
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDestabilization of the metal site as a hub for the pathogenic mechanism of five ALS-linked mutants of copper, zinc superoxide dismutase†
Raúl
Mera-Adasme‡
*,
Hannes
Erdmann
,
Tomasz
Bereźniak
and
Christian
Ochsenfeld
*
Chair of Theoretical Chemistry, Department of Chemistry, University of Munich (LMU), Munich 81377, Germany. E-mail: christian.ochsenfeld@cup.uni-muenchen.de; raul.mera@usach.cl
First published on 25th August 2016
Abstract
Amyotrophic lateral sclerosis (ALS) is a lethal neurodegenerative disease, with no effective pharmacological treatment. Its pathogenesis is unknown, although a subset of the cases is linked to genetic mutations. A significant fraction of the mutations occur in one protein, copper, zinc superoxide dismutase (SOD1). The toxic function of mutant SOD1 has not been elucidated, but damage to the metal site of the protein is believed to play a major role. In this work, we study the electrostatic loop of SOD1, which we had previously proposed to work as a “solvent seal” isolating the metal site from water molecules. Out of the five contact points identified between the electrostatic loop and its dock in the rest of the protein, three points were found to be affected by ALS-linked mutations, with a total of five mutations identified. The effect of the five mutations was studied using methods of computational chemistry. We found that four of the mutations destabilize the proposed solvent seal, while the fifth mutation directly affects the metal-site stability. In the two contact points unaffected by ALS-linked mutations, the side chains of the residues were not found to play a stabilizing role. Our results show that the docking of the electrostatic loop to the rest of SOD1 plays a role in ALS pathogenesis, in support of that structure acting as a solvent barrier for the metal site. The results provide a unified pathogenic mechanism for five different ALS-linked mutations of SOD1.
Significance to metallomicsOur work contributes to establishing and explaining a connection between transition metals and familial forms of the neurodegenerative disorder amyotrophic lateral sclerosis (ALS). Several mechanisms have been proposed for the pathogenesis of the familial forms of ALS that are caused by mutations of the protein copper, zinc superoxide dismutase (SOD1). Among them are protein misfolding, metal dyshomeostasis and pro-oxidant catalysis. Alterations in SOD1's metal site could be involved in all these mechanisms. By connecting five ALS-linked mutations of SOD1 to damage at the metal site, our work highlights the importance of zinc and copper ions in the pathogenesis of ALS. |
1 Introduction
ALS (amyotrophic lateral sclerosis) is a neurodegenerative disease affecting motor neurons. The syndrome consists of a progressive paralysis that kills most patients within 2–4 years after diagnosis,1 although several forms of the disease with faster and slower progression exist. As of now, there is no pharmacological treatment for ALS that can prolong the life of the patient by more than a few months.2The lack of effective treatment for ALS is linked to the limited knowledge available on the underlying causes of the disease.1,3 Among what is currently known is the existence of familial variants of ALS (fALS), with different survival times, characteristic of each mutation.4 The familial cases can often be traced to point mutations in specific proteins. In particular, an important fraction of the familial cases is caused by mutations in the enzyme copper, zinc superoxide dismutase (SOD1).5 A large amount of the current research on ALS pathogenesis focuses on SOD1, its mutants, and the familial variants of ALS linked to them. The aim is to obtain insight that can be applied to the understanding of the more prevalent sporadic form of ALS (sALS).
SOD1 is a family of proteins with members present in all eukaryota, and related proteins expressed in prokaryota.6,7 Its physiological function is to catalyse the dismutation of the potentially toxic superoxide ion into molecular oxygen and hydrogen peroxide. Thus, SOD1 is part of the cellular defenses against oxidative stress. In mammals, the enzyme forms a homodimeric structure with one catalytic site in each subunit. The catalytic site consists of a binuclear complex of copper and zinc, where copper is the catalytic metal, and zinc contributes to the catalysis in an indirect way, as well as to maintain the structural integrity of the protein.6,8
Along the protein's catalytic cycle, the copper coordinated to it fluctuates between its Cu(II) and its Cu(I) form. In the former case, the protein is said to be in an oxidized state. The Cu(II) ion is coordinated by four histidine residues and a weakly bound water molecule.9,10 The zinc ion is coordinated by three histidine and one aspartate residues. In the oxidized state, one of the histidine residues (H63 in the human sequence) is coordinated to both the copper ion and the zinc ion, in what is called an imidazolate bridge between the metals. When the protein is in its reduced state, the imidazolate bridge is lost, and H63 becomes exclusively a Zn(II) ligand. The water molecule is also lost from the copper center which assumes a distorted trigonal-planar coordination geometry. The distorted-tetrahedral coordination geometry of the Zn(II) center is unchanged by the oxidation state of SOD1.8,9
Apart from the mentioned imidazolate bridge, there is a second connection between both metal centers in SOD1. An aspartate residue, D124, is a second-sphere ligand to both metals. D124 forms a hydrogen bond to the H46 residue (a copper ligand) and another to the H71 residue (a zinc ligand). The resulting secondary bridge is not altered by the redox status of the protein8,9 and has been shown to be critical for SOD1's zinc binding.11,12 Computational results suggest that alterations in the secondary bridge cause the binding of H71 to the zinc center to weaken, potentially exposing the metal to the solvent.13
The over a hundred different mutations in SOD1 that lead to ALS are spread along the whole sequence of the protein.5 How such different mutations in distant and different parts of the enzyme cause similar clinical syndromes is poorly understood, but it is known that the effect does not depend on the loss of enzymatic activity by SOD1. Rather, the ALS-linked mutants of SOD1 would gain a new, pathological function responsible for the disease.14 Several hypotheses involving different structural elements of SOD1 exist.15 Even though many of the ALS-linked mutations occur far from the metal site, one of the hypotheses proposed is that the loss of the structural copper and/or zinc ion is a key-step towards the pathological gain of function. Demetallation of SOD1 could be toxic by altering the fold of the protein, to which it is closely related.16 The misfolded and metal deficient protein could then interact abnormally with cell structures such as the endoplasmic reticle.17 The lack of copper bound to G93A-SOD1 has recently been shown to be critical for the ALS-like syndrome in mice that overexpress it.18 The zinc-free protein could also present an altered redox specificity19,20 or produce dyshomeostasis in the concentrations of zinc.21 Abnormal zinc levels have been observed in animal models carrying the ALS-linked G93A mutant of SOD1.22 In addition, proteins involved in zinc homeostasis have been shown to play a role in animal models for the disease, as well as in human patients.23–27 The mechanism by which the metal site of SOD1 could be damaged by ALS-linked mutations has not been elucidated.
In a previous computational work,28 we suggested that the electrostatic loop, a structural element of SOD1 that contains the D124 residue, was important to seal the solvent away from the secondary bridge. The solvent seal would allow the low dielectric needed for D124 to form strong hydrogen bonds to H46 and H71. The proposed model is consistent with experimetal results suggesting that perturbations in the electrostatic loop are a common feature among ALS-linked SOD1 mutants. Molnar et al. used H/D exchange followed by proteolysis in order to measure the mobility of different structural elements of SOD1 on 13 different ALS-linked mutants of the protein, and found the electrostatic loop to be destabilized, as compared to the wild-type structure, in all the mutants studied.29 In the present work, we investigate the existence of interactions keeping the putative solvent seal in place, and the nature of such interactions, if any. For this purpose, we study models for the WT protein and for several fALS-linked mutants at the density functional and classical levels of theory. We employ density functional theory-based methods to obtain accurate interaction energies30 of isolated interaction points, while classical methods, thanks to their low computational requirements, are used for sampling the conformational space of the aminoacidic sidechains involved in the interactions, and to investigate dynamic effects that cannot be studied using single-point density-functional calculations. Classical methods are also appropiate for studying the interaction sites in the context of the whole protein. However, electronic effects are not explicitly accounted for in classical methods.
2 Materials and methods
2.1 Density functional theory calculations
The BLYP density functional31,32 was employed in all geometry optimizations and most density-functional theory (DFT) calculations. For validation of the interaction energies, single-point calculations were also performed using the TPSS33 and the MPW1B9534 functionals. Grimme's D3 dispersion correction35 was used in all DFT calculations. At the BLYP, BP86 and TPSS levels, the resolution of the identity (RI) approximation36 was used to speed up the computations. Calculations were performed using the NWChem program package, version 6.3.37Calculations were performed using Karlsruhe basis sets.38,39 Geometry optimizations were performed using the split-valence polarization basis sets (def2-SVP). For single point calculations we employed the triple-ζ quality basis sets augmented with polarization functions (def2-TZVPP). Basis-set convergence tests were performed using a quadruple-ζ, polarized basis set (def2-QZVPP).
The conductor-like screening model (COSMO)40 was used to model solvent effects. As the interactions studied occur in the outside of the protein, a dielectric constant of 80 was used. Tests were performed with other values for the dielectric constant in order to validate the methodology.
Computational efficiency considerations were not a priority when choosing the size of the reduced models. Rather, each model was constructed using as many residues as necessary in order to isolate each contact point. Because of the previous, the size of the models for different contact points varies considerably.
Two different schemes were used to cap aminoacidic residues included in the reduced models. The first scheme was used when atoms from the protein backbone were involved in the interaction, or where contiguous residues had to be included, while the second scheme was used for residues with only the side chain involved in the interaction and not contiguous to other residues included in the model.
In the first scheme used, the residues were included up to the backbone carboxyl group of the residue preceding the N-terminal residue of the fragment and up to the backbone nitrogen of the residue after the C-terminal residue in the fragment. The α carbons bonded to these carbon and nitrogen atoms were replaced with hydrogens, and these carbon and nitrogen atoms were kept fixed during geometry optimizations.
In the second scheme, only the side chains of the corresponding residues were considered. For each residue, the bond between the α and β carbons was cut, and the α carbons were replaced by hydrogens. The positions of the β carbons were kept fixed during geometry optimization.
The carbonyl group in the backbone of the residue T39 was added to point 2, in order to maintain the hydrogen bond between that residue and the H43 and R43 residues (the latter in the mutant structure). The group was added as a methyl amide, with its carbon and nitrogen atoms fixed during optimizations. In interaction points that included metal ligands (H120 in point 2 and H71 in point 5), the coordinating atom was kept fixed during geometry optimizations in order to keep the metal-bound orientation.
2.2 Molecular dynamics simulations
Molecular dynamics (MD) simulations were performed using the GROMACS package, version 4.6.44 The protein was described using the AMBER99sb-ildn force field.45,46 For the active site, we used the force field parameters previously developed by us,47 which are consistent with AMBER force fields. A 2 fs integration step and a 1 ps sampling step were used. Periodic boundary conditions were applied in all directions with a box size that ensured a distance of at least 2 nm between images. Long-range interactions were calculated using the particle mesh Ewald (PME) algorithm,48,49 while other nonbonded interactions were cut at 14 Å. To account for truncation, a dispersion correction was applied to the energy and pressure.Prior to the simulations, the proteins were solvated in simple point charge water,50 and 4 Na+ ions were added to neutralize the system. Further ions were added to a final concentration of 0.1 M NaCl.
The whole box was subjected to energy minimization using the steepest descent algorithm. The resulting structure was equilibrated for 0.1 ns in a NVT ensemble at 310 K and later a NPT ensemble at 310 K and 1 atm. The Berendsen thermostat and barostat were used for equilibration.51
The simulations were run for 27 ns. The Nosé–Hoover thermostat52,53 and the Parrinello–Rahman barostat54,55 were used to produce an NPT ensemble, at 310 K and 1 atm. The first 7 ns of each simulation were left for equilibration and not considered in the analyses.
3 Results and discussion
The contact points between the electrostatic loop and other structural elements of the SOD1 protein were identified by visual inspection of the high-resolution crystallographic structure for the reduced protein.8 3 out of 5 of these sites were found to contain residues whose substitutions are linked to fALS.5,56 The interaction sites are shown in Fig. 1. Detailed information about the found sites is shown in Table 1. Contact points between different parts of the electrostatic loop were also found. Some of these sites are the subject of ALS-linked mutations, and they appear to play a role in the structural integrity of the loop. As the present study focuses on the loop–protein interaction, the intra-loop interaction sites, some of which have been studied previously,28,57 were not considered.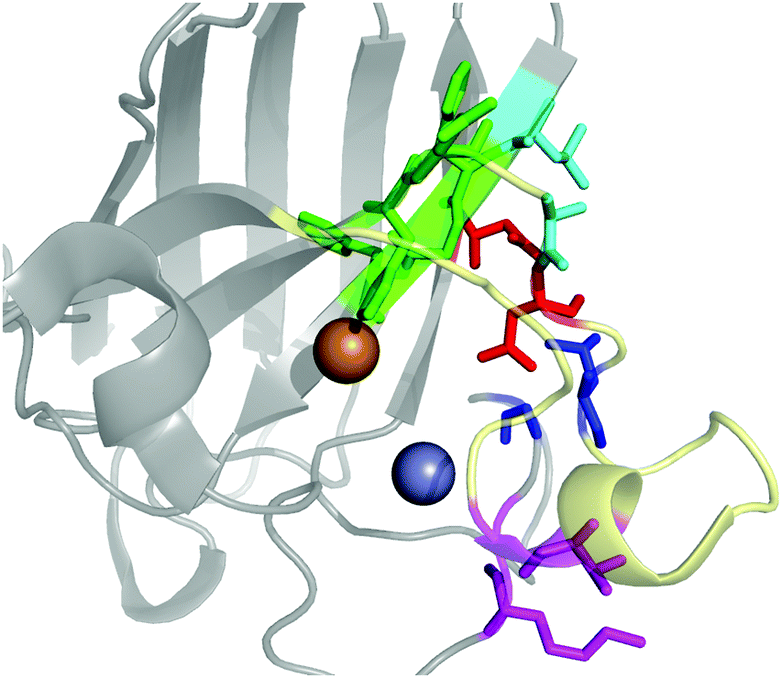 | ||
| Fig. 1 The electrostatic loop of the crystallographic structure of SOD1 and its surrounding structural elements. The loop is shown in pale yellow. Interaction sites between the electrostatic loop and the body of the protein are depicted in colors. | ||
| Site | Color | Body residues | E-loop residues | Mutations |
|---|---|---|---|---|
| Point 1 | Red | N86 | D124 | N86D, N86S, N86I, N86K |
| Point 2 | Green | H43, G44 | H120, E121, K122, A123 | H43R |
| Point 3 | Blue | G73 | L126 | L126S |
| Point 4 | Cyan | L42 | A123 | None |
| Point 5 | Magenta | K70, H71 | T135, K136 | None |
3.1 Quantum chemistry study of the contact points and their associated fALS-linked mutations
As the effect of each contact point needed to be isolated, the use of reduced models seems appropriate for the present study. The size of the models was determined by chemical, rather than computational considerations. The chosen criteria were to isolate distinct interactions, avoiding the overlap of different ones. The reduced models were produced for each contact point as described in Section 3, and interaction energies were calculated for each contact point and its related mutants. Quantum-chemical interaction energies for all the ALS-linked SOD1 mutants studied in this work are compiled in Table 2.| Contact point | Mutations | E (kcal mol−1) |
|---|---|---|
| Point 1 | WT | −8.5 |
| N86D | −0.9 | |
| N86S | −1.9 | |
| N86I | 4.2 | |
| N86K | −6.1 | |
| Point 2 | WT | −20.5 |
| H43R | −16.4 | |
| Point 3 | WT | −3.4 |
| L126S | −2.7 | |
| Point 4 | WT | −1.8 |
| Point 5 | WT | −5.8 |
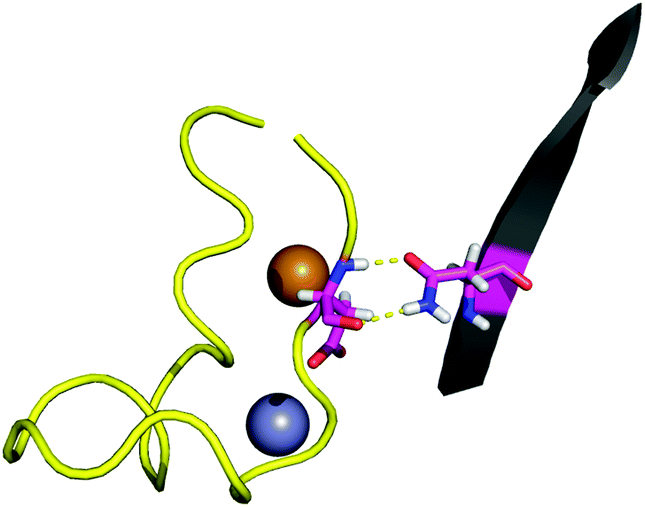 | ||
| Fig. 2 The interaction between the side-chain of N86 and the backbone of D124. | ||
The BLYP-D3/def2-TZVPP interaction energy for the wild-type (WT) point 1 is of −8.5 kcal mol−1. Since the D3 dispersion correction is a simple term added to the energy, it allows easy computation of its contribution. In this case −5.6 kcal mol−1 out of the total energy difference is given by dispersion forces. The interaction energy is over five times weaker than the interaction energy between D124 and the metal site of the protein,28 which also keeps D124 in its position. The small value of the interaction energy of the D124–N86 interaction in comparison to that of the D124–metal-site interactions suggests that the latter is mainly responsible for keeping D124 fixed. We have previously shown that D124–metal-site interaction in SOD1 is highly dependent on the dielectric constant of the environment.28 The electrostatic loop, of which D124 is a part, was proposed to have the role of blocking the access of solvent to the metal-site. The location of the D124–N86 interaction in an electrostatic-loop–protein contact point, as well as its strength, indicates that it plays a role in keeping the solvent barrier which maintains the low dielectric needed for the D124–metal-site interaction.
Although not very accurate for covalent bond energies, the BLYP-D3 density functional has been found reliable for weak interactions. Benchmark calculations report a mean absolute deviation (MAD) of 0.24 kcal mol−1 for the S22 set, which is highly relevant for the present case.30 The same work reports an average MAD of 1.1 kcal mol−1 for a meta-set including S22. For the meta-set, and especially for the S22 set, BLYP-D3 performance is comparable to MP2 methods and also to hybrid and double-hybrid functionals.30 To ensure that the calculated energies are not an artifact of a particular functional, we performed single-point energy calculations on point 1 using two additional density functionals, dielectric constants and basis sets. The results are shown in Table 3. The table shows that the results are largely independent of the chosen density functional. The energy obtained using a quadruple-ζ basis set indicates that basis set convergence is mostly reached at the triple-ζ level. Regarding structures, the interaction energy using def2-TZVPP-optimized structures shows no significant difference to that obtained using double-ζ-optimized structures. The table also indicates that this particular interaction, unlike the one between D124 and the metal site of SOD1, is not highly sensitive to the dielectric of the environment. BLYP-D3/def2-TZVPP with COSMOε=80 is considered an appropriate method for the calculation of interaction energies for the present work, while the use of the def2-SVP basis set is considered to be sufficient for geometry optimizations.
| Functional | Basis set | ε | E (kcal mol−1) |
|---|---|---|---|
| BLYP-D3 | def2-TZVPP | 80 | −8.5 |
| BLYP-D3 | def2-TZVPP(opt) | 80 | −8.8 |
| TPSS-D3 | def2-TZVPP | 80 | −8.4 |
| MPW1B95-D3 | def2-TZVPP | 80 | −7.0 |
| BLYP-D3 | def2-QZVPP | 80 | −7.8 |
| BLYP-D3 | def2-TZVPP | 20 | −8.9 |
| BLYP-D3 | def2-TZVPP | 4 | −10.7 |
An interesting ALS-linked mutant involving point 1 is the N86D mutant. The mutation is expected to strengthen the hydrogen bond between the residue 86 and the backbone nitrogen of D124. The second hydrogen bond present in the N86–D124, between the amide nitrogen in N86 and the backbone carboxylate of D124 is not only eliminated, but replaced by a repulsive interaction. The calculated interaction energy for the mutant pair D86–D124 is −0.9 kcal mol−1, i.e. within the expected error for the methodology.30 The attractive and repulsive interactions cancel each other and the interaction with D124 disappears. A different mutation, N86S, completely eliminates one of the hydrogen bonds, while the other is damaged due to the smaller size of the residue of serine in the mutant compared to the asparagine residue found in the WT variant. As expected, the interaction energy is also strongly reduced for the S86–D124 pair, to −1.9 kcal mol−1.
The mutation N86I has the obvious effect of eliminating both hydrogen bonds, and replacing glutamine with the more bulky and non-polar residue isoleucine. An unfavorable interaction dominated by steric tension is expected for this mutant. Indeed, calculations give an interaction energy of 4.2 kcal mol−1.
The mutation N86K differs from the other mutations in point 1 in that it has little effect in the DFT-calculated interaction energy of the contact point, with an energy of −6.1 kcal mol−1. Because of the larger size of the lysine residue in comparison to the asparagine present in the WT structure, we explored the possibility of steric clashes between K86 and residues that cannot be considered part of the contact point. Interaction energies were calculated for larger models of 111 and 124 atoms (compared to the original 35-atoms model for the mutant contact point). The effect of the larger models on the interaction energies was minor (see the ESI†), indicating that the interaction energy of point 1 is not significantly altered by the N86K mutation, and that no important steric effects arise from the mutation.
For all but one of the mutations analyzed, the interaction in point 1 is severely affected, being completely eliminated by the N86D and N86I mutations, and greatly reduced by the N86S mutation. On the other hand, assuming a favorable conformation for the interaction, the mutation N86K has a negligible effect in the interaction energy of point 1. The mutants N86D, N86K, and N86S have been reported by Byström et al. to produce alterations in the structure of apo-SOD1.58 Such alterations would be expected if the side chain of N86 was critical in keeping the electrostatic loop docked to the rest of the protein. While our results are consistent with Byström et al.'s findings for the N86D and N86S mutants, the quantum chemical interaction energies are unable to explain the effect of the role of N86K mutation in the structure of SOD1. It is possible that the effect of the N86K mutation is related to solvent or other effects not considered in the statical view provided by single-point QM calculations. Hence, the mutation was studied by classical molecular dynamics simulations, as discussed in Section 3.2.1.
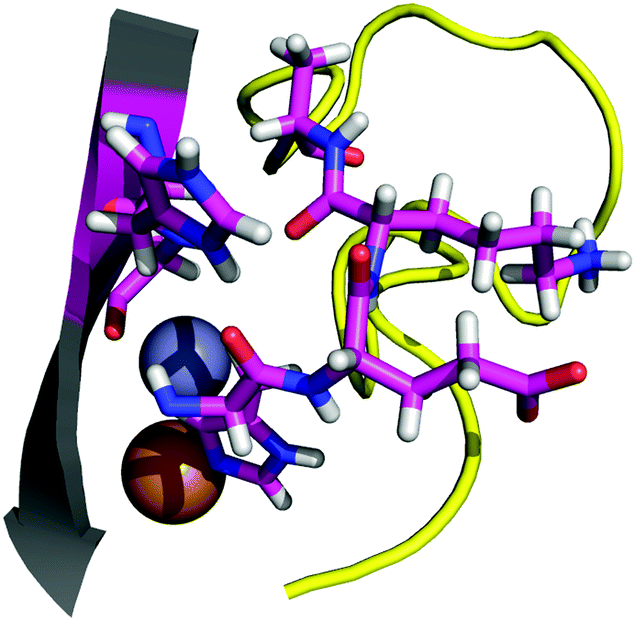 | ||
| Fig. 3 The interaction between the six aminoacidic residues involved in point 2. | ||
The total interaction energy calculated for point 2 is −20.5 kcal mol−1. The interaction is, as for the other cases, dominated by dispersion forces, as the removal of the D3 correction yields an unfavorable interaction of 10.8 kcal mol−1. For the H43R mutant, the interaction between the two fragments is weakened to −16.4 kcal mol−1. Relative to the wild-type energy, a variation of the interaction by about 4 kcal mol−1 seems a rather mild effect for the ALS-linked mutation. In absolute terms, however, the weakening of the interaction is comparable with the effect of the already studied N86S mutation. Considering the previous discussion, we modelled a system using the H43G mutation, which eliminates any side chain contribution from the residue 43 to the interaction. The interaction energy obtained for the H43G mutant is −12 kcal mol−1. The results indicate two things: first, the H43–H120 interaction is likely the main interaction in point 2, accounting for almost half of the total interaction energy for the contact point. Second, the ALS-linked H43R mutation greatly damages that interaction, reducing its interaction energy by almost half. Experimental studies by Kitamura et al.59 and Fujimaki et al.60 on the H43R mutant of SOD1 report conformations with altered metal binding. These conformations become pro-oxidant in the presence of copper, which appears to occupy the zinc binding site. Previous computational work at the classical level describes augmented mobility of the zinc-site and the electrostatic loop.61 Danielsson et al. studied the engineered SOD1 mutant H43F and found that the H43–H120 interaction is important for metal binding and for the dynamics of the protein.62 Our results are in line with the findings from previous studies, which point to alterations in the solvent seal and a loss of affinity for metals. Because the H120 residue is a copper ligand, and on the very corner of the electrostatic loop (normally not considered part of it ref. 6), it is not clear to what extent the breakage of the solvent seal is relevant in the H43R case, and to what extent the pathogenic effect on this mutant is linked to the destabilization of the H120 backbone, which would directly damage the metal site.
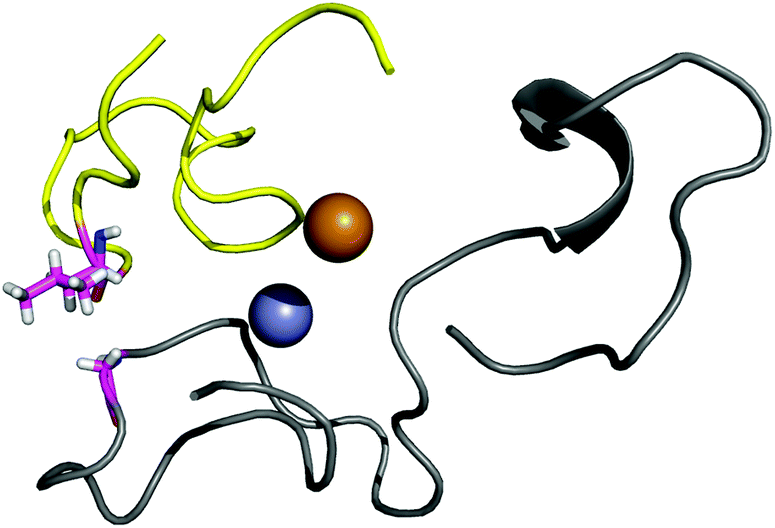 | ||
| Fig. 4 The interaction between the residues G73 and L126. | ||
 | ||
| Fig. 5 The interaction between the residues L42 and A123. | ||
The calculations indicate small interaction energies for both points: −3.4 kcal mol−1 for point 3 and −1.8 kcal mol−1 for point 4. While the interaction is stronger for the point affected by an ALS-mutation, the difference is very small.
For both contact points, only dispersive interactions are expected. In agreement with that, the interactions are unfavorable when the D3 dispersion correction is not considered (3.8 and 3.2 kcal mol−1 for points 3 and 4, respectively).
For point 3, the ALS-linked mutation has practically no effect in the interaction energy, with a total of −2.7 kcal mol−1 for the mutant structure. In point 4, the leucine can be replaced by a glycine, thus producing the in silico mutation L42G, without an effect in the interaction energy for that point. The mutated point 4 has an interaction energy of −1.3 kcal mol−1.
The results suggest that the effect of the L126S mutation in SOD1 is unrelated to the interaction between the electrostatic loop and the rest of the protein. The main difference between both contact points is their spacial location in the protein. The effect of the location of each interaction point cannot be treated with reduced models, so we analyze the effect of these mutants by employing classical methods in Section 3.2.2.
 | ||
| Fig. 6 The interaction between the four residues involved in point 5. | ||
The quantum chemistry studies performed indicate that the ALS-linked mutations in the contact points between the electrostatic loop and the rest of the SOD1 structure have the effect of significantly diminishing the interaction energy of those contact points. We have found that all residues with a side-chain that contributes to the interaction are subjects of ALS-linked mutations. The previous study indicates that the electrostatic loop's detachment to the rest of SOD1 does participate in the pathological function of ALS-linked SOD mutants.
For both the N86K and the L126S mutants, the pathological role of aminoacidic substitution could not be explained by the quantum-chemistry calculations performed. In the latter case, the interaction of the contact point is weak, and the contribution of the side chain of L126 to it is negligible.
3.2 Molecular dynamics simulations
In order to elucidate the effect of ALS-linked mutations N86K in point 1, and L126S in point 3, the mutations were studied using force-field molecular dynamics. For the wild-type and the two mutant structures, three MD simulations of 27 ns were performed, with 20 ns of production trajectories obtained from each of them. Thus, the total simulation time amounts to 60 ns for each structure.The RMSD values for the backbones shown in Fig. 7 and in the ESI,† indicate that the folding of both the mutant and the wild-type protein is very stable along the trajectories, including the initial 7 ns which were left for equilibration. The RMSD values also indicate that the time scales used in this work do not allow for the sampling of considerable backbone transitions, but only fast motions involving aminoacidic sidechains, such as those studied in this work.
 | ||
| Fig. 7 RMSD for the backbone of one of the three trajectories for the L126S mutant protein against the starting structure, after least-squares fit of the backbones. The RMSD for the other trajectories are given in the ESI.† | ||
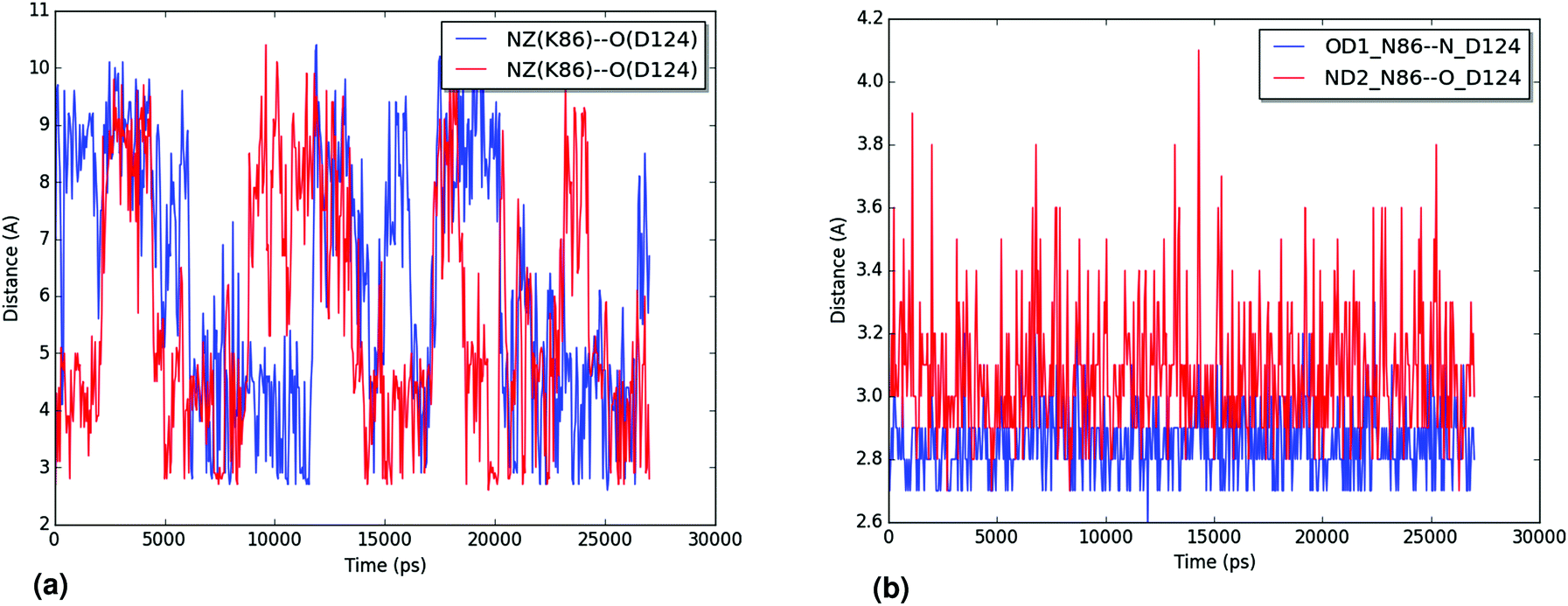 | ||
| Fig. 8 (a) Distances for the interaction between the ζ-nitrogen of K86 and the backbone oxygen of D124 along one of the trajectories for the N86K mutant structure. The distances are shown in blue and red for each monomer of the protein. (b) Distances for the interaction between the δ1 oxygen and δ2 nitrogen of N86 and the backbone nitrogen and oxygen of D124, in blue and red, respectively, along one of the trajectories for the WT structure. Only one of the monomers is shown. For both graphics, atoms are labeled following the PDB convention. | ||
The hydration of the relevant atoms of the 86 residue (the Nζ for K86 and the Oδ1 and Nδ2 for N86), shown for all trajectories in the ESI,† reveals a similar picture: the nitrogen in the K86 residue interacts more closely with the solvent than the heteroatoms in the sidechain of N86 do. Both analyses reveal the effect of the N86K mutation, which leads to an impaired interaction in the contact point 1. Our results, which agree with the experimental findings of Byström et al.,58 clarify that the impaired interaction caused by the N86K mutant is given by a more favorable interaction of K86 with the solvent, and/or by the increased degrees of freedom of the lysine residue, compared to the original asparagine. Additional studies are suggested in order to establish the exact contribution of both factors.
We analyzed the trajectories for events of breaking of the D124–H46 interaction in the wild-type and mutant trajectories, defining dissociation as a D124–H46 bond length of over 2.5 Å. In Table 4 it can be seen that there is a large difference in the number of dissociation events for the wild-type trajectories and that for the L126S trajectories. The mutant trajectory with the least events still over twice the amount of them as compared to the wild-type trajectory with the most events. Although the number of dissociation events is clearly higher in the mutant trajectories, it is important to also study for how long D124–H46 stays dissociated after each event, i.e. how long the events last. For all the trajectories, most of the dissociation events have a duration of 1 ps (one frame) or less. When we remove these extremely short dissociation events, a comparison of the durations is possible. A summary of the data is shown in Table 5, while detailed numbers are given in the ESI.† In Table 5 it can be seen that the wild-type trajectories do not have dissociation events longer than 1 ps, except for one event in trajectory WT-1, with 2 ps duration. The picture is very different for the mutant trajectories, where the trajectory with the least dissociation events contains 19 of them, and all of the trajectories contain events of at least 49 ps. As the duration of dissociation events does not follow a normal distribution (particularly for the WT trajectories), a statistical analysis of these quantities was not performed.
| Trajectory | Dissociation events |
|---|---|
| WT-1 | 23 |
| WT-2 | 10 |
| WT-3 | 16 |
| L126S-1 | 52 |
| L126S-2 | 79 |
| L126S-3 | 74 |
| Trajectory | Events | Longest event | Average duration |
|---|---|---|---|
| WT-1 | 1 | 2 | 2 |
| WT-2 | 0 | N/A | N/A |
| WT-3 | 0 | N/A | N/A |
| L126S-1 | 21 | 113 | 20.8 |
| L126S-2 | 19 | 49 | 11.8 |
| L126S-3 | 35 | 87 | 23.2 |
The analysis of the MD trajectories clearly shows that the mutation L126S has the effect of placing a serine residue in a place where it can compete with the D124–H46 interaction. Hence, unlike the other ALS-linked mutations studied in this work, the effect of the L126S mutation is to directly impair the secondary bridge, rather than indirectly damaging it by allowing the solvent to enter the metal site.
4 Conclusions
The pathogenic mechanism of the ALS-linked mutants of SOD1 has been for long considered an important step towards understanding the pathogenesis of both fALS and sALS.14 In this work, we provide a unified pathogenic mechanism for several ALS-linked SOD1 mutations. Our results show that contact-points between the electrostatic loop of SOD1 and the rest of the protein are affected by ALS-linked mutations, except for those contact points with no or minimal side-chain involvement in the interaction. The mutations were found to damage the loop–protein interaction. The one mutation in a residue with minimal side-chain involvement (L126S) was found to directly damage the secondary bridge by forming a hydrogen bond between S126 and D124.Every site where a mutation could affect the interaction between the electrostatic loop and the rest of SOD1 is indeed affected by an ALS-causing mutation, and those mutations are shown to have that effect. Hence, our results indicate that the interaction between the electrostatic loop and the rest of the SOD1 protein is important for the pathogenesis of SOD1-linked ALS in general. The conclusion is in agreement with our previously-proposed role of the electrostatic loop as a seal preventing solvent from accessing the metal site of SOD1. The solvent seal would prevent water from reaching the metal site and breaking the hydrogen bonds in the secondary bridge.28 A general role of the electrostatic loop in SOD1-linked ALS is in agreement with destabilization of the loop being a common feature of ALS-linked SOD1 mutants, measured as an increased H/D exchange in the electrostatic loop in 13 different ALS-linked mutants.29
Our results provide not only a pathogenic mechanism for five ALS-linked SOD1 mutants, but also a connection between a general structural feature of ALS-linked SOD1 mutants29 with the loss of affinity of the protein for metals, particularly zinc. A diminished affinity of SOD1 for metals is expected to affect the general stability of SOD1. Mulligan et al. report that, under denaturating conditions, zinc release and dimer dissociation occur simultaneously.16 In line with the latter results, the mutants N86S, N86D and N86K have been experimentally found to weaken the dimer interface.58 Zinc dishomeostasis, another probable consequence of the loss of affinity of SOD1 for the metal, has also been connected to familial and sporadic ALS pathogenesis.19–23
Acknowledgements
RM-A thanks Profs. Fernando Mendizábal and Sebastián Miranda-Rojas for useful discussion, as well as financial support under Conicyt-Aka-ERNC-001. Financial support by the Excellence Cluster EXC114 (CIPSM) and the Volkswagen Stiftung within the funding initiative of ‘New Conceptual Approaches to Modeling and Simulation of Complex systems’ is acknowledged. Kim Novacek and Jasmin Dums are grafetully acknowledged for help with some of the QM calculations.References
- M. C. Kiernan, S. Vucic, B. C. Cheah, M. R. Turner, A. Eisen, O. Hardiman, J. R. Burrell and M. C. Zoing, Lancet, 2011, 377, 942–955 CrossRef CAS.
- R. G. Miller, J. D. Mitchell, M. Lyon and D. H. Moore, Cochrane Db. Syst. Rev., 2007, CD001447 CAS.
- A. Bento-Abreu, P. Van Damme, L. Van Den Bosch and W. Robberecht, Eur. J. Neurosci., 2010, 31, 2247–2265 CrossRef PubMed.
- A. E. Renton, A. Chiò and B. J. Traynor, Nat. Neurosci., 2014, 17, 17–23 CrossRef CAS PubMed.
- O. Abel, ALS online genetic database, http://alsod.iop.kcl.ac.uk/.
- R. Rakhit and A. Chakrabartty, Biochim. Biophys. Acta, 2006, 1762, 1025–1037 CrossRef CAS PubMed.
- Y. Sakurai, I. Anzai and Y. Furukawa, J. Biol. Chem., 2014, 289, 20139–20149 CrossRef CAS PubMed.
- R. W. Strange, S. Antonyuk, M. A. Hough, P. A. Doucette, J. S. Valentine and S. S. Hasnain, J. Mol. Biol., 2006, 356, 1152–1162 CrossRef CAS PubMed.
- M. A. Hough and S. S. Hasnain, J. Mol. Biol., 1999, 287, 579–592 CrossRef CAS PubMed.
- R. J. F. Branco, P. A. Fernandez and M. J. Ramos, J. Phys. Chem. B, 2006, 110, 16754–16762 CrossRef CAS PubMed.
- S. V. Seetharaman, D. D. Winkler, A. B. Taylor, X. Cao, L. J. Whitson, P. A. Doucette, J. S. Valentine, V. Schirf, B. Demeler, M. C. Carroll, V. C. Culotta and P. J. Hart, Biochemistry, 2010, 49, 5714–5725 CrossRef CAS PubMed.
- L. Banci, I. Bertini, D. E. Cabelli, R. A. Hallewell, J. W. Tung and M. S. Viezzoli, Eur. J. Biochem., 1991, 196, 123–128 CrossRef CAS PubMed.
- R. Mera-Adasme, F. Mendizábal, M. Gonzalez, S. Miranda-Rojas, C. Olea-Azar and D. Sundholm, Inorg. Chem., 2012, 51, 5561–5568 CrossRef CAS PubMed.
- J. S. Valentine and P. J. Hart, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3617–3622 CrossRef CAS PubMed.
- J. Perry, D. Shin, E. Getzoff and J. Tainer, Biochim. Biophys. Acta, 2010, 1804, 245–262 CrossRef CAS PubMed.
- V. K. Mulligan, A. Kerman, S. Ho and A. Chakrabartty, J. Mol. Biol., 2008, 383, 424–436 CrossRef CAS PubMed.
- K. Homma, T. Fujisawa, N. Tsuburaya, N. Yamaguchi, H. Kadowaki, K. Takeda, H. Nishitoh, A. Matsuzawa, I. Naguro and H. Ichijo, Mol. Cell, 2013, 52, 75–86 CrossRef CAS PubMed.
- J. R. Williams, E. Trias, P. R. Beilby, N. I. Lopez, E. M. Labut, C. S. Bradford, B. R. Roberts, E. J. McAllum, P. J. Crouch, T. W. Rhoads, C. Pereira, M. Son, J. L. Elliott, M. C. Franco, A. G. Estévez, L. Barbeito and J. S. Beckman, Neurobiol. Dis., 2016, 89, 1–9 CrossRef CAS PubMed.
- J. B. Hilton, A. R. White and P. J. Crouch, J. Mol. Med., 2015, 93, 481–487 CrossRef CAS PubMed.
- S. Barber and P. J. Shaw, Nat. Rev. Neurosci., 2001, 2, 806–819 CrossRef PubMed.
- A. P. Smith and N. M. Lee, Amyotrophic Lateral Scler., 2007, 8, 131–143 CrossRef CAS PubMed.
- J. Kim, T.-Y. Kim, J. J. Hwang, J.-Y. Lee, J.-H. Shin, B. J. Gwag and J.-Y. Koh, Neurobiol. Dis., 2009, 34, 221–229 CrossRef CAS PubMed.
- M. Kaneko, T. Noguchi, S. Ikegami, T. Sakurai, A. Kakita, Y. Toyoshima, T. Kambe, M. Yamada, M. Inden, H. Hara, K. Oyanagi, T. Inuzuka, H. Takahashi and I. Hozumi, J. Neurosci. Res., 2015, 93, 370–379 CrossRef CAS PubMed.
- E. Tokuda, S.-I. Ono, K. Ishige, A. Naganuma, Y. Ito and T. Suzuki, Toxicology, 2007, 229, 33–41 CrossRef CAS PubMed.
- K. Puttaparthi, W. L. Gitomer, U. Krishnan, M. Son, B. Rajendran and J. L. Elliott, J. Neurosci., 2002, 22, 8790–8796 CAS.
- K. Hashimoto, Y. Hayashi, K. Watabe, T. Inuzuka and I. Hozumi, Neuroscience, 2011, 189, 293–298 CrossRef CAS PubMed.
- S. Nagano, M. Satoh, H. Sumi, H. Fujimura, C. Tohyama, T. Yanagihara and S. Sakoda, Eur. J. Neurosci., 2001, 13, 1363–1370 CrossRef CAS PubMed.
- R. Mera-Adasme, C.-M. Suomivuori, A. Fierro, J. Pesonen and D. Sundholm, JBIC, J. Biol. Inorg. Chem., 2013, 18, 931–938 CrossRef CAS PubMed.
- K. S. Molnar, N. M. Karabacak, J. L. Johnson, Q. Wang, A. Tiwari, L. J. Hayward, S. J. Coales, Y. Hamuro and J. N. Agar, J. Biol. Chem., 2009, 284, 30965–30973 CrossRef CAS PubMed.
- L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2004, 108, 6908–6918 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 242, 652–660 CrossRef CAS.
- M. Valiev, E. Bylaska, N. Govind, K. Kowalski, T. Straatsma, H. Van Dam, D. Wang, J. Nieplocha, E. Apra, T. Windus and W. de Jong, Comput. Phys. Commun., 2010, 181, 1477–1489 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- J. Word, S. C. Lovell, J. S. Richardson and D. C. Richardson, J. Mol. Biol., 1999, 285, 1735–1747 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Van der meersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 1–17 Search PubMed.
- R. Mera-Adasme, G. Savasci and J. Pesonen, goChem: A library for computational chemistry, 2013, http://www.gochem.org.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins, 2006, 65, 712–725 CrossRef CAS PubMed.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins, 2010, 78, 1950–1958 CAS.
- R. Mera-Adasme, K. Sadeghian, D. Sundholm and C. Ochsenfeld, J. Phys. Chem. B, 2014, 118, 13106–13111 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. Perkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8592 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren and J. Hermans, Interaction models for water in relation to protein hydration, Reidel, Dordrecht, The Netherlands, 1981, p. 331 Search PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef.
- S. Nosé and M. Klein, Mol. Phys., 1983, 50, 1055–1076 CrossRef.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- C. M. Lill, O. Abel, L. Bertram and A. Al-Chalabi, Amyotrophic Lateral Scler., 2011, 12, 238–249 CrossRef PubMed.
- S. P. Keerthana and P. Kolandaivel, RSC Adv., 2015, 5, 34630–34644 RSC.
- R. Byström, P. M. Andersen, G. Gröbner and M. Oliveberg, J. Biol. Chem., 2010, 285, 19544–19552 CrossRef PubMed.
- F. Kitamura, N. Fujimaki, W. Okita, H. Hiramatsu and H. Takeuchi, Biochemistry, 2011, 50, 4242–4250 CrossRef CAS PubMed.
- N. Fujimaki, F. Kitamura and H. Takeuchi, Biochemistry, 2013, 52, 5184–5194 CrossRef CAS PubMed.
- G. Muneeswaran, S. Kartheeswaran, K. Muthukumar, C. D. Dharmaraj and C. Karunakaran, Biophys. Chem., 2014, 185, 70–78 CrossRef CAS PubMed.
- J. Danielsson, W. Awad, K. Saraboji, M. Kurnik, L. Lang, L. Leinartaité, S. L. Marklund, D. T. Logan and M. Oliveberg, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3829–3834 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6mt00085a |
| ‡ Present address: Departamento de Ciencias del Ambiente, Facultad de Química y Biología, Universidad de Santiago de Chile, Chile. |
| This journal is © The Royal Society of Chemistry 2016 |