
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of 2D covalent organic frameworks by taking advantage of the variable orientation of imine bonds†
Shun-Qi
Xu‡
,
Rong-Ran
Liang‡
 ,
Tian-Guang
Zhan
,
Qiao-Yan
Qi
and
Xin
Zhao
*
,
Tian-Guang
Zhan
,
Qiao-Yan
Qi
and
Xin
Zhao
*
CAS Key Laboratory of Synthetic and Self-assembly Chemistry for Organic Functional Molecules, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: xzhao@sioc.ac.cn
First published on 30th January 2017
Abstract
A model system has been established to construct two-dimensional (2D) covalent organic frameworks (COFs) by taking advantage of the variable orientation of imine bonds. During the assembly process, the imine bonds adopt an unprecedented heterodromous orientation to facilitate the formation of the COFs.
Covalent organic frameworks (COFs), a class of periodic two-dimensional (2D) and three-dimensional (3D) crystalline organic porous materials, have drawn considerable attention due to their important applications in many fields, including gas storage,1 separation,2 catalysis,3 sensing,4 energy storage,5 and delivery,6 just like their analogs, metal organic frameworks (MOFs).7 Since the first two COFs reported by O. M. Yaghi and co-workers in 2005,8 a variety of COFs have been designed and constructed over the past 11 years.9 The design strategy for the construction of COFs includes two key points. The first one is the symmetries of building blocks, which dictate the topological structures of pores in COFs. For example, while a COF with hexagonal pores can be fabricated by the combination of a C3-symmetrical building block and a C2-symmetrical building block, the condensation of a C4-symmetrical monomer and a C2-symmetrical monomer leads to the formation of a COF with tetragonal pores. The second key point is the type of bonds (also known as linkages) which connect the building blocks. The typical reactions used for constructing COFs include boronic acid trimerization, boronate ester formation, Schiff base reaction and nitrile trimerization (high temperature required).9 Among them, an imine bond formed from the Schiff base reaction is one of the most popular linkages.10 A notable feature of the imine bond (and also hydrazone bond) is its variable orientation which theoretically can result in a homodromous or a heterodromous orientation of C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N linkages in COFs. Such a unique feature should endow the CN bond with a great advantage over the other bonds by providing variable direction of bond formation, which greatly facilitates the assembly of well-ordered frameworks from building blocks. However, so far, the usefulness of the variable orientation of CN bond has never been demonstrated for the construction of COFs. It could be attributed to the lack of a model suitable for conducting such an investigation. In this work, we report the first example of the construction of COFs by taking advantage of the variable orientation of the imine bond.
N linkages in COFs. Such a unique feature should endow the CN bond with a great advantage over the other bonds by providing variable direction of bond formation, which greatly facilitates the assembly of well-ordered frameworks from building blocks. However, so far, the usefulness of the variable orientation of CN bond has never been demonstrated for the construction of COFs. It could be attributed to the lack of a model suitable for conducting such an investigation. In this work, we report the first example of the construction of COFs by taking advantage of the variable orientation of the imine bond.
To demonstrate the crucial role that the variable orientation of the imine bond can play in the construction of COFs, a model system has been established by the combination of two C3-symmetrical monomers. While one monomer possesses three aldehyde groups located at the ends of the three branches (NTBA and NTBCA), the other peripherally bears six amine groups (HAT-6NH2)11 (Scheme 1). As can be seen in Scheme 1, in order to facilitate the formation of an extended two-dimensional framework with a periodic structure, the CN bonds in the COF must adopt a heterodromous orientation, that is, alternately clockwise and counterclockwise. In contrast, a homodromous orientation of imine bonds would result in incomplete bond formation (as indicated by the blue circles in Scheme 1) and thus lead to the formation of an amorphous polyimine with a disordered internal structure. It is totally different from the COFs fabricated from the combination of C6-symmetric and C3-symmetric monomers or the combination of two common C3-symmetric monomers, in which the CN linkages adopt a homodromous orientation (see Scheme S1 in the ESI† for comparison). Furthermore, it can also be clearly found that, if dynamic covalent bonds other than CN were used for the model system described in Scheme 1, no COFs could be generated, as a result of fixed orientations of those bonds.
