
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simplifying microfluidic separation devices towards field-detection of blood parasites†
S. H.
Holm
a,
J. P.
Beech
a,
M. P.
Barrett
b and
J. O.
Tegenfeldt
*a
aDivision of Solid State Physics, NanoLund, Lund University, PO Box 118, S-221 00 Lund, Sweden. E-mail: jonas.tegenfeldt@ftf.lth.se; Fax: +46 46 222 3637; Tel: +46 46 222 8063
bWelcome Trust Centre for Medical Parasitology, Institute of Infection, Immunity and Inflammation, College of Medical, Veterinary and Life Sciences, University of Glasgow, UK
First published on 8th March 2016
Abstract
With our sights set on a simple and inexpensive diagnostics device based on extraction and enrichment of parasites from human blood, we present a device design that relies on a combination of multiple different deterministic lateral displacement arrays. Our end goal is a microfluidic device that will be easy to use in the rural, resource-deprived areas where simple-to-use medical tools are crucially needed for rapid and accurate diagnosis. Here, we exemplify this in the application of blood parasite enrichment from a sample of blood. With trypanosomes as a model system we show a combination of functionalities designed into a single device based on several deterministic lateral displacement arrays of different depths arranged in series. With only one inlet and no expensive or complicated pumping mechanisms to run separations we ensure the level of simplicity necessary for field use.
Introduction
For many tropical diseases, identifying parasites or infected blood cells among a huge background of normal blood cells is the gold standard for diagnosis.1 Particle separation techniques such as acoustophoresis,2 margination3 and inertial focusing4,5 have been developed that show great promise as candidates for the field diagnosis of tropical diseases, however the gap between how microfluidics devices are run in labs and how they need to be run in the field is still considerable. In well-equipped labs there are pressure pumps, expensive pressure control units or syringe pumps plus advanced microscopes to monitor device function, giving information that is in turn used to make changes to flow rates and pressures. In the field however the lack of equipment, and in many cases maybe even access to a power grid, means that devices designed to work well in the lab, are doomed to failure. One approach would be to develop cheaper, less power consuming flow-control and optical systems. Another approach is to develop systems that require as little control as possible, which can perform complex operations with minimum requirements on external control. Paper-based microfluidics6 is the epitome of the second approach, where all functions are designed into the device and the only external control required is the application of a droplet of sample followed by a simple visual readout. While these systems are promising for many applications, they are not well-suited to high-resolution particle separations for which device structures often need to be much more well-defined on the microscopic scale than the random fibrous structure of paper. Here we describe a device that is designed to perform complex, high-resolution size and shape-based particle separation without expensive fluidic control equipment. The separation mechanism in our device is deterministic lateral displacement (DLD).7 The basic concept is to use simple and cheap pressure generation (a disposable syringe where air is compressed to give an over-pressure), simple sample input (one inlet only for the entire device), and sequential particle sorting units based on DLD to decomplex a mixture of particles. While applicable to any system containing particles that differ in size and/or shape we focus here on the separation of parasites from the genus Trypanosoma from blood cells. We have previously shown how trypanosomes and erythrocytes exhibit different behaviours in DLD devices as the device depth is changed, a fact that we previously used to perform separations.8 In the present work we use multiple array parameters, including depth, to realize complex functions in our device while simultaneously simplifying fluid control and use the decomplexing of a mixture of human blood and trypanosomes as a test system.Endemic to sub-Saharan Africa, human African trypanosomiasis (HAT) is a fatal vector-borne parasitic disease caused by the protozoan parasite Trypanosoma brucei, transmitted by tsetse flies.9 Its diagnosis relies to a great extent on tedious and laborious microscopic detection in blood smears or lymph node aspirates.10 The key bottleneck with conventional microscopic diagnosis of HAT is the relatively low number of parasites against the overwhelming background of blood cells. Consequently the detection threshold for normal microscopic diagnosis is relatively high (∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ml−1)11 in comparison to the parasitaemia (parasite number density in blood). This varies periodically during the course of the disease but can at times be as low as 100 ml−1, and consequently, there could be more than 50 million blood cells (predominantly erythrocytes) per parasite.11 Today, expensive and power-consuming technologies, such as ion exchange chromatography and centrifugation, are available at most modern facilities, and can be used for relatively efficient parasite detection (via enrichment relative to blood cell concentration). However, it is challenging to employ these tools in the geographical areas where the disease is prevalent, due to the lack of reliable power and storage facilities. Our aim is to use a microfluidic sorting scheme to extract the parasites for visual inspection, thus addressing the limitations of current technologies.
000 ml−1)11 in comparison to the parasitaemia (parasite number density in blood). This varies periodically during the course of the disease but can at times be as low as 100 ml−1, and consequently, there could be more than 50 million blood cells (predominantly erythrocytes) per parasite.11 Today, expensive and power-consuming technologies, such as ion exchange chromatography and centrifugation, are available at most modern facilities, and can be used for relatively efficient parasite detection (via enrichment relative to blood cell concentration). However, it is challenging to employ these tools in the geographical areas where the disease is prevalent, due to the lack of reliable power and storage facilities. Our aim is to use a microfluidic sorting scheme to extract the parasites for visual inspection, thus addressing the limitations of current technologies.
Deterministic lateral displacement
Our device, Fig. 1, can be fabricated in cheap materials, has no moving parts or power requirements and being no larger than a microscope slide, is highly portable. It is based on Deterministic Lateral Displacement (DLD), a method first shown in 2004 by Huang et al. that in its simplest configuration separates particles based on size.7 In brief, DLD uses an array of obstacles in a fluidic channel. Particles smaller than a critical size follow the overall flow direction, while larger particles are deflected at an angle relative to the flow, defined by the array. The critical size depends on the obstacle–obstacle distance (perpendicular to the flow direction, Gap) and the distance each row is shifted relative to the previous (Row shift), see Fig. 2A.12–16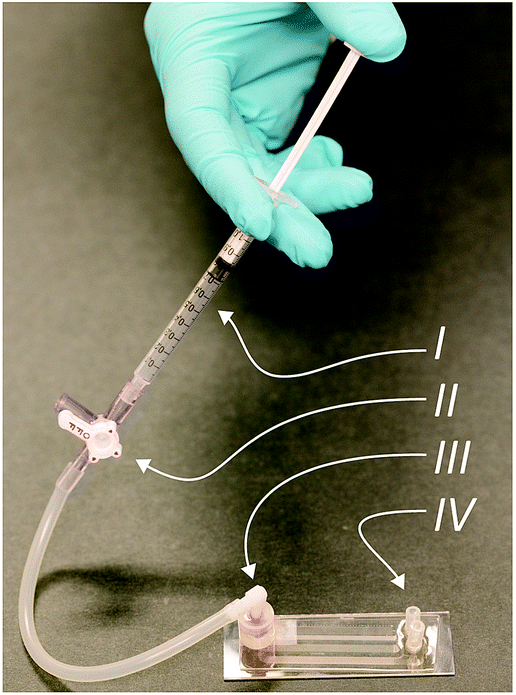 | ||
| Fig. 1 Complete device consisting of a syringe (I), a pressure valve (II), sample inlet (III) and outlets for sorted particles (IV). Here, the fluid is actuated by compressing air with a simple syringe. When the desired pressure is reached, the valve is closed allowing for a stable pressure throughout the measurement. The sample is injected into the single inlet reservoir prior to attaching the pressure system. Following an analysis, the sorted particles (erythrocytes, leucocytes and T. cyclops parasites) can be visualized in, or collected from the outlets for further processing. | ||
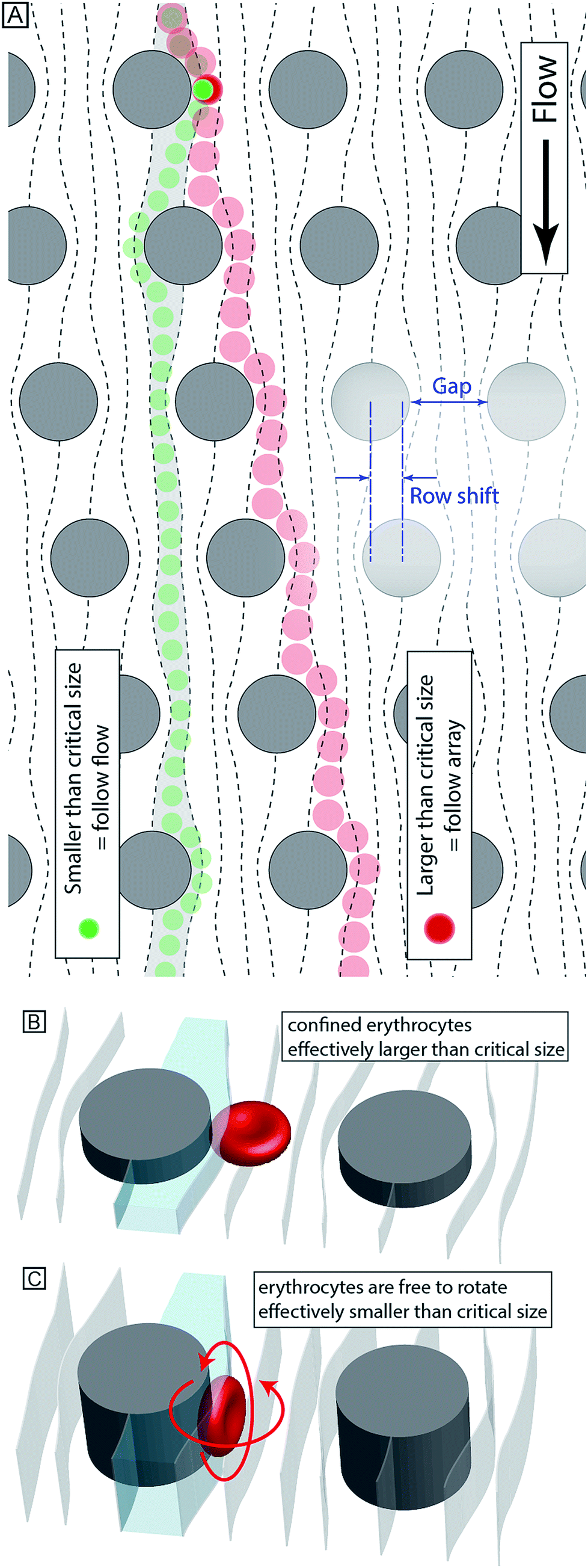 | ||
| Fig. 2 (A) Schematic illustration of size-based sorting in DLD devices. Particles smaller than the critical size follow the direction of fluid flow while particles larger than the critical size follow rows of posts. (B–C) Non-spherical particles (such as erythrocytes) behave very differently when confined in shallow devices compared to in deep devices where they are able to rotate. In a shallow device (B) erythrocytes have an effective size corresponding to their diameter. In a deep device (C) they are instead sorted according to their much smaller thickness. | ||
The method has been shown capable of size-fractionating a wide range of samples including blood components,17,18 cancer cells19 and synthetic particles such as droplets in two-phase flow.20 A recent review21 by Bridle et al. gives an expansive overview of this particle separation technique.
By controlling and exploiting shear forces our group recently added deformability22 to the list of parameters by which particles can be separated using DLD devices. Also, by controlling the orientation of particles as they pass through the device, we have been able to separate particles based on shape. As a proof of principle we showed that channel depth could be used to control the orientation of particles, and used this effect to accentuate the differences in shape of parasites and human blood cells8 making otherwise very difficult separations possible.
The work presented herein builds on these findings and involves a complete redesign of the device. The outcome is a more robust, easier to use device, with a significantly increased throughput. Our method provides a dramatically simpler and cheaper alternative to existing methods with the potential of enabling a fast and cheap point-of-care device that will have significant impact on field diagnosis.
Device design
In designing our device we prioritized simplicity and ease of use. We require the device to handle blood with low concentrations of parasites, with as little dilution as possible, and output close to all parasites in a sample stream free of blood cells. We designed a device with one inlet only, so that flow can be driven simply using a disposable syringe. To avoid pressure control systems that are both expensive and demanding with regards to user input, all functionality must be designed into the device: (1) Removal of leukocytes in order to avoid clogging in subsequent steps, (2) creation of cell-free plasma, and (3) transfer of parasites into the cell-free plasma. The functionality in each section of the device comes from a combination of the array spacing parameters and the depth of the channel (height of the posts). The final device consists of three DLD arrays in series, each with its own depth, optimized to carry out these three tasks, Fig. 3A–B.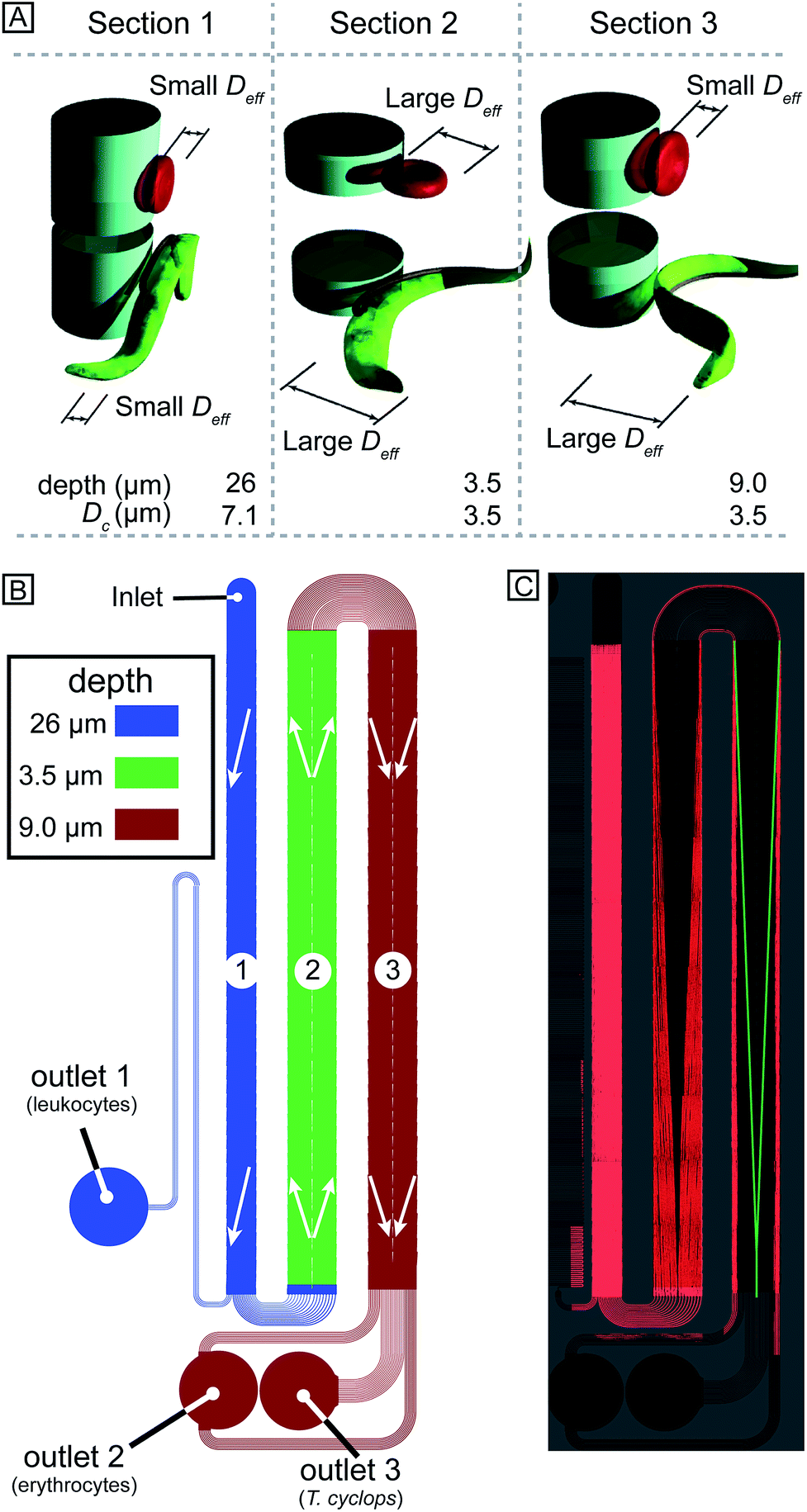 | ||
| Fig. 3 (A) Basic principle of how orientation changes the effective size of particles of different shape. (B) Schematic overview with the three different sections colour coded. Arrows represent the direction of displacement in each section. (C) False-coloured mosaic image consisting of 34 time-integrated micrographs showing the trajectories of erythrocytes and the parasites through the device. | ||
Section (1) is designed to function like a traditional size-based DLD with a depth larger than the diameter of all blood cells. As we have previously shown,8 in deep devices particles rotate due to shear forces and are consequently sorted according to their smallest dimension. Here, the spherical leukocytes, being in the range 8–15 μm, are displaced and removed from the sample via a side channel. Erythrocytes and trypanosomes, on the other hand, have a smallest dimension of ∼2.5 μm which is below the critical size in this section. As a consequence, they are not laterally displaced and mainly follow the flow to the subsequent section.
Section (2) is shallow (3.5 μm) to minimize the rotational freedom of the cells and consequently maximize their effective size. The array is designed so that all cells are laterally displaced towards the channel walls creating a cell-free stream in the centre. A mirrored design23 is employed to minimize the migration distance of cells to the nearest wall which results in a higher throughput.
Section (3) has the same post size, post gap and row shift as section (2) but is deeper, (9 μm from previous optimization8). The row shift direction is also mirrored such that displaced particles are focused into the centre of the channel. The key idea is that the depth of this section is sufficient for erythrocytes to rotate and behave like 2.5 μm particles, as decided by their thickness, and they consequently remain near the walls. At the same time the longer parasites are still partially confined and behave as effectively larger particles being displaced back into the cell-free plasma and into a separate outlet where a dense array of posts (8 μm in diameter with a separation of 2 μm while keeping the array wide, 4 mm, in order to minimize the effect of clogged particles) serves as a sieving structure to capture and concentrate the cells of interest once they have been sorted. The parasites can thereafter be detected (future work will focus on detection schemes for the enriched parasites).
Simple pump system
The single inlet design not only simplifies the sample handling and introduction to the device, but it also allows for a device which does not require advanced and expensive pressure controllers that would otherwise be necessary to fine-tune and balance the pressures applied to the different inlets. Instead, here a simple approach is employed where the air in a syringe is compressed in a controlled way, thereby resulting in a well-defined overpressure (see Fig. 4). The relatively large volume of air in the system compared to the small volume of fluid handled in the device, together with well-sealed connections ensures that the overpressure is maintained at a stable level over the course of the experiment.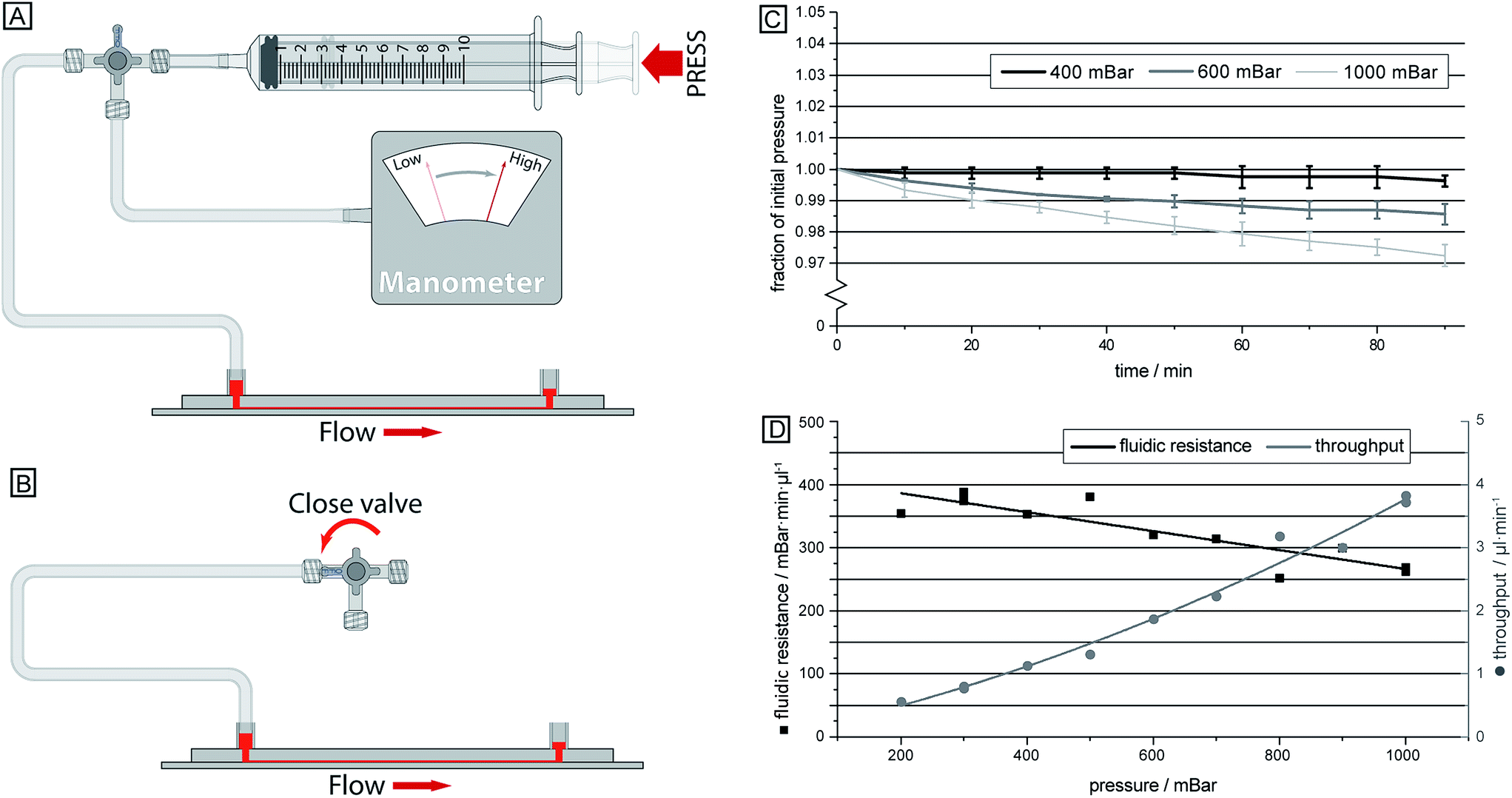 | ||
| Fig. 4 (A–B) Schematic overview of the device connected with a simple syringe that was used in all measurements to induce fluid flow. A manometer was initially used to relate the syringe compression with the resulting overpressure, ensuring that the correct pressure was achieved. Thereafter a valve is closed resulting in a consistent pressure over longer times. (C) Applied pressure measured over 90 minutes given as fraction of the initial pressure. (D) Throughput in terms of sample fed into the system measured as a function of the pressure difference across the device. | ||
Method
Device design and fabrication
L-Edit 11.02 (Tanner Research, Monrovia, CA USA) was used to draw the device design and photolithographic masks were manufactured by Delta Mask (Delta Mask, Enschede, The Netherlands). To fabricate a master consisting of a single continuous device three separate cycles of UV-lithography were performed which consisted of resist spinning, precisely aligned exposures (Karl Suss MJB4, Munich, Germany) and development using SU8 (SU8-2005, SU8-2007 and SU8-2015, Microchem Corp., MA, USA) on 3′′ silicon wafers. After treatment of the master with an anti-adhesion layer of 1H,1H,2H,2H-per-fluorooctyl-trichlorosilane (ABCR GmbH & Co. KG, Karlsruhe, Germany)24 to facilitate demoulding, replicas were created in PDMS (Sylgard 184, Dow Corning, Midland, MI, USA) by standard replica moulding.25 PDMS casts were oxygen plasma bonded to blank PDMS slides (Plasma Preen II-862, Plasmatic Systems Inc., North Brunswick, NJ, USA) to obtain devices entirely consisting of PDMS. To prevent sticking of cells in the devices, they were immediately filled with 0.2% (w/v) PLL(20)-g[3.5]-PEG(2) (SuSoS AG, Dubendorf, Switzerland) in deionized water and allowed to rest for at least 20 min before flushing with deionized water for another 20 min. Silicon inlets and outlets 12 mm and 5 mm outer diameter silicon tubing (228-0725 and 228-0707, VWR International LLC, Radnor, PA, USA) were glued (Elastosil A07, Wacker Chemie AG, Munich, Germany) onto the device. The sample was applied to the inlet reservoir, which was subsequently connected to the syringe to drive the sample through the device.To maximize the throughput, the device has been kept as wide as possible. This means however that, together with the relatively small angle of displacement, the device needs to be long. In fact, the final device is around 14 cm. To fit this onto a standard glass slide the device is split up into its three different sections with multiple channels connecting the end of each array with the start of the proceeding array, see Fig. 5A–B. To ensure that the particles maintain their relative lateral position while moving between the different sections, careful calculations and CFD simulations (COMSOL Multiphysics 4.3, Comsol AB, Stockholm, Sweden) have been conducted, the key being to keep a constant resistance across the width of the device such that the flow and particle profiles are preserved around the 180° bends. More info on this optimization can be found in ESI section (3).†
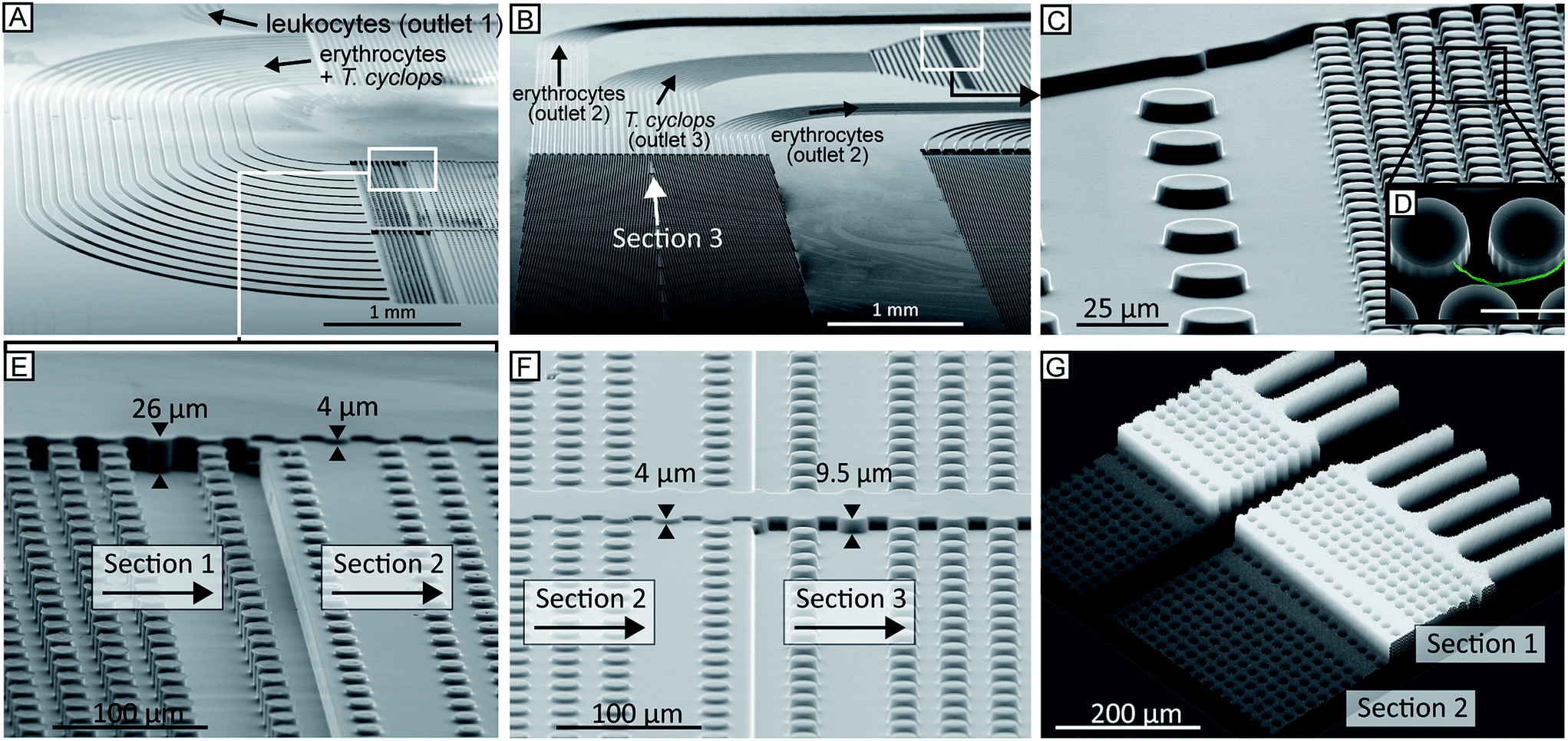 | ||
| Fig. 5 (A–F) SEM and (G) confocal images of the device. (A–B) The channels connecting the different sections of the device are designed to be of equal fluidic resistance, and therefore their individual widths are chosen to compensate for the varying lengths. Due to these channels, the flow symmetry is maintained and the relative positions of the particles are conserved between subsequent sections allowing for longer and more complex devices to be created. By precise alignment of the complementary photolithographic masks in three separate UV-lithographic cycles, sections of different channel depth could be fabricated into a single continuous device, (E–F). After the final section in the device, section (3), the trypanosomes have been focused to the centre and thus exit to the trypanosome outlet where a dense array of pillars (C–D) is defined with the purpose of capturing the parasites. Note the inset (D) where a parasite is highlighted (coloured green), scale bar is 10 μm. (G) Confocal micrograph of a fluorescent solution (0.1% rhodamine B in H2O) passing through the step-change in depth between section (1) and (2), same as (E). The grey scale is mapped to the fluorescence signal. | ||
Experimental setup
All experiments were performed in compliance with the relevant laws and institutional guidelines. Blood was extracted via finger pricking (Haemolance, MedCore AB, Kista, Sweden) of healthy volunteers from which informed consent was obtained. Prior to each experiment, the devices were flushed with sterile filtered Cunningham's medium26 with 20% fetal calf serum (Cat. No. F2442, Sigma-Aldrich, St. Louis, MO, USA), the medium used to culture the parasites, and 2 mM EDTA (Cat. No. E6758, Sigma-Aldrich) to inhibit coagulation of the blood. It is important to note that Cunningham's medium is only necessary to sustain our model system.In order to obtain good statistics on the device performance, measurements were often conducted with the same device for several hours. Consequently, to counteract particle sedimentation and ensure a homogenous sample, a small magnetic stir bar (length 5 mm, diameter 2 mm) was placed in the inlet reservoir and controlled by a magnet connected to a small electric motor. With a stir rate of ∼1 Hz and the dimensions of the stir bar and inlet reservoir as given above, the shear rate is estimated to be on the order of 10 s−1 to 100 s−1, which is less than the physiological shear rates of up to ∼104 s−1.27,28 Further, microscopic examination confirmed that the stir bar did not induce any morphological changes in the sample. For actual field use of the final device, the short time of analysis would eliminate the need for any stirring.
Measurements and analysis
The lateral distribution of cells was characterized at the end of each section in the device through an inverted Nikon Eclipse TE2000-U microscope (Nikon Corporation, Tokyo, Japan) using an EMCCD camera (iXon EM+ DU-897, Andor Technology Ltd, Belfast, UK) and various objective lenses (Nikon, 10/20x Plan Fluor and 60x Apo TIRF). Differential Interference Contrast (DIC) was used to ensure adequate contrast of blood cells and trypanosomes.Particle tracking software, based on available MATLAB code, was written in MATLAB R2014b (The MathWorks Inc, Natick, MA, USA) and optimized for accurate and efficient particle recognition. Detailed information together with the code can be found in ESI section (4).†
Blood and trypanosomes were analysed at concentrations, which allowed for automated particle tracking while the addition of anticoagulants opened up for measurements over several hours per device without any blood clotting affecting the flow behaviour.
For all measurements, the morphologically similar but much less dangerous (and less restricted) parasite T. cyclops was used instead of the human parasites T. brucei rhodesiense or T. brucei gambiense.8
Blood and parasites were initially analysed separately in order to better characterize how the output of the device varied with flow rate. Subsequently, with the optimum flow rate determined, spiked samples were separated. For the initial samples where blood was run separately, it was diluted 10× in autoMACS Running Buffer (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). This is an isotonic PBS solution containing 2 mM EDTA, 0.5% BSA and 0.09% azide. In addition to preventing blood clotting, a lower concentration decreases the particle–particle interaction. For field-diagnostics we envision the collection of blood in pre-filled vials (e.g. BD Vacutainer® 366450, BD Biosciences, Franklin Lakes, NJ, USA). For the spiked blood samples, the blood was washed twice in Cunningham's buffer to remove the lytic factors causing natural lysis of T. cyclops, which is not adapted to human physiology (this step would not be necessary for the T. brucei). The 10× diluted blood sample was then spiked with trypanosomes at a concentration of approximately 1000 μl−1. This is in most cases a higher concentration than what occurs in normal rare cell detection. However, it allows us to accurately determine the device performance in terms of sorting efficiency. Detailed protocols are available in the ESI section (7).†
Results and discussion
Multi-layered device characterization
During UV-lithographic fabrication of the master, the different sections of varying depths could be precisely aligned to each other with an estimated positioning error less than 3 μm. This is small enough to have a negligible effect on the flow, and consequently, the relative lateral position of the particles in the device. Fig. 5A–E shows SEM micrographs of a finalized continuous device in PDMS consisting of multiple depths. Here, three of the main features of the device are shown. First, the channels of equal resistance connecting the subsequent arrays can be seen in Fig. 5A–B. Due to the complexity of the device with multiple arrays in series it will be longer than a normal DLD device. Consequently, to fit the entire device on a normal glass slide the device design is folded, forming a serpentine. In order to maintain the relative particle positions between sections, multiple channels with identical fluidic resistances are used to connect subsequent sections. To accomplish this, the shorter channels are designed to be narrower than the longer ones. Between section (1) and (2), there are in total 16 channels while 28 channels connect section (2) and (3). A detailed description of these channels can be found in ESI section (3).† In Fig. 5C–D the sieving structure in the outlet is shown. This feature has been implemented to capture sorted cells of interest and to focus them to a smaller area allowing for easier detection. In Fig. 5E–G the two step changes in height of the device are shown. The first of these two SEM micrographs shows how the deep channels in section (1) transition to the shallow channels in section (2). The other micrograph shows the end of the same section but here the transition is from the shallow section (2) to the intermediate section (3). Fig. 5G further shows the transition from deep to shallow channel depth, here visualized after being filled with 1 mg ml−1 rhodamine B (#83689, Sigma) in H2O and imaged using confocal microscopy.Using a simple syringe to control flow rate
Compressing the syringe leads to a pressure drop across the device acting to drive the sample, which prior to this has been loaded into the sample inlet. Having a stable pressure is required as flow rate affects the sorting of deformable cells, as shown previously, allowing for sensitive measurements to take place over a longer time. Here we succeeded in achieving high pressure stability, Fig. 4C, by utilizing a large dead volume (∼12 ml) together with sealed connections. At an initially applied overpressure of 1 bar across the device the resulting pressure decreased less than 3% over 90 minutes. The volumetric throughput is ∼390 μl during this time, which according to Boyle's law explains the pressure decrease, indicating little to no leakages in the system.The results from the calibration of the flow speed in relation to the syringe compression are given in Fig. 4D. Here, it can be seen that the hydrodynamic resistance of the device decreases with increasing pressure, which is expected due to the elastic PDMS device becoming deformed, and inflated, at high pressures. Consequently, the flow rate is not linearly proportional to the applied driving pressure, instead it varies between 2.5 and 3.8 μl (min bar)−1. The volumetric flow rate here is given as the sample input rate. From this, approximately 20% of the total flow is separated out to outlet 1, enriched with leukocytes, while the sorting between erythrocytes and trypanosomes takes place in the remaining volume.
Erythrocytes and leukocytes at various flow rates
The behaviour of soft, deformable cells in a DLD device depends to a great extent on the flow rates. Increasing the flow rates leads to a larger force exerted on the cells by the fluid acting to deform them. Consequently their size and, as a result, the sorting in the device might change. Further, cells in channels deeper than their size can rotate freely under the influence of the shear field of the fluid, which as shown previously, acts to make the cells present their smallest dimension to the posts. An erythrocyte would due to this effect have an effective size the same as its thickness, ∼2.5 μm instead of its diameter, ∼7.5 μm. However, at low flow rates the shear stress exerted by the fluid becomes smaller, which acts to decrease the rotational effect. Another effect of higher flow rates is the increased pressure drop, which acts to inflate the deformable PDMS devices, something which in turn leads to an increase of the critical size.The erythrocytes and parasites were initially measured separately at various flow rates to allow for automated particle tracking of a large number of cells. Thereafter, spiked blood samples were analysed at the most promising flow rates.
The results from the separate measurements of erythrocytes and T. cyclops are presented in Fig. 6, with the sorting efficiency further analysed in Fig. 7.
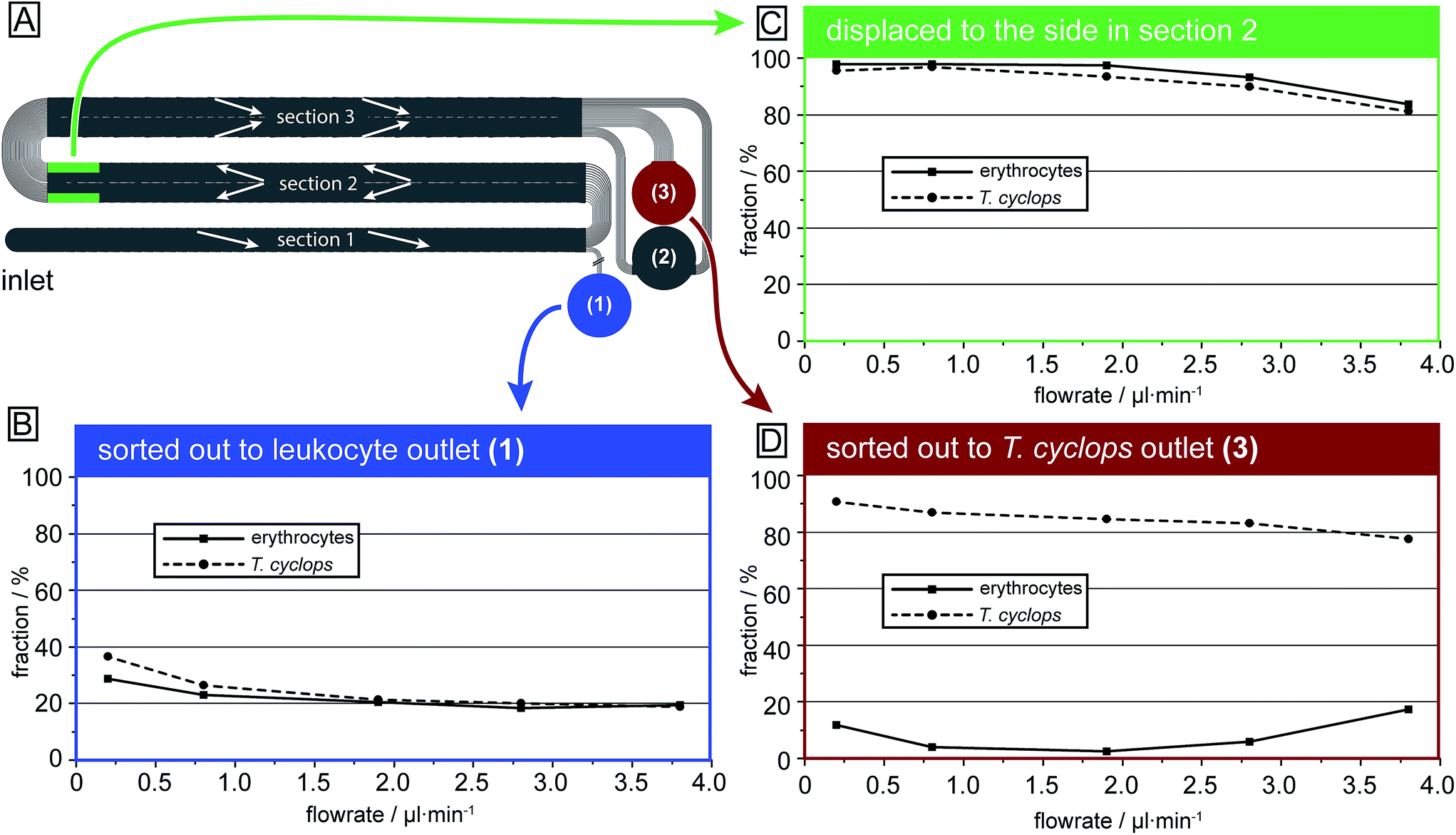 | ||
| Fig. 6 Sorting efficiencies as a function of flowrate (i.e. the fraction of cells in section (1) that is sorted out into outlet 1, the fraction in section (2) which is focused along the channel walls and the fraction in section (3) which are sorted out into outlet 3). The numbers are given as the fraction of that cell type in that specific section. (A) Schematic overview of the device highlighting the origin of the data in the graphs. (B) After the first section the erythrocytes and parasites are sorted out in proportion to the fluid flow going into the outlet. (C) In section (2) most erythrocytes and parasites are focused to the side. (D) For the separation of T. cyclops and erythrocytes intermediate flow rates give the highest separation efficiency. Each data point represents a minimum of 1000 counts. For the highest flow rates the number of counts was around 7000 for both erythrocytes and T. cyclops. | ||
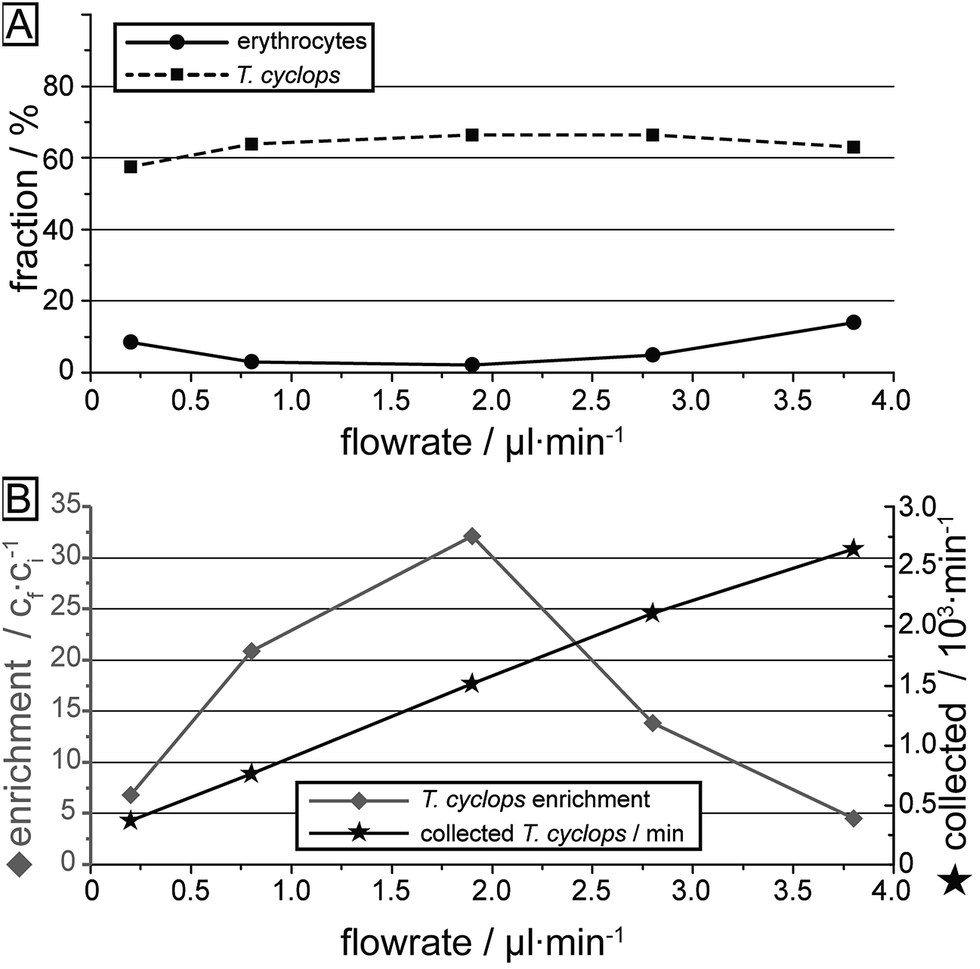 | ||
| Fig. 7 Overall sorting performance versus applied pressure. (A) Fraction of erythrocytes and T. cyclops which are sorted into outlet 3. The numbers are given as the fraction of each cell type injected into the device. (B) Concentration enrichment of T. cyclops (ratio between the number of T. cyclops and the total number of particles) and the rate of trypanosomes arriving at the collection reservoir. | ||
In Fig. 7 the output of each section is given for both erythrocytes and T. cyclops at several different flow rates as a fraction of the total number of that cell type in that specific section. It can be seen that in section (1), Fig. 6B, the fraction of cells separated out are larger at low flow rates but converges to ∼20% with increasing flow rates (and consequently ∼80% of the cells continue to section (2)). Section (1) is deeper than the largest dimensions of both cell types, the length of the T. cyclops and the diameter of the erythrocytes. As a result the cells can rotate freely as discussed previously, resulting in a decreased effective size. With the section designed with a critical size of 7.1 μm, the cells are not expected to be laterally displaced. However, at low flow rates the shear stress exerted on the particles by the fluid is limited which leads to a decrease in both the orientation of the cells and also the cell deformation. As a consequence, the fraction of cells sorted out into outlet 1 can be higher than the expected 20%.
In section (2), Fig. 6C, the cells are steered towards the sides of the channel to open up a cell-free stream in the centre. Due to the low depth of this section, the cells are restricted from rotating which leads them to display a larger effective size and consequently they are more easily laterally displaced into the side streams along the channel walls. Here, the side streams are defined as the outermost 25% on both sides of the channel while the centre stream constitutes the remaining 50%. The size of these streams is defined by how the end of section (3) is divided into outlet 2 and 3, meaning that any cells residing in the centre stream at the end of section (2) are likely to also be sorted into outlet 3. In the same way, any cells residing in the side streams will end up in outlet 2 unless they are laterally displaced in section (3).
At lower flow rates, around 95% of cells entering section (2) will at the end reside in the side streams. However, at high flow rates the soft erythrocytes will, by deforming due to shear forces, avoid being laterally displaced and will end up as a contaminant among the parasites in outlet 3. At 3.8 μl min−1 around 20% of the erythrocytes exiting this section do so in the centre stream and are expected to end up as a contamination in outlet 3, leading to poor separation sensitivity.
Section (3) is identical to section (2) except for the direction of displacement and the depth, Fig. 6D. By the increase in depth to 9.0 μm, larger than the diameter of the erythrocytes, there is no steric hindrance to their orientation, leading to a decrease in their effective size as previously discussed. They are therefore not laterally displaced in this section and, if focused into the side streams in section (2), are sorted into outlet 2. Similar to the situation in section (1), it can be seen that if the flow rate is too low, the shear forces are not high enough to allow the cells to rotate and adopt the smaller effective size. Instead they have a maximum effective size equal to their diameter leading to a significant lateral displacement. At low flow rates (0.2 μl min−1) 12% of the erythrocytes entering this section are sorted into outlet 3 even though they exit section (2) well-focused in the side streams. Consequently, the erythrocyte contamination in outlet 3 is here a result of unwanted lateral displacement in section (3). An increase of the flow rate leads to a decrease in the fraction of erythrocytes sorted out into outlet 3, however at too high pressure, the fraction once again increases. This is however believed to be an effect of erythrocytes not being well focused into the side streams when they exit section (2). This could potentially be avoided by increasing the width of section (2), leading to a decrease in the flow velocity (while maintaining the volumetric flow rate) and consequently the shear stress exerted on cells in this section.
For the T. cyclops the highest fraction of extracted cells is acquired at low pressures while the fraction decreases with increasing pressures. This shows how adjusting the depth of the channel can have a large impact on the behaviour of the cells, allowing for separation based on their differences in shape.
The measurements of T. cyclops and erythrocytes carried out separately at various flow rates are summarized in Fig. 7. Firstly, the fraction of the two cells types which ends up in outlet 3 is shown, here calculated based on the input sample. This figure is similar to Fig. 6D, but the cells removed in section (1) have also been taken into account. The most notable difference is the decreased capture rate of T. cyclops at low flow rates which is due to the increase in fraction of T. cyclops being sorted out into outlet 1. As a result, the separation efficiency is decreased at these flow rates. The largest enrichment (32 times) is reached at 1.9 μl min−1. At this flow rate the fraction of erythrocytes entering outlet 3 is kept at a minimum (2.1%) while a large fraction of the trypanosomes are retained (66.5%). As a consequence, this was chosen as the flow rate for all the remaining measurements. However, it can be noted that even though the enrichment is the highest at this flow rate, the number of collected T. cyclops per minute increases with increasing flow rate. At a flow rate of 3.8 μl min−1 the number of collected T. cyclops is almost twice that of 1.9 μl min−1. The optimum trade-off between throughput and purity will be determined by the choice of detection scheme in the specific application.
Blood samples spiked with T. cyclops
As discussed, a flow rate of 1.9 μl min−1 was chosen for the continued studies of spiked blood samples in order to receive the maximum enrichment of T. cyclops. In these measurements, the sorting of leukocytes was also considered in order to show the versatility of the device, and its potential for applications other than erythrocyte and T. cyclops separation.On the whole, the outcome, given in Fig. 8, agrees with the previous measurements. For the erythrocytes 21.7% are diverted to outlet 1 (leukocyte outlet). For the remaining erythrocytes which continue into section (2) the fraction successfully focused into the side stream amounts to 97.6%. From this number, a small fraction (0.6%) is not retained along the sides of the channel in section (3) but instead end up in outlet 3 (T. cyclops outlet) as a contamination. The remaining 97.0% which still remain in the side streams exit into the correct outlet which is close to the result of the separate measurements.
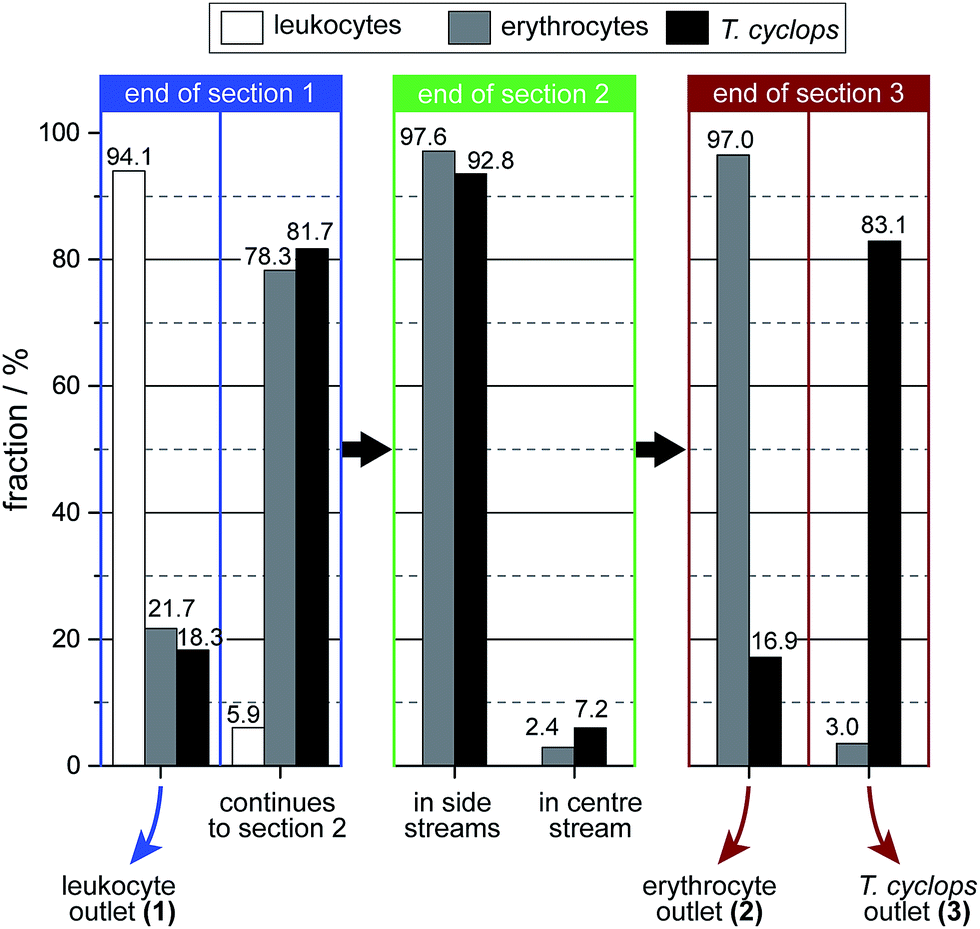 | ||
| Fig. 8 Distribution of the three different cell types at the end of each section. The numbers are given as the fraction of that cell type in that section. | ||
For the trypanosomes 18.3% is laterally displaced into outlet 1 while 92.8% of the remaining parasites are successfully focused into the side streams. This is, in agreement with previous measurements a smaller fraction than for the erythrocytes. But as the T. cyclops should exit into outlet 3 from the centre of section (3), this would lead to an increased sorting efficiency. In the end, 83.1% of the remaining T. cyclops exit into outlet 3, equivalent to a 30-fold enrichment in the number concentration. The leukocytes are, contrary to the other cells, laterally displaced to a great extent in section (1). The majority of them, 94.1%, are sorted out into outlet 1 while the remaining fraction continues to the shallower section (2). A large fraction of these leucocytes are not able to enter this shallow section, however over the time scale of our experiments the number of leucocytes filtered out was too small to adversely affect the device performance. This is believed to be an effect of the device's robustness to pressure changes, with section (2) being aimed at focusing all cells towards the wall. The result of this is that any smaller pressure variations will not have an effect on the device performance.
The total output from the device is summarized in Fig. 9, here we can see that 100% of the extracted leukocytes are found in outlet 1 owing to the large size and shape difference between leukocytes and the other cell types. There is a relatively large number of background cells present, around 20% of each cell type, as expected due to the design of section (1). The addition of a focusing section, similar to section (2), positioned before the sorting-out of leukocytes takes place, the number of background cells in outlet 1 could be greatly reduced. In outlet 2 the fraction of erythrocytes amounts to 76.0% (77.4%), while 67.9% (66.5%) of the T. cyclops are retained in outlet 3 (results of the earlier measurements on individual cell types are given in parenthesis). As can be seen, these fractions are in close agreement with the earlier measurements with a marginally smaller number of erythrocytes retained in outlet 2 while a slightly larger fraction of T. cyclops can be steered into outlet 3. It should however be noted that these numbers rely on the bifurcation of the flow into the outlets. The current device was designed to have, as described earlier, half the flow at the end of section (3) guided to outlet 2 and the other half to outlet 3. Changing these fractions would potentially lead to a better performance of the device. For example, decreasing the fraction of the flow in section (3) bifurcated into outlet 3 would remove more erythrocyte and would likely increase the specificity of the device.
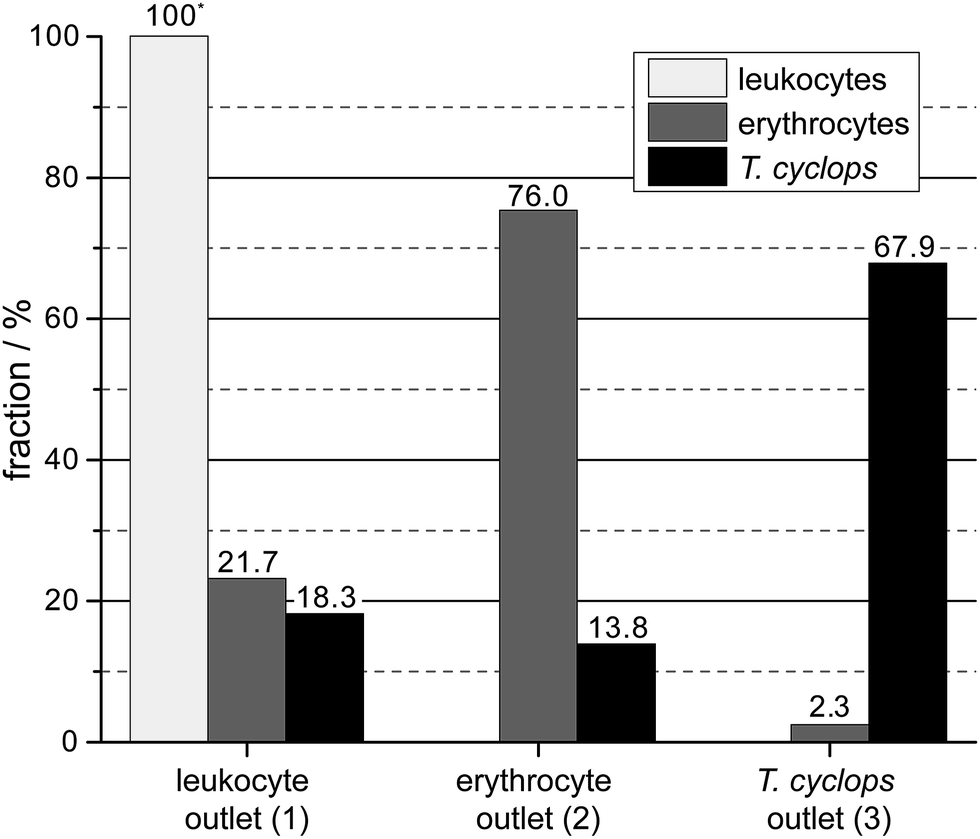 | ||
| Fig. 9 The output of the device given as the fractional distribution of the three measured cell types. | ||
Conclusions
We have demonstrated a proof-of-principle of a device that, while simple-to-use, can perform complex separations such as the fractionation of parasites, leukocytes and erythrocytes. Our method utilizes several steps along the device where differences in morphology and size between the particles are given different weights by varying the depth of the DLD array. The principle of using multiple successive arrays to focus and then separate particles could be applied to any of the particle parameters (or combinations thereof) that have been shown to affect behaviour in DLD devices such as size, shape, deformability and dielectric properties. What is more, the device functions without expensive pressure controllers and can be run using pressurized air from a single disposable syringe.We believe the approach that we have presented here shows great promise as a point-of-care test for a multitude of diseases. In addition to our test system with African trypanosomiasis other protozoan parasites could be considered such as, but not limited to, leishmaniasis, Chagas disease and malaria, where particle size, shape and deformability can be leveraged for the enrichment of hard to find objects. These diseases affect millions worldwide, not only via direct infection of human populations but also via the socioeconomic burden of livestock infection.29
While we believe the current device offers several advantages over current methods there is still work to be done towards a fully functioning field-ready diagnostic device. Most notably lacking is portable detection. One solution may be the devices developed by Ozcan et al.30 that have shown great promise for field-detection of pathogens combined with telemedicine. Another would be integrated electronic cell counting based on impedance spectroscopy.31
Further, the current experiments were performed in devices made using soft lithography. This is a rapid prototyping technique that is suitable for research, but for field applications large numbers of devices would be needed at lower cost. Foil-based fluidics32,33 and injection moulding approaches would provide the necessary basis for mass-produced devices and be fully compatible with our method.
Acknowledgements
This work was carried out within NanoLund at Lund University as part of LAPASO (EU FP7 project 607350) and was supported by the Swedish Research Council (VR) grants no. 2007-584, 2011-6035 and 2015-05426, the Crafoord Foundation grants no. 2005-1123, 2008-0841 and 2015-0007 and the Knut and Alice Wallenberg Foundation. All device processing were conducted within Lund Nano Lab. We would like to thank Zhen Li (Lund University, Sweden) for her help in acquiring confocal images.Notes and references
- P. Mitashi, E. Hasker, V. Lejon, V. Kande, J. J. Muyembe, P. Lutumba and M. Boelaert, PLoS Neglected Trop. Dis., 2012, 6, e1919 Search PubMed
.
- A. Lenshof, C. Magnusson and T. Laurell, Lab Chip, 2012, 12, 1210–1223 RSC
- H. Wei Hou, H. Y. Gan, A. A. Bhagat, L. D. Li, C. T. Lim and J. Han, Biomicrofluidics, 2012, 6, 24115–2411513 Search PubMed
- M. E. Warkiani, G. Guan, K. B. Luan, W. C. Lee, A. A. S. Bhagat, P. Kant Chaudhuri, D. S.-W. Tan, W. T. Lim, S. C. Lee, P. C. Y. Chen, C. T. Lim and J. Han, Lab Chip, 2014, 14, 128–137 RSC
- D. Di Carlo, Lab Chip, 2009, 9, 3038–3046 RSC
- D. M. Cate, J. A. Adkins, J. Mettakoonpitak and C. S. Henry, Anal. Chem., 2015, 87, 19–41 CrossRef CAS PubMed
- L. R. Huang, E. C. Cox, R. H. Austin and J. C. Sturm, Science, 2004, 304, 987–990 CrossRef CAS PubMed
- S. H. Holm, J. P. Beech, M. P. Barrett and J. O. Tegenfeldt, Lab Chip, 2011, 11, 1326–1332 RSC
- M. P. Barrett, R. J. S. Burchmore, A. Stich, J. O. Lazzari, A. C. Frasch, J. J. Cazzulo and S. Krishna, Lancet, 2003, 362, 1469–1480 CrossRef
- J. E. Rosenblatt, L. B. Reller and M. P. Weinstein, Clin. Infect. Dis., 2009, 49, 1103–1108 CrossRef PubMed
- F. Chappuis, L. Loutan, P. Simarro, V. Lejon and P. Büscher, Clin. Microbiol. Rev., 2005, 18, 133–146 CrossRef PubMed
- D. W. Inglis, J. A. Davis, R. H. Austin and J. C. Sturm, Lab Chip, 2006, 6, 655–658 RSC
- K. Loutherback, K. Chou, J. Newman, J. Puchalla, R. Austin and J. Sturm, Microfluid. Nanofluid., 2010, 9, 1143–1149 CrossRef
- S. Cerbelli, M. Giona and F. Garofalo, Microfluid. Nanofluid., 2013, 15, 431–449 CrossRef CAS
- M. Jiang, K. Budzan and G. Drazer, Microfluid. Nanofluid., 2015, 19, 427–434 CrossRef
- R. Devendra and G. Drazer, Microfluid. Nanofluid., 2014, 17, 519–526 CrossRef CAS
- J. A. Davis, D. W. Inglis, K. J. Morton, D. A. Lawrence, L. R. Huang, S. Y. Chou, J. C. Sturm and R. H. Austin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14779–14784 CrossRef CAS PubMed
- D. W. Inglis, K. J. Morton, J. A. Davis, T. J. Zieziulewicz, D. A. Lawrence, R. H. Austin and J. C. Sturm, Lab Chip, 2008, 8, 925–931 RSC
- K. Loutherback, J. D’Silva, L. Liu, A. Wu, R. H. Austin and J. C. Sturm, AIP Adv., 2012, 2, 042107 CrossRef PubMed
- H. N. Joensson, M. Uhlen and H. A. Svahn, Lab Chip, 2011, 11, 1305–1310 RSC
- J. McGrath, M. Jimenez and H. Bridle, Lab Chip, 2014, 14, 4139–4158 RSC
- J. P. Beech, S. H. Holm, K. Adolfsson and J. O. Tegenfeldt, Lab Chip, 2012, 12, 1048–1051 RSC
- Y. S. Lubbersen, R. M. Boom and M. A. I. Schutyser, Chem. Eng. Process., 2014, 77, 42–49 CrossRef CAS
- M. Beck, M. Graczyk, I. Maximov, E. L. Sarwe, T. G. I. Ling, M. Keil and L. Montelius, Microelectron. Eng., 2002, 61–62, 441–448 CrossRef CAS
- Y. Xia and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 550–575 CrossRef CAS
- I. Cunningham, J. Protozool., 1977, 24, 325–329 CrossRef CAS PubMed
- T. G. Papaioannou and C. Stefanadis, Hellenic Journal of Cardiology, 2005, 46, 9–15 Search PubMed
- O. Traub and B. C. Berk, Arterioscler., Thromb., Vasc. Biol., 1998, 18, 677–685 CrossRef CAS
- P. M. Kristjanson, B. M. Swallow, G. J. Rowlands, R. L. Kruska and P. N. de Leeuw, Agricultural Systems, 1999, 59, 79–98 CrossRef
- Z. Gorocs and A. Ozcan, IEEE Reviews in Biomedical Engineering, 2013, 6, 29–46 CrossRef PubMed
-
H. Morgan and D. Spencer, Chapter 10, Microfluidic Impedance Cytometry for Blood Cell Analysis, The Royal Society of Chemistry, 2015 Search PubMed
-
J. P. Beech, T. Mäkelä, P. Majander and J. O. Tegenfeldt, Presented in part at the μTAS - the 12th international conference on miniaturized systems for chemistry and life sciences, San Diego, CA, USA, 2008 Search PubMed
- A. Laurberg Vig, T. Mäkelä, P. Majander, V. Lambertini, J. Ahopelto and A. Kristensen, J. Micromech. Microeng., 2011, 21, 035006 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ay00443a |
| This journal is © The Royal Society of Chemistry 2016 |