
Promoted hydrogen release from alkali metal borohydrides in ionic liquids†
He
Fu
a,
Yong
Wu
a,
Jun
Chen
a,
Xiaojuan
Wang
b,
Jie
Zheng
*a and
Xingguo
Li
*a
aBeijing National Laboratory for Molecular Sciences (BNLMS), College of Chemistry and Molecular Engineering, Peking University, Beijing, China. E-mail: zhengjie@pku.edu.cn; xgli@pku.edu.cn
bAcademy for Advanced and Interdisciplinary Studies (AAIS), Peking University, Beijing, P. R. China
First published on 13th July 2016
Abstract
The use of alkali metal borohydrides for hydrogen storage has long been restricted by high dehydrogenation temperature and large endothermic dehydrogenation enthalpy. Here we report that the dehydrogenation properties of NaBH4 and LiBH4 can be significantly improved by the ionic liquid (IL) 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide (bmimNTf2). The borohydrides form homogeneous solutions in bmimNTf2, which release more than 70% of theoretical hydrogen below 180 °C, significantly lower than that in the solid state (370 °C for LiBH4 and 500 °C for NaBH4). The dehydrogenation reactions become highly exothermic in the IL, which is in contrast to the highly endothermic process in their solid states. The drastically changed dehydrogenation behaviour in IL is attributed to the destabilization of borohydrides due to the more favorable charge transfer from BH4− to the cation in the IL, which is in line with the established stability rule of metal borohydrides. The IL remains unchanged after dehydrogenation, which provides the possibility of its repeated use.
Introduction
Hydrogen is an outstanding next-generation energy source for its abundance, cleanliness and high energy capacity. However, safe and compact storage of hydrogen has long been a crucial and restrictive step in the widespread use of hydrogen energy.1–3 For on-board applications, many properties including gravimetric/volumetric capacity, (de)hydrogenation thermodynamics and kinetics and reversibility should be considered. Researchers have investigated many hydrogen storage materials such as metal hydrides,4 borohydrides,5 amides/imides,6 alanates,7 ammonia borane (AB)8 and their combinations.9 Unfortunately, these solid state materials or their combinations can hardly meet the requirement of practical application.Alkali metal borohydrides are promising candidates for hydrogen storage due to their high gravimetric hydrogen capacity (LiBH4, 18.5 wt%; NaBH4, 10.7 wt%). However, the use of these compounds through thermal dehydrogenation is predominantly hindered by the extraordinarily high dehydrogenation temperature.10,11 This drawback mainly originates from the high dehydrogenation enthalpy of alkali metal borohydrides, i.e., the strong endothermic effect during dehydrogenation (Table 1). Therefore, modifying the dehydrogenation enthalpy by either destabilizing the reactants or stabilizing the products is a crucial and effective way to lower the dehydrogenation temperature.
| Reactant | Product | T d (°C) | ΔrH (kJ mol−1 H2) | Ref. |
|---|---|---|---|---|
| a These dehydrogenation temperatures are given by the extrapolation of the Van't Hoff plot. b These reaction enthalpies are calculated using the thermodynamic data of reactants and products. | ||||
| NaBH4 | Na + B + 2H2 | 534a | 123,b 106.8 | 10 and 31 |
| NaBH4 | NaH + B + 3/2H2 | — | 133.32b | 10 |
| LiBH4 | LiH + B + 3/2H2 | 370a | 67,b 74 | 11 and 12 |
| 2LiBH4 + MgH2 | 2LiH + MgB2 + 4H2 | 270 | 40.5 | 15 |
| 6LiBH4 + CaH2 | 6LiH + CaB6 + 10H2 | 350 | 59 | 32 |
| 2NaBH4 + MgH2 | 2Na + MgB2 + 5H2 | 515 | 90.9 | 15 and 31 |
| 3LiBH4 + TiF3 | 3LiF + TiB2 + B + 6H2 | 269 | −157.1 | 17 |
| 3NaBH4 + YF3 | 3NaF + 3/4YB4 + 1/4YH2 + 23/4H2 | 423 | 51.2 | 19 |
| NaBH4 (in IL) | NaBH1.04 + 1.48H2 | 179 | −183.0 | This work |
| LiBH4 (in IL) | LiBH1.16 + 1.42H2 | 162 | −138.3 | This work |
There are many examples of the “stabilizing the products” strategy. By adding hydrides and/or other borohydrides containing alkali-earth or transition metals, stable borides can form, which significantly decreases the dehydrogenation enthalpy and lowers the dehydrogenation temperature. For instance, in the dehydrogenation reaction of a LiBH4 + MgH2 composite, a more stable phase, MgB2, is formed as the final product.12 As a result, the dehydrogenation temperature is lowered to 270 °C. In another example, LiBH4 is ball-milled with CaH2 and forms CaB6 as the final product upon dehydrogenation.13 Many similar composites are investigated including LiBH4 + CeH2,13 LiBH4 + Ca(BH4)2,14 NaBH4 + MgH2,15 NaBH4–CaH2/Ca(BH4)2,16 LiBH4 + TiF3,17 LiBH4 + AlF3,18 NaBH4–YH3,19etc. Although the “stabilizing the product” strategy shows certain effectiveness, the dehydrogenation temperature of most of these composites remains very high (typically >300 °C), with a dehydrogenation enthalpy larger than 40 kJ mol−1 H2. Moreover, the introduced additives significantly lower the hydrogen content and often cause complicated side reactions and formation of by-products.
An alternative strategy is to destabilize the reactants. This strategy requires thermodynamic modification on the borohydrides themselves. It is more difficult to apply as the thermodynamic stability of solid borohydrides is dominated by their intrinsic properties. High-energy ball-milling might cause some destabilization effects by introducing more defects. But the experimental investigation on ball-milled NaBH4–MgH2 and LiBH4–MgH2 composites shows little improvement in terms of both the dehydrogenation temperature and thermodynamics.20,21
Here we report a novel approach to destabilize the alkali borohydrides NaBH4 and LiBH4 by dissolving the borohydrides into an ionic liquid (IL). There have been a few studies on using ILs for hydrogen storage. The IL methylguanidinium borohydride has been studied as a hydrogen storage material dissolved in ethylene diglyme.22 In this case, the IL behaves just like other solid borohydrides, which is catalytically decomposed to give hydrogen and a solid product. Imidazolium-based ILs have been shown to have a catalytic effect on dehydrogenation from ammonia borane (AB)23–25 and ethylene diaminebisborane (EDB).26 However, as the dehydrogenation of AB and EDB is thermodynamically favourable, similar or even better promotion effects can also be achieved by a large variety of solid catalysts.27,28
For the thermodynamically highly stable alkali metal borohydrides, the effect of IL on the dehydrogenation is more drastic and unique. Dissolving in IL significantly lowers the dehydrogenation temperature and completely reverses dehydrogenation thermodynamics. Interestingly, NaBH4 and LiBH4 exhibit very similar dehydrogenation behaviour in the IL. Over 70% of theoretical hydrogen can be released from 160–180 °C. Moreover, the IL shows no structural changes after dehydrogenation and can be easily recycled, behaving as a pure catalyst in the very classic definition. The favourable dehydrogenation temperature of the liquid borohydride-IL hydrogen storage system will greatly advance the application of borohydrides as high capacity hydrogen storage materials.