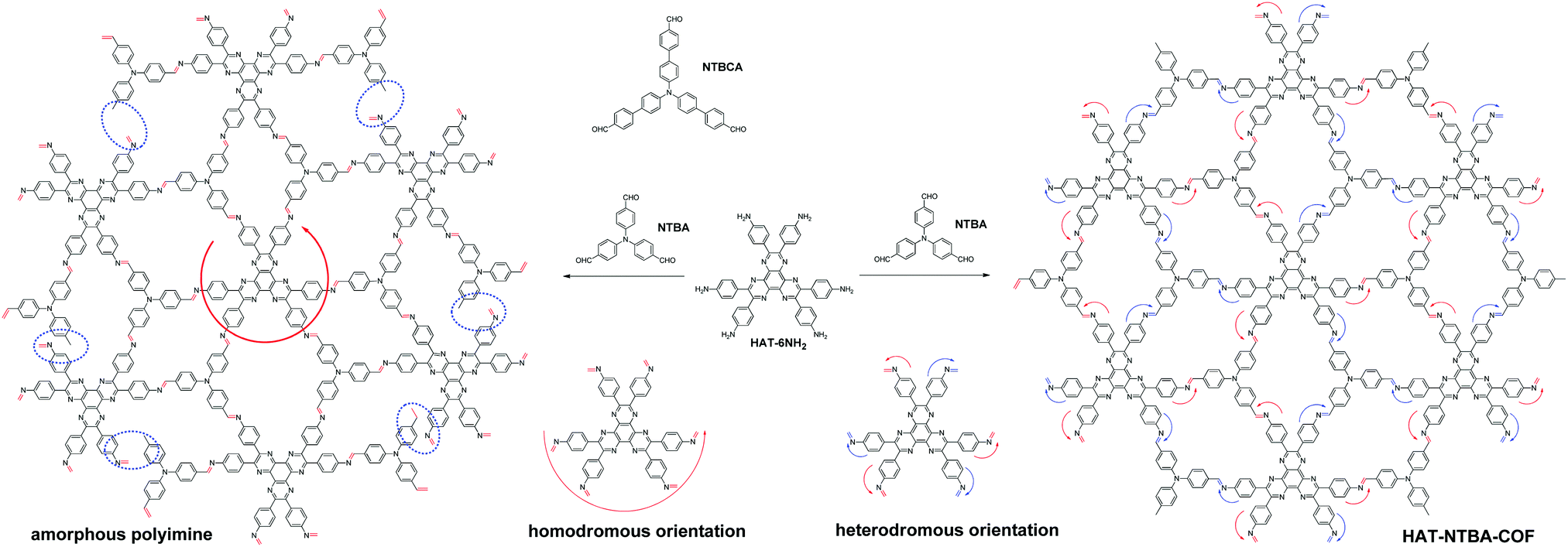 | ||
| Scheme 1 Construction of COFs by taking advantage of the variable orientation of CN linkages (HAT-NTBA-COF as a representative). | ||
A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (molar ratio) mixture of HAT-6NH2 and 4,4′,4′′-nitrilotribenzaldehyde (NTBA) in dimethylacetamide–mesitylene–acetic acid (aq., 6 M) (5/5/1, v/v/v) ternary solvent in a sealed glass ampoule was heated at 120 °C for 72 h, which afforded a yellow powder (HAT-NTBA-COF). Under the same conditions, another COF, called HAT-NTBCA-COF, was prepared from the condensation of HAT-6NH2 and 4′,4′′′,4′′′′′-nitrilotris(([1,1′-biphenyl]-4-carbaldehyde)) (NTBCA). Both the COFs are insoluble in common organic solvents and show high thermal stabilities (Fig. S1, ESI†). Fourier transform infrared spectroscopy (FTIR) was carried out to characterize the as-obtained powders (Fig. S2 and S3, ESI†). The band around 3337 cm−1, which was attributed to the vibrations of the –NH2 groups of HAT-6NH2, attenuated dramatically in the IR spectra of the COFs, indicating the high polymerization degree of the condensation reactions. Similarly, compared with the starting monomers NTBA and NTBCA, the intensity of CO stretching vibrations decreased significantly in the two COFs, also indicating the high polymerization degree. However, the peak corresponding to the vibrations of the newly formed imine bonds could not be distinguished from the band of aromatic CN in the HAT core. Both of them appear around 1600 cm−1. Solid-state cross-polarization with magic angle spinning (CP/MAS) 13C NMR spectroscopy was also performed for the two COFs. The resonance signals at 159 ppm in the spectrum of HAT-NTBA-COF and 157 ppm in the spectrum of HAT-NTBCA-COF could be attributed to the chemical shifts of the carbons in the CN bonds (Fig. S4 and S5, ESI†). Scanning electron microscopy (SEM) revealed spherical morphology for both the COFs (Fig. S6, ESI†).
