
Measurement of sample and plasma properties in solution-cathode glow discharge and effects of organic additives on these properties
Christian G.
Decker
and
Michael R.
Webb
*
Department of Chemistry and Biochemistry, University of North Carolina Wilmington, Wilmington, NC 28403, USA. E-mail: webbm@uncw.edu; Fax: +1 910 962 3013; Tel: +1 910 962 7285
First published on 27th July 2015
Abstract
Solution-cathode glow discharge was studied in an attempt to further elucidate the processes involved in the plasma. Spectroscopic and electrical properties were measured with and without the influence of three organic modifiers: formic acid, acetic acid, and ethanol. Degradation products (CO, C2, and CH) of the modifiers were detected in the plasma spectroscopically. Properties of solutions before and after exposure to the discharge were compared. The effects of organic additives on these solution properties were measured. A single, simple mechanism was not consistent with the data, so a multi-part mechanism is proposed for solution and analyte transport into the plasma. Evaporation from the cathode surface, droplet generation, and chemical generation of volatile species may each play a role.
Introduction
For just over two decades, electric discharges where one electrode is an electrolytic aqueous solution have been used in analytical atomic spectrometry. The first report of such system was from Cserfalvi et al.,1 who used electrolyte cathode discharge (ELCAD) in 1993 to detect metallic elements via optical emission spectrometry (OES). A slightly different design, introduced in 2005 by Webb et al.,2 was named solution-cathode glow discharge (SCGD). Jamroz et al.3–5 refer to a similar system as an atmospheric pressure glow discharge (APGD) generated in contact with a flowing liquid cathode. The present paper deals with a recently described descendent of the SCGD design,6 and is also referred to as an SCGD. Although there are differences among the versions of these discharges, they are similar enough that results can be cautiously translated among them. Results from more distantly related systems may also provide insights, but these results should be treated with more caution due to greater differences among the systems. These related discharges include APGD in contact with a flowing liquid cathode with a He or Ar jet,7 alternating-current electrolyte atmosphere liquid discharge (ac-EALD),8,9 and liquid-sampling – atmospheric pressure glow discharge (LS-APGD).10,11 Several reviews13–17 detail the development of all these discharges.SCGD and related discharges have several advantages over more established analytical atomic spectrometry instruments. Unlike flame atomic absorption spectrometry (FAAS) and inductively coupled plasma (ICP)-OES, they normally operate in atmospheric pressure air without compressed gasses. They use little power (∼70 W to power the discharge) compared to ICPs (1–1.5 kW to power the plasma). SCGDs produce relatively few (primarily atomic) analyte emission lines, so they require relatively low spectral resolution.6 The small size, low power requirements, lack of compressed gasses, and moderate spectral resolution mean that the instrument has a small footprint, which gives SCGDs potential as portable instruments or detectors incorporated into chromatographs.12 Some of the same attributes lead to low construction and operation costs.
Early studies focused on calibration with simple solutions and studies of the discharges' operating mechanisms. Recent studies have shifted to applications involving more complex samples. Since 2009, these discharges have been applied to tea infusions,3 tea leaves,3 mineral water,3 tuna fish,19–21 aquatic plant matter,19 oyster tissue,21 coal fly ash,21,22 groundwater,22 hepatitis-B vaccine,20 lake water,5 soil leachates,5 spruce needle leachates,5 colloidal silica,23 zirconium alloys,24,25 simulated natural water,8 honey,15 human hair,26,27 and stream sediment.26,27 This shift towards applications is vital for a technique to become truly relevant,18 but there is a continued need for more basic research. The mechanisms involved in solution and analyte transport are not entirely understood, and recent studies have shown that additives can affect analyte signals through processes that have only begun to be explained.
Concentration1 and identity28 of the sample solution's electrolyte have long been known to affect the strength of atomic emission. More recently, organic additives such as formic acid, acetic acid, ethanol, and non-ionic surfactants have been found to significantly enhance analyte emission. Before the effect was found in discharges with direct OES detection, Zhu et al.29 used SCGD to generate mercury vapour, which was then flowed into an ICP-OES for detection. They found that formic acid, acetic acid, or ethanol in the sample enhanced mercury emission in the ICP. Shekhar et al.19 later found that the same additives enhanced mercury signals when emission from the discharge itself was measured. Other studies9,24,25 have used this enhancement.
Doroski and Webb30 studied the effects of formic acid, acetic acid, and ethanol on a variety of metal analytes. Analyte elements were found to fall into two groups: those that are known to be amenable to chemical vapour generation (Hg, Se, Ag, and Pb) and those that do not undergo vapour generation as readily (Ca, Fe, Cu, Mg, and Ni). The vapour formers showed relatively large signal enhancements, and the non-vapour formers showed smaller signal enhancements or even suppression. The effects of 3% by volume of the additives on analyte signal varied even within each of the two groups, but the general trends found in that study will be a useful comparison for the results presented in the current work. Excluding Fe, which was not detected in ethanol at the analytical wavelength, the average changes in signal for the analytes that are not known to form volatile species were +10% for formic acid, +40% for acetic acid, and −30% for ethanol. For the vapour formers except Hg, the average changes in signal were +470% for formic acid, +210% for acetic acid, and +80% for ethanol. All three additives increased Hg emission by comparable amounts to each other – roughly +1000%. The present study compares the properties of solution before and after exposure to the plasma and measures how organic additives affect these properties. It also measures how additives affect the spectroscopic and electrical properties of the plasma. The results of all of these measurements will be used to gain insight into both the normal operation of solution-cathode glow discharge and the operation of the discharge with organic additives.
Similar enhancement has also been observed using non-ionic4,5,9,15,22 and ionic surfactants.27 That effect will not be studied directly here, but it seems likely that there is overlap between the enhancement mechanisms.
Experimental
In these experiments, a discharge was maintained in a 3 mm vertical gap between a tungsten rod and a solution emerging from a glass tube. The details of this arrangement have been described extensively in previous papers.6,30 The diameter of the tungsten anode was altered for the current study. A 4 mm diameter tungsten rod with a rounded point was used as the anode in place of the 2 mm diameter tungsten rod that was used previously. The voltage, which was provided to the anode through a 1 kΩ resistor, was adjusted until an average current of 70 mA was established through the discharge. The voltage was maintained at this level while the current was allowed to fluctuate. Approximately 1 kV was required, but the voltage varied slightly depending on the solution composition.Light was collected and directed to a spectrograph by a pair of 25 mm diameter, 50 mm focal length fused silica lenses and a fiber optic cable. Details of the optical arrangement can be found in an earlier paper.6 Two spectrographs were used. An previously described6 Ocean Optics Maya2000 Pro spectrograph covered wavelengths 189–413 nm with a resolution of approximately 0.35 nm. An Ocean Optics USB4000 spectrograph covered 177–890 nm with a resolution of approximately 2 nm. The USB4000 is a 9 × 6 × 3 cm spectrograph with a 3648 pixel linear CCD (Toshiba TCD1304AP). The configuration used in this study had a 600 line per mm grating blazed at 300 nm, and a 25 μm entrance slit. Although the USB4000 covers the ultraviolet range, it was only used at wavelength greater than 400 nm because it has lower resolution and lower sensitivity than the Maya2000 Pro.
The solution was provided as described previously6 using a Spectec Perimax peristaltic pump with Spectec Antipuls tubing. The peristaltic pump was used to provide a sample flow at 2.7 mL min−1 and an electrolyte flow at 1.8 mL min−1.
Conductivity was measure with a YSI Model 35 conductance meter. Solution pH was measured with a VWR SympHony probe attached to an Orion 520A pH meter. Current and voltage were measured by connecting a National Instruments USB-6009 to the voltage and current monitors of the power supply. The current and voltage data was collected using a National Instruments LabVIEW program that was written in-house. The program records 1 data point every second, where that data point is the average of 900 measurements.
All standards were prepared by dilution of high-purity 1000 ppm solutions (SPEX Certiprep for Cu, SPEX Certiprep or Ricca for Ni, Ricca for Hg, and BDH Aristar Plus for all others). Nitric acid (Optima grade, Fisher Scientific), formic acid (analysis grade, Acros Organics), acetic acid (Optima grade, Fisher Scientific), and ethanol (USP grade, AAPER) solutions were all prepared by diluting concentrated solutions using water filtered by a Milli-Q system.
Results and discussion
Sample consumption
Sample consumption was measured as the difference between the mass going to waste when the discharge was off and the mass going to waste when the discharge was on. Each measurement was the average of 5–10 individual 25 minute long collections of waste. The uncertainty of the balance was neglected in error propagation because it was generally about 500-fold smaller than the standard deviation of replicates of the same experiment. The measured masses were converted to rates. The rates with the plasma off were defined as solution delivery rates, and the differences between the solution delivery rates and the corresponding rates with the plasma off were defined as solution consumption rates. These rates can be seen in Table 1. Also in the table are the results of the same experiments with sample solutions containing 0.1 mol L−1 nitric acid and 3% formic acid, 3% acetic acid, or 3% ethanol. Here and throughout this paper, percent composition refers to percent by volume.| Additive | Solution delivery rate (g min−1) | Solution consumption rate (g min−1) | Voltage (V) | Current (mA) | Power (W) |
|---|---|---|---|---|---|
| None | 4.51 ± 0.02 | 0.474 ± 0.008 | 956.9 ± 0.7 | 69 ± 3 | 66 ± 3 |
| 3% formic acid | 4.51 ± 0.01 | 0.54 ± 0.03 | 989.7 ± 0.8 | 70 ± 4 | 69 ± 3 |
| 3% acetic acid | 4.52 ± 0.02 | 0.49 ± 0.03 | 1016.7 ± 0.7 | 69 ± 3 | 71 ± 3 |
| 3% ethanol | 4.43 ± 0.01 | 0.42 ± 0.01 | 954.2 ± 0.7 | 69 ± 3 | 66 ± 3 |
The changes in sample consumption for solutions modified with formic acid and ethanol qualitatively agree with the signal enhancement for non-vapour formers. Formic acid causes both a small increase in sample consumption and an increase in signal. Ethanol causes both a small decrease in sample consumption and a decrease in signal. Acetic acid does not fit the trend as well. It causes a smaller increase in sample consumption than formic acid does, but it causes the largest signal enhancement. One possible interpretation of these data is that multiple mechanisms are responsible for sample loss and that not all of them contribute to analyte signal.
Evaporation from the cathode surface would be expected to remove the volatile solvents disproportionately to the non-volatile solutes. Small concentrations of formic acid increase the boiling point of an aqueous solution only slightly. At 2.7× the formic acid concentration used here, the boiling point of a formic acid–water mixture is 102.3 °C.31 Small concentrations of acetic acid increase the boiling point by a greater degree. At 2.3× the acetic acid concentration used here, the boiling point of an acetic acid–water mixture is 116.5 °C.31 Small concentrations of ethanol decrease the boiling point slightly. At 1.9× the ethanol concentration used here, the boiling point of an ethanol–water mixture is 95.0 °C.32 Were this the only factor, formic acid would be expected to only slightly decrease sample consumption, acetic acid would be expected to decrease sample consumption by a greater amount, and ethanol would be expected to slightly increase sample consumption. Evaporation alone cannot explain the observed differences in sample consumption.
Schwartz et al.33 observed droplets ejected from the solution surface and proposed that an electrospray-like mechanism was responsible for their generation. Whatever their mechanism of creation, the droplets are responsible for carrying some of the sample solution into the plasma. Analyte is most likely also carried into the plasma in these droplets. Additives might affect the size and/or number of droplets. They might also affect how much analyte is incorporated into the droplets and/or how easily the solvent is removed from the droplets. This combination of factors means that droplet generation could account for the discrepancies in both signal and sample consumption relative to what is expected from a purely evaporative mechanism, but we have not yet been able to clearly establish a link between the observed droplets and sample consumption or analyte signal.
Electrical characteristics
The current and voltage of the power supply were measured with each solution. Measurements were made every 1 s for 10 minute intervals. The results can be found in Table 1. Somewhat higher voltages were required to maintain the same current when formic or acetic acid were used as additives. The measurements are not corrected for the power and voltage lost to the 1 kΩ ballast resistor or the solution itself, both of which are in series with the discharge. The resistor accounts for 4.8 W and 69 V at a current of 69 mA and 4.9 W and 70 V at a current of 70 mA. The resistance of the solution cannot be as easily accounted for because it depends not only on the conductivity of the solution (discussed later), but also on the length and cross-sectional area of the solution connecting the cathode to the graphite rod. The higher powers applied when formic acid and acetic acid are used could contribute to signal enhancement using these additives, but no corresponding decrease in power was observed that would explain signal suppression in ethanol. Applied power may be a factor in the enhancement mechanism, but it seems unlikely to be the predominant one.Sample properties
Several properties of the solutions used in the discharge were measured, and these properties were measured both for the solution entering the discharge and for the waste collected after the discharge. The results of these measurements for conductivity, pH, and density are shown in Table 2. Conductivity was slightly decreased by any of the additives. All else being equal, this conductivity change would increase the voltage that would need to be applied to maintain a certain current. Plasmas with formic acid or acetic acid do require more voltage, but plasmas with ethanol do not, even though ethanol results in the lowest conductivity. It seems unlikely that the solution conductivity causes a significant portion of the change in the electrical characteristics of the discharge. More notably, the conductivities of all of the post-discharge solutions are higher than their corresponding pre-discharge solutions. Jamroz et al.34 detected NH4+, NO3−, and NO2− generated in solutions after they flowed through a system similar to our SCGD. Their system used HCl as the supporting electrolyte, so the production mechanism of the N-containing species presumably involves N2 from the air. Estimating from their graphs, the concentrations generated at ∼3.3 mL min−1 were ∼0.05 mg L−1 NO2−, ∼1 mg L−1 NH4+, and ∼40 mg L−1 NO3−, and these concentrations showed a general decreasing trend as flow rate increased. Considering the 6 g L−1 concentration of nitric acid used in our discharge, these specific species seem unlikely to be the primary cause of the conductivity increase. The conductivity is primarily a function of the concentration of nitric acid, so this change suggests that water is lost at a greater rate than nitric acid.| Additive | Pre-discharge conductivity (mS cm−1) | Post-discharge conductivity (mS cm−1) | Pre-discharge pH | Post-discharge pH | Pre-discharge density (g mL−1) | Post-discharge density (g mL−1) |
|---|---|---|---|---|---|---|
| None | 37.5 | 40.5 | 1.09 | 1.08 | 1.001 ± 0.001 | 1.000 ± 0.002 |
| 3% formic acid | 36.8 | 40.7 | 1.03 | 1.00 | 1.012 ± 0.001 | 1.013 ± 0.002 |
| 3% acetic acid | 36.3 | 41.1 | 1.11 | 1.07 | 1.005 ± 0.002 | 1.005 ± 0.002 |
| 3% ethanol | 36.1 | 40.4 | 1.24 | 1.19 | 0.9967 ± 0.0009 | 0.997 ± 0.004 |
Additives affected the measured pH. Ethanol changes the junction potential in pH measurements, so meaningful comparisons cannot be easily made between solutions with different water–ethanol ratios.35 It seems likely that 3% formic acid or acetic acid could cause a similar effect. If the solvent composition is constant, some meaning can be drawn, so comparisons will only be made between pre- and post-discharge solutions with the same additive. In all cases, post-discharge pH was found to be lower than pre-discharge pH, although the changes were around the limit of measurable differences for the pH probe used. Jamroz et al.34 found a similar result, with an increase in acid concentration from 0.1 mol L−1 (added as hydrochloric acid) to ∼0.12 mol L−1 at a solution flow rate of ∼3.3 mL min−1. Similarly to the conductivity trend, the pH trend could be caused by water being lost faster than nitric acid. Plasma chemical processes may also affect the pH.
The densities of the various solutions were determined by measuring the mass of the solutions in 25 mL volumetric flasks at a temperature of approximately 20 °C. Six samples of each solution were measured, and the averages and standard deviations of these measurements are reported in Table 2. The 0.1 mol L−1 nitric acid solution had a density slightly higher than pure water. Adding formic acid or acetic acid shifted this density slightly upwards, and adding ethanol shifted this density slightly downwards. All of these trends are as would be expected from past measurements.36 The densities of the solutions collected from the discharge waste showed no clear difference from their corresponding initial solutions, but relatively large changes in the concentrations of nitric acid or the additives would have been necessary for it to have been detected in this way.
Surface tension and viscosity were not measured, but the effects of the additives on these properties were investigated in the literature because all three may affect solution vaporization. Surface tension was estimated based on data from two papers where Vazquez et al.37,38 report the surface tension of aqueous solutions with various concentrations of formic acid,37 acetic acid,37 and ethanol38 over a range of temperatures. The highest temperature at which measurements were made was 50 °C. Pure water was found to have a surface tension of 67.92 mN m−1 at this temperature. Interpolating to find the surface tension at the correct concentration, we estimate that at 50 °C, the surface tensions are 65.2 mN m−1 for 3% formic acid, 61.7 mN m−1 for 3% acetic acid, and 60.4 mN m−1 for 3% ethanol. Extrapolating to higher temperatures yields lower values, but with the same relative ranking of surface tensions. Aqueous solutions with small additions of formic acid, acetic acid, or ethanol are known to have higher viscosities than water.39,40 There are no clear correlations between signal enhancement and the surface tension or viscosity. Ethanol–water solutions have the lowest surface tension, but ethanol suppresses signal for many elements. All three additives increase viscosity, but ethanol suppresses signal while the others enhance it.
Spectral changes
Spectra were acquired for an aqueous solution of 0.1 mol L−1 nitric acid and for solutions of 0.1 mol L−1 nitric acid modified with 3% formic acid, 3% acetic acid, or 3% ethanol.Fig. 1 shows the spectra taken using the Maya2000 Pro spectrometer from 190–400 nm. Fig. 2 shows the spectra taken using the USB4000 spectrometer from 400–800 nm. The results for the unmodified nitric acid solution were in line with spectra from similar systems.1,2,41 A series of very faint bands from NO was observed between about 190 and 250 nm (the NO γ system).42 The most prominent spectral feature was OH emission, which consisted of a strong band starting at 306 nm and falling off towards longer wavelengths and two similar but weaker bands at 281 and 261 nm (the 3064 Å system).42 Emission from N2 had bandheads from 337 to 436 nm (the second positive system).42 Emission from H was detected at 486 and 656 nm. Emission from Na at 589 and 590 nm appeared as one peak with a slight dip in the center due to the approximately 0.35 nm resolution of the spectrograph. Three closely spaced O lines appeared as one line at 777 nm. A Hg line at 546 nm was detected whether the discharge was on or off, and is assumed to be from fluorescent lights.
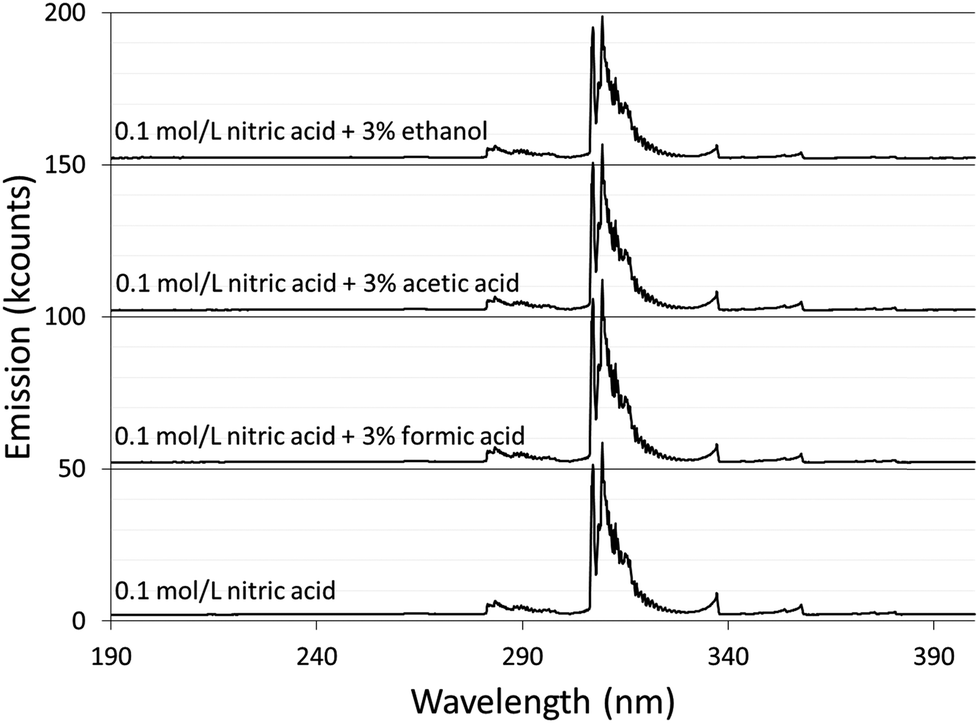 | ||
| Fig. 1 Background spectra from 190 to 400 nm of solution-cathode glow discharge with 0.1 mol L−1 nitric acid and modified with 3% by volume of formic acid, acetic acid, or ethanol. For visibility, each spectrum is offset 50k counts compared to the one below it. | ||
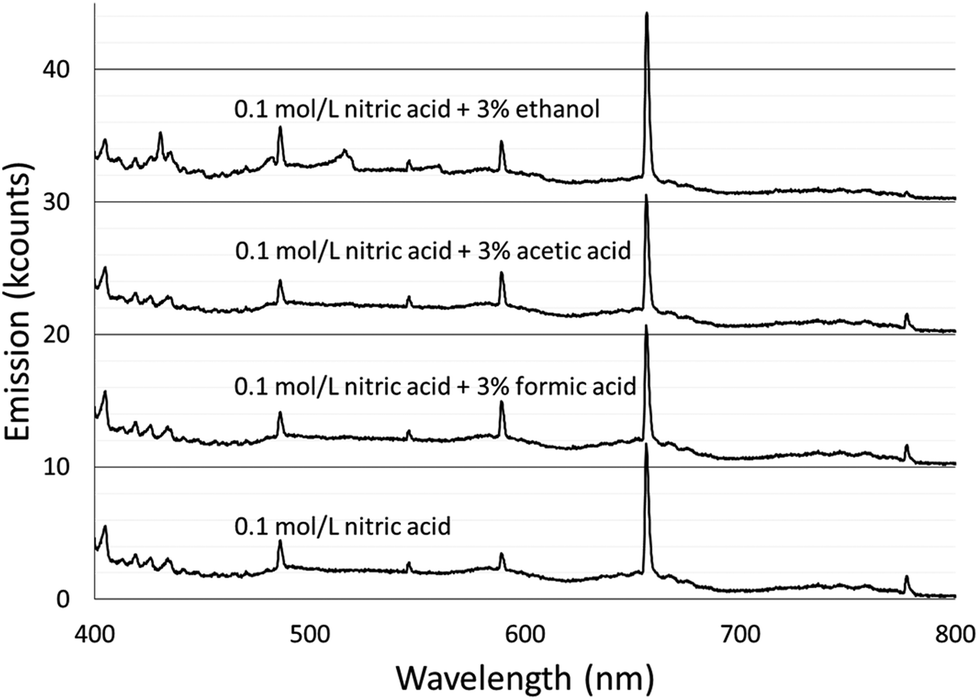 | ||
| Fig. 2 Background spectra from 400 to 800 nm of solution-cathode glow discharge with 0.1 mol L−1 nitric acid and modified with 3% by volume of formic acid, acetic acid, or ethanol. For visibility, each spectrum is offset 10k counts compared to the one below it. | ||
The majority of the spectral features described above showed little change with the addition of acetic or formic acid, but larger intensity changes were caused by the addition of ethanol. At the peak OH emission wavelength, 309 nm, observed intensities changed +6% with formic acid, −3% with acetic acid, and −17% with ethanol. Emission from H at 656 nm changed −9% with formic acid, −10% with acetic acid, and +20% with ethanol. At the peak N2 emission wavelength, observed intensities changed −13% with formic acid, −9% with acetic acid, and −31% with ethanol.
The additives also caused new bands to appear. Ethanol produced several weak bands from about 410 to 605 nm, as shown in Fig. 3. The low resolution of the USB4000 spectrograph, the weakness of the bands, and interference from N2 bands make some of the weaker bands difficult to confidently identify, but the largest bandhead at 431 nm matches CH (the 4300 Å system)42 and the second largest at 517 nm matches C2 (the Swan system).42 The general shapes and approximate wavelengths of the remainder of the bands appear to also be from C2. No CH or C2 bands were observed in the discharge with the unmodified nitric acid solution or the discharges with solutions modified by formic or acetic acid. All three additives introduce new bands in the 190–250 nm range. Fig. 4 shows the spectra for the unmodified solution as well as for solutions modified with formic acid or ethanol. The corresponding spectrum with acetic acid is nearly identical to the spectrum with formic acid. These bands can be seen weakly when formic acid or acetic acid is used as an additive, but they are interfered with by NO emission, which is of comparable strength. When ethanol is used as an additive, the new bands are much more prominent and can be clearly identified as CO bands (the fourth positive system).42,43
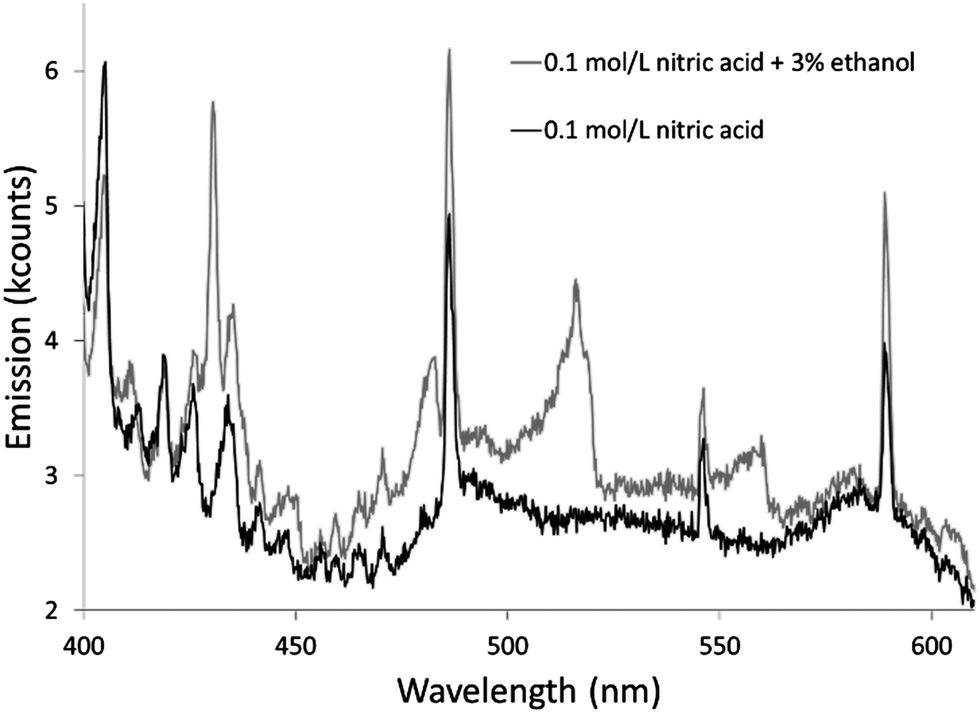 | ||
| Fig. 3 Background spectra from 400 to 610 nm of solution-cathode glow discharge with 0.1 mol L−1 nitric acid and modified with 3% by volume ethanol. Emission from CH and C2 causes the differences in the spectra. | ||
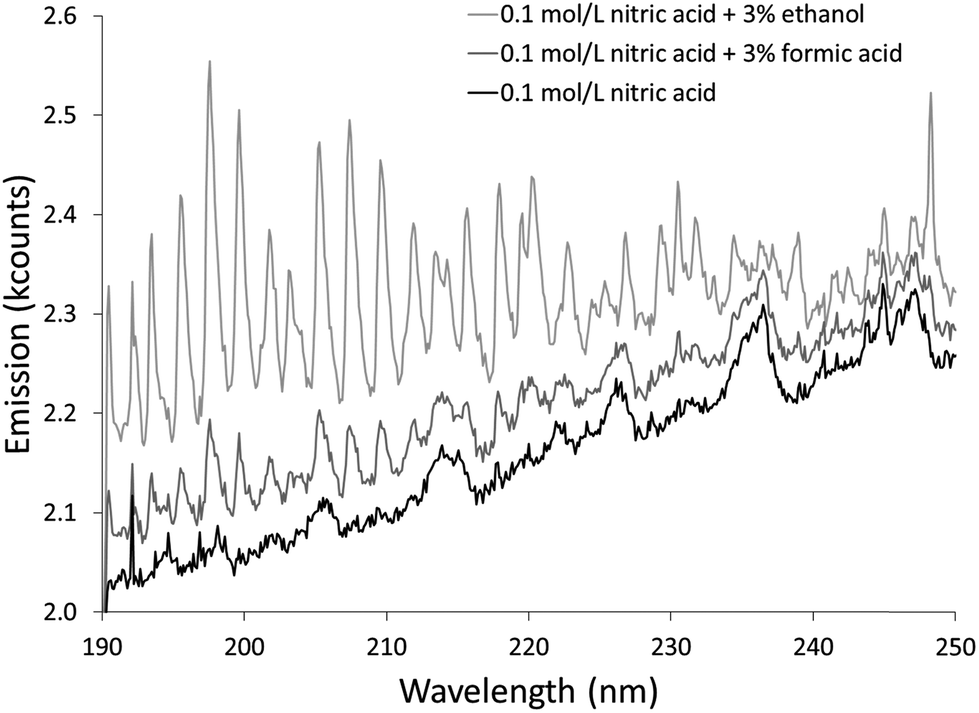 | ||
| Fig. 4 Background spectra from 190 to 250 nm of solution-cathode glow discharge with 0.1 mol L−1 nitric acid and modified with 3% by volume ethanol or formic acid. Emission from CO causes the differences in the spectra. | ||
The relatively low resolution of the spectrographs makes it difficult to find areas of continuum emission without band structure, but there was no obvious shift in the strength of the background continuum in these areas. For example, the average emission between 500 and 540 nm changed by −0.3% with formic acid and +1% with acetic acid. Ethanol caused an increase in this area, but that was due to the appearance of C2 emission.
These results can be compared to those found by Greda et al.4,5 They used non-ionic surfactants as additives and found that general background, NO, OH, and N2 emission decreased with any of the surfactants they tested (Triton x-45, Triton x-405, Triton x-705, and Triton x-100).5 They did not measure the effects of these surfactants on H emission. They noted that they could not find any C2, CH, or CO emission.4,5
All of the observed changes in the spectra cannot be assigned to one simple cause. Some changes may be due in part to changes in the amounts of certain chemicals in the plasma. For example, the amount of OH in the plasma is tied to the amount of water entering the plasma. In turn, the amount of water entering the plasma is linked to the amount of solution entering the plasma and the fraction of that solution that is water. Formic acid causes the largest increase in OH emission and also has the largest increase in sample consumption. Ethanol causes a decrease in OH emission and also causes a decrease in sample consumption. For all three, the additive slightly reduces the fraction of the sample solution that is water, but (as discussed earlier) the portion of the sample entering the plasma may not have identical composition to the original sample solution.
Emission from carbon containing species clearly results from an increase in these species (formed by degradation of the additives) in the plasma. This emission is highest with ethanol, which may mean that ethanol enters the plasma at a greater rate than formic acid or acetic acid, perhaps due to ethanol's much lower boiling point. It may also mean that ethanol is more easily decomposed in the plasma.
The evidence of decomposition products from ethanol and the organic acids is relevant to the proposed vapour generation mechanism. Photochemical vapour generation is known to be possible using low molecular weight organic acids (including formic acid and acetic acid),44 and the detection of CO from all three additives at least indicates that the additives are reacting, although it remains to be demonstrated that they react with the analytes. That ethanol shows the strongest emission from carbon-containing species but the weakest enhancement of vapour formers may at first seem at odds, but it can be at least qualitatively explained by considering the droplet generation mechanism that is believed to usually introduce the analyte. If the droplet generation mechanism and the vapour generation mechanisms operate independently, a suppression in the droplet generation mechanism could partly cancel out an enhancement due to vapour generation, resulting in a lesser total enhancement than would otherwise be expected. If the two mechanisms are coupled, the effects could be even more dramatic. That is, if vapour generation volatilizes analytes from droplets in the plasma, rather than from the solution surface, a suppression in droplet generation combined with a large degree of vapour generation could produce only a small overall enhancement of signal.
Other emission features are less easily explained by their concentrations in the plasma. H presumably primarily arises from water entering the plasma, but it is affected differently by the additives than OH is. N2 presumably enters the plasma from the atmosphere, and this process seems unlikely to be strongly affected by an additive. Emission from these species may be affected by their interactions with other species in the plasma. It may not be a coincidence that the largest changes in emission from these species occurs when the most emission from carbon-containing species is present.
In addition to the concentration of a species in the plasma, the energetics of the plasma also affect the amount of emission. Two measures of these energetics are described in the next section.
Temperature measurements
Features from emission spectra can also be used as thermal probes. The OH rotational temperature is commonly used as a proxy for the gas-kinetic temperature, but the OH formation process also influences the measured temperature.45 Although the OH rotational temperature can therefore not be treated as synonymous with the gas-kinetic temperature, it still may provide insight into the energetics of plasmas that are similar to each other.The OH emission band at 306 nm has been used to determine the OH rotational temperature of an earlier version of an SCGD.2 In this study, the relative intensities of individual lines within the OH band were used to generate a plot based on the Boltzmann distribution, and the slope of this plot was used to calculate the OH rotational temperature. This method requires higher resolution than the spectrographs we are using provide. Instead, we used LIFBASE46 to simulate spectra at different temperatures and compared these spectra to experimental spectra. Settings for the simulated spectra were: 305–330 nm wavelength range, 0.35 nm resolution, 1 atmosphere pressure. Spectra were simulated in increments 100 K, and the R2 of the fit to the experimental data was calculated. For each experimental spectrum, there was a clear smooth trend in R2 with an easily identifiable maximum. The spectrum of the discharge using the 0.1 mol L−1 aqueous nitric acid solution without additive fit a temperature of 4000 K with an R2 of 0.9975, as shown in Fig. 5. This temperature is higher than was found in an earlier SCGD study.2 In that study, the OH rotational temperature was found to vary vertically in the plasma between about 2800 and 3500 K, but was approximately 3000 K in the region we were observing for the present study. Complicating matters somewhat, the present study images an approximately 0.6 mm diameter spot, while the previous study limited the width of the observation area by using a narrow monochromator entrance slit. It is also likely that the difference between the ways that the temperatures were calculated here and in the previous paper contributed to the disparity, so a more controlled comparison would be needed before strong conclusions could be drawn. That said, there is at least one difference between the designs of the older SCGD and the newer SCGD that could affect the OH rotational temperature. The current system uses a smaller cathode than the earlier system did, which increases the power density. The increased power density may increase the rotational temperature. Adding credence to this supposition, a study of the original system found that another change to the system geometry – changing the distance between the anode and the cathode – affected the OH rotational temperature.
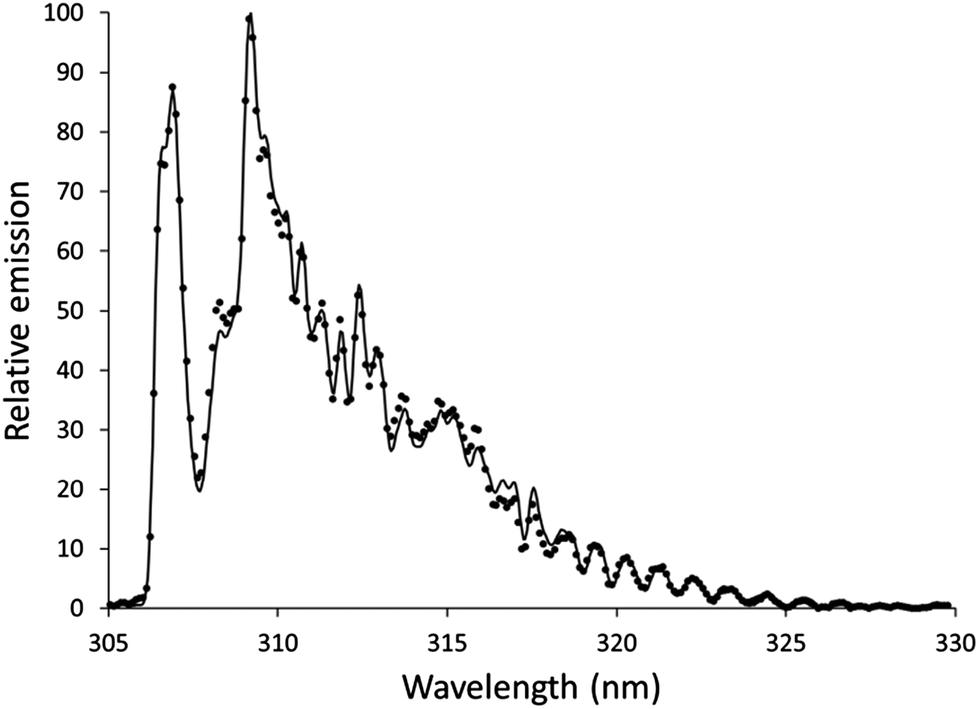 | ||
| Fig. 5 Example of an experimental spectrum (points) and a spectrum simulated using LIFBASE (line). The experimental spectrum is of solution cathode discharge using 0.1 mol L−1 nitric acid without additive, and the simulated spectrum is at a temperature of 4000 K. | ||
The procedure was repeated with the organic additives added to 0.1 mol L−1 nitric acid aqueous solution. With 3% formic acid, the best fit was found using a temperature of 3900 K, which gave an R2 of 0.9977. With 3% acetic acid, the temperature was 4000 K and the R2 was 0.9975. With 3% ethanol, the temperature was 4300 K and the R2 was 0.9974.
These temperatures do not correlate with the effects of the various additives on analyte signals. Formic acid and acetic acid cause little or no change in the observed temperature, even though they both cause measureable analyte signal enhancement. Ethanol causes an increase in the observed temperature, even though it causes analyte signal depression. To the degree that OH rotational temperature is related to gas-kinetic temperature, higher temperatures would be expected to cause greater desolvation and atomization, both leading to greater analyte signals. The lack of connection between the observed OH rotational temperature and the strength of analyte emission either means that the connection between the two temperatures is too weak for the OH rotational temperature to be a useful proxy here or that larger effects from other processes mask the influence of temperature on analyte signal.
The Fe excitation temperature was also measured spectroscopically. Each background-corrected spectrum was calculated based on the average of 4 spectra from solutions containing 20 mg L−1 Fe and the average of 4 spectra from blanks. Before taking the difference between the two averaged spectra, the blank was scaled to eliminate any offset. An example background-corrected spectrum is shown in Fig. 6. The spectra were used to calculate the Fe excitation temperature using a Boltzmann plot as described by Sung and Lim.47 The 371.993, 374.948, 382.043, and 385.991 nm lines were used in the plot. The 373.486 and 373.713 nm lines were not used because they were not sufficiently resolved. The Boltzmann plot (Fig. 7) had an R2 of 0.998 and its slope corresponded to a temperature of 2930 ± 100 K. This temperature is much lower than the 5930 K that was measured in the older SCGD system.48 As with the OH rotational temperature measurements, the area from which emission was collected is likely to have affected the measured temperature. It is also likely that the choice of emission lines accounts for at least part of this discrepancy. The Fe temperature of the older SCGD was measured using all the lines that were used in the present study plus 3 lines that could not be resolved well enough to be used here.48 As with the OH rotational temperature measurements, comparisons between Fe excitation temperature of the older SCGD and that of the newer one should be made with caution until a more controlled comparison can be made. Comparisons among temperatures measured for the current system under different conditions are likely to be more meaningful.
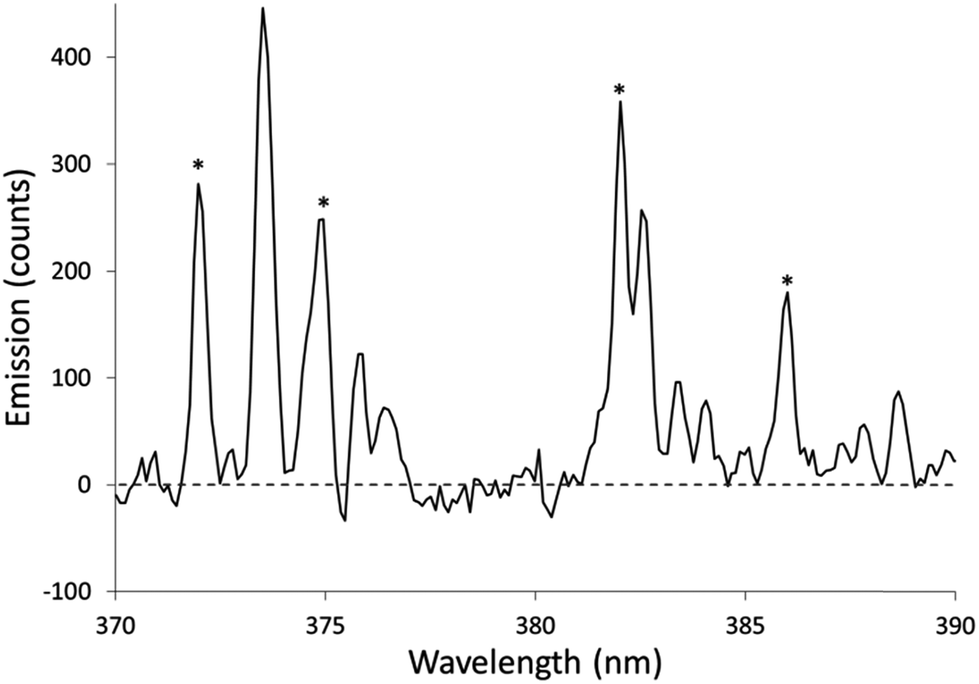 | ||
| Fig. 6 Example of a background-corrected spectrum used to calculate an Fe excitation temperature. The solutions used contained 0.1 mol L−1 nitric acid without additive. The sample solution contained 20 mg L−1 Fe. Asterisks (*) show the locations of lines used for temperature measurements. | ||
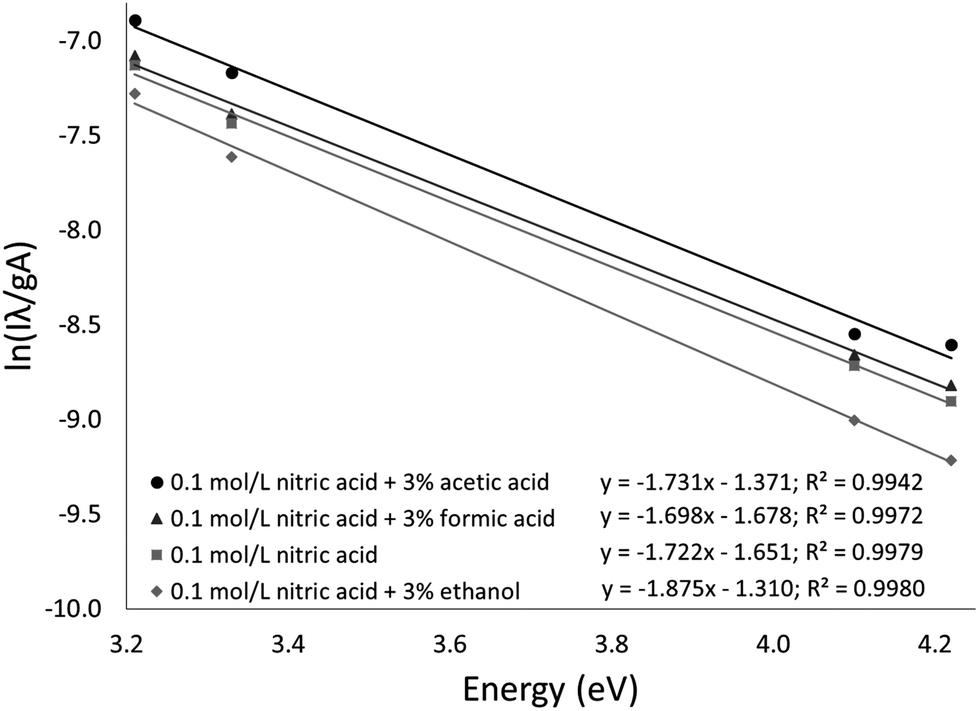 | ||
| Fig. 7 Boltzmann plots used to calculate Fe excitation temperatures. | ||
The procedure was repeated for solutions with 3% added formic acid, acetic acid, or ethanol. The resulting temperatures were 2970 ± 110 K for formic acid (R2 = 0.997), 2910 ± 160 K for acetic acid (R2 = 0.994), and 2690 ± 90 K for ethanol (R2 = 0.998). The Boltzmann plots used to calculate these temperatures are shown in Fig. 7. Formic acid and acetic acid do not appear to have a meaningful effect on the Fe excitation temperature. Ethanol is once again the outlier, but it decreases the Fe excitation temperature where it increased the OH rotational temperature. The decreased Fe excitation temperature may help to explain why emission of many elements is depressed by an ethanol additive. If all other factors were constant, a lower excitation temperature would lead to less emission. Overall, the rotational and excitation temperatures are not able to explain the observed signal enhancement, which suggests that transport (including atomization) plays a larger role than plasma energetics.
The effects of the various additives on the emission intensity of these Fe lines is also consistent with the data from other elements reported earlier30 and discussed above. The average changes in intensity of these Fe lines are +6% for formic acid, +20% for acetic acid, and −38% for ethanol.
Conclusions
We have measured electrical and spectroscopic properties of a solution-cathode glow discharge, and we have measured how these properties are altered by adding formic acid, acetic acid, or ethanol to the sample solution. We have also measured several properties of the samples solutions before and after contact with the discharge. The results do not point to a simple picture of either the normal operation of a SCGD or its operation with organic additives in the sample solution.The results are consistent with three coexisting mechanisms for sample consumption: (1) a mechanism (possibly evaporation from the cathode surface) that consumes sample while contributing little to the analyte emission; (2) a mechanism (possibly droplet generation) that contributes to the analyte signal and can be enhanced or suppressed by organic additives; and (3) a mechanism involving organic additives that produces volatile species containing certain elements. Although there is evidence for all three of these mechanisms, they have been difficult to observe, possibly because they are all present.
References
- T. Cserfalvi, P. Mezei and P. Apai, J. Phys. D: Appl. Phys., 1993, 26, 2184–2188 CrossRef CAS.
- M. R. Webb, F. J. Andrade, G. Gamez, R. McCrindle and G. M. Hieftje, J. Anal. At. Spectrom., 2005, 20, 1218–1225 RSC.
- P. Jamroz, P. Pohl and W. Zyrnicki, J. Anal. At. Spectrom., 2012, 27, 1032–1037 RSC.
- K. Greda, P. Jamroz and P. Pohl, J. Anal. At. Spectrom., 2013, 28, 134–141 RSC.
- K. Greda, P. Jamroz and P. Pohl, Talanta, 2013, 108, 74–82 CrossRef CAS PubMed.
- T. A. Doroski, A. M. King, M. P. Fritz and M. R. Webb, J. Anal. At. Spectrom., 2013, 28, 190–195 RSC.
- K. Greda, P. Jamroz and P. Pohl, J. Anal. At. Spectrom., 2013, 28, 1233–1241 RSC.
- R. M. Huang, Z. L. Zhu, H. T. Zheng, Z. F. Liu, S. C. Zhang and S. H. Hu, J. Anal. At. Spectrom., 2011, 26, 1178–1182 RSC.
- Q. Xiao, Z. Zhu, H. Zheng, H. He, C. Huang and S. Hu, Talanta, 2013, 106, 144–149 CrossRef CAS PubMed.
- W. C. Davis and R. K. Marcus, J. Anal. At. Spectrom., 2001, 16, 931–937 RSC.
- S. Konegger-Kappel, B. T. Manard, L. X. Zhang, T. Konegger and R. K. Marcus, J. Anal. At. Spectrom., 2015, 30, 285–295 RSC.
- A. J. Schwartz, Z. Wang, S. J. Ray and G. M. Hieftje, Anal. Chem., 2013, 85, 129–137 CrossRef CAS PubMed.
- M. A. Mottaleb, J. S. Yang and H. J. Kim, Appl. Spectrosc. Rev., 2002, 37, 247–273 CrossRef CAS.
- M. R. Webb and G. M. Hieftje, Anal. Chem., 2009, 81, 862–867 CrossRef CAS PubMed.
- K. Greda, P. Jamroz, A. Dzimitrowicz and P. Pohl, J. Anal. At. Spectrom., 2015, 30, 154–161 RSC.
- Q. He, Z. L. Zhu and S. H. Hu, Appl. Spectrosc. Rev., 2014, 49, 249–269 CrossRef CAS.
- P. Mezei and T. Cserfalvi, Appl. Spectrosc. Rev., 2007, 42, 573–604 CrossRef CAS.
- H. A. Laitinen, Anal. Chem., 1973, 45, 2305 CrossRef.
- R. Shekhar, Talanta, 2012, 93, 32–36 CrossRef CAS PubMed.
- R. Shekhar, D. Karunasagar, K. Dash and M. Ranjit, J. Anal. At. Spectrom., 2010, 25, 875–879 RSC.
- R. Shekhar, D. Karunasagar, M. Ranjit and J. Arunachalam, Anal. Chem., 2009, 81, 8157–8166 CrossRef CAS PubMed.
- R. Shekhar, K. Madhavi, N. N. Meeravali and S. J. Kumar, Anal. Methods, 2014, 6, 732–740 RSC.
- Z. Wang, A. J. Schwartz, S. J. Ray and G. M. Hieftje, J. Anal. At. Spectrom., 2013, 28, 234–240 RSC.
- R. Manjusha, M. A. Reddy, R. Shekhar and S. Jaikumar, J. Anal. At. Spectrom., 2013, 28, 1932–1939 RSC.
- R. Manjusha, M. A. Reddy, R. Shekhar and S. J. Kumar, Anal. Methods, 2014, 6, 9850–9856 RSC.
- Q. Li, Z. Zhang and Z. Wang, Anal. Chim. Acta, 2014, 845, 7–14 CrossRef CAS PubMed.
- Z. Zhang, Z. Wang, Q. Li, H. J. Zou and Y. Shi, Talanta, 2014, 119, 613–619 CrossRef CAS PubMed.
- P. Mezei, T. Cserfalvi, H. J. Kim and M. A. Mottaleb, Analyst, 2001, 126, 712–714 RSC.
- Z. L. Zhu, G. C. Y. Chan, S. J. Ray, X. R. Zhang and G. M. Hieftje, Anal. Chem., 2008, 80, 7043–7050 CrossRef CAS PubMed.
- T. A. Doroski and M. R. Webb, Spectrochim. Acta, Part A, 2013, 88, 40–45 CrossRef CAS.
- T. Ito and F. Yoshida, J. Chem. Eng. Data, 1963, 8, 315–320 CrossRef CAS.
- N. Kamihama, H. Matsuda, K. Kurihara, K. Tochigi and S. Oba, J. Chem. Eng. Data, 2012, 57, 339–344 CrossRef CAS.
- A. J. Schwartz, S. J. Ray, E. Elish, A. P. Storey, A. A. Rubinshtein, G. C. Y. Chan, K. P. Pfeuffer and G. M. Hieftje, Talanta, 2012, 102, 26–33 CrossRef CAS PubMed.
- P. Jamroz, K. Greda, P. Pohl and W. Zyrnicki, Plasma Chem. Plasma Process., 2014, 34, 25–37 CrossRef.
- R. G. Bates, M. Paabo and R. A. Robinson, J. Phys. Chem., 1963, 67, 1833–1838 CrossRef CAS.
- Perry's Chemical Engineers' Handbook, ed. R. H. Perry, D. W. Green and J. O. Maloney, McGraw-Hill, New York, 1999 Search PubMed.
- E. Alvarez, G. Vazquez, M. SanchezVilas, B. Sanjurjo and J. M. Navaza, J. Chem. Eng. Data, 1997, 42, 957–960 CrossRef.
- G. Vazquez, E. Alvarez and J. M. Navaza, J. Chem. Eng. Data, 1995, 40, 611–614 CrossRef CAS.
- P. B. David and H. C. Jones, J. Am. Chem. Soc., 1915, 37, 1194–1198 CrossRef.
- I. S. Khattab, F. Bandarkar, M. A. A. Fakhree and A. Jouyban, Korean J. Chem. Eng., 2012, 29, 812–817 CrossRef CAS.
- M. R. Webb, F. J. Andrade and G. M. Hieftje, Anal. Chem., 2007, 79, 7899–7905 CrossRef CAS PubMed.
- R. W. B. Pearse and A. G. Gaydon, The Identification of Molecular Spectra, John Wiley & Sons Inc., New York, 1963 Search PubMed.
- P. H. Krupenie, The Band Spectrum of Carbon Monoxide, National Bureau of Standards, Washington, D.C., 1966 Search PubMed.
- X. M. Guo, R. E. Sturgeon, Z. Mester and G. J. Gardner, Anal. Chem., 2003, 75, 2092–2099 CrossRef CAS PubMed.
- P. Bruggeman, D. C. Schram, M. G. Kong and C. Leys, Plasma Processes Polym., 2009, 6, 751–762 CrossRef CAS.
- J. Luque and D. R. Crosley, LIFBASE version 2.1.1, SRI International, Menlo Park, CA, 1999 Search PubMed.
- Y. I. Sung and H. B. Lim, J. Anal. At. Spectrom., 2003, 18, 897–901 RSC.
- M. R. Webb, F. J. Andrade and G. M. Hieftje, J. Anal. At. Spectrom., 2007, 22, 766–774 RSC.
| This journal is © The Royal Society of Chemistry 2016 |