Experimental section
Synthetic procedure
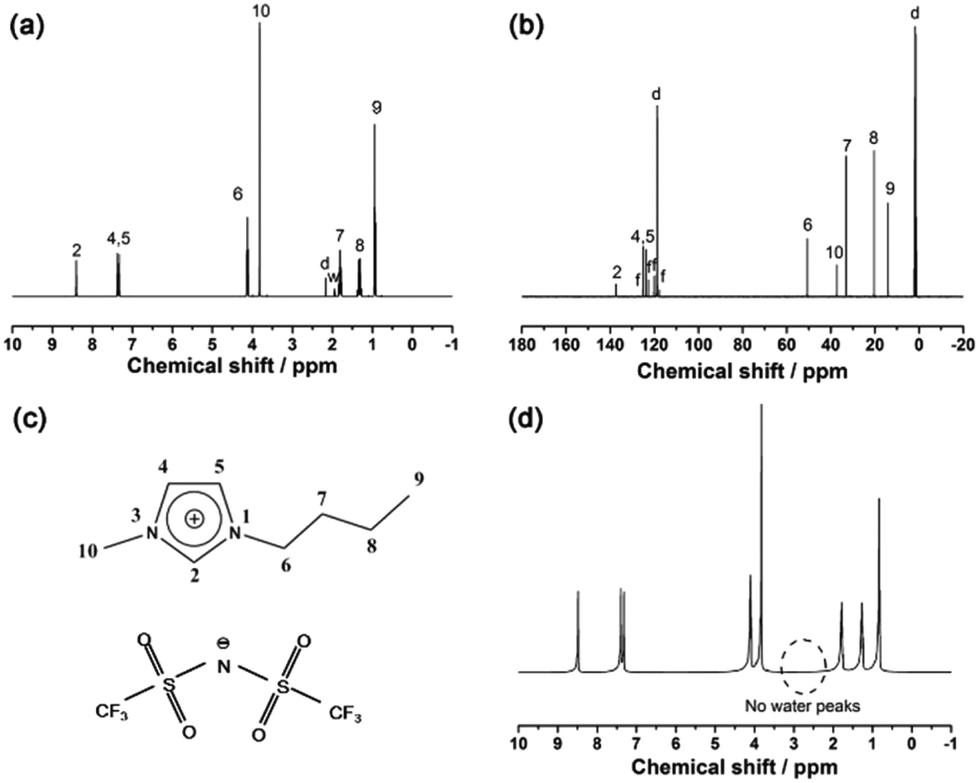 | ||
| Fig. 1 (a) 1H NMR spectrum of the as-prepared bmimNTf2 sample in CD3CN. (b) 13C NMR spectrum of the as-prepared bmimNTf2 sample in CD3CN. The digits in (a) and (b) are corresponding to the atoms labelled in (c). The letters d, w and f represent CD3CN, water and trifluoromethyl, respectively; (c) ionic structure of bmimNTf2; (d) 1H NMR spectrum of the as-prepared bmimNTf2 sample without deuterated solvent. | ||
Characterization techniques
Results and discussion
Characterization of bmimNTf2 and the borohydride-bmimNTf2 solution
The structure of the as-prepared bmimNTf2 sample is investigated by 1H and 13C NMR spectra (Fig. 1a and b). The resonance peaks on these spectra can be ascribed to the corresponding H and C atoms in the ion structure shown in Fig. 1c. However, a small resonance peak corresponding to water is found in the 1H NMR spectrum. To find out whether this water residue is from bmimNTf2 or deuterated solvent, we performed a 1H NMR experiment of pure bmimNTf2 without deuterated solvent (Fig. 1d). No peaks related to water are found in these spectra. Therefore, these results proved that the bmimNTf2 prepared is pure and dry. Therefore, it can be used in the follow-up experiments.After milling, the mixtures of MBH4 and IL are clear and colourless solutions, as shown in Fig. 2b. The disappearance of borohydride solids during milling indicates dissolution of borohydrides in bmimNTf2. The 1H NMR spectra of NaBH4-IL and LiBH4-IL samples are shown in Fig. 2a. The multiplets from −0.5 to 0.5 ppm prove the existence of BH4− anions in the samples. In the 11B NMR spectra (Fig. 2b), the quintets centred at −37.0 and −36.8 ppm can be ascribed to the resonance signals of BH4− in the samples. These spectroscopic features are very similar to those of BH4− in the solid state and other solvents.29,30
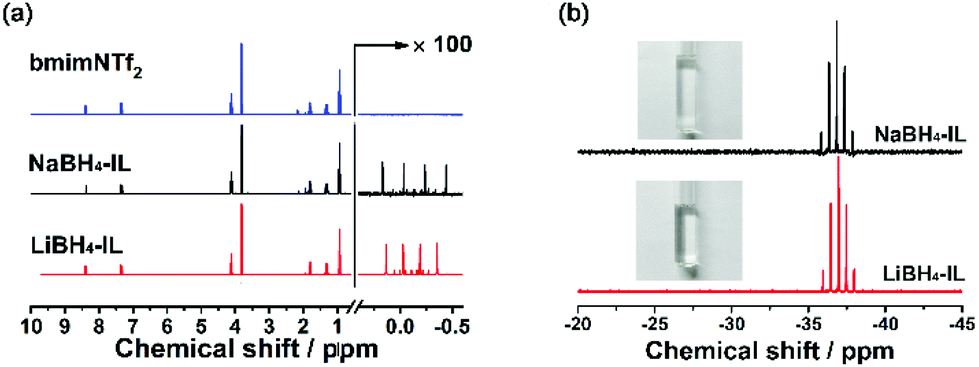 | ||
| Fig. 2 (a) 1H NMR spectra of pristine bmimNTf2, NaBH4-IL and LiBH4-IL samples; (b) 11B NMR spectra of the NaBH4-IL and LiBH4-IL samples. The inset pictures are the photographs of the corresponding samples. | ||
Furthermore, in both 1H (Fig. 2a) and 13C NMR spectra of NaBH4-IL and LiBH4-IL samples (Fig. S3, ESI†), the resonance peaks corresponding to bmimNTf2 do not change, indicating that the structure of IL does not change after dissolution of the borohydrides. These results indicate that the borohydrides can be dissolved into the IL without changing the structure of the IL or BH4− during the milling procedure.