:2 (molar ratio) mixture of HAT-6NH2 and 4,4′,4′′-nitrilotribenzaldehyde (NTBA) in dimethylacetamide–mesitylene–acetic acid (aq., 6 M) (5/5/1, v/v/v) ternary solvent in a sealed glass ampoule was heated at 120 °C for 72 h, which afforded a yellow powder (HAT-NTBA-COF). Under the same conditions, another COF, called HAT-NTBCA-COF, was prepared from the condensation of HAT-6NH2 and 4′,4′′′,4′′′′′-nitrilotris(([1,1′-biphenyl]-4-carbaldehyde)) (NTBCA). Both the COFs are insoluble in common organic solvents and show high thermal stabilities (Fig. S1, ESI†). Fourier transform infrared spectroscopy (FTIR) was carried out to characterize the as-obtained powders (Fig. S2 and S3, ESI†). The band around 3337 cm−1, which was attributed to the vibrations of the –NH2 groups of HAT-6NH2, attenuated dramatically in the IR spectra of the COFs, indicating the high polymerization degree of the condensation reactions. Similarly, compared with the starting monomers NTBA and NTBCA, the intensity of CO stretching vibrations decreased significantly in the two COFs, also indicating the high polymerization degree. However, the peak corresponding to the vibrations of the newly formed imine bonds could not be distinguished from the band of aromatic CN in the HAT core. Both of them appear around 1600 cm−1. Solid-state cross-polarization with magic angle spinning (CP/MAS) 13C NMR spectroscopy was also performed for the two COFs. The resonance signals at 159 ppm in the spectrum of HAT-NTBA-COF and 157 ppm in the spectrum of HAT-NTBCA-COF could be attributed to the chemical shifts of the carbons in the CN bonds (Fig. S4 and S5, ESI†). Scanning electron microscopy (SEM) revealed spherical morphology for both the COFs (Fig. S6, ESI†).
It should be noted that the orientations of the CN linkages in COFs are extremely hard to be directly observed by the available techniques. However, they can be deduced from the structures of the polymeric products. As stated above, in order to produce COFs from condensation reactions between HAT-6NH2 and the trialdehydes, a heterodromous orientation of CN linkages must be adopted; otherwise, amorphous polyimines with disordered internal structures will be generated. Powder X-ray diffraction (PXRD) patterns of the as-obtained powders were recorded. Their PXRD patterns displayed intense diffraction peaks, clearly indicating the formation of crystalline materials. In order to elucidate their crystal structures, the experimental PXRD patterns were compared with the simulated PXRD patterns generated by using the Accelarys Materials Studio 7.0 software. In the simulations, two possible stacked models, namely eclipsed (AA) stacking and staggered (AB) stacking, were established using the compass II force-field implemented in the Forcite module after geometry optimizations. In the PXRD profile of HAT-NTBA-COF, diffraction peaks at 2θ = 3.87°, 6.63°, 13.50° and 14.09° were observed (Fig. 1a), which could be assignable to the (100), (110), (220) and (130) facets. It was found that its simulated PXRD pattern with the AA stacking model reproduced well the experimental PXRD pattern of HAT-NTBA-COF, indicating that the polymer prepared from HAT-6NH2 and NTBA holds the structure of the COF as illustrated in Scheme 1. The formation of the COF strongly suggests that the CN bonds in HAT-NTBA-COF do adopt a heterodromous orientation. Pawley refinement was performed and it yielded unit cell parameters of a = b = 25.83 Å, and c = 4.10 Å, and α = β = 90° and γ = 118°, with Rwp = 2.63% and Rp = 2.08%. As revealed by the difference plot (Fig. 1b), the refined PXRD profile matches the experimental profile quite well. It should be noted that the interlayer distance could not be obtained from the experimental PXRD data because the (001) peak was hard to identify in the broad region of 2θ = 18–24°. Instead, it was set to be 4.10 Å by force-field-based molecular mechanics calculations (Table S1 and Fig. S7, ESI†). The twisted conformation of the phenyl units in HAT-6NH2 results in weak interlayer interactions between the COF sheets and thus decreases the degree of order of stacking.
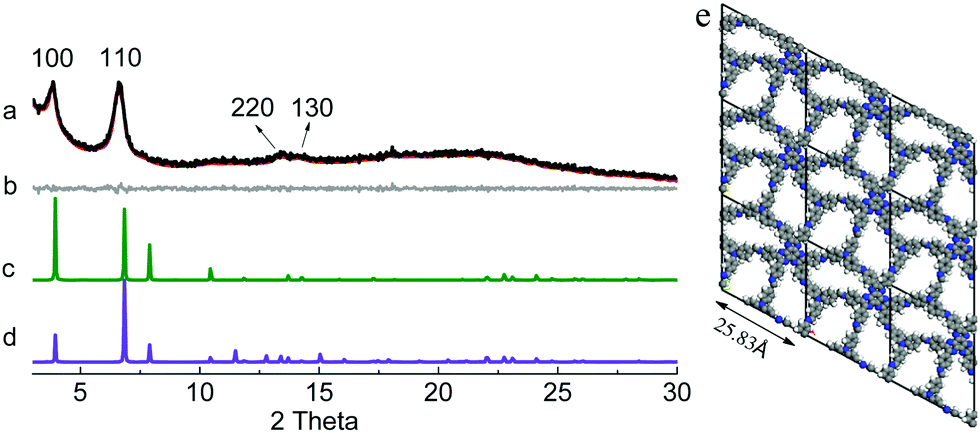 | ||
| Fig. 1 (a) Experimental (black) and refined (red) PXRD patterns of HAT-NTBA-COF, (b) difference plot between the experimental and refined PXRD patterns, and simulated PXRD patterns of the COF with (c) AA and (d) AB stacking, and (e) the unit cell structure of HAT-NTBA-COF in AA stacking. | ||
HAT-NTBCA-COF exhibited diffraction peaks at 3.03°, 5.26° and 6.04° (Fig. 2), corresponding to the (100), (110) and (200) facets, respectively. The comparison of the experimental PXRD pattern with the simulated ones also indicated that the as-prepared polymer from HAT-6NH2 and NTBCA possessed a COF structure similar to HAT-NTBA-COF, with AA stacking of the 2D layers. Pawley refinement gave rise to unit cell parameters of a = b = 33.03 Å, and c = 4.20 Å, and α = β = 90° and γ = 120°, with Rwp = 3.57% and Rp = 2.88%. And the PXRD pattern obtained through Pawley refinement matched well with its experimental pattern. The formation of crystalline HAT-NTBCA-COF demonstrates again that the variable orientation of the CN linkage has really played a crucial role in facilitating the formation of the COFs. Similar to that of HAT-NTBA-COF, its interlayer distance was also set by molecular mechanics calculations (Table S2 and Fig. S8, ESI†).
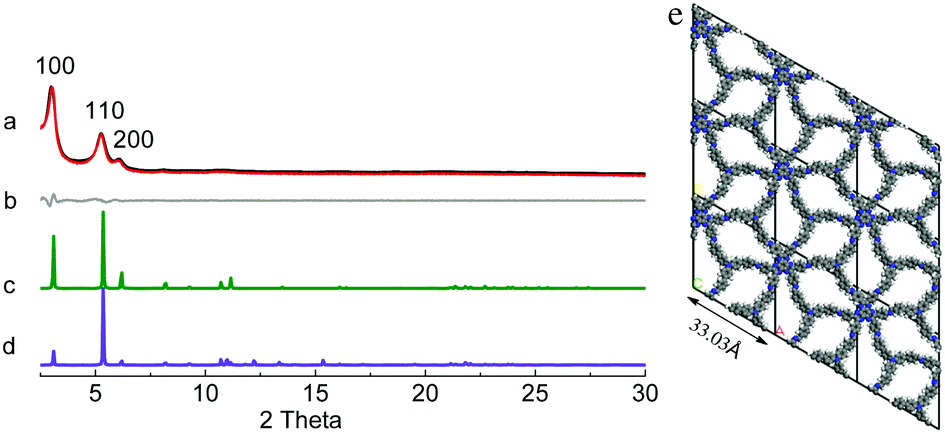 | ||
| Fig. 2 (a) Experimental (black) and refined (red) PXRD patterns of HAT-NTBCA-COF, (b) difference plot between the experimental and refined PXRD patterns, and simulated PXRD patterns of the COF with (c) AA and (d) AB stacking, and (e) the unit cell structure of HAT-NTBCA-COF in AA stacking. | ||
Nitrogen adsorption–desorption measurements were carried out for the two COFs at 77 K. Both the sorption curves display type I isotherm according to the IUPAC recommendation (Fig. 3),12 indicating the microporous feature of the COFs. Brunauer–Emmett–Teller (BET) surface areas were calculated to be 628.0 and 439.9 m2 g−1 for HAT-NTBA-COF and HAT-NTBCA-COF, respectively (Fig. S9 and S10, ESI†). And the total pore volume (at p/p0 = 0.99) of the former was estimated to be 0.68 cm3 g−1, while the latter had a value of 0.32 cm3 g−1. The pore size distributions of the two COFs were estimated using the nonlocal density functional theory (NLDFT), which exhibited main distributions centred around 9.5 Å for HAT-NTBA-COF and 12.7 Å for HAT-NTBCA-COF (Fig. 3). These values are close to the theoretical pore sizes estimated by PM3 calculations, which are 10.2 Å for HAT-NTBA-COF and 13.5 Å for HAT-NTBCA-COF. These results corroborate again the obtention of the COFs by taking advantage of the variable orientation of the CN bond. The CO2 storage capacities of the COFs have then been examined. Their CO2 adsorption isotherms were recorded at 273 K and 298 K, respectively (Fig. S11 and S12, ESI†). HAT-NTBA-COF exhibits 9.4 wt% and 5.3 wt% CO2 uptakes at 273 and 298 K, respectively. In the case of HAT-NTBCA-COF, CO2 uptakes of 6.0 wt% (273 K) and 3.7 wt% (298 K) are obtained.
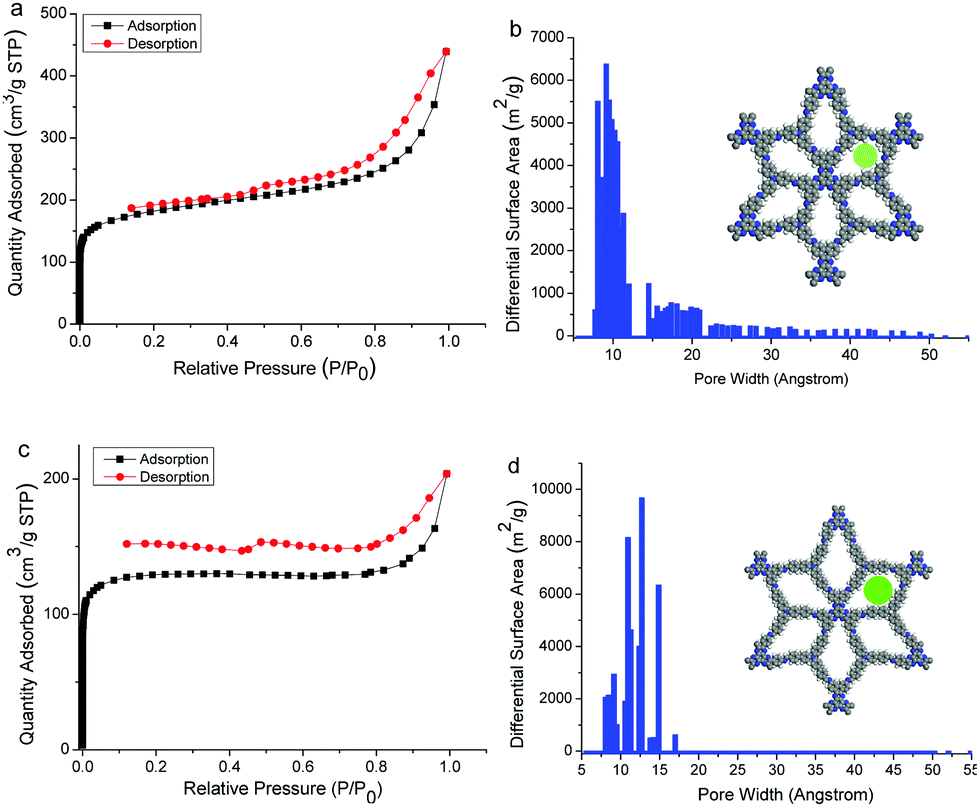 | ||
| Fig. 3 N2 adsorption–desorption isotherms (77 K) of (a) HAT-NTBA-COF and (c) HAT-NTBCA-COF, and pore size distribution profiles of (b) HAT-NTBA-COF and (d) HAT-NTBCA-COF. | ||
