
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Emerging investigators series: microbial communities in full-scale drinking water distribution systems – a meta-analysis†
Quyen M.
Bautista-de los Santos
a,
Joanna L.
Schroeder
a,
Maria C.
Sevillano-Rivera
b,
Rungroch
Sungthong
a,
Umer Z.
Ijaz
a,
William T.
Sloan
a and
Ameet J.
Pinto
*bc
aInfrastructure and Environment Research Division, School of Engineering, University of Glasgow, UK
bDepartment of Civil and Environmental Engineering, Northeastern University, Boston, MA, USA. E-mail: a.pinto@neu.edu
cDepartment of Bioengineering, Northeastern University, Boston, MA, USA
First published on 30th March 2016
Abstract
In this study, we co-analyze all available 16S rRNA gene sequencing studies from bulk drinking water samples in full-scale drinking water distribution systems. Consistent with expectations, we find that Proteobacteria, particularly Alpha- and Betaproteobacteria, dominate drinking water bacterial communities irrespective of origin of study and presence/absence of or disinfectant residual type. Microbial communities in disinfectant residual free systems are more diverse than in those that maintain a disinfectant residual. Further, we find positive associations between mean relative abundance and occurrence of bacteria within a disinfectant category group. The relative abundance and occurrence of key bacterial genera (e.g. Legionella, Mycobacterium, Pseudomonas) is influenced by the presence/absence of a disinfectant residual and the type of disinfectant residual used. Similarly, we find widespread distribution of bacterial genera that are of interest from both an ecological and process perspectives (e.g. nitrification, predation). By estimating the contribution of potential contaminating genera to published drinking water datasets, we recommend that routine sequencing of negative controls be included in drinking water studies. Finally, we test the utility of predicting the metabolic potential of drinking water communities using 16S rRNA gene data and recommend against this practice. Though data heterogeneity across available datasets is a major confounding factor in our meta-analysis, we recommend that efforts to standardize sample processing protocols to address it may not be optimal for the drinking water microbial ecology field at this juncture. Rather, we recommend standardizing data and meta-data reporting, starting with making all sequencing data publicly available, and sample sharing as means of supporting future efforts for comparative analyses across drinking water systems/studies.
Water impactMicrobial communities in drinking water systems can mediate wide-ranging impacts from biofiltration for pollutant removal to public health risks. Understanding their distribution and abundance across systems will enable improved microbial management strategies for the drinking water industry. We conducted a meta-analysis of microbial communities in bulk drinking water using all available 16S rRNA gene sequencing data to highlight differences and similarities across full-scale drinking water distribution systems with different microbial growth control strategies. |
1. Introduction
Drinking water distribution systems (DWDSs) are designed, built, and managed with the purpose of conveying potable and palatable water from drinking water treatment plants (DWTPs) to the consumer's taps. The transport of drinking water (DW) through the DWDS is accompanied by a mass migration of the microbial communities that are an inevitable component of this ecosystem and controlling their growth is paramount to the provision of safe DW. Minimizing undesirable microbial growth in the DWDS is currently achieved by managing two primary factors: ensuring low concentration of assimilable organic carbon (AOC)1 and other growth-rate limiting substrates (e.g. nitrogen, phosphorus)2 and/or applying residual disinfectants such as chlorine or chloramine. DWDSs without disinfectant residual typically aim to maintain AOC levels below 10 μg l−1, while residual disinfectant concentrations in disinfected DWDSs are typically maintained lower than the maximum guideline levels (chlorine: 5.0 mg l−1; monochloramine: 3.0 mg l−1)3 to avoid taste and odor issues. Despite these rigorous efforts, it is well documented that DWDSs harbor abundant and diverse microbial communities.4–7Recent advances in our understanding of the DW microbiome can in large part be attributed to the application of high-throughput and deep DNA sequencing based methods that target the 16S rRNA gene.8,9 These approaches have also highlighted the influence of process operations,5,10,11 disinfectant type,12–14 environmental conditions,7 hydraulic conditions,15,16 distribution system structure,7,17 premise plumbing (also referred to as building plumbing) characteristics,13,18etc. on the structure and composition of the DW microbiome. Emerging from these studies is a general consensus on the types of microorganisms that are typically encountered in DW samples. Bacteria within the phylum Proteobacteria,19 and in particular those within the classes of Alpha- and Betaproteobacteria, have been shown to be dominant in nearly every study published thus far. Nonetheless, studies have also reported differences in the dominance of these classes depending on a range of factors, including but not limited to seasons7,17 and disinfection strategy.6,12,20,21 Despite this emerging consensus about the composition of the DW microbiome, particularly the bacterial community, to our knowledge there has been no study that attempts a collective analysis (i.e. meta-analysis) of all publicly available DW datasets. Such efforts are particularly critical, as the study of the DW microbiome with high-throughput sequencing methods is nascent, compared to efforts to characterize microbiomes associated with other environments, e.g. human gut22 and even another engineered aquatic system, i.e. the wastewater/activated sludge system.23,24 Such an early-stage meta-analysis effort can reveal conserved features across DW systems and help identify targeted research questions and also highlight opportunities to improve future DW microbiome studies.
In this study, we systematically collate and compare all publicly available datasets involving bulk DW samples collected at the outlet of the DWTP (DWTPoutlet), in the DWDS, and at point-of-use (POU). We have focused our analysis on bulk DW samples for several reasons. First, bulk water represents the primary mode of customer exposure to DW microbial communities. Second, studies have clearly shown that bacterial communities in bulk water and biofilms on pipe walls are distinct,25,26 although biofilms influence the former27,28 and can have potential impacts on health.29 Finally, several studies have demonstrated that though there is temporal variation,7 the bulk DW bacterial community within a given distribution system is relatively stable irrespective of the sampling location6,7,30 over short time-scales and is even reproducible over annual time-scales.7 In contrast, biofilms are extremely spatially heterogeneous31 and are likely to develop over time-scales that are much longer than the residence time of water within a given DWDS. This spatial heterogeneity and uncertainty related to time-scales of community assembly results in a poor understanding of how a biofilm community at one location in the DWDS may relate to those at other locations within the same system. Therefore, the lack of rigorous characterization of biofilm heterogeneity even for a single DWDS, limits the utility of comparing biofilm communities across systems. The objectives of this study were to: (1) identify microbial populations that are detected across all publicly available bulk DW datasets; (2) evaluate the variation in the occurrence and relative abundance of target microbial groups, at the phylum/class and operational taxonomic unit (OTU) level, (3) evaluate the relationship between occurrence and relative abundance of taxa across systems, (4) determine the association between disinfection strategy and microbial community, and (5) provide insights into their functional potential across all samples and within disinfection strategy type, to the extent possible.
2. Methods
2.1. Data collection
We focused our efforts on published datasets that involved (1) collection of bulk water samples from either the DWTPoutlet, in the DWDS and/or at the POU, (2) extraction of DNA from the sample without an enrichment or cultivation step, (3) PCR amplification of any of the hypervariable regions of the 16S rRNA gene from the extracted DNA, and (4) sequencing of the PCR product on any high-throughput DNA sequencing platform (i.e. Illumina MiSeq, 454 pyrosequencing, and Ion Torrent). Further, we focus on differences across sampling locations, rather than temporal change at each sampling location. As a result, multiple temporally distinct samples collected from the same sampling location were collapsed into a single sample. Based on these criteria, we were able to identify 21 distinct studies with 6,5,4,2,2,1, and 1 datasets from USA,7,32–36 China,10,37–40 Netherlands,6,20,26,41 UK,15,28 Switzerland,11,42 Australia,43 and France,44 respectively. Of these 21 datasets, only 14 datasets were either publicly available or made available upon data request (Table S1†). Hence, only these 14 datasets comprising of 142 distinct sampling locations were included in this study.6,7,10,15,26,28,32–35,37–39,432.2. Data processing
The FASTA/FASTQ files from individual datasets were processed using a combination of tools and quality filtering criteria depending on sequencing platforms and hypervariable regions of the 16S rRNA gene sequenced. The FASTQ files containing single-end reads were quality filtered using sickle v.1.3345 with a minimum quality score of 28 and a minimum length of 150 bp after trimming and then converted to FASTA format using the fastq_to_fasta command in the FASTX-Toolkit v.0.0.13.2.46 The FASTQ files containing paired-end reads were processed using pear v.0.8.147 to make contigs, with a minimum quality score of 28 and a minimum length of 150 bp after assembly. The FASTA files were dereplicated in mothur,48 and unique sequences were matched against the SILVA 119 SSURef_Nr database49 using blastn50 with an identity ≥97% and an expect (e) value less than 0.000005. The best match 16S rRNA gene sequences from the SILVA 119 database were extracted and used for further analysis. Sequences that did not find a suitable match in the Silva 119 database were excluded from alpha and beta-diversity analysis. The best-match sequences corresponding to each sample were then aligned against the SILVA seed alignment available through mothur.48 The alignment was screened to remove poorly aligned sequences and filtered using the vertical = T and trump = . options in mothur.48 The filtered alignment was then clustered into OTUs at sequence similarity cutoff of 97% using the average neighbor clustering approach.48 All sequences were classified using the Naïve Bayesian classifier51 (80% confidence threshold) using SILVA taxonomy and consensus taxonomy of OTUs was estimated using 80% consensus cutoff.2.3. Data analyses and statistics
The number of sequences across the 142 sampling locations varied from 223 to 10.8 million. Given significant variability in sample size,52 we subsampled the data to normalize the dataset. In order to determine the appropriate subsampling depth, we estimated the Good's coverage for all sampling locations at sampling depths ranging from 200–2500 sequences. An appropriate sampling depth was determined by selecting subsampling depths that provided >80% Good's coverage for each sample while retaining the maximum number of sampling locations from the dataset. This presented the options of subsampling at 500 and 1000 sequences per sample, with the loss of 2 and 6 sampling locations at each of these subsampling depths, respectively (Fig. S1†). A Mantel test conducted using distances matrices constructed with Bray–Curtis metric at subsampling depths of 500 and 1000 sequences per sampling location showed significant correlations between the two distance matrices (Mantel's R = 0.995, p = 0), indicating that a small benefit from a higher subsampling depth was accompanied by the loss of 4 additional sampling locations. As a result, a subsampling depth of 500 was selected to maximize the number of sampling locations retained. All estimates of alpha and beta-diversity were performed at this subsampling depth.A subsampled OTU table was used as input for a range of diversity analyses using vegan53 and plots using the package ggplot254 in R.55 Specifically, we estimated richness (i.e. observed OTUs), Inverse Simpson index, Shannon index, and Pielou's evenness as measures of alpha-diversity. Beta-diversity analyses involved clustering of samples using the heatmap2 module in gplots56 using the Bray–Curtis distance metric, while overlap in membership between communities was estimate using the Jaccard index in mothur.48 The most abundant sequence in each OTU was used as the representative sequence (see results) and RAxML57 was used to construct a maximum likelihood phylogenetic tree with the generalized time reversible (GTR) substitution model and GAMMA distribution model using 1000 bootstraps using these representative sequences. The resultant phylogenetic tree and relevant OTU data were then visualized in EvolView.58 Permutational multivariate analysis of variance (PERMANOVA)53 tests were conducted to determine the effects of the study of origin, source water type, disinfectant strategy, and proportion of data retained after matching the SILVA database on differences between samples using the Bray–Curtis and Jaccard metrics.
We estimated the mean relative abundance (MRA) and occurrence of each OTU across all sampling locations and sampling locations grouped by disinfection strategy. For these calculations, we estimated the relative abundance of each OTU for a sampling location by using all reads in the sample and not just the subset of reads matching the SILVA database. These full-samples were also used to compare occurrence and MRA of key OTUs across disinfection strategies (see Results and discussion section). To check for the likelihood of contamination in DW studies, we extracted all OTUs classifying to the genus level that corresponded to the list of kit/reagent contamination genera identified by Salter et al.59 and estimated their contribution to the overall dataset. The subsampled OTU table was also used to predict functional potential of the bacterial and archaeal (where present) communities using Tax4Fun.60 Tax4Fun generates a relative abundance of KEGG61 orthology (KO) groups associated with each sampling location depending on matches of the representative sequence from each OTU to KEGG organisms, while also providing information on fraction of OTUs that do not match KEGG organisms, i.e. the FTU metric. Analysis of variance (ANOVA) was performed to assess whether FTU values were significantly different across the three disinfectant strategies. For comparisons of KO relative abundance in samples grouped by disinfection strategy, we picked a subset of samples from each disinfection strategy (as outlined in the results and discussion) such that the distribution of FTU values and mean FTU was not significantly different between disinfection strategies. Significantly different KO's across different disinfection strategies were identified using the Kruskal–Wallis with Benjamini–Hochberg62 correction with a false discovery rate of 0.05. A schematic outlining the workflow for all data-analyses in this manuscript is provided in the supplemental material (Fig. S2† with a summary).
3. Results and discussion
3.1. Data structure and composition
The 14 datasets consisted of 142 distinct sampling locations, with 79 and 63 sampling locations associated with systems with and without a disinfectant residual, respectively. Of the 79 sampling locations from systems with a disinfectant residual, 40 and 39 were from chlorinated and chloraminated systems, respectively. Data for a majority of these sampling locations was obtained on the 454 pyrosequencing platform (n = 103), with data for 25 and 14 locations obtained on the Illumina MiSeq and Ion Torrent sequencing platforms, respectively. The 16S rRNA gene hypervariable regions also varied depending on the datasets. Specifically, the hypervariable regions covered by the sequencing libraries for the 142 sampling locations included V1–V2 (n = 17), V1–V3 (n =7), V3 (n =14), V3–V4 (n = 2), V3–V5 (n = 2), V4 (n = 25), V4–V5 (n = 20), V4–V6 (n = 3), and V5–V6 (n = 52). Given the significant amount of data heterogeneity (sequencing platform and target 16S rRNA gene hypervariable region), we could not cluster sequences across studies directly into OTUs, a constraint highlighted by other recent meta-analysis efforts.63,64 Hence, we utilized a pre-processing step of sequence matching to the SILVA database as a means of being able to combine this highly heterogeneous data (i.e. a reference based approach). A limitation of this approach is that the analysis becomes database dependent, and the results will be constrained to the taxonomic groups present in the database used as reference.Given the reported dominance of Proteobacteria (a dominant phylum also in the reference 16S rRNA gene databases) in DW samples, it was surprising to discover a high level of variability in terms of the proportion of sequences for each sampling location matching a reference sequence in the SILVA database (Fig. 1), which ranged from 22.7% to as high as 99% across all locations. The low proportion of matches to the SILVA database was not specific to any particular study, but rather there was significant variability within studies themselves. For example, the proportion of sequences with SILVA matches was 28–85%, 23–84%, and 36–77% for samples from Holinger et al.,32 Pinto et al.,7 and Roeselers et al.,6 respectively. There were indicative trends suggesting that a greater proportion of sequences generated from systems without a disinfectant residual were less likely to find a match in the reference 16S rRNA gene database. The average proportion of data with matches to the SILVA database for chlorinated (Chl), chloraminated (Chm), and disinfectant residual-free (Drf) samples were 82.1 ± 13.9% (n = 40), 83.9 ± 16.1 (n = 39) and 52 ± 8.5% (n = 63), respectively (Fig. 1A, p < 0.0001 for Chl-Drf and Chm-Drf groups). This suggests that disinfectant residual-free DW systems harbor bacterial diversity that is not well represented in 16S rRNA gene reference database and will render reference based OTU picking approaches vulnerable to poorly capturing overall diversity. However, this observation should be treated with caution as a majority of the samples from the Drf dataset emerge from a single comprehensive study,6 and hence is heavily influenced by 16S rRNA gene primer choice.
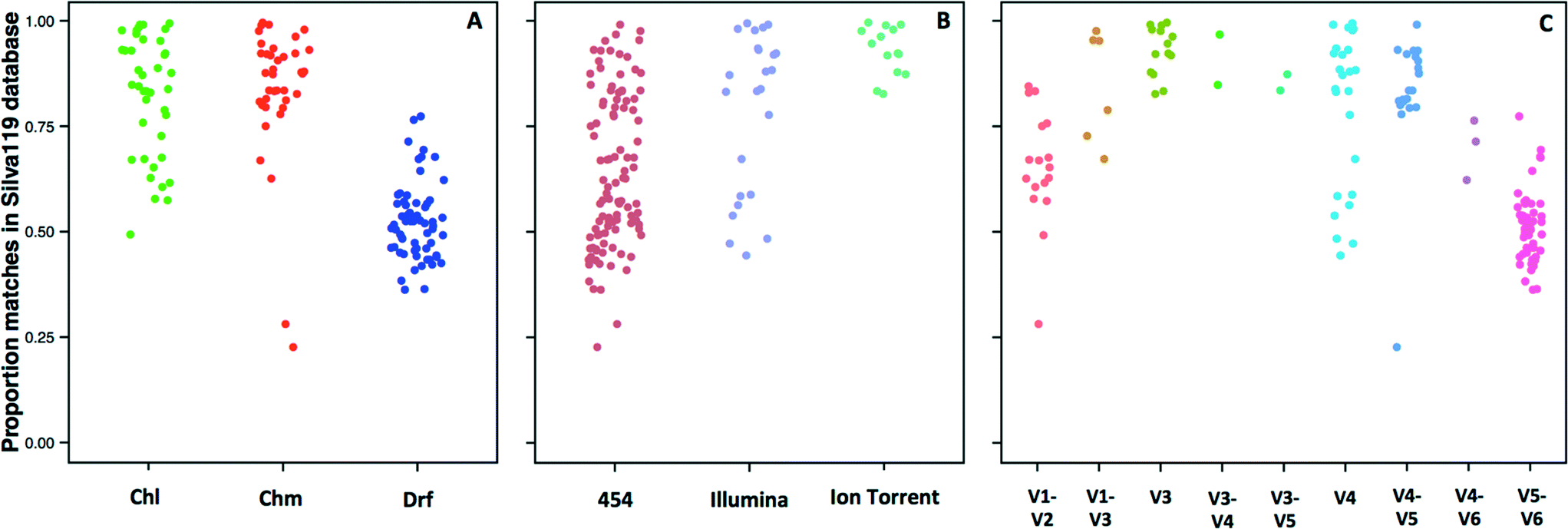 | ||
| Fig. 1 Proportion of reads from each sample location matching a reference sequence in the SILVA119 database with a minimum percent identity of 97% (e value < 0.000005) grouped by (A) disinfection group (Chl: chlorinated, Chm: chloraminated, Drf: disinfectant residual-free); (B) sequencing platform; and (C) 16S rRNA gene hypervariable region represented in the datasets utilized in this study. | ||
Significant differences in the proportion of data matching the SILVA database were observed according to the sequencing platform (454-Illumina and 454-Ion Torrent, p < 0.001) (Fig. 1B); however, the samples sequenced with Ion Torrent consist of only one hypervariable region amplified, therefore these results should be interpreted with caution. Similarly, significant differences in the proportion of data matching the SILVA database were observed according to the hypervariable region amplified, with p-values ranging from 6.1 × 10−14 to 0.001. The direct effect of the lack of matches in the reference database meant that a proportion of data from each sample was not used for alpha and beta-diversity analyses. Specifically, all alpha and beta-diversity analyses were based on 81.5% of the sequence data from 142 sampling locations, with the average sequence data retained per sampling location being 69.4 ± 19.9%. It is also important to note that our efforts to combine multiple datasets does not account for biases that arise from sample collection and handling protocols,65,66 DNA extraction,67 and PCR amplification68 approaches. As a result, this meta-analysis study does not provide a quantitative perspective on similarities and differences between the samples included in this study. Rather, we aim to highlight indicative differences that might be prime candidates for follow-up studies designed using standardized protocols across sample/system types.
3.2. Microbial community composition
Across all datasets, bacteria constituted a majority of the microbial community with the archaea being detected at very low levels, despite the fact that several studies used 16S rRNA gene primers that span bacterial and archaeal domains (e.g. V4 primer set used by Caporaso et al.).9 Specifically, archaeal sequences were detected in 9.5%, 19.5%, and 89% of the sampling locations from chlorinated, chloraminated, and disinfectant residual-free systems, respectively. Despite the widespread detection of archaeal sequences in disinfectant residual-free locations they contributed at a low level towards the overall community, with their MRA across Drf locations being 0.13 ± 3.3%. As has been reported in several previous DW studies, Proteobacteria were by far the most dominant bacterial phylum with their MRA for chlorinated, chloraminated, and disinfectant residual-free locations being 68 ± 42.7%, 75 ± 42.9%, and 54 ± 20.9%, respectively (Fig. 2A). Within Proteobacteria, Alpha- and Betaproteobacteria were dominant and constituted greater than 80% of the proteobacterial sequences across all locations. Actinobacteria was the second most abundant phyla in disinfected systems, constituting 11.7 ± 16.2 and 8.2 ± 10.7% of the data from chlorinated and chloraminated systems, respectively. In contrast, Acidobacteria was the second most dominant phyla for the disinfectant residual-free locations (MRA = 6.3 ± 4%), while it constituted less than 1% of the sequences in disinfected systems. These differences between disinfection strategies was not only limited to the abundance of the various phyla, but also with respect to their occurrence (Fig. 2B). For example, sequences from phyla Nitrospinae and Crenarchaeota were not detected in any of the disinfected samples, while being present in 29% and 46.7% of the samples without a disinfectant residual. Similarly, several low to medium abundance phyla were detected much more routinely in disinfectant residual-free systems compared to the systems with a disinfectant residual, indicating a greater taxonomic diversity of the bacterial community in absence of a disinfectant residual.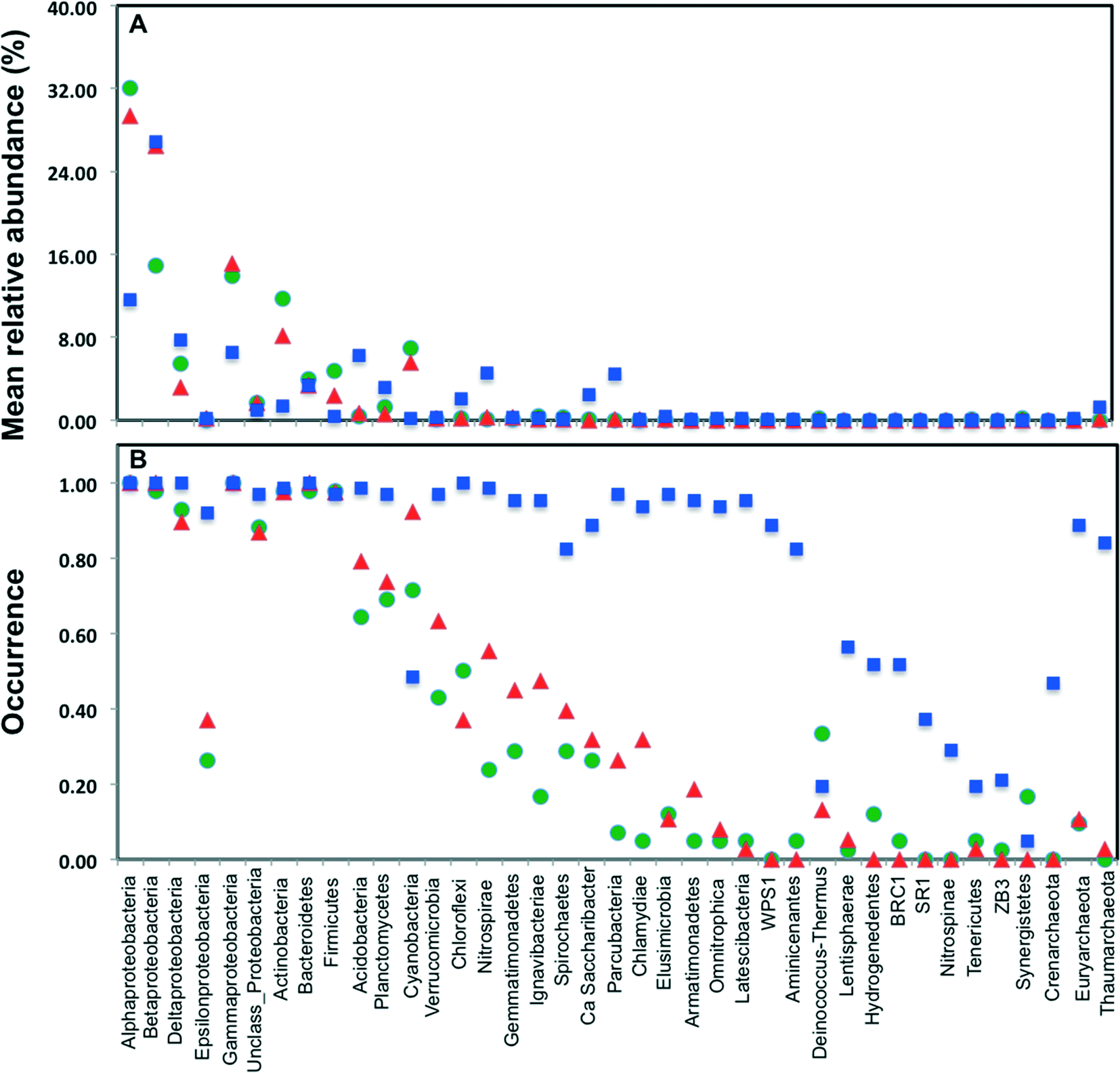 | ||
| Fig. 2 (A) Mean relative abundance of bacterial phyla/classes grouped by disinfection strategy (Chl: chlorinated, green; Chm: chloraminated, red; Drf: disinfectant residual-free, blue), estimated as the number of reads assigned to the phylum/class divided by the total number of reads in each sampling location averaged over the disinfection strategy; (B) occurrence of main bacterial phyla/classes per disinfection group, estimated as the proportion of sampling locations in which the phylum/class was detected. This figure only includes reads in each sampling location that mapped to the SILVA database. | ||
3.3. Alpha-diversity of bacterial communities
There were no significant differences in alpha-diversity between the sampling locations with chlorine and chloramine as the disinfectant residual (Fig. 3). The inverse Simpson index was slightly higher for the chlorinated (12.8 ± 15.4) as compared to the chloraminated (9.3 ± 6.4) systems, however they also showed higher variability across locations. Consistently, the samples from disinfectant residual-free systems were richer, more diverse, and more even as compared to the samples with a residual disinfectant (p < 0.0001). For example, the average number of OTUs in the disinfectant residual-free systems was 225 ± 60 as compared to 85 ± 60 and 87 ± 25 for chlorinated and chloraminated samples, respectively. Similarly, bacterial communities in disinfectant residual-free systems were significantly more even (0.84 ± 0.14) as compared to those in the chlorinated (0.64 ± 0.19) and chloraminated (0.64 ± 0.13) systems. This observation of higher diversity in disinfectant residual free sampling locations arises despite the fact that a smaller proportion of sequences from the non-disinfected samples were utilized for OTU construction due to fewer matches to the SILVA database (Fig. 1A). As a result, it is likely that the magnitude of difference in diversity between disinfectant residual-free and disinfected systems is much larger than depicted in Fig. 3. These consistent differences between samples with and without a disinfectant residual could in large part be attributable to the selective pressures exerted by the process of disinfection on the DW microbial community.6,33,35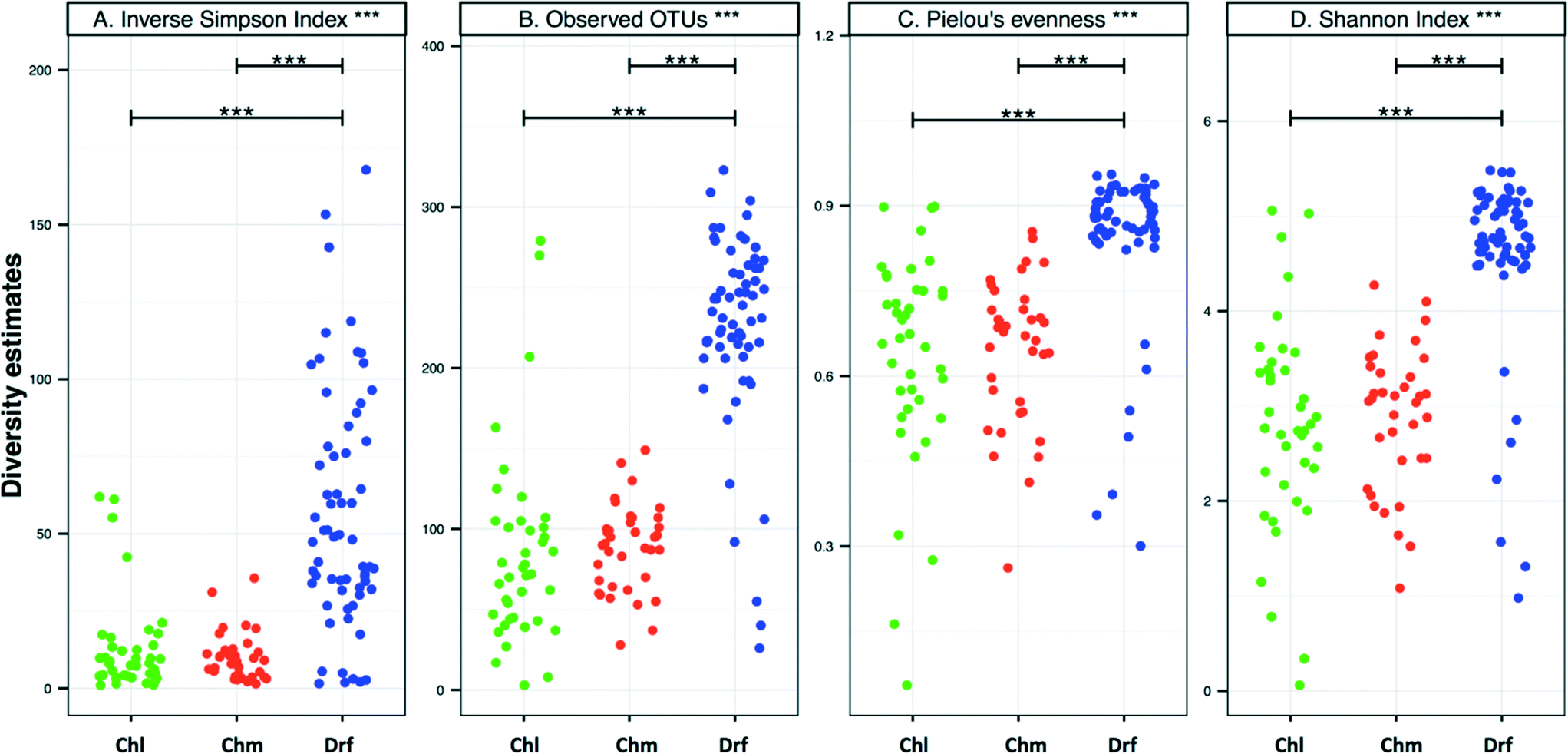 | ||
| Fig. 3 (A–D) Alpha-diversity per sample grouped by disinfection group (Chl: chlorinated, Chm: chloraminated, Drf: disinfectant residual-free). These analyses were done using the OTU table subsampled to 500 reads per sample. Significant differences between disinfection strategies were evaluated using ANOVA and are indicated by bars at the top of each figure panel (p values: * = <0.01, ** = <0.001, *** = <0.0001). | ||
3.4. Shared membership across disinfection strategies
The most commonly detected OTUs in chlorinated, chloraminated, and disinfectant residual-free systems were Porphyrobacter (class: Alphaproteobacteria) (MRA= 9.8 ± 22%, occurrence = 0.62), Bosea (class: Alphaproteobacteria) (MRA = 11.6 ± 45%, occurrence = 0.53), and Nitrospira (phylum: Nitrospirae) (MRA = 11 ± 14.1%, occurrence = 0.86), respectively. Table 1 provides an overview of the most commonly detected OTUs (occurrence >0.5) across the different disinfection strategies. Of the 7124 OTUs retained after subsampling, 6.6% (n = 470), 8.6% (n = 611), and 2.4% (n = 169) were shared (present in all samples under consideration) by: (i) chloraminated and chlorinated, (ii) chloraminated or chlorinated and disinfectant residual-free, and (iii) chlorinated, chloraminated, and disinfectant residual-free locations, respectively. Proteobacteria constituted a majority of the OTUs shared between samples emerging from all three disinfection strategies (n = 131) with 56, 41, and 22 OTUs classified as Alpha-, Beta, and Gammaproteobacteria, followed by OTUs within the phylum Bacteroidetes (n = 12) and Actinobacteria (n = 10) (Fig. 4A). Though there was no clear relationship between the abundance of an OTU at sampling locations with one disinfection strategy and its abundance or occurrence across the others, there was a clear and positive relationship between abundance and occurrence of an OTU within a disinfection strategy (Fig. 4B–D). This suggests that if an OTU is found to be abundant in a system within a microbial growth control strategy, it is likely to occur widely in similar systems. A similar relationship between relative abundance and occurrence of OTUs has also been reported recently,7,17 with proposals of the utility of occupancy-abundance based modeling approaches towards microbial management in DW systems.7| Chlorinated systems (Chl) | Chloraminated system (Chm) | Disinfectant residual free (Drf) | ||||||
|---|---|---|---|---|---|---|---|---|
| OTU | Classification genus level | Commonly detected in | MRA (stdev) | Occurrence | MRA (stdev) | Occurrence | MRA (stdev) | Occurrence |
| 4 | Porphyrobacter | Chl/Chm | 9.85(22.15) | 0.62 | 2.26(3.75) | 0.50 | 0.02(0.13) | 0.02 |
| 6 | Mycobacterium | Chl | 8.62(31.19) | 0.54 | 2.26(9.33) | 0.26 | 0.02(0.13) | 0.02 |
| 12 | Sphingomonas | Chl | 9.23(20.29) | 0.51 | 0.24(0.54) | 0.18 | 0.05(0.38) | 0.02 |
| 15 | Vampirovibrio | Chl | 15.54(29.82) | 0.51 | 1.47(2.6) | 0.45 | 0.14(0.4) | 0.13 |
| 30 | Bosea | Chm | 1.51(4.64) | 0.23 | 11.55(45.69) | 0.53 | 0.02(0.13) | 0.02 |
| 94 | Nitrospira | Drf | 0(0) | 0.00 | 0(0) | 0.00 | 11(14.14) | 0.86 |
| 162 | Parcubacteria | Drf | 0(0) | 0.00 | 0(0) | 0.00 | 6.25(10.85) | 0.71 |
| 189 | Bdellovibrio | Drf | 0.46(2.21) | 0.05 | 0.47(0.98) | 0.26 | 2.86(4.26) | 0.68 |
| 167 | Parcubacteria | Drf | 0(0) | 0.00 | 0.16(0.44) | 0.13 | 5.86(9.74) | 0.67 |
| 59 | Sideroxydans | Drf | 0(0) | 0.00 | 0(0) | 0.00 | 14.57(41.29) | 0.67 |
| 265 | Nitrospira | Drf | 0(0) | 0.00 | 0(0) | 0.00 | 2.21(2.82) | 0.67 |
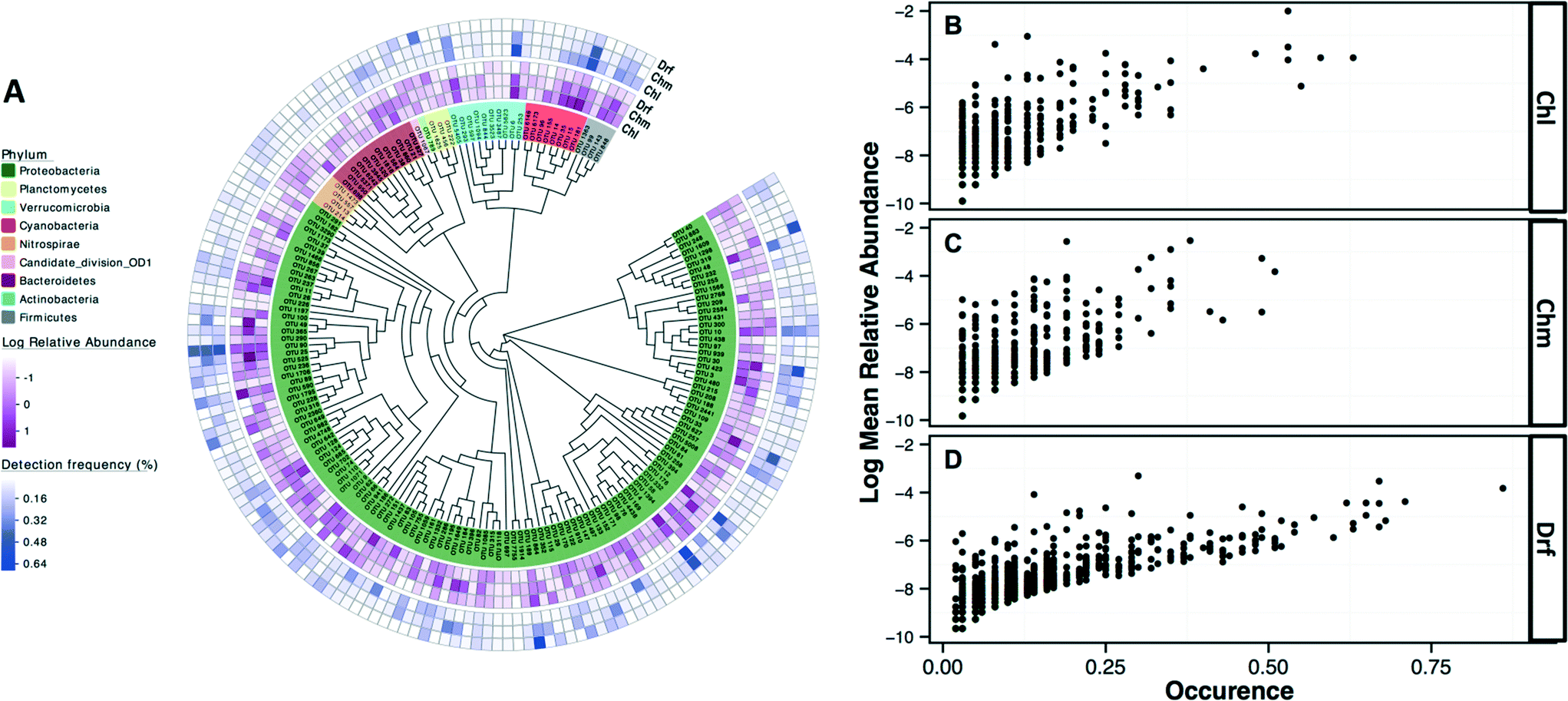 | ||
| Fig. 4 (A) A maximum likelihood phylogenetic tree of representative sequences from OTUs detected in samples across all three disinfection strategies. Color legends indicate the phylum of each OTU and the outer rings correspond to the log normalized relative abundance and occurrence within each disinfection strategy. Figure panels B–D highlight the positive relationship between the relative abundance and occurrence of all OTUs within a given disinfection strategy, irrespective of study origin. All data in these figures was constructed using the OTU table subsampled to 500 reads per sample. (Chl: chlorinated, Chm: chloraminated, Drf: disinfectant residual-free). | ||
3.5. Incidence of bacteria within Legionella, Mycobacterium, and Pseudomonas genera across disinfection strategies
Disinfectant residual-free systems showed significantly higher relative abundance and occurrence of OTUs classified as Legionella at the genus level as compared to chlorinated (p < 0.01) and chloraminated (p < 0.001) systems. The MRA of Legionella OTUs was 0.17 ± 0.68%, 0.18 ± 0.24%, and 0.58 ± 0.5%, while the occurrence of Legionella OTUs was 0.5, 0.59, and 0.97 in chlorinated, chloraminated, and disinfectant residual-free systems, respectively (Fig. 5). This higher MRA and occurrence of Legionella in disinfectant residual-free system was also accompanied by a greater diversity of OTUs. Specifically, chlorinated, chloraminated, and disinfectant residual-free systems harbored 2.2 ± 3.67, 7.21 ± 11.62, and 25.03 ± 13.55 OTUs that classified as Legionella, respectively. In contrast to Legionella, OTUs classified as Mycobacterium and Pseudomonas were more abundant and more frequently detected in disinfected systems as compared to disinfectant residual-free systems, with each of them exhibiting different trends when comparing chlorinated vs. chloraminated systems. For instance, mycobacterial OTUs were more abundant and frequent in chlorinated (MRA = 8.93 ± 15.37%, occurrence = 0.93) as compared to chloraminated systems (MRA = 2.84 ± 7.73%, occurrence = 0.79), though the difference between the two was not significantly different (Fig. 5). Similarly, OTUs classified as Pseudomonas were slightly more abundant in chloraminated systems (MRA = 3.17 ± 14.6%, occurrence = 0.87) as compared to chlorinated systems (MRA = 1.24 ± 3.28%, occurrence = 0.93) (Fig. 5), but this difference was also not significant. It is important to note that genus level classification though informative is not indicative of the presence of pathogens. For example, the genus Legionella contains in excess of 50 characterized species69 with less than half posing a health risk and even fewer species ever isolated from treated DW.70,71 The same is true for bacteria within the genera Mycobacterium and Pseudomonas. As a result, our findings should not be interpreted to suggest that one disinfection strategy is better than the other from the “pathogen” perspective. Rather, this finding should encourage rigorous follow-up studies that use standardized protocols with species-specific primers for quantitative assessment of the occurrence and absolute abundance of organisms of interest at DW systems that span the three disinfection strategies.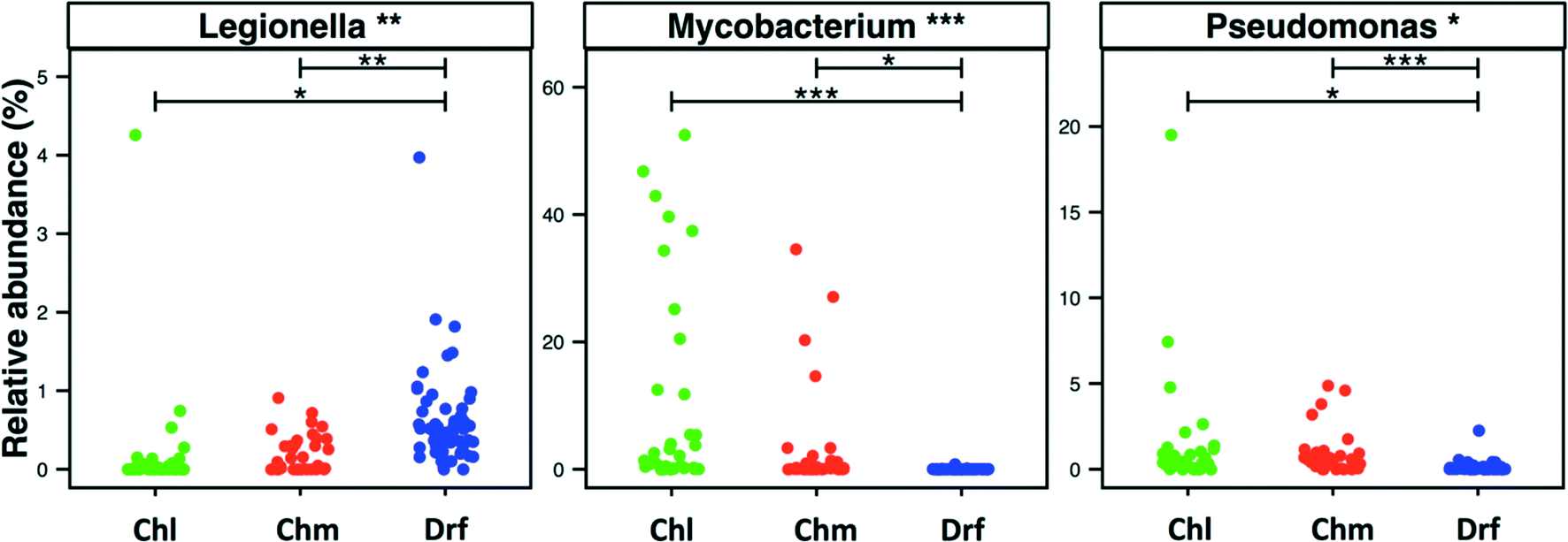 | ||
| Fig. 5 Relative abundance of OTUS classified as Legionella, Mycobacterium and Pseudomonas in each sample visualized by disinfection strategy type (Chl: chlorinated, Chm: chloraminated, Drf: disinfectant residual-free). One dataset from chloraminated samples from Shaw et al. 2015 though retained in the statistical analyses is removed from the figure due to high abundance of Pseudomonas (>80%) to allow for better visualization of the remaining data. Significant differences between groups, evaluated by ANOVA, are indicated by bars at the top of each figure panel (p-value legend: * = <0.01, ** = <0.001, *** = <0.0001). | ||
3.6. Detection of ecologically relevant OTU's across disinfection strategies
The broad detection of Nitrospira in disinfectant residual-free systems (Table 1) is particularly interesting given the (1) impact of nitrification in the DWDS on the stability of DW quality and its implications for infrastructure (e.g. corrosion)72 and (2) the recent discovery of complete ammonia oxidizing (comammox) Nitrospira bacteria,73–75 including in a DWTP.75 To this end, we evaluated the diversity and relative abundance of OTUs linked to nitrifying organisms. These nitrifying organisms were grouped as ammonia oxidizing archaea (AOA), ammonia oxidizing bacteria (AOB), nitrite oxidizing or comammox bacteria (NOB/CB), strict nitrite oxidizing bacteria (NOB), and anaerobic ammonia oxidizing bacteria (anammox) (Fig. 6). Disinfectant residual-free systems exhibited the greatest relative abundance of AOA (MRA = 0.48 ± 0.8%) and they were detected in 0.79 of the disinfectant residual-free locations. However, AOA were also consistently low abundance in disinfected systems with the maximum MRA being 3.3%, while being detected in only 0.50 of the chloraminated locations with no detection in chlorinated systems. Disinfectant residual-free samples also harbored higher abundance and greater diversity of AOB and NOB/CB (Fig. 6). For example, the MRA of AOB was 0.01 ± 0.02%, 0.19 ± 0.34% and 0.56 ± 1.62%, while the occurrence of AOB was 0.15, 0.36 and 0.9 in chlorinated, chloraminated and disinfectant residual-free systems, respectively. Strict NOB were extremely low in abundance and were detected in only 0.20 of the sampling locations across the three disinfection strategies with maximum MRA of 0.12%. Interestingly, OTUs classified as Nitrospira, a genus that includes both strict NOB and the newly discovered comammox73–75 bacteria were detected at a higher relative abundance and frequency than either AOB or NOB in disinfectant residual-free systems. For instance, while the NOB and AOB were detected in 0.20 and 0.54 of all sampling locations, NOB/CB were detected in 0.68 of sampling locations across all disinfection strategies, with their MRA nearly 4 fold higher than AOB and AOA combined. Given this finding, it is likely that comammox bacteria may play a significantly more important role in nitrification in DW systems (either DWTP or DWDS), as compared to strict AOB and NOB.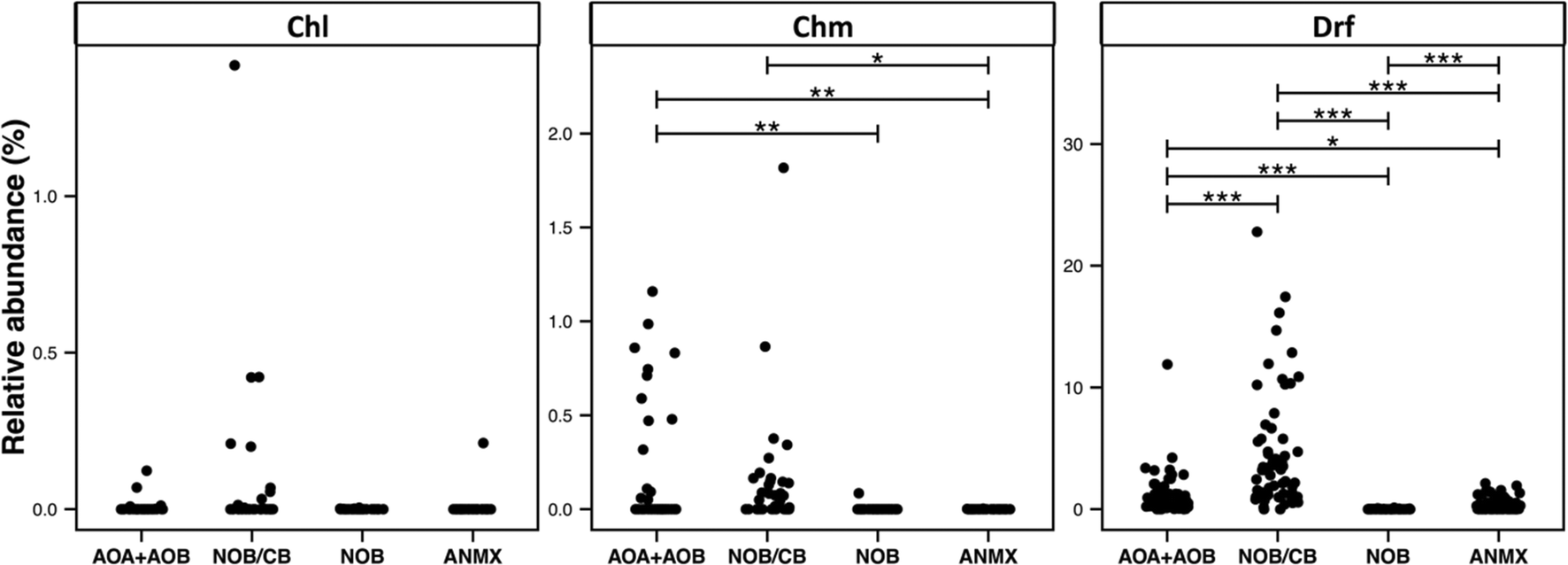 | ||
| Fig. 6 Relative abundance of nitrifier OTUs in each sample visualized by disinfection strategy type (Chl: chlorinated, Chm: chloraminated, Drf: disinfectant residual-free). X-axis labels correspond to: ammonia oxidizing archaea + bacteria (AOA + AOB), Nitrospira based nitrite oxidizing or comammox bacteria (NOB/CB), strict nitrite oxidizing bacteria (NOB), and anammox bacteria (ANMX). Significant differences between groups, evaluated by ANOVA, are indicated by bars at the top of each figure panel (p-value legend: * = <0.01, ** = <0.001, *** = <0.0001). | ||
Another broadly distributed class of OTUs that has thus far received little attention within DW studies involves predatory bacteria. Specifically, OTUs classified as Bdellovibiro (class: Deltaproteobacteria) and Vampirovibrio (phylum: Melainabacteria) were among the top 10 frequently detected OTUs across all three disinfection strategies (Table 1). This wide-scale detection of bacteria with a predatory lifestyle is particularly interesting as it highlights a poorly explored ecological dynamic within DW systems and may even provide an avenue for microbial growth control76 in the DWTP/DWDS. Predatory bacteria are phylogenetically diverse and genus level identification is not sufficient to ascertain the presence of bacteria with obligate or facultative predatory lifestyle. Nonetheless, OTUs classified to some genera can be categorized as emerging from predatory bacteria (e.g. Bdellovibrio). Specifically, we found several OTUs classified as Bdellovibrio (n = 114), Cystobacter (n = 10), Lysobacter (n = 46), Peredibacter (n = 13), and Vampirovibrio (n = 92), all of which can be functionally classified as obligate or non-obligate predatory bacteria. The three most frequently detected predatory OTUs (i.e. Bdellovibrio, Lysobacter, and Vampirovibrio), showed a significantly higher occurrence in disinfectant residual-free systems as compared to disinfected systems. For example, Bdellovibrio, Lysobacter, and Vampirovibrio were detected in 0.95, 0.52 and 0.98 of the locations from the disinfectant residual-free systems, respectively while the detection of the same predatory OTUs in chlorinated and chloraminated samples ranged from 0.25–0.38, 0.38 and 0.64–0.88, respectively. Further, though Bdellovibrio was significantly more abundant in disinfectant residual-free systems, both Lysobacter and Vampirovibrio exhibited a greater relative abundance in chlorinated systems. Specifically, Lysobacter and Vampirovibrio exhibited a relative abundance of 4.87 ± 13.69% and 5.21 ± 7.2% in chlorinated samples, respectively, while constituting less than 1% of the overall community for chloraminated and disinfectant residual-free samples. A possible explanation for the higher abundance and detection frequency of predatory bacteria in disinfectant residual-free systems could be the higher biomass present in these systems, as this provides a rich source of nutrients for predatory bacteria.
3.7. Potential for contamination affecting DW microbial studies
Studies involving low-biomass samples are particularly susceptible to contamination emerging from a range of potential sources – from sample handling to PCR/DNA extraction reagents to contaminants from the sequencing process itself (e.g. sequences from one sample being attributed to another). Recent studies have demonstrated that kit/reagent contamination can critically impair studies that rely on sequencing datasets59,77 with one study proposing an extended list of common kit-contamination genera.59 Though majority of studies include negative controls during the sample processing, DNA extraction, and PCR amplification step, these negative controls are rarely included during the sequencing process itself. To our knowledge, only one DW study has explicitly stated the inclusion of a negative control during the sequencing process.34 In this study though the number of sequences in the negative controls were significantly lower than the samples of interest, the classification of OTUs detected in negative controls was highly similar to those commonly detected in DW samples.Overall, 18.5 ± 23% of the sequencing data across all studies was associated with a list of potentially contaminating genera provided by Salter et al.59 Approximately 23.5 ± 19.8%, 29.6 ± 25.5%, and 8.5 ± 18.3% of data was associated with these genera for chlorinated, chloraminated, and disinfectant residual-free systems, with the proportions being significantly higher in disinfected as compared to disinfectant residual-free samples (Fig. 7), which typically have a significantly higher cell count.41,78 The lower proportion of potentially contaminating data in disinfectant residual-free datasets could be related to higher biomass concentration in these samples. It is important to note that we do not suggest that these numbers accurately reflect levels of contamination in published DW datasets. What this exercise emphasizes is the need to routinely sequence negative controls is particularly critical for DW studies, not only because of the low-biomass nature of these samples but also because bacteria associated with kit/reagent contamination genera are also commonly found in DW samples. As a result, a genuine contaminant might be passed of as belonging to the DW sample under consideration.
 | ||
| Fig. 7 Proportion of potential contaminating sequences in each dataset per disinfection group (Chl: chlorinated, Chm: chloraminated, Drf: disinfectant residual-free). Significant differences between groups, evaluated by ANOVA, are indicated by bars at the top of each figure panel (p-value legend: * = <0.01, ** = <0.001, *** = <0.0001). List of potentially contaminant genera obtained from Table 1 in Salter et al. (2014). | ||
3.8. Differences in community structure and composition across disinfection strategies
Clustering of samples showed a clear distinction between disinfected and disinfectant residual-free samples (Fig. S3†), but there was no clear clustering by the type of disinfectant residual (i.e. chloramine vs. chlorine). Nonetheless, multiple factors can confound such broad level clustering (Fig. S4†). As discussed above, the available DW sequencing data is highly heterogeneous. A majority of the factors that contribute to data heterogeneity (e.g. DNA extraction protocol, PCR primer choice, sequencing platform, etc.) can largely be collapsed into one major variable – origin of study. PERMANOVA tests conducted using distance matrices constructed (after subsampling) using Bray Curtis/Jaccard metrics indicated that origin of study had a strong impact on differences between samples (R2 = 0.34/0.24, p = 0.001) followed by type of source water (surface water, groundwater or mixed) (R2 = 0.02/0.02, p = 0.001) and disinfection type (R2 = 0.014/0.01, p = 0.01). Another variable that could affect the similarity between samples is the proportion of data used following the SILVA matching exercise (Fig. 1). However, this had a minor effect on the community membership and structure based clustering using Jaccard (R2 = 0.007, p = 0.049) and Bray Curtis distance metrics (R2 = 0.007, p = 0.04), respectively. This confounding aspect of variation between studies is a common theme across meta-analysis efforts.63,64,793.9. Predicting microbial community functional profiles
Increasingly 16S rRNA gene data is being utilized to leverage functional datasets to predict the metabolic characteristics of whole microbial communities using tools such as PiCrust,80 Tax4Fun,60etc. Such approaches rely on matching 16S rRNA gene sequences to organisms represented in functional databases and using the abundance of associated OTUs to predict the metabolic potential of a given microbial community. Though this is a rather cost-effective and hence, an attractive way to get more information for less resource (16S rRNA gene studies are significantly inexpensive as compared to metagenomic studies on a per sample basis) there is also potential for over or under-predicting the metabolic potential of the microbial community depending on the composition of these functional databases and the sample under consideration.To this end, we wanted to test the utility of Tax4Fun,60 which leverages the KEGG database,61 to capture differences in metabolic potential of microbial communities in disinfected and disinfectant residual-free systems. The OTU sequences from disinfectant residual-free samples exhibited significantly lower similarity to organisms in the KEGG database; this was despite the fact that only sequences matching the SILVA database were used for this exercise. Specifically, greater than 80% of the disinfectant residual-free sampling locations had less than 50% of sequences matching organisms in the KEGG database (Fig. 8), while for the disinfected group 35.3 ± 24% of the sequences per sample had no match. This clearly indicates that the metabolic potential of DW microbial communities will be vastly under-represented by function predictions tools that leverage 16S rRNA gene data, particularly for disinfectant residual-free systems. Despite this under-representation, we wanted to test the utility of this approach to detect relevant differences between samples that may be related to the presence and absence of a disinfectant residual. To adjust for this range of sample FTUs, we established a FTU threshold of 0.5, with 10 disinfectant residual-free sampling locations meeting this threshold. We then picked 5 chlorinated and 5 chloraminated sampling locations such that there was no significant difference in the FTUs between disinfected and disinfectant residual-free locations used for this exercise (p = 0.83). Using this subset of samples (n = 20), we tested for differences in relative abundance of KO's (i.e. gene level) between disinfected and disinfectant residual-free sampling locations. Of the 100 most abundant KO's returned by Tax4Fun, only 17 showed significant difference in relative abundance between disinfected and disinfectant residual-free locations (corrected p-value < 0.01) (data not shown). Surprisingly no genes involved in oxidative stress or detoxification5 were significantly different between disinfected and disinfectant residual-free locations. The majority of these significantly different KO's were associated with functions that are widely distributed across bacterial populations (e.g. carbohydrate metabolism, DNA repair, etc.). Further, though the difference in relative abundance of these KO's was significant, the magnitude of difference between disinfected and disinfectant residual-free samples was less than 2 fold for a majority and hence, may not necessarily provide informative insights about the selection pressure exerted by a disinfectant residual. Only one KO showed a significant difference (p = 0.0073) with a large effect size in terms of relative abundance to merit follow-up investigations. Specifically, K06994, a putative drug exporter gene within the resistance-nodulation-cell division (RND) superfamily was >30 times more abundant in disinfected locations as compared to disinfectant residual-free locations.
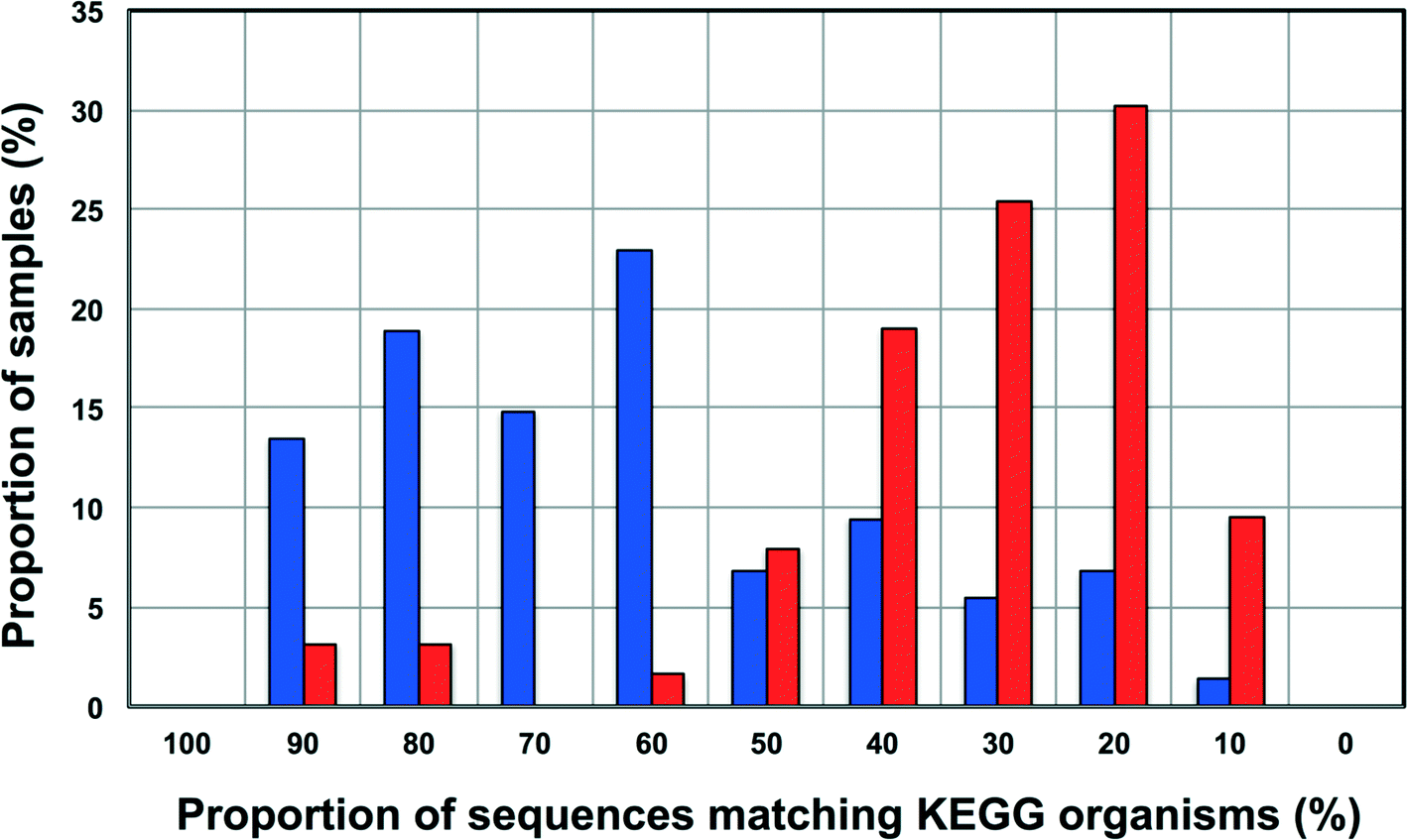 | ||
| Fig. 8 Proportion of sequences matching organisms in the KEGG database (%) versus proportion of samples (%) for disinfected (in blue) and disinfectant residual-free (in red) datasets in each category. The proportion of sequences matching KEGG organisms was estimated as (1-FTU)*100, where FTU= fraction of OTUs that could not be mapped to KEGG organisms as estimated by Tax4Fun. | ||
4. Conclusions and future directions
We provide a number of interesting insights into differences between disinfected and disinfectant residual-free systems by co-analyzing available 16S rRNA gene datasets from bulk DW samples. For example, the higher occurrence of Legionella OTUs in disinfectant residual-free systems and of Mycobacterium and Pseudomonas OTUs in disinfected systems is a prime candidate for follow-up investigations. Further, the broad detection of Nitrospira OTUs and OTUs linked to predatory bacteria may provide for exciting avenues for future research involving fundamental ecological questions with a significant practical impact (e.g. revisiting nitrification in drinking water systems in light of new findings regarding comammox Nitrospira, exploring the potential of predatory bacteria for biocontrol). Similarly, we clearly highlight the critical aspect of including negative controls in sequencing efforts for DW studies. However, as discussed above our meta-analysis effort is significantly confounded by data heterogeneity, particularly with respect to the ones we can identify based on the data (Fig. 1B and C). If all data included in this study was obtained from standardized protocols spanning sample collection, DNA extraction, PCR amplification, target hypervariable region of the 16S rRNA gene, and sequencing platform – undoubtedly the insights generated using a meta-analysis effort would not only be much more robust but the data would also lend itself to asking targeted and quantitative questions which is currently not possible. Thus making a case for standardized protocols across all DW studies as an attractive prospect. However, efforts to standardize protocols without appropriate resources to sustain and support them are likely to be more disruptive than beneficial. For example, it may “price-out” some researchers from collecting data that meets field-approved standards. Standardizing protocols in a rapidly changing methodological landscape presents the pitfalls of generating “kit monopolies” (i.e. one reagent or sample processing kit becomes the default), while also risking the creation of methodological inertia in a field that has only recently begun to exploit the power of high-throughput DNA sequencing. For example, consider the rate at which DNA sequencing approaches81–84 have changed over the last few years. Despite the fact that Sanger sequencing was widely used for DW microbial studies until 2010,85 we have not included that data in this study because of its low-throughput nature (low sequencing depth and sample diversity). Similarly, it is likely that with the advent of long-read sequencing technologies,86,87 a meta-analysis effort five years from now might choose to exclude data generated from currently popular sequencing platforms due to their short-read nature and hence, lower phylogenetic resolution of the data.Rather than devoting resources towards standardizing protocols across DW studies, we would suggest researchers choose sample/data collection and processing approaches that are (1) methodologically robust based on best-available information and (2) achievable given resource availability. Rather, efforts should be made to: (i) standardize data reporting approaches by depositing raw data in publicly available databases; and (ii) measure and provide supporting parameters as possible (temperature, water chemistry parameters, ATP, cell counts, TOC, AOC, etc.) along with sample metadata,88 in a format that can be easily integrated into sequence data processing approaches and diversity analyses. This would be a particularly good place to start, since our experience conducting this meta-analysis has shown that these standard practices are not yet commonplace within the DW community. And finally, another possible option to support comparative analyses across systems would be to make provisions for sample sharing, either DNA extract or filtered sample itself. Though, this still retains DNA extraction or sample collection variabilities, it will eliminate primer and sequencing platform biases and allow for robust de-novo clustering89 for microbial community analyses, with the ability to assess the aforementioned biases using statistical approaches.
Acknowledgements
This study was supported by the Engineering and Physical Sciences Research Council (Grant: EP/K035886/1 and EP/M016811/1) and the University of Glasgow. QM Baustista-de los Santos was supported by the University of Glasgow – James Watt Scholarship and by Scottish Water. Maria Sevillano-Rivera is supported by the Department of Civil and Environmental Engineering, Northeastern University. Rungroch Sungthong is supported by the Engineering and Physical Sciences Research Council (Grant: EP/M016811/1). Umer Zeeshan Ijaz is supported by a NERC Fellowship (NE/L011956/1).References
- D. v. d. Kooij, J. - Am. Water Works Assoc., 1992, 84, 57–65 Search PubMed.
- M. W. LeChevallier, W. Schulz and R. G. Lee, Appl. Environ. Microbiol., 1991, 57, 857–862 CAS.
- W. H. O., WHO, 2011, p. 541 Search PubMed.
- V. Gomez-Alvarez, R. P. Revetta and J. W. Santo Domingo, Appl. Environ. Microbiol., 2012, 78, 6096–6102 CrossRef PubMed.
- Y. Chao, L. Ma, Y. Yang, F. Ju, X. X. Zhang, W. M. Wu and T. Zhang, Sci. Rep., 2013, 3, 3550 Search PubMed.
- G. Roeselers, J. Coolen, P. W. J. J. van der Wielen, M. C. Jaspers, A. Atsma, B. de Graaf and F. Schuren, Environ. Microbiol., 2015, 17, 2405–2514 CrossRef PubMed.
- A. J. Pinto, J. Schroeder, M. Lunn, W. Sloan and L. Raskin, mBio, 2014, 5 CrossRef CAS PubMed.
- M. L. Sogin, H. G. Morrison, J. A. Huber, D. M. Welch, S. M. Huse, P. R. Neal, J. M. Arrieta and G. J. Herndl, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12115–12120 CrossRef CAS PubMed.
- J. G. Caporaso, C. L. Lauber, W. A. Walters, D. Berg-Lyons, C. A. Lozupone, P. J. Turnbaugh, N. Fierer and R. Knight, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4516–4522 CrossRef CAS PubMed.
- W. Lin, Z. Yu, H. Zhang and I. P. Thompson, Water Res., 2014, 52, 218–230 CrossRef CAS PubMed.
- K. Lautenschlager, C. Hwang, F. Ling, W.-T. Liu, N. Boon, O. Köster, T. Egli and F. Hammes, Water Res., 2014, 62, 40–52 CrossRef CAS PubMed.
- V. Gomez-Alvarez, R. P. Revetta and J. W. Santo Domingo, Appl. Environ. Microbiol., 2012, 78, 6095–6102 CrossRef CAS PubMed.
- H. Wang, S. Masters, M. A. Edwards, J. O. Falkinham and A. Pruden, Environ. Sci. Technol., 2014, 48, 1426–1435 CrossRef CAS PubMed.
- C. Hwang, F. Ling, G. L. Andersen, M. W. LeChevallier and W. T. Liu, Appl. Environ. Microbiol., 2012, 78, 7856–7865 CrossRef CAS PubMed.
- Q. M. Bautista-de los Santos, J. L. Schroeder, O. Blakemore, J. Moses, M. Haffey, W. Sloan and A. J. Pinto, Water Res., 2016, 90, 216–224 CrossRef CAS PubMed.
- I. Douterelo, R. L. Sharpe and J. B. Boxall, Water Res., 2013, 47, 503–516 CrossRef CAS PubMed.
- F. Ling, C. Hwang, M. W. LeChevallier, G. L. Andersen and W.-T. Liu, ISME J., 2015, 10, 582–595 CrossRef PubMed.
- J. Yu, D. Kim and T. Lee, Water Sci. Technol., 2010, 61, 163–171 CrossRef CAS PubMed.
- C. R. Proctor and F. Hammes, Curr. Opin. Biotechnol., 2015, 33, 87–94 CrossRef CAS PubMed.
- J. El-Chakhtoura, E. Prest, P. Saikaly, M. van Loosdrecht, F. Hammes and H. Vrouwenvelder, Water Res., 2015, 74, 180–190 CrossRef CAS PubMed.
- T.-H. Chiao, T. M. Clancy, A. Pinto, C. Xi and L. Raskin, Environ. Sci. Technol., 2014, 48, 4038–4047 CrossRef CAS PubMed.
- C. A. Lozupone, J. Stombaugh, A. Gonzalez, G. Ackermann, D. Wendel, Y. Vázquez-Baeza, J. K. Jansson, J. I. Gordon and R. Knight, Genome Res., 2013, 23, 1704–1714 CrossRef CAS PubMed.
- S. J. McIlroy, A. M. Saunders, M. Albertsen, M. Nierychlo, B. McIlroy, A. A. Hansen, S. M. Karst, J. L. Nielsen and P. H. Nielsen, Database, 2015 DOI:10.1093/database/bav062.
- A. M. Saunders, M. Albertsen, J. Vollertsen and P. H. Nielsen, ISME J., 2016, 10, 11–20 CrossRef CAS PubMed.
- K. Henne, L. Kahlisch, I. Brettar and M. G. Höfle, Appl. Environ. Microbiol., 2012, 78, 3530–3538 CrossRef CAS PubMed.
- G. Liu, G. L. Bakker, S. Li, J. H. G. Vreeburg, J. Q. J. C. Verberk, G. J. Medema, W. T. Liu and J. C. Van Dijk, Environ. Sci. Technol., 2014, 48, 5467–5476 CrossRef CAS PubMed.
- J. L. Schroeder, M. Lunn, A. J. Pinto, L. Raskin and W. T. Sloan, PLoS One, 2015, 10, e0117221 Search PubMed.
- I. Douterelo, S. Husband and J. B. Boxall, Water Res., 2014, 54, 100–114 CrossRef CAS PubMed.
- M. E. Schoen and N. J. Ashbolt, Water Res., 2011, 45, 5826–5836 CrossRef CAS PubMed.
- K. Lautenschlager, C. Hwang, W. T. Liu, N. Boon, O. Köster, H. Vrouwenvelder, T. Egli and F. Hammes, Water Res., 2013, 47, 3015–3025 CrossRef CAS PubMed.
- J. Wimpenny, W. Manz and U. Szewzyk, FEMS Microbiol. Rev., 2000, 24, 661–671 CrossRef CAS PubMed.
- E. P. Holinger, K. A. Ross, C. E. Robertson, M. J. Stevens, J. K. Harris and N. R. Pace, Water Res., 2014, 49, 225–235 CrossRef CAS PubMed.
- C. Hwang, F. Ling, G. L. Andersen, M. W. LeChevallier and W.-T. Liu, Appl. Environ. Microbiol., 2012, 78, 7856–7865 CrossRef CAS PubMed.
- P. Ji, J. Parks, M. A. Edwards and A. Pruden, PLoS One, 2015, 10, e0141087 Search PubMed.
- H. Wang, C. R. Proctor, M. A. Edwards, M. Pryor, J. W. Santo Domingo, H. Ryu, A. K. Camper, A. Olson and A. Pruden, Environ. Sci. Technol., 2014, 48, 10624–10633 CrossRef CAS PubMed.
- Y. Zhang and Q. A. He, Water Sci. Technol.: Water Supply, 2013, 13, 358–367 CrossRef CAS.
- K. Huang, X.-X. Zhang, P. Shi, B. Wu and H. Ren, Ecotoxicol. Environ. Saf., 2014, 109, 15–21 CrossRef CAS PubMed.
- S. Jia, P. Shi, Q. Hu, B. Li, T. Zhang and X.-X. Zhang, Environ. Sci. Technol., 2015, 49, 12271–12279 CrossRef CAS PubMed.
- D. N. Zeng, Z. Y. Fan, L. Chi, X. Wang, W. D. Qu and Z. X. Quan, World J. Microbiol. Biotechnol., 2013, 29, 1573–1584 CrossRef PubMed.
- X. Bai, X. Ma, F. Xu, J. Li, H. Zhang and X. Xiao, Sci. Total Environ., 2015, 533, 24–31 CrossRef CAS PubMed.
- E. I. Prest, J. El-Chakhtoura, F. Hammes, P. E. Saikaly, M. C. M. van Loosdrecht and J. S. Vrouwenvelder, Water Res., 2014, 63, 179–189 CrossRef CAS PubMed.
- K. Lautenschlager, C. Hwang, W. T. Liu, N. Boon, O. Koster, H. Vrouwenvelder, T. Egli and F. Hammes, Water Res., 2013, 47, 3015–3025 CrossRef CAS PubMed.
- J. L. A. Shaw, P. Monis, L. S. Weyrich, E. Sawade, M. Drikas and A. J. Cooper, Appl. Environ. Microbiol., 2015, 81, 6463–6473 CrossRef CAS PubMed.
- D. Costa, A. Mercier, K. Gravouil, J. Lesobre, V. Delafont, A. Bousseau, J. Verdon and C. Imbert, Water Res., 2015, 81, 223–231 CrossRef CAS PubMed.
- N. A. Joshi and J. N. Fass, 2011, available at https://github.com/najoshi/sickle.
- FASTX-Toolkit. http://hannonlab.cshl.edu/fastx_toolkit/.
- J. Zhang, K. Kobert, T. Flouri and A. Stamatakis, Bioinformatics, 2014, 30, 614–620 CrossRef CAS PubMed.
- P. Schloss, S. Westcott, T. Ryabin, J. Hall, M. Hartmann, E. Hollister, R. Lesniewski, B. Oakley, D. Parks, C. Robinson, J. Sahl, B. Stres, G. Thallinger, D. Van Horn and C. Weber, Appl. Environ. Microbiol., 2009, 75, 7537–7541 CrossRef CAS PubMed.
- E. Pruesse, C. Quast, K. Knittel, B. M. Fuchs, W. Ludwig, J. Peplies and F. O. Glöckner, Nucleic Acids Res., 2007, 35, 7188–7196 CrossRef CAS PubMed.
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, J. Mol. Biol., 1990, 215, 403–410 CrossRef CAS PubMed.
- Q. Wang, G. Garrity, J. Tiedje and J. Cole, Appl. Environ. Microbiol., 2007, 73, 5261–5267 CrossRef CAS PubMed.
- S. J. Weiss, Z. Xu, A. Amir, S. Peddada, K. Bittinger, A. Gonzalez, C. Lozupone, J. R. Zaneveld, Y. Vazquez-Baeza and A. Birmingham, Report 2167-9843, PeerJ PrePrints, 2015.
- K. Lautenschlager, C. Hwang, W. T. Liu, N. Boon, O. Köster, H. Vrouwenvelder, T. Egli and F. Hammes, Water Res., 2013, 47, 3015–3025 CrossRef CAS PubMed.
- H. Wickham, Ggplot2: Elegant graphics for data analysis, 2009, http://ggplot2.org Search PubMed.
- R Core Team, 2016, https://www.R-project.org.
- G. Warnes, B. Bolker and T. Lumley, gplots: Various R programming tools for plotting data. R package version 2.6.0, https://CRAN.R-project.org/package=gplots Search PubMed.
- A. Stamatakis, Bioinformatics, 2014 DOI:10.1093/bioinformatics/btu033.
- H. Zhang, S. Gao, M. J. Lercher, S. Hu and W. H. Chen, Nucleic Acids Res., 2012, 40, 569–572 CrossRef PubMed.
- S. J. Salter, M. J. Cox, E. M. Turek, S. T. Calus, W. O. Cookson, M. F. Moffatt, P. Turner, J. Parkhill, N. J. Loman and A. W. Walker, BMC Biol., 2014, 12, 87 CrossRef PubMed.
- K. P. Aßhauer, B. Wemheuer, R. Daniel and P. Meinicke, Bioinformatics, 2015, 31, 2882–2884 CrossRef PubMed.
- H. Ogata, S. Goto, K. Sato, W. Fujibuchi, H. Bono and M. Kanehisa, Nucleic Acids Res., 1999, 27, 29–34 CrossRef CAS PubMed.
- Y. Benjamini and Y. Hochberg, J. R. Stat. Soc. Series B. Stat. Methodol., 1995, 57, 289–300 Search PubMed.
- R. I. Adams, A. C. Bateman, H. M. Bik and J. F. Meadow, Microbiome, 2015, 3, 1–18 CrossRef PubMed.
- O. Koren, D. Knights, A. Gonzalez, L. Waldron, N. Segata, R. Knight, C. Huttenhower and R. E. Ley, PLoS Comput. Biol., 2013, 9, e1002863 CAS.
- C. L. Lauber, N. Zhou, J. I. Gordon, R. Knight and N. Fierer, FEMS Microbiol. Lett., 2010, 307, 80–86 CrossRef CAS PubMed.
- L. Cuthbertson, G. B. Rogers, A. W. Walker, A. Oliver, T. Hafiz, L. R. Hoffman, M. P. Carroll, J. Parkhill, K. D. Bruce and C. J. van der Gast, J. Clin. Microbiol., 2014, 52, 3011–3016 CrossRef CAS PubMed.
- L. M. Feinstein, W. J. Sul and C. B. Blackwood, Appl. Environ. Microbiol., 2009, 75, 5428–5433 CrossRef CAS PubMed.
- A. J. Pinto and L. Raskin, PLoS One, 2012, 7, e43093 CAS.
- D. Burstein, F. Amaro, T. Zusman, Z. Lifshitz, O. Cohen, J. A. Gilbert, T. Pupko, H. A. Shuman and G. Segal, Nat. Genet., 2016, 48, 167–175 CrossRef CAS PubMed.
- P. W. van der Wielen and D. van der Kooij, Appl. Environ. Microbiol., 2013, 79, 825–834 CrossRef CAS PubMed.
- B. A. Wullings, G. Bakker and D. van der Kooij, Appl. Environ. Microbiol., 2011, 77, 634–641 CrossRef CAS PubMed.
- Y. Zhang, N. Love and M. Edwards, Crit. Rev. Environ. Sci. Technol., 2009, 39, 153–208 CrossRef CAS.
- H. Daims, E. V. Lebedeva, P. Pjevac, P. Han, C. Herbold, M. Albertsen, N. Jehmlich, M. Palatinszky, J. Vierheilig, A. Bulaev, R. H. Kirkegaard, M. v. Bergen, T. Rattei, B. Bendinger, P. H. Nielsen and M. Wagner, Nature, 2015, 528, 504–509 CrossRef CAS PubMed.
- M. A. H. J. van Kessel, D. R. Speth, M. Albertsen, P. H. Nielsen, H. J. M. Op den Camp, B. Kartal, M. S. M. Jetten and S. Lücker, Nature, 2015, 528, 555–559 CrossRef CAS PubMed.
- A. J. Pinto, D. N. Marcus, U. Z. Ijaz, Q. M. Bautista-de lose Santos, G. J. Dick and L. Raskin, mSphere, 2016, 1 DOI:10.1128/mSphere.00054-15.
- R. E. Sockett and C. Lambert, Nat. Rev. Microbiol., 2004, 2, 669–675 CrossRef CAS PubMed.
- M. J. Cox, S. J. Salter, E. M. Turek, S. T. Calus, W. O. Cookson, M. F. Moffatt, P. Turner, J. Parkhill, N. J. Loman and A. W. Walker, in C21. Conflict or peaceful co-existence? The bacterial lung microbiome and host immunity, American Thoracic Society, 2015, p. A3955 Search PubMed.
- S. Gillespie, P. Lipphaus, J. Green, S. Parsons, P. Weir, K. Juskowiak, B. Jefferson, P. Jarvis and A. Nocker, Water Res., 2014, 65, 224–234 CrossRef CAS PubMed.
- A. Shade, J. Gregory Caporaso, J. Handelsman, R. Knight and N. Fierer, ISME J., 2013, 7, 1493–1506 CrossRef PubMed.
- M. G. I. Langille, J. Zaneveld, J. G. Caporaso, D. McDonald, D. Knights, J. A. Reyes, J. C. Clemente, D. E. Burkepile, R. L. Vega Thurber, R. Knight, R. G. Beiko and C. Huttenhower, Nat. Biotechnol., 2013, 31, 814–821 CrossRef CAS PubMed.
- T. C. Glenn, Mol. Ecol. Resour., 2011, 11, 759–769 CrossRef CAS PubMed.
- B. P. Hodkinson and E. A. Grice, Adv. Wound Care, 2015, 4, 50–58 CrossRef PubMed.
- E. Mardis, Annu. Rev. Genomics Hum. Genet., 2008, 9, 387–402 CrossRef CAS PubMed.
- M. L. Metzker, Nat. Rev. Genet., 2010, 11, 31–46 CrossRef CAS PubMed.
- P. Y. Hong, C. Hwang, F. Ling, G. L. Andersen, M. W. LeChevallier and W. T. Liu, Appl. Environ. Microbiol., 2010, 76, 5631–5635 CrossRef CAS PubMed.
- N. J. Loman and M. Watson, Nat. Methods, 2015, 12, 303–304 CrossRef CAS PubMed.
- R. Roberts, M. Carneiro and M. Schatz, Genome Biol., 2013, 14, 405 CrossRef PubMed.
- P. Yilmaz, R. Kottmann, D. Field, R. Knight, J. R. Cole, L. Amaral-Zettler, J. A. Gilbert, I. Karsch-Mizrachi, A. Johnston, G. Cochrane, R. Vaughan, C. Hunter, J. Park, N. Morrison, P. Rocca-Serra, P. Sterk, M. Arumugam, M. Bailey, L. Baumgartner, B. W. Birren, M. J. Blaser, V. Bonazzi, T. Booth, P. Bork, F. D. Bushman, P. L. Buttigieg, P. S. G. Chain, E. Charlson, E. K. Costello, H. Huot-Creasy, P. Dawyndt, T. DeSantis, N. Fierer, J. A. Fuhrman, R. E. Gallery, D. Gevers, R. A. Gibbs, I. S. Gil, A. Gonzalez, J. I. Gordon, R. Guralnick, W. Hankeln, S. Highlander, P. Hugenholtz, J. Jansson, A. L. Kau, S. T. Kelley, J. Kennedy, D. Knights, O. Koren, J. Kuczynski, N. Kyrpides, R. Larsen, C. L. Lauber, T. Legg, R. E. Ley, C. A. Lozupone, W. Ludwig, D. Lyons, E. Maguire, B. A. Methe, F. Meyer, B. Muegge, S. Nakielny, K. E. Nelson, D. Nemergut, J. D. Neufeld, L. K. Newbold, A. E. Oliver, N. R. Pace, G. Palanisamy, J. Peplies, J. Petrosino, L. Proctor, E. Pruesse, C. Quast, J. Raes, S. Ratnasingham, J. Ravel, D. A. Relman, S. Assunta-Sansone, P. D. Schloss, L. Schriml, R. Sinha, M. I. Smith, E. Sodergren, A. Spor, J. Stombaugh, J. M. Tiedje, D. V. Ward, G. M. Weinstock, D. Wendel, O. White, A. Whiteley, A. Wilke, J. R. Wortman, T. Yatsunenko and F. O. Glockner, Nat. Biotechnol., 2011, 29, 415–420 CrossRef CAS PubMed.
- S. L. Westcott and P. D. Schloss, PeerJ, 2015, 3, e1487 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ew00030d |
| This journal is © The Royal Society of Chemistry 2016 |