
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of thermal processing on the behaviour of peanut allergen peptide targets used in multiple reaction monitoring mass spectrometry experiments†
R. L.
Sayers
*a,
P. E.
Johnson‡
a,
J. T.
Marsh‡
a,
P.
Barran
b,
H.
Brown
c and
E. N. C.
Mills
a
aInstitute of Inflammation and Repair, Manchester Academic Health Science Centre, Manchester Institute of Biotechnology, The University of Manchester, UK. E-mail: rebekah.sayers@postgrad.manchester.ac.uk
bSchool of Chemistry, Michael Barber Centre, Manchester Institute of Biotechnology, The University of Manchester, UK
cCampden BRI (Chipping Campden) Limited - part of the Campden BRI group, Chipping Campden, Gloucestershire, UK
First published on 19th April 2016
Abstract
Mass spectrometry-based methods offer an alternative means of determining allergens in foods. Whilst targeted methods are likely to offer the most robust approach for detection and quantification, little is known about how food processing may affect the behaviour of peptide targets. A systematic study has been undertaken to investigate the effects of thermal processing (boiling, roasting, frying) on the behaviour of a suite of peanut peptide targets representing the major clinically-relevant allergens. Initially the effect of thermal processing on protein extractability was investigated and a mass spectrometry-compatible buffer identified comprising 50 mM Tris-HCl, pH 8.8 containing 50 mM dithiothreitol and 0.04% (w/v) acid labile detergent which was able to extract 45–100% of protein from raw, boiled, roasted and fried peanuts using sonication at 60 °C. Eight peptide targets were identified including two peptides from each cupin allergen, Ara h1 and Ara h3 and four peptides from the prolamin superfamily allergens Ara h2, 6 and 7. AQUA peptide standards were synthesised and used to undertake multiple-reaction monitoring experiments, giving assay sensitivities of 0.1–30 amoles of peptide on-column (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 signal:noise), calculated limits of quantification between 96–1343 amoles of peptide on-column and a linear dynamic range of 4–5 orders of magnitude. Absolute quantification of individual peanut allergens in thermally processed samples showed that peptide targets in the cupin allergens were more prone to processing-induced effects than those from Ara h2, 6 and 7. Targets flanked by arginine residues showed greater thermostability. Identification of processing-stable targets, coupled with more efficient extraction procedures and a wide dynamic range, shows that targeted mass spectrometry methods have great potential as an additional method for quantifying peanut allergens in complex food matrices.
:1 signal:noise), calculated limits of quantification between 96–1343 amoles of peptide on-column and a linear dynamic range of 4–5 orders of magnitude. Absolute quantification of individual peanut allergens in thermally processed samples showed that peptide targets in the cupin allergens were more prone to processing-induced effects than those from Ara h2, 6 and 7. Targets flanked by arginine residues showed greater thermostability. Identification of processing-stable targets, coupled with more efficient extraction procedures and a wide dynamic range, shows that targeted mass spectrometry methods have great potential as an additional method for quantifying peanut allergens in complex food matrices.
Introduction
IgE mediated food allergies have been estimated to affect around 1–2% of adults and between 5–6% of infants and young children.1 At present there is no accepted cure for food allergy and hence allergic individuals have to practice food avoidance, usually life-long. Those at risk of a severe reaction are given rescue medication, often in the form of self-administered adrenaline. In order to help support food allergic consumers avoid their problem food, labelling legislation has been enacted around the world which requires a list of priority allergenic foods to be labelled, irrespective of the level at which they might be included in a recipe. In the European Union this is enshrined in the Food Information for Consumers regulation EC/1169/2011 which lists 14 allergenic foods that must be labelled on pre-packaged and loose products (including catered foods).2 However, this legislation does not address the issues of cross-contact allergens, where traces of allergenic ingredients may find their way into non-allergenic food products through, for example, the use of common food processing lines or where cleaning processes cannot guarantee an allergen-free environment. Dose distribution modelling using threshold dose challenges in food allergic subjects has indicated that consumption of residual allergen of around 3–30 mg protein for most allergenic foods, apart from shrimp, is sufficient to cause a reaction in around 10% of the food allergic population.3–6 Concerns over the hazard posed by such traces have led to the use of precautionary labelling (PAL) to warn allergic consumers of the potential presence of allergens.7 However, recent surveys have indicated that PAL gives no clear indication of likely presence of allergen.8 This work relied on immunoassay test kits to provide data on the presence and quantity of allergens but a recent ring trial for analysis of egg and milk showed significant short comings in the technology with regards quantification4,9 Although assay sensitivities as low as 1 ppm have been reported, accurate quantification at such low levels remains a challenge.10 Food processing adversely affects detection of allergens partly by reducing their solubility in the simple buffers compatible with immunoassays, resulting in under-estimation of allergen content and false negative test results.11–14 The changes in three-dimensional structure induced by processing can also reduce recognition by the antibody preparations used in immunoassays, leading to falsely declared allergen-free foods.4,14 Such effects can be compounded by covalent modifications, notably sugars, through the formation of Maillard reaction products. Mass spectrometry (MS) based methods have the potential to address these short comings, providing an alternative and complementary approach to immunological assays.9,12 The technique can be compatible with extraction methods that employ denaturants, detergents, and sonication, to aid recovery from processed food products and therefore improve quantification.13,15 However, the inclusion of detergents can necessitate additional clean-up steps making methods more difficult to use in routine analysis and Maillard adducts can reduce their susceptibility to the proteolytic processes employed in proteomic workflows.16,17One of the most prevalent allergies in the UK is peanut, which is estimated to affect around 2% of children and accidental consumption has been known to cause fatalities.18–21 Of the 17 peanut allergens listed in the IUIS allergen database the most clinically important are the 2S albumins Ara h2 and 6, which belong to the prolamin superfamily.22–24 They comprise a conserved pattern of cysteine residues and adopt a compact α-helical bundle disulphide-bonded structure which makes them stable to thermal treatment and digestion. IgE responses towards these proteins are an important marker of clinical reactivity to peanut. The other clinically important allergens belong to the cupin superfamily, and comprise the seed storage globulins Ara h1, a 7S vicilin-type globulin and Ara h3, an 11S legumin-type globulin. These proteins share a common stable β-barrel structure and form large aggregates on heating.
Since these proteins represent the main allergenic hazard in peanuts, they are the target analytes of choice for any MS-based method. Peptides selected from these allergens need to be peanut-specific and resistant to food processing induced modifications. This is because peanuts are rarely consumed raw, usually undergoing some form of thermal processing most commonly roasting or frying, whilst boiling may reduce allergenicity of peanuts.25 Several targeted approaches using liquid chromatogram electrospray ionisation (LC/ESI) MS/MS methods with triple quadrapole mass analyser for determination of peanut have been described with limits of detection of 4–10 ppm of peanut in simple food matrices.26–28 However to date, there has been no systematic study on the impact of food processing on either the efficiency of MS-compatible extraction procedures, or on peptide targets used in MS methods. Therefore, we have investigated the effect of boiling roasting and frying on the extraction of proteins from peanuts and their allergen profiles determined by targeted MS analysis using stable isotope dilution multiple-reaction monitoring (SID-MRM) MS.
Materials and methods
Materials
All reagents and chemicals were of analytical grade unless otherwise stated. HPLC grade acetonitrile and water, urea, thiourea and tris(hydroxymethyl)aminomethane, were purchased from Fisher Scientific UK Ltd (Loughborough, UK). Iodoacetamide, dithiothreitol (DTT), proteomic-grade trypsin, formic acid and 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonate (CHAPS) were purchased from Sigma (Poole, Dorset, UK). RapiGest™ (Sodium 3-[(2-methyl-2-undecyl-1,3-dioxolan-4-yl)methoxy]-1-propanesulfonate) was the gift of Dr Lee Gethings (Waters Corporation, Wilmslow, UK). 2D Quant-Kit™ was obtained from GE Healthcare (Buckinghamshire, UK). Isotopologues of the target peptide sequences were synthesised with either 13C(6)15N(4) C-term R or 13(C)15N(2) C-term K by JPT Peptide Technologies GmbH (Berlin, Germany). Peptides were produced with trifluroacetic acid as a counter ion, target mass confirmed by LC-MS and purity confirmed as >95% by HPLC.Peanut samples
Peanuts (var. Runner) were the kind gift of Dr Sue O'Hagan (PepsiCo, Leicester, UK) and included raw samples with skins intact (raw 1), mechanically blanched (raw 2) and oil-fried (152 °C for 400 seconds) peanuts. Lightly roasted mechanically defatted peanut flour manufactured by the Golden Peanut Company (GPC), LLC, Alpharetta, GA 30022, USA was obtained from Byrd Mill (Byrd Mill Co., Ashland, VA 23005, USA). Peanuts (raw 1) were peeled and 10 g batches either boiled (0.5, 1, 2, 4 or 6 h, under reflux) or roasted in a domestic fan-assisted oven (15 min at 150, 160, 170 or 180 °C). Peanuts and peanut products were ground using a pestle and mortar and defatted by addition of hexane (1:10, w/v), once for 3 h and then repeated overnight. The total protein content of defatted samples was determined by Kjeldahl using the Dumas combustion method and a Nitrogen conversion factor of 5.46.29
Protein extraction
Protein extractability was assessed using five different buffers:(1) 0.01 M phosphate buffered saline (PBS), pH 7.4;
(2) 50 mM Tris-HCl, pH 8.8;
(3) 50 mM Tris-HCl, pH 8.8 containing 50 mM DTT;
(4) 50 mM Tris-HCl, pH 8.8 containing 50 mM DTT with 0.04% (w/v) RapiGest™;
(5) Chaotropic zwitterionic buffer, pH 8.8 (7 M urea, 2 M thiourea, 2% (w/v) CHAPS, 1% (w/v) DTT).
