
TEMP and copper cocatalyzed oxygenation of ketones with molecular oxygen: chemoselective synthesis of α-ketoesters†
Xiaoqiang
Huang
a,
Xinwei
Li
a,
Miancheng
Zou
a,
Jun
Pan
a and
Ning
Jiao
*ab
aState Key Laboratory of Natural and Biomimetic Drugs, Peking University, School of Pharmaceutical Sciences, Peking University, Xue Yuan Rd. 38, Beijing 100191, China. E-mail: jiaoning@pku.edu.cn; Fax: +86-010-8280-5297
bState Key Laboratory of Elemento-organic Chemistry, Nankai University, 94 Weijin Road, Tianjin 300071, China
First published on 11th February 2015
Abstract
A novel and practical method for the synthesis of α-ketoesters through TEMP and copper cocatalyzed chemoselective oxidative coupling of commercially available methyl ketones with alcohols is developed. Mild conditions and broad substrate scope make this chemistry an efficient tool for the late-stage modification of bioactive compounds. Detailed mechanistic studies demonstrate a novel mechanism involving an organocatalytic cycle and SET process. The oxygenation process with molecular oxygen enables this transformation.
To construct α-ketoesters is among the most important transformations in organic synthesis, not only because they are widely found in bioactive molecules1 but also because they are used as versatile building blocks for further transformations.2 So far, various procedures have been developed, such as esterification of α-ketoacyl derivatives,3 palladium-catalyzed carbonylation,4 and dehydrogenation of α-hydroxyl ester.5 However, these methods usually suffer from harsh reaction conditions, low atom economy, and the usage of a noble metal catalyst.6
Due to the cheapness and low toxicity of copper catalysts and natural, abundant, and environmentally friendly character of molecular oxygen, copper catalyzed aerobic oxidation7,8 and oxygenation7,9 represents an attractive strategy for chemists in view of green and sustainable chemistry. Accordingly, our group reported two approaches to α-ketoesters via copper catalyzed aerobic oxidative esterification of alcohols with 1,3-diones10 (eqn (a), Scheme 1) or α-carbonyl aldehydes11 (eqn (b), Scheme 1). However, the relatively low atom efficiency and the need for extra steps to prepare starting materials might limit their applications. To address these problems, we have now developed an efficient and practical approach to α-ketoesters directly starting from simple and commercially available methyl ketones (eqn (c), Scheme 1). Very recently, we reported a copper catalyzed aerobic oxidative coupling of simple ketones with alcohols to furnish esters via C–C bond cleavage employing 200 mol% pyridine.12 It is very interesting that when pyridine is replaced with TEMP (2,2,6,6-tetramethylpiperidine) which is more electron-rich and sterically demanding compound, a complete chemoselectivity switch happens, resulting in the generation of α-ketoesters (Scheme 2). During the preparation of this manuscript, Song and co-workers reported a copper catalyzed esterification of acetophenones with alcohols to α-ketoesters with the addition of pyridine (50 mol%) as ligand and TFA (50 mol%) as additive.13 Herein, we describe a novel TEMP and copper cocatalyzed oxygenation of ketones with molecular oxygen. In this chemistry: (1) A practical, atom and step economic method to α-ketoesters is achieved via direct coupling of readily available methyl ketones with alcohols; (2) The preference for chemoselective oxidative C–H bond functionalization over C–C bond cleavage is significantly controlled by reaction conditions; (3) The oxygenation process with molecular oxygen enables this transformation. (4) Detailed mechanistic studies indicate an organocatalytic cycle and SET (single electron transfer) process are involved in this reaction.
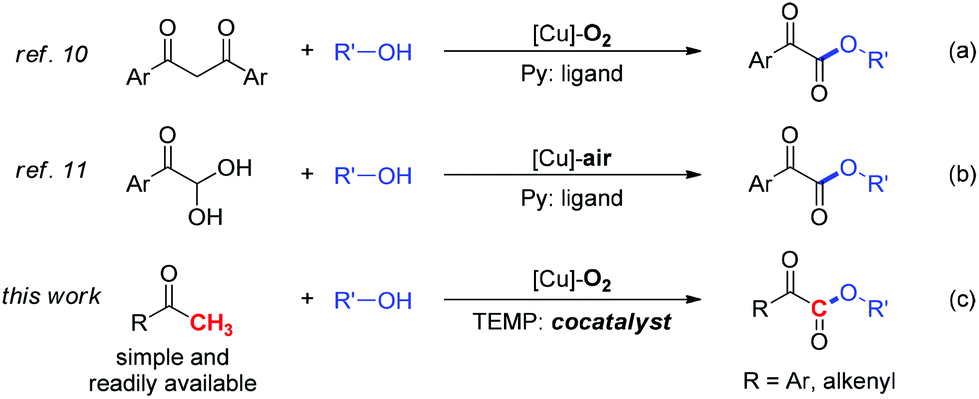 | ||
| Scheme 1 Approaches to α-ketoesters via Cu-catalyzed aerobic oxidative coupling reactions. | ||
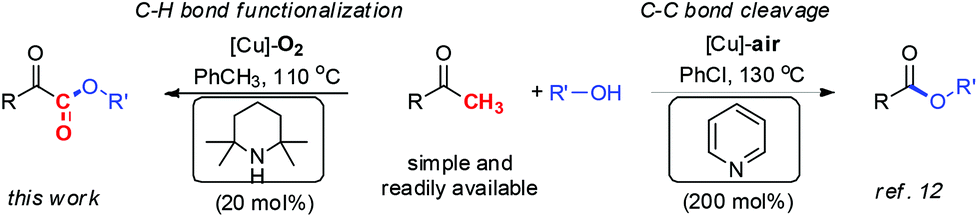 | ||
| Scheme 2 Reaction conditions controlled chemoselective coupling of methyl ketones with alcohols. | ||
We commenced our study by carrying out copper-catalyzed aerobic oxidative coupling of acetophenone (1a) with cyclohexanol (2a). Employing reaction conditions of our previous work,12 C–C bond cleavage product 3aa′ was obtained as the main product while the desired C–H bond functionalization product 3aa is produced in 11% yield (entry 1, Table 1). Only 4% yield of 3aa was detected under the reaction conditions of ref. 11 (entry 2). Encouraged by these results, a variety of ligands were screened (entries 3–13). To our delight, when 20 mol% of TEMP was used, the best results in yield and chemoselectivity for 3aa over 3aa′ were obtained (entry 6). As expected, no product was generated under argon atmosphere (entry 14). The reaction did not work without copper catalyst or TEMP (entries 15–16). After further evaluation of extensive reaction parameters (see ESI† for more information), the chemoselective aerobic oxidative coupling of 1a with 2a delivered α-ketoesters 3aa in 75% yield under the standard conditions: CuBr (10 mol%), TEMP (20 mol%) in PhCH3 under O2 at 110 °C (entry 6, Table 1).
|
|
|||
|---|---|---|---|
| Entry | Ligand | Yield of 3aab (%) | Yield of 3aa′b (%) |
| a Reaction conditions: 1a (0.40 mmol), 2a (0.80 mmol), CuBr (0.04 mmol), and ligand (0.08 mmol) in PhCH3 (2.0 mL) under O2 (balloon) was stirred at 110 °C for 10 h. b GC yields, isolated yield is listed in parentheses. c Conditions of ref. 12: pyridine (200 mol%) in PhCl (0.5 mL) under air at 130 °C. d Conditions of ref. 11: pyridine (50 mol%) under air at 90 °C for 18 h. e Under Ar. f Without CuBr. Py = pyridine; DMAP = N,N-dimethyl-4-aminopyridine; TEMP = 2,2,6,6-tetramethylpiperidine; 1,10-Phen = phenanthroline; 2,2′-Bpy = 2,2′-bipyridine. | |||
| 1c | Py | 11 | 52 |
| 2d | Py | 4 | 1 |
| 3 | Py | 27 | 3 |
| 4 | DMAP | 25 | 33 |
| 5 | 2,4-Di-Me-Py | 22 | 1 |
| 6 | TEMP | 75 (70) | 2 |
| 7 | Piperidine | 0 | 0 |
| 8 | Pyrrole | 0 | 0 |
| 9 | (iPr)2NH | 9 | 0 |
| 10 | (Cy)2NH | 6 | 1 |
| 11 | 1,10-Phen | 0 | 0 |
| 12 | 2,2′-Bpy | 4 | 0 |
| 13 | L-Proline | 0 | 0 |
| 14e | TEMP | 0 | 0 |
| 15f | TEMP | 0 | 0 |
| 16 | None | 0 | 0 |
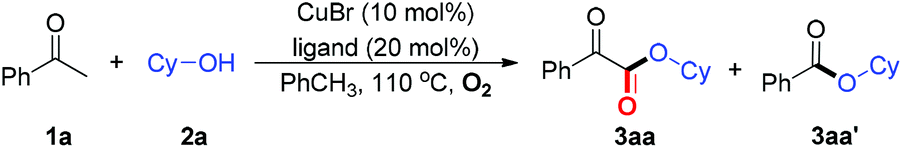
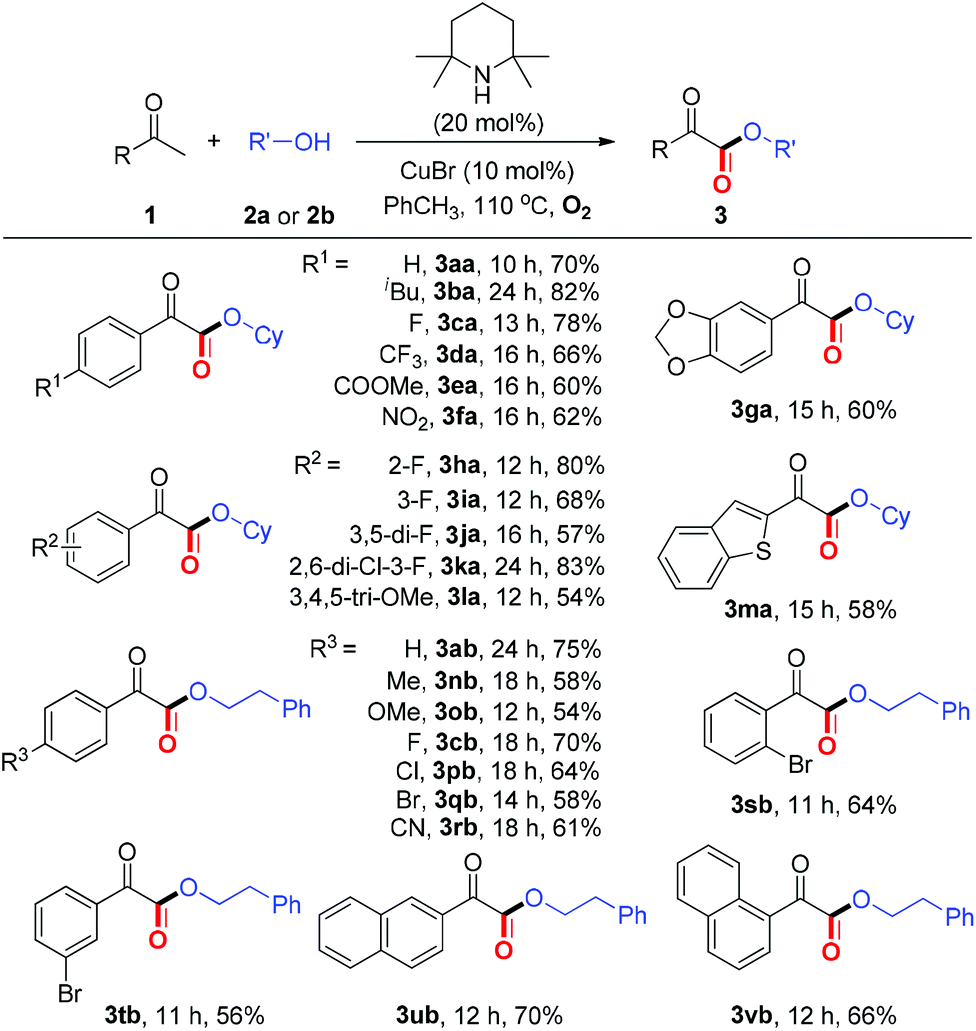
With the optimized reaction conditions in hand, we next examined the substrate scope of aryl methyl ketones. As summarized in Table 2, moderate to good yields, ranging from 54%–83%, were obtained for the coupling of ketones (1a–1v) bearing either electron-rich or electron deficient substituents with alcohols to form α-ketoesters 3. A wide range of functional groups including –halo, –CF3, –COOMe, –CN, were well tolerated under the present protocol. Furthermore, the reaction with benzothiophen-2-yl methyl ketones proceeded smoothly to generate the corresponding α-ketoester (3ma). It is noteworthy that all these ketones (1a–1v) are commercially available, which demonstrates the present method is very practical.
| a Reaction conditions: 1 (0.40 mmol), 2 (0.80 mmol), CuBr (0.04 mmol), and TEMP (0.08 mmol) in PhCH3 (2.0 mL) under O2 (balloon) was stirred at 110 °C. Isolated yields. |
|---|
|
Then, we turned our attention to investigating the reaction scope with respect to alcohols (Table 3). To our satisfaction, several secondary alcohols were found to be compatible under the current aerobic oxidative conditions (2c–2d). Various primary alcohols, regardless of the electronic variation and position of the substituents on the aromatic ring, performed well to afford the corresponding α-ketoesters (3ae–3as). Significantly, alcohols bearing an ester group, pyridine ring, azido group, and alkenyl group (2p–2s) were also well tolerated.
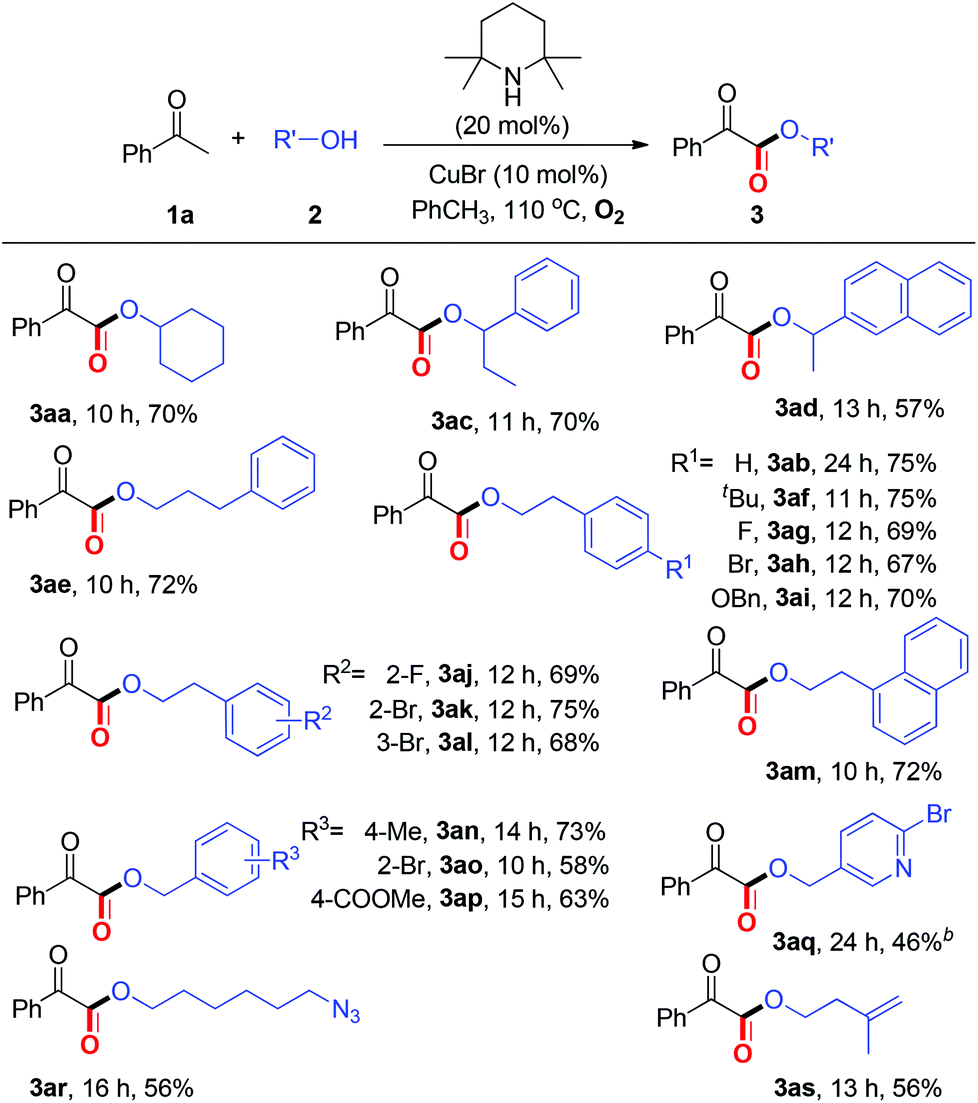
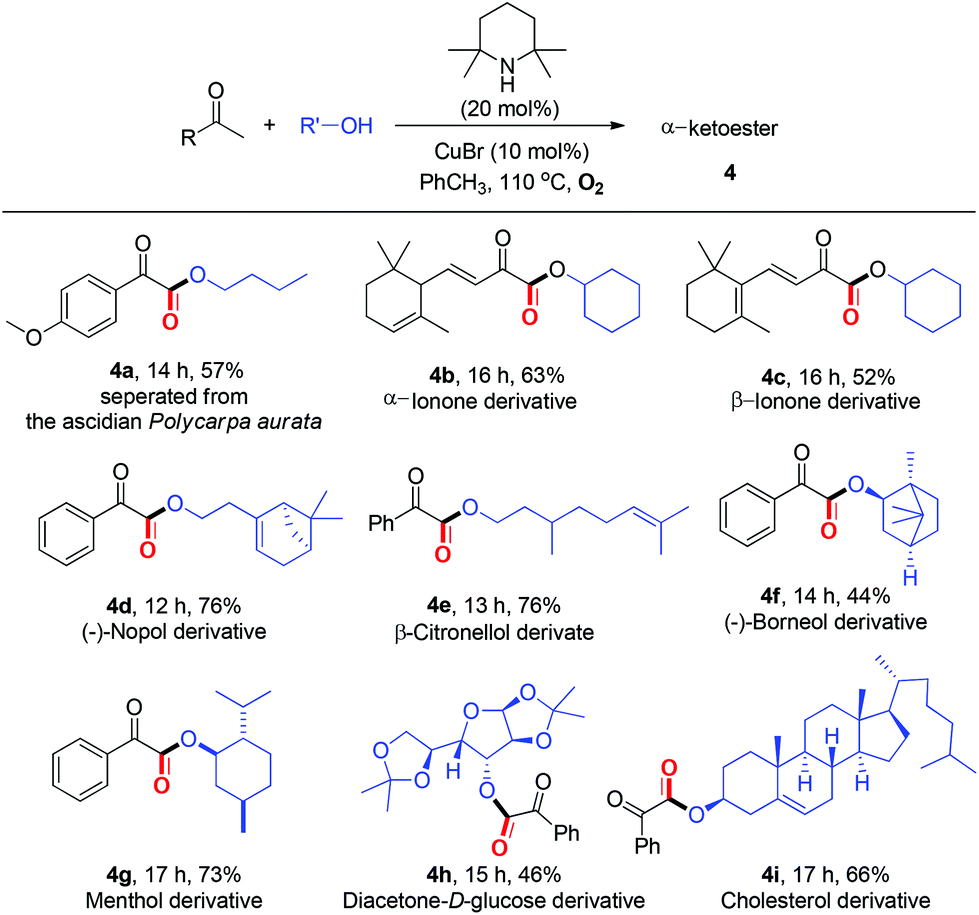
Within the broad substrate scope and the mild reaction conditions, we tested the possibility of the current esterification for the late-stage functionalization14 of natural ketones and alcohols (Table 4). Remarkably, natural product 4a could be constructed in 57% yield by one step directly from commercially available methyl ketone 1n and nBuOH under standard conditions. Alkenyl methyl ketones, α/β-ionones, which are important spices, were proved to be competent (4b–4c). Notably, several natural alcohols, such as β-citronellol, diacetone-D-glucose, and cholesterol, underwent the desired esterification to afford complex α-ketoesters (4d–4i). Especially in point of the fact that α-ketoester moieties are widely present in natural products, biologically active and synthetically useful compounds,1,2b the feasibility of the present protocol for late-stage functionalization is highly significant.
| a Reaction conditions: ketone (0.40 mmol), alcohol (0.80 mmol), CuBr (0.04 mmol), and TEMP (0.08 mmol) in PhCH3 (2.0 mL) under O2 (balloon) was stirred at 110 °C. Isolated yields. |
|---|
|
The first hint about the reaction mechanism was found when some potential intermediates were subjected to the standard conditions. Phenylglyoxal monohydrate 5a was converted into 3aa in 89% yield, which is slightly higher than the corresponding reaction with ketone 1a. Furthermore, no 3aa was obtained when 5a was reacted with 2a under conditions in the absence of copper, or TEMP or molecular oxygen (eqn (1)). In addition, 6 and 7 gave the desired product in much lower yields (eqn (2)–(3)). In combination with our previous research,11 a plausible mechanism might take the formation of a phenylglyoxal monohydrate intermediate into account.
 | (1) |
 | (2) |
 | (3) |
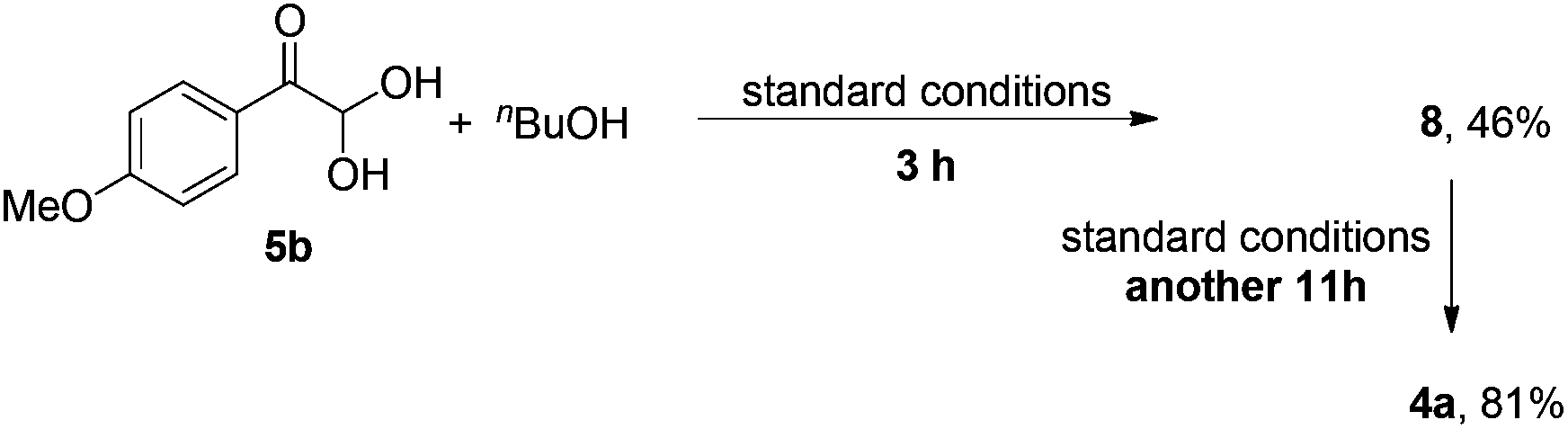
Although the isolation of phenylglyoxal monohydrate failed, α-carbonyl hemiacetal 8 was separated in 4% yield from the reaction mixture of 1o with nBuOH at 3 h (eqn (4)). In addition, when 5b was subjected to the standard conditions, 8 could be isolated in 46% yield at 3 h and complete formation of α-ketoester 4a occurred after stirring another 11 h (eqn (5)). These results demonstrate that the α-hydroxyl ester, generated from the addition of alcohol to phenylglyoxal monohydrate, might be a key intermediate in the process.
 | (4) |
 | (5) |
Furthermore, the esterification reaction was totally inhibited when TEMPO (2,2,6,6-tetramethyl-piperidinooxy), or BHT (2,6-di-tert-4-methylphenol) were employed as radical scavengers (eqn (6)). To further confirm whether a radical process is involved, EPR (electroparamagnetic resonance) experiments were carried out. Employing DMPO (5,5-dimethyl-1-pyrroline N-oxide) as a free radical spin trapping agent, signals with classic four peaks were observed, which were identified as signals correspond to DMPO-(OH) [g = 2.022, αN = αH = 15.0 G] (Scheme 3).15 These signals did not appear without ketone, copper or O2 (see ESI† for more information). These results imply a hydroxyl radical might be generated during the transformation.
 | (6) |
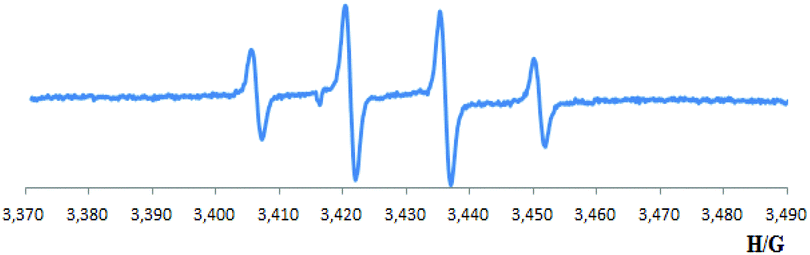 | ||
| Scheme 3 The EPR spectrum (X band, 9.7 GHz, RT) of DMPO-(OH). | ||
In addition, an intermolecular competition experiment was conducted by subjecting an equivalent amount of 1o and deuterated 1o-D3 to the standard conditions. After stopping the reaction at 3 h, the value of kH/kD was evaluated as 2.18, which suggested that Csp3–H bond cleavage might be involved in the rate-determining step (eqn (7)).16
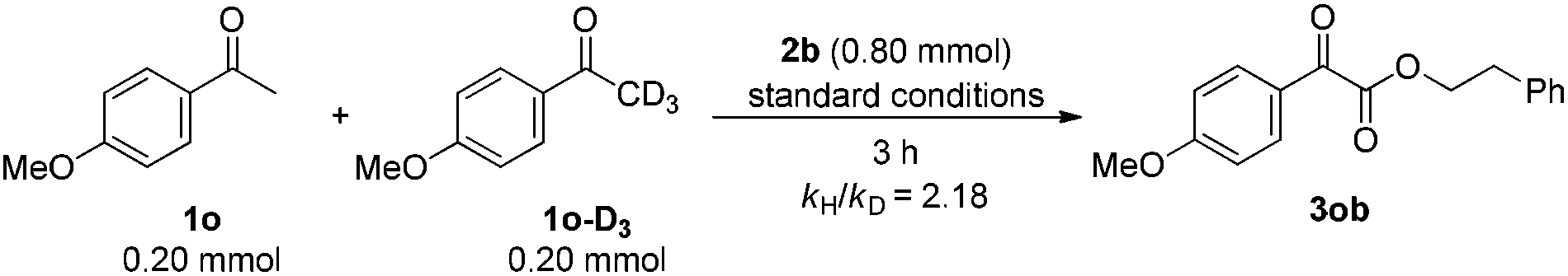 | (7) |
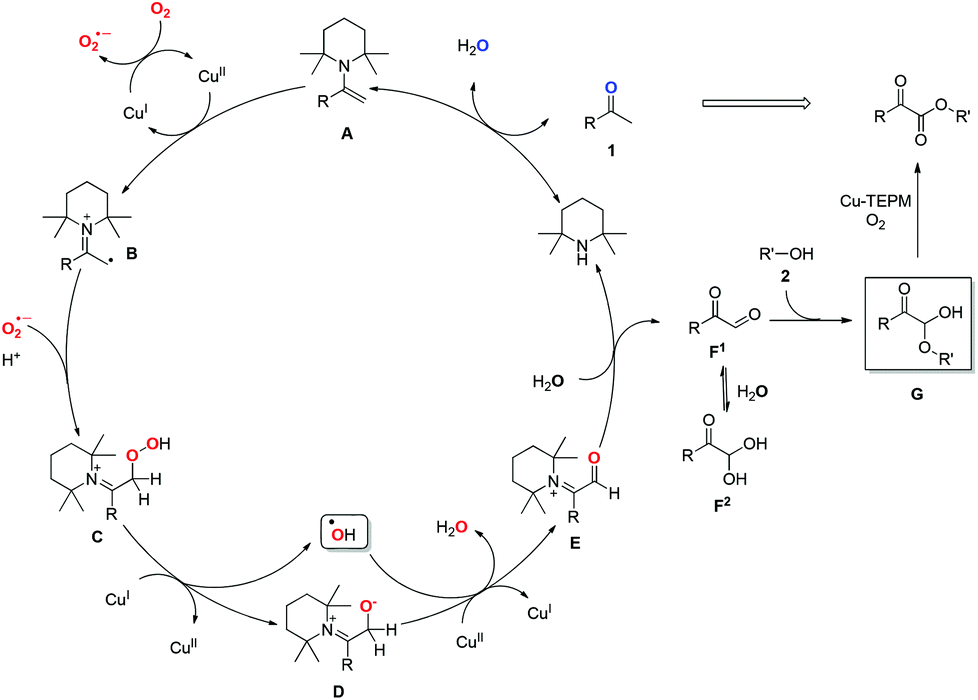
Based on the above results and the previous reports, mechanistic details for this transformation are proposed in Scheme 4. Initially, condensation of secondary amine catalyst TEMP with methyl ketone 1 generates an electron-rich enamine A.17 The facile single electron oxidation of A by CuII would afford iminyl radical cation B.18 Subsequently, coupling of B with superoxide radical anion (O2˙−), which is formed via the reaction of CuI and O2 in the presence of enamine A or N-ligand TEMP,19 leads to the formation of peroxide intermediate C. Then, the SET20 reduction of C by CuI generates anion intermediate D along with the release of hydroxyl radical, which has been trapped by EPR.10,15 Subsequent oxidation of D by CuII and hydroxyl radical affords α-carbonyl iminyl cation E. The following hydrolysis of E would release the key intermediate phenylglyoxal F1 and regenerate amine catalyst TEMP, thereby completing the organocatalytic cycle.21 The addition of alcohol to F would afford α-carbonyl hemiacetal G, which could be separated from reaction mixture. Finally, oxidation of G produces α-ketoester under the copper–TEMP–O2 system.11
 | ||
| Scheme 4 The proposed mechanism. | ||
Notably, TEMP is indispensable in this transformation. Firstly, it serves as an amine catalyst to activate the ketones, facilitating the selective C–H bond oxidation over C–C bond cleavage owing to its electron-rich and sterically demanding features. In addition, it plays the role of ligand, improving the oxidative ability of the copper catalyzed aerobic oxidation system.
To further probe the proposed mechanism, 18O isotope experiments were investigated. When the reaction was performed in the presence of H218O (200 mol%), the result that 3aa–16O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3aa-16O218O:3aa–16O18O2 = 2.8:1.2:1 was observed (eqn (8)) (calculated by HRMS, and the 18O labelling at keto-carbonyl group of product was detected by GC-MS (m/z = 107), see ESI† for more information), which supports the reversible condensation of ketone and amine, as well as the hydrolysis of E. The observation of 3aa–16O18O2 might be reasonably accounted for by the equilibrium between F1 and F2. In addition, no product could be obtained by adding 4 Å MS into the standard conditions, which further confirms the participation of water. Because oxygen atom exchange of H2O with ketones and phenylglyoxal monohydrate intermediates occurs, an isotope experiment with 18O2, which would generate H218O, could not give solid proof about the O-source of new forming ester-carbonyl group.
:3aa-16O218O:3aa–16O18O2 = 2.8:1.2:1 was observed (eqn (8)) (calculated by HRMS, and the 18O labelling at keto-carbonyl group of product was detected by GC-MS (m/z = 107), see ESI† for more information), which supports the reversible condensation of ketone and amine, as well as the hydrolysis of E. The observation of 3aa–16O18O2 might be reasonably accounted for by the equilibrium between F1 and F2. In addition, no product could be obtained by adding 4 Å MS into the standard conditions, which further confirms the participation of water. Because oxygen atom exchange of H2O with ketones and phenylglyoxal monohydrate intermediates occurs, an isotope experiment with 18O2, which would generate H218O, could not give solid proof about the O-source of new forming ester-carbonyl group.
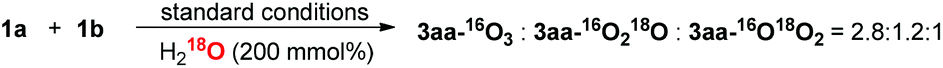 | (8) |
Conclusions
In conclusion, we have reported a novel TEMP and copper cocatalyzed aerobic oxidative coupling of methyl ketones with alcohols for the chemoselective synthesis of valuable α-ketoesters. Electron-rich and sterically demanding TEMP plays a key role in this transformation. Starting from commercially available substrates, a wide range of α-ketoesters are prepared under mild conditions. The oxygenation process with molecular oxygen enabled this transformation. On the basis of control experiments, EPR experiments, and isotopic labelling experiments, a proposed mechanism involving an organocatalytic cycle and SET process is illustrated. Further investigations on the synthetic applications are ongoing in our group.Acknowledgements
Financial support from National Basic Research Program of China (973 Program) (grant no. 2015CB856600) and National Natural Science Foundation of China (no. 21325206, 21172006), and National Young Top-notch Talent Support Program are greatly appreciated. We thank Zun Wang in this group for reproducing the results of 3ac and 4a.Notes and references
- (a) M. Wessels, G. M. König and A. D. Wright, J. Nat. Prod., 2001, 64, 1556 CrossRef CAS PubMed; (b) M. F. Elsebai, S. Kehraus, U. Lindequist, F. Sasse, S. Shaaban, M. Gutschow, M. Josten, H.-G. Sahl and G. M. König, Org. Biomol. Chem., 2011, 9, 802 RSC; (c) B. A. Boughton, L. Hor, J. A. Gerrard and C. A. Hutton, Bioorg. Med. Chem., 2012, 20, 2419 CrossRef CAS PubMed.
- (a) D. Zhu, Y. Yang, J. D. Buynak and L. Hua, Org. Biomol. Chem., 2006, 4, 2690 RSC; (b) M. Hayashi and S. Nakamura, Angew. Chem., Int. Ed., 2011, 50, 2249 CrossRef CAS PubMed; (c) B. Eftekhari-Sis and M. Zirak, Chem. Rev., 2014, 115, 151 CrossRef PubMed; (d) W. Zhao, P. K. Yan and A. T. Radosevich, J. Am. Chem. Soc., 2015, 137, 616 CrossRef CAS PubMed.
- (a) F. Babudri, V. Fiandanese, G. Marchese and A. Punzi, Tetrahedron, 1996, 52, 13513 CrossRef CAS; (b) M. A. de las Heras, J. J. Vaquero, J. L. García-Navio and J. Alvarez-Builla, J. Org. Chem., 1996, 61, 9009 CrossRef PubMed; (c) D. Enders, M. H. Bonten and G. Raabe, Angew. Chem., Int. Ed., 2007, 46, 2314 CrossRef CAS PubMed; (d) J. M. Xiang and B. L. Li, Chin. Chem. Lett., 2009, 20, 55 CrossRef CAS.
- (a) T. Sakakura, H. Yamashita, T. Kobayashi, T. Hayashi and M. Tanaka, J. Org. Chem., 1987, 52, 5733 CrossRef CAS; (b) F. Ozawa, N. Kawasaki, H. Okamoto, T. Yamamoto and A. Yamamoto, Organometallics, 1987, 6, 1640 CrossRef CAS.
- (a) M. Tanaka, T.-a. Kobayashi and T. Sakakura, Angew. Chem., Int. Ed. Engl., 1984, 23, 518 CrossRef; (b) E. V. Johnston, E. A. Karlsson, L.-H. Tran, B. Åkermark and J.-E. Bäckvall, Eur. J. Org. Chem., 2010, 1971 CrossRef CAS; (c) S. R. Reddy, S. Stella and A. Chadha, Synth. Commun., 2012, 42, 3493 CrossRef CAS.
- For other representative methods, see: (a) N. Thasana, V. Prachyawarakorn, S. Tontoolarug and S. Ruchirawat, Tetrahedron Lett., 2003, 44, 1019 CrossRef CAS; (b) Y. Su, L. Zhang and N. Jiao, Org. Lett., 2011, 13, 2168 CrossRef CAS PubMed; (c) A. Nagaki, D. Ichinari and J.-i. Yoshida, Chem. Commun., 2013, 49, 3242 RSC; (d) Z. Zhang, J. Su, Z. Zha and Z. Wang, Chem. – Eur. J., 2013, 19, 17711 CrossRef CAS PubMed; (e) S. H. Kim, K. H. Kim and J. N. Kim, Adv. Synth. Catal., 2011, 353, 3335 CrossRef CAS ; also see ref. 2c.
- For reviews on copper-catalyzed aerobic oxidative and oxygenation, see: (a) T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329 CrossRef CAS PubMed; (b) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed; (c) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC; (d) S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234 CrossRef CAS PubMed.
- For recent examples of copper catalyzed aerobic oxidation reactions, see: (a) S. Guo, B. Qian, Y. Xie, C. Xia and H. Huang, Org. Lett., 2010, 13, 522 CrossRef PubMed; (b) X. Li, L. Huang, H. Chen, W. Wu, H. Huang and H. Jiang, Chem. Sci., 2012, 3, 3463 RSC; (c) Y. Yang, W. Dong, Y. Guo and R. M. Rioux, Green Chem., 2013, 15, 3170 RSC; (d) J. Yu, Y. Jin, H. Zhang, X. Yang and H. Fu, Chem. – Eur. J., 2013, 19, 16804 CrossRef CAS PubMed; (e) L. Gu, J. Liu and H. Zhang, Chin. J. Chem., 2014, 32, 1267 CrossRef CAS; (f) C. Tang and N. Jiao, Angew. Chem., Int. Ed., 2014, 53, 6528 CrossRef CAS PubMed; (g) X. Li, X. Liu, H. Chen, W. Wu, C. Qi and H. Jiang, Angew. Chem., Int. Ed., 2014, 53, 14485 CrossRef CAS PubMed; (h) Y. Yan, Y. Yuan and N. Jiao, Org. Chem. Front., 2014, 1, 1176 RSC; (i) Z. Zhang and X. Jiang, Org. Lett., 2014, 16, 4400 CrossRef CAS PubMed.
- For recent examples of copper catalyzed oxygenation reactions with molecular oxygen, see: (a) C. Zhang and N. Jiao, J. Am. Chem. Soc., 2009, 132, 28 CrossRef PubMed; (b) S. Chiba, L. Zhang and J.-Y. Lee, J. Am. Chem. Soc., 2010, 132, 7266 CrossRef CAS PubMed; (c) H. Wang, Y. Wang, D. Liang, L. Liu, J. Zhang and Q. Zhu, Angew. Chem., Int. Ed., 2011, 50, 5678 CrossRef CAS PubMed; (d) Q. Liu, P. Wu, Y. Yang, Z. Zeng, J. Liu, H. Yi and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 4666 CrossRef CAS PubMed; (e) Y. Su, X. Sun, G. Wu and N. Jiao, Angew. Chem., Int. Ed., 2013, 52, 9808 CrossRef CAS PubMed; (f) C. J. Allpress, A. Miłaczewska, T. Borowski, J. R. Bennett, D. L. Tierney, A. M. Arif and L. M. Berreau, J. Am. Chem. Soc., 2014, 136, 7821 CrossRef CAS PubMed.
- C. Zhang, P. Feng and N. Jiao, J. Am. Chem. Soc., 2013, 135, 15257 CrossRef CAS PubMed.
- C. Zhang and N. Jiao, Org. Chem. Front., 2014, 1, 109 RSC.
- X. Huang, X. Li, M. Zou, S. Song, C. Tang, Y. Yuan and N. Jiao, J. Am. Chem. Soc., 2014, 136, 14858 CrossRef CAS PubMed.
- X. Xu, W. Ding, Y. Lin and Q. Song, Org. Lett., 2015, 17, 516 CrossRef CAS PubMed.
- (a) E. Lee, A. S. Kamlet, D. C. Powers, C. N. Neumann, G. B. Boursalian, T. Furuya, D. C. Choi, J. M. Hooker and T. Ritter, Science, 2011, 334, 639 CrossRef CAS PubMed; (b) D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 5767 CrossRef CAS PubMed; (c) T. Kang, Y. Kim, D. Lee, Z. Wang and S. Chang, J. Am. Chem. Soc., 2014, 136, 4141 CrossRef CAS PubMed; (d) S. H. Oh, Y. R. Malpani, N. Ha, Y.-S. Jung and S. B. Han, Org. Lett., 2014, 16, 1310 CrossRef CAS PubMed.
- (a) H. Zhao, J. Joseph, H. Zhang, H. Karoui and B. Kalyanaraman, Free Radical Biol. Med., 2001, 31, 599 CrossRef CAS; (b) C. Groussard, F. Rannou-Bekono, G. Machefer, M. Chevanne, S. Vincent, O. Sergent, J. Cillard and A. Gratas-Delamarche, Eur. J. Appl. Physiol., 2003, 89, 14 CrossRef CAS PubMed; (c) Y. Yan, P. Feng, Q.-Z. Zheng, Y.-F. Liang, J.-F. Lu, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2013, 52, 5827 CrossRef CAS PubMed ; also see ref. 10.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS PubMed.
- For reviews on enamine catalysis, see: (a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef CAS PubMed; For selected recent amine catalyzed reactions, see: (b) M. Kreis and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 3436 CrossRef PubMed; (c) J. A. Terrett, M. D. Clift and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 6858 CrossRef CAS PubMed; (d) K. S. Halskov, F. Kniep, V. H. Lauridsen, E. H. Iversen, B. S. Donslund and K. A. Jørgensen, J. Am. Chem. Soc., 2015, 137, 1685 CrossRef CAS PubMed; (e) W. Li, Z. Du, K. Zhang and J. Wang, Green Chem., 2015, 17, 781 RSC; (f) D. Wang, C. Xu, L. Zhang and S. Luo, Org. Lett., 2015, 17, 576 CrossRef CAS PubMed.
- For SOMO catalysis, see: (a) T. D. Beeson, A. Mastracchio, J.-B. Hong, K. Ashton and D. W. C. MacMillan, Science, 2007, 316, 582 CrossRef CAS PubMed; (b) S. Mukherjee and B. List, Nature, 2007, 447, 152 CrossRef CAS PubMed.
- For generation of O2˙− under aerobic conditions with CuI and amine, see: (a) H. Speisky, M. Gómez, C. Carrasco-Pozo, E. Pastene, C. Lopez-Alarcón and C. Olea-Azar, Bioorg. Med. Chem., 2008, 16, 6568 CrossRef CAS PubMed; (b) F.-T. Du and J.-X. Ji, Chem. Sci., 2012, 3, 460 RSC.
- For reviews on copper catalyzed SET processes see: (a) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC; For selected recent examples, see: (b) Q. Liu, P. Wu, Y. Yang, Z. Zeng, J. Liu, H. Yi and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 4666 CrossRef CAS PubMed; (c) C. Uyeda, Y. Tan, G. C. Fu and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 9548 CrossRef CAS PubMed.
- For generation of α-carbonyl aldehydes from methyl ketones, see: (a) Y.-P. Zhu, M. Lian, F.-C. Jia, M.-C. Liu, J.-J. Yuan, Q.-H. Gao and A.-X. Wu, Chem. Commun., 2012, 48, 9086 RSC; (b) L. Zhang, X. Bi, X. Guan, X. Li, Q. Liu, B. D. Barry and P. Liao, Angew. Chem., Int. Ed., 2013, 52, 11303 CrossRef CAS PubMed; (c) N. Mupparapu, S. Khan, S. Battula, M. Kushwaha, A. P. Gupta, Q. N. Ahmed and R. A. Vishwakarma, Org. Lett., 2014, 16, 1152 CrossRef CAS PubMed; (d) Q. Feng and Q. Song, Adv. Synth. Catal., 2014, 356, 2445 CrossRef CAS ; also see ref. 19b.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00028a |
| This journal is © the Partner Organisations 2015 |