In conclusion, having a deep understanding of the principles of formation of COFs is crucial for their construction and development of their functions. In this work, a model system has been established by a reasonable design to demonstrate the construction of COFs by taking advantage of the variable orientation of the imine bond. This process has never been observed before due to the lack of a suitable model. On the basis of the established model, COFs bearing a heterodromous orientation of CN linkages have been fabricated for the first time, demonstrating the crucial role that the variable orientation of the CN bond can play in the construction of COFs. This work offers a new insight into the fundamental understanding of the formation of COFs, which should provide useful guidance to the design of COFs with novel structures and functions.
We thank the National Natural Science Foundation of China (No. 21632004) and the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB20020000) for financial support.
Notes and references
-
(a) P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450 CrossRef CAS PubMed
; (b) H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875 CrossRef CAS PubMed
-
(a) H. Oh, S. B. Kalidindi, Y. Um, S. Bureekaew, R. Schmid, R. A. Fischer and M. Hirscher, Angew. Chem., Int. Ed., 2013, 52, 13219 CrossRef CAS PubMed
-
(a) Q. Fang, S. Gu, J. Zheng, Z. Zhuang, S. Qiu and Y. Yan, Angew. Chem., Int. Ed., 2014, 53, 2878 CrossRef CAS PubMed
-
(a) S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 17310 CrossRef CAS PubMed
-
(a) F. Xu, H. Xu, X. Chen, D. Wu, Y. Wu, H. Liu, C. Gu, R. Fu and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 6814 CrossRef CAS PubMed
-
(a) L. Bai, S. Z. F. Phua, W. Q. Lim, A. Jana, Z. Luo, H. P. Tham, L. Zhao, Q. Gao and Y. Zhao, Chem. Commun., 2016, 52, 4128 RSC
-
(a) M.-S. Yao, W.-X. Tang, G.-E. Wang, B. Nath and G. Xu, Adv. Mater., 2016, 28, 5229 CrossRef CAS PubMed
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef PubMed
-
(a) P. J. Waller, F. Gándara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053 CrossRef CAS PubMed
- J. L. Segura, M. J. Mancheño and F. Zamora, Chem. Soc. Rev., 2016, 45, 5635 RSC
- S.-Q. Xu, T.-G. Zhan, Q. Wen, Z.-F. Pang and X. Zhao, ACS Macro Lett., 2016, 5, 99 CrossRef CAS
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquérol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Preparation of COFs, FTIR spectra, solid state 13C NMR, and BET plots. See DOI: 10.1039/c6cc09906h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |