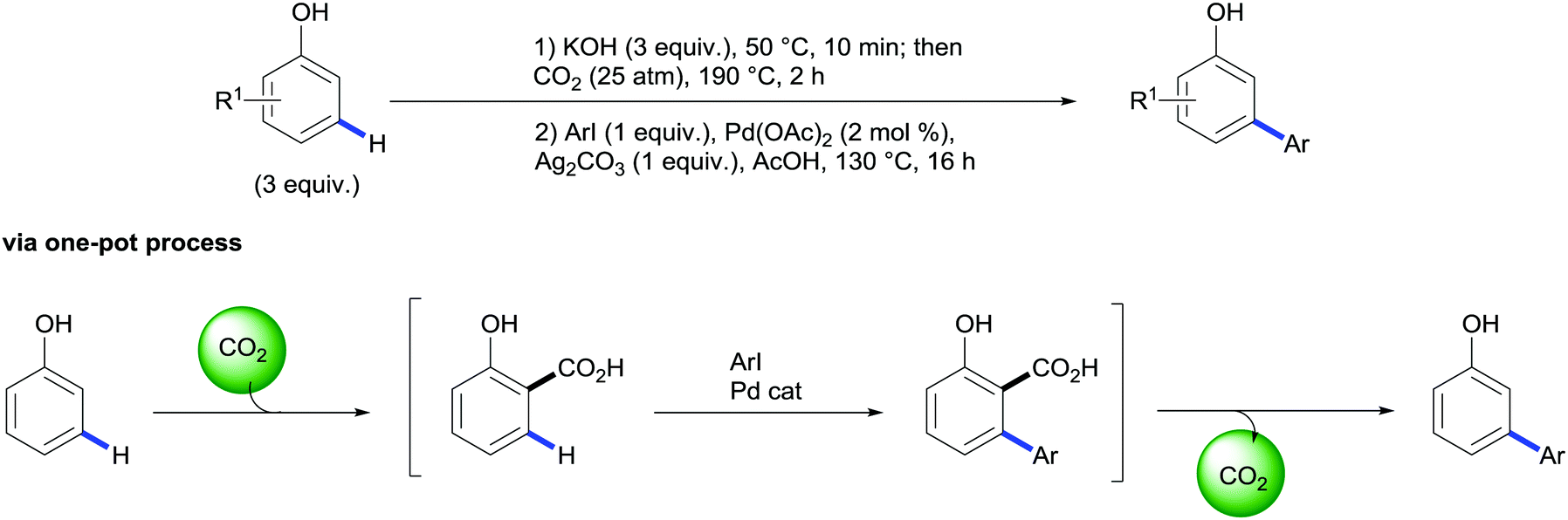 | ||
| Scheme 22 Synthesis of m-arylated phenols via one-pot sequential carboxylation followed by palladium-catalysed C–H bond arylation/decarboxylation of phenols. | ||
Very recently the ortho C–H bond arylation of benzoic acids and heteroaromatic carboxylic acids has been reported to take place under ruthenium catalysis by the groups of Ackermann,46 Gooßen,47 Larrosa48 and Weix.49 Gooßen's methodology also described that the addition of a copper catalyst and raising the reaction temperature up to 190 °C triggered the concomitant protodecarboxylation affording two examples of meta-substituted biaryls (Scheme 23).
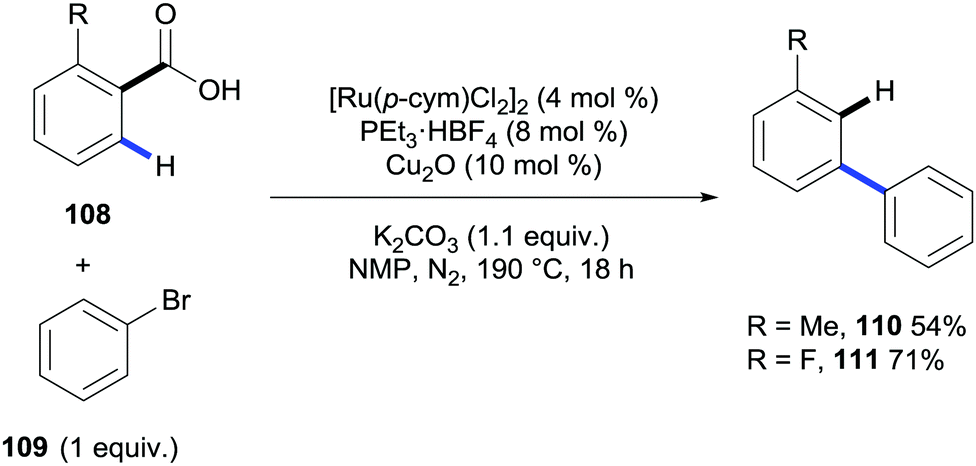 | ||
| Scheme 23 Ruthenium- and copper-catalysed system for the arylation/decarboxylation of benzoic acids. | ||
The Su and the You groups published in 2015 the rhodium-catalysed oxidative C2-(hetero)arylation of heteroaromatic moieties (i.e. (benzo)thiophenes, (benzo)furans, N-protected indoles, etc.) with aromatic carboxylic acids in the presence of a silver salt (Scheme 24).50,51 Forcing temperatures were needed when 2,4-dimethoxybenzoic acid is replaced for a less electron rich coupling partner, presumably to fully trigger the protodecarboxylation event.
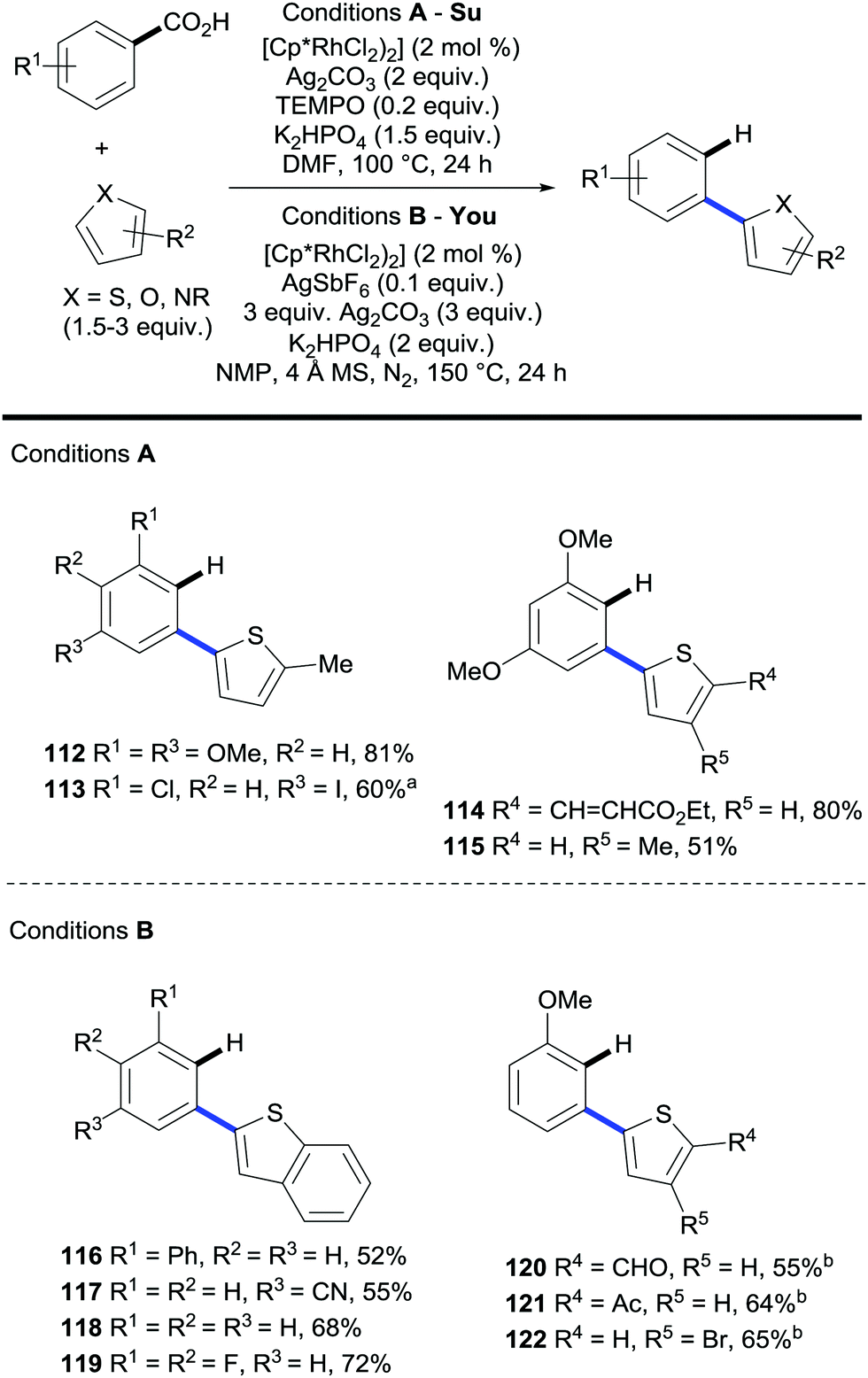 | ||
| Scheme 24 Rhodium-catalysed C2-arylation of thiophenes. a Reaction conditions: [(Cp*RhCl2)2] (4 mol%), Ag2CO3 (3 equiv.), Li2CO3 (1.5 equiv.), TEMPO (0.2 equiv.), DMF, 100 °C, 24 h, then 140 °C, 6 h. b AgSbF6 (0.2 equiv.), 48 h. | ||
Remarkably, these protocols involve double C–H activation processes obviating the need for prefunctionalised starting materials as aryl donors although large amounts of silver are needed. Also, they describe the regioselective cross-coupling between two heteroaromatic moieties, transformations that have traditionally proven to be very challenging.
The applicability of this strategy is not restricted to the formation of C–C bonds but C–heteroatom bond-forming protocols have recently started to flourish. Gooßen and co-workers developed the alkoxylation of potassium aryl benzoates with concomitant protodecarboxylation mediated by a copper/silver system and using trialkylborates as the alkoxide source (Scheme 25).52
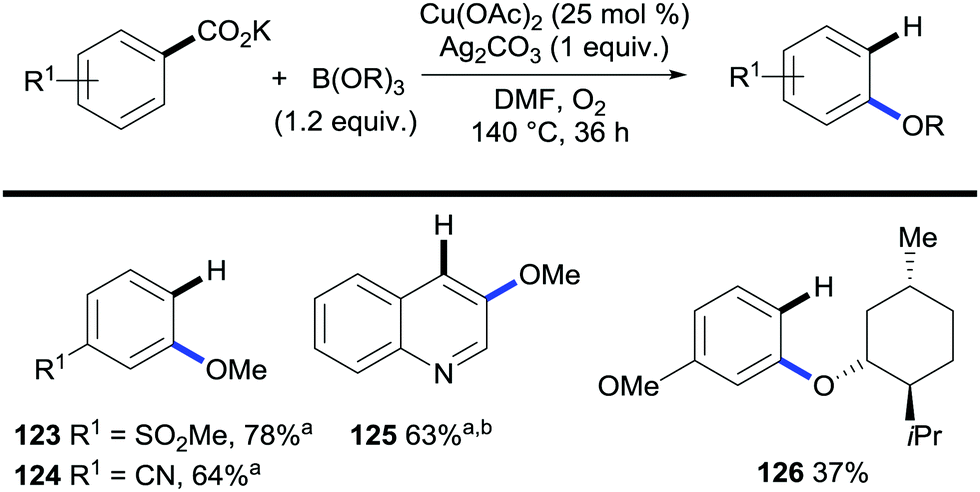 | ||
| Scheme 25 Copper-mediated C–H bond alkoxylation/decarboxylation of potassium aryl benzoates. a B(OR)3 (5 equiv.). b Quinoline (0.19 equiv.). | ||
N-Aryl benzamides have also been accessed through amidation/decarboxylation of benzoic acids under rhodium catalysis with isocyanates (Scheme 26).53 This method uses catalytic amounts of Cu2O to promote protodecarboxylation of the amidation product. However, the authors showed that in its absence the desired decarboxylated product is obtained and although lower yields were exhibited, this showed the ability of rhodium to promote protodecarboxylation. The authors subsequently reported that using very similar conditions the rhodium catalyst can be replaced by ruthenium thus eliminating the necessity of copper for the decarboxylation step.54
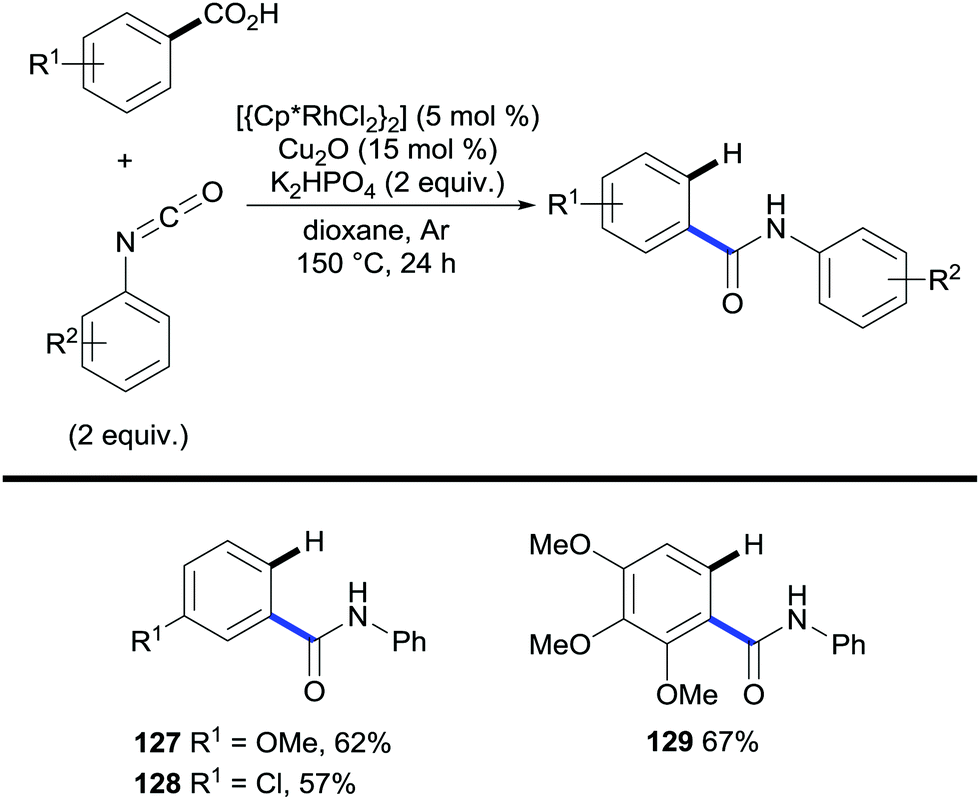 | ||
| Scheme 26 Rhodium-catalysed C–H bond amidation/decarboxylation of benzoic acids. | ||
Remarkably, the use of carboxylic acids as traceless directing groups has also inspired researchers in other fields to develop unprecedented transformations. In this regard, a series of 4-B-alkenylated o-carborane derivatives have been accessed regioselectively for the first time from their parent o-carborane-1-carboxylic acid via an iridium-catalysed carboxylate-directed B[4]–H alkenylation/protodecarboxylation sequence (Scheme 27).55
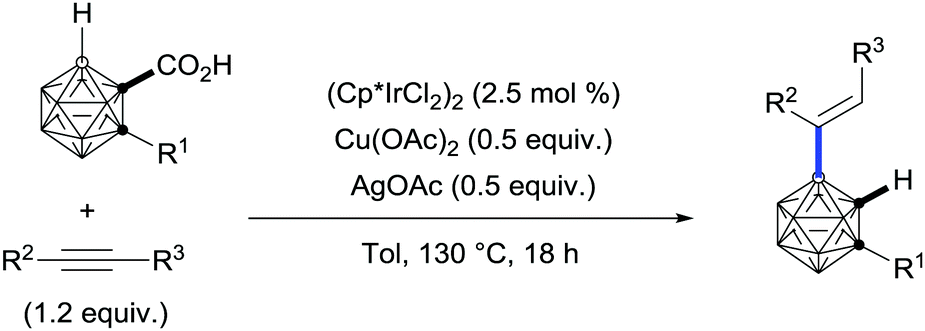 | ||
| Scheme 27 Iridium-catalysed B–H bond alkenylation/decarboxylation of o-carborane benzoic acids. | ||
Functionalisation/decarboxylation reaction involving one metals and one step
In these processes both the C–H functionalization and the protodecarboxylation steps are carried out by the same transition metal, thus avoiding the need for catalyst compatibility and/or additional reaction steps.The first reported method to find the appropriate choice of catalyst for such reactions dates back to 2012. Lee and co-workers, in a similar protocol to Miura's vinylations of heteroaromatic carboxylic acids discussed in the previous section (Schemes 16 and 17),35,38 designed and prepared a carbene-based Pd(0) catalyst (cat3, Scheme 28) which was applied to both the arylation/decarboxylation and decarboxylative arylation of various heteroaromatic systems using aryl bromides and aryl chlorides as the coupling partners. The authors showed that C2-arylation/decarboxylation of N-alkylindole-3-carboxylic acid occurred whilst C2-decarboxylative arylation took place when N-alkylindole-2-carboxylic acid was the substrate of choice (Scheme 28). More recently, ruthenium has proven to be an exceptional catalyst at promoting protodecarboxylation events after ortho-C–H bond functionalisation reactions.
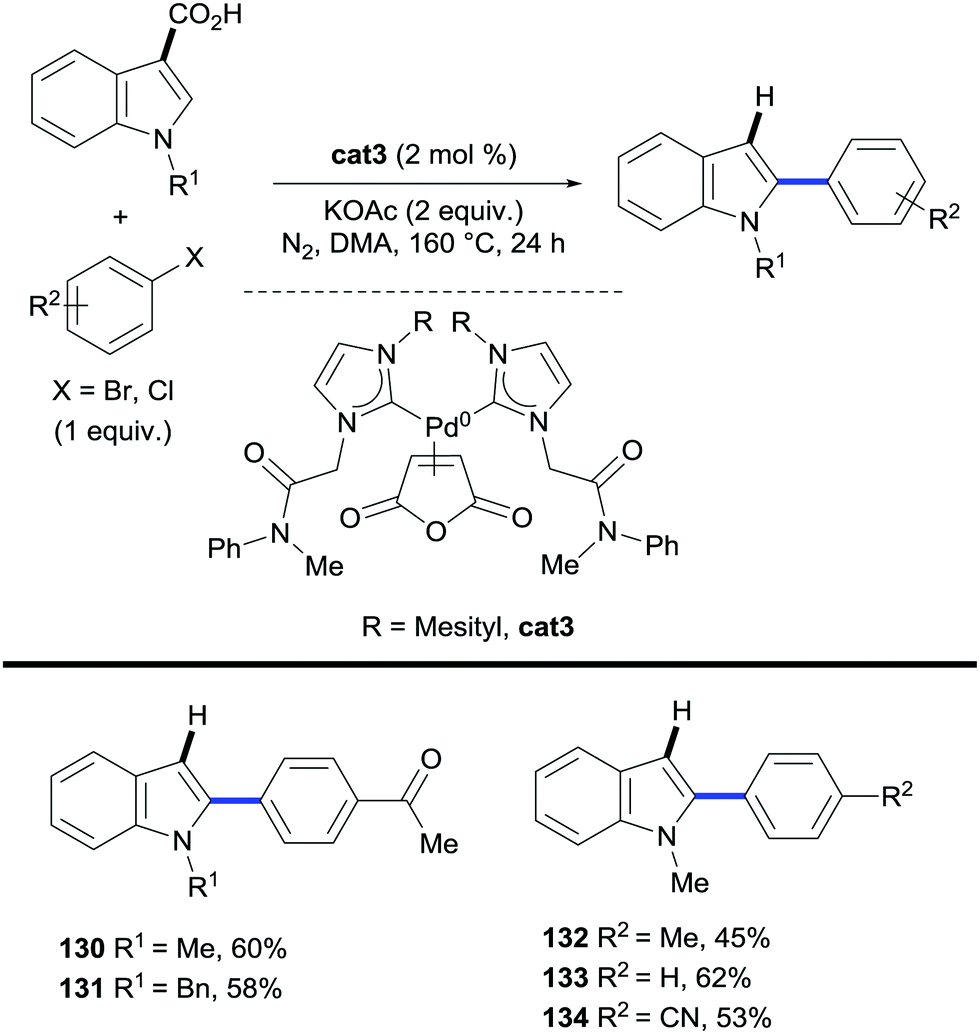 | ||
| Scheme 28 Palladium-catalysed C2-arylation/decarboxylation of N-alkylindole-3-carboxylic acids. | ||
Further to the ruthenium-catalysed amidation/decarboxylation discussed in the previous section,54 the Ackermann,56 Gooßen,57,58 Hartwig and Zhao59 laboratories have independently demonstrated the ability of ruthenium to catalyse C–H bond olefination/decarboxylation sequences to yield alkenylarenes under remarkably mild conditions.60
These articles feature similar redox-neutral processes via stereoselective syn-hydroarylation reactions employing biaryl alkynes and aryl–alkyl alkynes as coupling partners (Scheme 29).
 | ||
Scheme 29 Ruthenium-catalysed C–H bond alkenylation/decarboxylation of benzoic acids via hydroarylation of alkynes. E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z ratios shown in parenthesis. :Z ratios shown in parenthesis. | ||
In addition, Ackermann also reports that acrylates can engage in oxidative C–H bond alkenylation/decarboxylation reactions (Scheme 30). Importantly, these methods provide routes to access both meta- and para-alkenylarenes depending on the substitution pattern on the parent benzoic acid.