Peanut samples were extracted in triplicate using either 20 mg of sample in 1 ml of buffers (1–3) and (5), or 5 mg in 250 μl of buffer (4). Extractions were performed either using a Stuart SB3 rotator (Bibby Scientific Ltd, Staffordshire, UK) for 1 h at 19–21 °C for buffers (1) and (2), or for 15 min at 60 °C in an ultrasonic bath for buffers (3)–(5). Samples were clarified by centrifugation (10000g, 20 °C, 10 min), the supernatants removed and stored at −20 °C until required. Extracted protein was quantified using a 2D Quant-Kit™ (GE Healthcare Life Sciences, Buckinghamshire, UK), which can determine protein in the presence of interfering substances such as detergents and reducing agents, using bovine serum albumin (BSA) as the standard. The extraction efficiency was calculated as the amount of extracted protein relative to the total protein in the defatted peanut samples.
Sample preparation for MS analyses
Extracts for untargeted MS analysis were prepared using buffer (5). The extracted protein was then precipitated using precipitants in 2D Quant-Kit™ and re-solubilised in 50 mM Tris-HCl, pH 8.8 containing 50 mM DTT and 0.04% (w/v) RapiGest™. Extracts for targeted MS analysis were prepared using buffer (4). Samples were then diluted to 1 mg protein per mL with 50 mM ammonium bicarbonate prior to addition of fresh 50 mM DTT to give a final concentration of 5 mM DTT and heated to 80 °C for 10 min. Proteins were then alkylated by addition of 150 mM idoacetamide to give a final concentration of 15 mM and incubated for 30 min in the dark at (19–21 °C). Subsequently proteins were digested by addition of trypsin (0.1 mg mL−1, 1:10 (w/w)) (≥10000 benzoyl arginine ethyl ester (BAEE) units per mg protein) for 3 h at 37 °C and after a second addition of trypsin (0.1 mg mL−1) and incubated for 15 h at 31 °C. The resulting digests were passed through spin column (Pierce™ C-18 spin columns, Thermo Fisher Scientific, UK.), and allowed to dry by air before being re-suspended buffer a (0.1% (v/v) formic acid in HPLC grade water), and stored at −20 °C.
Untargeted MS analysis
Untargeted data acquisition was carried out by the Biological Mass Spectrometry Facility of the University of Manchester. Briefly two samples of raw peanut were separated using a gradient from 92% A (0.1% (v/v) formic acid in HPLC grade water) and 8% B (0.1% (v/v) formic acid in HPLC grade acetonitrile) to 33% B, in 44 min at 300 nL min−1, using a 250 μm × 75 mm i.d. 1.7 μM BEH C18, analytical column (Waters). Samples were analysed in singlet by LC-MS/MS using an UltiMate® 3000 Rapid Separation LC (RSLC, Dionex Corporation, Sunnyvale, CA) coupled to an Orbitrap Elite (Thermo Fisher Scientific, Waltham, MA) mass spectrometer (tune parameters in Table S-1†). Peptides were selected for fragmentation automatically by data dependant analysis; +2 or +3 precursor ions and previously observed ions were excluded from fragmentation for a 30 minute period. Raw data was analysed using PEAKS™ version_6 (Bioinformatics Solutions Inc., Waterloo, Ontario, Canada), and protein identification performed using a database of curated peanut sequences from UniProt (08.07.2015).Targeted MRM experiments
| Protein family | Allergen | UniProt ID | Subunit Mr (kDa) | Peptide target residues | Peptide target sequence | Peptide target name | Previous identification | |
|---|---|---|---|---|---|---|---|---|
| Cupins | 7S vicillin-globulin | Ara h1 | P43237 | 61.72 | 329–342 | VLLEENAGGEQEER | Ara h1(P43237)329–342 | 29, 32, 33 |
| 555–577 | DLAFPGSGEQVEK | Ara h1(P43237)555–577 | 29, 32, 34, 35 | |||||
| 11S legumin-globulin | Ara h3 | Q647H4 | 59.64 | 25–41 | QQPEENACQFQR | Ara h3(Q647H4)25–41 | 32 | |
| 372–384 | SPDIYNPQAGSLK | Ara h3(Q647H4)372–384 | 27, 32, 34, 36–39, 40 | |||||
| Prolamins | 2S albumins | Ara h2 | Q6PSU2 | 17.99 | 103–115 | CCNELNEFENNQR | Ara h2(Q6PSU2)103–115 | 14, 32, 36,40 |
| 147–155 | NLPQQCGLR | Ara h2(Q6PSU2)147–155 | 32,40 | |||||
| Ara h6 | Q647G9 | 14.85 | 136–144 | CDLDVSGGR | Ara h6(Q647G9)136–144 | None | ||
| Ara h7 | B4XID4 | 17.38 | 143–151 | NLPQNCGFR | Ara h7(B4XID4)143–151 | None | ||
A stock solution, containing 1 mM of each labelled peptide, was used to prepare serial dilutions from 100 and 33.3 nM, 1:10 (v/v) in 0.1% (v/v) formic acid in HPLC grade-water and analysed using 6 replicate injections. A second series of dilutions was prepared in a reduced, alkylated and trypsin digested peanut protein extract (1:10 (v/v) roasted peanut flour protein extract in 0.1% (v/v) formic acid in HPLC grade-water) and analysed using 3 replicate injections. Peanut samples were diluted 1:20 (v/v) in 0.1% (v/v) formic acid in HPLC grade-water and spiked with stable isotope mix to give a final concentration of 5 nM, equivalent to 15 fmoles of each peptide loaded onto the column.
Samples were randomised and analysed using 3 replicate injections with a blank sample included every 5 injections. Every tenth samples was an extract prepared from the lightly roasted mechanically defatted peanut flour and was included as a quality control sample to check instrument base-line variability. Samples were analysed on a Waters nanoACQUITY® Ultra Performance LC™ (Waters, Milford, MA, USA) interfaced to a Thermo Scientific TSQ Vantage triple stage quadrupole Mass Spectrometer (Thermo Fisher Scientific, Warrington, UK). Three microliters of each sample was loaded onto an analytical reverse-phase C18 column (75 μm × 15 cm) with 1.8 μm ACQUITY UPLC HSS (High Strength Silica) T3 particle packing (Waters, Milford, MA, USA) heated to 40 °C and equilibrated in 98% 0.1% formic acid in HPLC-grade water (buffer A) and 2% 0.1% formic acid in acetonitrile (buffer B). At a flow rate of 250 nL−1, the gradient was ramped from 2 to 23% (v/v) B in 5 min and then to 45% (v/v) B in 30 min followed by column rise at 90% (v/v) B. The mass spectrometer was operated in positive ESI mode using a NSI ion source, peak width set to 0.70 FWHM in Q1 and Q3 with auto selected dwell time <0.05 ms. over a spectral acquisition time of 35 min. Targeted analysis was performed using the list of predetermined transitions and collision energies generated by Skyline.
Data analysis
:1. The LLOD required a minimum of 2 transitions to be visible above the noise for reliable detection. Using the linear regression analyses performed on SID in extract, limits of detection (LOD) and quantification (LOQ) were calculated using the calibration plot method (eqn (S-1)†).51
Results and discussion
Effect of thermal processing on protein solubility
The defatted raw peanut samples comprised 43–45% (w/w) wet weight protein (Table S-3†) with the mechanically blanched peanuts (raw 2) containing slightly less protein than the hand peeled peanuts (raw 1) although both were within the range normally expected for peanuts. Boiling reduced the protein content to about 25% (w/w) as a consequence of progressive leaching into the cooking water, and increased the moisture content of the seeds. In contrast moisture was lost during roasting causing a slight increase in protein content at temperatures above 160 °C. It is also possible that extensive boiling and roasting conditions chemically modified the protein and affected the nitrogen determination further contributing to these changes.Protein extractability from the raw peanut samples was broadly similar, irrespective of the buffer and extraction conditions employed (Fig. 1 and Table S-3†). However, processing reduced protein extractability using simple buffers and gentle extraction conditions (buffers 1 and 2). Buffer 1 was inefficient at solubilising protein from processed peanuts, extracting only 1–14% of the protein from all samples. Whilst protein extraction by buffer 2 was less affected by processing, it was still reduced by ∼50% after 30 min boiling, and by an additional ∼25% after 1 h boiling. After this time no further reduction in extractability was observed even after 6 h boiling. Extraction by buffer 2 was also reduced by ∼50% following roasting at 150 °C, and decreased progressively with roasting temperature to only 13% at 180 °C. Protein extractability from the roasted peanut flour and fried peanuts was similar, with ∼50% of the total protein being extracted by buffer 2. The addition of reducing agents to buffer 2 and use of sonication at elevated temperatures (buffer 3) did not generally enhance protein extractability but had the advantage of reducing extraction time. The addition of an acid labile anionic detergent, RapiGest™, (buffer 4) greatly improved the extractability of the protein from the severely heat treated peanut samples although it was still reduced to less than 50% for the extensively boiled peanuts and those roasted at 180 °C. The chaotropic buffer (buffer 5) was the most efficient buffer and whilst protein extractability was reduced to 75% after 1 h boiling it then increased to >100% after prolonged boiling times. In contrast, protein extractability with buffer 5 was reduced as a function of roasting temperature to ∼50% after roasting at 180 °C for 15 min. The over estimation in protein observed in certain samples is likely to be a result of using a mammalian protein, BSA, as a standard. BSA has a significantly different amino acid composition to plant proteins and will give a different response in the copper reaction that is used in the 2D Quant-Kit™. This maybe further compounded by thermally induced modifications to the peanut proteins affecting responsiveness in the assay.
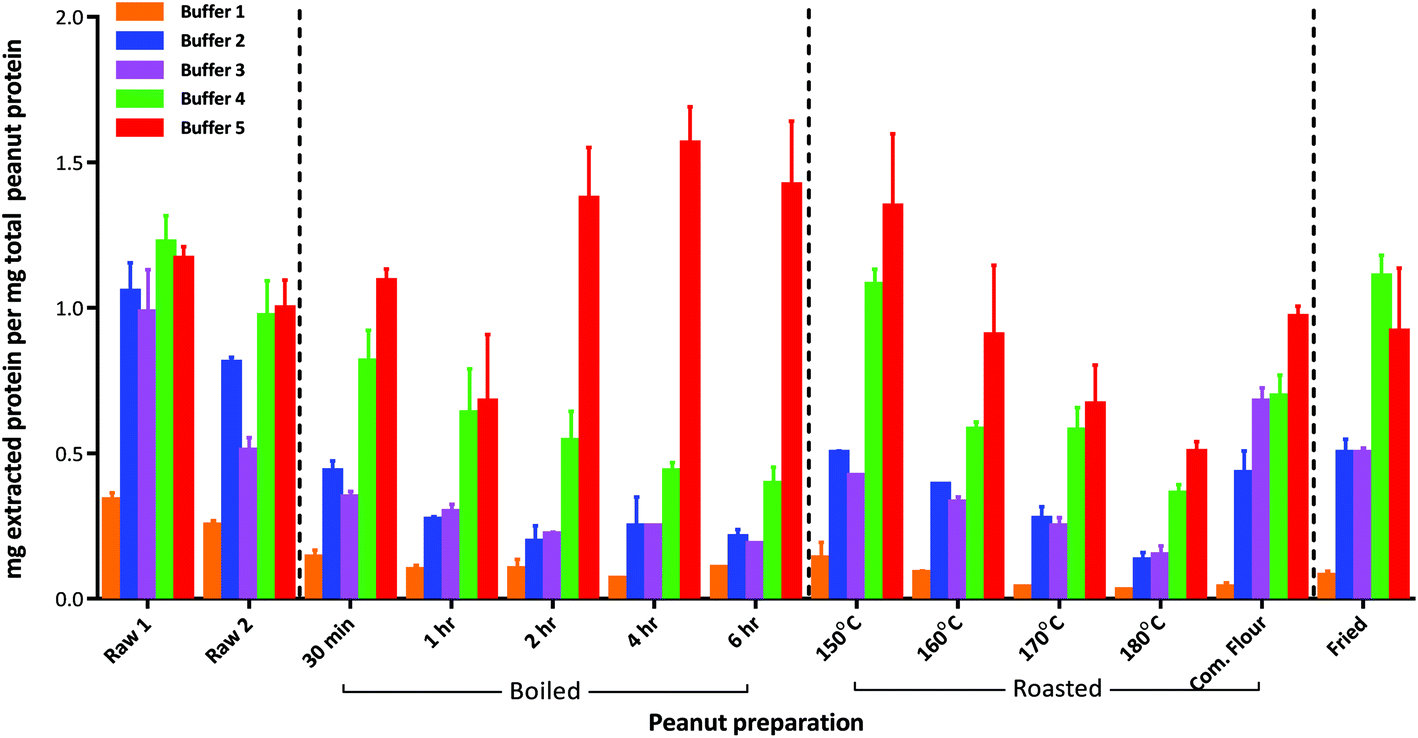 | ||
| Fig. 1 Effect of thermal processing conditions on extractability of peanut proteins using five different buffer conditions. The extraction efficiency was calculated as the amount of protein extracted (mg) determined by the 2D Quant-Kit™ divided by the amount of protein present in the defatted peanut flour (mg) calculated from the protein Nitrogen content determined using the Kjeldahl method. Results are displayed as mean value and error bars corresponding to the SD over 3 replicate samples. Buffers were as follows: (1) 0.01 M Phosphate Buffered Saline (PBS), pH 7.4; (2) 50 mM Tris-HCl, pH 8.8; (3) 50 mM Tris-HCl, pH 8.8, 50 mM DTT; (4) 50 mM Tris-HCl, pH 8.8, 50 mM DTT with 0.04% (w/v) RapiGest™; (5) Chaotropic zwitterionic buffer, pH 8.8 (7 M urea, 2 M thiourea, 2% (w/v) CHAPS, 1% (w/v) DTT). Com. Flour – commercially available lightly roasted mechanically defatted peanut flour. | ||
An overall comparison of protein extractability using the five different buffer conditions (Fig. 2) showed that the use of a shorter extraction time (15 min) combined with the use of sonication and inclusion of a reducing agent (buffer 3) gave equivalent protein extraction to a more gentle extraction undertaken over 1 h (buffer 2). The addition of an acid labile detergent (buffer 4) enhanced extraction and was found to be statistically significant using a 2-way ANOVA with Sidak's multiple comparison test, for all except the raw samples and the lightly roasted mechanically defatted commercial flour. Extraction of raw peanuts was optimal without the need for addition of reducing agents, detergent or chaotropes. The lack of effect on protein extractability from the roasted peanut flour when an acid labile detergent was included may be due to its fine particle size. The most effective buffer for solubilising highly processed samples was the chaotropic buffer 5. However, as this is not compatible with MS analysis and in order to minimise clean-up steps, which may introduce additional errors in quantitation, buffer 4 was chosen for targeted MS analysis.
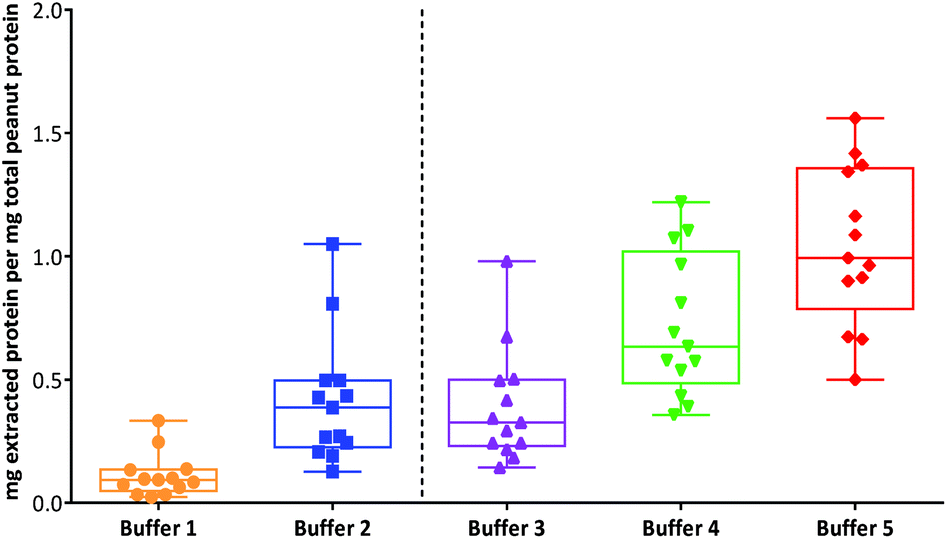 | ||
| Fig. 2 Box-whisper plots showing the comparison of different buffers for extraction of protein from thermally processed peanuts. The box represents the 25th to 75th percentiles with whiskers extending from the smallest to largest values and a line at the median. All extractions were performed in triplicate. Buffers were as follows: (1) 0.01 M Phosphate Buffered Saline (PBS), pH 7.4; (2) 50 mM Tris-HCl, pH 8.8; (3) 50 mM Tris-HCl, pH 8.8, 50 mM DTT; (4) 50 mM Tris-HCl, pH 8.8, 50 mM DTT with 0.04% (w/v) RapiGest™; (5) chaotropic zwitterionic buffer, pH 8.8 (7 M urea, 2 M thiourea, 2% (w/v) CHAPS, 1% (w/v) DTT). | ||
Peptide target selection and verification
A suite of eight peptide targets was identified from the major allergen families in peanut using an informatics pipeline (Fig. S-1†). A curated list of non-redundant peanut allergen sequences from Ara h1, 3, 2, 6 and 7 was first interrogated using CONSeQuence, a machine learning algorithm which takes account of factors such as charge and hydrophobicity.41 A second analysis of peptide secondary structure and candidate peptides was performed using MCPRED, an algorithm that assesses the likelihood of having missed cleavages.42 The list of candidates was then assessed for uniqueness by undertaking BLAST searching against UNIPROT. Peptide candidates longer than 20 residues were excluded. Peptides containing methionine or tryptophan were avoided and preference was given to those flanked by arginine residues rather than lysine in an effort to minimise processing-induced modifications. The final candidate list was then assessed using data from untargeted MS analysis of raw peanut samples to ensure the presence and abundance of peptides in raw peanut samples (Table S-4†). Six of the peptide targets selected for the main peanut allergens Ara h1, 2 and 3 had previously been identified as MRM candidates with two additional peptides identified from prolamin proteins Ara h6 and 7 (Table 1 and Fig. S-2†). Peptides were mapped on to the 3D structures available for the cupin allergens, Ara h1, and Ara h3 together with the prolamin superfamily allergen Ara h6.Ara h1: when synthesised in the seed Ara h1 contains a precursor which is then post translationally cleaved generating ragged N-termini, two of which dominate and are arrowed in Fig. 3A.16 Ara h1 exists as a homotrimer, each monomer held together by hydrophobic interactions and usually glycosylated.43 Within each monomer the N-terminal and C-terminal modules both contain a β-barrel core domain. The target peptides are located on opposite extended loop domains at the monomer–monomer contact during trimer formation (Fig. 3B). This is also the strategic location for IgE epitopes as it may provide some protection from digestion.44 Ara h1(P43237)329–342 is known to overlap an immunologically active epitope EQEERGQRRW and Ara h1(P43237)555–577 overlaps two different immunologically active epitopes IDQIEKQAKD and KDLAFPGSGE.45 Ara h1(P43237)329–342 has been cited in the literature and is flanked by arginine residues making it less susceptible to Maillard modifications.31,32 A second peptide Ara h1(P43237)329–342, has previously been identified as a peptide target.26,28,31,33,34 This peptide is flanked by lysine residues and is therefore less robust to processing-induced modifications. Mueller et al. detected the Maillard reaction products pyrraline and carboxymethyllysine (CML). Ara h1(P43237)329–342 is located in a flexible region of the protein which was not fully resolved in the crystal structure.
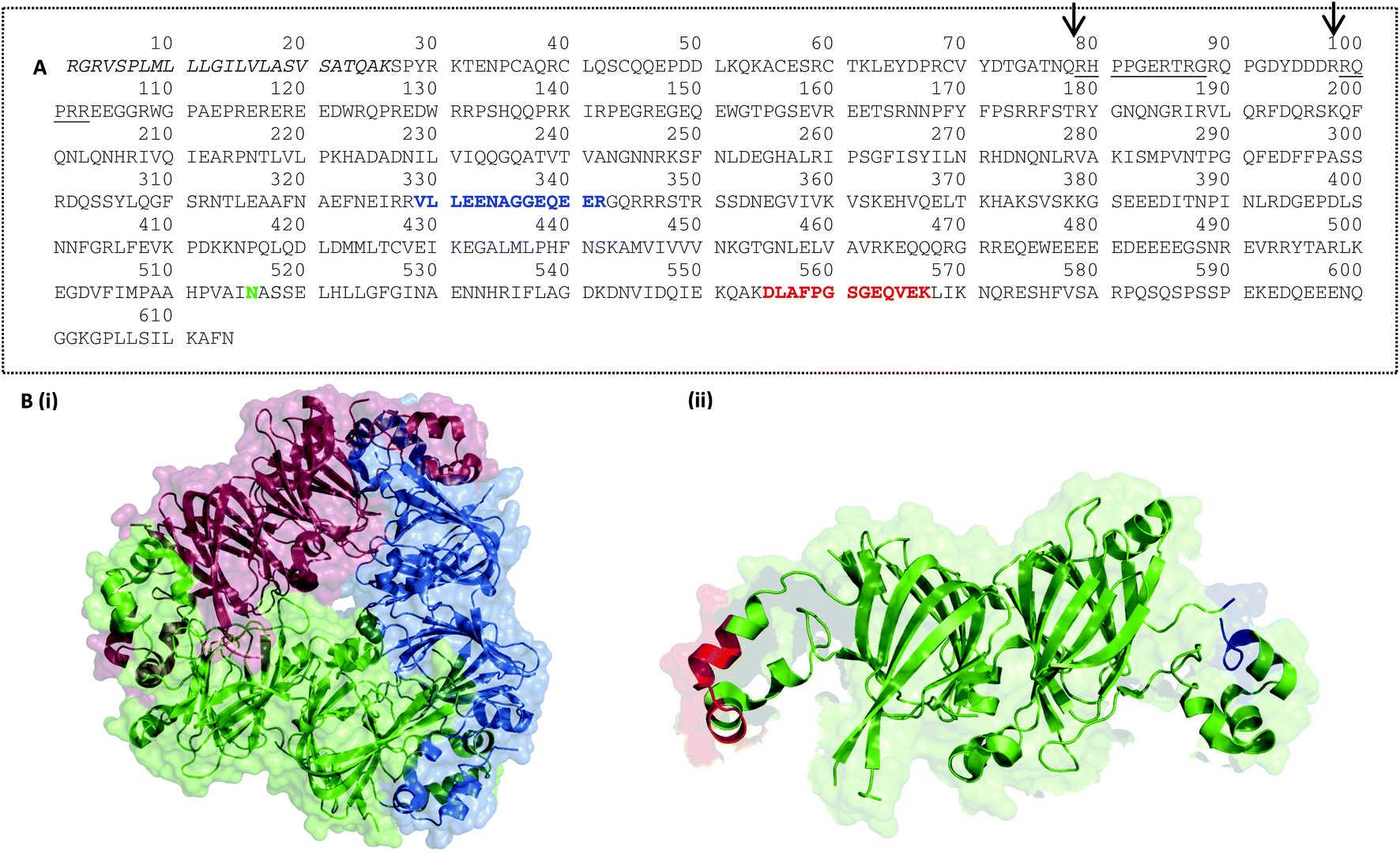 | ||
| Fig. 3 Peptide targets for Ara h1 were mapped on to the protein sequence (A) and its 3D structure (B). (A) Ara h1 isoform sequence P43237 (UniProt) is shown with the signal peptide sequence in italics, possible mature N-termini underlined and glycosylation site in green. Target peptides Ara h1(P43237)329–342 and Ara h1(P43237)555–577 are shown in red and blue respectively (B) 3D structure of Ara h1 showing (i) Ara h1 trimer (DOI: 10.2210/pdb3smh/pdb), coloured by monomer and (ii) Ara h1 monomer with target peptides Ara h1(P43237)329–342 and Ara h1(P43237)555–577 marked in red and blue respectively. Cartoons show van der Waal's space-filling at 60 or 80% transparency and cartoon structures modelled using the PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC. | ||
Ara h3: Ara h3 is a multigenic protein, which undergoes posttranslational processing to yield an acidic a basic subunit covalently linked by an intermolecular disulphide bond forming each monomer (Fig. S-4†).46 The monomers assemble into heterotrimers, which in the mature protein associate to form a hexamer.47 Peptide Ara h3(Q647H4)25–41 has only been mentioned previously by Chassaigne et al. but was chosen as a representative peptide from the acidic subunit and is flanked by arginine residues. The second peptide Ara h3(Q647H4)372–384 has been published in several literary sources but has an N-terminal lysine that is more likely to become modified.13,26,35–39 Hebling et al. have previously identified Maillard modifications of lysine, CML and pyrraline, in the related peptide SPDIYNPQAGSLKTANDLNLLILR.
Ara h2, 6 and 7: the 2S-albumins allergens Ara h2 and 6 share a high degree of sequence homology (59%) and have been shown to be potent allergens.48,49 These allergens share a common 3D structure cross-linked by intramolecular disulphide bonds which cause them to be both thermally stable and resistant to digestion. The 3D structure has only been determined for Ara h6 (Fig. S-5†) and shows the target Ara h6 peptide sequence, which is flanked by arginine residues and lies on flexible loop regions of the protein, outside the protease resistant core. Both Ara h2 peptide targets Ara h2(Q6PSU2)103–115 and Ara h2(Q6PSU2)147–155 have been previously described as peptide targets.13,31,35,38,39,50 Ara h6(Q647G9)136–144 and Ara h7(B4XID4)143–151 are both novel peptide targets and are flanked by arginine residues.
Development of multiple-reaction monitoring (MRM) experiments for peanut allergens
A 12 point SID series of the synthesised AQUA peptides was used to generate standard curves for each target in either buffer or in reduced, alkylated and digested roasted peanut flour extract (buffer 4) in order to assess matrix interference. Transitions are listed in Table S-2.† Peak areas corresponding to the total ion intensities, for 3 transitions, were used when constructing calibration curves and calculating LOD and LOQ values. The ratio between the 3 monitored transitions remained constant across the processed samples with <20% variation. Concentration values were adjusted using the theoretical peptide content calculated by adjusting the dry weight to account for the contribution of counter-ions and calculating the percentage purity of target in the peptide content from the peak area in the HPLC trace. Corrected stock solutions were between 0.77–0.86 mM.Cupin allergens
Ara h1: the contribution of each monitored MRM transition to the total ion intensity was assessed for each of the peptide targets using a mid-range concentration. Extracted ion chromatograms (XIC) are displayed with fragmentation patterns in Fig. S-6i-ii.† The signal intensity for all transitions associated with Ara h1 targets were strong, and although fragmentation of Ara h1(P43237)555–577 is directed towards formation of a N-terminal proline y9 ion improving the signal of this product ion, others responded more equivalently.28 Comparison of the ion chromatograms for Ara h1(P43237)329–342 and Ara h1(P43237)555–577 at the same concentration showed a 6-fold difference in the signal intensity most likely due to variations in ionisation efficiency. The SID series evaluated in peanut extract demonstrated a decrease in the analytical response compared to SID performed in buffer (Fig. 4(ii)). Using log-transformed data a clear response plateau was observed at levels where peptides were detected but not quantifiable as they were outside of the linear range of the assay.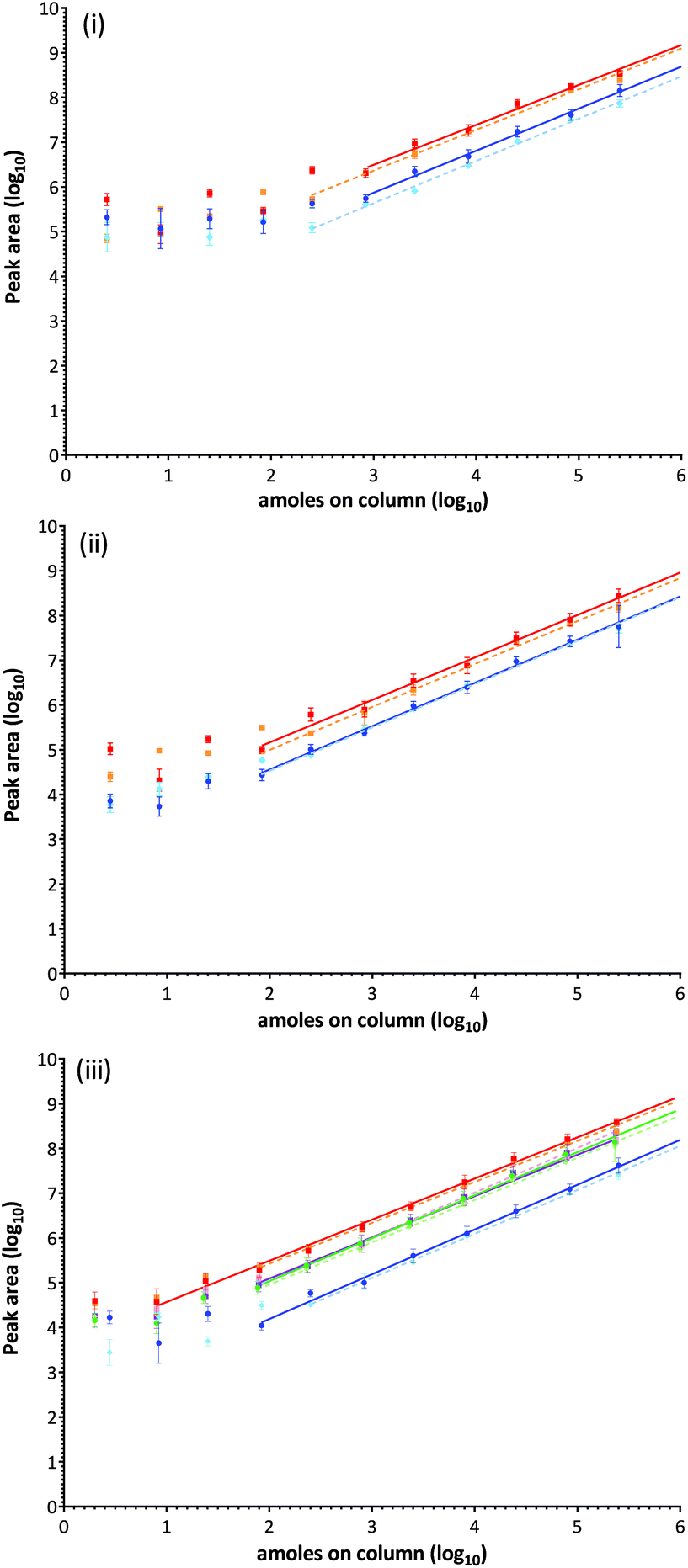 | ||
| Fig. 4 Calibration curves for each isotopically labelled peptide target in buffer (solid line) and in a peanut extract (dashed line) constructed from log-transformed data. (i) Ara h1(P43237)329–342 in blue/light blue and Ara h1(P43237)555–577 in red/orange. (ii) Ara h3(Q647H4)25–41 in blue/light blue and Ara h3(Q647H4)372–384 in red/orange. (iii) Ara h2(Q6PSU2)103–115 in blue/light blue, Ara h2(Q6PSU2)147–155 in red/orange, Ara h6(Q647G9)136–144 in green/light green and Ara h7(B4XID4)143–151 in purple/pink. | ||
Ara h3: XIC's and fragmentation patterns are shown in Fig. S-6iii-iv.† The SID calibration curves performed in buffer and matrix are presented in Fig. 4(ii). The y10 ion fragment was highest for peptide Ara h3(Q647H4)25–41, with the y7 and y6 ions being readily identified. The y7, y8 and y9 ions were readily detected in peptide Ara h3(Q647H4)372–384, the y7 ion being the most intense.
Ara h2, 6 and 7: XIC's and fragmentation patterns are shown in Fig. S-6v-viii.† The SID calibration curves performed in buffer and matrix are presented in Fig. 4(iii). All transitions monitored for Ara h2(Q6PSU2)103–115 were comparable but less intense than Ara h2(Q6PSU2)147–155 in which the y7 fragment ion was most abundant. Ara h6Q647G9136–144 and Ara h7(B4XID4)143–151 both have a least 2 good transitions. Ara h7(B4XID4)143–151 was dominated by y7 ion. The Ara h7 peptide was the only target where an improved response in matrix over buffer was observed.
Statistical analysis using a pairwise t-test showed no significance between SID performed in buffer and matrix for any of the target peptides. Differences between resulting linear regression analyses for the same target peptide were therefore the result of instrument variation over the course of the experiments. The LLOD was evaluated by visual inspection of the XIC's (Table 2). The majority of peptides were reproducibly detected between 1–30 amoles on column, except Ara h1(P43237)329–342 which was detected at the lowest concentration injected (0.1 amoles on the column). At these levels peanut could be confidently identified but not accurately quantified. Accurate detection and quantification was defined by the linear dynamic range of the given peptide and was typically achieved over 4–5 orders of magnitude. Calculated LOD's were determined in the range of 32–448 amoles and LOQ's between 96–1343 amoles on column.51 Calculated LOD's were much higher than LLOD values for the cupin allergens but were similar for the 2S-albumins targets. As expected LOQ's were lower for the 2S albumin peptides from Ara h2, 6 and 7, with Ara h2(Q6PSU2)147–155, Ara h6(Q647G9)136–144 and Ara h7(B4XID4)143–151 performing especially well with LOQ's of 96–126.89 amoles on column.
:1 for minimum of 2 transitions
| Target peptide | Linear regression analysis | Assay sensitivities (amoles on column) | |||||
|---|---|---|---|---|---|---|---|
| r 2 | Line equation | Sy|x | y (log10) | LOD | LOQ | LLOD | |
| Ara h1(P43237)329–342 | 0.9931 | Y = 0.9460 × X + 2.797 | 0.08 | 5.09 | 248.38 | 745.15 | 0.1 |
| Ara h1(P43237)555–577 | 0.9863 | Y = 0.9098 × X + 3.636 | 0.11 | 5.73 | 125.93 | 377.80 | 1 |
| Ara h3(Q647H4)25–41 | 0.9938 | Y = 0.9705 × X + 2.587 | 0.09 | 4.40 | 104.26 | 312.79 | 30 |
| Ara h3(Q647H4)372–384 | 0.9902 | Y = 0.9604 × X + 3.075 | 0.11 | 5.50 | 447.71 | 1343.13 | 1 |
| Ara h2(Q6PSU2)103–115 | 0.9925 | Y = 0.9829 × X + 2.160 | 0.09 | 4.24 | 208.07 | 624.21 | 10 |
| Ara h2(Q6PSU2)147–155 | 0.9935 | Y = 0.9143 × X + 3.596 | 0.09 | 5.15 | 32.10 | 96.29 | 30 |
| Ara h6(Q647G9)136–144 | 0.9953 | Y = 0.9456 × X + 3.067 | 0.08 | 4.63 | 42.30 | 126.89 | 30 |
| Ara h7(B4XID4)143–151 | 0.9939 | Y = 0.9225 × X + 3.246 | 0.09 | 4.84 | 40.04 | 120.11 | 30 |
Effect of thermal processing on peptide target quantification
To overcome fluctuations in signal intensity, quantification was performed on normalised data corresponding to the total ion intensity of all monitored transitions. Results are presented as nmoles peptide per mg of extracted protein (Fig. 5). These were converted to protein amount (mg) for each allergenic protein using molecular weights for mature protein sequences (Fig. S-7†). Values appear to be underestimated by a ∼10-fold, since Ara h1 and Ara h3 together account for around 50% of peanut seed proteins. This most likely due to loss of peptides following the C-18 spin column clean-up which has previously been shown to reduce signal intensity by 50–90%.38 After clean-up solvent was allowed to evaporate and peptides were re-suspended in buffer. During this process further peptide could be lost if it was not fully re-solubilised. The contribution from each of the 3 monitored MRM transitions for each target remained consistent independent of thermal processing. The majority of CV's relating to the normalised peak area intensities were <10% with less than 13% of results having a CV between 10–20%. Variation in protein extraction was responsible for the range in samples observed (Fig. S-7†).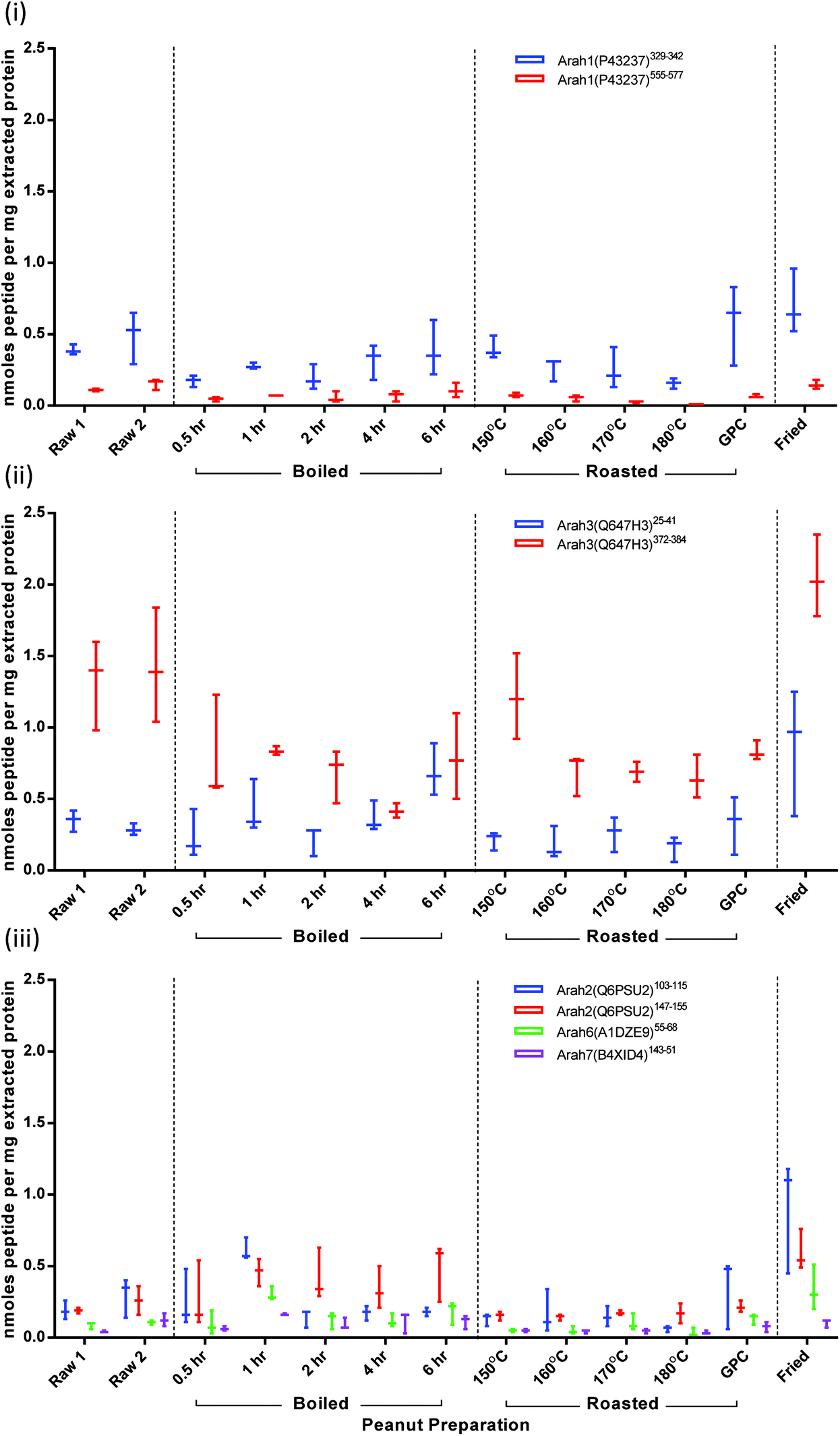 | ||
| Fig. 5 Effect of thermal processing on quantification of peanut allergen peptide targets. Results are displayed according to protein family; Cupin superfamily (i) 7S-vicillin type protein Ara h1, (ii) 11S-legumin type protein Ara h3 and prolamin superfamily (iii) 2S ablumins Ara h2, 6, 7. Results are expressed as nmoles of peptide per mg of extracted protein. | ||
Ara h1: peptide Ara h1(P43237)329–342 was present at double the intensity of peptide Ara h1(P43237)555–577 in both raw and processed samples (Fig. 5(i)). The difference in the abundance of these peptide targets could be due to differences in the corresponding ionisation efficiencies or fragmentation patterns. Ara h1(P43237)555–577 is flanked by lysine residues which are less efficiently cleaved by trypsin than arginine.52 Such effects may be compounded the formation of modified amino acids as a consequence of thermal treatment as lysine is susceptible to processing-induced modifications. It has been reported that this sequence frequently carries a Maillard modification at the C-terminal K.34 Modified peptides would not be detected using the current MRM targets. The roasting series shows how such a processing modification could explain the reduction in abundance, although this reduction is not statistically significant compared to raw. It is evident that the Ara h1 comprises a smaller proportion of the extracted protein from all the boiled and roasted samples but was represented in the extracts of the roasted peanut flour and fried peanuts at a level equivalent to the raw peanuts (Fig. S-7†).
Ara h3: the basic subunit peptide Ara h3(Q647H3)372–384 was reported around 2–5-fold higher than the acidic subunit peptide Ara h3(Q647H3)25–41 in the raw peanut samples (Fig. 5(ii)). As peptide Ara h3(Q647H4)25–41 is located close to the N-terminus of the acidic subunit it may be susceptible to missed cleavages and hence give lower than expected responses.53 Furthermore these peptides are not representative of the acidic or basic subunits of all Ara h3 isoforms. For example, neither target represents isoforms corresponding to Ara h3.02 (formerly Ara h4).54 Peptide abundance appeared to be reduced as a function of processing time or temperature. This could be the result of the formation of Maillard reaction products during, for example, roasting. Ara h3(Q647H3)372–384 was more susceptible to processing induced effects and was reduced in all the thermally processed samples apart from the roasted peanut flour. This was found to be statistically significant by 2-way ANOVA when comparing target levels in raw peanuts compared to all processed samples, except the sample roasted at 150 °C. Boiling also gave statistically significant differences when compared to raw for peptide Ara h3(Q647H3)372–384. The intramolecular disulphide bond which links acidic and basic subunits in Ara h3 is thought to be metastable and readily hydrolyses during boiling. The resulting separated basic subunit may have the propensity to aggregate and may not re-solubilise effectively during the extraction procedure, affecting recoveries of peptide target. Whilst Ara h3(Q647H3)372–384 could be a good marker of processing procedures, it may not be reliable for allergen quantification in processed foods. The Ara h3(Q647H3)25–41 peptide was less affected by thermal processing such that in the 6 h boiled samples levels were equivalent to those of the Ara h3(Q647H3)372–384 peptide.
Ara h2: peptides from Ara h2, Ara h2(Q6PSU2)103–115 and Ara h2(Q6PSU2)147–155, behaved quite similarly and were often found in equivalent amounts in all samples analysed, apart from peanuts boiled for ≥2 h (Fig. 5(iii)). In those samples Ara h2(Q6PSU2)147–155 appeared to be less affected than Ara h2(Q6PSU2)103–115. Reduction of ion abundances in roasted compared to raw or boiled could be partially explained by associated function as a trypsin inhibitor which is reported to increase during roasting.55
Ara h6 and Ara h7: Ara h6(Q647G9)136–144 and Ara h7(B4XID4)143–151 were present in similar amounts in all samples analysed (Fig. 5(iii)).
If one considers the relationship in molar terms the 2S-albumin fraction was found to be present in equivalent amounts to Ara h1, which may be a reflection of its greater solubility. This may help to explain the clinical significance of allergens belonging to 2S-albumins such as Ara h2. Allergens that retain their solubility following processing will have greater availability for interaction with IgE, contributing to their higher allergenic potential.
Conclusions
Any analytical method for determining the presence of allergens in foods requires an effective extraction methodology. The inefficiency of simple salt buffers to extract processed protein underlies, in part, the lack of reliability of immunological assays for quantification.11 The recovery of highly processed protein is greatly improved using MS compatible buffers but for more complete extraction the use of harsh denaturing conditions employing chaotropic buffers is required56,57 However, the addition of sample clean-up step to remove any interfering contaminants may result in sample loss through binding to the plastics and stationary phase of the column.58 Further studies are required to assess whether the inclusion of acid labile detergents at higher levels (such as 2% (w/w)) and the use of spin filters can further improve extraction efficiency and sample processing.56,57 It was evident that variations in sample extraction adversely affected the reproducibility of the MRM analysis. This might be improved by increasing sample size used for extraction and would in any case, be a requirement for application of such methods to the analysis of complex food matrices.We have identified a set of peptide targets found in the major peanut allergens thought to be responsible for the allergenic activity of peanuts. The selectivity and sensitivity of MRM experiments allowed detection of target peptides at low attomole levels. Low level LOQ's were also achieved between atto-femtomole levels. When the impact of thermal processing on the behaviour of the peptide targets was assessed those from the cupin allergens Ara h1 and Ara h3 showed more complex behaviour than those from the 2S albumin allergens Ara h2, 6 and 7. Differences in the behaviour of peptide targets from the same protein were evident which may relate to differences in efficiency of trypsin digestion, variations in the abundance of different isoforms and processing-induced modifications. Ara h1 and 3 form complex aggregated structures following thermal processing, impairing both their extractability and hence their reliability as targets for quantification in thermally processed peanuts.59,60 The prolamin proteins which are heat-stable appeared to be less affected and in this study the peptide targets from these proteins proved more stable to the effects of thermal processing.59,61 Trypsin cleavage yields targets flanked by lysine or arginine which can become modified through Maillard-type reactions affecting digestion and detection. Targets flanked by arginine residues appeared to be more thermostable.
This report shows that the effect of food processing procedures on peptide target behaviour is unpredictable and hence an assessment of the robustness of any allergen target should be a pre-requisite for development of a MS method for detection of allergens in food. These data will support development of targeted MS methods to the analysis of peanut in foods. The ability to couple sensitive detection methodology with a wide dynamic range, to harsh methods of extraction required for processed foods will allow methods able to quantifying peanut allergens in complex food matrices to be developed in future.
Acknowledgements
This work was funded through a BBSRC CASE awarded to R. L. Sayers. We would like to thank Sue O'Hagan at PepsiCo for peanut samples, Lee Gethings and Antonietta Wallace at Waters for RapiGest™ and helpful discussions, Frances Smith for helpful discussions and Cunyu Yan of the Michael Barber Centre for support with MS analysis.References
- B. I. Nwaru, L. Hickstein, S. S. Panesar, A. Muraro, T. Werfel, V. Cardona, A. E. Dubois, S. Halken, K. Hoffmann-Sommergruber, L. K. Poulsen, G. Roberts, R. Van Ree, B. J. Vlieg-Boerstra, A. Sheikh and E. F. A. A. G. Group, Allergy, 2014, 69, 62–75 CrossRef CAS PubMed.
- Official Journal of the European Union, L 304, 22 November 2011.
- B. K. Ballmer-Weber, M. Fernandez-Rivas, K. Beyer, M. Defernez, M. Sperrin, A. R. Mackie, L. J. Salt, J. O. B. Hourihane, R. Asero, S. Belohlavkova, M. Kowalski, F. de Blay, N. G. Papadopoulos, M. Clausen, A. C. Knulst, G. Roberts, T. Popov, A. B. Sprikkelman, R. Dubakiene, S. Vieths, R. van Ree, R. Crevel and E. N. C. Mills, J. Allergy Clin. Immunol., 2015, 135, 964–971 CrossRef CAS PubMed.
- S. L. Taylor, S. L. Hefle, C. Bindslev-Jensen, S. A. Bock, A. W. Burks, L. Christie, D. J. Hill, A. Host, J. O. B. Hourihane, G. Lack, D. D. Metcalfe, D. A. Moneret-Vautrin, P. A. Vadas, F. Rance, D. J. Skrypec, T. A. Trautman, I. M. Yman and R. S. Zeiger, J. Allergy Clin. Immunol., 2002, 109, 24–30 CrossRef PubMed.
- S. L. Taylor, D. A. Moneret-Vautrin, R. W. R. Crevel, D. Sheffield, M. Morisset, P. Dumont, B. C. Remington and J. L. Baumert, Food Chem. Toxicol., 2010, 48, 814–819 CrossRef CAS PubMed.
- S. L. Taylor, R. W. R. Crevel, D. Sheffield, J. Kabourek and J. Baumert, Food Chem. Toxicol., 2009, 47, 1198–1204 CrossRef CAS PubMed.
- A. DunnGalvin, C. H. Chan, R. Crevel, K. Grimshaw, R. Poms, S. Schnadt, S. L. Taylor, P. Turner, K. J. Allen, M. Austin, A. Baka, J. L. Baumert, S. Baumgartner, K. Beyer, L. Bucchini, M. Fernandez-Rivas, K. Grinter, G. F. Houben, J. Hourihane, F. Kenna, A. G. Kruizinga, G. Lack, C. B. Madsen, E. N. Clare Mills, N. G. Papadopoulos, A. Alldrick, L. Regent, R. Sherlock, J. M. Wal and G. Roberts, Allergy, 2015, 70, 1039–1051 CrossRef CAS PubMed.
- O. N. Robertson, J. O. B. Hourihane, B. C. Remington, J. L. Baumert and S. L. Taylor, Food Addit. Contam., Part A, 2013, 30, 1467–1472 CrossRef CAS PubMed.
- P. E. Johnson, S. Baumgartner, T. Aldick, C. Bessant, V. Giosafatto, J. Heick, G. Mamone, G. O'Connor, R. Poms, B. Popping, A. Reuter, F. Ulberth, A. Watson, L. Monaci and E. N. Mills, J. AOAC Int., 2011, 94, 1026–1033 CAS.
- S. Jayasena, M. Smits, D. Fiechter, A. de Jong, J. Nordlee, J. Baumert, S. L. Taylor, R. H. Pieters and S. J. Koppelman, J. Agric. Food Chem., 2015, 63, 1849–1855 CrossRef CAS PubMed.
- R. E. Poms, C. Capelletti and E. Anklam, Mol. Nutr. Food Res., 2004, 48, 459–464 CAS.
- A. Gomaa, S. Ribereau and J. Boye, J. Nutr. Food Sci., 2012, 52(2), 483–489 Search PubMed.
- C. M. Hebling, M. A. McFarland, J. H. Callahan and M. M. Ross, J. Agric. Food Chem., 2012, 61(24), 5638–5648 CrossRef PubMed.
- T.-J. Fu and N. Maks, J. Agric. Food Chem., 2013, 61, 5649–5658 CrossRef CAS PubMed.
- S. M. Albillos, F. Al-Taher and N. Maks, Food Chem., 2011, 127, 1831–1834 CrossRef CAS.
- S. Khuda, A. Slate, M. Pereira, F. Al-Taher, L. Jackson, C. Diaz-Amigo, E. C. Bigley 3rd, T. Whitaker and K. M. Williams, J. Agric. Food Chem., 2012, 60, 4195–4203 CrossRef CAS PubMed.
- R. A. Kopper, N. J. Odum, M. Sen, R. M. Helm, J. S. Stanley and A. W. Burks, Int. Arch. Allergy Immunol., 2005, 136, 16–22 CrossRef CAS PubMed.
- R. S. H. Pumphrey and M. H. Gowland, J. Allergy Clin. Immunol., 2007, 119, 1018–1019 CrossRef PubMed.
- N. Nicolaou, M. Poorafshar, C. Murray, A. Simpson, H. Winell, G. Kerry, A. Harlin, A. Woodcock, S. Ahlstedt and A. Custovic, J. Allergy Clin. Immunol., 2010, 125(1), 191–197 CrossRef CAS PubMed , e1–13.
- P. J. Turner, M. H. Gowland, V. Sharma, D. Ierodiakonou, N. Harper, T. Garcez, R. Pumphrey and R. J. Boyle, J. Allergy Clin. Immunol., 2015, 135(4), 956–963 CrossRef PubMed , e1.
- C. Venter, S. Hasan Arshad, J. Grundy, B. Pereira, C. Bernie Clayton, K. Voigt, B. Higgins and T. Dean, Allergy, 2010, 65, 103–108 CrossRef CAS PubMed.
- N. Nicolaou and A. Custovic, Curr. Opin. Allergy Clin. Immunol., 2011, 11, 222–228 CrossRef CAS PubMed.
- S. Hazebrouck, B. Guillon, M.-F. Drumare, E. Paty, J.-M. Wal and H. Bernard, Mol. Nutr. Food Res., 2012, 56, 548–557 CAS.
- N. Nicolaou, C. Murray, D. Belgrave, M. Poorafshar, A. Simpson and A. Custovic, J. Allergy Clin. Immunol., 2011, 127, 684–685 CrossRef PubMed.
- P. J. Turner, S. Mehr, R. Sayers, M. Wong, M. H. Shamji, D. E. Campbell and E. N. C. Mills, J. Allergy Clin. Immunol., 2014, 134, 751–753 CrossRef CAS PubMed.
- L. Monaci, E. De Angelis, S. L. Bavaro and R. Pilolli, Food Addit. Contam., Part A, 2015, 1–10 Search PubMed.
- G. Mamone, G. Picariello, S. Caira, F. Addeo and P. Ferranti, J. Chromatogr. A, 2009, 1216, 7130–7142 CrossRef CAS PubMed.
- K. J. Shefcheck, J. H. Callahan and S. M. Musser, J. Agric. Food Chem., 2006, 54, 7953–7959 CrossRef CAS PubMed.
- J. B. Misra, Peanut Sci., 2001, 28, 48–51 CrossRef CAS.
- B. MacLean, D. M. Tomazela, N. Shulman, M. Chambers, G. L. Finney, B. Frewen, R. Kern, D. L. Tabb, D. C. Liebler and M. J. MacCoss, Bioinformatics, 2010, 26, 966–968 CrossRef CAS PubMed.
- H. Chassaigne, J. V. Nørgaard and A. J. van Hengel, J. Agric. Food Chem., 2007, 55, 4461–4473 CrossRef CAS PubMed.
- J. Sealey-Voyksner, J. Zweigenbaum and R. Voyksner, Food Chem., 2016, 194, 201–211 CrossRef CAS PubMed.
- J. Heick, M. Fischer and B. Pöpping, J. Chromatogr. A, 2011, 1218, 938–943 CrossRef CAS PubMed.
- G. A. Mueller, S. J. Maleki, K. Johnson, B. K. Hurlburt, H. Cheng, S. Ruan, J. B. Nesbit, A. Pomes, L. L. Edwards, A. Schorzman, L. J. Deterding, H. Park, K. B. Tomer, R. E. London and J. G. Williams, Allergy, 2013, 68, 1546–1554 CrossRef CAS PubMed.
- M. Careri, L. Elviri, M. Maffini, A. Mangia, C. Mucchino and M. Terenghi, Rapid Commun. Mass Spectrom., 2008, 22, 807–811 CrossRef CAS PubMed.
- C. Bignardi, L. Elviri, A. Penna, M. Careri and A. Mangia, J. Chromatogr. A, 2010, 1217, 7579–7585 CrossRef CAS PubMed.
- C. Bignardi, M. Mattarozzi, A. Penna, S. Sidoli, L. Elviri, M. Careri and A. Mangia, Food Anal. Methods, 2013, 6, 1144–1152 CrossRef.
- M. Careri, A. Costa, L. Elviri, J. B. Lagos, A. Mangia, M. Terenghi, A. Cereti and L. P. Garoffo, Anal. Bioanal. Chem., 2007, 389, 1901–1907 CrossRef CAS PubMed.
- C. H. Parker, S. E. Khuda, M. Pereira, M. M. Ross, T. J. Fu, X. Fan, Y. Wu, K. M. Williams, J. DeVries, B. Pulvermacher, B. Bedford, X. Zhang and L. S. Jackson, J. Agric. Food Chem., 2015, 63, 10669–10680 CrossRef CAS PubMed.
- P. E. Johnson, R. L. Sayers, L. A. Gethings, A. Balasundaram, J. T. Marsh, J. I. Langridge and E. N. C. Mills, Anal. Chem., 2016 DOI:10.1021/acs.analchem.5b04466.
- C. E. Eyers, C. Lawless, D. C. Wedge, K. W. Lau, S. J. Gaskell and S. J. Hubbard, Mol. Cell. Proteomics, 2011, 10(11), 1–12 Search PubMed.
- C. Lawless and S. J. Hubbard, OMICS: J. Integr. Biol., 2012, 16, 449–456 CrossRef CAS PubMed.
- C. Cabanos, H. Urabe, M. R. Tandang-Silvas, S. Utsumi, B. Mikami and N. Maruyama, Mol. Immunol., 2011, 49, 115–123 CrossRef CAS PubMed.
- M. Chruszcz, S. J. Maleki, K. A. Majorek, M. Demas, M. Bublin, R. Solberg, B. K. Hurlburt, S. Ruan, C. P. Mattison, H. Breiteneder and W. Minor, J. Biol. Chem., 2011, 286, 39318–39327 CrossRef CAS PubMed.
- D. S. Shin, J. Biol. Chem., 1998, 273, 13753–13759 CrossRef CAS PubMed.
- S. R. Piersma, M. Gaspari, S. L. Hefle and S. J. Koppelman, Mol. Nutr. Food Res., 2005, 49, 744–755 CAS.
- T. Jin, F. Guo, Y. W. Chen, A. Howard and Y. Z. Zhang, Mol. Immunol., 2009, 46, 1796–1804 CrossRef CAS PubMed.
- K. Otsu, R. Guo and S. C. Dreskin, Clin. Exp. Allergy, 2015, 45, 471–484 CrossRef CAS PubMed.
- T. Kleber-Janke, R. Crameri, U. Appenzeller, M. Schlaak and W. Becker, Int. Arch. Allergy Immunol., 1999, 119, 265–274 CrossRef CAS PubMed.
- R. Pedreschi, J. Norgaard and A. Maquet, Nutrients, 2012, 4, 132–150 CrossRef CAS PubMed.
- D. R. Mani, S. Abbatiello and S. Carr, BMC Bioinf., 2012, 13, S9 CrossRef CAS PubMed.
- T. Glatter, C. Ludwig, E. Ahrné, R. Aebersold, A. J. R. Heck and A. Schmidt, J. Proteome Res., 2012, 11, 5145–5156 CrossRef CAS PubMed.
- J. Marsh, N. Rigby, K. Wellner, G. Reese, A. Knulst, J. Akkerdaas, R. van Ree, C. Radauer, A. Lovegrove, A. Sancho, C. Mills, S. Vieths, K. Hoffmann-Sommergruber and P. R. Shewry, Mol. Nutr. Food Res., 2008, 52(Suppl 2), S272–S285 Search PubMed.
- P. Rabjohn, E. Helm, J. Stanley, C. West, H. Sampson, A. Burks and G. Bannon, J. Clin. Invest., 1999, 103, 535–542 CrossRef CAS PubMed.
- S. J. Maleki, O. Viquez, T. Jacks, H. Dodo, E. T. Champagne, S.-Y. Chung and S. J. Landry, J. Allergy Clin. Immunol., 2003, 112, 190–195 CrossRef CAS PubMed.
- J. R. Wisniewski, A. Zougman, N. Nagaraj and M. Mann, Nat. Methods, 2009, 6, 359–362 CrossRef CAS PubMed.
- L. L. Manza, S. L. Stamer, A.-J. L. Ham, S. G. Codreanu and D. C. Liebler, Proteomics, 2005, 5, 1742–1745 CrossRef CAS PubMed.
- H. John, M. Walden, S. Schäfer, S. Genz and W.-G. Forssmann, Anal. Bioanal. Chem., 2004, 378, 883–897 CrossRef CAS PubMed.
- F. Blanc, Y. M. Vissers, K. Adel-Patient, N. M. Rigby, A. R. Mackie, A. P. Gunning, N. K. Wellner, P. S. Skov, L. Przybylski-Nicaise, B. Ballmer-Weber, L. Zuidmeer-Jongejan, Z. Szepfalusi, J. Ruinemans-Koerts, A. P. Jansen, H. Bernard, J. M. Wal, H. F. Savelkoul, H. J. Wichers and E. N. Mills, Mol. Nutr. Food Res., 2011, 55, 1887–1894 CAS.
- E. L. van Boxtel, L. A. M. van den Broek, S. J. Koppelman and H. Gruppen, Mol. Nutr. Food Res., 2008, 52, 674–682 CAS.
- Y. M. Vissers, F. Blanc, P. S. Skov, P. E. Johnson, N. M. Rigby, L. Przybylski-Nicaise, H. Bernard, J. M. Wal, B. Ballmer-Weber, L. Zuidmeer-Jongejan, Z. Szepfalusi, J. Ruinemans-Koerts, A. P. Jansen, H. F. Savelkoul, H. J. Wichers, A. R. Mackie, C. E. Mills and K. Adel-Patient, PLoS One, 2011, 6, e23998 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6an00359a |
| ‡ Current address: The Food Allergy Research and Resource Programme, The University of Nebraska, Lincoln, USA. |
| This journal is © The Royal Society of Chemistry 2016 |