Dehydrogenation properties
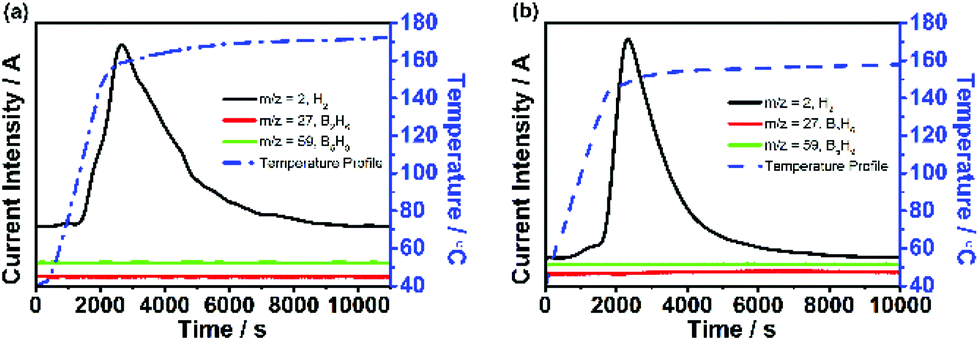
Fig. 3a and b show the dehydrogenation profile of NaBH4-IL and LiBH4-IL monitored by TPD/MS, respectively. The onset dehydrogenation temperature is 104 °C for NaBH4-IL and 85 °C for LiBH4-IL, while the peak temperature is 179 °C for NaBH4-IL and 162 °C for LiBH4-IL, respectively. No gaseous impurities are found under these conditions. The IL decomposes and emits some gaseous impurities above 200 °C. To avoid the decomposition of IL, the dehydrogenation temperature is limited to 180 °C for NaBH4 and 160 °C for LiBH4, respectively.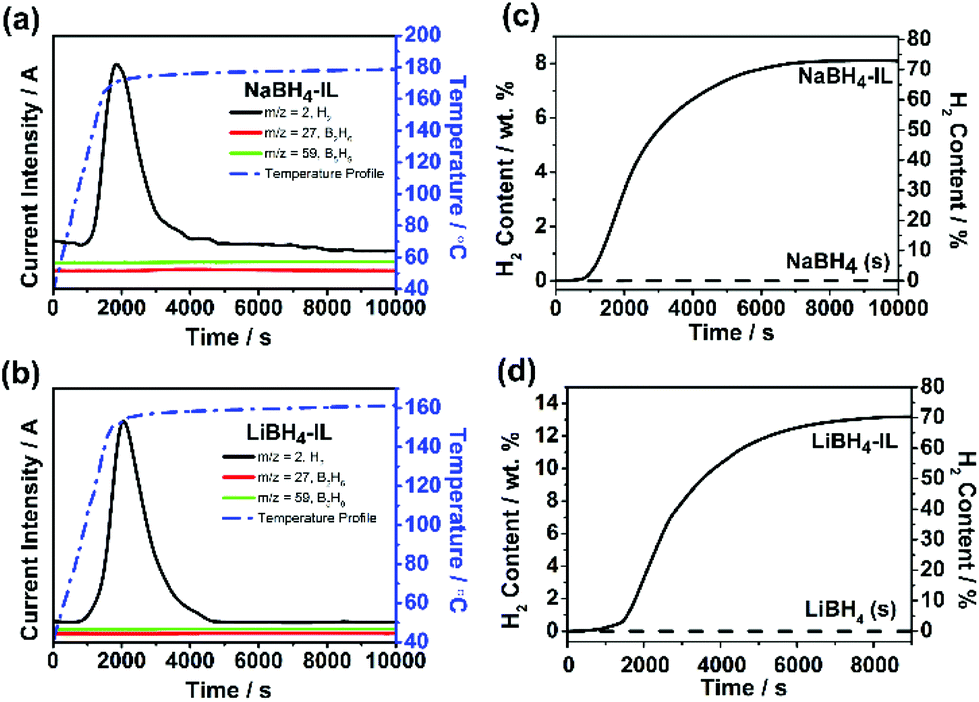 | ||
| Fig. 3 TPD/MS curves of NaBH4-IL (a) and LiBH4-IL (b) samples in their dehydrogenation reaction. The MS signals for H2 (m/z = 2, black), B2H6 (m/z = 27, red), and B5H9 (m/z = 59, green) are demonstrated in solid lines. The blue dashed curves show the temperature profile; isothermic dehydrogenation profiles of NaBH4-IL (c) and LiBH4-IL (d). For comparison, the hydrogen released at the same temperature from solid NaBH4 and LiBH4 is also shown. | ||
Fig. 3c and d show the amount of H2 released from both samples determined by TCD measurement. For the NaBH4-IL sample, the dehydrogenated hydrogen content is 8.1% based on NaBH4, covering 74% of the hydrogen in NaBH4. For the LiBH4-IL sample, the dehydrogenated content is 13.2 wt% based on LiBH4, covering 71% of the hydrogen in LiBH4. At the dehydrogenation temperature of NaBH4-IL and LiBH4-IL samples, the solid state NaBH4 and LiBH4 show no evidence of dehydrogenation. Such dehydrogenation properties are definitely not from a hydrolysis reaction since no residual water is detected in bmimNTf2 (Fig. 1d).
The dehydrogenation products
After dehydrogenation, the samples appear as a clear solution with some precipitates at the bottom of the reaction tube. The solution is studied using 11B, 1H and 13C NMR spectra. 11B NMR spectra of the solution indicate that the BH4− anion in borohydrides completely decomposes in the dehydrogenation reaction (Fig. 4a and b), which is in agreement with the absence of the B–H resonance peak in the 1H NMR spectra in the 0.5 to −0.5 ppm region (Fig. 4c). The full range 11B NMR spectra are shown in Fig. S4 (see the ESI†), showing no soluble boron-containing species in the solution phase. It means that all the boron-containing species are enriched in the solid phase.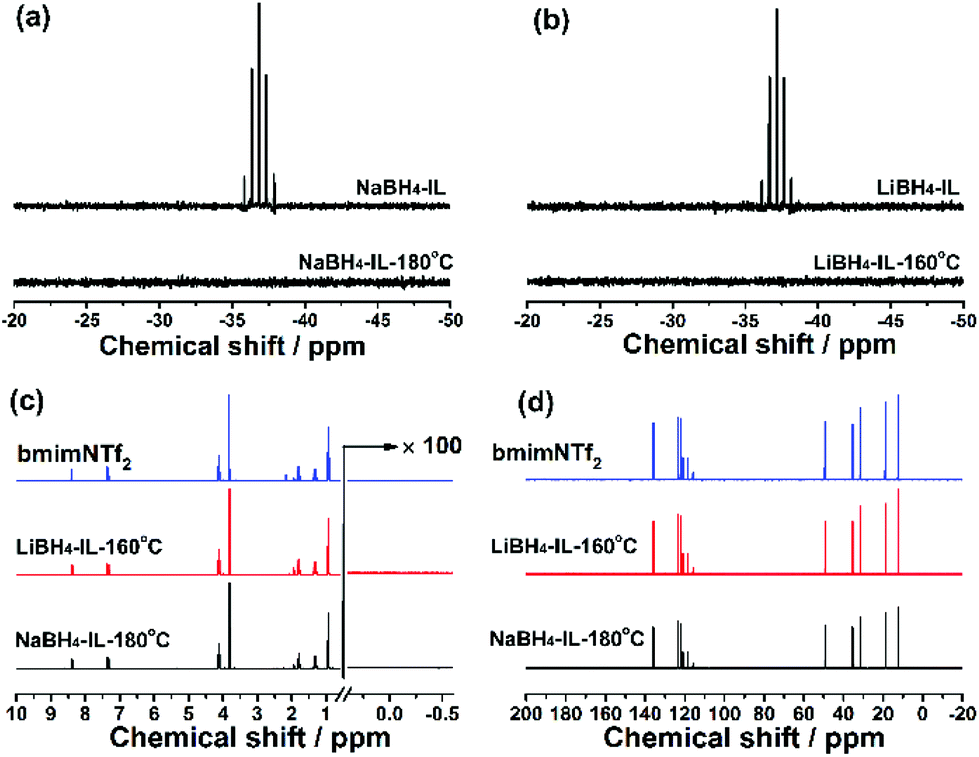 | ||
| Fig. 4 11B NMR spectra of the NaBH4-IL (a) and LiBH4-IL (b) samples before and after dehydrogenation. 1H NMR (c) and 13C NMR (d) spectra of pristine bmimNTf2 and dehydrogenated samples. The 13C NMR spectra are recorded without deuterated solvents. | ||
Regarding the structure of IL after dehydrogenation, the 1H NMR (Fig. 4c) spectra suggest that the chemical bonding of H atoms is not changed by heating or chemical reactions. Meanwhile, no evidence for the formation of C–B bonds or any other new C-related bonds is found in 13C NMR spectra (Fig. 4d). The above results indicate that the IL bmimNTf2 is chemically stable and serves as a pure solvent in the dehydrogenation. Therefore, we may conclude that the dehydrogenated samples consist of a boron-containing solid phase and a liquid phase of pure IL. The stability of the IL during dehydrogenation makes recycling of the IL after dehydrogenation for repeated use possible.
The precipitate is also characterized using XRD (Fig. S5, ESI†) and FT-IR (Fig. S6, ESI†). The XRD patterns show broad peaks for both NaBH4-IL and LiBH4-IL samples, indicating that the dehydrogenation products are amorphous. In the IR spectra, only very weak peaks are detected in the B–H stretching region (2250–2400 cm−1), indicating little residual B–H bond in the dehydrogenation products. This indicates rather complete elimination of the B–H bonds at moderate temperature in IL. According to the dehydrogenation capacity (Fig. 3c and d), the nominal dehydrogenation reaction of NaBH4-IL and LiBH4-IL can be described by eqn (1) and (2), respectively.
| NaBH4-IL → NaBH1.04 + IL + 1.48H2 | (1) |
| LiBH4-IL → LiBH1.16 + IL + 1.42H2 | (2) |
In their solid states, borohydrides usually exhibit multiple dehydrogenation steps. A stable intermediate species, M2B12H12, is found to be the key intermediate in the dehydrogenation reaction pathway of alkali metal borohydrides.33–35 However, in MBH4-IL samples, the dehydrogenation capacity is much higher (70–75%) compared to the partial dehydrogenation of MBH4 to M2B12H12. The stoichiometry of the dehydrogenation products in the IL cannot be attributed to any known alkali metal hydroborates. Rather, it best matches a mixture of boron and metal hydride, which is the dehydrogenation product at high temperature for solid state LiBH4 and NaBH4.10,11 This is also in agreement with the very low residual B–H bonding in the solid precipitate. Further characterization of the solid dehydrogenation product will be our next research focus.
Another interesting observation is the highly similar dehydrogenation behaviour in terms of both dehydrogenation temperature and the dehydrogenation capacity of NaBH4-IL and LiBH4-IL. In the solid state, there is a significant difference in the dehydrogenation temperature and dehydrogenation mechanism.5 The nearly metal independent dehydrogenation behaviour has important implications for the dehydrogenation mechanism, as will be discussed later.
Thermodynamic and kinetic investigation
Since the dehydrogenation temperatures of borohydride-IL samples are far lower than those calculated from thermodynamic parameters of solid state materials, the thermodynamic property of this system is very interesting. Therefore, we perform DSC measurements to compare the thermodynamic behaviour of NaBH4 and LiBH4 in IL and their solid states. In their crystalline states, NaBH4 and LiBH4 show very strong endothermic behaviour during dehydrogenation at high temperature (370 °C for LiBH4 and 500 °C for NaBH4) (Fig. 5b and d). The solid state LiBH4 experiences an orthorhombic to tetragonal phase change (∼130 °C) and melting (∼280 °C) before dehydrogenation.36 The dehydrogenation enthalpy is reported to be 108 kJ mol−1 and 74 kJ mol−1 for pristine NaBH4 and LiBH4, respectively.10,11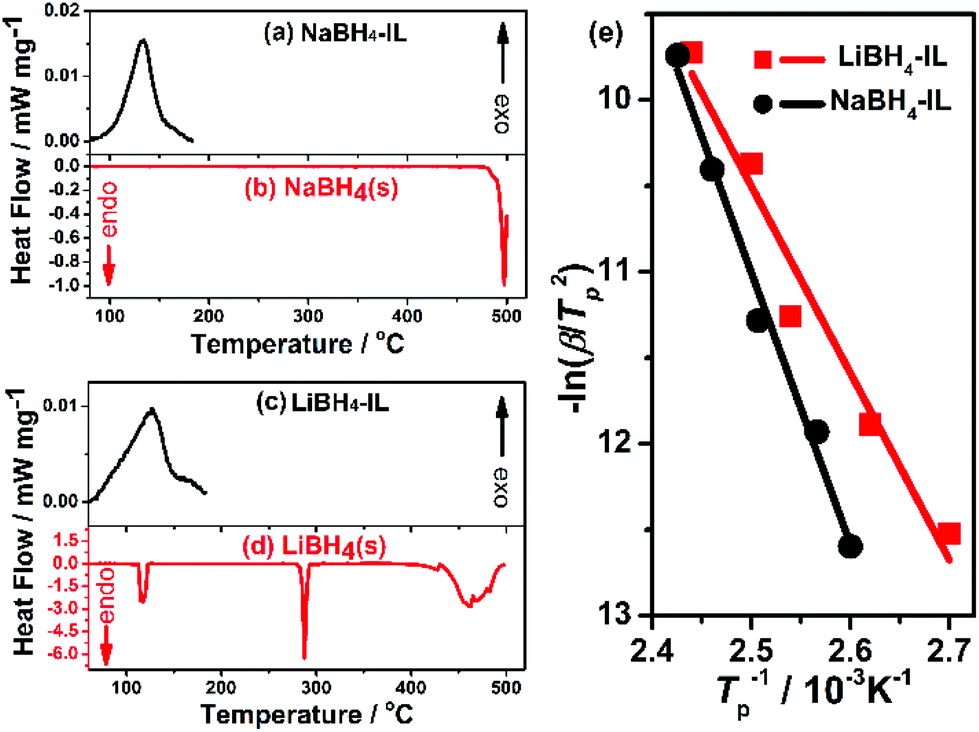 | ||
| Fig. 5 DSC curves of NaBH4-IL and solid state NaBH4 (a, b) and LiBH4-IL and solid state LiBH4 (c, d) at a heating rate of 2 K min−1. The Kissinger's plots of NaBH4-IL (black) and LiBH4-IL (red) are demonstrated in (e). The apparent activation energy (Ea) is calculated from the slope of the fitted lines (the solid lines) according to the Kissinger's equation (eqn (3)). | ||
Surprisingly, the dehydrogenation of NaBH4-IL and LiBH4-IL samples becomes highly exothermic (Fig. 5a and c). The NaBH4-IL sample exhibits an exothermic peak at 134 °C, and the dehydrogenation enthalpy is −183.0 kJ mol−1 H2. The LiBH4-IL sample also shows an exothermic peak at 127 °C, and the dehydrogenation enthalpy is −138.3 kJ mol−1 H2. In contrast to their very similar dehydrogenation temperature and dehydrogenation capacity, the dehydrogenation enthalpy of NaBH4-IL and LiBH4-IL is quite different. The dehydrogenation enthalpies are lower than those in most of the systems listed in Table 1,12,15,31,32 but are similar to those observed in the borohydride–fluoride composites like LiBH4–YF3,17 which has an exothermic dehydrogenation enthalpy due to the formation of highly thermodynamically stable products such as LiF and TiB2. However, the exothermic dehydrogenation is rather surprising for MBH4-IL, as the IL is unchanged after dehydrogenation. The favourable dehydrogenation of MBH4-IL, therefore, is a result of destabilized reactants. It is a much advantageous approach because no reactive additives are involved in the dehydrogenation reaction.
The activation energy of the dehydrogenation process is evaluated using the Kissinger's equation by measuring multiple DSC curves (Fig. S7 and S8, ESI†) at different heating rates (eqn (3)).37
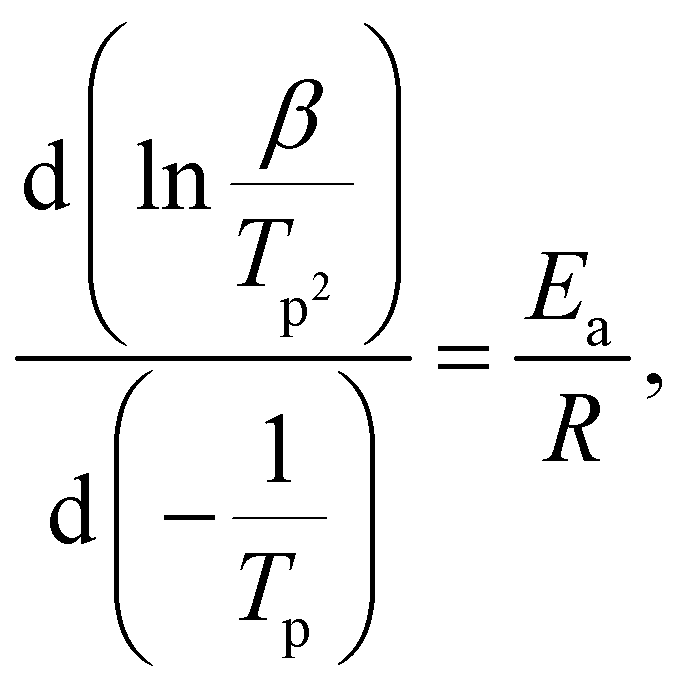 | (3) |
The apparent activation energies for NaBH4-IL and LiBH4-IL samples are calculated to be 131.3 kJ mol−1 and 91.4 kJ mol−1, respectively (Fig. 5e). The high apparent activation energy shows that both NaBH4-IL and LiBH4-IL are thermodynamically unstable systems stabilized by kinetic barriers. It also provides the possibility to further lower the dehydrogenation temperature by introducing suitable catalysts in the MBH4-IL system, which will be a very interesting field deserving further exploration.
Dehydrogenation mechanism study
| ΔrH(MBH4, s) = ΔfH(MBH) − ΔfH(MBH4, s) > 0 | (4) |
The solid dehydrogenation product is represented by “MBH”, which can be considered as a mixture of metal hydride and elemental boron, the high temperature dehydrogenation product of NaBH4 and LiBH4.
In contrast, DSC results also suggest that dehydrogenation of MBH4 in the IL (eqn (1) and (2)) is highly exothermic:
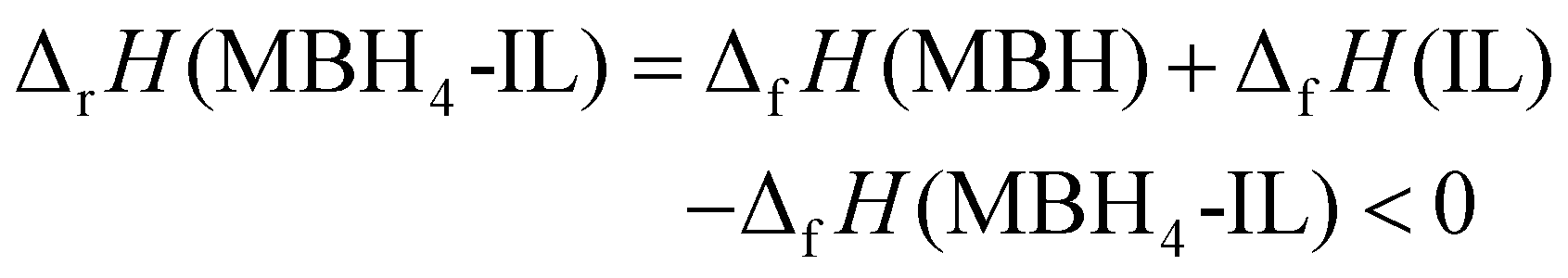 | (5) |
Here the solid dehydrogenation product precipitated from the IL is also represented by “MBH”, according to the stoichiometry derived from the dehydrogenation capacity. The term ΔfH(MBH4-IL) describes the formation enthalpy of the MBH4-IL samples. This term can be approximated as the sum of the inherent formation enthalpy of IL (ΔfH(IL)) and the energy of the dissolved MBH4 (ΔfH(MBH4 in IL)).
| ΔfH(MBH4-IL) ≈ ΔfH(MBH4 in IL) + ΔfH(IL) | (6) |
This is a rather good approximation for a dilute MBH4-IL solution. In this case we can treat the IL as an infinitely large bath which is little affected by the dissolved borohydrides.
According to eqn (4), (5), and (6), the borohydride in IL becomes less stable compared to that in the solid state (eqn (7)),
| ΔfH(MBH4 in IL) ≫ ΔfH(MBH4, s) | (7) |
Both NaBH4 and LiBH4 are typical ionic crystals.38,39 Using the gas phase ions M+(g) and BH4−(g) as the reference state, ΔfH(MBH4, s) is virtually the lattice enthalpy of MBH4 (ΔUMBH4). On the other hand, ΔfH(MBH4 in IL) is the solvation enthalpy (Es) in the IL, determined by the interaction between the borohydrides and the IL. The destabilization effect (eqn (7)), therefore, is a result of the large difference between ΔUMBH4 and Es:
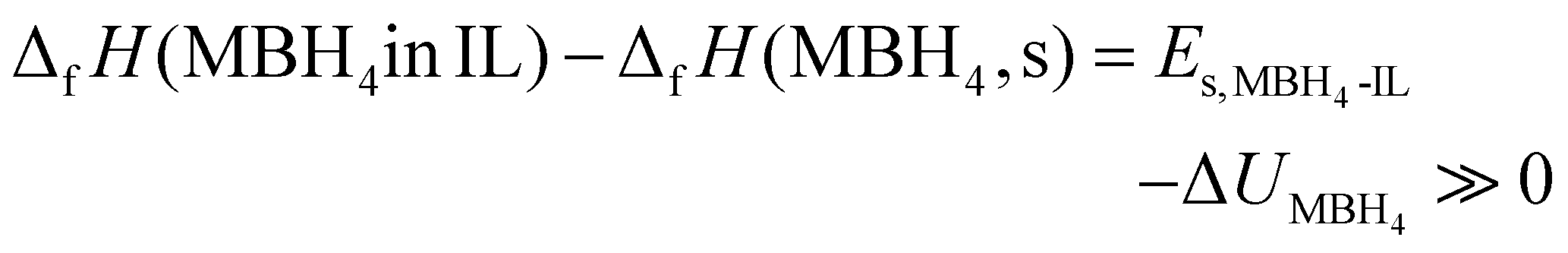 | (8) |
The energetics of the M–B–H species are summarized in Scheme 1. In this diagram, two approximations are made except that in deriving eqn (6): (1) in the case of pristine LiBH4, we neglect the energy change due to phase transition and melting. However, these terms are small compared to the lattice enthalpy of LiBH4 and can be included as a slight upshift of the position of LiBH4 in the energy diagram. (2) We use the same dehydrogenation product “MBH” for both pristine MBH4 and MBH4-IL. This could be complicated by other dehydrogenation products, particularly in the case of low temperature dehydrogenation in the solid state. Nevertheless, the MBH4 is the most stable M–B–H species compared to all the dehydrogenation products composed of M–B–H. For instance, one well known stable dehydrogenation intermediate M2B12H12 is also an endothermic product from MBH4.34 Therefore, other M–B–H type solid state dehydrogenation products can be regarded as a band around the representative “MBH” species.
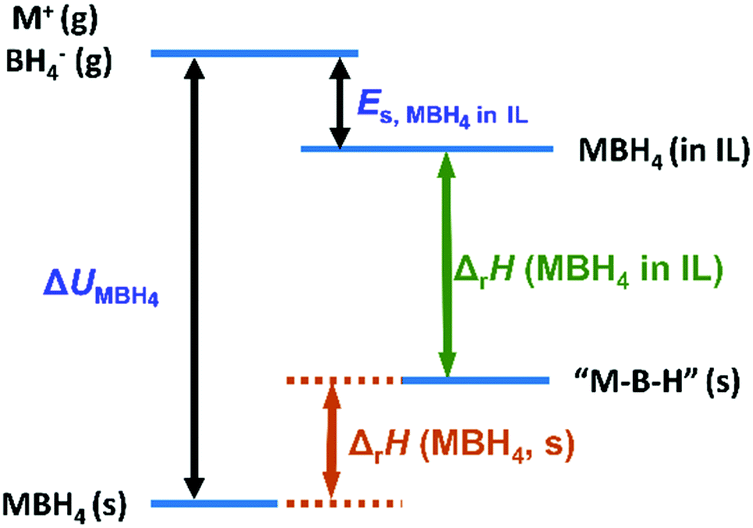 | ||
| Scheme 1 A schematic energy diagram of the dehydrogenation of borohydrides in the solid state and IL solution. | ||
The above approximations may cause slight adjustments on the energetic diagram, as shown in Fig. S9 (see the ESI†). However, the simplified diagram (Scheme 1) is sufficiently illustrative. The stability increases in the order: MBH4 in IL < the dehydrogenation product (represented by “MBH”) < MBH4(s), as suggested by the DSC results. This diagram clearly shows that the reversed dehydrogenation thermodynamics in the IL is caused by destabilization of MBH4. This diagram also explains the limitation of thermodynamic modulation in the solid state. In pristine MBH4, the thermodynamics is dominated by the large lattice enthalpy (ΔUMBH4). Consequently, the dehydrogenation is always highly endothermic for M–B–H type products, as suggested by previous studies.34,40
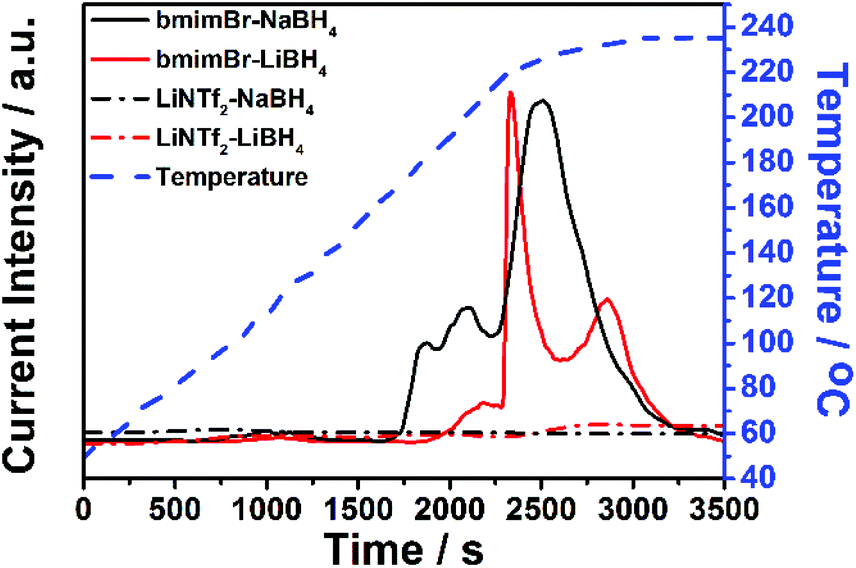 | ||
| Fig. 6 TPD curves of bmimBr-NaBH4 (black, solid), bmimBr-LiBH4 (red, solid), LiNTf2–NaBH4 (black, dash dot) and LiNTf2–LiBH4 (red, dash dot) samples. The temperature profile is demonstrated as blue dashed line. | ||
At this stage, it is illustrative to recall the highly similar dehydrogenation temperature and capacity for LiBH4 and NaBH4 in the IL (Fig. 3), while a large difference is observed in their solid states. In dilute IL solution of borohydrides, BH4− is mostly surrounded by bmim+ cations rather than the metal cations. The almost cation independent dehydrogenation temperature and capacity, therefore, are a natural consequence of the new dehydrogenation mechanism in IL, i.e., dehydrogenation dominated by the bmim+/BH4− interaction.
Destabilization of borohydrides due to the bmim+/BH4− interaction is in well accordance with the stability rule observed in solid metal borohydrides.41 Nakamori et al. reported that the stability of borohydride is determined by the electronegativity of the metal in the metal borohydride. Metals with lower electronegativity (e.g. alkali metals) form more stable borohydrides.42 This rule is well explained by charge transfer between BH4− and metal cations.41,43 Charge transfer from BH4− to the cations will lower the negative charge on BH4− and consequently destabilize the borohydride. In alkali metal borohydride such as NaBH4 and LiBH4, an ionic picture has been proposed by ab initio calculations.39 The charge on the alkali metal cation is close to +1. In stable borohydrides such as LiBH4, NaBH4 and Ca(BH4)2, the charge transfer between Mn+ and BH4− is relatively low, while in unstable borohydrides such as Zn(BH4)2 and CuBH4, the charge transfer is relatively large.44
The charge transfer strongly depends on the nature of the cation. Cations that are more inclined to accept negative charges favour the charge transfer and consequently destabilize the borohydride. For simple metal cations, this trend can be well described by the electronegativity of the corresponding metal. In our study, bmim+ is a large cation with a conjugated system. Such a structure is more favourable to accommodate extra negative charges, as the electron delocalization effect can stabilize the extra electron density. Indeed, both experimental and theoretical studies suggest that there is a notable charge transfer effect from NTf2− to bmim+ in bmimNTf2 or similar ILs.45 Such a charge transfer effect is expected to be more favourable from the less electron pulling BH4− (compared to NTf2−). Therefore, the bmim+/BH4− interaction destabilizes the BH4− due to the enhanced electron transfer to bmim+, which explains the significantly lower dehydrogenation temperature and exothermic nature of the dehydrogenation process.
To further verify the charge transfer based destabilization mechanism, the dehydrogenation of borohydrides in another IL bmmimNTf2 (bmmim = 1-butyl-2,3-dimethylimidazolium) is studied. The cation bmmim+ differs from bmim+ by the methyl group on the 2-C of the imidazolium ring. The 2-H in bmim+ may be deprotonated by strong bases. Methyl substitution eliminates this potentially active H species. Fig. 7 shows the TPD/MS curves of NaBH4-bmmimNTf2 and LiBH4-bmmimNTf2 samples. They show a very similar dehydrogenation temperature to that of NaBH4-bmimNTf2 and LiBH4-bmimNTf2 samples. Destabilization of BH4−, therefore, is not associated with the 2-H of bmim+. Furthermore, this result can be well explained by the charge transfer mechanism, as the methyl substitution for H has little effect on the electron affinity of the imidazolium cation. The bulky imidazolium cation with the conjugated structure may stabilize an extra negative charge and thus cause more favourable charge transfer from BH4−, which is little affected by the alkyl groups on the imidazolium ring.
 | ||
| Fig. 7 TPD/MS curves of NaBH4-bmmimNTf2 (a) and LiBH4-bmmimNTf2 (b) samples. The MS signals for H2 (m/z = 2, black), B2H6 (m/z = 27, red), and B5H9 (m/z = 59, green) are demonstrated in solid lines. The blue dashed lines show the temperature profile of the dehydrogenation reaction. | ||
The MBH4-IL system represents a new type of liquid hydrogen storage system. Similar to the well-studied liquid organic hydrogen storage systems,46,47 the MBH4-IL shares many advantages in transport, storage and handling arising from the liquid form. Liquid hydrogen storage systems based on ILs are rare but not totally absent. Previous reports show that ILs can promote the dehydrogenation kinetics of B–N–H type compounds such as NH3BH3 and BH3NH2CH2CH2NH2BH3.23–26 However, as the first step dehydrogenation of these materials is thermodynamically favourable, the role of the IL is mainly a kinetic promoter and toxic gas inhibitor. In fact, similar or even better promotion effects have also been achieved by many other solid catalysts.8,27
On the other hand, the role of the IL for LiBH4 and NaBH4 is truly unique and indispensable. The promotion effect in terms of dehydrogenation temperature is unrivalled by any other additive reported so far. The strong ion pair interaction in MBH4-IL dramatically changes the dehydrogenation temperature and thermodynamics of the borohydrides, which is an important advantage towards the application of NaBH4 and LiBH4 as high capacity hydrogen storage materials. Although inclusion of IL reduces the hydrogen capacity in one dehydrogenation cycle, repeated use of the IL is possible as the IL is unaffected during the dehydrogenation and can be easily separated from the solid state dehydrogenation residue. With engineering efforts to recycle the IL, high hydrogen capacity is attainable for the MBH4-IL system.
Conclusions
The dehydrogenation of alkali metal borohydrides NaBH4 and LiBH4 in the ionic liquid bmimNTf2 is studied. Three important observations are noted. (1) The dehydrogenation in IL occurs at 160–180 °C, which is similar for NaBH4-IL and LiBH4-IL and is much lower than that in their solid states. (2) The dehydrogenation process in the IL is strongly exothermic while it is endothermic in the solid state. (3) The IL remains unaffected after the dehydrogenation process and can be easily separated from the solid dehydrogenation residue of the borohydrides.The distinct dehydrogenation behaviour originates from the interaction between BH4− and the cation in the IL which destabilizes BH4−. The destabilization mechanism involves a more favourable charge transfer from BH4− to the bulky imidazolium cation, which is in line with the well-established stability rule of metal borohydrides. The borohydride-IL system is a promising liquid hydrogen storage system with high attainable hydrogen capacity at moderate temperature.
Acknowledgements
This study is supported by National Natural Science Foundation of China (no. U1201241, 11375020, 51431001 and 21321001). The authors acknowledge Prof. Taiwei Chu from Peking University for his assistance in synthesizing the ionic liquids.Notes and references
- L. Schlapbach and A. Zuttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- J. Graetz, Chem. Soc. Rev., 2009, 38, 73 RSC.
- S.-i. Orimo, Y. Nakamori, J. R. Eliseo, A. Zuettel and C. M. Jensen, Chem. Rev., 2007, 107, 4111–4132 CrossRef CAS PubMed.
- B. Sakintuna, F. Lamaridarkrim and M. Hirscher, Int. J. Hydrogen Energy, 2007, 32, 1121–1140 CrossRef CAS.
- A. Zuettel, A. Borgschulte and S.-I. Orimo, Scr. Mater., 2007, 56, 823–828 CrossRef CAS.
- P. Chen, Z. T. Xiong, J. Z. Luo, J. Y. Lin and K. L. Tan, Nature, 2002, 420, 302–304 CrossRef CAS PubMed.
- B. Bogdanovic and M. Schwickardi, J. Alloys Compd., 1997, 253, 1–9 CrossRef.
- B. Peng and J. Chen, Energy Environ. Sci., 2008, 1, 479–483 CAS.
- J. H. Luo, X. D. Kang and P. Wang, Int. J. Hydrogen Energy, 2013, 38, 4648–4653 CrossRef CAS.
- P. Martelli, R. Caputo, A. Remhof, P. Mauron, A. Borgschulte and A. Zuettel, J. Phys. Chem. C, 2010, 114, 7173–7177 CAS.
- P. Mauron, F. Buchter, O. Friedrichs, A. Remhof, M. Bielmann, C. N. Zwicky and A. Zuettel, J. Phys. Chem. B, 2008, 112, 906–910 CrossRef CAS PubMed.
- J. J. Vajo, S. L. Skeith and F. Mertens, J. Phys. Chem. B, 2005, 109, 3719–3722 CrossRef CAS PubMed.
- S.-A. Jin, Y.-S. Lee, J.-H. Shim and Y. W. Cho, J. Phys. Chem. C, 2008, 112, 9520–9524 CAS.
- J. Y. Lee, D. Ravnsbaek, Y.-S. Lee, Y. Kim, Y. Cerenius, J.-H. Shim, T. R. Jensen, N. H. Hur and Y. W. Cho, J. Phys. Chem. C, 2009, 113, 15080–15086 CAS.
- J. F. Mao, X. B. Yu, Z. P. Guo, H. K. Liu, Z. Wu and J. Ni, J. Alloys Compd., 2009, 479, 619–623 CrossRef CAS.
- J. F. Mao, Z. P. Guo, X. B. Yu and H. K. Liu, J. Phys. Chem. C, 2011, 115, 9283–9290 CAS.
- Y. H. Guo, X. B. Yu, L. Gao, G. L. Xia, Z. P. Guo and H. K. Liu, Energy Environ. Sci., 2010, 3, 465–470 CAS.
- P. P. Yuan, B. H. Liu, H. P. Zhu, W. Y. Pan and Z. P. Li, J. Alloys Compd., 2013, 557, 124–129 CrossRef CAS.
- J. Zou, L. Li, X. Zeng and W. Ding, Int. J. Hydrogen Energy, 2012, 37, 17118–17125 CrossRef CAS.
- S. Garroni, C. Pistidda, M. Brunelli, G. B. M. Vaughan, S. Surinach and M. D. Baro, Scr. Mater., 2009, 60, 1129–1132 CrossRef CAS.
- K. Crosby and L. L. Shaw, Int. J. Hydrogen Energy, 2010, 35, 7519–7529 CrossRef CAS.
- A. Doroodian, J. E. Dengler, A. Genest, N. Roesch and B. Rieger, Angew. Chem., Int. Ed., 2010, 49, 1871–1873 CrossRef CAS PubMed.
- D. W. Himmelberger, L. R. Alden, M. E. Bluhm and L. G. Sneddon, Inorg. Chem., 2009, 48, 9883–9889 CrossRef CAS PubMed.
- R. K. Ahluwalia, J. K. Peng and T. Q. Hua, Int. J. Hydrogen Energy, 2011, 36, 15689–15697 CrossRef CAS.
- M. E. Bluhm, M. G. Bradley, R. Butterick, U. Kusari and L. G. Sneddon, J. Am. Chem. Soc., 2006, 128, 7748–7749 CrossRef CAS PubMed.
- S. Sahler, S. Sturm, M. T. Kessler and M. H. G. Prechtl, Chem. – Eur. J., 2014, 20, 8934–8941 CAS.
- A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef CAS PubMed.
- B. Peng and J. Chen, Energy Environ. Sci., 2008, 1, 479–483 CAS.
- Y. Yan, A. Remhof, D. Rentsch and A. Zuettel, Chem. Commun., 2015, 51, 700–702 RSC.
- Y. Yan, A. Remhof, D. Rentsch, Y.-S. Lee, Y. W. Cho and A. Zuettel, Chem. Commun., 2013, 49, 5234–5236 RSC.
- C. Milanese, S. Garroni, A. Girella, G. Mulas, V. Berbenni, G. Bruni, S. Suriñach, M. D. Baró and A. Marini, J. Phys. Chem. C, 2011, 115, 3151–3162 CAS.
- F. E. Pinkerton and M. S. Meyer, J. Alloys Compd., 2008, 464, L1–L4 CrossRef CAS.
- S. J. Hwang, R. C. Bowman, J. W. Reiter, J. Rijssenbeek, G. L. Soloveichik, J. C. Zhao, H. Kabbour and C. C. Ahn, J. Phys. Chem. C, 2008, 112, 3164–3169 CAS.
- Y. Yan, A. Remhof, S.-J. Hwang, H.-W. Li, P. Mauron, S.-i. Orimo and A. Zuettel, Phys. Chem. Chem. Phys., 2012, 14, 6514–6519 RSC.
- Y. Liu, S. Giri, J. Zhou and P. Jena, J. Phys. Chem. C, 2014, 118, 28456–28461 CAS.
- J. P. Soulie, G. Renaudin, R. Cerny and K. Yvon, J. Alloys Compd., 2002, 346, 200–205 CrossRef CAS.
- H. E. Kissinger, Anal. Chem., 1957, 29, 1702–1706 CrossRef CAS.
- P. C. Aeberhard, K. Refson and W. I. F. David, Phys. Chem. Chem. Phys., 2013, 15, 8081–8087 RSC.
- Z. Łodziana and M. J. van Setten, Phys. Rev. B: Condens. Matter, 2010, 81, 024117 CrossRef.
- Y. S. Zhang, E. Majzoub, V. Ozolins and C. Wolverton, J. Phys. Chem. C, 2012, 116, 10522–10528 CAS.
- Y. Nakamori, H. W. Li, K. Kikuchi, M. Aoki, K. Miwa, S. Towata and S. Orimo, J. Alloys Compd., 2007, 446, 296–300 CrossRef.
- Y. Nakamori, K. Miwa, A. Ninomiya, H. W. Li, N. Ohba, S. I. Towata, A. Zuttel and S. I. Orimo, Phys. Rev. B: Condens. Matter, 2006, 74, 045126 CrossRef.
- K. Miwa, N. Ohba, S.-i. Towata, Y. Nakamori and S.-i. Orimo, Phys. Rev. B: Condens. Matter, 2006, 69, 245120 CrossRef.
- Y. Nakamori, H. Li, M. Matsuo, K. Miwa, S. Towata and S. Orimo, J. Phys. Chem. Solids, 2008, 69, 2292–2296 CrossRef CAS.
- O. Holloczki, F. Malberg, T. Welton and B. Kirchner, Phys. Chem. Chem. Phys., 2014, 16, 16880–16890 RSC.
- R. H. Crabtree, Energy Environ. Sci., 2008, 1, 134–138 CAS.
- D. Teichmann, W. Arlt, P. Wasserscheid and R. Freymann, Energy Environ. Sci., 2011, 4, 2767–2773 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures including illustrations, calibration curves, energetic diagrams, detailed NMR spectra and characterization of precipitations. See DOI: 10.1039/c6qi00167j |
| This journal is © the Partner Organisations 2016 |