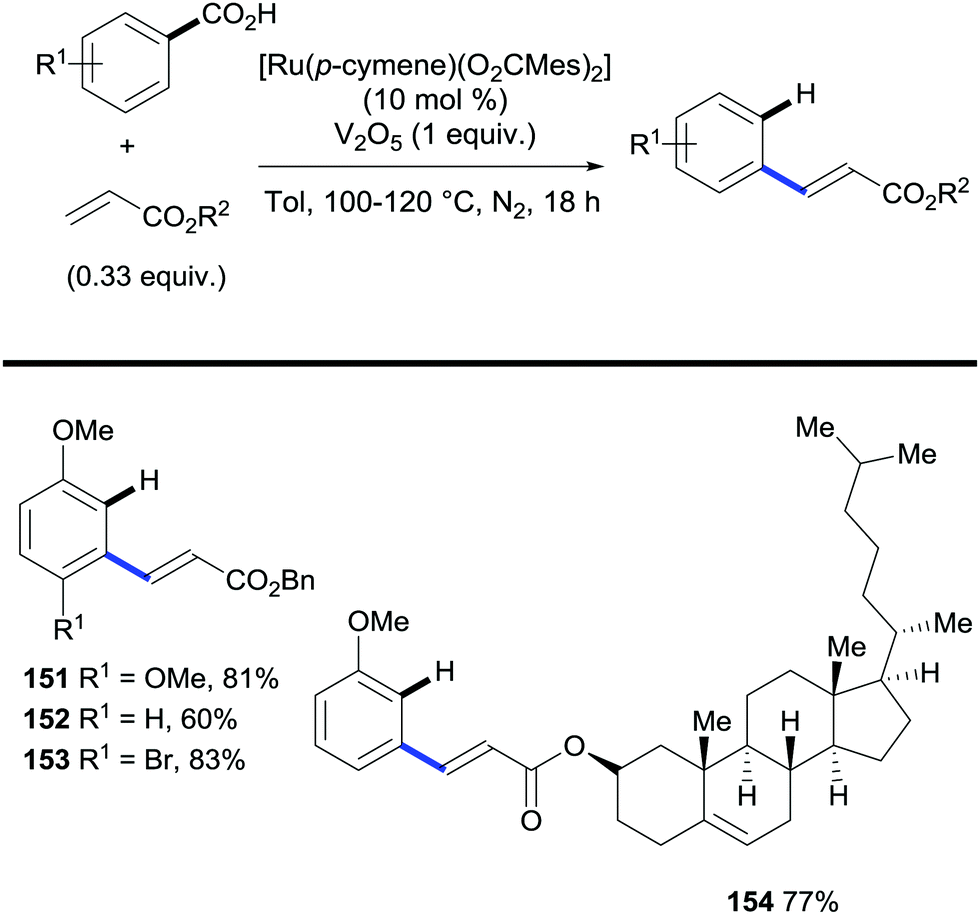 | ||
| Scheme 30 Ruthenium-catalysed C–H bond alkenylation/decarboxylation of benzoic acids via oxidative alkenylation. | ||
Opposite to most methods, these reactions do not operate through a functionalisation/protodecarboxylation sequence. This was demonstrated by submitting the alkenylated benzoic acid product to the standard reaction conditions; this afforded no protodecarboxylation product (Scheme 31A). Thus, the decarboxylation takes place simultaneously to the alkenylation reaction. The authors proposed the mechanism depicted in (Scheme 30B) that is initiated by carboxylate-directed ruthenium C–H bond metalation to generate intermediate II. Subsequently the alkyne moiety is inserted into the C–Ru bond forming intermediate species III that upon extrusion of CO2 releases the product and regenerates the ruthenium catalyst that restarts the catalytic cycle. This mechanism completely prevents a second alkenylation at the other ortho position of the carboxylate.
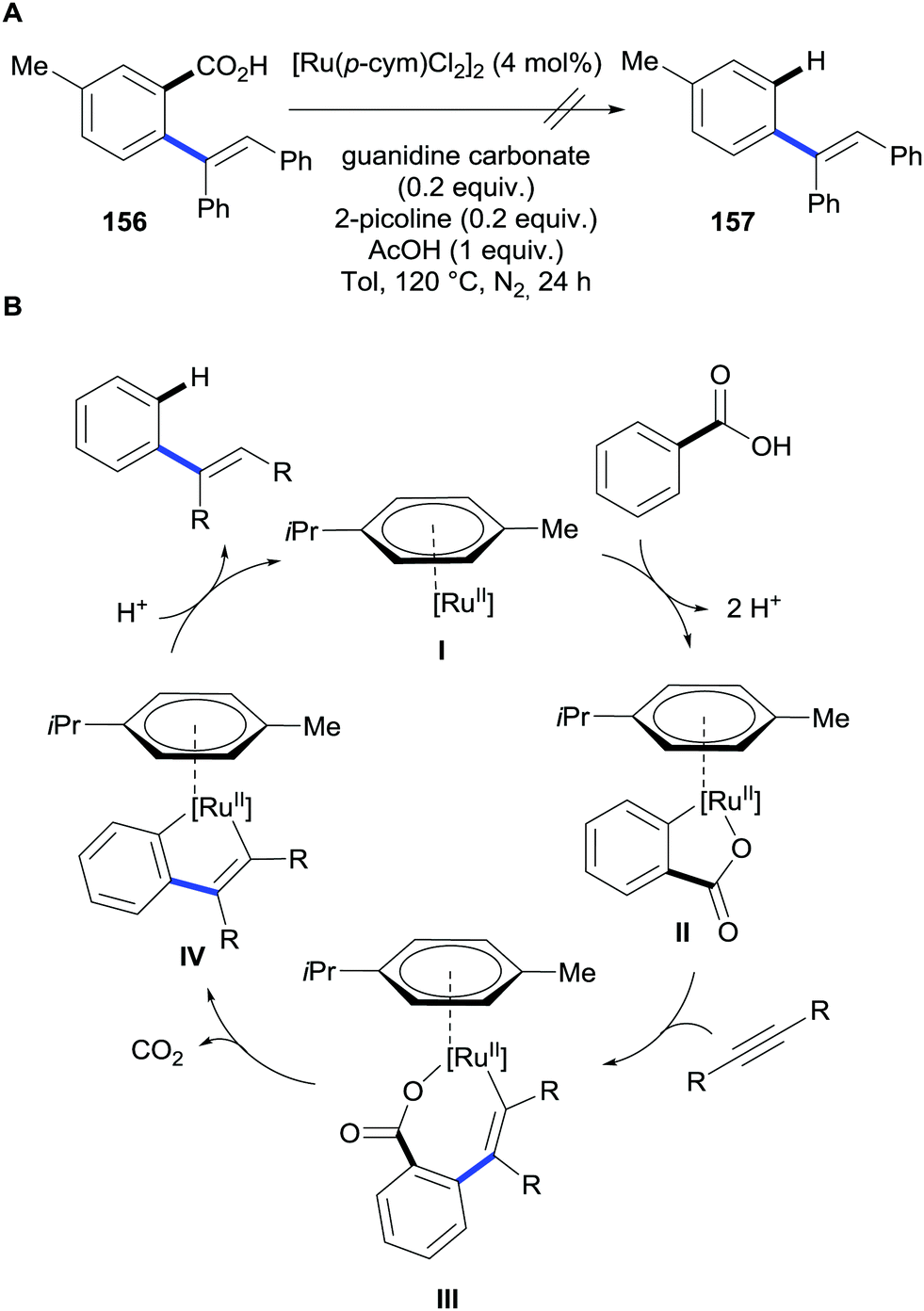 | ||
| Scheme 31 (A) Attempts for the ruthenium-catalysed decarboxylation of alkenyl benzoic acids. (B) Proposed mechanism for the C–H bond ruthenium-catalysed alkenylation/decarboxylation of benzoic acids via hydroarylation of alkynes. | ||
Conclusions
In a field largely dominated by non-removable directing groups, the use of carboxylic acids as traceless directing groups for C–H functionalisation has emerged as a potent new concept in synthetic organic chemistry. By putting this strategy to use, not only new routes for accessing a wide variety of challenging substitution patterns have been discovered but new reactivities have been unveiled that may influence complimentary transformations. These methodologies are especially valuable due to the fact that carboxylic acids are cheap and readily available. During the last decade this strategy has witnessed major advances; from methods that needed two steps promoted by two different transition metals and purification of the material between steps to protocols using a catalyst able to promote tandem functionalisation/decarboxylation sequences on its own. However, the possibilities encompassed by this strategy are immense and only a few classes of transformations on a small range of substrates have been explored to date (Scheme 32). The field of traceless directing groups needs to evolve towards broadening the reactivity allowed by such methods, and in line with the latest developments in the field, accelerating the synthetic routes towards target molecules bearing challenging substitution patterns in a more sustainable manner.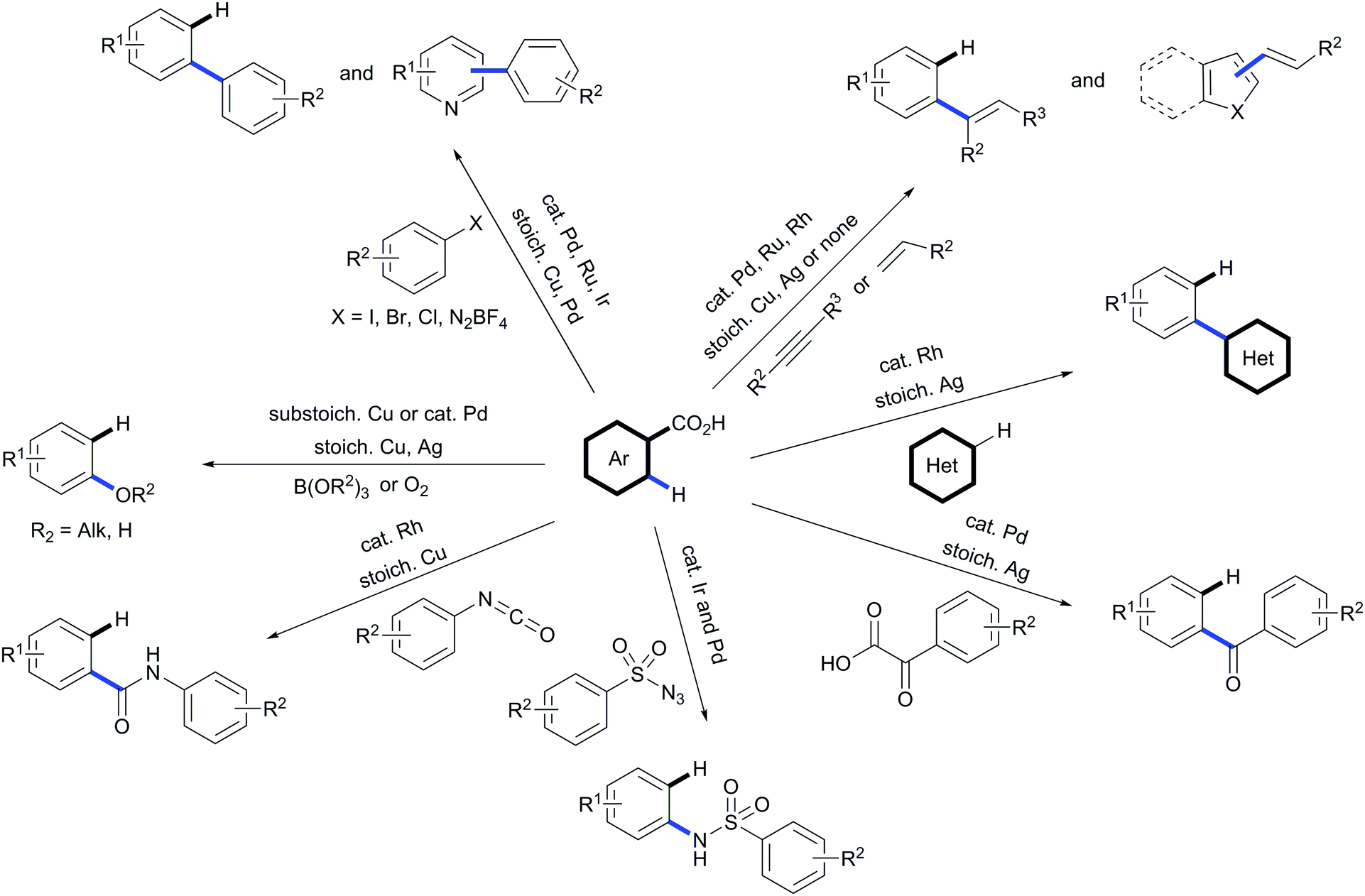 | ||
| Scheme 32 Scope of available transformations by using carboxylic acids as traceless directing groups. | ||
Acknowledgements
We gratefully acknowledge the Engineering and Physical Sciences Research Council (EPSRC, EP/L014017/2) and the Marie Skłodowska Curie actions (IF to MF) for funding.Notes and references
- L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed.
- X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed.
- R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC.
- J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC.
- O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902–4911 RSC.
- D.-G. Yu, B.-J. Li and Z.-J. Shi, Tetrahedron, 2012, 5130–5136 CrossRef CAS.
- M. Zhang, Y. Zhang, X. Jie, H. Zhao, G. Li and W. Su, Org. Chem. Front., 2014, 1, 843–895 RSC.
- Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC.
- G. Rousseau and B. Breit, Angew. Chem., Int. Ed., 2011, 50, 2450–2494 CrossRef CAS PubMed.
- H. Sun, N. Guimond and Y. Huang, Org. Biomol. Chem., 2016, 14, 8389–8397 CAS.
- Y. Wei, P. Hu, M. Zang and W. Su, Chem. Rev. DOI:10.1021/acs.chemrev.6b00516.
- M. Pichette Drapeau and L. J. Gooßen, Chem. – Eur. J., 2016, 22, 18654–18677 CrossRef CAS PubMed.
- I. I. Harrison and S. Harrison, Compendium of Organic Synthetic Methods, John Wiley & Sons, Inc, Hoboken, NJ, 1971, pp. 16–74 Search PubMed.
- M. Brill, F. Lazreg, C. S. J. Cazin and S. P. Nolan, Carbon Dioxide in Organometallics, Springer International Publishing, 2015, pp. 225–278 Search PubMed.
- L. J. Goossen, N. Rodríguez and K. Goossen, Angew. Chem., Int. Ed., 2008, 47, 3100–3120 CrossRef CAS PubMed.
- J. Cornella and I. Larrosa, Synthesis, 2012, 653–676 CAS.
- L. J. Gooßen, W. R. Thiel, N. Rodríguez, C. Linder and B. Melzer, Adv. Synth. Catal., 2007, 349, 2241–2246 CrossRef.
- Z. Li, Z. Fu, H. Zhang, J. Long, Y. Song and H. Cai, New J. Chem., 2016, 40, 3014–3018 RSC.
- L. J. Goossen, F. Manjolinho, B. A. Khan and N. Rodríguez, J. Org. Chem., 2009, 74, 2620–2623 CrossRef CAS PubMed.
- L. Xue, W. Su and Z. Lin, Dalton Trans., 2011, 40, 11926–11936 RSC.
- L. J. Gooßen, N. Rodríguez, C. Linder, P. P. Lange and A. Fromm, ChemCatChem, 2010, 2, 430–442 CrossRef.
- L. J. Gooßen, C. Linder, N. Rodríguez, P. P. Lange and A. Fromm, Chem. Commun., 2009, 7173–7175 RSC.
- J. Cornella, C. Sanchez, D. Banawa and I. Larrosa, Chem. Commun., 2009, 7176–7178 RSC.
- P. Lu, C. Sanchez, J. Cornella and I. Larrosa, Org. Lett., 2009, 11, 5710–5713 CrossRef CAS PubMed.
- R. Grainger, J. Cornella, D. C. Blakemore, I. Larrosa and J. M. Campanera, Chem. – Eur. J., 2014, 20, 16680–16687 CrossRef CAS PubMed.
- J. S. Dickstein, C. A. Mulrooney, E. M. O’Brien, B. J. Morgan and M. C. Kozlowski, Org. Lett., 2007, 9, 2441–2444 CrossRef CAS PubMed.
- Z.-M. Sun, J. Zhang and P. Zhao, Org. Lett., 2010, 12, 992–995 CrossRef CAS PubMed.
- H. A. Chiong, Q.-N. Pham and O. Daugulis, J. Am. Chem. Soc., 2007, 129, 9879–9884 CrossRef CAS PubMed.
- L. Huang, D. Hackenberger and L. J. Gooßen, Angew. Chem., Int. Ed., 2015, 54, 12607–12611 CrossRef CAS PubMed.
- S. Pan, B. Zhou, Y. Zhang, C. Shao and G. Shi, Synlett, 2015, 277–281 Search PubMed.
- C. Arroniz, A. Ironmonger, G. Rassias and I. Larrosa, Org. Lett., 2013, 15, 910–913 CrossRef CAS PubMed.
- J. Miao and H. Ge, Org. Lett., 2013, 15, 2930–2933 CrossRef CAS PubMed.
- Y.-H. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 14654–14655 CrossRef CAS PubMed.
- S. Mochida, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2010, 4–7 Search PubMed.
- S. Mochida, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2011, 76, 3024–3033 CrossRef CAS PubMed.
- D. Lee and S. Chang, Chem. – Eur. J., 2015, 21, 5364–5368 CrossRef CAS PubMed.
- A. J. S. Johnston, K. B. Ling, D. Sale, N. Lebrasseur and I. Larrosa, Org. Lett., 2016, 18, 6094–6097 CrossRef CAS PubMed.
- A. Maehara, H. Tsurugi, T. Satoh and M. Miura, Org. Lett., 2008, 10, 1159–1162 CrossRef CAS PubMed.
- S. Agasti, A. Dey and D. Maiti, Chem. Commun., 2016, 52, 12191–12194 RSC.
- K. Kim, D. Vasu, H. Im and S. Hong, Angew. Chem., Int. Ed., 2016, 55, 8652–8655 CrossRef CAS PubMed.
- J. Cornella, M. Righi and I. Larrosa, Angew. Chem., Int. Ed., 2011, 50, 9429–9432 CrossRef CAS PubMed.
- J. Luo, S. Preciado and I. Larrosa, Chem. Commun., 2015, 51, 3127–3130 RSC.
- C. Arroniz, J. G. Denis, A. Ironmonger, G. Rassias and I. Larrosa, Chem. Sci., 2014, 5, 3509–3514 RSC.
- J. Luo, S. Preciado and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 4109–4112 CrossRef CAS PubMed.
- J. Luo, S. Preciado, S. O. Araromi and I. Larrosa, Chem. – Asian J., 2016, 11, 347–350 CrossRef CAS PubMed.
- R. Mei, C. Zhu and L. Ackermann, Chem. Commun., 2016, 52, 13171–13174 RSC.
- A. Biafora, T. Krause, D. Hackenberger, F. Belitz and L. J. Gooßen, Angew. Chem., Int. Ed., 2016, 55, 14752–14755 CrossRef CAS PubMed.
- M. Simonetti, D. M. Cannas, A. Panigrahi, S. Kujawa, M. Kryjewski, P. Xie and I. Larrosa, Chem. – Eur. J., 2017, 23, 549–553 CrossRef CAS PubMed.
- L. Huang and D. J. Weix, Org. Lett., 2016, 18, 5432–5435 CrossRef CAS PubMed.
- Y. Zhang, H. Zhao, M. Zhang and W. Su, Angew. Chem., Int. Ed., 2015, 54, 3817–3821 CrossRef CAS PubMed.
- X. Qin, D. Sun, Q. You, Y. Cheng, J. Lan and J. You, Org. Lett., 2015, 17, 1762–1765 CrossRef CAS PubMed.
- S. Bhadra, W. I. Dzik and L. J. Gooßen, Angew. Chem., Int. Ed., 2013, 52, 2959–2962 CrossRef CAS PubMed.
- X.-Y. Shi, K.-Y. Liu, J. Fan, X.-F. Dong, J.-F. Wei and C.-J. Li, Chem. – Eur. J., 2015, 21, 1900–1903 CrossRef CAS PubMed.
- X.-Y. Shi, X.-F. Dong, J. Fan, K.-Y. Liu, J.-F. Wei and C.-J. Li, Sci. China: Chem., 2015, 58, 1286–1291 CrossRef CAS.
- Y. Quan and Z. Xie, J. Am. Chem. Soc., 2014, 136, 15513–15516 CrossRef CAS PubMed.
- N. Y. P. Kumar, A. Bechtoldt, K. Raghuvanshi and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 6929–6932 CrossRef CAS PubMed.
- L. Huang, A. Biafora, G. Zhang, V. Bragoni and L. J. Gooßen, Angew. Chem., Int. Ed., 2016, 55, 6933–6937 CrossRef CAS PubMed.
- A. Biafora, B. A. Khan, J. Bahri, J. M. Hewer and L. J. Gooßen, Org. Lett., 2017, 19, 1232–1235 CrossRef CAS PubMed.
- J. Zhang, R. Shrestha, J. F. Hartwig and P. Zhao, Nat. Chem., 2016, 8, 1144–1151 CrossRef CAS PubMed.
- M. Simonetti and I. Larrosa, Nat. Chem., 2016, 8, 1086–1088 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |