
Emerging investigators series: virus mitigation by coagulation: recent discoveries and future directions
Joe
Heffron
and
Brooke K.
Mayer
*
Department of Civil, Construction and Environmental Engineering, Marquette University, Milwaukee, WI 53233, USA. E-mail: Brooke.Mayer@marquette.edu; Tel: 414 288 2161
First published on 12th April 2016
Abstract
Waterborne viruses are widespread and persistent in the environment. Coagulation is an effective process for mitigating viruses in drinking water. This review examines recent studies of virus mitigation by coagulation processes in the context of the latest scientific advances. Virus sorption is impacted by electrostatic forces, as well as the hydrophobic effect, steric hindrance, hydrodynamics and interactions with the water matrix. Organic matter in the water may hinder or enhance sorption, depending on virus structure and environmental factors. In addition to physical separation in flocs, coagulation processes have been shown to inactivate viruses. This review evaluates reports of virus inactivation due to coagulation processes from both a process and experimental perspective. The use of bacteriophages as surrogates for human viruses is discussed, and future research needs relevant to virus coagulation are identified.
 Joe Heffron | Joe Heffron is a PhD student at Marquette University. He earned his M.S. in civil engineering from Marquette and his B.A. in environmental science and policy from Drake University. Joe served as an environmental education volunteer in Peace Corps - Armenia from 2008–2010. Joe's interests include electrocoagulation for the mitigation of pathogens and heavy metals in drinking water and decentralized water treatment and reuse. |
 Brooke Mayer | Dr. Brooke Mayer is an Assistant Professor in the Department of Civil, Construction and Environmental Engineering at Marquette University. She graduated from the Environmental Engineering program at Arizona State University with her B.S. in 2004, M.S. in 2006, and Ph.D. in 2008. Brooke's research interests primarily relate to physicochemical water and wastewater treatment processes, with specific areas of emphasis in virus quantification and mitigation, removal/degradation of emerging chemical contaminants, and the waste-to-resource paradigm embodied by phosphorus recovery. |
Water impactViruses are ubiquitous contaminants in both surface and groundwaters. Mitigation of viruses by coagulation is an effective method to pre-treat water for disinfection or membrane filtration and thus reduce energy or chemical disinfection requirements. Understanding virus sorption and inactivation by coagulation has broader implications for water/wastewater treatment, as well as virus fate and persistence in the environment. |
1. Introduction
Waterborne viruses account for an estimated 30 to 40% of infectious diarrhea in the U.S.1 Associated with both acute gastric and respiratory diseases and chronic conditions,2 some viruses can persist for several months in the environment3 and travel up to 100 m in groundwater.4 Several families/genera of waterborne viruses are included on the U.S. Environmental Protection Agency's (EPA) Contaminant Candidate List (both CCL 3 and draft CCL 4) for drinking water, indicating the need for further research into occurrence and treatment.5 Likewise, the World Health Organization's (WHO) Guidelines for Drinking Water Quality6 cites eight virus categories that are of concern for drinking water, all of which have high persistence and infectivity relative to other pathogens. Although many viruses are only moderately tolerant of conventional water treatment,6,7 adenoviruses show high resistance to emerging treatment technologies such as UV disinfection.8 Coagulation can be used to reduce virus loads and minimize the required dose for disinfection. Coagulation is also an effective pre-treatment for virus removal by filtration systems.9–15This review assesses current research of virus reduction by coagulation. Future avenues for research are discussed in light of evidence of the forces influencing virus sorption, as well as recent findings of virus inactivation in coagulation processes. Three coagulation processes are considered: conventional chemical coagulation, enhanced coagulation and electrocoagulation. In conventional chemical coagulation, metal hydrolytes are formed by dissolution of a metal salt in water. Aluminum and iron salts such as Al2(SO4)3 and FeCl3 are commonly used in water treatment,16 although novel coagulants like polyaluminum chlorides (PACls) have gained particular attention for virus mitigation.9,10,17–22 Polymeric iron coagulants have also been developed,23,24 but these coagulants have not been evaluated for virus mitigation.
The EPA's Disinfectants and Disinfection Byproduct Rule (DBPR) promotes enhanced coagulation prior to disinfection of drinking water to prevent the formation of potentially carcinogenic disinfection byproducts.25 Enhanced coagulation uses high doses of chemical coagulant and/or pH adjustment for effective removal of humic acids, fulvic acids and other dissolved and suspended organic material (collectively called natural organic matter, NOM). For this reason, enhanced coagulation has been evaluated for virus mitigation in waters with high NOM concentrations.12,26,27
Electrocoagulation is the in situ production of coagulant by electro-oxidation of a sacrificial electrode. Both iron and aluminum sacrificial electrodes have been tested for virus mitigation.14,28–30 Some researchers consider aluminum electrodes preferable to iron electrodes, because iron is released as soluble ferrous ions and may not be fully oxidized to form insoluble coagulant.28,29,31 While the coagulant hydrolytes formed by iron electrocoagulation have been characterized,32,33 less is known about the species formed during aluminum coagulation.
In all variations of coagulation, metal cations are introduced in water to form hydrolyte species. These hydrolytes destabilize colloids by overcoming the repulsive forces between colloidal particles. Colloidal destabilization commonly occurs by 1) neutralizing surface charges or forming bridges between particles to allow incorporation into a developing floc (charge neutralization or inter-particle bridging mechanism) or 2) by sorption of the colloid to a pre-formed floc (sweep flocculation mechanism). The resulting flocs may then be separated by gravity and/or filtration. Colloids are thus physically removed from the water matrix. Waterborne viruses are typically less than 100 nm in diameter,4 making them among the smallest colloids removable by coagulation. Physical removal is usually considered the primary mechanism for virus mitigation due to coagulation, although recent research shows that coagulation can also render viruses noninfectious (inactivation mechanism).
This review examines recent studies of virus mitigation by coagulation processes in the context of the latest scientific advances in understanding virus sorption and inactivation. To begin, the forces influencing virus sorption and subsequent physical removal are described, including the role of electrostatic forces, the hydrophobic effect, steric hindrance, hydrodynamics, and cation bridging. Consideration is given to discussion of environmental matrix effects, e.g., the influence of organic matter and divalent cations. Next, the phenomenon of virus inactivation during coagulation processes is addressed, including approaches and challenges to quantifying virus inactivation exclusive of physical removal. Critical analysis of recent discoveries and findings is used to inform recommendations for new research directions for mitigating waterborne viruses by coagulation, including appropriate selection and use of virus surrogates in laboratory and field studies.
2. Physical removal of virions
Physical removal is generally considered to be the dominant form of virus mitigation in coagulation processes. Therefore, many sources do not distinguish between physical removal and overall virus reduction. A summary of virus coagulation studies is provided in Tables 1 and 2. Table 1 summarizes results of studies using a single virus for testing, while Table 2 summarizes studies testing multiple viruses in tandem to facilitate comparison of results among viruses. As shown in the Tables, chemical coagulation has been shown to reduce viruses by 0.5 to 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log10 (i.e., 90% to 99.99999% reduction), with a typical reduction of approximately 3log10. In studies of chemical coagulation with post-treatment microfiltration, virus concentrations were reduced up to 8log10, with a typical reduction of 5log10. Enhanced coagulation has been shown to reduce virus concentrations by up to 4.5log10 (ref. 27) and up to 7log10 (ref. 12) with post-treatment microfiltration. However, enhanced coagulation has not been studied as thoroughly as conventional chemical coagulation and is used for more challenging water sources. Electrocoagulation with post-treatment microfiltration has shown promising results in mitigating bacteriophage MS2, surpassing the EPA's Surface Water Treatment Rule (SWTR) of 4log10 reduction of viruses.14,29,30,34
log10. Reduction values are based on cultural assays. Unless otherwise noted, pH values indicate conditions after coagulant addition. Reduction ranges indicate temporal variation (e.g., in filtration systems)
log10 (i.e., 90% to 99.99999% reduction), with a typical reduction of approximately 3log10. In studies of chemical coagulation with post-treatment microfiltration, virus concentrations were reduced up to 8log10, with a typical reduction of 5log10. Enhanced coagulation has been shown to reduce virus concentrations by up to 4.5log10 (ref. 27) and up to 7log10 (ref. 12) with post-treatment microfiltration. However, enhanced coagulation has not been studied as thoroughly as conventional chemical coagulation and is used for more challenging water sources. Electrocoagulation with post-treatment microfiltration has shown promising results in mitigating bacteriophage MS2, surpassing the EPA's Surface Water Treatment Rule (SWTR) of 4log10 reduction of viruses.14,29,30,34
log10. Reduction values are based on cultural assays. Unless otherwise noted, pH values indicate conditions after coagulant addition. Reduction ranges indicate temporal variation (e.g., in filtration systems)
| Coagulation process | Coagulant | Dose (mg Al L−1 or mg Fe L−1) | Bacteriophage | log10 virus reduction | pH | NOM (mg L−1 DOC) | Source |
|---|---|---|---|---|---|---|---|
| NOM = natural organic matter, DOC = dissolved organic carbon, TOC = total organic carbon. CC = chemical coagulation, EnC = enhanced coagulation, ElC = electrocoagulation, MF = microfiltration, UF = ultrafiltration. a Initial pH value (before coagulant addition). | |||||||
| CC | PACl | 1 | Qβ | 4 | 7 | 0 | 22 |
| AlCl3 | 3 | ||||||
| PACl | 4 | 2.2 (TOC) | |||||
| AlCl3 | 1.5 | ||||||
| CC + MF | PACl | 1.1 | Qβ | 6–7 | 6.8 | 1.1 (TOC) | 13 |
| CC + MF | FeCl3 | 10 | MS2 | 4.5 | 6.3 | 0 | 98 |
| 3.5 | 8.3 | ||||||
| CC + MF | PACl | 0.5 | Qβ | 4–5 | 6.8 | 0.6 | 11 |
| CC + MF | PACl | 1.1 | Qβ | 7 | 6.8 | 0.9 | 9 |
| 7 | 7.8 | ||||||
| Alum | 1.1 | 6.5 | 6.8 | ||||
| 0.5 | 7.8 | ||||||
| CC + UF | PACl, alum | 3 | MS2 | 7 | 7a | 2.5 (TOC) | 15 |
| EnC + MF | PACl | 4 | MS2 | 7 | 5.5a | 4.2 | 12 |
| 3–4 | 6.5a | ||||||
| FeCl3 | 3–4 | 5a | |||||
| 3–4 | 6.5a | ||||||
| ElC | Aluminum electrodes | 30 | MS2 | 3 | 6.2 | 0 | 28 |
| ElC + MF | Iron electrodes | 10 | MS2 | 5 | 6.3a | 0 | 14 |
| 4.5 | 7.3–8.3a | ||||||
| ElC + MF | Iron electrodes | 13 | MS2 | 6.5 | 6.4a | 0 | 30 |
| 1.5 | 4.9 | ||||||
| 11.5 | 4.5 | 7.5a | 0 | ||||
| 1 | 4.9 | ||||||
| ElC + MF | Aluminum electrodes | 10 | MS2 | 4 | 6.4a | 5.2 | 29 |
log10. Reported inactivation below 1log10 is shown as “<1”, while negligible inactivation is shown as “≪1”. Darker shading indicates comparatively greater reduction efficiencies for a given experiment. Unless otherwise noted, reduction values are based on cultural assays, and pH values indicate conditions after coagulant addition. Reduction and inactivation ranges indicate temporal variation (e.g., in filtration systems)
| n.r. = not reported for the given data set, NOM = natural organic matter, DOC = dissolved organic carbon, VLP = virus-like particle. a Quantified by quantitative reverse transcription polymerase chain reaction (qRT-PCR).bInitial pH value (before coagulant addition).cQuantified by enzyme-linked immunosorbent assay (ELISA). |
|---|
|
The physical incorporation of viruses into flocs most likely happens in one of two ways: incorporation in the developing floc (charge neutralization or inter-particle bridging), and/or sorption to surfaces of formed flocs (sweep flocculation). Shirasaki et al.35 found negligible (<0.5log10) reduction of poliovirus on preformed flocs, compared to approximately 3log10 reduction during floc formation. In another study, Shirasaki et al.20 found that the physical removal of two bacteriophages occurred during rapid mixing, with little or no additional removal during flocculation and after settling. Kreißel et al.19 similarly found that significant virus mitigation occurred only during floc formation. In a study of virus removal by electrocoagulation, Tanneru et al.29 concluded that sweep flocculation was the dominant mechanism of virus removal based on fluorescent microscopy and virus recovery from flocs. This discrepancy may be due to differences in floc formation between electrocoagulation and chemical coagulation. Chemical coagulation is limited by reaction kinetics and forms dense flocs, while electrocoagulation is limited by coagulant ion diffusion and forms sparse flocs.36 During electrocoagulation, coagulant is continuously released, so treatment cannot be separated into rapid-mix coagulation and flocculation stages. Therefore, it is difficult to determine whether viruses are incorporated into growing flocs or sorbed to the floc surface.
In the absence of coagulant, viruses at high concentrations may also destabilize to form aggregates due to environmental conditions. Aggregate formation is important from both a theoretical and an experimental perspective. Due to larger size and lower surface charge, aggregates are easier to remove than monodispersed virions. In addition to being more susceptible to treatment processes, aggregates lead to artificially low results when quantifying viruses by cultural methods.37 Unfortunately, aggregation is often a result of laboratory methods and does not necessarily represent natural conditions.
Numerous factors may influence physical removal of virions in flocs, as described in the following sections. These include the electrostatic and van der Waals forces modeled by the Derjaguin, Landau, Verwey and Overbeek (DLVO) theory, as well as non-DLVO factors such as the hydrophobic effect, structural incompatibility between viruses and sorbents (steric hindrance), and interactions with one another (aggregation) and constituents in the water matrix. The impact of these factors depends on the virus itself (e.g., its structure, surface charge, or degree of permeability), the nature of the sorbent (floc characteristics), and the composition of the water matrix. The influence of these phenomena on virus sorption has been studied more extensively for virus transport through porous media.38–42 Still, many of the lessons learned apply to coagulation as well.
2.1. Electrostatic interactions
Electrostatic forces affect the sorption of virions to surfaces like flocs. Investigators use multiple measures to describe electrostatic forces. Isoelectric point (pI) is the pH at which a particle or surface has a neutral charge in the electrolyte solution. At pH levels above the pI, the surface is negatively charged in solution; below the pI, the surface has a positive charge. Electrophoretic mobility is a measure of particle movement in the presence of an electric field and can be used to infer the electric potential near the particle surface. Electrostatic forces often govern virion sorption due to the long range of electrostatic influence, as well as the low pI of most enteric viruses, indicative of a strong, negative potential near the particle surface at neutral pH. However, electrostatic forces are attenuated at high ionic strength due to screening of the electric field.Bacteriophage MS2 has been offered as an example of the effect of the viral genome on pI. The theoretical pI of MS2 based on total charged capsid moieties is approximately 7 (ref. 49) or 9,50 while the pI of MS2’s RNA genome is approximately 3.50 The measured pI of MS2 is generally accepted to be between 3 and 4, closer to the RNA pI than the capsid pI.51 Using another method to calculate the capsid pI, Penrod et al.38 accurately predicted MS2's measured pI by evaluating only those charged structures exposed on the surface of the capsid. However, Schaldach et al.43 found better correlation with experimental electrophoretic mobility data when allowing for capsid permeability than using the Penrod method.
However, a study by Dika et al.46 comparing MS2 bacteriophages and virus-like particles (VLPs) seems to support Penrod et al.'s model for predicting pI. VLPs are assembled by expression of the viral coat proteins in a bacterial host but lack the viral genome. Instead of having a pI between 7 and 9 as predicted, MS2 VLPs had a measured pI between pH 3 and 4. Dika et al. hypothesize that negatively-charged host material was trapped within the VLPs during propagation. Considering the intricate, optimized packing of the viral genome into the capsid during normal bacteriophage propagation,52,53 as well as evidence from electron micrographs,46 VLPs likely do not contain enough host material in their interior to constitute a negative charge density comparable to whole virions. To accept the interpretation of viruses as soft colloids, we should see at least some increase in the pI of VLPs compared to bacteriophages to reflect the influence of the genome. The permeability model may also be more applicable to some virions than others.
Researchers have also investigated the impact of the genome on virus–virus sorption phenomena responsible for aggregation. Dika et al.57 found that MS2 aggregates formed at pH 4 did not re-disperse when the solution was then acidified to pH 2. Other experiments of MS2 aggregation using pH titration confirm this trend.37,46 By contrast, MS2 VLPs aggregated only near the pI value and dispersed at lower pH. The team hypothesized that the difference in aggregation reversibility was due to the attractive influence of the genome. However, VLPs did not aggregate at any pH at high ionic strength, whereas entire virions did. At high ionic strength, the effective distance of electrostatic forces decreases, so VLPs and entire virions should behave more similarly if permeability impacts surface charge. In this study, the MS2 and MS2 VLPs behaved more similarly at low ionic strength. It is also unclear why the relative absence of RNA in VLPs would explain aggregation in this study, while the presence of residual RNA in VLPs was used to explain the VLP pI measured in Dika et al.'s previous study.46
Other tests found similar patterns of irreversible aggregation for somatic bacteriophages PRD1 and ΦX174 and F-specific bacteriophages Qβ and GA.48,58,59 Bacteriophages PRD1, Qβ and GA all have measured pI values between 2 and 4 reported in the literature,51 so aggregation in this pH range is not unusual. From an evolutionary standpoint, enteric viruses and bacteriophages may gain a selective advantage by aggregating to avoid inactivation by proteases in the stomach (pH < 4 (ref. 60)), and dispersing in the near-neutral pH of the intestines for the greatest chance of infection. Aggregation has been shown to inhibit virus inactivation by chemical disinfectants.61
However, aggregation below pH 4 is unexpected for ΦX174, which has a generally accepted pI of 6.6 from capillary isoelectric focusing, chromatofocusing and aggregation studies.51 If the pI of ΦX174 was indeed higher than pH 4, aggregation occurred solely in a pH range where virions should have a positive net charge. Chrysikopoulos and Syngouna41 and Aronino et al.58 recently reported lower pI values for ΦX174 (4.4 and 2.6, respectively) based on electrokinetic measurements. However, in an extensive review of virus pIs, Michen and Graule51 discounted Aronino et al.'s finding because Aronino et al. did not report purifying their bacteriophages for testing. Chrysikopoulos et al. also did not report purifying virus stocks.41,62 Discrepancies in reported pI values for viruses are common in the literature, as differences in electrolyte concentration, composition and purity, and the strain of virus used, may all affect the measured pI.51 The interpretation of previous experimental results must be based on electrophoretic mobility measurements made under the conditions of a given experiment, rather than on values reported in the literature.
2.2. Non-electrostatic sorption phenomena
When electrostatic interactions are repulsive or neutral, van der Waals and non-DLVO phenomena like the hydrophobic effect, steric hindrance and interactions with constituents in the water matrix may lead to differences in virion sorption. As detailed below, van der Waals and non-DLVO forces tend to modify the effects of electrostatic forces, especially when electrostatic forces are minimized (e.g., by electrostatic screening or at pH values near the pI of the particle).Arising from electronic resonance between surfaces, van der Waals interactions create an attractive force proportional to the polarizability of the virion and the abiotic surface.16 In practice, van der Waals forces cannot be measured independently of electrostatic and non-DLVO phenomena.59 In an extensive study of interactions influencing bacteriophage adsorption to surfaces, Armanious et al.63 found minimal impact of surface polarizability on bacteriophage adsorption. However, the two surfaces compared also differed in hydrophobicity.
The hydrophobic effect arises from hydrogen bonds that preferentially form between water molecules to the exclusion of nonpolar molecules. The hydrophobic effect results in the tendency of nonpolar substances to partition out of the aqueous phase. Armanious et al.63 found that the hydrophobic effect moderated electrostatic repulsion to allow adsorption of bacteriophages fr, GA, MS2 and Qβ to nonpolar surfaces. Armanious et al. also established a method for quantifying hydrophobicity based on the size and number of nonpolar patches on the capsid surface, and predicted a pattern of decreasing hydrophobicity: Qβ ≫ fr > GA ≫ MS2. Armanious et al.'s method was able to explain broad trends in bacteriophage sorption to hydrophobic surfaces, although completely isolating the hydrophobic effect from other phenomena is not possible. Other researchers37,48 experimentally determined a relative hydrophobicity of GA > Qβ > MS2.
In two separate studies,48,59 Dika et al. found that surface hydrophobicity could explain differences in the sorption of bacteriophages. Using chemical force microscopy, the hydrophobicity of bacteriophages MS2, Qβ and GA were compared to known hydrophobic and hydrophilic surfaces.48 Hydrophobicity influenced virus sorption to surfaces even in low ionic strength solution (1 mM NaNO3), where electrostatic forces are expected to dominate.48 Bacteriophages MS2, PRD1 and ΦX174 were also compared. Despite varying charge densities among the three bacteriophages in low ionic strength electrolyte, they demonstrated similar electrophoretic mobility at high ionic strength (100 mM).59 Nevertheless, the bacteriophages differed in their affinities for membranes of varying hydrophobicity. Hydrophobicity has also been determined to favorably impact virus sorption to finely powdered activated carbon.64
The molecular-level structure of virus capsids and the sorbent surface may also hinder virion adsorption at close range. This steric hindrance occurs when interactions between the adsorbent and adsorbate are limited by the spatial orientation of their molecular structures. Several studies have found evidence of steric hindrance in virus sorption. Penrod et al.38 found that steric interactions (here considering all non-electrostatic repulsion to be steric in nature) may lead to increased MS2 mobility in porous media when electrostatic forces are screened (i.e., at high ionic strength). Armanious et al.63 also suggested that the variable topography of bacteriophage fr and MS2 capsids, as determined by X-ray crystallography, may have resulted in poor adsorption to a gold surface in comparison to bacteriophages Qβ and GA. Dika et al.48 found that bacteriophages preferentially sorbed to stainless steel over glass, despite similar surface hydrophobicity. This trend was more apparent at high ionic strength, which fits with the theory that surface roughness impacts electrostatic interactions when the roughness is on a scale comparable to the Debye length (a measure of the effective range of electrostatic forces).48 In all cases, steric hindrance appeared to moderate sorption in conditions of similar electrostatic charge and hydrophobicity, rather than broadly define sorption behavior.
2.3. Impact of water matrix composition on virus sorption
Suspended and dissolved materials in the water matrix, like NOM and dissolved salts, can dramatically impact virion sorption. Because of the heterogeneous charge distribution and polarity of organic matter in the environment, the effect of NOM on virus sorption involves electrostatic forces, hydrophobicity and steric interactions. Generally, NOM contains both polar and nonpolar moieties and has a negative charge at neutral and high pH due to deprotonation of carboxyl and phenyl groups.39,63 In porous media filtration tests, Zhuang and Jin39 found that MS2 breakthrough was more rapid in the presence of sorbed or dissolved organic material, while ΦX174 breakthrough was relatively unaffected. MS2 has been shown to be relatively hydrophilic37,63 and typically more negatively charged at neutral pH than ΦX174.59 Therefore, MS2 sorption may be expected to be governed by electrostatic forces rather than hydrophobicity, while the hydrophobic effect may mediate ΦX174 sorption. Zhuang and Jin concluded that NOM both competes for sorption sites on the media and enhances sorption of nonpolar virions by creating hydrophobic sorption sites. Armanious et al.63 found high sorption of bacteriophages GA and Qβ at pH 6 on a NOM-coated surface, while MS2 and fr sorption was negligible. GA and Qβ sorption decreased significantly from pH 6 to pH 8, likely due to electrostatic repulsion arising from deprotonation of carboxyl groups on the NOM and capsid surfaces. When ionic strength was increased from 10 mM to 100 mM to screen electrostatic forces, Qβ sorption was high even at pH 8, while MS2 sorption was measureable, though low. These results again illustrate that the hydrophobic effect predominates only when electrostatic forces are weak. Yuan et al.55 found that MS2 deposition on silica was greater than on NOM-coated surfaces, even at ionic strengths high enough to effectively screen electrostatic charges. The team concluded that the results may be due to steric hindrance, by which NOM surface structures prevent binding in contrast to the even surface of silica.Deposition experiments have shown that cation bridging can significantly increase virion sorption to like-charged surfaces.54,65,66 In cation bridging, divalent cations (e.g., Ca2+ and Mg2+) complex with negatively-charged moieties on both the capsid and the solid surface. The presence of Ca2+ and Mg2+ ions has been shown to dramatically increase sorption of viruses to repulsive surfaces in comparison to monovalent ions, beyond the expected increase due to screening of electrostatic forces.54,66 By contrast, rotavirus adsorption to an oppositely-charged (non-repulsive) surface was shown to be independent of Ca2+ or Mg2+ concentrations.54 The effect of cation bridging can be significant at Ca2+ and Mg2+ concentrations typical of drinking water sources.54 For the bacterium Pseudomonas aeruginosa, cation bridging has been shown to significantly enhance sorption to repulsive surfaces at concentrations as low as 10−5 M Ca2+ or Mg2+.67 Interactions between MS2 virions have been shown to transition from repulsive to attractive between 10 mM Ca2+ and 50 mM Ca2+.65
Ca2+ ions have been shown to have a greater positive influence on virus sorption to repulsive surfaces than Mg2+ ions.54,66 Ca2+ ions are large and have weakly bound spheres of hydration that allow inner-sphere complexation with carboxyl groups on the virus capsid and the solid surface.54,66 By contrast, Mg2+ ions have tightly-bound spheres of hydration that may allow only outer-sphere complexation. The mechanism for the relatively weak sorption observed in the presence of Mg2+ may not be bridging, but rather charge neutralization by complexation with negatively-charged moieties on either the virion or the surface.66 The ability to form bonds with carboxyl groups makes cation bridging particularly important in the sorption of negatively charged viruses to NOM. In an experiment conducted by Pham et al.,66 Ca2+ improved deposition of MS2 on a NOM-coated silica surface to a far greater extent than on a bare silica surface, although the bare silica was more negatively charged than the NOM-coated surface. For comparison, using NOM from the same source but in a monovalent electrolyte, Yuan et al.55 found poorer adsorption of MS2 on a NOM-coated surface than a silica surface. Mylon et al.65 found that a lower concentration of Ca2+ was required to destabilize MS2 in the presence of NOM (10 mg L−1 total organic carbon; TOC).
2.4. Implications of electrostatic and non-electrostatic phenomena for virus aggregation
Electrostatic repulsion contributes to virion stability, so aggregation typically occurs at high ionic strength or pH ranges near the virion pI.37 Non-DLVO forces may also impact virus aggregation. Some investigators have suggested that protein loops extending from the capsid surface may contribute to the high stability of virions by steric hindrance.56,65 Virus aggregation has been shown to be higher in the presence of divalent cations, although not in the typical range of Ca2+ and Mg2+ concentrations in drinking water.54,56,65 Hydrodynamic forces may also influence aggregation. Langlet et al.37 suggest that the low electrophoretic mobility of virion aggregates may be due to hydrodynamic drag. Aggregates may show greater hydrodynamic drag due to permeability. Because of this drag, aggregates would tend to stay aggregated once formed.37 From another perspective, the hydrodynamic drag of individual virions due to capsid permeability may counteract the repulsive electrostatic forces of surfaces and neighboring virions, leading to aggregation.2.5. Implications of electrostatic and non-electrostatic phenomena for coagulation
While porous media studies provide valuable insights, not all lessons can be assumed to apply to virus coagulation. Unlike sorption to solid surfaces, coagulation may occur by sorption to solid flocs and/or complexation of the virion surface by dissolved coagulant (charge neutralization or inter-particle bridging). In addition, metal oxide flocs differ in structure, charge and polarity from porous media. The challenge lies in determining which parameters are necessary and/or sufficient to describe virion sorption during coagulation/flocculation.Hydrophobicity is unlikely to have a strong effect on coagulation in many cases, as aluminum and iron hydroxides are polar.68 However, in the presence of NOM, hydrophobicity may be an important partitioning factor for some viruses. Tanneru et al.29 found that aluminum flocs became more hydrophobic following sorption of NOM. Rebhun et al.69 enhanced removal of hydrophobic (logKow > 4.5) polyaromatic compounds by adding dissolved humic acid. Therefore, NOM may actually enhance sorption of very hydrophobic virions. Due to the rough, fractal structure of aluminum and iron flocs,70 steric hindrance may also play a role in sorption to flocs.
To our knowledge, conclusive evidence of the effect of divalent cations on virus sorption to metal hydroxide flocs (as opposed to electrostatically repulsive and/or nonpolar surfaces) does not exist. In 1958, Chang et al.71 concluded that Ca2+ and Mg2+ may have inhibited virus mitigation. However, the comparison was made between tests using synthetic versus raw water sources, so the difference in virus mitigation cannot be conclusively attributed to divalent cations, as opposed to, e.g., NOM. Chaudhuri and Engelbrecht72 later showed that alum coagulation of bacteriophage T4 was not affected by Ca2+ and Mg2+ concentrations up to 330 mg L−1 as CaCO3. However, Chaudhuri and Engelbrecht used a synthetic water free of NOM. The hypothesis of cation bridging between NOM and anionic coagulants/polymers has not been tested for viruses. Inhibition of coagulation by NOM has been documented for bacteriophages MS2 (ref. 30) and Qβ.22 However, neither the influence of divalent cations nor the impact of phage hydrophobicity was tested in these studies. Microbalance experiments of virus deposition on aluminum or iron hydroxide surfaces, similar to those conducted on silica and NOM coated surfaces, could better identify the importance of surface charge, hydrophobicity, and roughness, as well as divalent cations and NOM concentrations.
3. Inactivation during coagulation processes
In addition to physical removal by sorption and co-precipitation, some studies have investigated bacteriophage inactivation by coagulation processes. Viruses may be inactivated by damage to the virion protein capsid and/or the viral genome. Damage to viral proteins manifests as an inability of the virus to attach to the host cell and/or inject the genome, while genomic damage prevents replication and proliferation of the virus in the host.73 Whether viruses are physically removed or inactivated is not simply a Talmudic question. Coagulation processes increase sludge production, and sludge must be properly handled. If high levels of virus inactivation can be achieved, sludge treatment and handling will be safer and more cost-effective. Safe sludge handling is critical for decentralized water treatment, especially in developing countries that lack the infrastructure for proper disposal. If people risk contact with the sludge, we must be sure coagulation does not just concentrate pathogens.Evidence of inactivation has been documented for both chemical coagulation and electrocoagulation, as summarized in Table 3. In the case of aluminum coagulants, polynuclear Al13 and Al30 species are thought to chemically oxidize virions.19,35 While soluble, monomeric aluminum species are predominantly anionic above pH 6,16 soluble Al13 and Al30 species are cationic near neutral pH.19,35 Since most virions have negative surface charges,51 the polynuclear cations may interact with and oxidize virions to a greater extent than monomeric anions. PACls produce more polynuclear hydrolytes in solution than simple aluminum salts.9,35 Correspondingly, the most evidence for virus inactivation has been observed with PACl coagulation.9,10,17–22 Coagulation with simple aluminum and iron salts (e.g., Al2(SO4)3, AlCl3, FeCl3 and Al(NO3)3) has shown only limited virus inactivation.17,22,74 Polynuclear iron coagulants have also been developed,23,24 but these coagulants have not been evaluated for virus mitigation to our knowledge.
