
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing H2 evolution performance of an immobilised cobalt catalyst by rational ligand design†
Janina
Willkomm
,
Nicoleta M.
Muresan
and
Erwin
Reisner
*
Christian Doppler Laboratory for Sustainable SynGas Chemistry, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: reisner@ch.cam.ac.uk; Web: http://www-reisner.ch.cam.ac.uk/
First published on 2nd February 2015
Abstract
The catalyst [CoIIIBr((DO)(DOH)(4-BnPO3H2)(2-CH2py)pn)]Br, CoP3, has been synthesised to improve the stability and activity of cobalt catalysts immobilised on metal oxide surfaces. The CoP3 catalyst contains an equatorial diimine–dioxime ligand, (DOH)2pn = N2,N2′-propanediyl-bis(2,3-butanedione-2-imine-3-oxime), with a benzylphosphonic acid (4-BnPO3H2) group and a methylpyridine (2-CH2py) ligand covalently linked to the bridgehead of the pseudo-macrocyclic diimine–dioxime ligand. The phosphonic acid functionality provides a robust anchoring group for immobilisation on metal oxides, whereas the pyridine is coordinated to the Co ion to enhance the catalytic activity of the catalyst. Electrochemical investigations in solution confirm that CoP3 shows electrocatalytic activity for the reduction of aqueous protons between pH 3 and 7. The metal oxide anchor provides the catalyst with a high affinity for mesostructured Sn-doped In2O3 electrodes (mesoITO; loading of approximately 22 nmol cm−2) and the electrostability of the attached CoP3 was confirmed by cyclic voltammetry. Finally, immobilisation of the catalyst on ruthenium-dye sensitised TiO2 nanoparticles in aqueous solutions in the presence of a hole scavenger establishes the activity of the catalyst in this photocatalytic scheme. The advantages of the elaborate catalyst design in CoP3 in terms of stability and catalytic activity are shown by direct comparison with previously reported phosphonated Co catalysts. We therefore demonstrate that rational ligand design is a viable route for improving the performance of immobilised molecular catalysts.
Introduction
Solar fuels generation through artificial photosynthesis requires a well-balanced combination of light harvesting and charge separation with proton reduction and water oxidation catalysis, preferentially in a photoelectrochemical (PEC) cell.1 As for H2 evolution, molecular synthetic catalysts based on 3d transition metals like Fe,2 Co3 or Ni4 are currently under intensive investigation as an alternative to the current benchmark H2 evolving catalysts: scarce and expensive Pt5 and fragile enzymes known as hydrogenases.6 However, the use of catalysts in a PEC cell requires their stable integration into electrodes, which is particularly challenging for molecular catalysts.7An advantage of synthetic molecular catalysts compared to solid-state materials or enzymes is the relative ease to control and characterise their composition and to study their mechanisms and kinetics in great detail. This strength provides a rational route to elaborated and improved catalyst design through mechanistic understanding and often by adopting hydrogenase-related principles.8 For example, bio-inspired nickel bis(diphosphine) catalysts were reported to generate H2 photo-9 and electrocatalytically9,10 in aqueous solution. These Ni complexes remain electroactive when heterogenised on carbon-based electrodes,11 and immobilisation on metal oxide nanoparticles9 and on carbon nitride12 has allowed for their exploitation for photocatalytic H2 production in heterogeneous schemes. Synthetic mimics of the [FeFe]-hydrogenase active site evolve H2 from water when combined with CdTe quantum dots as a photosensitiser13 and when incorporated into a protective environment, e.g. a metal organic framework14 or a micellar system.15
Cobalt catalysts with a bis(dimethylglyoximato) equatorial ligand (dmgH−)2 and an activity enhancing axial pyridine ligand,3h,16 [CoCl(dmgH)2(py)] (Fig. 1A), have long been identified as one of the most active molecular catalysts for the reduction of aqueous protons and a wealth of experimental and theoretical information is available.17 These catalysts belong to the class of cobaloximes and they are also among the very few synthetic catalysts reported as O2-tolerant during catalysis, which is an important consideration for their use in full water splitting systems.16a,18 Cobaloximes have been integrated into photocatalytic systems by wiring the catalyst to a light absorber. For example, supramolecular homogeneous systems with a dye covalently linked to the Co catalyst,19 colloidal systems containing dye-sensitised titania (with CoP1, R = PO3H2; Fig. 1A)20 or carbon nitride21 and their immobilisation on photocathodes7b,22 have been reported. However, these assemblies suffer from the drawback of anchoring the cobaloxime to the light absorber via the monodentate axial pyridine ligand. The Co–pyridine bond becomes labile during catalysis, which may result in the loss of the Co(dmgH)2 core from the light absorber unit during irradiation.19a,23 Consequently, the stability and performance of these photocatalytic systems are limited.
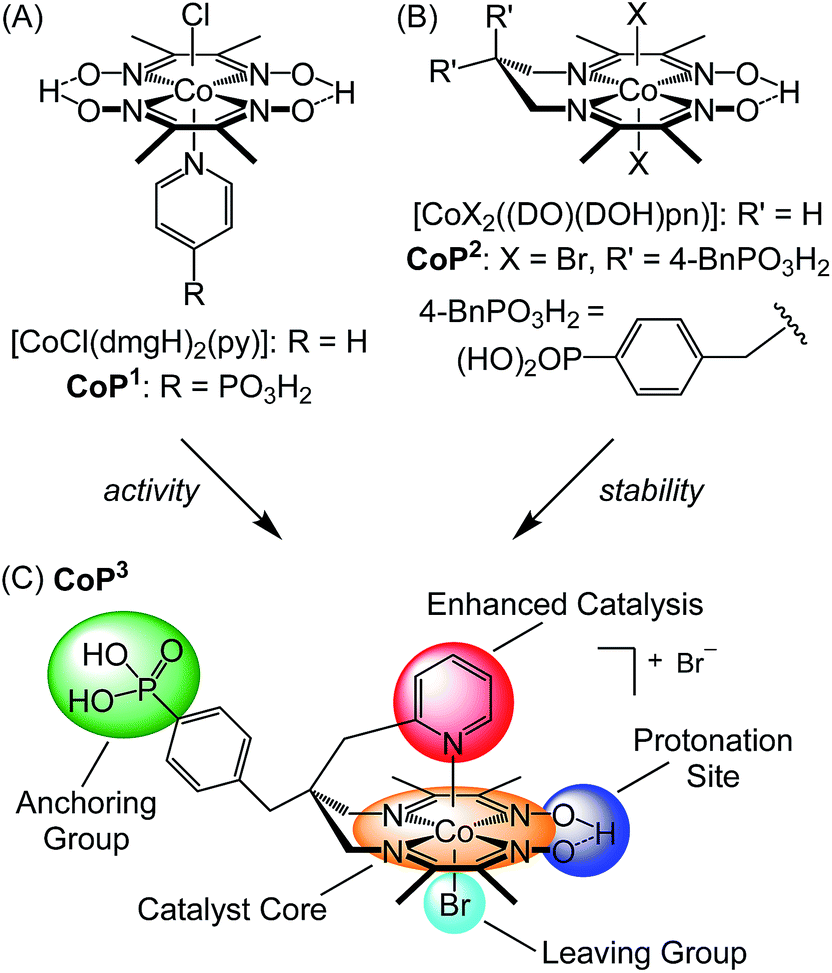 | ||
| Fig. 1 Chemical structures of (A) cobaloximes with an axial pyridine ligand, (B) cobalt diimine–dioxime catalysts, and (C) catalyst CoP3 reported in this study. CoP3 was designed to incorporate the activity enhancing pyridine of CoP1 (A),20b and the stable catalyst core and anchoring functionality of CoP2 (B).24 | ||
A more robust class of cobalt catalysts, [CoX2((DO)(DOH)pn)] with X = bromide or chloride and the tetradentate ligand (DOH)2pn = N2,N2′-propanediyl-bis(2,3-butanedione-2-imine-3-oxime) (R′ = H; Fig. 1B),3d,3i,25 was recently integrated into electrodes. This Co catalyst was immobilised on a carbon-based electrode via click chemistry (X = Cl, R′ = H, N3)26 and on a conducting metal oxide electrode via a phosphonic acid linker (CoP2, X = Br, R′ = 4-BnPO3H2; Fig. 1B).24,27 Anchoring of the Co catalyst through the propanediyl bridgehead of the pseudo-macrocyclic equatorial ligand provides a substantially more stable anchoring to an electrode than immobilisation via the axial pyridine in cobaloximes.
In this work, we present a cobalt catalyst for H2 evolution, which does not only display good stability when anchored onto metal oxide surfaces, but also enhanced catalytic activity compared to the previously reported immobilised Co catalyst CoP2. The novel cobalt catalyst, CoP3, contains a pendant pyridine and a dangling phosphonic acid group linked to the bridgehead of the equatorial diimine–dioxime ligand (Fig. 1C). The axial pyridine ligand coordinates to the metal centre and enhances the activity of the cobalt catalyst. Covalent linkage to the equatorial ligand framework ensures that the pyridine does not diffuse away from the catalyst core during turnover. The phosphonic acid group allows for attachment to metal oxide surfaces and is also tightly bound to the ligand framework. The electrochemistry of CoP3 in solution and when immobilised on mesoporous indium–tin oxide electrodes (ITO|mesoITO), as well as the photocatalytic activity of CoP3 in Ru-dye sensitised systems is reported and the results are directly compared with previously reported cobalt catalysts CoP1 and CoP2 (Fig. 1).
Results and discussion
Synthesis and characterisation of CoP3
Complex CoP3 was synthesised in six steps from commercially available starting materials with an overall yield of approximately 10% (Scheme 1 and ESI† for experimental details).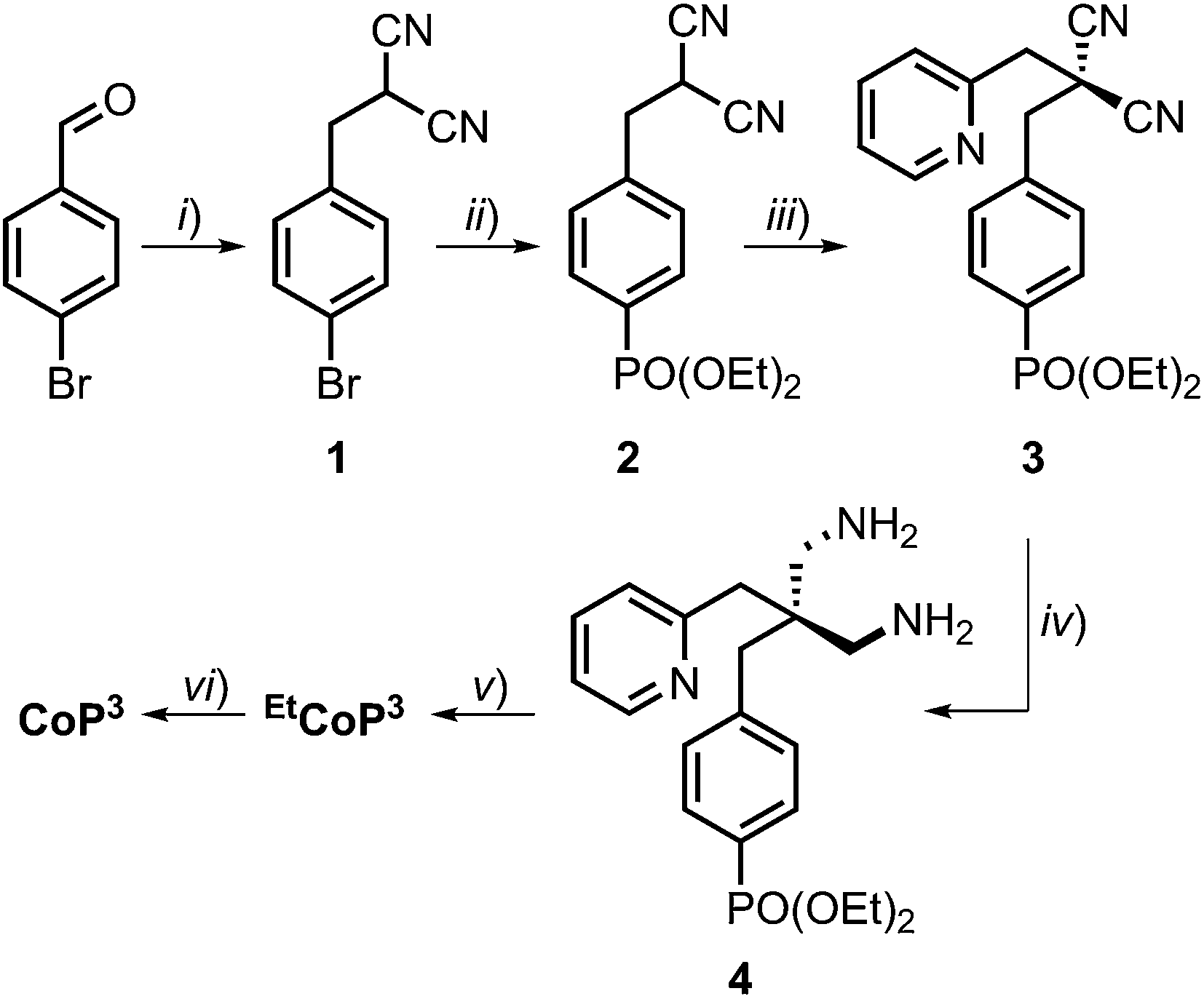 | ||
| Scheme 1 (i) Malononitrile, NaBH4, ethanol/water (95/5), 3 h, r.t., 80%; (ii) HPO(OEt)2, Et3N, Pd(PPh3)4, PPh3, tetrahydrofuran, 48 h, reflux, 73%; (iii) 2-(bromomethyl)pyridine·HBr, K2CO3, acetone, 3d, r.t., 58%; (iv) borane, tetrahydrofuran, 24 h, r.t., 99%; (v) 2,3-butanedione monoxime, CoBr2·6H2O, air, methanol, 5d, r.t., 45%; (vi) bromotrimethylsilane, dichloromethane, 48 h, r.t., 65% yield. The chemical structure of CoP3 is shown in Fig. 1C. | ||
Compound 1 was prepared via condensation of 4-bromobenzaldehyde with malononitrile and reduction by NaBH4.28 The phosphonate ester derivative 2 was synthesised from 1 in a Pd-catalysed cross-coupling reaction with diethyl phosphite. Introduction of the pendant pyridine was achieved by alkylation of 2 with 2-(bromomethyl)pyridine. The resulting malononitrile derivative 3 was reduced to the diamine 4 by treatment with borane. Complex EtCoP3 was obtained from a one-pot, three-step condensation–complexation–oxidation reaction:24,25c the diimine–dioxime ligand was prepared via condensation of 4 and 2,3-butanedione monoxime, followed by addition of CoBr2·6H2O and oxidation of the CoII ion in air to form EtCoP3. Hydrolysis of the phosphonate ester using bromotrimethylsilane yielded the target complex CoP3. 1H, 13C and 31P NMR spectra of the compounds are shown in Fig. S1 to S11.†
The final complex CoP3 was characterised by 1H, 13C, 31P and NOE NMR spectroscopy, UV-vis and ATR-IR spectroscopy, mass spectrometry and elemental analysis. The 31P NMR spectra of the phosphonate ester compounds 2–4 and EtCoP3 feature a signal at approximately 19 ppm, which is shifted to 13 ppm in CoP3 as expected upon hydrolysis of the phosphonate ester. Both cobalt complexes, EtCoP3 and CoP3 display a characteristic 1H NMR signal at approximately 19 ppm, which is assigned to the bridge proton of the equatorial (DO)(DOH)pn ligand.24,291H NMR signals of the methylene protons on the propanediyl bridgehead of diamine 4 exhibit a downfield shift from 2.5 ppm to 3.7 and 4.1 ppm upon formation of the cobalt diimine–dioxime complex EtCoP3. Moreover, these diastereotopic methylene protons (2J(H,H) = 15 Hz) show a significantly different chemical shift (for CoP3: Δδ = 0.6 ppm in DMSO-d6). This difference is presumably due to two different axial ligands in the octahedral coordination sphere and is an indication of coordination of the pendant pyridine ligand to the metal centre in EtCoP3 and CoP3. Evidence for coordination is also given by a 0.7 ppm upfield shift of the signal of the pyridine proton in 6-position upon formation of the cobalt complexes (H6, Table S1†).29 In addition, a NOE response was observed for this proton after saturation of the oxime proton signal at 19.2 ppm (Fig. S12†) revealing that both protons have to be in close proximity to each other.29 When trifluoroacetic acid (TFA) was added to a solution of CoP3 in DMSO-d6, no shift of the pyridine proton signals was observed (Fig. S13†). If protonated, new signals would be expected in the range of 8 to 9 ppm.30 Thus, the pyridine remains ligated to the cobalt centre and is not protonated even in the presence of a strong acid.
The 1H NMR spectrum of CoP3 in D2O shows a similar upfield shift for the pyridine proton in 6-position as in DMSO-d6 (7.8 ppm in CoP3vs. 8.5 ppm in diamine 4) and the spectrum remained unchanged for at least three weeks (Fig. S14†). Electronic absorption spectra of CoP3 in water show a strong π–π* absorption at λ = 259 and 219 nm (ε = 1.864 × 104 L mol−1 cm−1 and 2.774 × 104 L mol−1 cm−1; Fig. S15†). Similar absorption features are obtained in pH 7 phosphate buffer and pH 4.5 acetate buffer and no changes in the UV-vis spectrum were apparent when the solution was acidified with TFA (Fig. S15†), demonstrating the good stability of the catalyst in aqueous solutions.
Electrochemical studies in solution
The electrochemical response of CoP3 was investigated in organic as well as aqueous electrolyte solutions using a three-electrode set-up with a glassy carbon working electrode (0.07 cm2). A cyclic voltammogram (CV) of CoP3 recorded in DMF/TBABF4 electrolyte solution (TBABF4 = tetrabutylammonium tetrafluoroborate, 0.1 M) exhibits two reversible one-electron reduction waves at E1/2 = −0.67 V and −1.07 V vs. Fc+/Fc, which are assigned to the CoIII/CoII and CoII/CoI redox couples, respectively (Fig. S16A†).3i,24 Upon addition of 1 to 10 equivalents of TFA, a catalytic proton reduction wave appeared close to the potential of the initial CoII/CoI redox couple at a half-wave potential, Ecat/2, of −1.06 V vs. Fc+/Fc, (Fig. S16B†). Thus, an overpotential (η) of approximately 110 mV is required to reduce TFA protons (E0(H+/H2) = −0.95 V vs. Fc+/Fc for 10 mM TFA in DMF)31 with CoP3, which is comparable to previously reported [Co(DO)(DOH)pn]-type complexes.3i,24CVs recorded in aqueous Britton–Robinson buffer (pH 3 to 7) feature a reversible CoIII/CoII redox couple and quasi-reversible CoII/CoI reduction (Fig. 2A). When scanning towards more cathodic potential, a third reduction wave is observed which is attributed to catalytic proton reduction by the complex.3d Comparable electrochemical responses were obtained when a pH 7 triethanolamine (TEOA)/Na2SO4 electrolyte solution and pH 4.5 acetate or ascorbic acid (AA) solution were used (Fig. S17†), except that no CoIII/CoII reduction wave can be observed in cathodic scans in AA solution, presumably due to the chemical reduction of CoIIIP3 to CoIIP3 (Fig. S18†). The onset of a weak wave, tentatively assigned to CoII/CoIII oxidation, is observed at approximately 0.05 V vs. NHE before AA oxidation starts at 0.2 V vs. NHE.
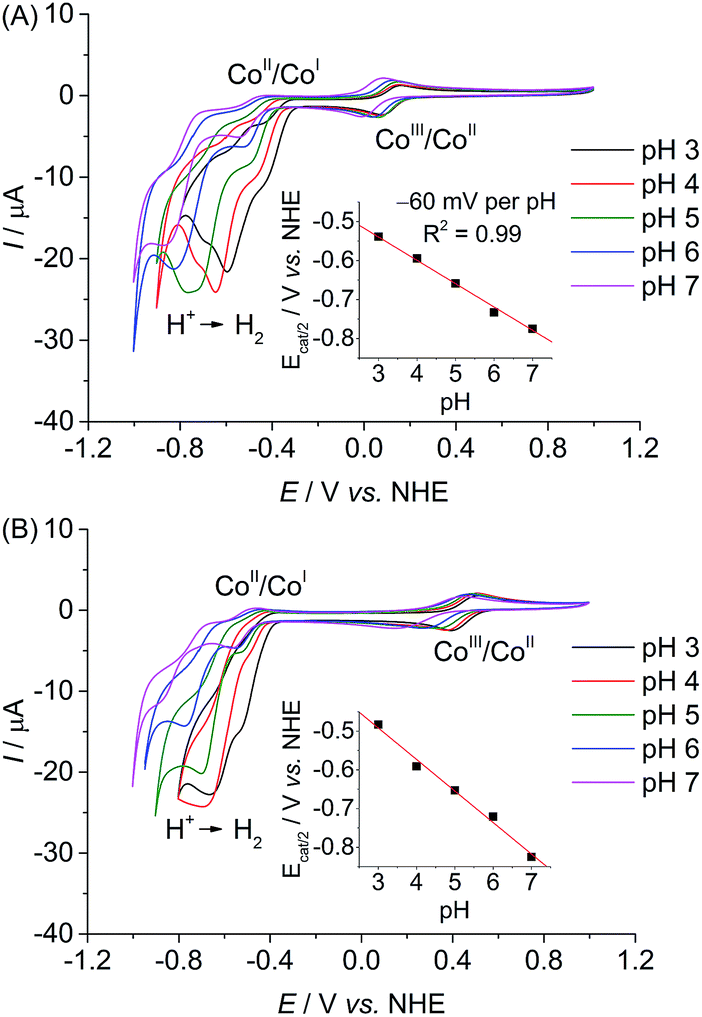 | ||
| Fig. 2 CVs with dissolved (A) CoP3 and (B) CoP2 (0.8 mM) recorded in an aqueous Britton–Robinson buffer at different pH values on a glassy carbon working electrode at 20 mV s−1. The insets show the correlation between the half-wave potential of the catalytic reduction wave, Ecat/2, and the pH value. The red traces represent the linear fit of the data points. | ||
The pH-dependent investigation also revealed that the half-wave potential of the catalytic reduction wave of CoP3, Ecat/2, shifts by approximately −60 mV per pH unit increase (Fig. 2A); in agreement with a one proton–one electron coupled process according to the Nernst equation. This was previously attributed to protonation of the oxime functionality in [Co(DO)(DOH)pn]-type complexes.3i,25a
Comparison of the electrochemical response of CoP3 to the previously reported complex CoP2 allows us to elucidate any beneficial effect of the additional axial pyridine ligand on the proton reduction activity. CVs of CoP2 recorded in the pH range from 3 to 7 are shown in Fig. 2B. A shift in redox potential is observed for the CoIII/CoII redox couple in CoP3 compared to CoP2 (ΔE1/2 = −0.24 V at pH 7), which is consistent with a coordinated pyridine in CoP3. For both cobalt diimine–dioxime catalysts, the catalytic reduction wave decreases with increasing pH indicating a higher proton reduction activity under more acidic conditions, which has been previously observed for (DO)(DOH)pn-type cobalt catalysts.26 Peak currents of the catalytic reduction wave, Icat, and Icat/Ip ratios taking into account the non-catalytic CoIII/CoII reduction peak currents, Ip, are similar for both complexes at pH 3 and 4 (Table S2†). But, CoP3 features higher Icat and Icat/Ip ratios at pH values above 4 revealing a higher activity of CoP3 under more pH neutral conditions (Table S2†). Moreover, the half wave potential Ecat/2 of CoP3 is observed at less negative potentials than for CoP2 under pH neutral conditions (−0.83 V for CoP2vs. −0.78 V vs. NHE for CoP3).
The half-wave potential, E1/2, of the CoII/CoI reduction wave in CoP3 shifts with about −33 mV per pH at pH values below 6 and becomes almost pH independent above pH 6 (Fig. S19A†). Such a change in slope was not observed for E1/2(CoII/CoI) in CoP2 (Fig. S19B†), suggesting an alteration in the coordination sphere specific to CoP3, e.g. a ligated and non-ligated, probably protonated pendant pyridine ligand. The pH-dependencies of E1/2 of the CoIII/CoII reduction wave change in a similar manner for CoP2 and CoP3 (Fig. S20†) and are ascribed to protonation/deprotonation occurring at moieties present in both complexes, e.g. at phosphonic acid groups9 or aquo ligands. Due to a different number of those functionalities the slopes differ for both complexes.
Based on these findings, we suggest that the enhanced catalytic activity of CoP3 under near neutral conditions is due to coordination of the pyridine to the cobalt centre during the catalytic cycle. The electron donating ability of the pyridine ligand would allow for the formation of a more basic Co-hydride species in the rate limiting step of the catalytic cycle, thereby improving proton reduction catalysis.16a,32 A similar increase of catalytic current and decrease in overpotential has previously been observed when an axial pyridine ligand was introduced to the coordination sphere of cobaloxime complexes at neutral pH.16a Addition of one and four equivalents of pyridine to a CoP2-containing electrolyte solution at pH 7 did not result in any increase of the catalytic reduction wave, which demonstrates that the covalent integration of the pyridine as achieved in CoP3 is also critical to enhance the activity of the cobalt diimine–dioxime catalyst (Fig. S21†).25c
The comparable pH-dependent shifts of E1/2(CoII/CoI) for CoP2 and CoP3 below pH 6 suggest a temporary non-coordinated pyridine in CoP3 upon reduction. Although the axial pyridine in CoP3 is coordinated to the cobalt centre in the initial CoIII state even in the presence of a strong acid (see above), reduction to CoII or a formal CoI species results in a labile Co–pyridine bond and subsequent release of the pyridine from the Co ion. However, the covalently linked pyridine ligand remains in close proximity to the cobalt centre and could improve catalysis in two distinct ways. It could be partially protonated under acidic conditions (pKa of 2-picoline: 5.96)33 and consequently act as a proton relay in the catalytic cycle or it could readily re-coordinate and enhance activity as described above.16a The fully reversible CoIII/CoII redox couple indicates that the pyridine re-coordinates to the Co centre upon oxidation of the complex.
Finally, both Co diimine–dioxime catalysts were compared to the phosphonated cobaloxime catalyst CoP1. Among the series of phosphonated cobalt catalyst, CoP1 is the most active proton reduction catalyst at neutral pH, featuring a large proton reduction wave at more positive potential than CoP2 and CoP3 (Fig. S17A†). Under more acidic conditions, no CoII to CoIII oxidation wave was observed for CoP1 in the anodic reverse scans (Fig. S17B and S18A†) indicating catalyst decomposition due to hydrolysis of the equatorial (dmgH−)2 ligand.34
Electrochemical studies with heterogenised catalysts
The phosphonic acid anchoring groups in CoPn (n = 1 to 3) allow for the grafting of the complexes onto metal oxide surfaces.20,24 The electrochemical response of the three cobalt catalysts immobilised onto ITO|mesoITO electrodes was compared to determine the loading of the Co catalysts to the metal oxide surface and the stability during voltammetry, specifically when cycling between the CoIII, CoII and CoI oxidation states. The electrodes were prepared from ITO nanoparticles as described previously24 and were loaded with catalysts by immersing a cleaned slide into a 6 mM catalyst solution in dry DMF for 15 h. The ITO|mesoITO|CoPn electrodes were gently rinsed with fresh DMF, dried under N2 and studied in a CoPn-free DMF/TBABF4 electrolyte solution (0.1 M).CVs of the ITO|mesoITO|CoP3 electrode in DMF/TBABF4 are shown in Fig. 3. A linear correlation between the peak current density, JP, of the reversible CoII/CoI reduction at E1/2 = −1.03 V vs. Fc+/Fc and the scan rate, v, confirms that CoP3 is immobilised on the ITO|mesoITO surface. The disappearance of the CoIII/CoII redox couple for the immobilised complex at E1/2 = −0.69 V vs. Fc+/Fc with the concomitant appearance of a new wave at E1/2 = −0.43 V vs. Fc+/Fc during consecutive scans is presumably due to a cathodically induced replacement of the axial bromido ligand by DMF.16b,35 CVs of ITO|mesoITO|CoP2 show comparable features in DMF/TBABF4 (Fig. S22 and S23B†) with E1/2 = −0.59 V and −1.17 V vs. Fc+/Fc for CoIII/CoII and CoII/CoI, respectively. The determination of any JP–v correlation was not possible for ITO|mesoITO|CoP1 due to the poor stability of the immobilised CoP1 on ITO and subsequent rapid decrease of the redox waves within the first few scans (Fig. S23A;† see below).
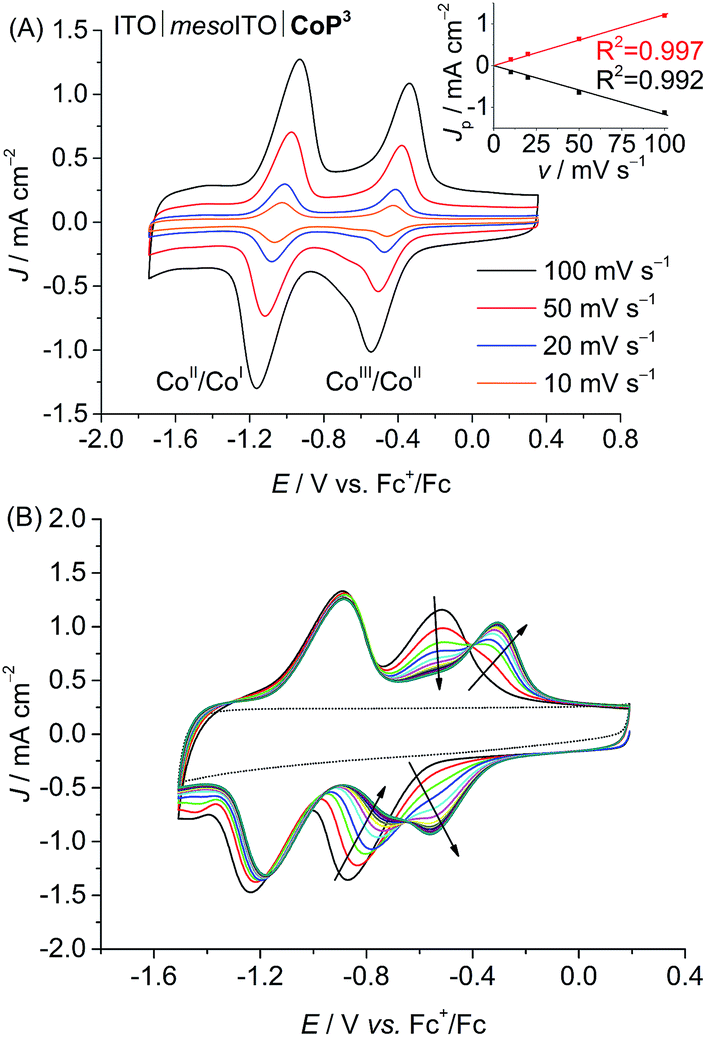 | ||
| Fig. 3 (A) CVs of ITO|mesoITO|CoP3 in DMF/TBABF4 electrolyte (0.1 M) at different scan rates (10, 20, 50, 100 mV s−1). Inset: The correlation between the peak current density, Jp (CoII/CoI), and scan rate, v, is shown. The black and red traces represent linear fits to the data points. (B) Consecutive CVs of ITO|mesoITO|CoP3 in DMF/TBABF4 (0.1 M) at a scan rate of 100 mV s−1 showing cathodically induced replacement of the axial bromido ligand by DMF. The background of ITO|mesoITO without catalyst is shown as dotted line. Note that ligand exchange has already occurred in the CVs shown in (A). | ||
The amounts of catalyst immobilised onto the mesoporous ITO electrodes were estimated by integration of the redox waves (reduction and oxidation) from the first CV scans in DMF/TBABF4 (Table 1). Loadings between 22 and 28 nmol cm−2 (referenced to the geometrical surface area of the electrode) were determined for the three ITO|mesoITO|CoPn electrodes. We only observed small differences in the loadings, which might be due to different spatial demands of the catalysts. Comparable results and trends were obtained when the integration of the redox waves was performed with CV scans recorded in aqueous electrolyte solution (Table S3, Fig. S24 and S25†) and loadings are comparable to a previously reported Ru-based compound on mesostructured ITO.36 The results show that CoP3 binds well and with a comparable loading to CoP2 to the metal oxide electrode despite only having one anchoring group.
After 10 consecutive scans at v = 100 mV s−1 practically no desorption of CoP3 and CoP2 was observed, whereas approximately 80% of CoP1 was lost from the ITO|mesoITO electrode (Table 1). As discussed above, reduction of low spin CoIII results in a labile CoII and CoI species, which leads to the loss of the Co(dmgH)2 core from the ITO-anchored phosphonated pyridine in CoP1.7b This instability was not observed for CoP2 and CoP3, demonstrating the much improved robustness when anchoring the cobalt catalysts with one (CoP3) or two (CoP2) phosphonic acid groups on the tetradendate equatorial (DO)(DOH)pn ligand to the ITO electrode (Fig. 3 and S23B†).24CoP3 therefore displays much higher stability on an electrode than CoP1 and is significantly more active as a proton reduction catalyst than CoP2 as shown by electrochemical investigations in solution.
Photocatalytic studies
The photocatalytic activity of the CoPn catalysts was studied in solution and in heterogeneous suspension systems containing either TiO2 or ZrO2 nanoparticles with TEOA (0.1 M, pH 7) or AA (0.1 M, pH 4.5) as buffer and sacrificial electron donor (SED). [RuII(2,2′-bipyridine)2(2,2′-bipyridine-4,4′-bisphosphonic acid)]Br2 (RuP, Fig. 4A) was used as photosensitiser. Photoexcited RuP (RuP*) can operate through an oxidative (E0(RuP+/RuP*) = −0.95 V vs. NHE)37 or reductive quenching mechanism (E0(RuP*/RuP−) = 1.07 V vs. NHE),38 which would generate RuP− (E0(RuP/RuP−) = −1.05 V vs. NHE).9,38,39 Photoinduced electron transfer from RuP to the CoPn catalyst can occur either directly (homogeneous system; Fig. 4B) or via the injection of electrons into the conduction band (CB) of the semiconductor TiO2 (ECB = −0.70 V at pH 7; ECB = −0.55 V vs. NHE at pH 4.5)40 by a ‘through particle’ mechanism (Fig. 4C).9RuP* and RuP− are unable to transfer electrons into the more negative CB of ZrO2 (ECB = −1.40 V vs. NHE at pH 7, ECB = −1.26 V vs. NHE at pH 4.5),41 which only allows for direct electron transfer from photoexcited RuP to the catalyst as in the homogeneous system in RuP|ZrO2|CoPn (Fig. 4C). A comparison of the electrocatalytic onset potentials for proton reduction of the CoPn catalysts with the thermodynamic driving force from photogenerated RuP* and RuP−, and the semiconductors is summarised in Fig. 4D. It illustrates that photo-H2 evolution is thermodynamically possible with all three catalysts, but kinetic factors may have a detrimental effect on some of the systems.42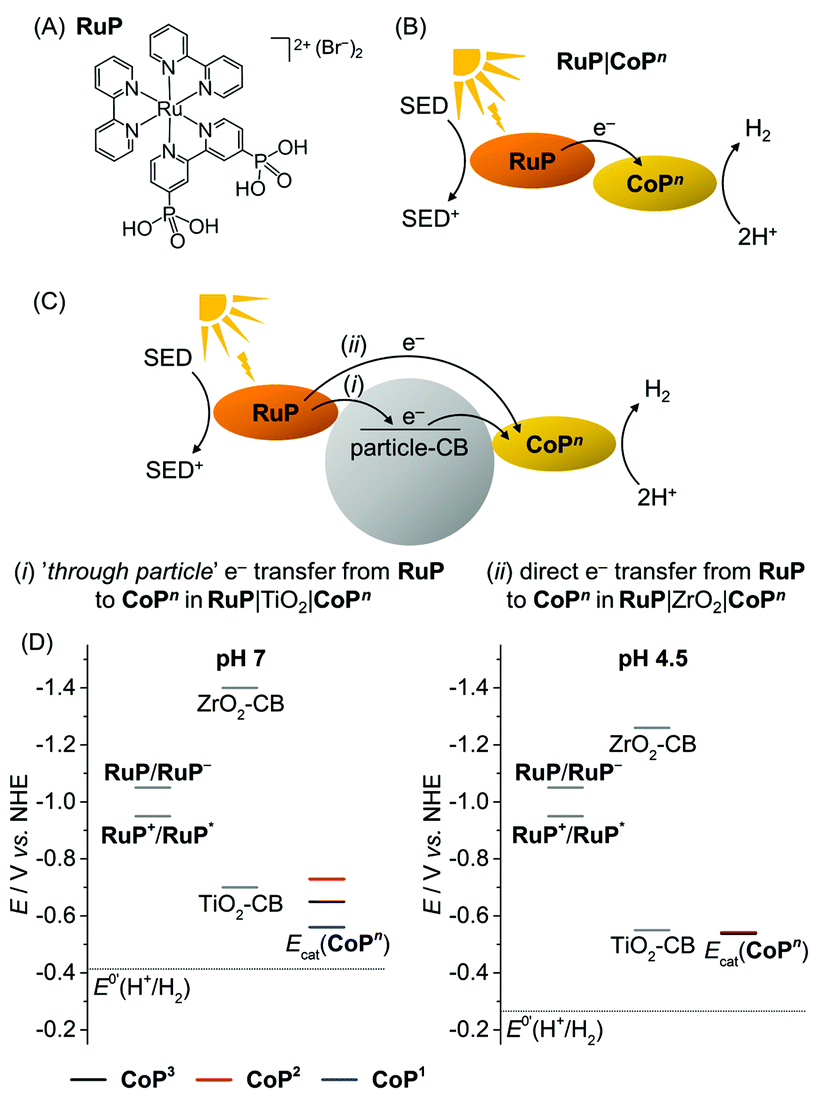 | ||
| Fig. 4 (A) Chemical structure of the photosensitiser RuP. (B and C) Electron transfer mechanisms from the photoexcited RuP dye to the catalyst CoPn in the homogenous and heterogeneous suspension systems with TiO2 and ZrO2 particles. The ‘through particle’ electron transfer pathway proceeds through oxidative quenching of RuP and is only accessible in RuP|TiO2|CoPn (see text). (D) Schematic energy diagram with the redox potentials of RuP+/RuP* and RuP/RuP− generated upon photoexcitation, conduction band potentials of the semiconductor particles (TiO2-CB and ZrO2-CB), the thermodynamic redox potential for proton reduction, E0’(H+/H2), and the catalytic proton reduction onset potentials, Ecat, of the CoPn catalysts determined from CVs in TEOA/Na2SO4 (0.1 M each, pH 7) and acetate electrolyte solution (0.1 M, pH 4.5). | ||
In a standard experiment, 0.1 μmol CoPn and 0.1 μmol RuP were used in 2.25 mL of aqueous solution containing the SED (homogeneous RuP|CoPn system) and 5 mg of metal oxide nanoparticles were added for the particle systems (RuP|TiO2|CoPn or RuP|ZrO2|CoPn). The samples were kept at 25 °C and irradiated with visible light from a solar light simulator equipped with an AM 1.5G, IR and UV filter (λ > 420 nm). The activity is expressed as Co-based turnover number, TONCo (mol H2 per mol CoPn), which was obtained after four hours of visible light irradiation (Table 2). At this point, all systems had lost their photoactivity under these standard conditions.
| TOFCob (1 h)/h−1 | TONCoc (4 h) | n (H2)/μmol (4 h) | |
|---|---|---|---|
| a The following standard conditions were employed unless otherwise noted: AM 1.5G, 100 mW cm−2, λ > 420 nm irradiation, 0.1 μmol of CoPn and 0.1 μmol of RuP in homogenous solution or in suspensions with TiO2 or ZrO2 nanoparticles (5 mg) in aqueous TEOA or AA solution (2.25 mL, 0.1 M). Mean values ± standard deviation (σ) given from at least three different reaction vessels. b TOF based on CoPn for the first hour of irradiation. c TON based on CoPn and total of headspace H2 accumulated after four hours irradiation. d Below the limit of detection by gas chromatography. e Particles were loaded with the catalyst and the dye, centrifuged and re-suspended in fresh buffer solution prior to use. f n.d. = not defined (no CoP3 present or amount of CoP3 not precisely known). g TOF is based on the maximum H2 evolution rate after the initial lag period. | |||
| pH 7 (TEOA) | |||
| RuP|CoP3 | — | — | <0.03d |
| RuP|ZrO2|CoP3 | — | — | <0.03d |
| RuP|TiO2|CoP3 | 10.3 ± 0.4 | 12.3 ± 0.3 | 1.23 ± 0.03 |
| RuP|TiO2|CoP3centr.e | n.d. | n.d. | 0.74 ± 0.27 |
| RuP|TiO2|CoP2 | 0.6 ± 0.1 | 2.4 ± 0.1 | 0.24 ± 0.01 |
| RuP|TiO2|CoP1 | 44.0 ± 0.9 | 56.6 ± 2.2 | 5.66 ± 0.22 |
| RuP|TiO2, no CoP3 | n.d. | n.d. | 0.14 ± 0.07 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| pH 4.5 (AA) | |||
| RuP|CoP3 | 2.1 ± 0.6 | 3.1 ± 0.4 | 0.31 ± 0.04 |
| RuP|ZrO2|CoP3 | 8.1 ± 2.2g | 9.9 ± 0.2 | 0.99 ± 0.02 |
| RuP|TiO2|CoP3 | 12.8 ± 0.6g | 18.4 ± 0.5 | 1.84 ± 0.05 |
| RuP|TiO2|CoP2 | 1.2 ± 0.2 | 1.2 ± 0.1 | 0.12 ± 0.01 |
| RuP|TiO2|CoP1 | — | — | <0.03d |
| RuP|TiO2, no CoP3 | — | — | <0.03d |
| RuP|ZrO2, no CoP3 | n.d. | n.d. | 0.09 ± 0.02 |
| RuP, no CoP3 | — | — | <0.03d |
We first investigated the photocatalytic activity of CoP3 in pH 7 TEOA solution. No H2 was generated in the RuP|CoP3 and RuP|ZrO2|CoP3 systems, but RuP|TiO2|CoP3 produced a TONCo of 12.3 ± 0.3 (Fig. 5A). No H2 or only trace amounts of H2 were detectable when omitting CoP3, RuP, SED or light from this system or when CoBr2 was added instead of CoP3 (Table S5†). Increasing the concentration of CoP3 in RuP|TiO2|CoP3 to 0.2 μmol resulted in a slight enhancement in the overall TONCo (16.5 ± 0.5; Fig. S26A†). The highest TONCo of 22.0 ± 1.5 was observed when the amount of RuP was increased to 0.2 μmol (Table S4 and Fig. S26B†).
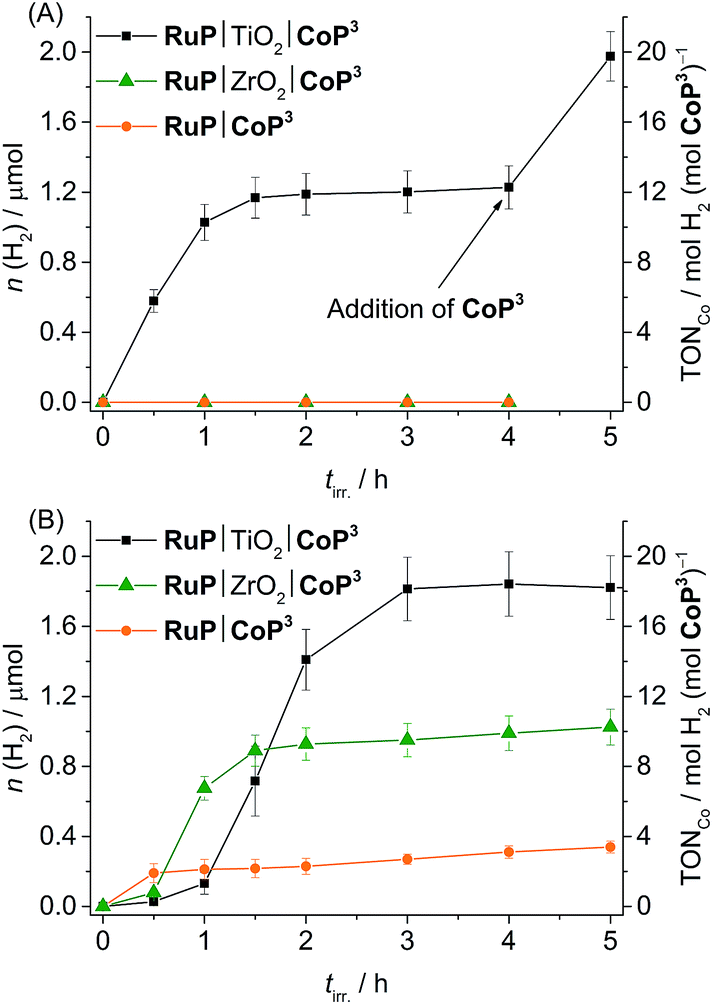 | ||
| Fig. 5 Photoactivity of CoP3 expressed as total amount of headspace H2 over irradiation time and TONCo (AM 1.5G, 100 mW cm−2, λ > 420 nm) in different systems (RuP|TiO2|CoP3, RuP|ZrO2|CoP3 and RuP|CoP3) in (A) pH 7 TEOA (2.25 mL, 0.1 M) and (B) pH 4.5 AA solution (2.25 mL, 0.1 M). A 1:1 ratio of CoP3 and RuP (0.1 μmol each) was used and either 5 mg of TiO2 or ZrO2 were added in case of particle systems. | ||
The lack of photo-H2 evolution in the homogeneous and ZrO2-containing systems suggests that RuP* is not capable of reducing CoP3 directly to initiate proton reduction which is in agreement with the previously reported inactivity of RuP|ZrO2|CoP1, RuP|CoP1 and a [CoBr2((DO)(DOH)pn)] complex in combination with a Ru-dye and triethylamine as SED in solution.20a,25b,39 A possible explanation may be that the photoexcited state life-time of RuP* is too short-lived and the more reducing RuP− is not generated in aqueous TEOA solution.43 Addition of TiO2 facilitates oxidative quenching of RuP* and charge separation, which allows for efficient electron transfer from RuP to CoP3via its CB in a ‘through particle’ mechanism, thereby triggering photoactivity of this system.20a,39 A comparable, surface-linker free cobalt diimine–dioxime catalyst with a pendant pyridine ligand was studied in solution using a Re photosensitiser and TEOA as sacrificial agent. A Co-based TONCo of approximately 15 has been reported for this homogeneous photocatalytic system under near neutral conditions (pH 7.7).25c The cobalt diimine–dioxime catalyst with a pendant pyridine ligand therefore keeps the full activity when immobilised on a semiconductor as is evident from the maximum TONCo of 22.0 ± 1.5 observed with RuP|TiO2|CoP3.
Photo-H2 evolution activity of the deactivated RuP|TiO2|CoP3 system was fully recovered by addition of fresh CoP3 to the suspension (Fig. 5A), indicating complete degradation of CoP3 within the first few hours of photocatalysis. To date, no detailed studies on possible degradation products of [Co(DO)(DOH)pn] catalysts are available, but partial regeneration of the catalyst by addition of fresh (DOH)2pn ligand to a deactivated system was reported, which suggests ligand degradation, most likely through hydrogenation.25b,44 The reduction of CoP3 could also lead to a ligand radical species (CoIIL˙, L = ligand) instead of the formal CoI species.24 Reductive coupling of two CoIIL˙ radical species might result in the formation of catalytically inactive dimer complexes.45 The formation of a Co-containing solid-state deposit would be another possible degradation pathway.46 The absence of photocatalytic activity after several hours of irradiation, the recovery of activity by addition of fresh CoP3 and the lack of activity when replacing CoP3 with CoBr2 support that a molecular Co species is the catalyst in the RuP|TiO2|CoP3 system.
When stirring CoP3 (0.1 μmol) with 5 mg TiO2 in an aqueous pH 7 TEOA solution, approximately 60% of the catalyst was attached to the particles as determined by spectrophotometry following λ = 259 nm (Fig. S27A†). RuP binds well to TiO2 and approximately 80% (λmax = 288 and 455 nm) were adsorbed in the presence of 0.1 μmol CoP3 (Fig. S27B†). The overlap of the strong absorption bands in RuP prevented the accurate determination of the CoP3 loading in the presence of RuP. Approximately 60% of photocatalytic activity remained (0.74 ± 0.27 μmol H2) when unbound CoP3 and RuP were removed from the pre-loaded particles by centrifugation and RuP|TiO2|CoP3 was resuspended in a fresh TEOA buffer solution (Table 2). This observation agrees well with the loading of CoP3 and shows that the majority of attached CoP3 remained on the particle surface and was not replaced by the dye (5 mg P25 TiO2 nanoparticles have a loading capacity of approximately 0.25 μmol RuP).6d
Full spectrum irradiation (AM 1.5G, 100 mW cm−2, no UV filter) of dye-free TiO2|CoP3 resulted in a TONCo of 17.2 ± 1.3 in pH 7 TEOA solution, demonstrating that conduction band electrons can be transferred to CoP3. The photo-H2 production activity decreased by 97% when phosphate buffer (50 mM, pH 7) was added to the system (Fig. S28†). The phosphate anions and the phosphonic acid group in CoP3 compete for surface binding sites on TiO2. This experiment demonstrates that binding of CoP3 to the TiO2 nanoparticle via the (–PO3H2) anchoring group is essential for effective electron transfer from the TiO2 conduction band to the catalyst20a and further supports that a molecular catalyst rather than a solid state deposit is active on TiO2.
Finally, an unoptimised external quantum efficiency (EQE) of 0.35 ± 0.02% was determined for the RuP|TiO2|CoP3 system (0.1 μmol RuP, 5 mg TiO2, 0.2 μmol CoP3) in an aqueous pH 7 TEOA solution (0.1 M) after 1 h irradiation at λ = 465 nm (I = 22 mW cm−2), which is close to the absorption maximum of RuP (λmax = 455 nm). This value is comparable to the previously reported EQE for RuP|TiO2|CoP1 (1.0 ± 0.2%)39 and colloidal systems containing carbon nitrides and molecular Ni catalysts (0.37 and 1.51%).12,47
In pH 4.5 AA solution, a TONCo of 18.4 ± 0.5, 9.9 ± 0.2 and 3.1 ± 0.4 was observed with RuP|TiO2|CoP3, RuP|ZrO2|CoP3 and RuP|CoP3, respectively (Table 2 and Fig. 5B). The three systems were completely deactivated after 4 h of visible light irradiation. Control experiments with CoBr2 instead of CoP3 and in the absence of CoP3, RuP, electron donor or light showed no or only trace amounts of H2 (Table S8†). The different activity of the three systems can be explained by two different mechanisms occurring under these experimental conditions (pH 4.5, AA). Previous studies have shown that RuP* is readily quenched oxidatively on TiO2 by electron transfer to the TiO2 conduction band in the picosecond time-scale,9,48 whereas RuP* undergoes reductive quenching by AA to generate RuP− in solution or in the ZrO2 system.9 Inefficient photocatalytic H2 evolution has been previously reported for [CoX2(DO)(DOH)pn] complexes in combination with a Ru-dye in AA.49 The oxidative quenching pathway in the TiO2-containing system provides a possible explanation for the improved photocatalytic activity of RuP|TiO2|CoP3.
The initial lag period of photo-H2 evolution in AA was dependent on the ratio of CoP3 to RuP and is presumably due to the slow accumulation of CoI species, which is required to enter the catalytic cycle. An increased lag phase with enhanced photostability and a higher final TONCo was observed in all three photocatalytic systems when changing the CoP3:RuP ratio from 1:1 to 2:1. At a CoP3:RuP ratio of 1:2, a reduced lag phase with a shorter lifetime of photocatalysis and a somewhat lower final TONCo was achieved (Table S7 and Fig. S29†). Recovery of the photocatalytic activity of RuP|TiO2|CoP3 by addition of either fresh CoP3 or RuP was not successful suggesting simultaneous degradation of both, dye and catalyst. By providing new CoP3 and RuP, the initial photocatalytic activity of the RuP|TiO2|CoP3 system could be regained (Fig. S30†). Photo-degradation of RuP in AA has been observed previously.9 Similar pathways as discussed above might account for degradation of the Co catalyst in an aqueous AA solution.
Finally, the photocatalytic activity of the colloidal RuP|TiO2|CoP3 system was compared to the activity of CoP1 and CoP2 using standard conditions (0.1 μmol CoPn and 0.1 μmol RuP on 5 mg TiO2). In TEOA solution (0.1 M, pH 7), a TONCo of 56.6 ± 2.2 was obtained for CoP1,20b whereas the RuP|TiO2|CoP2 system only produced small amounts of H2 (TONCo = 2.4 ± 0.1; Table 2 and Fig. S31A†). In AA at pH 4.5, only traces of H2 were produced with CoP1, which is catalytically unstable under acidic conditions (see above). A TONCo of approximately 1 was achieved for CoP2 during 4 h visible light irradiation in AA (Table 2, Fig. S31B†).
The results from photocatalytic experiments are in agreement with trends observed during electrochemical investigation of the three catalysts: CoP1 shows the fastest turnover rate at neutral pH, whereas CoP3 is the most active catalyst in an aqueous acidic solution. However, CoP3 is the best and most suitable catalyst when activity and stability on the metal oxide surface are taken into account. CoP2 displays strong attachment to metal oxides, but it shows overall modest catalytic activity. CoP1 is not stable during turnover in a pH 4.5 AA solution and can therefore not act as a catalyst under acidic conditions. The high photoactivity of CoP1 at pH 7 despite its labile anchoring to RuP|TiO2 particles in the colloidal suspension can be explained as follows: the Co(dmgH)2 core of CoP1 is released during catalysis but can re-coordinate to a TiO2-anchored pyridine ligand by a ‘hop-on, hop-off’ mechanism through a high probability of collision in the bulk of the suspension. When CoP1 is immobilised on an electrode such as ITO|mesoITO, however, the Co(dmgH)2 core will be released from the surface and will diffuse into the bulk solution, where it will not readily diffuse back to the electrode surface.
Conclusions
In summary, a new cobalt diimine–dioxime H2 evolution catalyst (CoP3) is described that features a stable binding site for attachment to metal oxide surfaces and a pendant pyridine ligand to enhance the catalytic activity. CoP3 was prepared in six steps and characterised by NMR, UV-vis and ATR-IR spectroscopy, mass spectrometry and elemental analysis. Electrochemical investigation of the new catalyst revealed that it is electrocatalytically active for proton reduction in aqueous solution over a wide pH range. CoP3 attaches with high loading and good stability to a mesostructured Sn-doped In2O3 electrode. We demonstrate that CoP3 produces H2 photocatalytically in dye-sensitised systems under visible light irradiation at neutral and acidic pH with different sacrificial reagents and showed that H2 evolution is improved in the presence of TiO2 particles compared to homogeneous systems. CoP3 displays significant advantages over previously reported immobilised Co catalysts as it shows a higher catalytic proton reduction activity and provides a strong and more stable anchoring to metal oxides surfaces on electrodes.Overall, our work emphasises the necessity for elaborated molecular catalyst design with regard to the assembly of efficient metal oxides–molecular catalyst hybrids and their application in (photo-)electrochemical cells. The availability of thorough experimental and theoretical studies for cobaloxime and cobalt diimine–dioxime catalysts enabled us to rationally design a catalyst with improved activity and stability on electrodes.
Acknowledgements
Support by the Christian Doppler Research Association (Austrian Federal Ministry of Science, Research and Economy and National Foundation for Research, Technology and Development), the OMV Group and the EPSRC (EP/H00338X/2) is gratefully acknowledged. We also thank Miss Manuela Gross for samples of the Ru dye used in this study.References
- (a) C.-Y. Lin, D. Mersch, D. A. Jefferson and E. Reisner, Chem. Sci., 2014, 5, 4906–4913 RSC; (b) J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed; (c) J. Barber and P. D. Tran, J. R. Soc., Interface, 2013, 10, 20120984 CrossRef PubMed; (d) C.-Y. Lin, Y.-H. Lai, D. Mersch and E. Reisner, Chem. Sci., 2012, 3, 3482–3487 RSC.
- (a) G. P. Connor, K. J. Mayer, C. S. Tribble and W. R. McNamara, Inorg. Chem., 2014, 53, 5408–5410 CrossRef CAS PubMed; (b) M. J. Rose, H. B. Gray and J. R. Winkler, J. Am. Chem. Soc., 2012, 134, 8310–8313 CrossRef CAS PubMed; (c) S. Kaur-Ghumaan, L. Schwartz, R. Lomoth, M. Stein and S. Ott, Angew. Chem., Int. Ed., 2010, 49, 8033–8036 CrossRef CAS PubMed; (d) A. M. Kluwer, R. Kapre, F. Hartl, M. Lutz, A. L. Spek, A. M. Brouwer, P. W. N. M. van Leeuwen and J. N. H. Reek, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10460–10465 CrossRef CAS PubMed; (e) G. A. N. Felton, A. K. Vannucci, J. Chen, L. T. Lockett, N. Okumura, B. J. Petro, U. I. Zakai, D. H. Evans, R. S. Glass and D. L. Lichtenberger, J. Am. Chem. Soc., 2007, 129, 12521–12530 CrossRef CAS PubMed; (f) C. Tard, X. Liu, S. K. Ibrahim, M. Bruschi, L. De Gioia, S. C. Davies, X. Yang, L.-S. Wang, G. Sawers and C. J. Pickett, Nature, 2005, 433, 610–613 CrossRef CAS PubMed.
- (a) L. Chen, M. Wang, K. Han, P. Zhang, F. Gloaguen and L. Sun, Energy Environ. Sci., 2014, 7, 329–334 RSC; (b) W. T. Eckenhoff, W. R. McNamara, P. Du and R. Eisenberg, Biochim. Biophys. Acta, 2013, 1827, 958–973 CrossRef CAS PubMed; (c) M. Guttentag, A. Rodenberg, C. Bachmann, A. Senn, P. Hamm and R. Alberto, Dalton Trans., 2013, 42, 334–337 RSC; (d) C. C. L. McCrory, C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 3164–3170 CrossRef CAS PubMed; (e) V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS PubMed; (f) Y. Sun, J. P. Bigi, N. A. Piro, M. L. Tang, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 9212–9215 CrossRef CAS PubMed; (g) J. P. Bigi, T. E. Hanna, W. H. Harman, A. Chang and C. J. Chang, Chem. Commun., 2010, 46, 958–960 RSC; (h) P. Du, J. Schneider, G. Luo, W. W. Brennessel and R. Eisenberg, Inorg. Chem., 2009, 48, 4952–4962 CrossRef CAS PubMed; (i) P.-A. Jacques, V. Artero, J. Pécaut and M. Fontecave, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20627–20632 CrossRef CAS PubMed; (j) X. Hu, B. S. Brunschwig and J. C. Peters, J. Am. Chem. Soc., 2007, 129, 8988–8998 CrossRef CAS PubMed; (k) P. Connolly and J. H. Espenson, Inorg. Chem., 1986, 25, 2684–2688 CrossRef CAS; (l) B. J. Fisher and R. Eisenberg, J. Am. Chem. Soc., 1980, 102, 7361–7363 CrossRef CAS.
- (a) P. Zhang, M. Wang, Y. Yang, D. Zheng, K. Han and L. Sun, Chem. Commun., 2014, 50, 14153–14156 RSC; (b) Z. Han, L. Shen, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2013, 135, 14659–14669 CrossRef CAS PubMed; (c) O. R. Luca, S. J. Konezny, J. D. Blakemore, D. M. Colosi, S. Saha, G. W. Brudvig, V. S. Batista and R. H. Crabtree, New J. Chem., 2012, 36, 1149–1152 RSC; (d) U. J. Kilgore, J. A. S. Roberts, D. H. Pool, A. M. Appel, M. P. Stewart, M. Rakowski DuBois, W. G. Dougherty, W. S. Kassel, R. M. Bullock and D. L. DuBois, J. Am. Chem. Soc., 2011, 133, 5861–5872 CrossRef CAS PubMed; (e) A. D. Wilson, R. H. Newell, M. J. McNevin, J. T. Muckerman, M. Rakowski DuBois and D. L. DuBois, J. Am. Chem. Soc., 2006, 128, 358–366 CrossRef CAS PubMed; (f) J.-P. Collin, A. Jouaiti and J.-P. Sauvage, Inorg. Chem., 1988, 27, 1986–1990 CrossRef CAS.
- (a) D. V. Esposito and J. G. Chen, Energy Environ. Sci., 2011, 4, 3900–3912 RSC; (b) R. B. Gordon, M. Bertram and T. E. Graedel, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1209–1214 CrossRef CAS PubMed; (c) M. Kirch, J.-M. Lehn and J.-P. Sauvage, Helv. Chim. Acta, 1979, 62, 1345–1384 CrossRef CAS.
- (a) T. Sakai, D. Mersch and E. Reisner, Angew. Chem., Int. Ed., 2013, 52, 12313–12316 CrossRef CAS PubMed; (b) P. D. Tran and J. Barber, Phys. Chem. Chem. Phys., 2012, 14, 13772–13784 RSC; (c) H. Krassen, A. Schwarze, B. Friedrich, K. Ataka, O. Lenz and J. Heberle, ACS Nano, 2009, 3, 4055–4061 CrossRef CAS PubMed; (d) E. Reisner, D. J. Powell, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 18457–18466 CrossRef CAS PubMed; (e) W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- (a) Y. Gao, X. Ding, J. Liu, L. Wang, Z. Lu, L. Li and L. Sun, J. Am. Chem. Soc., 2013, 135, 4219–4222 CrossRef CAS PubMed; (b) A. Krawicz, J. Yang, E. Anzenberg, J. Yano, I. D. Sharp and G. F. Moore, J. Am. Chem. Soc., 2013, 135, 11861–11868 CrossRef CAS PubMed; (c) G. F. Moore and I. D. Sharp, J. Phys. Chem. Lett., 2013, 4, 568–572 CrossRef CAS; (d) L. Alibabaei, M. K. Brennaman, M. R. Norris, B. Kalanyan, W. Song, M. D. Losego, J. J. Concepcion, R. A. Binstead, G. N. Parsons and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20008–20013 CrossRef CAS PubMed.
- (a) M. Wang, L. Chen, X. Li and L. Sun, Dalton Trans., 2011, 40, 12793–12800 RSC; (b) C. J. Curtis, A. Miedaner, R. Ciancanelli, W. W. Ellis, B. C. Noll, M. Rakowski DuBois and D. L. DuBois, Inorg. Chem., 2003, 42, 216–227 CrossRef CAS PubMed.
- M. A. Gross, A. Reynal, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2014, 136, 356–366 CrossRef CAS PubMed.
- A. Dutta, S. Lense, J. Hou, M. H. Engelhard, J. A. S. Roberts and W. J. Shaw, J. Am. Chem. Soc., 2013, 135, 18490–18496 CrossRef CAS PubMed.
- (a) A. K. Das, M. H. Engelhard, R. M. Bullock and J. A. S. Roberts, Inorg. Chem., 2014, 53, 6875–6885 CrossRef CAS PubMed; (b) A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Métayé, A. Fihri, S. Palacin and M. Fontecave, Science, 2009, 326, 1384–1387 CrossRef CAS PubMed.
- C. A. Caputo, M. A. Gross, V. W. Lau, C. Cavazza, B. V. Lotsch and E. Reisner, Angew. Chem., Int. Ed., 2014, 53, 11538–11542 CrossRef CAS PubMed.
- F. Wang, W.-G. Wang, X.-J. Wang, H.-Y. Wang, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2011, 50, 3193–3197 CrossRef CAS PubMed.
- S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997–17003 CrossRef CAS PubMed.
- F. Quentel, G. Passard and F. Gloaguen, Energy Environ. Sci., 2012, 5, 7757–7761 CAS.
- (a) D. W. Wakerley and E. Reisner, Phys. Chem. Chem. Phys., 2014, 16, 5739–5746 RSC; (b) M. Razavet, V. Artero and M. Fontecave, Inorg. Chem., 2005, 44, 4786–4795 CrossRef CAS PubMed.
- (a) B. H. Solis, Y. Yu and S. Hammes-Schiffer, Inorg. Chem., 2013, 52, 6994–6999 CrossRef CAS PubMed; (b) B. H. Solis and S. Hammes-Schiffer, Inorg. Chem., 2011, 50, 11252–11262 CrossRef CAS PubMed; (c) J. T. Muckerman and E. Fujita, Chem. Commun., 2011, 47, 12456–12458 RSC; (d) B. H. Solis and S. Hammes-Schiffer, J. Am. Chem. Soc., 2011, 133, 19036–19039 CrossRef CAS PubMed; (e) T. Lazarides, T. McCormick, P. Du, G. Luo, B. Lindley and R. Eisenberg, J. Am. Chem. Soc., 2009, 131, 9192–9194 CrossRef CAS PubMed; (f) J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed; (g) P. Du, K. Knowles and R. Eisenberg, J. Am. Chem. Soc., 2008, 130, 12576–12577 CrossRef CAS PubMed; (h) C. Baffert, V. Artero and M. Fontecave, Inorg. Chem., 2007, 46, 1817–1824 CrossRef CAS PubMed; (i) X. Hu, B. M. Cossairt, B. S. Brunschwig, N. S. Lewis and J. C. Peters, Chem. Commun., 2005, 4723–4725 RSC.
- F. Lakadamyali, M. Kato, N. M. Muresan and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 9381–9384 CrossRef CAS PubMed.
- (a) B. S. Veldkamp, W.-S. Han, S. M. Dyar, S. W. Eaton, M. A. Ratner and M. R. Wasielewski, Energy Environ. Sci., 2013, 6, 1917–1928 RSC; (b) A. Fihri, V. Artero, A. Pereira and M. Fontecave, Dalton Trans., 2008, 5567–5569 RSC; (c) A. Fihri, V. Artero, M. Razavet, C. Baffert, W. Leibl and M. Fontecave, Angew. Chem., Int. Ed., 2008, 47, 564–567 CrossRef CAS PubMed.
- (a) F. Lakadamyali, M. Kato and E. Reisner, Faraday Discuss., 2012, 155, 191–205 RSC; (b) F. Lakadamyali and E. Reisner, Chem. Commun., 2011, 47, 1695–1697 RSC.
- X.-W. Song, H.-M. Wen, C.-B. Ma, H.-H. Cui, H. Chen and C.-N. Chen, RSC Adv., 2014, 4, 18853–18861 RSC.
- (a) A. Krawicz, D. Cedeno and G. F. Moore, Phys. Chem. Chem. Phys., 2014, 16, 15818–15824 RSC; (b) Z. Ji, M. He, Z. Huang, U. Ozkan and Y. Wu, J. Am. Chem. Soc., 2013, 135, 11696–11699 CrossRef CAS PubMed.
- T. M. McCormick, Z. Han, D. J. Weinberg, W. W. Brennessel, P. L. Holland and R. Eisenberg, Inorg. Chem., 2011, 50, 10660–10666 CrossRef CAS PubMed.
- N. M. Muresan, J. Willkomm, D. Mersch, Y. Vaynzof and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 12749–12753 CrossRef CAS PubMed.
- (a) A. Bhattacharjee, E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave, M. J. Field and V. Artero, Chem.–Eur. J., 2013, 19, 15166–15174 CrossRef CAS PubMed; (b) P. Zhang, P.-A. Jacques, M. Chavarot-Kerlidou, M. Wang, L. Sun, M. Fontecave and V. Artero, Inorg. Chem., 2012, 51, 2115–2120 CrossRef CAS PubMed; (c) B. Probst, M. Guttentag, A. Rodenberg, P. Hamm and R. Alberto, Inorg. Chem., 2011, 50, 3404–3412 CrossRef CAS PubMed.
- E. S. Andreiadis, P.-A. Jacques, P. D. Tran, A. Leyris, M. Chavarot-Kerlidou, B. Jousselme, M. Matheron, J. Pécaut, S. Palacin, M. Fontecave and V. Artero, Nat. Chem., 2013, 5, 48–53 CrossRef CAS PubMed.
- M. R. J. Scherer, N. M. Muresan, U. Steiner and E. Reisner, Chem. Commun., 2013, 49, 10453–10455 RSC.
- F. Tayyari, D. E. Wood, P. E. Fanwick and R. E. Sammelson, Synthesis, 2008, 279–285 CAS.
- L. G. Marzilli, A. Gerli and A. M. Calafat, Inorg. Chem., 1992, 31, 4617–4627 CrossRef CAS.
- J. Xie, Q. Zhou, C. Li, W. Wang, Y. Hou, B. Zhang and X. Wang, Chem. Commun., 2014, 50, 6520–6522 RSC.
- V. Fourmond, S. Canaguier, B. Golly, M. J. Field, M. Fontecave and V. Artero, Energy Environ. Sci., 2011, 4, 2417–2427 CAS.
- J. L. Dempsey, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2010, 132, 1060–1065 CrossRef CAS PubMed.
- J. J. Spivey and K. M. Dooley, Catalysis, The Royal Society of Chemistry, Cambridge, 2007 Search PubMed.
- A. Adin and J. H. Espenson, Inorg. Chem., 1972, 11, 686–688 CrossRef CAS.
- (a) E. Reisner, V. B. Arion, M. F. C. Guedes da Silva, R. Lichtenecker, A. Eichinger, B. K. Keppler, V. Y. Kukushkin and A. J. L. Pombeiro, Inorg. Chem., 2004, 43, 7083–7093 CrossRef CAS PubMed; (b) A. B. P. Lever, Inorg. Chem., 1990, 29, 1271–1285 CrossRef CAS.
- P. G. Hoertz, Z. Chen, C. A. Kent and T. J. Meyer, Inorg. Chem., 2010, 49, 8179–8181 CrossRef CAS PubMed.
- H. Park, E. Bae, J.-J. Lee, J. Park and W. Choi, J. Phys. Chem. B, 2006, 110, 8740–8749 CrossRef CAS PubMed.
- V. Balzani, G. Bergamini, F. Marchioni and P. Ceroni, Coord. Chem. Rev., 2006, 250, 1254–1266 CrossRef CAS PubMed.
- F. Lakadamyali, A. Reynal, M. Kato, J. R. Durrant and E. Reisner, Chem.–Eur. J., 2012, 18, 15464–15475 CrossRef CAS PubMed.
- (a) Y. Xu and M. A. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CAS; (b) J. M. Bolts and M. S. Wrighton, J. Phys. Chem., 1976, 80, 2641–2645 CrossRef CAS.
- K. Sayama and H. Arakawa, J. Phys. Chem., 1993, 97, 531–533 CrossRef CAS.
- A. Reynal, J. Willkomm, N. M. Muresan, F. Lakadamyali, M. Planells, E. Reisner and J. R. Durrant, Chem. Commun., 2014, 50, 12768–12771 RSC.
- (a) H. Sun and M. Z. Hoffman, J. Phys. Chem., 1994, 98, 11719–11726 CrossRef CAS; (b) K. Kalyanasundaram, J. Kiwi and M. Grätzel, Helv. Chim. Acta, 1978, 61, 2720–2730 CrossRef CAS.
- L. I. Simándi, Z. Szeverényi and É. Budó-Záhonyi, Inorg. Nucl. Chem. Lett., 1975, 11, 773–777 CrossRef.
- E. B. Hulley, P. T. Wolczanski and E. B. Lobkovsky, J. Am. Chem. Soc., 2011, 133, 18058–18061 CrossRef CAS PubMed.
- S. Cobo, J. Heidkamp, P.-A. Jacques, J. Fize, V. Fourmond, L. Guetaz, B. Jousselme, V. Ivanova, H. Dau, S. Palacin, M. Fontecave and V. Artero, Nat. Mater., 2012, 11, 802–807 CrossRef CAS PubMed.
- J. Dong, M. Wang, X. Li, L. Chen, Y. He and L. Sun, ChemSusChem, 2012, 5, 2133–2138 CrossRef CAS PubMed.
- A. Reynal, F. Lakadamyali, M. A. Gross, E. Reisner and J. R. Durrant, Energy Environ. Sci., 2013, 6, 3291–3300 CAS.
- S. Varma, C. E. Castillo, T. Stoll, J. Fortage, A. G. Blackman, F. Molton, A. Deronzier and M.-N. Collomb, Phys. Chem. Chem. Phys., 2013, 15, 17544–17552 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and tables, synthetic procedures, experimental details for NMR and UV-vis spectroscopy, electrochemistry and photocatalytic experiments. See DOI: 10.1039/c4sc03946g |
| This journal is © The Royal Society of Chemistry 2015 |