log10. Reported inactivation below 1log10 is shown as “<1,” while negligible inactivation is shown as “≪1”
| Coagulation process | Coagulant | Dose (mg Al L−1 or mg Fe L−1) | Bacteriophage | log10 virus inactivation/aggregation | Inactivation quantification method | Aggregation determination method | Source |
|---|---|---|---|---|---|---|---|
| n.r. = not reported for the given data set. CC = chemical coagulation, EnC = enhanced coagulation, ElC = electrocoagulation, MF = microfiltration. DLS = dynamic light scattering, qRT-PCR = quantitative reverse transcription polymerase chain reaction. | |||||||
| CC | PACl | 1 | Qβ | 3.5 | Plaque assay (with recovery) | Phage recovery efficiency | 22 |
| T4 | 2 | ||||||
| MS2 | 2.5 | ||||||
| P1 | 3 | ||||||
| Alum | Qβ | 2 | |||||
| T4 | <1 | ||||||
| MS2 | 1.5 | ||||||
| P1 | 1 | ||||||
| CC | PACl | 10 | Qβ | 2 | Plaque assay (with recovery) | Phage recovery efficiency | 17 |
| PACl, alum, AlCl3, AlSO4 | 1 | MS2 | <1 | ||||
| P1 | <1 | ||||||
| Qβ | 1–1.5 | ||||||
| T4 | <1 | ||||||
| CC | PACl | 1.1 | MS2 | <1 | qRT-PCR (with recovery), plaque assay (with recovery) | DLS | 20 |
| Qβ | 4 | ||||||
| CC | Alum | 1.1 | MS2 | 1 | qRT-PCR, plaque assay | DLS | 18 |
| Qβ | 2 | ||||||
| PACl | MS2 | 2 | |||||
| Qβ | 5 | ||||||
| CC | PACl | 1.9 | MS2 | 3 | qRT-PCR, plaque assay | n.r. | 100 |
| 2.2 | Qβ | 2.5 | |||||
| Alum | ≪1 | ||||||
| CC | PACl | 10 | MS2 | 4 | qRT-PCR, plaque assay | DLS | 19 |
| Qβ | 6 | ||||||
| ΦX174 | ≪1 | ||||||
| EnC | PACl | 4 | MS2 | 1–2 | Plaque assay (with recovery) | n.r. | 12 |
| FeCl3 | 8 | 1–2 | |||||
| CC + MF | PACl | 1.1 | MS2 | <1 | qRT-PCR, plaque assay | Electron micrography | 21 |
| Qβ | 2.5 | ||||||
| CC + MF | PACl | 1.1 | MS2 | 2 | qRT-PCR, plaque assay | n.r. | 10 |
| Qβ | 4 | ||||||
| ElC | Aluminum electrodes | 30 | MS2 | < 1 | Plaque assay (with recovery) | Phage recovery efficiency | 28 |
| 30, 4 h flocculation | 3.5 | ||||||
| ElC + MF | Iron electrodes | 10 | MS2 | 1–2 | Plaque assay (with recovery) | Phage recovery efficiency | 30 |
As in sorption studies, multiple investigators found that inactivation by chemical coagulation occurred concurrently to floc formation, with little to no inactivation when viruses were spiked in a solution with pre-formed flocs.19,22 Kreißel et al.19 found that inactivation was greatest when viruses were exposed to soluble PACl at pH 4.5, indicating that inactivation may be related to soluble species rather than insoluble flocs. Other researchers have suggested alternate mechanisms of inactivation, such as deformation of virions by forces at the interphase boundary,18 and inhibition of infection by irreversible adsorption of coagulant polymers to the capsid surface (e.g., at binding sites).17
Electrocoagulation has been shown to disinfect algae and bacteria, although researchers often do not discern between physical removal and inactivation in their results.75–78 Few studies have investigated virus mitigation by electrocoagulation,14,28–30 and to date only Tanneru et al.28,30 have specifically examined virus inactivation by electrocoagulation. Disinfection occurs in electrocoagulation primarily by the oxidation of chloride to free chlorine.28,79 For this reason, electrocoagulation has only been shown to inactivate viruses in the presence of chloride ions. Because generation of free chlorine is an oxidative process occurring at the anode,79 chlorine production is a secondary and competing reaction to the oxidative dissolution of the anode itself. Tanneru et al.28 noted that bacteriophage inactivation required a prohibitively long contact time due to the low concentrations of free chlorine generated in the study (<0.1 mg L−1). Inclusion in flocs also shields viruses from inactivation by free chlorine.28
Tanneru et al.28 were able to detect damage to both the MS2 genome and proteins after electrocoagulation with extended flocculation times. The team detected conformational changes to proteins and an increase in the concentration of protein oxidation byproducts by attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR). Tanneru et al. also used quantitative reverse transcription polymerase chain reaction (qRT-PCR) amplification to directly investigate damage to the MS2 genome. A 77 bp section of the maturation protein coding region80 was amplified and compared between treated and initial samples. The short length of the amplicon likely makes this method a conservative indicator of total RNA damage. Tanneru et al. found a rapid decline in copy number in the first hour after electrocoagulation, comparable to the reduction found using a culture-based plaque assay. Tanneru et al. did not find increased genome inactivation with longer contact time, although overall inactivation continued to increase. These results suggest that for electrocoagulation, the mechanism of inactivation may vary over time. Varying mechanisms of inactivation can be at play, even for a given disinfectant. For example, Wigginton et al.73 found that free chlorine attacked MS2 proteins and genome, and inactivation manifested as an inability to inject the viral genome into the host cell. The method used by Wigginton et al. may offer a less conservative estimate of RNA damage because approximately half of the viral genome was analyzed.
Due to the different hypothesized inactivation mechanisms (i.e., production of free chlorine during electrocoagulation versus polynuclear cations in chemical coagulation), Tanneru et al.'s results cannot be extended to chemical coagulation. Wigginton et al.73 established that different chemical oxidants (i.e., 1O2, free chlorine, ClO2) vary in their mechanisms of inactivation. Likewise, the inactivation mechanism likely differs between free chlorine and the large, polynuclear cations suggested to be responsible for inactivation due to chemical coagulation (e.g., Al13 and Al30). Use of an approach similar to that of Wigginton et al. or Tanneru et al.28 would help to clarify the mechanism of inactivation by chemical coagulation. As a preliminary hypothesis, polynuclear aluminum species may predominately attack capsid surface proteins, because access to the internal structure would be limited by size and charge (especially as compared to free chlorine).
The following sections discuss difficulties in assessing virus inactivation in the laboratory. Due to the cost and duration of cultural assays, molecular methods seem to be an attractive option. However, research is required to prove the validity of molecular methods for quantifying inactivation. In addition, a given level of inactivation may be important to treatment efficiency, yet difficult to quantify due to its relative insignificance compared to other treatment fates. Virus aggregation also frustrates attempts to quantify inactivation, and no satisfactory method is available to ensure against aggregation of treated samples.
3.1. Quantification of virus inactivation
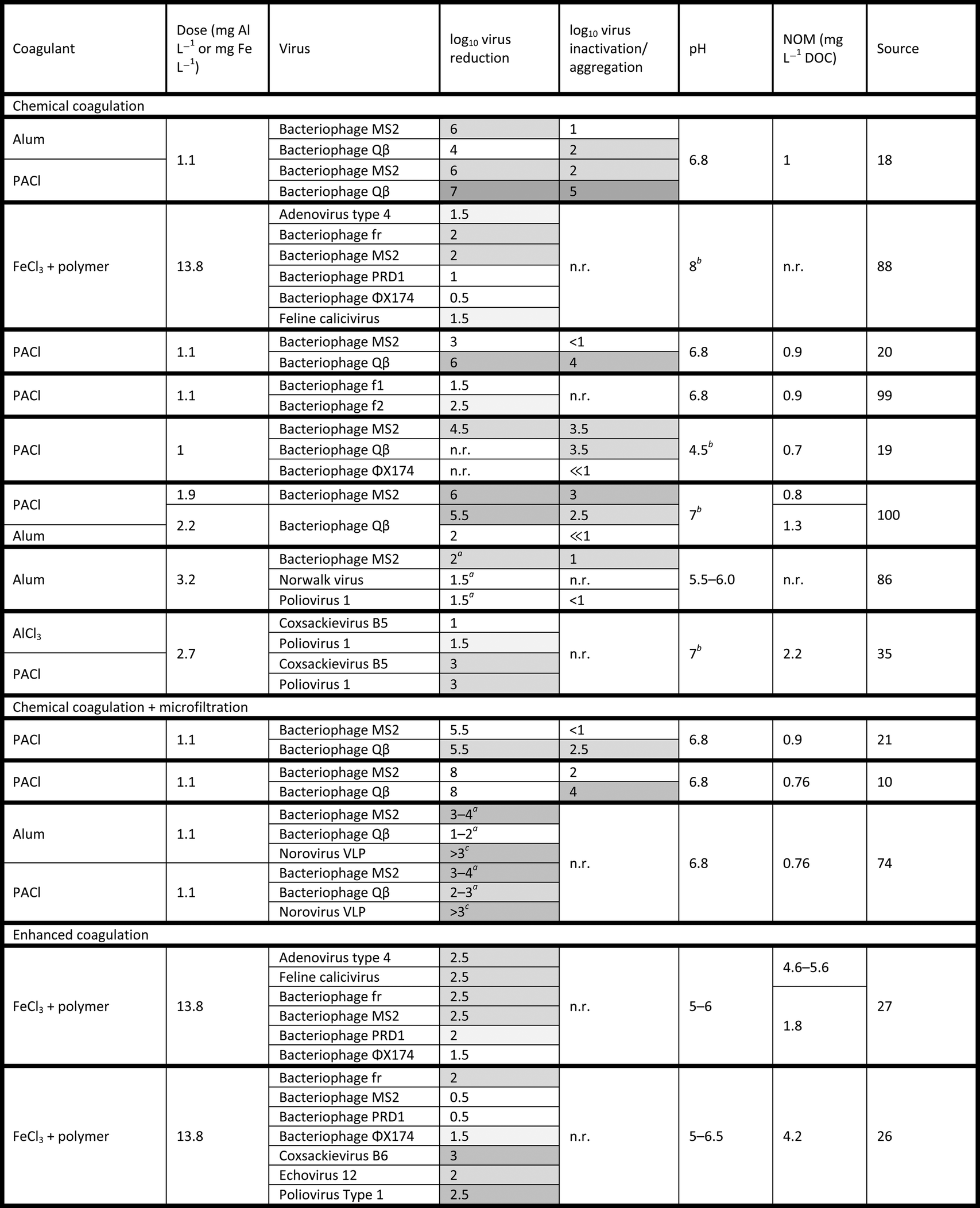
Quantification of virus inactivation presents an experimental challenge. Some authors17,22,28 have employed a cultural plaque assay to quantify the number of infectious viruses in solution and those sorbed to solids using a recovery protocol. This method may be thought of as a “plaque-forming unit (PFU) balance.” After gravitational separation, viruses are both sampled in the supernatant and recovered from the floc. The total virus recovery is compared to the untreated, control sample to determine inactivation. A PFU balance is distinct from a mass balance in that it is a discrete count of PFUs, not a continuous measure of mass. Like any plaque assay, the PFU balance can discriminate between infectious and inactive viruses but not between a single virus and an aggregate.81 Comparison of recovered PFUs to the initial concentration allows determination of virus inactivation, within the expected method recovery efficiency.28 However, the PFU balance approach requires twice the number of plaque assays to analyze the concentrations of viruses in solution and adsorbed to flocs, thereby increasing cost and time inputs.Other investigators9,10,18,19,21 have used qPCR (qRT-PCR for RNA viruses) to compare declines in copy number to declines in PFU counts. Compared to plaque assays, qPCR is comparatively rapid, and aggregation does not affect qPCR results. In contrast to plaque assays, qPCR assesses the total number of intact viral genomes in the sample, regardless of infectivity. This allows a comparison between plaque assay and qPCR results to assess total inactivation. However, there are several concerns with comparing molecular and cultural techniques. For one, molecular methods cannot differentiate between physical removal and inactivation due to genome damage. The copy number of even short amplicons can decrease during inactivation, as described by Tanneru et al.28 When assessing chemical oxidation by soluble PACl, Kreißel et al.19 also showed a decline in copy number of approximately 1log10 from the initial concentration. Whether the depressed recovery is an artifact of the method or indicative of genome destruction is unclear. In a coagulation study, this reduction in copy number would be indistinguishable from a reduction due to physical removal, as shown in Fig. 1. Therefore, qPCR may overstate the importance of physical removal and understate that of inactivation.
 | ||
| Fig. 1 The most detailed theoretical categorization of fate possible using three different quantification methods independently and in combination. The relative values shown in stacked columns were chosen only for visual clarity. In practice, resolving many of these quantities may be impossible, because quantities may differ in value and variability by several orders of magnitude. | ||
Similarly, some fraction of inactivated viruses are likely removed with the flocs and therefore counted as physical removal. Inactivated viruses could even be disproportionately removed. Destruction of viral proteins can dramatically alter virion structure and genome packing.82 The effect of morphological changes on sorption cannot be assumed to be negligible. If infectious viruses are more readily removed in flocs, then qPCR analysis of treated water would provide an appropriate means of quantifying total viruses (infectious + inactive). If inactive viruses are readily removed in the floc phase, qPCR analysis would again systematically underreport inactivation.
In one study, Shirasaki et al.20 analyzed both the liquid phase and dissolved floc of treated water by qPCR and plaque assay – essentially performing both a PFU balance and a copy number balance. Despite significant reductions in amplicons in the liquid phase, Shirasaki et al. recovered approximately all MS2 and Qβ amplicons from the floc (confidence intervals including 100% efficiency). The high recovery indicates that MS2 and Qβ inactivation by genome destruction was below detection in this study. This lack of genome inactivation may reflect the inactivation mechanism of PACl. However, Qβ inactivation was evident in both the floc and liquid phases, indicating the removal of inactivated viruses in flocs. For both bacteriophages, greater discrepancy between molecular and cultural quantification was observed in the liquid phase than in the floc phase. The greater discrepancy in the liquid phase could possibly be due to aggregation, especially considering that the liquid phase was only centrifuged (2000 × g, 10 min), not dissolved and agitated for resuspension like the floc phase. Regardless, Shirasaki et al.'s results suggest that genome inactivation may not significantly impact qPCR results for some bacteriophages and treatment processes. However, using qPCR without recovery from flocs may under-represent inactivation due to sorption of inactivated viruses in the floc, as in the case of Qβ. Shirasaki et al. do not report using this same approach in subsequent papers. Application-specific research is required to establish a firm methodological basis before using qPCR and plaque assays without recovery from flocs.
Fig. 1 summarizes the extent of information that could theoretically be learned using the quantification methods discussed above. The combination of plaque assay with recovery from flocs provides the most detailed account of virus fate; however, one or more of these fates are likely to be undetectable in practice. A plaque assay with recovery also provides more relevant information (i.e., the concentration of infectious viruses in the sludge) than qPCR and plaque assay without recovery. If some fates could be considered inconsequential for a particular application (esp., inactivation due to genome damage), the combination of qPCR and plaque assay without recovery would be comparable to plaque assay with recovery.
3.2. Detecting low levels of inactivation
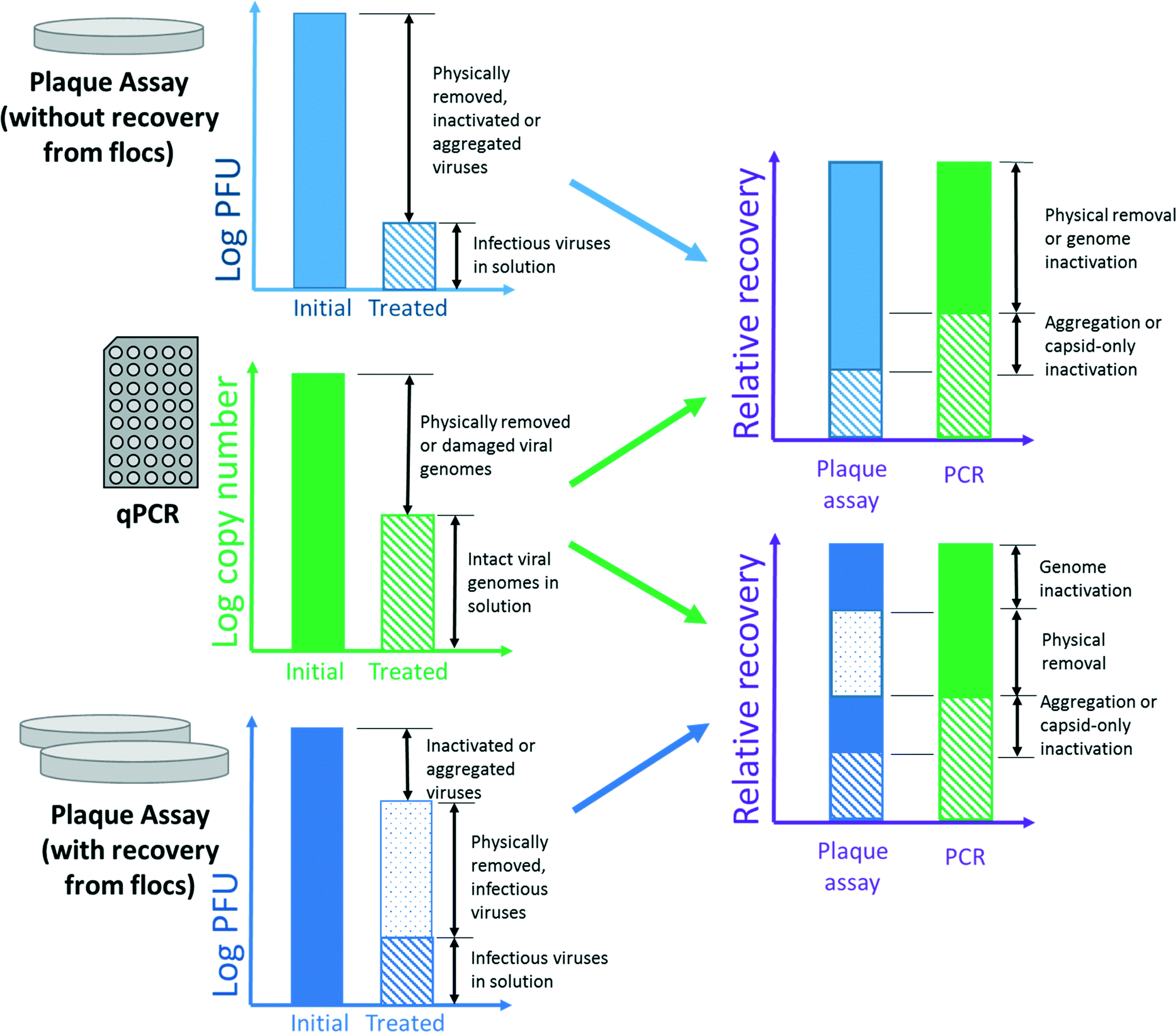
Working with high levels of virus reduction, such that logarithmic representations are customary, also presents an interesting conundrum. Whether quantified by molecular or cultural methods, inactivation is determined by subtracting a concentration of recovered PFUs or amplicons from an initial concentration that may be several orders of magnitude higher. Since the error of each quantity is relative to the concentration, inactivation can only be determined to a statistical degree of certainty when inactivation is a primary mechanism of reduction, as illustrated in Fig. 2. | ||
| Fig. 2 Confidence in quantifying decreasing amounts of inactivation. In this theoretical case, recovery of bacteriophages from the supernatant remains constant for all bars (104 PFU mL−1), while the number of bacteriophages recovered from the floc increases from 105 to 5 × 106 PFU mL−1. The standard error of the mean for all measurements is set as 30% (error bars, inset). The quantity recovered from the supernatant has no significant impact on inactivation. However, as inactivation decreases to near 0.5log10 reduction, the confidence intervals begin to overlap, and inactivation cannot be distinguished from the analytical uncertainty. | ||
In addition, inactivation is usually treated as independent from physical removal, rather than additive. If inactivation works as a polishing step, small numbers of inactivated viruses could have a great impact. For example, if an additional 0.09% of the initial virus concentration is inactivated beyond the 99.9% that can be removed in flocs, that minimal reduction means the difference between satisfying the EPA's SWTR requirements or not.34 The limited information available suggests inactivation might have this polishing effect. As discussed in section 3.1, Shirasaki et al.20 found greater inactivation of MS2 and Qβ in the liquid phase than the floc phase, which indicates that inactivation may contribute to virus reduction beyond the capacity of physical removal alone. In other words, inactivated viruses would not necessarily have been physically removed were they not inactivated. The ‘polishing’ effect of inactivation would significantly reduce the concentration of viruses remaining in the treated water after physical removal. However, this small virus reduction would be lost on the scale of the original, spiked concentration. Therefore, the amount of inactivation, although important for disinfection, would be difficult to discern experimentally. If the inactivation cannot be accurately assessed, conditions for inactivation cannot be optimized.
3.3. Determination of virus aggregation
In all methods, aggregation remains quantitatively indistinguishable from at least some inactivation, as shown in Fig. 1. Aggregation leads to artificially low plaque counts, because each plaque originates from many viruses instead of one. Based on aggregate size, Langlet et al.81 found that aggregation could be responsible for more than 4log10 reduction in PFUs (from an initial concentration of approximately 1011 PFU mL−1). However, an additional control can provide some ‘insurance’ against aggregation for the plaque assay with recovery method. The method recovery efficiency can be tested under conditions of minimal inactivation (e.g., adsorbing viruses to pre-formed flocs or quenching oxidants with sodium thiosulfate). The recovery efficiency shows not only that viruses can be recovered from the flocs, but also that the viruses in the treated water are no more aggregated than in the initial virus solution. Some investigators21,46 have also relied on electron micrographs to qualitatively show the presence or absence of aggregates.
Dynamic light scattering (DLS) is a method for determining both electrokinetic response of colloids and the size distribution of particles in solution. Many studies18–20,59,81 have used DLS analysis to determine conditions in which virions aggregate. However, DLS analysis requires very high virus concentrations (greater than 109 PFU mL−1)37 – higher than even the typical spiking concentrations used for testing (usually 107–108 PFU mL−1). Therefore, researchers cannot directly assess aggregation in the same samples to be tested by plaque assay and/or qPCR. Instead, researchers must try to show whether or not aggregation occurs in conditions similar to those tested.
A significant quantity of virus stock solution is necessary to achieve the required concentrations for DLS. These stock solutions may have higher ionic strengths and differ greatly in composition from natural waters. Electrolyte composition can significantly affect electrokinetic responses like aggregation.51 Aggregation has been shown to be greater in phosphate-buffered saline (PBS), commonly used for virus stocks, than in deionized water or bicarbonate solution.55 Preferably, virus stocks should be purified and spiked into the same water matrix used for coagulation tests. However, the method of virus purification may also significantly affect virion properties. As previously mentioned, Dika et al.57 compared three methods of MS2 purification: polyethylene glycol (PEG) precipitation, successive dialyses in deionized water and 1 mM NaNO3, and ultracentrifugation in a CsCl gradient. PEG precipitation resulted in a larger hydrodynamic radius of unaggregated viruses, with aggregation observed at pH 6. Dialysis resulted in aggregation at pH 4, while viruses separated in a CsCl gradient did not aggregate at any pH. Dika et al. note that each method has drawbacks: PEG appears to adhere to the capsid surface, dialysis retains viral and non-viral particles based only on membrane exclusion, and cesium ions may permanently deform protein structures. The experiment does not clarify which purification best approximates virus behavior in the environment, however. In addition, Dika et al. did not use a solvent wash after PEG precipitation to promote monodispersion, as used by many investigators.26,27,83–85 A solvent like chloroform or Vertrel™ may be able to strip adhered PEG from the capsid surface. If not, dialysis may best reproduce virus behavior in the environment, because foreign compounds are not introduced. To date, the best course of action is to control against aggregation by testing at pH values and ionic strengths where virions are likely to be stable. Aggregation may then be measured under similar conditions using DLS or qualitatively assessed by electron microscopy.
4. Use of virus surrogates in coagulation studies
Understanding the basis of virion sorption and inactivation is necessary not only for predicting and explaining virus reduction by coagulation technologies, but also for choosing appropriate bacteriophage surrogates for evaluating unit processes. Bacteriophage surrogates are most often used in lab and pilot studies for safety considerations, and because bacteriophage assays require significantly less time and resources than mammalian virus assays. In addition, human viruses are often difficult to propagate in large enough concentrations to show required reductions, e.g., the 4log10 reduction required by the SWTR.34
Very few studies have directly compared the effect of coagulation on both bacteriophage surrogates and the human viruses of interest, as shown in Table 2. Among those studies, there is no common consensus on the relative performance of bacteriophage surrogates and human viruses. For example, Mayer et al.26 found MS2 to be a conservative surrogate for poliovirus 1, while Shin and Sobsey86 found MS2 reduction to be greater than that of poliovirus 1. However, too many parameters differ between these studies to draw firm conclusions. Numerous factors, including the type and dose of coagulant, ionic strength, pH and composition of the water matrix, play a significant role in the absolute and relative reduction of different viruses.
Clear trends in removal and inactivation between bacteriophages are also difficult to discern, as shown in Table 2. MS2 and Qβ have been compared in the greatest number of coagulation studies. MS2 and Qβ are both commonly used as surrogates in water treatment studies as well. Both bacteriophages infect the F-pilus of E. coli, i.e., they are F-specific.87 MS2 and Qβ are of similar size (20–30 nm),37 with similar pI values (2–4) reported in the literature.51 Both bacteriophages have single-stranded RNA genomes, although Qβ's genome (4217 nt) is about 18% longer than that of MS2 (3569 nt).87 Out of 8 direct comparisons of reduction of MS2 and Qβ by coagulation (see Table 2), MS2 was reduced to a greater extent in 4 studies, with a negligible overall difference in reduction. However, the mechanism of mitigation was likely different for each bacteriophage. Qβ was inactivated to a greater extent than MS2 in a majority (5 of 7) of direct comparisons, with an average of 1.5log10 greater inactivation. In two tests using alum as the coagulant, treatment efficiency of MS2 was greater in both, while tests with PACl showed greater average reduction of Qβ. Thus, the suitability of MS2 or Qβ as a surrogate may depend both on the mechanism of reduction and the coagulant used. The variability of these well-studied bacteriophages illustrates the great need for additional head-to-head comparison studies.
Even tests performed using nearly identical conditions can differ significantly. Some tests comparing MS2 and Qβ used the same coagulant type and dose (1.1 mg L−1 PACl) with varying results,10,20–22,74 as shown in Table 2. Similarly, Mayer et al.26 and Abbaszadegan et al.27 used the same methods and source water (though the source water composition varied over time). Mayer et al. found relative virus reductions following the order of: fr > ΦX174 > MS2 ≥ PRD1. Abbaszadegan et al. found greater reduction for all four bacteriophages with the same coagulant dose, and bacteriophage reduction followed the order of: fr ≥ MS2 > PRD1 > ΦX174. The only clear difference between the conditions of the two studies was that Mayer et al.'s source water for bacteriophage tests had a higher NOM concentration, as shown in Table 2. In both tests, the four bacteriophages were considered to be either conservative or representative surrogates for the human viruses of interest. However, in a third test by Abbaszadegan et al.,88 bacteriophages fr and MS2 were reduced to a greater extent than the target viruses, while PRD1 and ΦX174 were conservative surrogates.
When inactivation plays a significant role in virus reduction, the biology of the virus – its morphology and infectious pathway – must be considered in addition to its physical properties. Sigstam et al.89 explain that the easily-oxidized amino acids cysteine and methionine are potential indicators of virus capsid degradation, although protein conformation determines the exposure of those amino acids to disinfectants. Sigstam et al. therefore used a complex model to explain cleavage at a particular methionine using the thermodynamics of oxidation in relation to the bonds between capsid proteins, and the “solvent accessible surface area” of that site. The team found that when genome inactivation was the primary disinfection mechanism, inactivation of F-specific bacteriophages fr, GA and MS2 was similar. However when capsid destruction was the primary disinfection mechanism, subtle variations in the protein coat of the four bacteriophages led to differing inactivation.
As with pI, even closely related virus strains can differ in susceptibility to inactivation. For example, Engelbrecht et al.90 measured significantly different free chlorine inactivation rates between serotypes of coxsackievirus (A9 and B5), echovirus (1 and 5) and poliovirus (1 and 2). Virus strains may differ in inactivation depending on the disinfectant as well. In a study by Cromeans et al.,91 echoviruses 1 and 11 had similar free chlorine inactivation rates, but the rate of echovirus 11 inactivation by monochloramine was two orders of magnitude slower than that of echovirus 1. Cromeans et al. also found significant differences in inactivation between serotypes of both human adenovirus (2 and 40, 41) and echovirus (1 and 11), although trends were similar between free chlorine and chloramine. Sigstam et al.89 suggest that closely-related viruses may be appropriate surrogates when the primary inactivation mechanism is genome destruction, but not necessarily for capsid-based inactivation. For chemical coagulation, the charge and large size of polynuclear cations may restrict oxidation to only the capsid surface. In this case, conservative surrogates must be chosen by direct comparison with the viruses of interest. For the generation of free chlorine by electrocoagulation, the inactivation mechanism is likely less specific, because both the viral proteins and genome are oxidized by free chlorine.28,73
Regarding bacteriophage surrogates, Kreißel et al.19 advance the hypothesis that F-specific bacteriophages may be uniquely susceptible to inactivation. F-specific bacteriophages have only one copy of the maturation protein responsible for binding to and infecting the F-pilus structure. The MS2 maturation protein (A protein) is known to be exposed near one of the five-fold vertices, possibly beneath a pore.92 Qβ maturation protein (A2 protein) is similarly located near the five-fold vertex, although conformation may be slightly less rigid than in MS2.92 This orientation may be similar in other F-specific bacteriophages. Kreißel et al. argue that the single maturation protein might be easily blocked or damaged, and F-specific bacteriophages would therefore show greater inactivation than somatic coliphages, which have multiple binding sites. Most studies of inactivation by coagulation have used F-specific bacteriophages, as shown in Table 3. Therefore, the use of these surrogates may overstate virus inactivation.
Kreißel et al. recommend using the somatic coliphage ΦX174 as a more conservative surrogate for coagulation studies. In their study, ΦX174 was insensitive to inactivation by PACl. The pI values for ΦX174 reported in the literature are higher than the pH 4.5 used in the Kreißel et al. study. If polyaluminum cations were responsible for virus inactivation, the coagulant should show little or no attraction to positively charged ΦX174 virions. However, Kreißel et al. measured a negative electrophoretic mobility for ΦX174 by DLS. From a practical perspective, ΦX174 forms large, ill-defined plaques if allowed to incubate overnight19,93 and is therefore not an ideal model virus.
If F-specific bacteriophages are more structurally fragile than somatic bacteriophages, they should also be more susceptible to indiscriminate disinfectants. However, experimental results are inconclusive. ΦX174 may be more sensitive than MS2 to chemical disinfection by HO˙.49 In a study of iodine inactivation,94 MS2 was slightly more sensitive than ΦX174, although neither was nearly as resistant to disinfection as bacteriophage GA, another F-specific bacteriophage. GA also has a relatively high resistance to temperature and pH.95 In contrast, Sigstam et al.89 found GA to be comparable or more susceptible than MS2 and fr to a range of disinfectants, including free chlorine. Regardless, GA's low susceptibility to some disinfectants and high persistence in the environment casts some doubt on the hypothesis that F-specific bacteriophages are structurally more sensitive to inactivation. In the case of free chlorine oxidation, Wigginton et al.73 noted that extensive damage to the MS2 A protein did not cause an inability to bind to host pili. Rather, damage to the capsid protein was likely responsible for inactivation. Therefore, the A protein may be robust to chemical oxidation. More importantly, the case of bacteriophage GA illustrates that a surrogate may be conservative in one application and overly susceptible in another.
In addition, a conservative surrogate for coagulation must be robust to both physical removal and disinfection. ΦX174 is less hydrophobic than MS2 (ref. 41 and 59) and may therefore be a more conservative surrogate for physical removal in the presence of NOM. ΦX174 and MS2 reduction in raw waters due to FeCl3 coagulation was compared in three studies,26,27,88 but the results conflicted as to which bacteriophage was reduced to a greater extent. GA has been shown to persist to a far greater degree than MS2 and Qβ in a pilot coagulation/ultrafiltration treatment plant.96 Jofre et al.97 found that in three water treatment plants, somatic coliphages were found in slightly more samples after prechlorination–flocculation–sedimentation and post-chlorination than F-specific coliphages, but phages of Bacteriodes fragilis were yet more resistant. Suffice it to say that not enough is known about common bacteriophage surrogates, let alone the countless other possible bacteriophages available for research.
Resistance to inactivation or physical removal does not automatically make for an excellent surrogate – the surrogate must be tested alongside the actual virus of interest. A good surrogate should be conservative compared to the human virus, but more importantly it must be representative. A ‘worst-case scenario’ is only valuable insofar as it is remotely possible. By using a surrogate that is especially insensitive, researchers may over-design treatment systems at great cost. Insensitive surrogates may also lead researchers to miss a potential treatment strategy that could be optimized for insensitive targets. This is particularly true in the case of inactivation due to coagulation. As illustrated in section 3.2, inactivation of less than 1log10 may be statistically indiscernible when physical removal is dominant. Therefore, the best surrogate is one tailored to the application. If possible, research should employ more than one surrogate, with different electrostatic charge, hydrophobicity and/or resistance to inactivation, as appropriate. Of course, additional research is required to assess these properties for the many possible bacteriophage surrogates.
5. Conclusions
At this stage in coagulation research, viruses can no longer be assumed to be inert nanoparticles. Both the complexity of viruses as bioparticles and the phenomenon of virus inactivation must be embraced. In particular, the role of permeability in virus sorption and aggregation remains unclear. Virion permeability has been estimated by interpreting empirical electrophoretic mobility data.50 However, to our knowledge, no empirical measures of virion permeability exist, and a clear link between permeability and virion composition and morphology has not been advanced. Furthermore, the direct influence of inner virion structures on surface charge or sorption has not been conclusively demonstrated.Non-DLVO forces must also be considered to explain and predict virus sorption behavior. Research shows that hydrophobicity is an important contributor to sorption, especially for nonpolar virions. Other forces, such as steric interactions and hydrodynamics, are likely to play a significant role when electrostatic forces are repulsive or minimal (e.g., at high ionic strength or near the virus or floc pI). In addition, the composition of the water matrix is also likely to play a strong role for many viruses. NOM may compete for sorption sites on flocs when repulsive electrostatic charges govern NOM-virion interactions, or NOM may act as a sorbent to enhance flocculation of hydrophobic virions. Ca2+ and Mg2+ enhance sorption of viruses to similarly-charged species like NOM, either by cation bridging or surface complexation. Most importantly, current research demonstrates that sorption varies by both virion and environmental conditions.
The potential for inactivation in coagulation processes is both a source of frustration and a promising avenue for water treatment research. Inactivation muddles unit treatment performance testing with artificially high reduction rates. However, future coagulation systems could be optimized for inactivation. Applied research should include at least two bacteriophage surrogates with varying susceptibility to physical removal and inactivation. To inform surrogate selection, and to enable design of improved treatment systems, the mechanism of inactivation by chemical coagulation must be determined. If viruses are inactivated by capsid protein damage, determining a surrogate by physical similarities may be inappropriate. This highlights the need for basic research into coagulation that directly compares human viruses of interest and bacteriophages. More comparisons between bacteriophages are also needed. With more systematic comparisons of multiple bacteriophages, researchers could begin to hypothesize about the variability in bacteriophage performance between experiments.
Plaque assays with recovery from flocs remains the gold standard for quantifying inactivation. More research is required to confirm the validity of using a combination of qPCR and plaque assay without recovery from flocs. The combination of qPCR and plaque assays may prove to be both acceptable and cost-saving for some viruses, but only if future research can show that the method does not underreport inactivation. Furthermore, continued research is needed to determine how inactivation impacts total virus reduction by coagulation. If inactivation of viruses acts as a polishing step for coagulation, seemingly insignificant inactivation would be critical for meeting treatment goals. Further studies comparing the recovery of viruses from flocs by both plaque assay and qPCR could help delineate the relationship between coagulation and inactivation. In addition, inactivation must be separated from aggregation, but quantitative assessment of virus aggregation in treated samples is currently not possible.
Ideally, continued research into the physico-chemical properties of viruses will allow us to predict sorption and inactivation behavior. This type of modeling would help to better identify bacteriophage surrogates as well. Currently, surrogates are often selected based on qualities like size and pI. Unfortunately, the complexity of virus sorption and inactivation eludes such simple measures. Therefore, it is essential to begin to draw connections between virus morphology and physical chemistry. Important strides in this direction have been referenced in this review, such as Langlet et al.'s model of virus electrokinetics,44 Sigstam et al.'s model of virus capsid susceptibility to inactivation89 and Armanious et al.'s method for assessing hydrophobicity from virion surface structure.63 However, these models are still under investigation and cannot yet confidently predict behavior of viruses. Through comparisons of morphologically similar bacteriophages, we can learn more about how minor changes in structure impact sorption and inactivation properties. In the future, we may be able to predict virus behavior and identify new bacteriophage surrogates based on subtle aspects like protein structures or genome size and conformation. The benefits of this work would extend far beyond use in coagulation – from filtration systems, to inactivation by nanoparticles, to modeling virus fate and persistence in the environment and the gastrointestinal tract.
Acknowledgements
This research was supported by the National Science Foundation (NSF) under Grant No. 1433003. All opinions expressed in the paper are the authors' and do not necessarily reflect the views of NSF.References
- N. R. Blacklow and H. B. Greenberg, N. Engl. J. Med., 1991, 325, 252–264 CrossRef CAS PubMed.
- T.-T. Fong and E. K. Lipp, Microbiol. Mol. Biol. Rev., 2005, 69, 357–371 CrossRef CAS PubMed.
- R. Kocwa-Haluch, Pol. J. Environ. Stud., 2001, 10, 485–487 Search PubMed.
- M. Abbaszadegan, M. Lechevallier and C. P. Gerba, J. - Am. Water Works Assoc., 2003, 95, 107–120 CAS.
- United States Environmental Protection Agency, 2015, http://www.epa.gov/ccl.
- WHO guidelines for drinking-water quality, ed. World Health Organization, Geneva, 4th edn, 2011 Search PubMed.
- Centers for Disease Control and Prevention, 2012, http://www.cdc.gov/safewater/effectiveness-on-pathogens.html.
- C. P. Gerba, D. M. Gramos and N. Nwachuku, Appl. Environ. Microbiol., 2002, 68, 5167 CrossRef CAS PubMed.
- N. Shirasaki, T. Matsushita, Y. Matsui, T. Urasaki, M. Kimura and K. Ohno, Water Sci. Technol.: Water Supply, 2014, 14, 429–437 CrossRef CAS.
- N. Shirasaki, T. Matsushita, Y. Matsui, M. Kobuke and K. Ohno, J. Water Supply: Res. Technol.--AQUA, 2010, 59, 501–511 CrossRef CAS.
- N. Shirasaki, T. Matsushita, Y. Matsui and K. Ohno, J. Water Supply: Res. Technol.--AQUA, 2008, 57, 501 CrossRef.
- T. Meyn and T. Leiknes, AIChE J., 2012, 58, 2270–2281 CrossRef CAS.
- T. Matsushita, Y. Matsui, N. Shirasaki and Y. Kato, Desalination, 2005, 178, 21–26 CrossRef CAS.
- B. Zhu, D. A. Clifford and S. Chellam, Water Res., 2005, 39, 3098–3108 CrossRef CAS PubMed.
- L. Fiksdal and T. Leiknes, J. Membr. Sci., 2006, 279, 364–371 CrossRef CAS.
- J. C. Crittenden, R. R. Trussell, D. W. Hand, K. J. Howe and G. Tchobanoglous, MWH's Water Treatment: Principles and Design, John Wiley & Sons, Inc., Hoboken, New Jersey, 3rd edn, 2012 Search PubMed.
- T. Matsushita, Y. Matsui and T. Inoue, Water Sci. Technol., 2004, 50, 201–206 CAS.
- T. Matsushita, N. Shirasaki, Y. Matsui and K. Ohno, Chemosphere, 2011, 85, 571–576 CrossRef CAS PubMed.
- K. Kreißel, M. Bösl, M. Hügler, P. Lipp, M. Franzreb and B. Hambsch, Water Res., 2014, 51, 144–151 CrossRef PubMed.
- N. Shirasaki, T. Matsushita, Y. Matsui, T. Urasaki and K. Ohno, Water Res., 2009, 43, 605–612 CrossRef CAS PubMed.
- N. Shirasaki, T. Matsushita, Y. Matsui, M. Kobuke and K. Ohno, J. Membr. Sci., 2009, 326, 564–571 CrossRef CAS.
- Y. Matsui, T. Matsushita, S. Sakuma, T. Gojo, T. Mamiya, H. Suzuoki and T. Inoue, Environ. Sci. Technol., 2003, 37, 5175–5180 CrossRef CAS PubMed.
- A. I. Zouboulis, P. A. Moussas and F. Vasilakou, J. Hazard. Mater., 2008, 155, 459–468 CrossRef CAS PubMed.
- G. Lei, J. Ma, X. Guan, A. Song and Y. Cui, Desalination, 2009, 247, 518–529 CrossRef CAS.
- United States Environmental Protection Agency, Stage 1 Disinfectants and Disinfection Byproducts Rule, EPA 816-F-01-014, 2001 Search PubMed.
- B. K. Mayer, H. Ryu and M. Abbaszadegan, Environ. Sci. Technol., 2008, 42, 6890–6896 CrossRef CAS PubMed.
- M. Abbaszadegan, B. K. Mayer, H. Ryu and N. Nwachuku, Environ. Sci. Technol., 2007, 41, 971–977 CrossRef CAS PubMed.
- C. T. Tanneru, N. Jothikumar, V. R. Hill and S. Chellam, Environ. Sci. Technol., 2014, 48, 14590–14598 CrossRef CAS PubMed.
- C. T. Tanneru and S. Chellam, Environ. Sci. Technol., 2013, 47, 4612–4618 CrossRef CAS PubMed.
- C. T. Tanneru and S. Chellam, Water Res., 2012, 46, 2111–2120 CrossRef CAS PubMed.
- D. Ghernaout, B. Ghernaout, A. Boucherit, M. W. Naceur, A. Khelifa and A. Kellil, Desalin. Water Treat., 2009, 8, 91–99 CrossRef CAS.
- K. L. Dubrawski and M. Mohseni, Water Res., 2013, 47, 5371–5380 CrossRef CAS PubMed.
- H. A. C. Moreno, D. L. Cocke, J. A. Gomes, P. Morkovsky, J. R. Parga, E. Peterson and C. Garcia, ECS Trans., 2007, 3, 67–78 Search PubMed.
- United States Environmental Protection Agency, Microbial and Disinfection Byproduct Rules Simultaneous Compliance Guidance Manual, EPA 815-R-99-015, 1999 Search PubMed.
- N. Shirasaki, T. Matsushita, Y. Matsui and T. Marubayashi, Chem. Eng. J., 2016, 284, 786–793 CrossRef CAS.
- T. Harif, M. Khai and A. Adin, Water Res., 2012, 46, 3177–3188 CrossRef CAS PubMed.
- J. Langlet, F. Gaboriaud, J. F. L. Duval and C. Gantzer, Water Res., 2008, 42, 2769–2777 CrossRef CAS PubMed.
- S. L. Penrod, T. M. Olson and S. B. Grant, Langmuir, 1996, 12, 5576–5587 CrossRef CAS.
- J. Zhuang and Y. Jin, J. Environ. Qual., 2003, 32, 816–823 CrossRef CAS PubMed.
- R. C. Bales, S. Li, K. M. Maguire, M. T. Yahya and C. P. Gerba, Water Resour. Res., 1993, 29, 957–963 CrossRef.
- C. V. Chrysikopoulos and V. I. Syngouna, Colloids Surf., B, 2012, 92, 74–83 CrossRef CAS PubMed.
- S. E. Dowd, S. D. Pillai, S. Wang and Y. Corapcioglu, Appl. Environ. Microbiol., 1998, 64, 405–410 CAS.
- C. M. Schaldach, W. L. Bourcier, H. F. Shaw, B. E. Viani and W. D. Wilson, J. Colloid Interface Sci., 2006, 294, 1–10 CrossRef CAS PubMed.
- J. Langlet, F. Gaboriaud, C. Gantzer and J. F. L. Duval, Biophys. J., 2008, 94, 3293–3312 CrossRef CAS PubMed.
- J. F. L. Duval and H. Ohshima, Langmuir, 2006, 22, 3533–3546 CrossRef CAS PubMed.
- C. Dika, J. F. L. Duval, H. M. Ly-Chatain, C. Merlin and C. Gantzer, Appl. Environ. Microbiol., 2011, 77, 4939–4948 CrossRef CAS PubMed.
- J. Langlet, L. Ogorzaly, J. C. Schrotter, C. Machinal, F. Gaboriaud, J. F. L. Duval and C. Gantzer, J. Membr. Sci., 2009, 326, 111–116 CrossRef CAS.
- C. Dika, M. H. Ly-Chatain, G. Francius, J. F. L. Duval and C. Gantzer, Colloids Surf., A, 2013, 435, 178–187 CrossRef CAS.
- B. K. Mayer, Y. Yang, D. W. Gerrity and M. Abbaszadegan, Microbiol. Insights, 2015, 8, 15–28 CrossRef PubMed.
- J. F. L. Duval and F. Gaboriaud, Curr. Opin. Colloid Interface Sci., 2010, 15, 184–195 CrossRef CAS.
- B. Michen and T. Graule, J. Appl. Microbiol., 2010, 109, 388–397 CAS.
- A. Borodavka, R. Tuma and P. G. Stockley, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15769–15774 CrossRef CAS PubMed.
- P. G. Stockley, R. Twarock, S. E. Bakker, A. M. Barker, A. Borodavka, E. Dykeman, R. J. Ford, A. R. Pearson, S. E. V. Phillips, N. A. Ranson and R. Tuma, J. Biol. Phys., 2013, 39, 277–287 CrossRef CAS PubMed.
- L. Gutiérrez, S. E. Mylon, B. Nash and T. H. Nguyen, Environ. Sci. Technol., 2010, 44, 4552–4557 CrossRef PubMed.
- B. Yuan, M. Pham and T. H. Nguyen, Environ. Sci. Technol., 2008, 42, 7628–7633 CrossRef CAS PubMed.
- T. H. Nguyen, N. Easter, L. Gutiérrez, L. Huyett, E. Defnet, S. E. Mylon, J. K. Ferri and N. A. Viet, Soft Matter, 2011, 7, 10449–10456 RSC.
- C. Dika, C. Gantzer, A. Perrin and J. F. L. Duval, Phys. Chem. Chem. Phys., 2013, 15, 5691–5700 RSC.
- R. Aronino, C. Dlugy, E. Arkhangelsky, S. Shandalov, G. Oron, A. Brenner and V. Gitis, Water Res., 2009, 43, 87–96 CrossRef CAS PubMed.
- C. Dika, J. F. L. Duval, G. Francius, A. Perrin and C. Gantzer, J. Colloid Interface Sci., 2015, 446, 327–334 CrossRef CAS PubMed.
- T. Patrick, Dan. Med. Bull., 1999, 46, 183–196 Search PubMed.
- M. J. Mattle, B. Crouzy, M. Brennecke, K. R. Wigginton, P. Perona and T. Kohn, Environ. Sci. Technol., 2011, 45, 7710–7717 CrossRef CAS PubMed.
- V. I. Syngouna and C. V. Chrysikopoulos, J. Contam. Hydrol., 2012, 129-130, 11–24 CrossRef CAS.
- A. Armanious, M. Aeppli, R. Jacak, D. Refardt, T. Sigstam, T. Kohn and M. Sander, Environ. Sci. Technol., 2015, 50, 732–743 CrossRef PubMed.
- T. Matsushita, H. Suzuki, N. Shirasaki, Y. Matsui and K. Ohno, Sep. Purif. Technol., 2013, 107, 79–84 CrossRef CAS.
- S. E. Mylon, C. I. Rinciog, N. Schmidt, L. Gutiérrez, G. C. L. Wong and T. H. Nguyen, Langmuir, 2010, 26, 1035–1042 CrossRef CAS PubMed.
- M. Pham, E. A. Mintz and T. H. Nguyen, J. Colloid Interface Sci., 2009, 338, 1–9 CrossRef CAS PubMed.
- A. J. De Kerchove and M. Elimelech, Langmuir, 2008, 24, 3392–3399 CrossRef CAS PubMed.
- S. A. Banwart, in Chemistry in Aquatic Systems: Local and Global Perspectives, ed. G. Bidoglio and W. Stumm, Springer Netherlands, 1st edn, 1994, pp. 337–374 Search PubMed.
- M. Rebhun, S. Meir and Y. Laor, Environ. Sci. Technol., 1998, 32, 981–986 CrossRef CAS.
- F. Xiao, P. Yi, X. R. Pan, B. J. Zhang and C. Lee, Desalination, 2010, 250, 902–907 CrossRef CAS.
- S. Chang, R. Stevenson, A. Bryant, R. Woodward and P. Kabler, Am. J. Public Health Nations Health, 1958, 48, 159–169 CrossRef CAS PubMed.
- M. Chaudhuri and R. S. Engelbrecht, J. - Am. Water Works Assoc., 1970, 62, 563–567 CAS.
- K. R. Wigginton, B. M. Pecson, T. Sigstam, F. Bosshard and T. Kohn, Environ. Sci. Technol., 2012, 46, 12069–12078 CrossRef CAS PubMed.
- N. Shirasaki, T. Matsushita, Y. Matsui, T. Urasaki, A. Oshiba and K. Ohno, Water Sci. Technol., 2010, 61, 2027–2034 CrossRef CAS PubMed.
- D. Ghernaout, A. Badis, A. Kellil and B. Ghernaout, Desalination, 2008, 219, 118–125 CrossRef CAS.
- A. C. Ndjomgoue-Yossa, C. P. Nanseu-Njiki, I. M. Kengne and E. Ngameni, Int. J. Environ. Sci. Technol., 2015, 12, 2103–2110 CrossRef CAS.
- K. A. Vakil, M. K. Sharma, A. Bhatia, A. A. Kazmi and S. Sarkar, Sep. Purif. Technol., 2014, 130, 160–166 CrossRef CAS.
- E. M. Symonds, M. M. Cook, S. M. McQuaig, R. M. Ulrich, R. O. Schenck, J. O. Lukasik, E. S. Van Vleet and M. Breitbart, Sci. Rep., 2015, 5, 9380 CrossRef CAS PubMed.
- S. Gao, M. Du, J. Tian, J. Yang, J. Yang, F. Ma and J. Nan, J. Hazard. Mater., 2010, 182, 827–834 CrossRef CAS PubMed.
- K. O. O'Connell, J. R. Bucher, P. E. Anderson, C. J. Cao, A. S. Khan, M. V. Gostomski and J. J. Valdes, Appl. Environ. Microbiol., 2006, 72, 478–483 CrossRef PubMed.
- J. Langlet, F. Gaboriaud and C. Gantzer, J. Appl. Microbiol., 2007, 103, 1632–1638 CrossRef CAS PubMed.
- D. A. Kuzmanovic, I. Elashvili, C. Wick, C. O'Connell and S. Krueger, J. Mol. Biol., 2006, 355, 1095–1111 CrossRef CAS PubMed.
- H. Ryu, B. K. Mayer and M. Abbaszadegan, J. Water Health, 2010, 8, 101–108 CrossRef CAS PubMed.
- J. Bae and K. J. Schwab, Appl. Environ. Microbiol., 2008, 74, 477–484 CrossRef CAS PubMed.
- J. A. Thurston-Enriquez, C. N. Haas, J. Jacangelo, K. R. Riley and C. P. Gerba, Appl. Environ. Microbiol., 2003, 69, 577–582 CrossRef CAS PubMed.
- G.-A. Shin and M. D. Sobsey, Water Sci. Technol.: Water Supply, 2015, 15, 158–163 CrossRef CAS.
- N. H. Acheson, Fundamentals of Molecular Virology, John Wiley & Sons, Inc., Hoboken, New Jersey, 1st edn, 2007 Search PubMed.
- M. Abbaszadegan, P. Monteiro, N. Nwachuku, A. Alum and H. Ryu, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2008, 43, 171–177 CrossRef CAS PubMed.
- T. Sigstam, G. Gannon, M. Cascella, B. M. Pecson, K. R. Wigginton and T. Kohn, Appl. Environ. Microbiol., 2013, 79, 3455–3467 CrossRef CAS PubMed.
- R. S. Engelbrecht, M. J. Weber, B. L. Salter and C. A. Schmidt, Appl. Environ. Microbiol., 1980, 40, 249–256 CAS.
- T. L. Cromeans, A. M. Kahler and V. R. Hill, Appl. Environ. Microbiol., 2010, 76, 1028–1033 CrossRef CAS PubMed.
- K. Toropova, P. G. Stockley and N. A. Ranson, J. Mol. Biol., 2011, 408, 408–419 CrossRef CAS PubMed.
- N. K. Beck, K. Callahan, S. P. Nappier, H. Kim, M. D. Sobsey and J. S. Meschke, J. Rapid Methods Autom. Microbiol., 2009, 17, 455–464 Search PubMed.
- G. M. Brion, N. B. O'Banion and G. L. Marchin, J. Water Health, 2004, 2, 261–266 Search PubMed.
- Y. Yang and M. W. Griffiths, Appl. Environ. Microbiol., 2013, 79, 4564–4567 CrossRef CAS PubMed.
- N. Boudaud, C. Machinal, F. David, A. Fréval-Le Bourdonnec, J. Jossent, F. Bakanga, C. Arnal, M. P. Jaffrezic, S. Oberti and C. Gantzer, Water Res., 2012, 46, 2651–2664 CrossRef CAS PubMed.
- J. Jofre, E. Ollé, F. Ribas, A. Vidal and F. Lucena, Appl. Environ. Microbiol., 1995, 61, 3227–3231 CAS.
- B. Zhu, D. A. Clifford and S. Chellam, Water Res., 2005, 39, 5153–5161 CrossRef CAS PubMed.
- N. Shirasaki, T. Matsushita, Y. Matsui, T. Urasaki and K. Ohno, Water Sci. Technol.: Water Supply, 2012, 12, 666–673 CrossRef CAS.
- N. Shirasaki, T. Matsushita, Y. Matsui, A. Oshiba, T. Marubayashi and S. Sato, Water Res., 2014, 48, 375–386 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |